

Abstract
Sporulation mutants of Streptomyces coelicolor appear white because they are defective in the synthesis of the gray polyketide spore pigment, and such white (whi) mutants have been used to define 13 sporulation loci. whiN, one of five new whi loci identified in a recent screen of NTG (N-methyl-N′-nitro-N-nitrosoguanidine)-induced whi strains (N. J. Ryding et al., J. Bacteriol. 181:5419–5425, 1999), was defined by two mutants, R112 and R650. R650 produced frequent spores that were longer than those of the wild type. In contrast, R112 produced long, straight, undifferentiated hyphae, although rare spore chains were observed, sometimes showing highly irregular septum placement. Subcloning and sequencing showed that whiN encodes a member of the extracytoplasmic function subfamily of RNA polymerase sigma factors and that the sigma factor has an unusual N-terminal extension of approximately 86 residues that is not present in other sigma factors. A constructed whiN null mutant failed to form aerial mycelium (the “bald” phenotype) and, as a consequence, whiN was renamed bldN. This observation was not totally unexpected because, on some media, the R112 point mutant produced substantially less aerial mycelium than its parent, M145. The bldN null mutant did not fit simply into the extracellular signaling cascade proposed for S. coelicolor bld mutants. Expression of bldN was analyzed during colony development in wild-type and aerial mycelium-deficient bld strains. bldN was transcribed from a single promoter, bldNp. bldN transcription was developmentally regulated, commencing approximately at the time of aerial mycelium formation, and depended on bldG and bldH, but not on bldA, bldB, bldC, bldF, bldK, or bldJ or on bldN itself. Transcription from the p1 promoter of the response-regulator gene bldM depended on bldN in vivo, and the bldMp1 promoter was shown to be a direct biochemical target for ςBldN holoenzyme in vitro.
Filamentous bacteria of the genus Streptomyces have a complex developmental cycle. At the start of differentiation, an aerial mycelium is formed, consisting of hyphae that grow out of the aqueous environment of the substrate mycelium into the air. These multigenomic aerial hyphae, which give the developing colonies a characteristic fuzzy appearance, subsequently differentiate to form chains of exospores (8, 20).
The isolation of bld mutants that lack aerial mycelium and, therefore, have a shiny, “bald” appearance has facilitated study of the development of aerial hyphae in Streptomyces coelicolor. Unlike a second class of developmental mutations (whi), which appear only to affect the differentiation of aerial hyphae into spores, bld mutations have pleiotropic effects which often cause defects in carbon catabolite repression and in cell-cell signaling and sometimes cause loss of antibiotic production, in addition to blocking differentiation (6, 20, 31, 38, 43, 53).
The behavior of bld mutants on different media suggests that aerial mycelium formation can occur by at least two different pathways. The aerial mycelium of S. coelicolor, when grown on rich media such as R2YE, is associated with the production of a small, hydrophobic peptide called SapB, a putative morphogen that coats the surface of aerial hyphae (52). SapB is a surfactant that allows aerial hyphae to break the surface tension of the aqueous environment of the substrate mycelium and grow into the air (51). On R2YE, SapB production and aerial mycelium formation depend on bldA, bldB, bldC, bldD, bldF, bldG, bldH, bldI, bldJ (formerly bld261 [38]), and bldK. However, aerial mycelium formation in these mutants can be restored by the exogenous addition of purified SapB protein or by growing them close to SapB-producing colonies (52). But, via a second pathway, aerial mycelium formation and sporulation can be restored to most bld mutants (an exception being bldB) simply by growing them on a minimal medium containing mannitol as the sole carbon source: conditions where no SapB is detectable (52).
A complex extracellular signaling cascade has been proposed to initiate the formation of aerial hyphae in S. coelicolor grown on rich media (36, 37, 38, 53). When some pairs of bld mutants are grown on R2YE in close proximity to each other, one mutant induces the other both to synthesize SapB and to erect aerial hyphae and sporulate. This “extracellular complementation” is always unidirectional, with one bld mutant acting as a “donor” and the other as a “recipient.” Experiments with the whole range of bld mutants showed that most could be arranged into the following hierarchy, in which each mutant can rescue the developmental defect in all the mutants to the left, but not to the right:
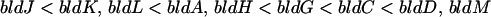 |
These data have led to a model in which aerial mycelium formation on rich media is initiated by a signaling cascade involving at least five different extracellular signals. Each signal causes the synthesis and/or release of the next signal, eventually causing the bldD-dependent production of SapB, and perhaps other morphogens, which allow aerial hyphae to overcome surface tension and grow into the air (36, 37, 38, 53). To date, there is biochemical evidence only for one of these putative extracellular signaling molecules: a covalently modified oligopeptide has been identified that rescues the mutant phenotype of the bldJ mutant in a bldK-dependent manner (36).
whiN is one of five new whi loci identified after NTG (N-methyl-N′-nitro-N-nitrosoguanidine) mutagenesis and was defined by two mutants, R112 and R650, which had strikingly different phenotypes (46). On minimal medium, colonies of R650 were medium gray and produced frequent spores that were longer than those of the wild type. In contrast, colonies of R112 were white and produced long, straight, undifferentiated hyphae, although rare spore chains were observed, sometimes showing highly irregular septum placement. The R112 mutation also showed clear signs of pleiotropic effects; in addition to the defects in sporulation, on some media R112 produced substantially less aerial mycelium than did the parental strain M145.
Here we show that WhiN is a member of the extracytoplasmic function (ECF) subfamily of RNA polymerase sigma factors and that it has an unusual N-terminal extension of approximately 86 residues that is not found in other sigma factors. We show that a constructed whiN null mutant is bald and, as a consequence, whiN was renamed bldN. bldN did not fit simply into the extracellular signaling cascade proposed for S. coelicolor (53). We show that bldN transcription is developmentally regulated, commencing approximately at the time of aerial mycelium formation, and that bldN transcription depends on bldG and bldH. Finally, we identify the p1 promoter of the response-regulator gene bldM as a direct biochemical target for ςBldN holoenzyme.
MATERIALS AND METHODS
Bacterial strains, plasmids, growth conditions, and conjugal plasmid transfer from Escherichia coli to Streptomyces spp.
The S. coelicolor strains used here are summarized in Table 1 and were cultured on R2YE, minimal medium (MM) containing 0.5% (wt/vol) mannitol as a carbon source, or MS (mannitol plus soya flour) agar (23). To bypass the methyl-specific restriction system of S. coelicolor, unmethylated plasmids were transferred by conjugation from the dam dcm hsdS E. coli strain ET12567 (30) as described by Ryding et al. (46). The plasmids used were pSET152 (3) and pIJ6650, a derivative of pKC1132 that has a 1.3-kb glkA fragment inserted at the BglII site (23).
TABLE 1.
Derivatives of S. coelicolor A3(2) used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| J1915 | ΔglkA119 SCP1− SCP2− | 21 |
| J2177 | ΔglkA119 bldN::hyg SCP1− SCP2− | This work |
| R112 | bldN112 SCP1− SCP2− | 46 |
| R650 | bldN650 SCP1− SCP2− | 46 |
| J1700 | bldA39 hisA1 uraA1 strA1 SCP1− SCP2− | 24 |
| J669 | bldB43 mthB2 cysD18 agaA7 SCP1NF SCP2* | 31 |
| J660 | bldC18 mthB2 cysD18 agaA7 SCP1NF SCP2* | 31 |
| J774 | bldD53 cysA15 pheA1 mthB2 strA1 SCP1NF SCP2* | 31 |
| 161 | bldF hisD3 pheA1 strA1 SCP1+ SCP2+ | 44 |
| J1501 | hisA1 uraA1 strA1 Pgl− SCP1− SCP2− | 9 |
| WC103 | bldG103 hisA1 uraA1 strA1 Pgl− SCP1− SCP2− | 6 |
| WC109 | bldH109 hisA1 uraA1 strA1 Pgl− SCP1− SCP2− | 6 |
| HU261 | bldJ261 hisA1 uraA1 strA1 Pgl− SCP1NF SCP2* | 53 |
| NS17 | bldK::aadA SCP1− SCP2− | 37 |
Sequencing.
