

Abstract
MicroRNA-21 (miRNA-21) is highly expressed in various tumors. Small-molecule inhibition of miRNA-21 is considered to be an attractive novel cancer therapeutic strategy. In this study, fluoroquinolone derivatives A1–A43 were synthesized and used as miRNA-21 inhibitors. Compound A36 showed the most potent inhibitory activity and specificity for miRNA-21 in a dual-luciferase reporter assay in HeLa cells. Compound A36 significantly reduced the expression of mature miRNA-21 and increased the protein expression of miRNA-21 target genes, including programmed cell death protein 4 (PDCD4) and phosphatase and tensin homology deleted on chromosome ten (PTEN), at 10 μM in HeLa cells. The Cell Counting Kit-8 assay (CCK-8) was used to evaluate the antiproliferative activity of A36; the results showed that the IC50 value range of A36 against six tumor cell lines was between 1.76 and 13.0 μM. Meanwhile, A36 did not display cytotoxicity in BEAS-2B cells (lung epithelial cells from a healthy human donor). Furthermore, A36 significantly induced apoptosis, arrested cells at the G0/G1 phase, and inhibited cell-colony formation in HeLa cells. In addition, mRNA deep sequencing showed that treatment with A36 could generate 171 dysregulated mRNAs in HeLa cells, while the expression of miRNA-21 target gene dual-specificity phosphatase 5 (DUSP5) was significantly upregulated at both the mRNA and protein levels. Collectively, these findings demonstrated that A36 is a novel miRNA-21 inhibitor.
Keywords: Quinolone derivatives, Small-molecule miRNA-21 inhibitor, Antitumor agent, Drug design
Graphical abstract

Highlights
-
•
Fluoroquinolone derivatives A1–A43 were synthesized and evaluated as miRNA-21 small-compound inhibitors.
-
•
The quinolone derivative A36 was validated as an active and specific miRNA-21 small-compound inhibitor.
-
•
A36 displayed differential anti-cell proliferation activity between normal and miRNA-21-overexpressing cancer cells.
1. Introduction
MicroRNAs (miRNAs) are a type of endogenous non-coding RNAs that play crucial roles in post-transcriptional regulation [1,2]. Biogenesis of most miRNAs starts with a primary miRNA transcript (pri-miRNA) from the genome [3], and then the pri-miRNA is processed by the enzyme Drosha in the nucleus, which cleaves pri-miRNA into a stem-loop precursor miRNA (pre-miRNA) [4,5]. The pre-miRNA is exported into the cytoplasm, where it is processed by an additional endonuclease Dicer into an approximately 22-nucleotide mature miRNA [6,7]. Mature miRNA is then loaded into the effector complex called the RNA-induced silencing complex (RISC), where it plays its regulatory function by partially binding to the complementary sequences of the 3′ untranslated regions (3′ UTR) of the target mRNAs, leading to translational repression or degradation of target mRNA [8,9]. Aberrant expression of these regulatory miRNAs has been strongly implicated in a wide range of human diseases, most notably cancers [[10], [11], [12]]. Among multiple disease-associated miRNAs, microRNA-21 (miRNA-21) has been extensively studied as it is overexpressed in many human cancers including cervical, breast, colorectal, liver, ovarian, and lung cancers [13]. The overexpression of miRNA-21 results in the downregulation of some important tumor-suppressor proteins such as programmed cell death protein 4 (PDCD4) [14], dual-specificity phosphatase 5 (DUSP5) [15], and phosphatase and tensin homology deleted on chromosome ten (PTEN) [16]. Impairment of miRNA-21 biogenesis or function can inhibit tumor growth and rescue chemosensitivity of tumors to chemotherapeutic drugs [17,18]. Therefore, miRNA-21 is considered as a potential target for cancer therapy [19].
Because of the inherent stability, better bioavailability, and cost-efficiency of small-molecule drugs, an increasing number of miRNA small molecule modulators have been identified [20,21]. To date, only a handful of miRNA-21 small-molecule inhibitors have been reported (Fig. 1). Recently, the natural product bPGN (Fig. 1) has been found to be capable of inhibiting Dicer-mediated processing of pre-miRNA-21 via binding to pre-miRNA-21 [22]. It was reported that methacycline (Fig. 1) is an inhibitor of Dicer-mediated processing of miRNAs and the study revealed that metal chelation plays a principal role in its inhibitory function [23]. By screening various classes of clinically approved drugs using the AbsorbArray enabled 2-dimensional combinatorial screening approach [24], mitoxantrone (Fig. 1) was found to bind to the A bulge motif of pre-miRNA-21 and inhibit the maturation of miRNA-21. In addition, diazobenzene derivative 4 [25], streptomycin [26], AC1MMYR2 [27], benzamide derivative 7 [28], 3,6-dibromo carbazole derivative 8 [29], and oxadiazole derivative 9 [18] have also been discovered as inhibitors of miRNA-21 (Fig. 1). Although these small-molecule compounds can potentially be utilized to develop new strategies for tumor treatment, some showed poor activities, and their pharmacophore, structure-activity relationship, and targets remain unclear. Therefore, it is of great significance to design and synthesize miRNA-21 small-molecule inhibitors with novel structures and high activities.
Fig. 1.

Structures of previously reported small-molecule inhibitors of miRNA-21.
We previously identified compound A (Fig. 2) as a selective small-molecule inhibitor of miRNA-21 [30]. To discover structurally diverse miRNA-21 small-molecule inhibitors, we designed novel derivatives using A as a lead compound. Converting an arylamide into a fused ring system is a frequently used ring-closure strategy in drug discovery [31]. Here, we closed the arylamide group in A to form a quinoline scaffold. The skeletons of fluoroquinolone and quinoline are similar, and compared with quinoline scaffold, fluoroquinolone skeleton has more sites that can be directly modified. Thus, the quinoline scaffold may be transformed into a fluoroquinolone skeleton (Fig. 2). Fluoroquinolones (such as norfloxacin, ciprofloxacin, and enoxacin) are inexpensive and commercially available; therefore, we introduced different substituents directly to the carboxyl group and the piperazine ring of fluoroquinolones to synthesize a series of target compounds. Herein, we report the synthesis and biological evaluation of fluoroquinolone derivatives as miRNA-21 inhibitors.
Fig. 2.

The design strategy of target compounds.
