

Abstract
Recombinant adeno-associated virus (rAAV) vectors have been developed for therapeutic treatment of genetic diseases. Current rAAV vectors administered to affected individuals often contain vector DNA-related contaminants. Here we present a thorough molecular analysis of the configuration of non-standard AAV genomes generated during rAAV production using single-molecule sequencing. In addition to the sub-vector genomic-size particles containing incomplete AAV genomes, our results showed that rAAV preparations were contaminated with multiple categories of subgenomic particles with a snapback genome (SBG) configuration or a vector genome with deletions. Through CRISPR and nuclease-based modeling in tissue culture cells, we identified that a potential mechanism leading to formation of non-canonical genome particles occurred through non-homologous end joining of fragmented vector genomes caused by genome lesions or DNA breaks present in the host cells. The results of this study advance our understanding of AAV vectors and provide new clues for improving vector efficiency and safety profiles for use in human gene therapy.
Keywords: MT: Oligonucleotides, Therapies and Applications, recombinant adeno-associated virus (rAAV) vectors, subgenomic particles, DNA lesion/break, non-homologous end joining, snapback genomes
Graphical abstract
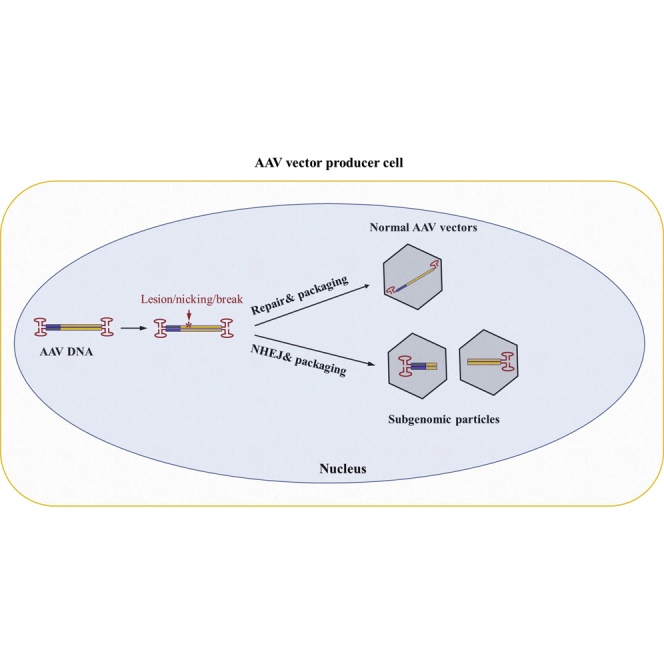
Zhang et al. revealed that vector DNA lesion and NHEJ are a potential mechanism leading to subgenomic AAV particle generation in the rAAV vector production process. This finding will provide an alternate pathway for discovering novel methodology to improve the safety and efficacy of AAV vectors.
Introduction
Recombinant adeno-associated virus (rAAV) vectors have been widely adopted as gene delivery tools for basic research as well as pharmaceutical drug vectors for human gene therapy.1 The vector genome is constructed by inserting the desired expression cassette and regulatory elements between two flanking copies of inverted terminal repeats (ITR). An ITR functions as the replication origin for AAV vectors and as a packaging signal for the rAAV production process. rAAV vectors are typically produced with a helper virus-free transfection method in mammalian cells with three different plasmids: an AAV cis-plasmid carrying the gene of interest and ITR, an AAV trans-plasmid encoding AAV structural genes and non-structural genes, and a helper plasmid supplying helper virus functions for AAV replication and packaging.2 Alternatively, rAAV vectors may also be produced using a non-adenovirus helper or a non-mammalian system with recombinant baculoviruses.1
Although rAAV vector preparation can be performed following a standard procedure, it does not result in production of a homogeneous population, even for vectors produced using good manufacturing practice (GMP) that are used clinically. Previously identified vector-related impurities include AAV particles containing plasmid backbone sequences and even host genomic sequences.3 Although defective interference particles are known to exist in wild-type AAV populations,4, 5, 6 similar particles found in rAAV vector preparations have never been fully characterized because of technical challenges in obtaining the detailed vector DNA sequences from the entire population. Previously, next generation sequencing (NGS) has been used to profile the rAAV genomic configuration and perform transcriptomics analysis.7 The Helicos-based sequencing platform has been used to profile the 3′ end of rAAV genomes.8 However, all of these data have only partial genomic information on the rAAV system. In a separate study, PacBio sequencing has been used to produce more long reads and has covered some special categories of rAAV genomes in the vector population.9 Here we systemically characterized the molecular state of rAAV vector genomes at the single-virus level. Through CRISPR-Cas9-based modeling in tissue culture cells, we identified DNA lesion/break as a potential cause of AAV vector subgenomic particle formation.