The nucleotide sequence of the 2.07-kb BspEI-XbaI fragment carrying whiN/bldN has been deposited in the DDBJ, EMBL, and GenBank databases under accession number AJ010584. The mutant alleles that originally defined the whiN locus were amplified from the chromosomes of R650 and R112 by PCR and sequenced directly without cloning. The oligonucleotides used were 5′-AGCGCGGTCGTCCTCGTCCGG-3′ and 5′-GTCAACGGACTTTCACCAGTG-3′ for the coding region and 5′-CTGCACCGCCGCCGCCGTTGAC-3′ and 5′-CGGACGAGGACGACCGCGCTCG-3′ for the upstream sequence. PCR reaction conditions were 20 cycles of 96°C for 30 s, 62°C for 50 s, and 72°C for 50 s, followed by an extension reaction at 72°C for 10 min. The reaction was performed in 100 μl 1× PCR buffer (Boehringer Mannheim) containing 200 μM concentrations of each of the four deoxynucleoside triphosphates, 5% (vol/vol) dimethyl sulfoxide, 50 pmol of each primer, and 50 ng of chromosomal DNA.
Construction of a whiN null mutant.
A whiN null mutant derivative of J1915, a plasmid-free, ΔglkA derivative of the wild-type strain, was constructed using the method of Buttner et al. (5). This method makes use of the counterselectable glucose kinase gene (glkA), which allows a positive selection to be made for gene replacement, provided that the mutations are constructed in a strain carrying a deletion of glkA.
A 4.05-kb AatII fragment, isolated from pIJ6706, was cloned into pUC19 digested with AatII to give pIJ6718, and a 1.8-kb hyg cassette (56) was blunt-end cloned into pIJ6718 as a replacement for a 66-bp XhoI fragment internal to whiN, yielding pIJ6719. The constructed whiN::hyg null mutant allele was removed from pIJ6719 as a 6.1-kb SspI-NdeI fragment and blunt-end cloned into the counterselectable delivery vector pIJ6650 digested with EcoRV to give pIJ6720.
pIJ6720 was introduced into S. coelicolor J1915 (ΔglkA119) by mating from E. coli and exconjugants in which the plasmid had presumptively integrated at the whiN locus by single crossover homologous recombination were selected with apramycin. Because the two intervals flanking the hyg cassette in pIJ6720 were of unequal size (2.75 and 1.3 kb), 12 apramycin-resistant colonies were screened using Southern hybridization to look for isolates in which the first crossover had occurred in the smaller, 1.3-kb interval; one such strain was identified (J2176). After two rounds of nonselective growth of J2176, isolates from which the delivery plasmid had been lost were selected on MM containing 100 mM 2-deoxyglucose. These 2-deoxyglucose-resistant isolates were a mixture of bald and sporulating colonies. Southern hybridization showed that in the bald colonies the plasmid had excised leaving the whiN::hyg mutant allele in the chromosome, and that in the sporulating colonies the plasmid had excised leaving the wild-type whiN gene in the chromosome; a representative whiN null mutant was designated J2177. (Note that the level of hygromycin resistance conferred by whiN::hyg was too low to permit effective selection of the null mutant allele in these experiments.)
Isolation of RNA and S1 nuclease mapping.
For RNA preparation, S. coelicolor strains were grown on cellophane disks on R2YE medium, and RNA was extracted as described by Kelemen et al. (22). For each S1 nuclease reaction, 30 μg of RNA was hybridized to a 32P-end-labeled probe at 45°C for 3 to 15 h, following denaturation at 65°C for 15 min. S1 nuclease (Boehringer) digestions and analysis of RNA-protected fragments were performed as described by Kieser et al. (23). Uniquely end-labeled probes were generated by PCR as follows. For low-resolution S1 nuclease mapping of bldN, the oligonucleotide 5′-GACGAGGACGACCGCGCTCGTC-3′ was 5′ end labeled using [γ-32P]ATP and T4 polynucleotide kinase (Pharmacia) and used in a PCR reaction with unlabeled universal primer and pIJ6714 as the template, generating a 1,070-bp product. The 904-bp probe used for high-resolution S1 nuclease mapping of bldN was generated in the same way but using the radiolabeled oligonucleotide 5′-AGCCAGGCCCGAGGCGTCAAC-3′. The 1.168-kb bldM probe was generated using the radiolabeled oligonucleotide 5′-GGCTGGTACGAAATCGTCAC-3′, unlabeled universal primer, and pIJ6626 (34) as a template. The 343-bp hrdB probe was generated using the radiolabeled oligonucleotide 5′-GCCATGACAGAGACGGACTCGGCG-3′, the unlabeled oligonucleotide 5′-CGGCCGCAAGGTACGCGTTGATGA-3′, and pIJ2034 (5) as the template. Sequencing ladders for high-resolution mapping were generated with the Taq Track kit (Promega) and the same oligonucleotide that was used to generate the S1 nuclease mapping probe.
Overproduction and purification of ςBldN.
A 1-kb ApaI-BamHI fragment carrying most of the bldN gene but lacking the 5′ end was isolated from pIJ6715 and cloned into pRSET (Invitrogen) digested with NdeI and BamHI using two complementary adapter oligonucleotides 5′-TATGATGGAACTGGTTGAACGGGCC-3′ and 5′-CGTTCAACCAGTTCCATCA-3′. These oligonucleotides introduced an NdeI restriction site at the 5′ end of the bldN gene and replaced the third, fifth, and sixth codons with synonymous codons commonly associated with genes expressed at high levels in E. coli. The cassette replacement was verified by sequencing the resulting plasmid, pIJ6721. Because of low expression levels, pIJ6721 was subsequently modified using PCR-based site-directed mutagenesis to replace the seventh codon, CGG, which is also rare in E. coli, with CGT. In this method two abutting oligonucleotides were used to amplify the entire pIJ6721 plasmid, simultaneously introducing the single-base-pair change. The two oligonucleotides were 5′-GCCCAGGCCGGCGAGGCCGAC-3′ and 5′-ACGTTCAACCAGTTCCATCAT-3′, and the PCR program was 10 cycles of 1 min at 96°C, 45 s at 60°C, and 7 min at 72°C, followed by 10 cycles of 1 min at 96°C, 45 s at 60°C, and 10.5 min at 72°C, with a final incubation at 72°C for 15 min. The reaction was performed in 50 μl of 1× PCR buffer (Promega) containing 200 μM concentrations each of the four deoxynucleoside triphosphates, 10% (vol/vol) glycerol, 50 pmol of each primer, 10 ng of template DNA, and 2.5 U of high-fidelity Pfu DNA polymerase (Promega). The PCR product was self-ligated to create pIJ6722, and the resulting bldN allele was sequenced over its entire length to ensure that only the desired mutation had been introduced.
pIJ6722 was introduced into E. coli BL21λDE3(pLysS) (50), and bldN expression was induced in exponentially growing cells (optical density 0.5 at 600 nm) by the addition of 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG), followed by a further 3 h of growth.