2. Experimental
2.1. Chemistry
2.1.1. Reagents and chemicals
Norfloxacin, ciprofloxacin, N-methylaniline, 3-methylaniline, benzylamine, 4-aminotetrahydropyran hydrochloride, acryloyl chloride, pivalic acid, cyclopentane carboxylic acid, cyclohexane carboxylic acid, tetrahydropyran-4-formic acid, bromomethyl cyclopropane, bromomethyl cyclopentane, and trifluoroacetic acid (TFA) were obtained from Energic Chemical Co., Ltd. (Shanghai, China). Enoxacin (purity≥99%) was supplied by Shanghai Macklin Biochemical Co., Ltd. (Shanghai, China). Levofloxacin (purity≥99%) was purchased from Tianjin Heowns Biochemical Technology Co., Ltd. (Tianjin, China). Gatifloxacin (purity≥99%) was obtained from Dalian Meilune Biotechnology Co., Ltd. (Dalian, China). (Boc)2O, cyclopropane carboxylic acid, methylsulfonyl chloride, cyclopropyl sulfonyl chloride, acetyl chloride, and diisopropylethylamine (DIPEA) of analytical grade were supplied by Nanjing Duodian Chemical Co., Ltd. (Nanjing, China). Benzyl bromide, tetrahydrofuran (THF), methylamine hydrochloride, ethylamine hydrochloride, cyclohexylamine, piperidine, 1-ethylpiperazine, aniline, K2CO3, dimethyl formamide (DMF), SOCl2, and 3-picolinic acid of analytical grade were purchased from Sinopharm (Beijing, China). Analytical grade propylamine was obtained from Jiuding Chemical Co., Ltd. (Shanghai, China). Proparynamine and cyclopropylamine (purity≥99%) were purchased from Accela ChemBio Co., Ltd. (Shanghai, China). Benzoic acid (purity≥99%) was supplied by Guangzhou Chemical Reagent Factory (Guangzhou, China). 3-methoxy benzoic acid, 3-fluorobenzoic acid, p-fluorobenzoic acid, and 3-hydroxybenzoic acid (purity≥99%) were purchased from Bide Pharmatech Ltd. (Shanghai, China). Phenylacetyl chloride and Boc-l-alanine (purity≥99%) were obtained from Bepharm Ltd. (Shanghai, China). 4-(2-chloroethyl) morpholine hydrochloride (purity≥99%) was purchased from Alfa Aesar Chemical Co., Ltd. (Tianjin, China). 2-(7-Azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate (HATU; purity≥99%) was supplied by Chembee Chemical Co., Ltd. (Shanghai, China). Analytical grade dichloromethane (DCM) and EtOH were purchased from Tianjin Fuyu Chemicals Co., Ltd. (Tianjin, China).
2.1.2. Synthesis of target compounds
Forty-three target compounds were synthesized. All the compounds were characterized and validated by 1H nuclear magnetic resonance (NMR), 13C NMR, and mass spectrum (MS). The details are documented in Synthetic experiments part and Figs. S1–99 in the Supplementary data.
2.2. Cell culture
Seven selected human cell lines, namely, HeLa, HCT-116, A549, MCF-7, U87, HCC827, and BEAS-2B, were incubated in a humidified atmosphere containing 5% CO2 at 37 °C. A549 and HCC827 cell lines were maintained in RPMI-1640 medium (Hyclone, Hackensack, NJ, USA) supplemented with 10% fetal bovine serum (FBS; Hyclone, Hackensack, NJ, USA). HeLa, HCT-116, MCF-7, U87, and BEAS-2B cell lines were cultured in Dulbecco's modified eagle medium (Hyclone, Hackensack, NJ, USA) supplemented with 10% FBS.
2.3. Functional assays
The inhibitory activities of the target compounds against miRNA-21 were evaluated using a dual-luciferase reporter assay. The detailed method is described in our previous study [32]. Other molecular biology assays including Topo I and Topo IIa inhibitory activity assay, reserve transcription-quantitative polymerase chain reaction (RT-qPCR), Western blotting assay, Cell Counting Kit-8 (CCK-8) assay, cell apoptosis analysis, cell cycle assay, colony formation assay in HeLa cell line, deep sequencing assay, and statistics are described in Functional assays part, and the details are documented in Synthetic experiments part, and Figs. S100–102 in the Supplementary data.
3. Results and discussions
3.1. Chemistry
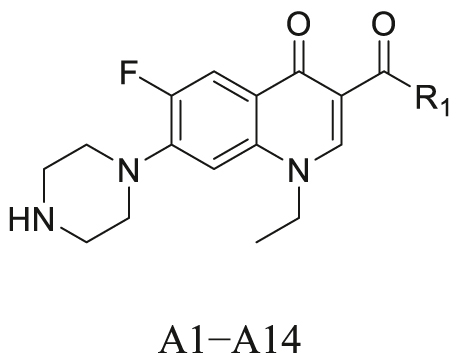
The synthetic route for A1–A14 is outlined in Scheme S1. Norfloxacin was used as the starting material. The 4-position of the piperazine ring of norfloxacin was protected with Boc to obtain intermediate 1a. Subsequently, the reaction of intermediate 1a with commercially available amines gave intermediates 2a–2m, which were deprotected by TFA to generate A1–A13. Norfloxacin and SOCl2 were refluxed in ethanol to obtain A14.
To explore the influence of the substituted group at the 4-position of the piperazine ring on the activity, A15–A36 were synthesized according to Scheme S2. Compounds A15–A32 were obtained from A6 via N-acylation or N-sulfonylation. Compound A6 was converted to A33–A36 by a nucleophilic substitution reaction with 4-(2-chloroethyl) morpholine, bromomethyl cyclopropane, bromomethyl cyclopentane, or benzyl bromide, respectively.
To further clarify the effect of enoxacin, ciprofloxacin, gatifloxacin, and levofloxacin derivatives on activity, A37–A43 were synthesized according to the synthetic procedures shown in Scheme S3. Intermediate 5 was obtained from enoxacin by Boc protection, acylation with cyclohexylamine, and Boc deprotection in three steps, followed by acylation of 5 with cyclopropane carboxylic acid to afford A37, which was alkylated with benzyl bromide to give A38. Compounds A39 and A40 were similarly synthesized from ciprofloxacin. Using the same synthesis method, A41 and A42 were prepared from gatifloxacin without Boc protection and deprotection, respectively. The reaction of levofloxacin with cyclohexylamine afforded A43.