Results
The molecular configurations of subgenomes in an rAAV vector population suggest non-homologous end joining (NHEJ) events during AAV replication and packaging
To reveal the molecular state of individual AAV genomes in a population produced by the typical triple-plasmid transfection method, we took advantage of the long reads and high accuracy of the PacBio Single-Molecule, Real-Time (SMRT) Sequencing Platform. From analyzing viral genomes at the single-virus level from multiple rAAV preparations, including single-stranded DNA genome vector as well as self-complementary DNA vectors, the highly heterogeneous rAAV population with only AAV genome DNA sequences was classified into the following categories based on our analysis of thousands of vector genomes (Figure 1). (1) Standard rAAV genomes contained the complete vector sequences, including the transgene expression cassette and flanking AAV ITRs. (2) Snapback genomes (SBGs) had the left or right moiety of standard duplex rAAV genomes. SBGs were classified as symmetric SBGs (sSBGs) or asymmetric SBGs (aSBGs) according to the DNA complementary state of the top and bottom strands. For sSBGs, the top and bottom strands complemented each other. Unlike sSBGs, DNA at the bottom strand of aSBGs did not match the top strand completely and, therefore, promoted loop formation in the middle region (Figures S1 and S2). (3) Incomplete rAAV genomes (ICGs) had an intact 3' ITR and partial AAV genome. These were presumably formed by an aborted packaging process. (4) In genome deletion mutants (GDMs), the middle region of the AAV genomes was deleted. (5) Secondary derivative genomes (SDGs) ere formed by using class 2–4 molecules as the template and through the same mechanism to generate the next generation of subgenomic vector molecules of classes 2–4 (Figures S3 and S4). Although the typical sSBG configuration may have been the product of a template switch, the existence of aSBGs, GDMs, and SDGs could not be explained by a template switch of the DNA polymerase during AAV replication. Because there were remnant signs of multiple DNA fragments in the aSBG, GDM, and SDG, we proposed that NHEJ events had occurred during the AAV replication and packaging processes.
Figure 1.

Molecular configurations of DNA genomes in rAAV vectors
AAV genomes were sequenced using the PacBio platform and compared with the reference sequences at the top. Besides the standard-sized AAV vector genomes (1), four typical categories of subgenomic rAAV genomes were found in the rAAV vectors: symmetric snapback genomes (sSBGs) and asymmetric SBGs (aSBGs) (2), genome deletion mutants (GDMs; 3), incomplete genomes (ICGs; 4), and secondary derivative genomes (SDGs; 5).
NHEJ as a mechanism for generating subgenomic particles in an rAAV population
An NHEJ reaction requires the presence of corresponding DNA fragments. Dissection of genomic configurations from subgenomic AAV particles suggested the existence of such fragments. First, we tested whether NHEJ events could lead to generation of SBGs. We transfected HEK293 cells with linear rAAV DNA fragments (Figure 2A) that were generated through restriction enzyme digestion in the presence of trans elements that complement AAV replication and packaging (Figure 2B). The parent vector plasmid pCB-EGFP-6.4k was oversized for AAV capsids. DNA recovered from vectors prepared using this oversized plasmid primarily consisted of smaller fragments that were less than 6.4 kb in size. In contrast, vectors prepared from the smaller vector plasmid pCB-EGFP-3.4k, which falls within the packaging limits for the AAV capsid, mainly produced viral particles with a 3.4-kb DNA genome. When linear fragments derived from pCB-EGFP-6.4k, ranging from 0.6–3.1 kb, were used for transfection, the most prominent genomes recovered from the prepared vectors appeared to be resulting from inter-molecular NHEJ (Figure 2). Even though intra-molecular DNA joining of the 5′ end and 3′ end was supposed to be more efficient, the vectors resulting from such a reaction were in relatively lower yield. This was most likely because their size was larger than the inter-molecular NHEJ products. More specifically, when a vector was prepared using fCB-GFP-2.3k, the vector DNA sizes from two ITRs to the breakpoints were 1.8 kb and 2.3 kb, respectively (Figure 2A). As shown in Figure 2B, the main vector size was 1.8 kb (3.6 kilonucleotides [knt] in single-stranded form), 2.3 kb (4.6-knt single-stranded DNA [ssDNA]), along with a faint 4.1-kb band (which was annealed from the plus strand and minus strand), which suggested intra-molecular joining. Similar observations were obtained for vectors prepared using fCB-GFP-0.6k (0.6-kb to 1.2-knt ssDNA, 1.8-kb to 3.6-knt ssDNA), fCB-GFP-1.0k (1.0-kb to 2.0-knt ssDNA, 1.8-kb to 3.6-knt ssDNA), fCB-GFP-1.6k (1.6-kb to 3.2-knt ssDNA, 1.8-kb to 3.6-knt ssDNA), and fCB-GFP-1.8k (1.8-kb to 3.6-knt ssDNA). The exception was for vectors prepared using fCB-GFP-3.1k, in which we only observed a 1.8-kb genome fragment. This is likely because the 3.1-kb SBG molecule, which was 6.2-knt ssDNA, was over the packaging size limit for AAV vectors.
