

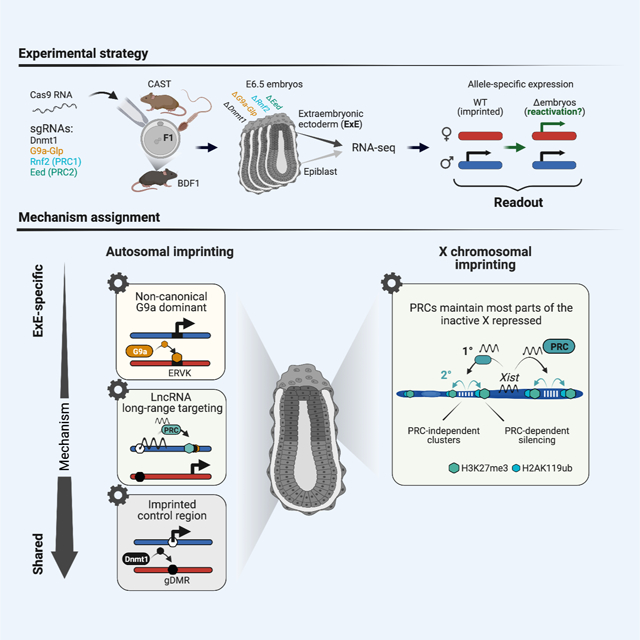
SUMMARY
Genomic imprinting and X chromosome inactivation (XCI) require epigenetic mechanisms to encode allele-specific expression, but how these specific tasks are accomplished at single loci or across chromosomal scales remains incompletely understood. Here, we systematically disrupted essential epigenetic pathways within polymorphic embryos to examine canonical and non-canonical genomic imprinting as well as XCI. We find that DNA methylation and Polycomb group repressors are indispensable for autosomal imprinting, albeit at distinct gene sets. Moreover, the extraembryonic ectoderm relies on a broader spectrum of imprinting mechanisms, including non-canonical targeting of maternal endogenous retrovirus (ERV)-driven promoters by the H3K9 methyltransferase G9a. We further identify Polycomb dependent and independent gene clusters on the imprinted X chromosome, which appears to reflect distinct domains of Xist-mediated suppression. From our data, we assemble a comprehensive inventory of the epigenetic pathways that maintain parent-specific imprinting in eutherian mammals, including an expanded view of the placental lineage.
Graphical Abstract
INTRODUCTION
Mammals have two autosomal gene copies, one inherited from each parent. The vast majority of genes are biallelically expressed, while a small subset is expressed in a parent-specific fashion (Barlow and Bartolomei, 2014; Monk et al., 2019; Tucci et al., 2019). Initially identified when uniparental diploid zygotes failed to produce viable offspring (McGrath and Solter, 1984; Surani et al., 1984), subsequent translocation analyses and mapping efforts confirmed that the lack of equivalence between parental genomes reflected the discrete activities of individual loci (Cattanach and Kirk, 1985). This phenomenon was later shown to be the result of gametic control of coding and non-coding gene expression in offspring, including the discoveries of the first imprinted genes Igf2r, Igf2 and H19 within the same year (Barlow et al., 1991; Bartolomei et al., 1991; DeChiara et al., 1991; Ferguson-Smith et al., 1991). As currently understood, genomic imprinting is maintained via distinct epigenetic mechanisms to propagate information from the oocyte or sperm into the next generation, and often regulates the expression of nearby genes as a secondary mechanism in cis (Barlow and Bartolomei, 2014). Multiple loci have been thoroughly dissected in great detail to explore how this process is carried out molecularly (Barlow, 2011; Peters, 2014). For example, classic epigenetic modifications, such as DNA methylation, can repress long non-coding RNAs (lncRNAs) that otherwise target and repress nearby genes (Mancini-DiNardo et al., 2006; Sleutels et al., 2002; Williamson et al., 2011).