3.2. The dual-luciferase reporter assay
The dual-luciferase reporter vector carrying the wild-type complementary sequence of miRNA-21 (named pmir-GLO miR-21 cs wt vector) (introduced in our previous publication [32]) was used to evaluate and screen any potential miRNA-21 inhibitors. The luminescence (Luc/Rlu) fold-changes of HeLa cells treated with the target compounds at 10 μM for 24 h were calculated and documented in detail. Compound A (Fig. 2), a selective small-molecule inhibitor of miRNA-21 we had identified previously [30], was used as a positive control. Norfloxacin, enoxacin, ciprofloxacin, gatifloxacin, and levofloxacin did not inhibit miRNA-21 (Table 1). Off note, enoxacin has been identified as a small-molecule enhancer of RNA interference (RNAi), and is capable of restoring normal levels of miRNAs by upregulating some miRNAs and simultaneously downregulating other miRNAs [[33], [34], [35], [36]]; however, its inhibitory effect on miRNA-21 was not obvious (Table 1). To investigate the effect of the quinolone 3-position substituents on the activity, we first introduced modifications to the carboxyl group of norfloxacin (Table 1). Cyclohexylamino (A6), tetrahydropyran-4-amino (A8), phenylamino (A10), 3-methylaniline (A12), and benzylamino (A13) substituents at the R1 position improved inhibitory activities compared to unmodified norfloxacin. Among them, A6 and A10 were more active than the positive control A, and A12 displayed similar activity as that of A. These results suggested that the cyclohexyl amino group at the R1 position was the most beneficial for the inhibitory activity against miRNA-21.
Table 1.
Structures and activities of selected floxacin compounds and A1-A14.
| Compounds | R1 | Luc/Rlu (fold change) a |
|---|---|---|
| 0.1% DMSO | – | 1.00 ± 0.03 |
| A | – | 1.51 ± 0.24 |
| norfloxacin | – | 1.10 ± 0.14 |
| enoxacin | – | 0.99 ± 0.05 |
| ciprofloxacin | – | 0.89 ± 0.13 |
| gatifloxacin | – | 1.07 ± 0.15 |
| levofloxacin | – | 0.94 ± 0.07 |
| A1 | 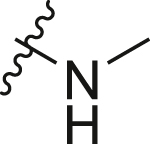 |
1.17 ± 0.11 |
| A2 | 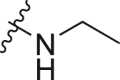 |
1.14 ± 0.10 |
| A3 | 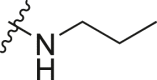 |
1.17 ± 0.03 |
| A4 | 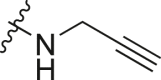 |
1.20 ± 0.17 |
| A5 | 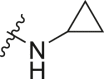 |
0.97 ± 0.01 |
| A6 | 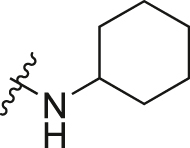 |
1.81 ± 0.13 |
| A7 |  |
0.96 ± 0.17 |
| A8 | 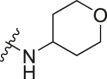 |
1.31 ± 0.26 |
| A9 |  |
0.95 ± 0.24 |
| A10 | 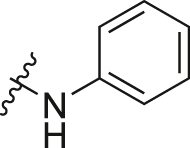 |
1.66 ± 0.29 |
| A11 | 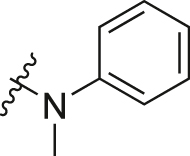 |
0.92 ± 0.03 |
| A12 | 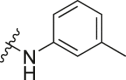 |
1.53 ± 0.31 |
| A13 | 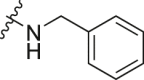 |
1.31 ± 0.02 |
| A14 | 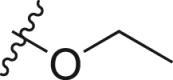 |
1.02 ± 0.09 |
Luc/Rlu values represent fold changes in firefly luciferase against the renilla luciferase control and relative to a 0.1% dimethyl sulfoxide (DMSO) control.
Data represent the mean ± SEM from at least three independent experiments.
Next, to explore the influence of the substituents at the 4-position of piperazine in the norfloxacin scaffold, A15–A36 were synthesized from A6. As shown in Table 2, the activities of most compounds were improved compared to that of A6, indicating that the introduction of a substituent at this position was beneficial for the inhibitory activities. First, when fatty acyl groups were linked, the activities of A15–A20 were slightly improved or retained compared to that of A6, especially A18 with a cycloproformyl exhibited potent activity. Second, the replacement of cyclohexanoyl (A20) with 4-tetrahydropyranoyl (A21) increased the activity. Benzoyl (A24) and 3-pyridine formyl (A25) at the R2 position resulted in a complete loss of activity. However, the introduction of electron-donating groups such as methoxy (A26) and hydroxyl (A29) on the benzene ring of A24 rescued the activity. Third, the compound with a cyclopropyl sulfonyl group (A32) displayed an improved activity compared to the compound with a methylsulfonyl group (A31). Finally, by comparing the activities of A36 and A24, A35, and A19, it was found that the activity was significantly improved when the carbonyl group was converted to an alkyl group. The most potent compound, A36, with a benzyl moiety, exhibited a 3.41-fold increase in luminescence signal, indicating that the benzyl group at the 4-position of piperazine in the norfloxacin scaffold was beneficial for activity.
Table 2.
Structures and activities of A15-A36.