Figure 2.

Inter-molecular NHEJ is a mechanism leading to formation of SBG molecules
(A) The parent plasmid pCB-GFP-6.4k was linearized with varying restriction enzymes to obtain linear fragments. The plasmid backbone is depicted with a dotted line. HEK293 cells with rAAV packaging helper functions were transfected with DNA fragments, plasmid pCB-GFP-6.4k, or pCB-GFP-3.4k. (B) The resulting rAAV vectors in the medium were harvested, and the DNA in the vectors was extracted and analyzed for genome status using a 1% agarose gel. For simplicity, fragments such as fCB-GFP-0.6k are referred to as 0.6 kb at the top of the gel. Red arrows indicate key fragments. The vectors recovered were quantified by qPCR using primers specific for poly(A) or GFP. (C) The ratio of vectors containing poly(A) or GFP. In pCB-GFP-3.4k, the vector size is 3.4 kb. For DNA fragments, the size of 5′ ITR-GFP is 1.8 kb, and the size of the poly(A) to 3′ ITR is indicated as the last three letters in the name.
When these fragments were used to supply AAV production, we noticed that the relative abundance differed among vectors produced. Vectors containing the poly(A) moiety or vectors containing the GFP moiety represented different NEHJ reactions; their relative ratio is graphed in Figure 2C. Based on these results, it was evident that the smaller subgenomic particles became more dominant. This suggested that subsequent DNA replication and packaging favor smaller genomes, which may be a major mechanism dictating the abundance of rAAV subgenomic particles.
To generate GDMs, the 5′ ITR moiety and the 3′ ITR moiety should be linked together. To demonstrate that NHEJ is the underlying mechanism for generating such molecules, we transfected HEK293 cells with a 5′ ITR fragment carrying the chicken ß–actin promoter with a CMV enhancer promoter and a 3′ ITR fragment carrying the GFP gene along with helper plasmids for AAV replication and packaging. Combination of these two fragments efficiently regenerated functional GFP expression (Figure 3A). Upon harvesting the packaged AAV vectors, rAAV GFP vectors were also regenerated, as shown in the transduction assay (Figure 3A). This experiment demonstrated that the GDM molecules, as shown in Figure 3B, were produced through the same mechanism that generated the SBG vector.
Figure 3.

Inter-molecular NHEJ is a mechanism leading to AAV GDMs
(A) HEK293 cells were transfected with ITR fragments containing the CB promoter or GFP gene alone or combined with supplemental helper plasmids for rAAV replication and packaging. The positive control was a 2.3-kb intact pCB-GFP-2.3k plasmid. Three days after transfection, GFP expression was monitored by fluorescence microscopy (center panel; scale bar, 50 μM). There are autofluorescent cells or possibly cryptic promoter activity in transfection panels 1 and 2. The harvested vectors were used to transduce GM16095 cells, and GFP expression was monitored 24 h after transduction (bottom panel). (B) The vector DNA recovered from (A) was electrophoresed in 1% agarose gel, and AAV genomes were detected by Southern blot using an ITR-specific probe. Δ indicates key fragments of 0.9 kb, 1.4 kb, and 2.3 kb.
We then used CRISPR-Cas9 system to generate the DNA breaks that may occur in tissue culture cells. In the vector production system, the Cas9 expression plasmid was co-transfected along with the vector plasmid pCB-EGFP-3.4k. In contrast to the control without guide RNA (gRNA), transfection with gRNA produced two distinct vectors with a size of 2.3 kb and 1.1 kb in a native gel (Figure 4A), which corresponded to the cutting site. In the denaturing gel, the original vector was present as a 3.4-kb ssDNA. In contrast, the vectors produced in the presence of gRNA appeared as ssDNA 4.6 knt or 2.2 knt in size (Figure 4B). Subsequently we recovered the 2.2-knt ssDNA from the gel, renatured the DNA, and then identified its size in the native gel. The 2.2-knt DNA fragment appeared as 1.1-kb double-stranded DNA (Figure 4C). These results suggested that the SBG molecules of both ends were formed in the presence of CRISPR-Cas9-induced digestion in tissue culture cells.
Figure 4.

Intra-host cell vector DNA break is a mechanism for AAV subgenomic particle formation
HEK293 cells were transfected with AAV plasmid pCB-GFP-3.4k along with Cas9-expressing plasmids with or without corresponding gRNA. (A) The resulting vector DNA was electrophoresed in native agarose gel. (B) The resulting vector DNA was electrophoresed in denaturing agarose gel. (C) The denatured fragments of (B) (indicated as ① and ②) were collected, renatured, and electrophoresed again in the native gel.
