

Abstract
The big-head schizothorcin (Aspiorhynchus laticeps) is an endemic and near-extinction freshwater fish in Xinjiang, China. In this study, a chromosome-scale genome assembly of A. laticeps was generated using PacBio and Hi-C techniques. The PacBio sequencing data resulted in a 1.58 Gb assembly with a contig N50 of 1.27 Mb. Using Hi-C scaffolding approach, 88.38% of the initial assembled sequences were anchored and oriented into a chromosomal-scale assembly. The final assembly consisted of 25 pseudo-chromosomes that yielded 1.37 Gb of sequence, with a scaffold N50 of 44.02 Mb. BUSCO analysis showed a completeness score of 93.7%. The genome contained 48,537 predicted protein-coding genes and 58.31% of the assembly was annotated as repetitive sequences. Whole genome duplication events were further confirmed using 4dTv analysis. The genome assembly of A. laticeps should be valuable and important to understand the genetic adaptation and endangerment process of this species, which could lead to more effective management and conservation of the big-head schizothorcin and related freshwater fish species.
Subject terms: Ichthyology, Taxonomy
| Measurement(s) | Genome Assembly Sequence |
| Technology Type(s) | Next Generation Sequencing |
| Sample Characteristic - Organism | Aspiorhynchus laticeps |
| Sample Characteristic - Location | Xinjiang Autonomous Region |
Background & Summary
Freshwater fish can not only provide sufficient food resources for people living in inland region, but also play crucial roles in maintaining ecological balance. However, limited knowledge of freshwater biodiversity has largely constrained effective conservation efforts and public concern1, especially for places considered as biodiversity hotspots2. Family Cyprinidae (minnows and carps) is a group of common freshwater fish family with abundant and diversified species (more than 1600 species), which dominated flowing waters and lakes worldwide3. Cyprinids are known to the public due to some small fish species or farmed carps, yet whether some large and elusive species are under proper management and conservation attentions is uncertain1.
The big-head schizothorcin (Aspiorhynchus laticeps) (Fig. 1), also known as Xinjiang datou fish, is a large-size cyprinid (maximum total length 200 cm) endemic to Xinjiang Uygur Autonomous Region of China1,4. A. laticeps was one of the main targets in local fishery industry with high commercial value1,5. As one of the top predators in local environment, this species also has high ecological value for maintaining ecosystem stability6. Apart from high commercial and ecological value, with an evolutionary history of nearly 300 million years, A. laticeps is also a unique and ideal candidate for phylogeny and evolution studies of schizothorcins5. However, due to slow growth rate, long life span and low reproductive rate, as well as the adverse impacts of overexploitation and habitat degradation, the population resources of A. laticeps were drastically declined in the 1980s1,4. As a result, this species was listed as an endangered species in the China Red Book of endangered animals in 19987. In recent years, based on distribution survey information, regional fish biologists believed that this species might be near extinction4. Effective management and conservation strategies are urgently needed for prosperity of A. laticeps.
Fig. 1.

A picture of the big-head schizothorcin used in the genome sequencing and assembly (age: 4 Years, total length: 48.60 cm).
Previous studies generally focused on biology and physio-ecology of A. laticeps5,8–10, yet limited genetic resources and a lack of genomic information have largely constrained conservation genetics of this species. Therefore, genetic information such as the degree of genetic diversity, evolutionary history and genetic basis of endangerment process, which could provide valuable reference information for resource management and conservation of A. laticeps, are still unknown. The development of sequencing techniques and genome-scale analytical approaches have greatly broadened our understanding of genetic adaptation and endangerment process of threatened animals11 including panda12, Baiji dolphin13 and finless porpoise14, leading to more effective management and conservation of these species.