| Compounds | R2 or R3 | Luc/Rlu (fold change) a |
|---|---|---|
| A15 | CH3 | 2.20 ± 0.16 |
| A16 |  |
2.84 ± 0.41 |
| A17 | 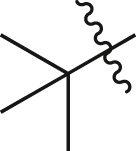 |
1.41 ± 0.07 |
| A18 | 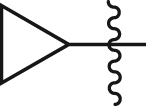 |
3.17 ± 0.46 |
| A19 | 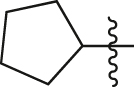 |
1.63 ± 0.06 |
| A20 | 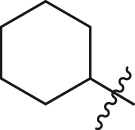 |
1.89 ± 0.14 |
| A21 | 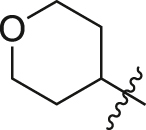 |
2.74 ± 0.45 |
| A22 |  |
2.25 ± 0.34 |
| A23 |  |
2.47 ± 0.06 |
| A24 | 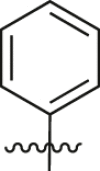 |
0.95 ± 0.25 |
| A25 | 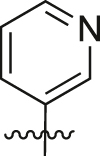 |
0.88 ± 0.27 |
| A26 | 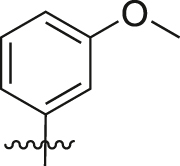 |
2.72 ± 0.31 |
| A27 | 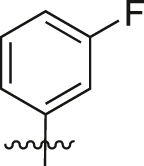 |
1.24 ± 0.29 |
| A28 | 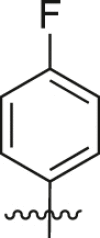 |
1.67 ± 0.10 |
| A29 | 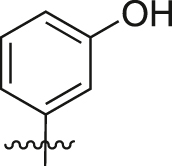 |
1.87 ± 0.30 |
| A30 | 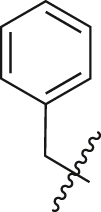 |
2.33 ± 0.51 |
| A31 | 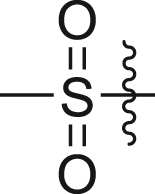 |
0.86 ± 0.11 |
| A32 | 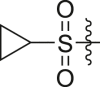 |
2.06 ± 0.24 |
| A33 | 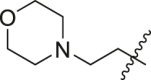 |
2.52 ± 0.46 |
| A34 | 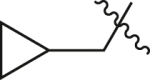 |
0.93 ± 0.13 |
| A35 | 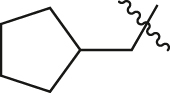 |
2.45 ± 0.17 |
| A36 | 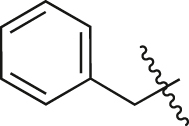 |
3.41 ± 0.25 |

Luc/Rlu values represent fold changes in firefly luciferase against the renilla luciferase control and relative to control. Data represent the mean ± SEM from at least three independent experiments.
To further study the structure-activity relationship of the compounds, we replaced the norfloxacin scaffold of A18 or A36 with enoxacin, ciprofloxacin, gatifloxacin, and levofloxacin to afford A37–A43. The activities of A37–A43 are summarized in Table 3. Compared to A18 and A36, these compounds showed a significant decline in activity, indicating that the norfloxacin scaffold was suitable for the activity.
Table 3.
Activities of A37-A43.
| Compounds | Luc/Rlu fold change a |
|---|---|
| A37 | 1.13 ± 0.20 |
| A38 | 1.81 ± 0.10 |
| A39 | 0.87 ± 0.17 |
| A40 | 1.80 ± 0.45 |
| A41 | 1.10 ± 0.21 |
| A42 | 1.50 ± 0.25 |
| A43 | 1.42 ± 0.15 |
Luc/Rlu values represent fold-changes in firefly luciferase against the renilla luciferase control and relative to control. Data represent the mean ± SEM from at least three independent experiments.
Among all the target compounds, A36 (Table 2) was selected for its potent inhibitory activity against miRNA-21, and hence became the compound of interest for further validation. Compound A36 displayed a concentration-dependent effect at 1–100 μM for 24 h (Fig. 3A). To assess the specificity of A36 towards miRNA-21, the vector control (pmir-GLO linker vector) and two different mutants of pmir-GLO-miR-21cs wt plasmid (named pmir-GLO-miR-21 cs mut1/mut2 vector) we had constructed previously [32] were transfected into HeLa cells. The results showed that A36 did not affect the luciferase signal in HeLa cells expressing the pmir-GLO linker vector (Fig. 3B), indicating that A36 did not interact with the luciferase reporter. Since the binding of miRNA and its target is highly sequence-dependent, the DNA linkers of the pmir-GLO-miR-21cs mut1/mut2 vector were designed with three consecutive point mutations compared to the wild-type complementary binding sequence of miRNA-21. The results showed that no change in the intensity of the luciferase signal was detected in HeLa cells transfected with the pmir-GLO-miR-21cs mut1/mut2 vector (Fig. 3B) after treatment with A36, indicating that A36 specifically acted on miRNA-21 rather than on other miRNAs. Because the pmir-GLO-miR-21 cs wt vector responded to the high concentration of endogenous miRNA-21 in HeLa cells, the expression of the luciferase reporter gene was largely suppressed (Fig. 3B). These results suggest that A36 was a specific miRNA-21 inhibitor.
Fig. 3.

Concentration-response assay and specificity of A36 as determined by dual-luciferase reporter assay. (A) Concentration-response assay of A36 using the pmir-GLO-miR-21cs wt plasmid in HeLa cells. (B) Specificity of A36 using the pmir-GLO-miR-21 linker plasmid and pmir-GLO-miR-21cs mut1/mut2 plasmid in HeLa cells. Statistical significance was determined using the Mann-Whitney U test (∗P < 0.05 vs. 0.1% DMSO).
3.3. Topo I and Topo IIa inhibitory activities
According to a previous report [37], lomefloxacin derivatives at 100 μM inhibited tumor growth by targeting Topo II. Topo I is also a pivotal target of antitumor drugs. To determine whether the target compounds could inhibit topoisomerase, we selected six structurally representative compounds to determine their Topo I and Topo IIα inhibitory activities at 10 μM. 10-hydroxycamptothecin (HCPT) and vepesid (VP-16) were used as positive control drugs. The results showed that none of the tested compounds had an inhibitory effect on Topo I (Fig. 4A). Most compounds were also inactive against Topo IIα except for A12 (Fig. 4B).
Fig. 4.

Topo I and IIα inhibitory activities of A6, A10, A12, A13, A36, and A40. All compounds were examined at a final concentration of 10 μM. (A) Topo I inhibitory activities. Lane 1: relaxed DNA marker; lane 2: supercoiled DNA (scDNA) pBR322; lanes 3–12: Topo I reaction products with various substrates: lane 3: 1 μL H2O; lane 4: 0.1% DMSO; lane 5: A10, 10 μM; lane 6: A12, 10 μM; lane 7: A13, 10 μM; lane 8: A6, 10 μM; lane 9: A36, 10 μM; lane 10: A40, 10 μM; lane 11: HCPT, 1 μM; lane 12: HCPT, 10 μM. (B) Topo IIα inhibitory activities. Lane 1: relaxed marker; lane 2: linear kDNA; lane 3: catenated kDNA; lanes 4–12: Topo IIα reaction product with various substrates: lane 4: 1 μL H2O; lane 5: 0.1% DMSO; lane 6: A10, 10 μM; lane 7: A12, 10 μM; lane 8: A13, 10 μM; lane 9: A6, 10 μM; lane 10: A36, 10 μM; lane 11: A40, 10 μM; lane 12: VP-16, 1 μM. HCPT: 10-hydroxycamptothecin; VP-16: vepesid.