DNA lesion/nicking is sufficient for generating AAV subgenomic particles
To investigate whether a DNA lesion was sufficient to generate SBG molecules, CRISPR-Cas9 nickase activity was introduced into the AAV production system (Figure 5). As presented in Figure 5B, in tissue culture cells, digestion with Cas9 at various positions generated two major SBG molecules corresponding to the cleavage sites. Using gRNA9 as an example, Cas9 digestion generated two vector genomes at 1.5 kb and 1.9 kb, respectively. However, nicking at the top strand by gRNA9 and Cas9-H840A only yielded a 1.5-kb vector genome. Nicking at the bottom strand by gRNA9 and Cas9-D10A yield a 1.9-kb vector genome. In contrast, digestion of the vector DNA by gRNA4 and Cas9 yielded two predominant vector genomes 0.6 kb and 1.2 kb in size; the 2.8-kb (5.6-knt ssDNA) SBG vector that should have appeared was not observed because it exceeded the packaging capacity of the AAV particle. Nicking with gRNA4 and Cas9-H840A yielded a 0.6-kb vector genome along with its dimer at 1.2 kb. On the other hand, nicking at the bottom strand by gRNA4 and Cas9-D10A yielded no major bands because the theoretical 2.8-kb (5.6-knt ssDNA) SBG vectors are oversized for AAV capsids. Similar results were obtained from gRNA13-induced nicking or cutting. Because NHEJ product replication was favoring smaller fragments, the corresponding larger DNA was not competitive and was greatly diminished; i.e., gRNA5 and gRNA10. This result suggested that DNA lesion/nicking was sufficient to generate DNA fragments that can lead to creation of subgenomic particles. There was clear plus strand or minus strand selection in which the nicking site and its 3′ end ITR formed snapback molecules.
Figure 5.

Intra-host cells vector DNA lesion is sufficient for SBG formation
(A) Illustration of gRNA sites in pCB-EGFP-3.4k for Cas9 nicking or digestion. HEK293 cells were transfected with plasmid pCB-EGFP-3.4k for vector production in the presence of Cas9 or Cas9 mutants (H840A or D10A) and corresponding gRNA. (B) The resulting vector DNA was separated by native agarose gel electrophoresis with Ethidium Bromide staining. C, Cas9 double cut; D, D10A nicking; H, H840A nicking. H stands for Cas9-H840A nicking. D stands for Cas9-D10A nicking. C stands for Cas9 cutting. (C) The table summarizes the potential DNA sizes that can be generated by nicking or cutting. The actual observed bands are summarized in the brackets. − indicates “not observed.”
Cellular DNA damage leads to subgenomic molecule formation
We further hypothesize that intracellular DNA damage events may lead to subgenomic DNA formation. As shown in Figure 6, hydrogen peroxide (H2O2) was added to investigate the effects of this DNA damage reagent on AAV production. Corresponding to an increased concentration of H2O2, the recovered rAAV vectors appeared as smears that were smaller in size compared with the standard AAV vectors. At 200 μM H2O2, the majority of vector DNA detected was small subgenomic DNA particles (Figure 6A). We prepared a library for the recovered DNA from these vectors and performed DNA sequencing using the PacBio platform. More than 50,000 genomes were sequenced. The majority of these sequences were not from AAV vector-related DNA and appeared as short DNA fragments. Sequences aligned to the initial vector appeared to be heavily fragmented (Figure 6B). SBGs produced from the initial vector were recovered in the sequencing as well (Figure 6B). Some of these molecules were found to contain portions of the plasmid backbone. These results showed that global DNA damage events can ruin rAAV production and lead to production of subgenomic particles.
Figure 6.

DNA-damaging conditions in the host cells promoted subgenomic particle formation
H2O2 at varying concentrations was added to the rAAV production system after transfection. (A) The resulting rAAV vectors were purified by CsCl gradient, and the vector DNA was analyzed by gel analysis. (B) Partially recovered vector genomes were sequenced and aligned to the reference sequence. The coverage is marked by blue lines. Example DNA configurations of AAV subgenomic particles are illustrated at the bottom.
Discussion
The heterogeneity in wild-type (WT) AAV virus and rAAV vectors has been well documented.4,10 Similar to what has been observed for WT AAVs,11 as shown in Figure 1, the subgenomic particles in rAAV vectors have similar molecular conformations: SBG, GDM, and ICG. In addition to these three major categories, a fourth category of subgenomic particles was identified as SDG particles arising from damage to the SBG, GDM, and IDG forms, followed by a second round of NHEJ events (Figure S3 and S4). The amount of subgenomic particles, such as SBGs, determined by SMRT sequencing has been published in our previous study; the amount of SBGs varied depending on the size of SBGs, which may range from 1%–40% of total AAV particles.12 The unique molecular configuration of GDM molecules prompted us to explore NHEJ events as a potential cause of subgenomic DNA particle formation. Although a DNA polymerase template switch mechanism may explain formation of sSBGs,4,10 the existence of GDM molecules, especially the large GDMs, which exceed the size of the parental AAV vector and have partial duplication of vector genome sequences in the junction (Figure 1), strongly favors NHEJ as the primary mechanism that led to formation of subgenomic particles.