In this study, we assembled a chromosome-scale genome sequence of A. laticeps using Illumina short reads, PacBio long reads and Hi-C techniques (Table 1; Fig. 2). The initial genome assembly had a total length of 1,582.9 Mb with 4,133 contigs and a contig N50 of 1.27 Mb (Table 2). After Hi-C scaffolding approach, 88.38% of the initial assembled sequences were anchored to 25 pseudo-chromosomes (according to our results of karyotype analysis), and the total length of the final genome assembly was 1,366.83 Mb, with 3,067 scaffolds and a scaffold N50 of 44.02 Mb (Table 2). In our assembled sequence, a total of 923.02 Mb of repetitive sequences were annotated, representing 58.31% of the genome assembly (Table 2). The repetitive sequences (Table 3) were dominated by DNA transposons (283.13 Mb, 17.89%), long interspersed elements (LINEs, 152.00 Mb, 9.60%) and long terminal repeats (LTRs, 93.17 Mb, 5.89%). In addition, combining ab initio, homology-based and Iso-Seq assisted gene prediction approaches, a total of 48,537 protein-coding genes were predicted, among which 47,211 (97.27%) were annotated (Table 2). Such large number of protein-coding genes suggested potential whole genome duplication (WGD) event of A. laticeps. Subsequently, analysis of fourfold synonymous third-codon transversion (4dTv) confirmed two major WGD events of A. laticeps (Fig. 3). The assembled genome sequences provide useful and valuable information for elucidating the genetic adaptation and underlying molecular basis of endangerment process of A. laticeps, which can facilitate to establish more effective management and conservation strategies of this species. These genomic data can be also used in future comparative genomics and phylogenomics studies to investigate genomic evolution and phylogeny of schizothorcins.
Table 1.
Sequencing data for the A. laticeps genome assembly.
| Sequencing technology | Illumina | PacBio | Hi-C | Iso-Seq |
|---|---|---|---|---|
| Library size (bp) | 350 | 20,000 | 350 | 3000 |
| Raw data (Gb) | 173.68 | 327.44 | 155.7 | 90.67 |
| Clean data (Gb) | 169.69 | — | 143.01 | — |
| Coverage (X)† | 106.06 | 204.65 | 89.38 | — |
| Mean read length (bp) | 144 | 18,225.38 | 144 | 1955.65 |
†The coverage was calculated using an estimated genome size of 1.6 Gb.
Fig. 2.

The Hi-C contact map of the A. laticeps genome. chr 1–25 represented for the 25 pseudo-chromosomes. The color bar showed the contact density from white (low) to black (high).
Table 2.
Assembly and annotation statistics of the A. laticeps genome.
| Total length (Mb) | N_contig | Contig N50 | N_scaffold | Scaffold N50 | |
|---|---|---|---|---|---|
| PacBio sequencing | 1,584,292,485 | 4,133 | 1,266,850 | — | — |
| Hi-C sequencing | 1,366,832,638 | — | — | 3,067 | 44,016,701 |
| Genome annotation | |||||
| Protein-coding gene | 48,537 (47,211 annotated, 97.27%) | ||||
| Repetitive sequence | 58.31% | ||||
| GC content | 37.99% | ||||
Note: N_contig and N_scaffold denote number of contig and number of scaffold respectively.
Table 3.
Statistics of repetitive sequences in the A. laticeps genome.
| Repeat size (bp) | Percentage of genome (%) | |
|---|---|---|
| Identification method | ||
| RepeatMasker | 479,782,511 | 30.31 |
| ProteinMask | 227,512,931 | 14.37 |
| De novo | 804,212,276 | 50.81 |
| TRF | 97,644,119 | 6.17 |
| Total | 923,021,458 | 58.31 |
| Biological classification | ||
| DNA | 283,131,445 | 17.89 |
| LINE | 151,999,356 | 9.6 |
| SINE | 8,091,895 | 0.51 |
| LTR | 93,174,051 | 5.89 |
| Unknown | 516,278,379 | 32.62 |
| Other | 115,288,798 | 7.28 |
| Total | 903,156,224 | 57.06 |
Fig. 3.
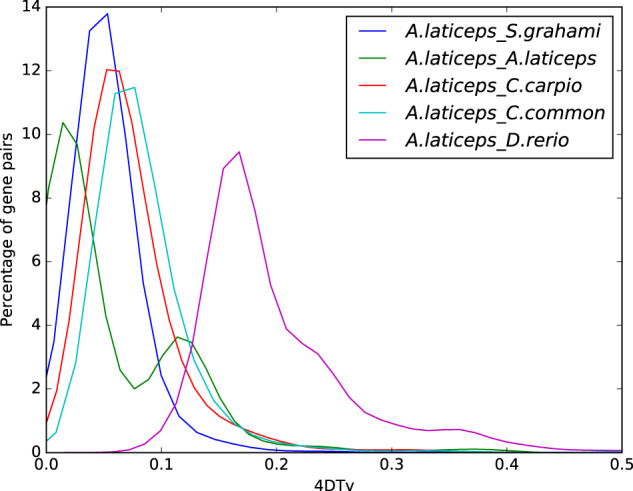
Analysis of fourfold synonymous third-codon transversion (4dTv) indicated whole genome duplication events.