3.4. Antiproliferative effects of selected compounds against various cancer cells
Because of the Topo IIα inhibitory activity of A12, it can be used as a reference compound; hence, A12 and A36 were selected for further investigation. To determine whether inhibition of miRNA-21 via A36 could have therapeutic effects in cancer cells that overexpress miRNA-21, we evaluated the antiproliferative effects of A12 and A36 on six tumor cell lines that overexpress miRNA-21 and the toxic effect on BEAS-2B (human normal lung epithelial cells) using the CCK-8 assay. HCPT was used as a positive control. The results are summarized in Table 4. The indicated cells were treated with increasing concentrations of A12 and A36 for 72 h. The results showed that the compounds (A12 and A36) exhibited low micromolar potency (IC50 1–13.0 μM) in all the tumor cells. Compound A12 showed good inhibitory activity in BEAS-2B cells (IC50 2.38 μM), this being consistent with the Topo IIα inhibitory activity resulting from its cytotoxic effects. However, A36 displayed an IC50 value of >100 μM against BEAS-2B cells, indicating that A36 displayed a selective inhibitory effect on tumor cells.
Table 4.
Antiproliferative activities of seven cell lines as determined via Cell Counting Kit-8 (CCK-8) assay (IC50, μM, mean ± SEM).
| Cell line | Cell type | IC50 (μM) |
||
|---|---|---|---|---|
| A12 | A36 | HCPT | ||
| HeLa | Cervical carcinoma | 1.07 ± 0.02 | 5.06 ± 0.61 | 0.92 ± 0.03 |
| HCT-116 | Colorectal carcinoma | 1.94 ± 0.35 | 1.76 ± 0.24 | 0.02 ± 0.01 |
| A549 | Lung carcinoma | 2.96 ± 0.64 | 7.59 ± 2.10 | 0.07 ± 0.01 |
| MCF-7 | Breast carcinoma | 1.49 ± 0.01 | 11.32 ± 0.87 | 0.20 ± 0.03 |
| U87 | Glioblastoma | 2.39 ± 0.76 | 9.10 ± 0.84 | 0.30 ± 0.10 |
| HCC827 | Lung carcinoma | 1.77 ± 0.39 | 13.0 ± 0.56 | 0.12 ± 0.04 |
| BEAS-2B | Normal lung epithelial cells | 2.38 ± 0.49 | >100 | 0.24 ± 0.01 |
HCPT: 10-hydroxycamptothecin, positive control.
3.5. Compound A36 inhibited the expression of mature miRNA-21 in HeLa cells
To further demonstrate the action of A36 on miRNA-21, reverse-transcription quantitative PCR (RT-qPCR) was employed to determine pri-, pre-, and mature miRNA-21 levels in HeLa cells following treatment with 0.1% dimethyl sulfoxide (DMSO), A12, and A36 at 10 μM for 24 h. Compound A36 significantly decreased the expression of mature miRNA-21, while the expressions of pri-miRNA-21 and pre-miRNA-21 were increased (Fig. 5A). According to previous reports [27,29,38,39], compound A36 might inhibit the processing of pre-miRNA-21 to mature miRNA-21. As the processing of pre-miRNA-21 to mature miRNA-21 was inhibited by A36, levels of pri-miRNA-21 and pre-miRNA-21 increased cumulatively. Compound A12 reduced the expression of pri-miRNA-21 due to its inhibitory effect on Topo IIα but had no apparent effect on the expressions of pre-miRNA-21 and mature miRNA-21.
Fig. 5.

Effects of A12 and A36 on the expression levels of pri-, pre-, and mature miRNA-21 as well as protein expressions of PDCD4 and PTEN in HeLa cells. (A) RT-qPCR analysis of pri-miRNA-21, pre-miRNA-21, and mature miRNA-21 levels in HeLa cells after treatment with 10 μM of A12 and A36 for 24 h. (B) Rescue of miRNA-21 target PDCD4 and PTEN protein expressions on treatment with A12 and A36. Western blotting results of PDCD4 and PTEN protein expressions in HeLa cell after treatment with 10 μM of A12, A36, and 0.1% DMSO control for 48 h. One representative plot of three independent experiments of Western blotting is shown. (C) Quantitative data of the three independent experiments. Statistical significance was determined using the Mann-Whitney U test (∗P < 0.05 vs. 0.1% DMSO).
3.6. Compound A36 rescued PDCD4 and PTEN protein expressions in HeLa cells
PDCD4 and PTEN are pivotal tumor suppressor proteins; their mRNAs are direct targets of miRNA-21. Since A36 could reduce the level of mature miRNA-21 in cells, the expression levels of its target genes were expected to increase. We determined the expression levels of PDCD4 and PTEN by Western blotting. Compound A36 significantly rescued both PDCD4 (1.74-fold increase) and PTEN (2.04-fold increase) protein expressions in HeLa cells, but A12 did not (Figs. 5B and C).
3.7. Compound A36 induced significant tumor cell apoptosis in HeLa cells in vitro
Apoptosis resistance is also one of the major characteristics of cancer cells; hence, we determined whether A12 and A36 could induce apoptosis in HeLa cells. The data showed that the compounds at 10 μM potently induced apoptosis in HeLa cells compared to the 0.1% DMSO control (Fig. 6A). The percentages of apoptotic cells determined after treatment with A12 and A36 at 10 μM were 50.8% and 17.9%, respectively (Fig. 6B). These results suggested that A12 and A36 significantly induced apoptosis in HeLa cells.
Fig. 6.

Effects of A12 and A36 on apoptosis, cell cycle distribution, and colony formation of HeLa cells. (A) The apoptosis activity of A12 and A36 on HeLa cells. After treatment with 0.1% DMSO (control) and 10 μM of A12 and A36 for 24 h, the HeLa cells were harvested, stained with Annexin V and propidium iodide (PI), and analyzed by flow cytometry. The upper left quadrant represents necrotic cells, the upper right quadrant represents late apoptotic cells, the lower left quadrant represents live cells, and the lower right quadrant represents early apoptotic cells. Results are representative of three independent experiments. (B) The apoptosis rate is the sum of the upper right and lower right quadrants. Data are the means ± SEM of values from three independent experiments. Statistical significance was determined using the Mann-Whitney U test (∗P < 0.05 vs. 0.1% DMSO). (C) Effects of A12 and A36 on the cell cycle distribution of HeLa cells. HeLa cells were treated with 0.1% DMSO or 10 μM of A12 and A36 for 24 h. FACS result representing the average value from three independent experiments is shown. (D) Quantitative analysis of cell cycle distribution in HeLa cells. Data are the mean ± SEM of values from three independent experiments (∗P < 0.05). (E) The effects of A12 and A36 on colony formation of HeLa cells. Representative photographs of colony formation of HeLa cells treated with 0.1% DMSO and 0.1 μM, 1.0 μM, 10 μM of A12 or A36 for 14 days. (F) Quantitative analysis of colony formation rate of HeLa cells. Data are the means ± SEM of values from three independent experiments (∗P < 0.05).