The essence of the NHEJ mechanism is ligation of various DNA fragments. We were able to regenerate those SBG and GDM molecules using DNA fragments derived from AAV vector genomes by straight in vitro restriction endonuclease digestion or CRISPR-Cas9 in-tissue culture cell digestion. Generation of SBG molecules was quite efficient. Often they were the dominant vector molecules produced (Figures 2, 4, and 5). When two fragments were introduced into the AAV packaging system, formation of GDMs could be confirmed as well (Figure 3). This mechanism can also explain why AAV vectors often contain host genomic DNA sequences as well as materials used for AAV production.
Another key point explored in this study was how the AAV fragments originated. The nickase experiment showed that simple nicking of AAV DNA in plasmid or replication forms was sufficient to generate corresponding SBGs (Figure 5). This suggests that a double-stranded AAV DNA break is not necessary. Even more interesting was that SBG formation is related to nicking DNA strands. The resulting SBG contained DNA from the nicking site to its 3′ ITR. This indicated that generation of such fragments leading to SBG formation was closely coupled to DNA replication.
Nicking/lesion of DNA in the rAAV genomes suggest that any host/viral factors that cause AAV genome damage could lead to subgenomic AAV particle formation. H2O2 is an oxidizer that can cause global DNA damage in tissue culture cells. Our study showed that, when H2O2 was present at a high concentration, rAAV production was completely disrupted and resulted in generation of primarily subgenomic particles (Figure 6A). SBGs and GDMs can be observed in the sequencing analysis (Figure 6B). We must also note that H2O2 is an extreme example of DNA damage. It may not represent what is happening at more physiologic levels of DNA damage.
Unlike the DNA template switch model, which only explains formation of largely symmetric SBGs, the NHEJ mechanism seamlessly explained formation of SBGs and GDMs. Therefore, we proposed a comprehensive model of subgenomic AAV particle formation in WT AAVs and rAAVs (Figure 7). When fragments with only one ITR are produced, it will undergo self-ligation or ligate to another fragment with only one ITR. In turn, this will create recombinant molecules with two ITRs. In case of molecules that are larger than the standard AAV size, they will not be packaged. Nicking of the AAV genome (DNA lesion) and break of AAV DNA leads to formation of various DNA fragments. We hypothesize that host factors and AAV proteins may cause nicking and breaks in rAAV genomes. Ligation of these fragments generates SBG and GMD molecules. Although it is also possible that such ligation will pick up any genomes from the host cells, the main products will be SBGs and GMDs because of their abundance in the replication center and proximity of these fragments. The subsequent DNA replication will favor SBGs or GMDs when they have small genomes. Alternatively, the replication dimer of rAAVs has breakpoints flanking the double-D ITR (Figure 6B), and, therefore, it will efficiently self-ligate and generate SBGs.
Figure 7.

A model of subgenomic AAV particle formation
The key point is that varying DNA fragments with only one ITR were generated from the lesion/break on the monomer or dimer of the replication form of AAV genomes. NHEJ then rejoins these fragments, and the resulting products restore two ITRs in a molecule, which can be replicated and packaged in an AAV capsid. This mechanism readily led to generation of SBGs, GDMs, and various other forms that are not illustrated in the figure.
Because oversized AAV vectors generally result in heterogeneous vector populations,13 it is clear that vector size is one of the determining factors leading to formation of subgenomic particles such as SBGs, GDMs, and ICGs. DNA elements with special secondary structure also increase the likelihood of these undesirable products, such as AAVs carrying gRNA and shRNA sequences.9
Although our data support the hypothesis that NHEJ can lead to generation of subgenomic particles, this study does not eliminate that other mechanisms, such as template switch, may give rise to generation of SBG particles. Based on the model we proposed, investigation of high-frequency break points that occur during rAAV vector genome replication is an area that may help find the solution for reducing subgenomic genomic particle formation in rAAV production. Such studies can minimize production of subgenomic particles in rAAV vectors, which cannot be removed by downstream processing, and improve the safety profile of AAV vectors. The present study is fundamental for understanding the basic biology of AAVs and development of the next generation of AAV vectors for human gene therapy.
Materials and methods
Cell lines and transfection
HEK293 cells (human embryonic kidney [HEK] cells transformed by DNA from human adenovirus type 5, CRL-1573, ATCC) and GM16095 cells (a human fibroblast cell line purchased from the Coriell Institute, Camden, NJ) were cultured in DMEM supplemented with 10% fetal bovine serum, 100 μg/mL penicillin, and 100 U/mL streptomycin (Invitrogen, Carlsbad, CA, USA). All cells were maintained in a humidified 37°C incubator with 5% CO2. PolyJet DNA in vitro transfection reagent (catalog number SL100688, SignaGen Laboratories, Frederick, MD, USA) was used to deliver DNA into HEK293 cells. Cells were seeded into six-well plates or 10-cm-diameter culture dishes 18–24 h prior to transfection so that the monolayer cell density reached the optimal 70%–80% confluency at the time of transfection. Complete culture medium with serum was freshly added to each plate 30 min before transfection. PolyJet-DNA complex for transfection was prepared according to the ratio of 3 μL PolyJet to 1 μg DNA using serum-free DMEM to dilute DNA and PolyJet reagent. This was incubated for 10–15 min at room temperature, and then the PolyJet/DNA mixture was added to the medium. The PolyJet/DNA complex-containing medium was then removed and replaced with fresh serum-free DMEM 12 h after transfection.