ςBldN was recovered from inclusion bodies by a minor modification of the method of Nguyen et al. (35). The cell pellet was resuspended in 20 ml of lysis buffer (50 mM Tris-HCl, pH 8.0; 10 mM EDTA, 1 mM dithiothreitol [DTT], 50 mM NaCl, 0.2% [wt/vol] sodium deoxycholate [NaDOC], 5% [vol/vol] glycerol, 200 μg of lysozyme per ml]) and incubated on ice for 30 min before lysis was completed by three 30-s cycles of sonication. The cell lysate was centrifuged for 20 min at 15,000 rpm in an SS34 rotor, and the supernatant was discarded. The inclusion bodies were purified by resuspension in 20 ml of wash buffer, which was TGED (50 mM Tris-HCl, pH 8.0; 5% [vol/vol] glycerol; 0.1 mM EDTA, 0.1 mM DTT) containing 50 mM NaCl and 2% (wt/vol) NaDOC, followed by stirring at 4°C for 1 h and repeated sonication as before. The inclusion bodies were recovered by centrifugation, and the washing procedure was repeated once again. The purified inclusion bodies were solubilized by resuspension in 20 ml of solubilisation buffer (TGED containing 50 mM NaCl and 0.25 [wt/vol] Sarkosyl [N-lauroylsarcosine]) and stirred for 1 h at 4°C. The solubilized material was dialyzed for at least 24 h against 2 liters of TGED containing 50 mM NaCl, with several changes of buffer to attempt the complete removal of Sarkosyl, followed by centrifugation and filtration through a 0.2-μm (pore size) cellulose acetate filter (Sartorius GmbH). Finally, the purified protein was dialyzed against TGED containing 50 mM NaCl and 50% (vol/vol) glycerol before storage at −20°C.
In vitro transcription.
Runoff transcription assays were performed using [α-32P]CTP (New England Nuclear) at 600 Ci/mmol as described by Buttner et al. (4). Transcription from the bldMp1 promoter region was assayed using two PCR-derived templates of 1.194 kb (template 1) and 1.168 kb (template 2), generated using the template pIJ6626 (31), the universal primer, and the bldM-specific oligonucleotide primers 5′-CTCTTGCGCGTCACGTTGAGC-3′ (template 1) and 5′-GGCTGGTACGAAATCGTCAC-3′ (template 2). Transcripts were analyzed on 6% polyacrylamide–7 M urea gels using a heat-denatured, 32P-labeled HinFI digest of ΦX174 as the size standards. E. coli core RNA polymerase was purchased from Epicentre Technologies (Madison, Wis.).
Scanning electron microscopy.
Scanning electron microscopy was performed as described by Ryding et al. (46).
RESULTS
whiN encodes an extracytoplasmic function RNA polymerase sigma factor.
We previously isolated two SCP2*-based clones, pIJ6705 and pIJ6706, that complemented the two NTG-generated whiN mutants R112 and R650 (46). Further analysis showed that pIJ6705 and pIJ6706 carried 4- and 9-kb inserts, respectively, and that the two inserts overlapped. Subcloning from the 4-kb insert of pIJ6705 into the conjugative vector pSET152, which integrates site specifically into the S. coelicolor chromosome at the phage ΦC31 attB site, identified a 2.07-kb BspEI-XbaI fragment that restored wild-type levels of sporulation to both mutants (pIJ6713; Fig. 1). The nucleotide sequence of this 2.07-kb fragment was determined and one partial (orf1; Fig. 1) and one complete (orf2; Fig. 1) protein-coding sequence were identified with the aid of the FRAME program (2). The incomplete coding sequence, orf1, encoded the 266 C-terminal residues of a protein showing 25% identity to E. coli phosphoserine phosphatase. When the unique KpnI site lying in the noncoding region between orf1 and orf2 was used to generate two 1-kb subclones, pIJ6716 and pIJ6717 (Fig. 1), neither subclone complemented the whiN mutants (for an explanation of this result, see the section below on promoter mapping). However, a slightly larger FokI fragment (pIJ6715; Fig. 1) carrying all of orf2 and more sequence upstream of the KpnI site fully complemented both whiN mutants. As a consequence, orf2 was designated whiN.
FIG. 1.

Genetic organization of the 4.1-kb DNA fragment carrying whiN/bldN. The positions of the protein coding regions are indicated by arrows, and restriction sites referred to in the text are marked. The extent of the subclones described in the text and their ability to complement the whiN mutants R112 and R650 are shown below. XbaI sites are derived from the cloning vector, pIJ698. The DNA sequence of the lefthand 2-kb XbaI-BspEI fragment is taken from the ongoing S. coelicolor genome sequence (accession number AL079345).
Global similarity searches of the EMBL databases showed that WhiN is a member of the ECF subfamily of RNA polymerase sigma factors (27, 32), showing, for example, 27% identity to Bacillus subtilis ςX (14, 15), and 30% identity to Myxococcus xanthus ςCarQ (11, 29) (Fig. 2). The whiN start codon cannot be unambiguously assigned from FRAME analysis and sequence inspection. There are potential GTG start codons at positions 133 and 262 and two adjacent potential ATG start codons at positions 391 and 394 (Fig. 3B and 4A). Of these, only the upstream GTG codon at position 133 is preceded by an appropriately positioned potential ribosome binding site (GGAG). The discovery that the NTG-induced mutation in the more severe whiN mutant, R112, is a change in this putative ribosome binding site (Fig. 3B; see below) strongly implicates GTG-133 as an in vivo translation start codon. The sigma factor arising from translation initiation at GTG-133 would carry an unusual N-terminal extension of approximately 86 amino acids that is not present in other sigma factors (Fig. 2). The DNA encoding this extension has somewhat unusual codon usage, as can be seen from the whiN FRAME plot (Fig. 4A). Analysis of this extension using the TMPRED program (European Molecular Biology Network–Swiss node, http://www.ch.embnet.org/software/TMPRED_form.html) (Fig. 4B) revealed a stretch of 20 hydrophobic amino acids (YAVPALAAAAVPAGPCYALA) from positions 34 to 53 (Fig. 3B) that might cause WhiN to interact with the membrane. If translation initiation were also to occur from either of the adjacent ATG start codons at positions 391 and 394, it would give rise to a protein lacking the unusual extension that would be approximately co-N-terminal with most other members of the ECF subfamily of sigma factors (Fig. 2 and 3B).
FIG. 2.

Amino acid alignment of WhiN/BldN with other members of the ECF subfamily of sigma factors. The proteins and their corresponding amino acid sequence accession numbers are WhiN_Strco, S. coelicolor WhiN (CAB55345); SigX_Cloac, Clostridium acetobutylicum SigX (AAC12856); SigX_Bacsu, B. subtilis SigX (P35165); CarQ_Myxxa, M. xanthus CarQ (S39877) and SigL_Myctu, Mycobacterium tuberculosis SigL (CAA17502). The amino acid substitution carried by the whiN point mutant, R650, and the location of the hyg cassette (resulting from the replacement of a 66-bp XhoI fragment) in the whiN::hyg null mutant are shown.
FIG. 3.

(A) Scanning electron micrographs showing the phenotypes of the whiN/bldN point mutants, R112 and R650. (B) Nucleotide sequence of the promoter region and the 5′ end of the whiN/bldN gene showing the transcription start point, the putative ribosome binding site (RBS), the potential start codons (boxed), the nucleotide substitutions carried by the whiN point mutants, R112 and R650, and the KpnI site and the stretch of hydrophobic residues (underlined) discussed in the text.
FIG. 4.

(A) FRAME plot (2) of the whiN/bldN gene showing the unusual codon usage at the 5′ end. The GTG and ATG potential translation start codons are marked. The window size was 40 codons, and the step size was five codons. The sequence analyzed begins immediately after the stop codon of the upstream gene, and the numbering is that used in Fig. 3B. (B) TMPRED hydrophobicity plot of WhiN/BldN showing possible transmembrane helices. Predictions for both inside-to-outside helices (i → o) and outside-to-inside helices (o → i) are shown. Scores above 500 are considered significant.