3.8. Compound A36 arrested HeLa cells at G0/G1 phase
To further understand the mechanism of the antiproliferative activity of A12 and A36, their effects on cell cycle distribution were studied. HeLa cells were treated with 10 μM of A12 and A36 for 24 h and then stained with propidium iodide (PI). DNA content was measured by flow cytometry, and the results are shown in Figs. 6C and D. The control group treated with 0.1% DMSO had approximately 63.7% cells in the G0/G1 phase, 10.1% in the S phase, and 26.2% in the G2/M phase. After treatment with A12 and A36, the percentage of cells in the G0/G1 phase increased to 66.5% and 67.9%, and the percentage of cells in the G2/M phase decreased to 25.2% and 22.3%, respectively. These results indicated that A36 arrested HeLa cells at the G0/G1 phase.
3.9. Compound A36 inhibited the colony formation in HeLa cell line
We extended the observation period of the antiproliferative activity, and the effects of A12 and A36 on the colony formation of HeLa cells was investigated. HeLa cells were treated with A12 and A36 at final concentrations of 0.1, 1, and 10 μM. As shown in Figs. 6E and F, A12 and A36 significantly inhibited the formation of HeLa cell colonies. At a concentration of 10 μM, the formation of HeLa cell colonies was almost 100% inhibited. The colony formation inhibition rates of A36 at 0.1 μM and 1.0 μM were 39.6% and 49.7%, respectively. However, due to the cytotoxic effects of A12, the colony formation inhibition rate at 1.0 μM was 82.3%, while it had almost no effect at 0.1 μM.
3.10. Compound A12 and A36 affected gene expressions of several key regulators of mitogen-activated protein kinase (MAPK) signaling pathway in HeLa cells
Gene expression alterations after A12 or A36 treatment in HeLa cells were detected using mRNA deep sequencing. The box plot shows that the distribution of the normalized data signal was highly similar among various samples (Fig. 7A), indicating that our data were reproducible and hence suitable for further analysis. Volcano plots for A12 vs. control group and A36 vs. control group, as well as the combined heatmap, showed that there were 731 and 171 significantly differentially expressed mRNAs (fold change beyond ±2 and P < 0.05) from 57,906 detected human mRNA transcripts in A12 and A36 treated HeLa cells, respectively (Figs. 7B and C, Tables S1 and S2). These data support the conclusion that A12 displays broad cytotoxicity effects, while A36 targets miRNA-21 expression without inducing larger-scale mRNA abnormalities, with greatly reduced cytotoxic effects. Producing fewer side effects is one of the biggest advantages of targeted drugs. The Venn diagram further shows that there were 66 dysregulated mRNAs shared in common between A12 and A36 treated HeLa cells, covering approximately 40% of the entire list of dysregulated mRNAs after A36 treatment (Fig. 7D). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis further illustrated that multiple signaling pathways were altered after A12 or A36 treatment (Figs. 7E and F). Taking the A36 treated group as an example, it was demonstrated that the MAPK signaling pathway was the most significantly altered among all. MAPK signaling is a potent and overactive signaling pathway for cell proliferation and apoptosis resistance during oncogenesis. DUSP4, DUSP5, and DUSP6 are negative regulators of MAPK signaling, and the dataset from mRNA deep sequencing showed that all of them were up-regulated in A12 treated Hela cells. Besides, DUSP4 and DUSP5 were also upregulated in A36 treated Hela cells (Table 5). And their upregulation suppressed tumor growth and induced tumor cell apoptosis [40]. Since a large number of mRNAs (4.3 times of genes from the A36 group) were altered in the A12 treated group, mounting-related signaling was found to be altered. We further combined the Tarbase database (archiving present experimentally validated miRNA-21 target genes) and our data. Interestingly, the results showed that among all miRNA-21 target genes, the protein expression of DUSP5 (important negative regulator in MAPK signaling) was found to be significantly upregulated in A36 and A12 treated HeLa cells. Three representative dysregulated mRNAs from the MAPK signaling pathway in A12 and A36 treated HeLa cells (including DUSP4, DUSP5, and DUSP6) were further validated using RT-qPCR (Fig. 7G). The RT-qPCR results showed that mRNA expressions of DUSP4, DUSP5, and DUSP6 were upregulated after A12 or A36 treatment, consistent with the deep sequencing data. As expected, the expression of DUSP5 protein was upregulated by both compounds (especially A36) at 10 μM for 48 h, as detected by Western blotting (Figs. 7H and I).
Fig. 7.

Treatment with 10 μM of A12 or A36 for 24 h led to generation of multiple dysregulated mRNAs in HeLa cells. (A) The box plots show similar distributions of normalized mRNA signal values from each sample. The central line represents the median of mRNA signal value, while the tails represent the upper and lower quartiles. (B) The volcano plot illustrates the differentially expressed mRNAs between control and A12 or between the control and A36 groups. (C) Heatmap displays the expression profiles and the fold change among these three groups. (D) Venn diagram displays the dysregulated mRNAs shared in common between A12 and A36 groups. (E and F) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis for dysregulated signaling in A12 or A36 group. (G) RT-qPCR validation of three representative altered mRNAs from array data (including DUSP4, DUSP5, and DUSP6). Bar represents mean ± SEM from three independent experiments, n = 3 for each experiment, ∗P < 0.05). (H) Rescue of DUSP5 protein expression on treatment with A12 or A36 at 10 μM, as analyzed by Western blotting after 48 h. (I) Quantitative results of the three independent Western blotting experiments. Data are the means ± SEM of values from three independent experiments (∗P < 0.05).
Table 5.