rAAV transduction
GM16095 cells were seeded into 12-well plates 24 h prior to transduction so that the monolayer cell density reached the optimal 70%–80% confluency at the time of transduction. The cells were washed with DMEM culture medium without serum twice, 3 min each time, before transduction. 10 μL of cell culture medium containing rAAV virions was added to the plate and incubated at the indicated time points. GFP fluorescence expression was observed using fluorescence microscopy (Leica D3000 B).
Plasmid construction
rAAV vector production plasmids include the vector plasmids for pAAV-CB-EGFP (4.3 kb), pAAV-CB-Cluc, pAAV-CMV-Cluc, pAAV-hAAT-hLC, pAAV-hAAT-hFVIII, and pAAV-ß-actin-hFVIII; the rep-cap plasmid pH22, pH28 contained the AAV2 rep and cap or AAV8 cap coding sequences and the mini-adenovirus helper pFΔ6. Plasmid pCB-GFP (2.3 kb) was digested by PvuII and AgeI to generate 0.9-kb and 1.4-kb DNA fragments. To construct plasmid pCB-GFP-6.4k, a 4.3-kb fragment from pAAV-ß-actin-hFVIII digested by SacI was inserted into the backbone plasmid pCB-GFP-3.4k with MluI digestion, generating the pCB-GFP-FVIII-7.7k plasmid. Subsequently the pCB-GFP-FVIII-7.7k plasmid was digested with KpnI to generate the pCB-GFP-6.4k plasmid. This plasmid was subjected to a series of restriction digestions to produce a series of DNA fragments with different lengths: fCB-GFP-0.6k, fCB-GFP-1.0k, fCB-GFP-1.6k, fCB-GFP-1.8k, fCB-GFP-2.3k, and fCB-GFP-3.1k.
For the CRISP-Cas9 expression system, the plasmids pCI-BN-spCas9, pCI-BN-Cas9H840A, and pCI-BN-Cas9D10A were constructed. The two backbone plasmids used were lentiSpCas9-Blast LV-MS2-zeo-tdt-1. The SpCas9 open reading frame (ORF) was concatenated with an Nuclear localization signals sequence by amplification using lentiSpCas9-Blast as a template and the three primers pCI-AAV2-VP2-SpCas9-Core-F1, pCI-AAV2-VP2-SpCas9-Core-F2, and pCI-AAV2-VP2-SpCas9-R2. After restriction digestion with NheI, the amplicon was inserted into the pCI plasmid backbone prepared previously in our lab, leading to pCI-BN-SpCas9. The D10A mutation fragment was obtained by two rounds of amplification. In the first round, two fragments were amplified by using pCI-BN-SpCas9 as a template and the following four primers: pCI-AAV2-VP2-SpCas9-Core-F1, SpD10A-R, SpD10A-F, and SpD10A-APAI-R. These two amplicons were used as templates to generate D10A mutation fragments by next-round amplification by the primers pCI-AAV2-VP2-SpCas9-Core-F1 and SpD10A-APAI-R. The mutation fragments were inserted into the pCI-BN-SpCas9 backbone by digestion of NheI and ApaI, leading to pCI-BN-SpCas9-D10A. pCI-BN-SpCas9-H840A was constructed with a similar strategy; the three primers used were SpD10A-APAI-F, H840A-F, and H840A-R. The gRNA target sequences in the pCB-GFP-3.4k rAAV genome were designed using the Broad Institute gRNA designer tool (https://www.broadinstitute.org/rnai/public/analysis-tools/sgrna-design). The plasmid LV-MS2-zeo-tdt-1 was digested with Apal and then self-ligated as the backbone plasmid. These gRNA sequences were constructed into the backbone plasmid LV-MS2-zeo-tdt-1 following the protocol described in this study.14 All sequences, including the primers used and gRNA targets, are listed in Table S1.