Methods
Sample collection and sequencing
A 4 years old A. laticeps individual was sampled from Scientific Observing and Experimental Station of Fishery Resources and Environment in Northwest China in April 2020. All experimental methods were performed according to relevant guidelines and regulations established by the Institutional Animal Care and Use Committee of Xinjiang Fisheries Research Institute and Xinjiang Uygur Autonomous Region Aquatic Bureau. The muscle tissue below the dorsal fin was taken and stored in the liquid nitrogen until DNA extraction. Genomic DNA was isolated using the cetyltrimethylammonium bromide (CTAB) method. High-quality DNA was used for library preparation and high-throughput sequencing.
Illumina short-insert (350 bp) libraries were prepared according to the protocol and paired-end (PE150) sequenced on the Illumina Novaseq 6000 platform (Illumina, Inc., San Diego, CA, USA). Long-read sequencing was performed using the PacBio Sequel II sequencer (Pacific Biosciences, Menlo Park, CA, USA). For Hi-C sequencing, fresh muscle was fixed with formaldehyde in a concentration of 1% and the fixation was terminated using 0.2 M glycine. A Hi-C library was prepared following the Hi-C library protocol15 and then sequenced using an Illumina Novaseq 6000 sequencing platform. The heart, liver and muscle tissues were pooled for full-length Iso-Seq on the PacBio Sequel II sequencing platform.
Genome assembly
A total of 173.68 Gb Illumina short-read data were generated. After quality control by using HTQC v1.92.316, clean data were utilized for genome size estimation (Table 1). K-mer analysis was conducted using Jellyfish v2.2.1017. The k value was set to 21 and the genome size was estimated to be 1676.07 Mb, with a heterozygosity ratio of 0.78% and repeat sequence ratio of 76.67%. A total of 327.44 Gb PacBio long-read data (Table 1) were used for de novo genome assembly using Wtdbg218 and the draft contigs were corrected using Arrow v2.2.119 with the same PacBio dataset. The Illumina short reads (clean data 169.69 Gb, Table 1) from the same individual were further used to polish the initial genome assembly using Pilon v1.2320 (parameters:–frags;–fix snp,indels;–vcf). These sequencing data resulted in a 1,582.9 Mb assembly with 4,133 contigs and a contig N50 of 1.27 Mb (Table 2). The draft genome contigs were then anchored and oriented into a chromosomal-scale assembly using the Hi-C data. A total of 143.01 Gb clean data (Table 1) were aligned to the draft genome assembly using BWA v0.7.1021. Duplication removal, sorting, and quality control were performed using HiC-Pro v2.8.022. Only uniquely mapped valid read pairs were used for further analysis. LACHESIS23 was then used to cluster, order, and orient the contigs into chromosomal-scale assembly. Finally, 88.38% of the initial assembled sequences were anchored to 25 pseudo-chromosomes (Fig. 2) with lengths ranging from 34.35 to 100.46 Mb, and the total length of the genome assembly was 1,366.83 Mb, with 3,067 scaffolds and scaffold N50 of 44.02 Mb (Table 2).
Repetitive sequence annotation
A combined strategy based on homology alignment and de novo search was applied in our repeat annotation pipeline. A de novo repetitive elements database was built by LTR_FINDER24, RepeatScout25, RepeatModeler (www.repeatmasker.org/RepeatModeler.html) with default parameters. Tandem repeats were also ab initio extracted using TRF v4.0926. Then all repeat sequences with lengths >100 bp and gap ‘N’ less than 5% constituted the raw transposable element (TE) library. The homolog-based predictions were searched against Repbase27 database employing RepeatMasker v3.3.028 software and its in-house scripts RepeatProteinMask (v3.2.2) with default parameters. The combination of Repbase and our de novo TE library was processed by uclust29 to yield a non-redundant library and RepeatMasker was used to identify DNA-level repeat. The results of repetitive sequence annotation are listed in Table 3.