The NTG-induced whiN mutations affect region 2.1 and the putative ribosome binding site.
The two whiN alleles that originally defined the locus were amplified from the chromosomes of R650 and R112 and sequenced. The “weak” mutant, R650 (Fig. 3A), carries a GGC to GAC change, giving rise to a glycine to aspartate (G103D) substitution in region 2.1 of WhiN (Fig. 2 and 3B). In other sigma factors, region 2.1 has been implicated in the interaction with core RNA polymerase. Deletion of region 2.1 in ς70 and ς32 of E. coli (25, 26) and point mutations in region 2.1 of E. coli ς70 (47) and B. subtilis ςE (47) all reduce binding of the sigma factor to core RNA polymerase. In addition, the RAP30 subunit of the human heteromeric general transcription initiation factor RAP30/74 contains a region that is similar in amino acid sequence to region 2.1 of bacterial sigma factors, and RAP30/74 binds E. coli core RNA polymerase and is displaced by ς70 (28).
The more “severe” mutant, R112 (Fig. 3A), has a wild-type coding sequence but carries a GGAG to GGAA change in the putative ribosome binding site upstream of GTG-133, reducing complementarity to the 3′ end of the 16S rRNA to 3 bp (Fig. 3B). Presumably, as a consequence, whiN mRNA is poorly translated in R112. Unlike the other whi mutants described by Chater (7) and Ryding et al. (46), R112 frequently throws off colonies that sporulate much more efficiently than R112 itself. Perhaps these isolates contain suppressor mutations that affect the ribosome and allow increased translation of the mutant whiN mRNA in the absence of the wild-type ribosome binding site.
A constructed whiN null mutant cannot erect aerial hyphae.
A whiN null mutant allele was constructed in vitro by replacing a 66-bp XhoI fragment internal to whiN with a hygromycin-resistance gene (hyg) (Fig. 1 and 2). This mutant allele was used to replace the wild-type allele in J1915 as described in Materials and Methods. The chromosomal structure of a representative whiN mutant was confirmed by Southern blot analysis (data not shown), and the strain was designated J2177.
J2177 was found to have a bald phenotype, being unable to erect aerial hyphae on MS agar or R2YE (Fig. 5). This observation was not totally unexpected. As previously noted (46), the more severe NTG-induced whiN mutant, R112, showed clear signs of pleiotropic effects; in addition to the defects in sporulation, on some media such as MM, R112 produced significantly less aerial mycelium than its parental strain, M145. In this respect, R112 did not fit the classical definition of a whi mutant, which should be solely defective in the differentiation of aerial hyphae into spores. Unlike some S. coelicolor bld mutants (31, 38), J2177 was not blocked in the production of actinorhodin or undecylprodigiosin, the two pigmented antibiotics made by this strain. For most of the bld mutants (an exception being bldB), mycelium formation and sporulation can be restored simply by growing them on MM containing mannitol as the sole carbon source. However, J2177 showed no signs of aerial mycelium formation on MM containing mannitol. As a consequence of the phenotype of the null mutant, whiN was renamed bldN. The bldN null mutant was fully complemented by pIJ6715, the pSET152 derivative carrying bldN (Fig. 5).
FIG. 5.

Scanning electron micrographs and photographs showing the bald phenotype of the constructed bldN null mutant, J2177 (A and B), and its complementation by pIJ6715, the pSET152 derivative carrying bldN (see Fig. 1) (C and D).
bldN does not fit simply into the extracellular signaling hierarchy proposed for S. coelicolor.
To see if bldN could be positioned in the extracellular complementation hierarchy proposed by Willey et al. (53), the bldN null mutant J2177 was grown on R2YE close to each bld mutant in turn. bldN restored a fringe of aerial mycelium formation to bldJ (formerly referred to as bld261 [38]), bldK and, to a very limited degree, bldH. It was not, however, able to restore aerial mycelium formation to any of the remaining bld mutants tested (bldA, bldC, bldD, and bldG), and none of the bld mutants restored aerial mycelium formation to bldN.
bldN is transcribed from a single promoter.
High-resolution S1 nuclease mapping of the bldN promoter region was performed using a PCR-generated probe and RNA isolated from J1915 grown on R2YE solid medium. A single promoter (bldNp) was identified, initiating transcription 83 to 84 bp upstream of the bldN GTG-133 start codon (Fig. 3B and 6A). The position of the promoter explains the results of the subcloning complementation experiments. Although the 1-kb KpnI-XbaI fragment contains the complete bldN coding sequence (pIJ6716; Fig. 1), it failed to complement R112 and R650 because the KpnI site used in the subcloning lies downstream of the bldN promoter (Fig. 3B), whereas the complementing FokI fragment encompasses both the bldN coding sequence and the promoter (pIJ6715; Fig. 1).
FIG. 6.

Transcriptional analysis of bldN. (A) High-resolution S1 nuclease mapping of the 5′ end of the bldN transcript in S. coelicolor J1915. The most likely transcription start points are indicated by the asterisks. Lanes C, T, A, and G represent a dideoxy sequencing ladder generated using the same oligonucleotide that was used to make the S1 mapping probe. (B) S1 nuclease protection analysis of bldN transcription during development of S. coelicolor J1915 on R2YE solid medium. For each time point, the presence of vegetative mycelium, aerial mycelium, and spores, as judged by microscopic examination, is shown. (C) S1 nuclease protection analysis of bldN and hrdB transcription during development of S. coelicolor WC103 (bldG) and WC109 (bldH) and their congenic parent J1501 (bldG+ bldH+) on R2YE solid medium. For each time point, the presence of vegetative mycelium, aerial mycelium, and spores, as judged by microscopic examination, is shown.
bldN transcription is temporally regulated.
bldN transcription was monitored by S1 nuclease protection analysis during development of S. coelicolor J1915 on R2YE solid medium. The bldN promoter was found to be temporally regulated; the bldN transcript was almost undetectable during vegetative growth, but its abundance increased dramatically during aerial mycelium formation and remained at a high level during sporulation (Fig. 6B). Because no attempt was made to fractionate the harvested cell material used for RNA preparation (thus, for example, the late sample contained vegetative and aerial mycelium as well as spores), no conclusions about the spatial location of bldN transcription within the colony could be drawn.
bldN transcription depends on bldG and bldH.
To see if bldN transcription depended on any of the other bld genes required for aerial mycelium formation, RNA was isolated from representative bldA, bldB, bldC, bldF, bldG, bldH, bldJ, and bldK mutants, as well as from the bldN null mutant itself. Each strain was grown on R2YE, a solid medium on which all of the bld mutants fail to produce aerial mycelium. These bld mutations exist in a complicated variety of genetic backgrounds (Table 1). Therefore, a preliminary experiment was conducted looking for any striking effects, keeping in mind the possible influence of the variable genetic background. bldN transcripts were readily detected in the bldA, bldB, bldC, bldF, bldK, and bldJ mutants, as well as in the bldN null mutant (data not shown), showing that aerial mycelium formation per se is not required for bldN transcription. Strikingly, however, bldN transcripts were undetectable in the bldG mutant, WC103, and the bldH mutant, WC109 (data not shown). Therefore, we repeated these experiments after the isolation of time courses of RNA samples from WC103, WC109, and their congenic parent, J1501 (bldG+ bldH+), again grown on R2YE solid medium. As in J1915 (Fig. 6B), bldN transcripts were almost undetectable in J1501 during vegetative growth but were readily detected during aerial mycelium formation and sporulation (Fig. 6C). However, bldN transcripts were again undetectable in the bldG and bldH mutants (Fig. 6C). In the absence of a bldN signal in the bldG and bldH mutants, as a positive internal control we examined the level of the transcript for hrdB, which encodes the principal, essential sigma factor of S. coelicolor (5). hrdB transcripts were readily detected at all time points in the bldG and bldH mutants and in J1501 (Fig. 6C). Therefore, bldN transcription depends, directly or indirectly, on bldG and bldH.