rAAV vector production and purification
The vectors rAAV-hM4D.b and rAAV-luc.b were ordered from Virovek. All other rAAV vectors, listed in a previous study,12 were produced using the triple-plasmid transfection system in HEK293 cells. PolyJet DNA in vitro transfection reagent was used to deliver DNA into HEK293 cells. The three plasmids were transfected at a molar ratio of 1:1:1. To produce CRISPR-digested AAV genome vectors, pCI-BN-spCas9, pCI-BN-Cas9H840A, or pCI-BN-Cas9D10A and the plasmid containing the corresponding gRNA were co-transfected with the vector plasmid, the rep-cap plasmid, and the helper plasmid into HEK293 cells at a molar ratio of 1:1:1:1:1. 72 h after transfection, medium was collected and precipitated with 40% of Polyethylene Glycol (final concentration 8%) overnight at 4°C. After centrifugation, the pellets were resuspended and treated with DNase I. AAVs of different densities were separated using CsCl gradient ultracentrifugation. AAVs of different densities were extracted and dialyzed against 5% sorbitol in phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4 [pH 7.2]). Vector genome titers were determined by quantitative real-time PCR with vector titers expressed as viral genome/mL. To obtain vectors representative of all viral particles, the gradient centrifugation step was skipped. Three days after transfection, the medium was collected and precipitated into a concentrated solution of rAAV particles. rAAV genomic DNA was purified and analyzed using agarose gel electrophoresis and quantitative real-time PCR.
DNA agarose gel electrophoresis
The rAAV genome was extracted and purified as follows. Viral vectors were treated with DNase I (1 U/mL) for 30 min at 37°C. Then 1 μL of 0.5 M EDTA was added (to a final concentration of 5 mM) and heated for 10 min at 75°C to stop DNase I activity. One half volume of lysis buffer (Direct PCR Tail, Viagen) containing proteinase K (40 μg/mL) was added and incubated for 1 h at 56°C and finally heated for 10 min at 95°C. One volume of phenol:chloroform:isoamyl alcohol (25:24:1) was added to the samples and vortexed thoroughly for approximately 20 s. This was then centrifuged at 4°C for 30 min at 16,000 × g. The upper aqueous phase was carefully removed and transferred to a fresh tube. 200 μL of 70% ethanol was added, and the tubes were centrifuged at 4°C for 10 min at 16,000 × g. The supernatant was carefully removed, and the pellet was allowed to air dry at room temperature. 20 μL of Tris-EDTA buffer was added to dissolve DNA. DNA concentration was measured using Nanodrop. 100 ng DNA was loaded on 1% of native gel and run at 120 V for 50 min in 400 mM Tris buffer [pH 7.5]. Equal DNA was loaded on 1% of denaturing gel and run at 60 V for 100 min in alkaline buffer of 30 mM NaOH and 2 mM EDTA. Gels were stained using 1× SYBR Safe DNA gel stain (Invitrogen), and a photo taken at a wavelength of 365 nm using a ChemiDOC MP Imaging System (Bio-Rad).
H2O2 treatment
HEK 293 cells were seeded in 20 15-cm dishes and incubated for 18 h. The old culture medium was replaced with free FBS DMEM containing a final concentration of 0 μM, 50 μM, 100 μM, and 200 μM H2O2 60 min prior to transfection. Three plasmids, pH22, pFΔ6, and pssAAV-CB-GFP-4.7k, were transfected into HEK293 cells using PolyJet DNA in vitro transfection reagent. 72 h after transfection, medium was collected, precipitated with 40% of PEG (final concentration 8%), and purified by CsCl gradient method. rAAV DNA was extracted and purified using the phenol:chloroform:isoamyl alcohol (25:24:1) method. 500 ng of rAAV DNA was subjected to sequencing by the PacBio SMRT platform. 30 ng of DNA was loaded on a 1% agarose gel and run at 120 V for 50 min.
Quantitative real-time PCR
Viral vectors (1 × 1010 vg, 1 μL) in solution containing DNase I (1 U/mL) were incubated for 30 min at 37°C. 1 μL of 0.5 M EDTA was added to a final concentration of 5 mM and subsequently heated for 10 min at 75°C to stop DNase I activity. Control samples received lysis buffer (Direct PCR Tail, Viagen) containing proteinase K (40 μg/mL), and were incubated for 1 h at 56°C and finally heated for 10 min at 95°C. Samples intended for thermal treatment were directly heated after heat inactivation of DNase I treatment at the indicated temperatures. The copy numbers of viral genomes subsequently released were quantified by real-time PCR and expressed in vg/mL. The primers targeting the GFP gene and poly(A) sequence used are listed in Table S1.
AAV genome sequencing and data analysis
For long-read PacBio SMRT sequencing, AAV samples were prepared according to SMRTbell procedures. DNA was extracted and purified by AMPure PB Beads and then repaired by a SMRTbell damage repair kit. The adaptor ligation reaction was performed, and then ExoIII and ExoVII were added to remove failed ligation products. AMPure PB Beads cleaning step was performed three times.
SMRT subread filtering and the high-quality circular consensus sequences corresponding to the rAAV library were generated using the SMRT analysis portal (minimum accuracy of 0.99 and minimum of 3 Circular Consensus Sequence passes) and considered for further analysis. Filtered reads were mapped to the reference rAAV genome using Minimap2 and processed alignments to illustrate configuration categories of molecules in the rAAV population. All sequencing data were deposited into and can be accessed at https://github.com/PacificBiosciences/pbmm2/Accession/PRJNA680507.