Protein-coding gene prediction and annotation
We employed ab initio, homology-based and Iso-Seq assisted prediction to detect the protein-coding genes. For homology-based prediction, protein sequences of Cyprinus carpio, Carassius auratus common and C. auratus red were downloaded from GenBank and Ensembl database30. The protein sequences were aligned against the genome assembly using TBLASTN v2.2.2631 (E-value ≤ 1e-5), and then matching proteins were aligned to the homologous genome sequences for accurate spliced alignments with GeneWise v2.4.132. The ab initio prediction was performed using Augustus v3.2.333, GeneID v1.434, GENESCAN v1.035, GlimmerHMM v3.0436, and SNAP v2013-11-2937 based on the repeat masked genome sequences. The Iso-Seq data were processed using SMRTlink v5.0 (PacBio, Menlo Park, CA) (parameters: min_length 200; max_drop_fraction 0.8; no_polish TRUE; min_zscore −9999; min_passes 1; min_predicted_accuracy 0.8; max_length 18000) to obtain full-length non-chimeric (FLNC) reads. The FLNC reads were then aligned to the genome using GMAP38 with parameters (–no-chimeras;–cross-species;–expand-offsets 1; -B 5; -K 50000; -f samse; -n 1), and then coding regions were predicted using PASA39 and GeMoMa v1.7.140. Finally, genes predicted by the above three methods were merged into a non-redundant reference gene set with EvidenceModeler v1.1.141 with identical weights, leading to a total of 48,537 protein-coding genes (Table 2).
Protein-coding genes were annotated by aligning the gene sequences to the SwissProt, NT, NR, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases using BLAST + v2.2.2842 with an e-value threshold of 1e-5. InterProScan v5.3143 was used to predict protein function based on conserved domains and motif by searching against ProDom, PRINTS, Pfam, SMRT, PANTHER and PROSITE. Ultimately, 47,211 (97.27%) predicted genes were successfully annotated (Table 2).
The predicted gene number was comparable with C. carpio and C. auratus, which was almost twice that detected in zebrafish Danio rerio44. Such large number of protein-coding genes suggested potential whole genome duplication (WGD) events of A. laticeps. To verify the WGD events, we performed analysis of fourfold synonymous third-codon transversion (4dTv). Syntenic blocks were identified using MCscan v0.845 and homologous protein sequences from these syntenic blocks were aligned using MUSCLE46, and the 4dTv values were calculated in PAML package47. The 4dTv results also confirmed two major WGD events of A. laticeps (Fig. 3).
Data Records
The sequencing dataset and genome assembly were deposited in public repositories. Illumina, PacBio, Hi-C and RNA-seq sequencing data used for Genome assembly have been deposited in the Genome Sequence Archive (GSA) at the National Genomics Data Center (NGDC)/China National Center for Bioinformation (CNCB) under accession number CRA00660448. The whole genome sequence data reported in this paper have been deposited in the National Center for Biotechnology Information (NCBI) GenBank database under the accession JALXFT000000000.149. Moreover, the genomic annotation results have been deposited at the Figshare database50.
Technical Validation
Evaluation of the quality of genomic DNA and RNA
In our DNA extraction section, the DNA quality and concentration were measured using agarose gel electrophoresis (1%), pulse field gel electrophoresis (1%) and Qubit 3.0 (Thermo Fisher Scientific, Inc., Carlsbad, CA, USA), respectively. For RNA, the integrity and quantity was evaluated using the Agilent 2100 Bioanalyzer (Agilent, USA). Subsequently, high-quality DNA and RNA were used for library preparation and high-throughput sequencing.
Evaluation of the completeness of genome assembly
The contamination evaluation of assembled genome sequence was performed against the NT database using BLAST+ v2.2.2841 with an e-value threshold of 1e-5. The results showed that no bacterial or artificial contaminants in our assembled genome. The completeness of the assembled genome sequence was evaluated using BUSCO v3.0.151. The BUSCO analysis against the vertebrata_odb10 database found that 95.7% of the conserved single copy orthologue genes, including 93.7% of the complete and 2.0% fragmented genes, were found in the genome assembly (Table 4). Also, the mapping rate of Illumina short reads from same individual were further used to evaluate the quality of the initial genome assembly using BWA v0.7.1021. By using a total of 169.69 Gb Illumina sequencing data from the same individual, the mapped read rate was 97.88% (Table 4), showing high genome assembly quality.