The bldMp1 promoter depends on bldN in vivo.
Promoters regulated by ECF sigma factors are similar in their −10 and −35 sequences (14, 16, 17, 27, 32, 40, 41). Visual inspection of the promoter regions of published bld gene sequences revealed a possible ECF consensus-like promoter (Fig. 7B) upstream of bldM. bldM encodes an apparently typical member of the FixJ subfamily of response regulators; however, aspartate-54, the putative site of phosphorylation, is not required for BldM function (33). The bldM null mutant fits into the extracellular signaling cascade proposed for S. coelicolor and is a member of the bldD extracellular complementation group (33). To determine if the sequence identified upstream of bldM functioned as a promoter, high-resolution S1 nuclease mapping was performed using a PCR-generated probe and RNA isolated from J1915. Two promoters were identified, initiating transcription 119 bp (bldMp1) and 167 to 168 bp (bldMp2) upstream of the bldM ATG start codon (Fig. 7A and B). The bldMp1 promoter corresponded to the putative ECF sigma factor consensus-like sequence (Fig. 7B).
FIG. 7.

Transcriptional analysis of bldM. (A) High-resolution S1 nuclease mapping of the 5′ ends of the bldMp1 and bldMp2 transcripts in S. coelicolor J1915. The most likely transcription start points are indicated by the asterisks. Lanes G, A, T, and C represent a dideoxy sequencing ladder generated using the same oligonucleotide that was used to make the S1 mapping probe. (B) Nucleotide sequence of the bldM promoter region showing the bldMp1 and bldMp2 transcription start points, the putative −10 and −35 sequences of the bldMp1 promoter, the putative ribosome binding site (RBS), and the start of the bldM coding sequence. (C) S1 nuclease protection analysis of bldM transcription during development of S. coelicolor J2177 (bldN) and its congenic parent J1915 (bldN+) on R2YE solid medium. For each time point, the presence of vegetative mycelium, aerial mycelium, and spores, as judged by microscopic examination, is shown. (D) S1 nuclease protection analysis of bldM and hrdB transcription during development of S. coelicolor WC103 (bldG) and WC109 (bldH) and their congenic parent J1501 (bldG+ bldH+) on R2YE solid medium. The RNA samples used were those described in Fig. 6C. For each time point, the presence of vegetative mycelium, aerial mycelium, and spores, as judged by microscopic examination, is shown.
To determine if bldMp1 depended on bldN, RNA was isolated from the constructed bldN null mutant, J2177, and its congenic parent, J1915 (bldN+). bldMp2 transcripts were detectable in J1915 and in the bldN null mutant (Fig. 7C). In contrast, bldMp1 transcripts were abundant in J1915 but were absent from the bldN null mutant, showing that bldMp1 transcription depended, directly or indirectly, on bldN (Fig. 7C).
Given that bldMp1 transcription depended on bldN and that bldN transcription in turn depended on bldG and bldH, it followed that bldMp1 transcription should be undetectable in bldG and bldH mutants. To determine if this were true, we examined the transcription of bldM in WC103 (bldG), WC109 (bldH), and J1501 (bldG+ bldH+), using the same time courses of RNA samples that were used to examine the transcription of bldN. Like bldN transcription (Fig. 6B and C), bldMp1 transcription was temporally regulated, being undetectable during vegetative growth but readily detectable during aerial mycelium formation and sporulation in J1501 (Fig. 7D). In contrast, bldMp1 transcripts were undetectable in the bldG and bldH mutants, confirming that bldMp1 transcription does indeed depend on bldG and bldH in vivo (Fig. 7D).
The bldMp1 promoter is a direct biochemical target for ςBldN.
To determine if the bldMp1 promoter is a direct biochemical target for ςBldN-containing RNA polymerase holoenzyme (EςBldN), we performed in vitro transcription assays using reconstituted EςBldN. A truncated form of ςBldN lacking the N-terminal extension (starting at Met-87) was overexpressed in E. coli and purified to near homogeneity, as described in Materials and Methods. In addition, two different bldMp1 promoter templates, designed to give runoff transcripts of 104 and 78 nucleotides, were amplified by PCR. ςBldN (1 pmol) was added to core RNA polymerase (1 pmol) to reconstitute EςBldN holoenzyme and then incubated separately in the presence of each bldMp1 promoter template. A runoff product of the predicted size was produced on each template (Fig. 8) in the presence of EςBldN but not in the presence of core enzyme alone, showing that bldMp1 is recognized by EςBldN in the absence of other transcriptional activators. The bldMp1 promoter is therefore a direct biochemical target for EςBldN holoenzyme.
FIG. 8.

In vitro transcription of the bldMp1 promoter by reconstituted EςBldN holoenzyme. Transcripts were generated from templates 1 and 2 (see Materials and Methods) using core RNA polymerase alone or core enzyme plus ςBldN. The expected sizes of the runoff transcripts from the bldMp1 promoter were 104 nucleotides (template 1) and 78 nucleotides (template 2). The size markers (M) were a 32P-end-labeled HinfI digest of ΦX174.
DISCUSSION
Different bldN alleles arrest S. coelicolor development at distinct stages.
In the first genetic screen for whi loci, conducted by Chater (7), most of the mutants assigned to a given locus were very similar in phenotype, as would be expected if most of the alleles were null, or close to null. In contrast, in the second screen there was wide phenotypic variation among the alleles of both whiK and whiN (46). Like whiN/bldN, disruption of whiK also gives rise to a bld phenotype (causing whiK to be renamed bldM [33]). Therefore, the phenotypic variation observed among the whiN and whiK mutants arises because the NTG-induced alleles retain various degrees of activity (the original screen specifically looked for white mutants and so would not have identified null mutants at either of these loci). In the case of whiN/bldN, R112 carries a mutation in the ribosome binding site and presumably produces reduced amounts of wild-type ςBldN, while in R650 the ςBldN produced carries an amino acid substitution in region 2.1 and is therefore likely to interact less efficiently with core RNA polymerase than the wild-type protein.
Are there further whi loci to be identified in S. coelicolor?