Representative dysregulated MAPK signaling related mRNAs in A12 or A36 treated HeLa cells (based on KEGG pathway analysis).
| Dysregulated mRNAs | A12 vs. DMSO control (14 mRNAs) |
A36 vs. DMSO control (6 mRNAs) |
||||
|---|---|---|---|---|---|---|
| Regulation | P value | Log2 (fold change) | Regulation | P value | Log2 (fold change) | |
| DUSP4 | Up | 1.12 × 10−6 | 3.76 | Up | 0.002 | 2.31 |
| DUSP5 (miRNA-21 target) | Up | 0.025 | 1.55 | Up | 0.020 | 1.59 |
| DUSP6 | Up | 0.017 | 1.75 | Not dysregulated | ||
| IL1A | Not dysregulated | Up | 0.0075 | 3.18 | ||
| CD14 | Not dysregulated | Down | 0.028 | −3.30 | ||
| DDIT3 GADD153 |
Up | 7.68 × 10−5 | 2.80 | Up | 0.034 | 1.45 |
| CACNG3 | Down | 0.035 | −5.82 | Down | 0.035 | −5.83 |
| PDGFRB | Down | 0.0011 | −2.41 | Not dysregulated | ||
| VEGFA | Up | 0.015 | 1.64 | Not dysregulated | ||
| CACNA1G | Down | 0.020 | −1.90 | Not dysregulated | ||
| ATF4 | Up | 0.034 | 1.43 | Not dysregulated | ||
| FGFR3 | Down | 0.046 | −1.36 | Not dysregulated | ||
| CACNG4 | Down | 0.05 | −1.42 | Not dysregulated | ||
| RP11-32B5.1(PAK2) | Up | 0.016 | 2.52 | Not dysregulated | ||
| HSPA2 | Down | 0.036 | −1.44 | Not dysregulated | ||
| PTPRR | Up | 0.0072 | 2.17 | Not dysregulated | ||
4. Conclusions
Forty-three fluoroquinolone derivatives were synthesized. Compound A36 exhibited the most potent inhibitory activity against miRNA-21. This significantly reduced the expression of mature miRNA-21 in the HeLa cells. At the cellular level, A36 displayed moderate antiproliferative activity against the six tumor cell lines. Furthermore, A36 significantly induced apoptosis, arrested cells at the G0/G1 phase, and inhibited colony formation of HeLa cells. It might exert anti-tumor effects by inhibiting the expression of miRNA-21 and upregulating the expressions of its target proteins PTEN, PDCD4, and DUSP5. In addition, A36 could generate 171 dysregulated mRNAs in HeLa cells. Collectively, these findings demonstrate that A36 is a novel miRNA-21 inhibitor.
CRediT author statement
Yuan-Yuan Hei: Investigation, Writing - Original draft preparation, Data curation; Si Wang: Investigation; Xiao-Xiao Xi: Investigation; Hai-Peng Wang: Validation; Yuanxu Guo: Investigation, Visualization; Minhang Xin: Validation; Congshan Jiang: Project administration, Conceptualization, Methodology, Writing - Reviewing and Editing; Shemin Lu: Conceptualization, Writing - Reviewing and Editing; San-Qi Zhang: Funding acquisition, Supervision, Conceptualization, Writing - Reviewing and Editing.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
Financial support from the National Natural Science Foundation of China (Grant No.: 81673354) is gratefully acknowledged.
Footnotes
Peer review under responsibility of Xi'an Jiaotong University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2021.12.008.
Contributor Information
Congshan Jiang, Email: jiangcongshan@xjtu.edu.cn.
San-Qi Zhang, Email: sqzhang@xjtu.edu.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Friedman R.C., Farh K.K., Burge C.B., et al. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hammond S.M. An overview of microRNAs. Adv. Drug Deliv. Rev. 2015;87:3–14. doi: 10.1016/j.addr.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cullen B.R. Transcription and processing of human microRNA precursors. Mol. Cell. 2004;16:861–865. doi: 10.1016/j.molcel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Denli A.M., Tops B.B., Plasterk R.H., et al. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432:231–235. doi: 10.1038/nature03049. [DOI] [PubMed] [Google Scholar]
- 5.Gregory R.I., Yan K.-P., Amuthan G., et al. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 6.Bernstein E., Caudy A.A., Hammond S.M., et al. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 7.Grishok A., Pasquinelli A.E., Conte D., et al. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell. 2001;106:23–34. doi: 10.1016/s0092-8674(01)00431-7. [DOI] [PubMed] [Google Scholar]
- 8.Hammond S.M., Bernstein E., Beach D., et al. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature. 2000;404:293–296. doi: 10.1038/35005107. [DOI] [PubMed] [Google Scholar]
- 9.Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hammond S.M. MicroRNAs as oncogenes. Curr. Opin. Genet. Dev. 2006;16:4–9. doi: 10.1016/j.gde.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 11.Peng Y., Croce C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target Ther. 2016;1:15004–15012. doi: 10.1038/sigtrans.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jafri M.A., Al-Qahtani M.H., Shay J.W. Role of miRNAs in human cancer metastasis: implications for therapeutic intervention. Semin. Cancer Biol. 2017;44:117–131. doi: 10.1016/j.semcancer.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Feng Y.H., Tsao C.J. Emerging role of microRNA-21 in cancer, Biomed. For. Rep. 2016;5:395–402. doi: 10.3892/br.2016.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X., Xin S., He Z., et al. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor PDCD4 and promotes cell transformation, proliferation, and metastasis in renal cell carcinoma. Cell. Physiol. Biochem. 2014;33:1631–1642. doi: 10.1159/000362946. [DOI] [PubMed] [Google Scholar]
- 15.Rushworth L.K., Kidger A.M., Delavaine L., et al. Dual-specificity phosphatase 5 regulates nuclear ERK activity and suppresses skin cancer by inhibiting mutant Harvey-Ras (HRasQ61L)-driven SerpinB2 expression. Proc. Natl. Acad. Sci. USA. 2014;111:18267–18272. doi: 10.1073/pnas.1420159112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lou Y., Yang X., Wang F., et al. MicroRNA-21 promotes the cell proliferation, invasion and migration abilities in ovarian epithelial carcinomas through inhibiting the expression of PTEN protein. Int. J. Mol. Med. 2010;26:819–827. doi: 10.3892/ijmm_00000530. [DOI] [PubMed] [Google Scholar]