Table 4.
Genome quality assessment statistics of the A. laticeps genome.
| Genomic characteristic | Percentage |
|---|---|
| Illumina reads mapping rate | 97.88% |
| Illumina reads coverage | 98.73% |
| BUSCO evaluation | n = 3,354 |
| Complete BUSCOs | 3,144 (93.7%) |
| Complete and single-copy BUSCOs | 2,289 (68.2%) |
| Complete and duplicated BUSCOs | 855 (25.5%) |
| Fragmented BUSCOs | 68 (2.0%) |
| Missing BUSCOs | 142 (4.3%) |
Acknowledgements
This work was funded by Investigation on fishery resources and environment in key waters of Northwest China and Youth Innovation Promotion Association CAS (No.2020211).
Author contributions
J.N., R.M., H.Z. and W.X. conceived the study. J.H. and T.Z. collected the samples. H.L. and M.M. extracted the genomic DNA and conducted sequencing. J.N., R.M., H.Z. and W.X. performed bioinformatics analysis. J.N. and H.Z. wrote the manuscript. All authors read and approved the final manuscript. H.Z. is the lead contact for this paper.
Code availability
All software used in this study are in the public domain, with parameters being clearly described in Methods. If no detail parameters were mentioned for the software, default parameters were used as suggested by developer.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Jiangong Niu, Renming Zhang.
Contributor Information
Hui Zhang, Email: zhanghui@qdio.ac.cn.
Weiwei Xian, Email: wwxian@qdio.ac.cn.
References
- 1.Bain MB. The conservation status of large migratory cyprinids including Aspiorhynchus laticeps of Xinjiang China. J Appl Ichthyol. 2011;27:80–85. doi: 10.1111/j.1439-0426.2011.01857.x. [DOI] [Google Scholar]
- 2.Dudgeon D, et al. Freshwater biodiversity: importance, threats, status and conservation challenges. Biol Rev. 2006;81:163–182. doi: 10.1017/S1464793105006950. [DOI] [PubMed] [Google Scholar]
- 3.Froese, R., Pauly, D. FishBase. www.fishbase.org (accessed on 25 March 2022), (2022).
- 4.Bain MB, Zhang S. Threatened fishes of the world: Aspiorhynchus laticeps (Day, 1877) (Cyprinidae) Environ Biol Fish. 2001;61:380. doi: 10.1023/A:1011673801865. [DOI] [Google Scholar]
- 5.Han, J. J., et al. Observation on embryonic development, morphology and growth of larvae and juveniles of Aspiorhynchus laticeps. South China Fish Sci17, 59–66. (2021). (In Chinese with English abstract)
- 6.Guo, Y., et al. Ichthyology of Xinjiang. Xinjiang Science and Technology Press, Urumchi, China. Pp 122 (2012).
- 7.Yue, P., Chen, Y. China red book of endangered animals, Volume 2: Pisces. Science Press, Beijing, China. Pp 244 (1998).