It is now clear that, in the cases of whiN/bldN and whiK/bldM at least, the whi mutant screen of Ryding et al. (46) identified special alleles of bld loci rather than null mutations at whi loci. However, there is reason to believe that there are further whi loci to identify in S. coelicolor. First, we have recently shown that whiL null mutants are white (M. J. Bibb and M. J. Buttner, unpublished data). Second, it is probable that a variety of potentially novel mutants were discarded by Ryding et al. (46) because they were partially suppressed by previously cloned whi genes. Of the 115 whi mutants chosen for study, 25 were excluded from further analysis because the introduction of either whiA, whiB, whiG, whiH, or whiJ (and sometimes more than one of these genes) increased gray pigmentation of the colonies, even though the morphological defects of the strains were not complemented (46). It seems very likely that at least some of these 25 strains will define novel whi loci. Until these experiments, suppression effects had not been reported in developmental work in Streptomyces spp., although the ability of an additional copy of whiG partially to suppress the spore pigment defect of whiH mutants without affecting their morphological phenotype has recently been noted (K. Flärdh, personal communication).
bldN and the S. coelicolor extracellular signaling hierarchy.
bldN does not fit simply into the extracellular signaling cascade proposed by Willey et al. (53):
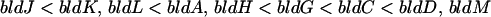 |
The bldN null mutant restored a fringe of aerial mycelium formation to bldJ (formerly referred to as bld261) and bldK, mutants that define the first two extracellular complementation groups in the cascade, and seemed to have some limited effect on bldH, which belongs to the third extracellular complementation group, but not on bldA, which also belongs to the third group. The bldN mutant was not, however, able to restore aerial mycelium formation to bld mutants that define the remaining three extracellular complementation groups in the cascade (bldG, bldC, and bldD), and none of the bld mutants restored aerial mycelium formation to bldN. Two other bld mutants have been described that do not fit cleanly into the hierarchy. bldI appears to be a member of the bldAH complementation group except that it is not complemented by bldC, and bldB appears to be in the same extracellular complementation group as bldC except that it fails to complement bldA and bldH (53). The extracellular signaling cascade was proposed in the light of experiments performed using bld mutants isolated by Merrick (31) and Champness (6). Recently, a large number of new bld mutants have been isolated, and the vast majority of these mutants fit into the cascade hierarchy (33, 38). Nevertheless, the behavior of bldB, bldI, and bldN emphasizes the need for further elaboration of the model.
Transcriptional dependence between bld genes.
Transcription of bldN depends, directly or indirectly, on bldG and bldH. bldH has not been characterized, but bldG (database accession number AF134889) encodes a homologue of SpoIIAA and RsbV, proteins that function as anti-anti-sigma factors in the regulation of ςF and ςB, respectively, in B. subtilis (1, 10, 49, 55). The genes encoding SpoIIAA, the anti-sigma factor SpoIIAB, and ςF lie in the same operon, as do the genes encoding RsbV, the anti-sigma factor RsbW, and ςB. In contrast, there is an anti-sigma factor gene adjacent to bldG, but no sigma factor gene. Since ςBldN does not direct transcription of its own gene, the effects of bldG on bldN transcription must be indirect, implying the involvement of a further sigma factor in the control of aerial mycelium formation in S. coelicolor.
In turn, transcription of the bldMp1 promoter depends on bldN, and this dependence arises because bldMp1 is a direct biochemical target for EςBldN holoenzyme. The second bldM promoter, bldMp2, however, does not depend on ςBldN.
What is the functional significance of the N-terminal extension of ςBldN?
Although the bldN start codon cannot be unambiguously assigned from FRAME analysis and sequence inspection, the discovery that the NTG-induced mutation in R112 is a change in the putative ribosome binding site upstream of GTG-133 strongly implies that ςBldN is synthesized with an N-terminal extension of approximately 86 residues that is absent from other sigma factors. During B. subtilis development, the mother cell-specific sigma factors ςE and ςK are synthesized as inactive pro-ς factors that are subsequently activated by proteolysis of the N-terminal 29 and 20 amino acids, respectively (10, 49), by membrane-localized proteases (42, 45). In both cases, the activation of this processing event is triggered by signals derived from the forespore, and this “cross talk” serves to coordinate the divergent programs of gene expression between the two cellular compartments within the sporangium (10, 49). By analogy, it is possible that ςBldN is synthesized as an inactive pro-ς factor, and it was for this reason that we overexpressed a truncated form of ςBldN that was approximately co-N-terminal with other ECF sigma factors for the in vitro transcription studies on the bldMp1 promoter. Given the stretch of 20 hydrophobic residues within the N-terminal extension of ςBldN, it is interesting to note that in B. subtilis the pro-sequences of both pro-ςE and pro-ςK promote membrane association (12, 18, 19, 57). Upon subcellular fractionation, the majority of pro-ςE and pro-ςK are present in the membrane fraction, whereas the majority of ςE and ςK are associated with core RNA polymerase in the cytoplasm (12, 57). In agreement with these observations, immunofluorescence microscopy showed that pro-ςE is associated with the cytoplasmic membrane in the predivisional sporangium and with the sporulation septum at the stage of assymmetric division (12, 18, 19). Similarly, immunofluorescence microscopy showed that pro-ςK is localized to the mother cell membranes that surround both the mother cell and the forespore (57).
The orthologue of ςBldN in Streptomyces griseus is also required for aerial mycelium formation and is under the control of the A-factor cascade.
In an accompanying study, Yamazaki et al. (54) show that the orthologue of ςBldN in S. griseus also plays an important role in differentiation. In S. griseus, the γ-butyrolactone signaling molecule A-factor (2-isocapryloyl-3R-hydroxymethyl-γ-butyrolactone) triggers a regulatory cascade required for both aerial mycelium formation and production of the antibiotic streptomycin (13). A-factor causes expression of a transcriptional activator called AdpA, which induces streptomycin biosynthesis by activating transcription of strR, the gene encoding the pathway-specific activator of the streptomycin cluster (39). Until now, no targets for AdpA have been identified to explain the morphological defects of an adpA mutant. However, Yamazaki et al. (54) report the isolation of new AdpA binding sites from S. griseus chromosomal DNA, one of which is the promoter of an ECF sigma factor gene they have named adsA (AdpA-dependent sigma factor), the S. griseus orthologue of bldN. As is true for S. coelicolor bldN, transcription of S. griseus adsA begins approximately at the time of aerial mycelium formation, and disruption of adsA also results in loss of aerial mycelium formation. Neither S. coelicolor bldN nor S. griseus adsA is required for antibiotic production.
The predicted AdpA binding site is not clearly conserved in the promoter region of S. coelicolor bldN. However, there is a likely orthologue of adpA in the emerging S. coelicolor genome sequence (Streptomyces coelicolor Genome Project, http://www.sanger.ac.uk/Projects/S_coelicolor/ [Sanger Centre, Wellcome Trust Genome Campus, Hinxton, Cambridge, United Kingdom]). It will be interesting to see the extent to which the regulatory pathways governing aerial mycelium formation are conserved between these two distantly related Streptomyces species.
ACKNOWLEDGMENTS
We thank Mark Paget and Gabriella Kelemen for helpful discussion, Kim Findlay for taking the scanning electron micrographs of R112 and R650, and Mervyn Bibb, Keith Chater, and David Hopwood for their comments on the manuscript.
This work was supported by BBSRC grant 83/P07658 (to M. J. Buttner), by a John Innes Foundation studentship (to V. Molle), by a Lister Institute Research Fellowship (to M. J. Buttner), and by a grant-in-aid to the John Innes Centre from the BBSRC.