Statistical analysis
All data were presented as means ± SD. Statistical analysis was performed by Student’s unpaired t test in SPSS software v.1.0.0.1406. p < 0.05 was considered statistically significant.
Acknowledgments
This work was supported by grants from the National Institutes of Health (HL142019, HL114152, and HL130871). D.A.F. is supported by NIH T32HL007910.
Author contributions
Conceptualization, J.Z. and W.X.; investigation, J.Z, P.G., X.Y., and W.X.; data interpretation/analysis, J.Z., P.G., X.Y., D.A.F., and W.X.; writing – original draft, J.Z. and W.X.; software and data curation, W.X., X.Y., and D.P.; manuscript editing, J.Z., D.A.F., A.K.L., P.L.M., M.C., J.F., R.W.H., Y.D., N.S., and W.X.; supervision, J.Z., R.W.H., and W.X. All authors have read and agreed to the published version of the manuscript.
Declaration of interests
W.X. holds equity in Ivygen Corporation and Nikegen LLC.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.08.027.
Supplemental information
Data availability
All AAV genome sequencing data are accessible at https://www.ncbi.nlm.nih.gov/bioproject/680507.
References
- 1.Samulski R.J., Muzyczka N. AAV-mediated gene therapy for research and therapeutic purposes (2014) Annu. Rev. Virol. 2014;1:427–451. doi: 10.1146/annurev-virology-031413-085355. [DOI] [PubMed] [Google Scholar]
- 2.Ferrari F.K., Xiao X., Mccarty D., Samulski R.J. New developments in the generation of Ad-free, high-titer rAAV genetherapy vectors. Nat. Med. 1997;3:1295–1297. doi: 10.1038/nm1197-1295. [DOI] [PubMed] [Google Scholar]
- 3.Wright J.F. Product-related impurities in clinical-grade recombinant AAV vectors: characterization and risk assessment. Biomedicines. 2014;2:80–97. doi: 10.3390/biomedicines2010080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laughlin C.A., Myers M.W., Risin D.L., Carter B.J. Defective-interfering particles of the human parvovirus adeno-associated virus. Virology. 1979;94:162–174. doi: 10.1016/0042-6822(79)90446-X. [DOI] [PubMed] [Google Scholar]
- 5.de la Maza L.M., Carter B.J. Heavy and light particles of adeno-associated virus. J. Virol. 1980;33:1129–1137. doi: 10.1128/jvi.33.3.1129-1137.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de la Maza L.M., Carter B.J. Molecular structure of adeno-associated virus variant DNA. J. Biol. Chem. 1980;255:3194–3203. doi: 10.1016/S0021-9258(19)85870-2. [DOI] [PubMed] [Google Scholar]
- 7.Lecomte E., Tournaire B., Cogné B., Dupont J.B., Lindenbaum P., Martin-Fontaine M., Broucque F., Robin C., Hebben M., Merten O.W., et al. Advanced characterization of dna molecules in raav vector preparations by single-stranded virus next-generation sequencing. Mol. Ther. Nucleic Acids. 2015;4 doi: 10.1038/mtna.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kapranov P., Chen L., Dederich D., Dong B., He J., Steinmann K.E., Moore A.R., Thompson J.F., Milos P.M., Xiao W. Native molecular state of adeno-associated viral vectors revealed by single-molecule sequencing. Hum. Gene Ther. 2012;23:46–55. doi: 10.1089/hum.2011.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie J., Mao Q., Tai P.W.L., He R., Ai J., Su Q., Zhu Y., Ma H., Li J., Gong S., et al. Short DNA hairpins compromise recombinant adeno-associated virus genome homogeneity. Mol. Ther. 2017;25:1363–1374. doi: 10.1016/j.ymthe.2017.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tai P.W.L., Xie J., Fong K., Seetin M., Heiner C., Su Q., Weiand M., Wilmot D., Zapp M.L., Gao G. Adeno-associated virus genome population sequencing achieves full vector genome resolution and reveals human-vector chimeras. Mol. Ther. Methods Clin. Dev. 2018;9:130–141. doi: 10.1016/j.omtm.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J., Yu X., Guo P., Firrman J., Pouchnik D., Diao Y., Samulski R.J., Xiao W. Satellite subgenomic particles are key regulators of adeno-associated virus life cycle. Viruses. 2021;13:1185. doi: 10.3390/v13061185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J., Yu X., Herzog R.W., Samulski R.J., Xiao W. Flies in the ointment: AAV vector preparations and tumor risk. Mol. Ther. 2021;29:2637–2639. doi: 10.1016/j.ymthe.2021.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu Z., Yang H., Colosi P. Effect of genome size on aav vector packaging. Mol. Ther. 2010;18:80–86. doi: 10.1038/mt.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All AAV genome sequencing data are accessible at https://www.ncbi.nlm.nih.gov/bioproject/680507.