- 17.Yu X., Chen Y., Tian R., et al. miRNA-21 enhances chemoresistance to cisplatin in epithelial ovarian cancer by negatively regulating PTEN. Oncol. Lett. 2017;14:1807–1810. doi: 10.3892/ol.2017.6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Naro Y., Ankenbruck N., Thomas M., et al. Small molecule inhibition of MicroRNA miR-21 rescues chemosensitivity of renal-cell carcinoma to topotecan. J. Med. Chem. 2018;61:5900–5909. doi: 10.1021/acs.jmedchem.7b01891. [DOI] [PubMed] [Google Scholar]
- 19.Markou A., Zavridou M., Lianidou E.S. miRNA-21 as a novel therapeutic target in lung cancer. Lung Cancer. 2016;7:19–27. doi: 10.2147/LCTT.S60341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Meter E.N., Onyango J.A., Teske K.A. A review of currently identified small molecule modulators of microRNA function. Eur. J. Med. Chem. 2020;188 doi: 10.1016/j.ejmech.2019.112008. [DOI] [PubMed] [Google Scholar]
- 21.Costales M.G., Childs-Disney J.L., Haniff H.S., et al. How we think about targeting RNA with small molecules. J. Med. Chem. 2020;63:8880–8900. doi: 10.1021/acs.jmedchem.9b01927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matarlo J.S., Krumpe L.R.H., Heinz W.F., et al. The natural product butylcycloheptyl prodiginine binds pre-miR-21, inhibits dicer-mediated processing of pre-miR-21, and blocks cellular proliferation. Cell Chem. Biol. 2019;10 doi: 10.1016/j.chembiol.2019.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garner A.L., Lorenz D.A., Sandoval J., et al. Tetracyclines as inhibitors of pre-microRNA maturation: a disconnection between RNA binding and inhibition. ACS Med. Chem. Lett. 2019;10:816–821. doi: 10.1021/acsmedchemlett.9b00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Velagapudi S.P., Costales M.G., Vummidi B.R., et al. Approved anti-cancer drugs target oncogenic non-coding RNAs. Cell Chem. Biol. 2018;25 doi: 10.1016/j.chembiol.2018.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gumireddy K., Young D.D., Xiong X., et al. Small-molecule inhibitors of microrna miR-21 function. Angew Chem. Int. Ed. Engl. 2008;47:7482–7484. doi: 10.1002/anie.200801555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bose D., Jayaraj G., Suryawanshi H., et al. The tuberculosis drug streptomycin as a potential cancer therapeutic: inhibition of miR-21 function by directly targeting its precursor. Angew Chem. Int. Ed. Engl. 2012;51:1019–1023. doi: 10.1002/anie.201106455. [DOI] [PubMed] [Google Scholar]
- 27.Shi Z., Zhang J., Qian X., et al. AC1MMYR2, an inhibitor of dicer-mediated biogenesis of Oncomir miR-21, reverses epithelial-mesenchymal transition and suppresses tumor growth and progression. Cancer Res. 2013;73:5519–5531. doi: 10.1158/0008-5472.CAN-13-0280. [DOI] [PubMed] [Google Scholar]
- 28.Naro Y., Thomas M., Stephens M.D., et al. Aryl amide small-molecule inhibitors of microRNA miR-21 function. Bioorg. Med. Chem. Lett. 2015;25:4793–4796. doi: 10.1016/j.bmcl.2015.07.016. [DOI] [PubMed] [Google Scholar]
- 29.Connelly C.M., Boer R.E., Moon M.H., et al. Discovery of inhibitors of MicroRNA-21 processing using small molecule microarrays. ACS Chem. Biol. 2017;12:435–443. doi: 10.1021/acschembio.6b00945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang C.S., Wang X.M., Zhang S.Q., et al. Discovery of 4-benzoylamino-N-(prop-2-yn-1-yl)benzamides as novel microRNA-21 inhibitors, Bioorg. Med. Chem. 2015;23:6510–6519. doi: 10.1016/j.bmc.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 31.Sun H., Tawa G., Wallqvist A. Classification of scaffold-hopping approaches. Drug Discov. Today. 2012;17:310–324. doi: 10.1016/j.drudis.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hei Y.Y., Guo Y.X., Jiang C.S., et al. The dual luciferase reporter system and RT-qPCR strategies for screening of MicroRNA-21 small-molecule inhibitors. Biotechnol. Appl. Biochem. 2019;66:755–762. doi: 10.1002/bab.1756. [DOI] [PubMed] [Google Scholar]
- 33.Felicetti T., Cecchetti V., Manfroni G. Modulating microRNA processing: enoxacin, the progenitor of a new class of drugs. J. Med. Chem. 2020;63:12275–12289. doi: 10.1021/acs.jmedchem.0c00510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melo S., Villanueva A., Moutinho C., et al. Small molecule enoxacin is a cancer-specific growth inhibitor that acts by enhancing TAR RNA-binding protein 2-mediated microRNA processing. Proc. Natl. Acad. Sci. U S A. 2011;108:4394–4399. doi: 10.1073/pnas.1014720108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sousa E., Graca I., Baptista T., et al. Enoxacin inhibits growth of prostate cancer cells and effectively restores microRNA processing. Epigenetics. 2013;8:548–558. doi: 10.4161/epi.24519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shan G., Li Y., Zhang J., et al. A small molecule enhances RNA interference and promotes microRNA processing. Nat. Biotechnol. 2008;26:933–940. doi: 10.1038/nbt.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou Y., Xu X., Sun Y., et al. Synthesis, cytotoxicity and topoisomerase II inhibitory activity of lomefloxacin derivatives. Bioorg. Med. Chem. Lett. 2013;23:2974–2978. doi: 10.1016/j.bmcl.2013.03.037. [DOI] [PubMed] [Google Scholar]
- 38.Velagapudi S.P., Gallo S.M., Disney M.D. Sequence-based design of bioactive small molecules that target precursor microRNAs. Nat. Chem. Biol. 2014;10:291–297. doi: 10.1038/nchembio.1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maiti M., Nauwelaerts K., Herdewijn P. Pre-microRNA binding aminoglycosides and antitumor drugs as inhibitors of Dicer catalyzed microRNA processing. Bioorg. Med. Chem. Lett. 2012;22:1709–1711. doi: 10.1016/j.bmcl.2011.12.103. [DOI] [PubMed] [Google Scholar]
- 40.Meeusen B., Janssens V. Tumor suppressive protein phosphatases in human cancer: emerging targets for therapeutic intervention and tumor stratification. Int. J. Biochem. Cell Biol. 2018;96:98–134. doi: 10.1016/j.biocel.2017.10.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.