REFERENCES
- 1.Alper S, Dufour A, Garsin D A, Duncan L, Losick R. Role of adenosine nucleotides in the regulation of a stress-response transcription factor in Bacillus subtilis. J Mol Biol. 1996;260:165–177. doi: 10.1006/jmbi.1996.0390. [DOI] [PubMed] [Google Scholar]
- 2.Bibb M J, Findlay P R, Johnson M W. The relationship between base composition and codon usage in bacterial genes and its use for the simple and reliable identification of protein-coding sequences. Gene. 1984;30:157–166. doi: 10.1016/0378-1119(84)90116-1. [DOI] [PubMed] [Google Scholar]
- 3.Bierman M, Logan R, O'Brien K, Seno E T, Rao R N, Schoner B E. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene. 1992;116:43–49. doi: 10.1016/0378-1119(92)90627-2. [DOI] [PubMed] [Google Scholar]
- 4.Buttner M J, Fearnley I M, Bibb M J. The agarase gene (dagA) of Streptomyces coelicolor A3(2): nucleotide sequence and transcriptional analysis. Mol Gen Genet. 1987;209:101–109. doi: 10.1007/BF00329843. [DOI] [PubMed] [Google Scholar]
- 5.Buttner M J, Chater K F, Bibb M J. Cloning, disruption and transcriptional analysis of three RNA polymerase sigma factor genes of Streptomyces coelicolor A3(2) J Bacteriol. 1990;172:3367–3378. doi: 10.1128/jb.172.6.3367-3378.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Champness W C. New loci required for Streptomyces coelicolor morphological and physiological differentiation. J Bacteriol. 1988;170:1168–1174. doi: 10.1128/jb.170.3.1168-1174.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chater K F. A morphological and genetic mapping study of white colony mutants of Streptomyces coelicolor. J Gen Microbiol. 1972;72:9–28. doi: 10.1099/00221287-72-1-9. [DOI] [PubMed] [Google Scholar]
- 8.Chater K F. Taking a genetic scalpel to the Streptomyces colony. Microbiology. 1998;144:1465–1478. doi: 10.1099/00221287-144-6-1465. [DOI] [PubMed] [Google Scholar]
- 9.Chater K F, Bruton C J, King A A, Suarez J E. The expression of Streptomyces and Escherichia coli drug resistance determinants cloned into the Streptomyces phage ΦC31. Gene. 1982;19:21–32. doi: 10.1016/0378-1119(82)90185-8. [DOI] [PubMed] [Google Scholar]
- 10.Errington J. Determination of cell fate in Bacillus subtilis. Trends Genet. 1996;12:31–34. doi: 10.1016/0168-9525(96)81386-2. [DOI] [PubMed] [Google Scholar]
- 11.Gorham H C, McGowan S J, Robson P R H, Hodgson D A. Light-induced carotogenesis in Myxococcus xanthus: light-dependent membrane sequestration of ECF sigma factor CarQ by anti-sigma factor CarR. Mol Microbiol. 1996;19:171–186. doi: 10.1046/j.1365-2958.1996.360888.x. [DOI] [PubMed] [Google Scholar]
- 12.Hofmeister A. Activation of the proprotein transcription factor pro-ςE is associated with its progression through three patterns of subcellular localization during sporulation in Bacillus subtilis. J Bacteriol. 1998;180:2426–2433. doi: 10.1128/jb.180.9.2426-2433.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horinouchi S, Beppu T. A-factor as a microbial hormone that controls cellular differentiation and secondary metabolism in Streptomyces griseus. Mol Microbiol. 1994;12:859–864. doi: 10.1111/j.1365-2958.1994.tb01073.x. [DOI] [PubMed] [Google Scholar]
- 14.Huang X, Helmann J D. Identification of target promoters for the Bacillus subtilis ςX factor using a consensus-directed search. J Mol Biol. 1998;279:165–173. doi: 10.1006/jmbi.1998.1765. [DOI] [PubMed] [Google Scholar]
- 15.Huang X, Decatur A, Sorokin A, Helmann J D. The Bacillus subtilis ςX protein is an extracytoplasmic function ς factor contributing to survival at high temperature. J Bacteriol. 1997;179:2915–2921. doi: 10.1128/jb.179.9.2915-2921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang X, Fredrick K L, Helmann J D. Promoter recognition by Bacillus subtilis ςW: autoregulation and partial overlap with the ςX regulon. J Bacteriol. 1998;180:3765–3770. doi: 10.1128/jb.180.15.3765-3770.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang X, Gaballa A, Cao M, Helmann J D. Identification of target promoters for the Bacillus subtilis extracytoplasmic function ς factor ςW. Mol Microbiol. 1999;31:361–371. doi: 10.1046/j.1365-2958.1999.01180.x. [DOI] [PubMed] [Google Scholar]
- 18.Ju J, Haldenwang W G. The “pro” sequence of the sporulation-specific ς transcription factor ςE directs it to the mother cell side of the sporulation septum. J Bacteriol. 1999;181:6171–6175. doi: 10.1128/jb.181.19.6171-6175.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ju J, Luo T, Haldenwang W G. Bacillus subtilis pro-ςE fusion protein localises to the forespore septum and fails to be processed when synthesized in the forespore. J Bacteriol. 1997;179:4888–4893. doi: 10.1128/jb.179.15.4888-4893.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelemen G H, Buttner M J. Initiation of aerial mycelium formation in Streptomyces. Curr Opin Microbiol. 1998;1:656–662. doi: 10.1016/s1369-5274(98)80111-2. [DOI] [PubMed] [Google Scholar]
- 21.Kelemen G H, Plaskitt K A, Lewis C G, Findlay K, Buttner M J. Deletion of DNA lying close to the glkA locus induces ectopic sporulation in Streptomyces coelicolor A3(2) Mol Microbiol. 1995;17:221–230. doi: 10.1111/j.1365-2958.1995.mmi_17020221.x. [DOI] [PubMed] [Google Scholar]
- 22.Kelemen G H, Brown G L, Kormanec J, Potúčková L, Chater K F, Buttner M J. The positions of the sigma factor genes, whiG and sigF, in the hierarchy controlling the development of spore chains in the aerial hyphae of Streptomyces coelicolor A3(2) Mol Microbiol. 1996;21:593–603. doi: 10.1111/j.1365-2958.1996.tb02567.x. [DOI] [PubMed] [Google Scholar]
- 23.Kieser T, Bibb M J, Buttner M J, Chater K F, Hopwood D A. Practical Streptomyces genetics. Norwich, United Kingdom: The John Innes Foundation; 2000. [Google Scholar]
- 24.Lawlor E J, Baylis H A, Chater K F. Pleiotropic morphological and antibiotic deficiencies result from mutations in a gene encoding a tRNA-like product in Streptomyces coelicolor A3(2) Genes Dev. 1987;1:1305–1310. doi: 10.1101/gad.1.10.1305. [DOI] [PubMed] [Google Scholar]
- 25.Lesley S A, Burgess R R. Characterization of the Escherichia coli transcription factor ς70: localization of a region involved in the interaction with core RNA polymerase. Biochemistry. 1989;28:7728–7734. doi: 10.1021/bi00445a031. [DOI] [PubMed] [Google Scholar]
- 26.Lesley S A, Brow M A, Burgess R R. Use of in vitro protein synthesis from polymerase chain reaction-generated templates to study interaction of Escherichia coli transcription factors with core RNA polymerase and for epitope mapping of monoclonal antibodies. J Biol Chem. 1991;266:2632–2638. [PubMed] [Google Scholar]
- 27.Lonetto M A, Brown K L, Rudd K E, Buttner M J. Analysis of the Streptomyces coelicolor sigE gene reveals the existence of a subfamily of eubacterial RNA polymerase ς factors involved in the regulation of extracytoplasmic functions. Proc Natl Acad Sci USA. 1994;91:7573–7577. doi: 10.1073/pnas.91.16.7573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCracken S, Greenblatt J. Related RNA polymerase-binding regions in human RAP30/74 and Escherichia coli ς70. Science. 1991;253:900–902. doi: 10.1126/science.1652156. [DOI] [PubMed] [Google Scholar]
- 29.McGowan S J, Gorham H C, Hodgson D A. Light-induced carotogenesis in Myxococcus xanthus: DNA sequence of the carR region. Mol Microbiol. 1993;10:713–735. doi: 10.1111/j.1365-2958.1993.tb00943.x. [DOI] [PubMed] [Google Scholar]