- 8.Han J, Hu J, Shi C, Zhang R. Effects of 2-phenoxyethanol as anaesthetics on juvenile Aspiorhynchus laticeps under different conditions. J Shanghai Ocean Univ. 2019;28:211–218. [Google Scholar]
- 9.Xie C, Zhang R, Tur X, Guo Y, Ma Y. Acute toxicity test of seven kinds of chemicals to young fish of Aspiorhynchus laticeps. Arid Zone Res. 2010;27:104–108. doi: 10.3724/SP.J.1148.2010.00104. [DOI] [Google Scholar]
- 10.Zhang T, et al. Acute toxicity of alizarin red S to Aspiorhynchus laticeps. J. Fish Res. 2019;41:157. [Google Scholar]
- 11.Wei FW, Ma TX, Hu YB. Research advances and perspectives of conservation genetics of threatened mammals in China. Acta Theriol Sin. 2021;41:571–580. [Google Scholar]
- 12.Zhao S, et al. Whole-genome sequencing of giant pandas provides insights into demographic history and local adaptation. Nat Genet. 2013;45:67–71. doi: 10.1038/ng.2494. [DOI] [PubMed] [Google Scholar]
- 13.Zhou X, et al. Baiji genomes reveal low genetic variability and new insights into secondary aquatic adaptations. Nat Commun. 2013;4:2708. doi: 10.1038/ncomms3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou X, et al. Population genomics of finless porpoises reveal an incipient cetacean species adapted to freshwater. Nat Commun. 2018;9:1276. doi: 10.1038/s41467-018-03722-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rao SS, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang X, et al. HTQC: a fast quality control toolkit for Illumina sequencing data. BMC Bioinform. 2013;14:1–4. doi: 10.1186/1471-2105-14-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marçais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruan J, Li H. Fast and accurate long-read assembly with wtdbg2. Nat Methods. 2020;17:155–158. doi: 10.1038/s41592-019-0669-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chin CS, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10:563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 20.Walker BJ, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Servant N, et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. doi: 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burton JN, et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol. 2013;31:1119–1125. doi: 10.1038/nbt.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35:W265–268. doi: 10.1093/nar/gkm286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21:i351–358. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- 26.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bao W, Kojima KK, Kohany O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA. 2015;6:11. doi: 10.1186/s13100-015-0041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinform. 2004;5:4.10.1–4.10.14. doi: 10.1002/0471250953.bi0410s05. [DOI] [PubMed] [Google Scholar]
- 29.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 30.Cunningham F, et al. Ensembl 2019. Nucleic Acids Res. 2019;47:D745–D751. doi: 10.1093/nar/gky1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gertz EM, et al. Composition-based statistics and translated nucleotide searches: Improving the TBLASTN module of BLAST. BMC Biol. 2006;4:41. doi: 10.1186/1741-7007-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doerks T, Copley RR, Schultz J, Ponting CP, Bork P. Systematic identification of novel protein domain families associated with nuclear functions. Genome Res. 2002;12:47–56. doi: 10.1101/gr.203201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19:ii215–225. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- 34.Blanco E, Parra G, Guigó R. Using geneid to identify genes. Curr Protoc Bioinform. 2007;18:4.3.1–4.3.28. doi: 10.1002/0471250953.bi0403s18. [DOI] [PubMed] [Google Scholar]
- 35.Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- 36.Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open-source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20:2878–2879. doi: 10.1093/bioinformatics/bth315. [DOI] [PubMed] [Google Scholar]
- 37.Korf I. Gene finding in novel genomes. BMC Bioinform. 2004;5:59. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- 39.Haas BJ, et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003;31:5654–5666. doi: 10.1093/nar/gkg770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keilwagen J, et al. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinform. 2018;19:189. doi: 10.1186/s12859-018-2203-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haas BJ, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9:R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGinnis S, Madden TL. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004;32:W20–25. doi: 10.1093/nar/gkh435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mulder N, Apweiler R. InterPro and InterProScan: tools for protein sequence classification and comparison. Methods Mol Biol. 2007;396:59–70. doi: 10.1007/978-1-59745-515-2_5. [DOI] [PubMed] [Google Scholar]
- 44.Xu P, et al. Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat Genet. 2014;46:1212–1219. doi: 10.1038/ng.3098. [DOI] [PubMed] [Google Scholar]
- 45.Wang Y, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40:e49. doi: 10.1093/nar/gkr1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 48.2022. NGDC/CNCB Genome Sequence Archive. https://ngdc.cncb.ac.cn/gsa/browse/CRA006604
- 49.2022. GenBank. JALXFT000000000.1
- 50.Zhang H. 2022. Genome annotation data for the big-head schizothorcin (Aspiorhynchus laticeps) figshare. [DOI] [PMC free article] [PubMed]
- 51.Waterhouse RM, et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol. 2018;35:543–548. doi: 10.1093/molbev/msx319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- 2022. NGDC/CNCB Genome Sequence Archive. https://ngdc.cncb.ac.cn/gsa/browse/CRA006604
- 2022. GenBank. JALXFT000000000.1
- Zhang H. 2022. Genome annotation data for the big-head schizothorcin (Aspiorhynchus laticeps) figshare. [DOI] [PMC free article] [PubMed]
Data Availability Statement
All software used in this study are in the public domain, with parameters being clearly described in Methods. If no detail parameters were mentioned for the software, default parameters were used as suggested by developer.