- 30.MacNeil D J, Occi J L, Gewain K M, MacNeil T, Gibbons P H, Ruby C L, Danis S L. Complex organization of the Streptomyces avermitilis genes encoding the avermectin polyketide synthase. Gene. 1992;115:119–125. doi: 10.1016/0378-1119(92)90549-5. [DOI] [PubMed] [Google Scholar]
- 31.Merrick M J. A morphological and genetic mapping study of bald mutants of Streptomyces coelicolor. J Gen Microbiol. 1976;96:299–315. doi: 10.1099/00221287-96-2-299. [DOI] [PubMed] [Google Scholar]
- 32.Missiakas D, Raina S. The extracytoplasmic function sigma factors: role and regulation. Mol Microbiol. 1998;28:1059–1066. doi: 10.1046/j.1365-2958.1998.00865.x. [DOI] [PubMed] [Google Scholar]
- 33.Molle V, Buttner M J. Different alleles of the response regulator gene bldM arrest Streptomyces coelicolor development at distinct stages. Mol Microbiol. 2000;36:1265–1278. doi: 10.1046/j.1365-2958.2000.01977.x. [DOI] [PubMed] [Google Scholar]
- 34.Molle V, Palframan W J, Findlay K C, Buttner M J. WhiD and WhiB, homologous proteins required for different stages of sporulation in Streptomyces coelicolor A3(2) J Bacteriol. 2000;182:1286–1295. doi: 10.1128/jb.182.5.1286-1295.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen L H, Jensen D B, Burgess R R. Overexpression and purification of ς32, the Escherichia coli heat shock transcription factor. Protein Expr Purif. 1993;4:425–433. doi: 10.1006/prep.1993.1056. [DOI] [PubMed] [Google Scholar]
- 36.Nodwell J R, Losick R. Purification of an extracellular signalling molecule involved in production of aerial mycelium by Streptomyces coelicolor. J Bacteriol. 1998;180:1334–1337. doi: 10.1128/jb.180.5.1334-1337.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nodwell J R, McGovern K, Losick R. An oligopeptide permease responsible for the import of an extracellular signal governing aerial mycelium formation in Streptomyces coelicolor. Mol Microbiol. 1996;22:881–893. doi: 10.1046/j.1365-2958.1996.01540.x. [DOI] [PubMed] [Google Scholar]
- 38.Nodwell J R, Yang M, Kuo D, Losick R. Extracellular complementation and the identification of genes involved in aerial mycelium formation in Streptomyces coelicolor. Genetics. 1999;151:569–584. doi: 10.1093/genetics/151.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohnishi Y, Kameyama S, Onaka H, Horinouchi S. The A-factor regulatory cascade leading to streptomycin biosynthesis in Streptomyces griseus: identification of a target gene for the A-factor receptor. Mol Microbiol. 1999;34:102–111. doi: 10.1046/j.1365-2958.1999.01579.x. [DOI] [PubMed] [Google Scholar]
- 40.Paget M S B, Kang J-G, Roe J-H, Buttner M J. ςR, an RNA polymerase sigma factor that modulates expression of the thioredoxin system in response to oxidative stress in Streptomyces coelicolor A3(2) EMBO J. 1998;17:5776–5782. doi: 10.1093/emboj/17.19.5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paget M S B, Chamberlin L, Atrih A, Foster S J, Buttner M J. Evidence that the extracytoplasmic function sigma factor, ςE, is required for normal cell wall structure in Streptomyces coelicolor A3(2) J Bacteriol. 1999;181:204–211. doi: 10.1128/jb.181.1.204-211.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peters H K, Haldenwang W G. Synthesis and fractionation properties of SpoIIGA, a protein essential for pro-ςE processing in Bacillus subtilis. J Bacteriol. 1991;173:7821–7827. doi: 10.1128/jb.173.24.7821-7827.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pope M K, Green B D, Westpheling J. The bld mutants of Streptomyces coelicolor are defective in the regulation of carbon utilisation, morphogenesis and cell-cell signalling. Mol Microbiol. 1996;19:747–756. doi: 10.1046/j.1365-2958.1996.414933.x. [DOI] [PubMed] [Google Scholar]
- 44.Puglia A-M, Capelletti E. A bald, superfertile, UV-resistant strain in Streptomyces coelicolor A3(2) Microbiologica. 1984;7:263–266. [PubMed] [Google Scholar]
- 45.Rudner D Z, Fawcett P, Losick R. A family of membrane-embedded metalloproteases involved in regulated proteolysis of membrane-associated transcription factors. Proc Natl Acad Sci USA. 1999;96:14765–14770. doi: 10.1073/pnas.96.26.14765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ryding N J, Bibb M J, Molle V, Findlay K C, Chater K F, Buttner M J. New sporulation loci in Streptomyces coelicolor A3(2) J Bacteriol. 1999;181:5419–5425. doi: 10.1128/jb.181.17.5419-5425.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schuler M F, Tatti K M, Wade K H, Moran C P. A single amino acid substitution in ςE affects its ability to bind core RNA polymerase. J Bacteriol. 1995;177:3687–3694. doi: 10.1128/jb.177.13.3687-3694.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharp M M, Chan C L, Lu C Z, Marr M T, Nechaev S, Merritt E W, Severinov K, Roberts J W, Gross C A. The interface of ς with core RNA polymerase is extensive, conserved, and functionally specialised. Genes Dev. 1999;13:3015–3026. doi: 10.1101/gad.13.22.3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stragier P, Losick R. Molecular genetic analysis of sporulation in Bacillus subtilis. Annu Rev Genet. 1996;30:297–341. doi: 10.1146/annurev.genet.30.1.297. [DOI] [PubMed] [Google Scholar]
- 50.Studier F W, Moffatt B A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 51.Tillotson R D, Wösten H A B, Richter M, Willey J M. A surface active protein involved in aerial mycelium formation in the filamentous fungus Schizophillum commune restores the capacity of a bald mutant of the filamentous bacterium Streptomyces coelicolor to erect aerial structures. Mol Microbiol. 1998;30:595–602. doi: 10.1046/j.1365-2958.1998.01093.x. [DOI] [PubMed] [Google Scholar]
- 52.Willey J, Santamaria R, Guijarro J, Geistlich M, Losick R. Extracellular complementation of a developmental mutation implicates a small sporulation protein in aerial mycelium formation by S. coelicolor. Cell. 1991;65:641–650. doi: 10.1016/0092-8674(91)90096-h. [DOI] [PubMed] [Google Scholar]
- 53.Willey J, Schwedock J, Losick R. Multiple extracellular signals govern the production of a morphogenetic protein involved in aerial mycelium formation by Streptomyces coelicolor. Genes Dev. 1993;7:895–903. doi: 10.1101/gad.7.5.895. [DOI] [PubMed] [Google Scholar]
- 54.Yamazaki H, Ohnishi Y, Horinouchi S. An A-factor-dependent extracytoplasmic function sigma factor (ςAdsA) that is essential for morphological development in Streptomyces griseus. J Bacteriol. 2000;182:4596–4605. doi: 10.1128/jb.182.16.4596-4605.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang X, Kang C M, Brody M S, Price C W. Opposing pairs of serine protein kinases and phosphatases transmit signals of environmental stress to activate a bacterial transcription factor. Genes Dev. 1996;10:2265–2275. doi: 10.1101/gad.10.18.2265. [DOI] [PubMed] [Google Scholar]
- 56.Zalacaín M, González A, Guerrero M C, Mattaliano R J, Malpartida F, Jiménez A. Nucleotide sequence of the hygromycin B phosphotransferase gene from Streptomyces hygroscopicus. Nucleic Acids Res. 1986;14:1565–1581. doi: 10.1093/nar/14.4.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang B, Hofmeister A, Kroos L. The prosequence of pro-ςK promotes membrane association and inhibits RNA polymerase core binding. J Bacteriol. 1998;180:2434–2441. doi: 10.1128/jb.180.9.2434-2441.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]