For decades, DNA methylation was considered the only epigenetic modification that could be transmitted from the germline into subsequent progeny. Recently, oocyte-specific trimethylation of H3 on lysine 27 (H3K27me3) was found to transiently imprint several loci within preimplantation embryos, some of which transition to a more permanent DNA methylated state to silence maternal alleles after implantation (Chen et al., 2019; Hanna et al., 2019; Inoue et al., 2017a; Matoba et al., 2018). Non-canonical imprinting by H3K27me3 appears to mostly function in placental development and regulates genes with critical functions in this lineage (Hanna and Kelsey, 2021). This surprising alternative strategy implies that mammals may use other mechanisms beyond DNA methylation to instruct parent-specific regulation. Oocyte-specific H3K27me3 also serves as a maternal imprint for the lncRNA Xist, which triggers paternal X chromosome inactivation (XCI) in mouse preimplantation embryos and extraembryonic tissues (Inoue et al., 2017b). Although similar to other methods of H3K27-based imprinting, X inactivation is capable of deploying local Xist expression status to suppress an entire chromosome and does so in combination with several classic epigenetic suppression pathways (Żylicz and Heard, 2020). Failure to establish or maintain either XCI or genomic imprinting results in embryonic lethality, emphasizing the developmental importance of these interrelated processes (Marahrens et al., 1997; Monk et al., 2019). However, the molecular relationship between imprinted XCI and other H3K27me3 regulated loci, or what distinguishes these strategies from canonical, DNA methylation-based imprinting, remains unresolved.
To compare and contrast the developmental roles of epigenetic regulators in these processes, we investigated allele-specific expression in early post-implantation mouse embryos generated from polymorphic crosses. We specifically examine transcriptomic information derived from cohorts of matched embryonic day (E)6.5 epiblast and extraembryonic ectoderm (ExE). With this data, we comprehensively mapped imprinted genes as well as genes that escape imprinting on the inactive X chromosome in ExE. Then, we systematically perturbed epigenetic pathways, including DNA methylation, Polycomb-based repression, and Histone 3 Lysine 9 methylation (H3K9me), to understand their primary contributions to parent-specific expression. We find that DNA methylation primarily functions at previously described canonical Imprint Control Regions (ICRs) within both lineages, whereas the early placenta exhibits a greater diversity of imprinting mechanisms. For example, we find that G9a recognizes and suppresses ERV-driven promoters exclusively at maternal loci, presumably through H3K9 methylation. Furthermore, our observations pinpoint explicit X chromosomal territories that depend on the Polycomb repressive complexes (PRC 1 and/or 2 (Almeida et al., 2020; Cerase et al., 2015)) rather than on Xist recruitment alone and suggest independent functions for PRC2 in Xist imprinting and chromosome-wide silencing. Finally, our data set enabled us to inventory parent-specific expression signatures as they depend on epigenetic pathways.
RESULTS
Capturing allele-specific expression in the early embryo and placenta
To explore parent-specific expression within the early embryonic and extraembryonic lineages, we conducted reciprocal crosses between CAST/EiJ (CAST) and B6D2F1/J (BDF1) strain animals, isolated E6.5-stage epiblast (Epiblast) and extraembryonic ectoderm (ExE), and performed RNA-seq (Figure 1A, Table S1 sheet A). Our RNA-seq analysis considers long polyadenylated transcripts and excludes many non-coding small RNA classes, such as mature miRNAs, that also exhibit imprinting behaviors. Nonetheless, we are able to assess the allelic state of unprocessed host transcripts using our current method. On average, we observe allele-specific expression for 9,271 and 9,226 genes for the Epiblast and ExE, respectively (29.8% and 29.6% of 31,159 genes with at least one detectable SNP and ≥10 overlapping reads, with variation between 9,001–9,810 and 8,874–9,659 genes per replicate, see STAR Methods).
Figure 1. An inventory of parental-specific expression from single mouse embryos.
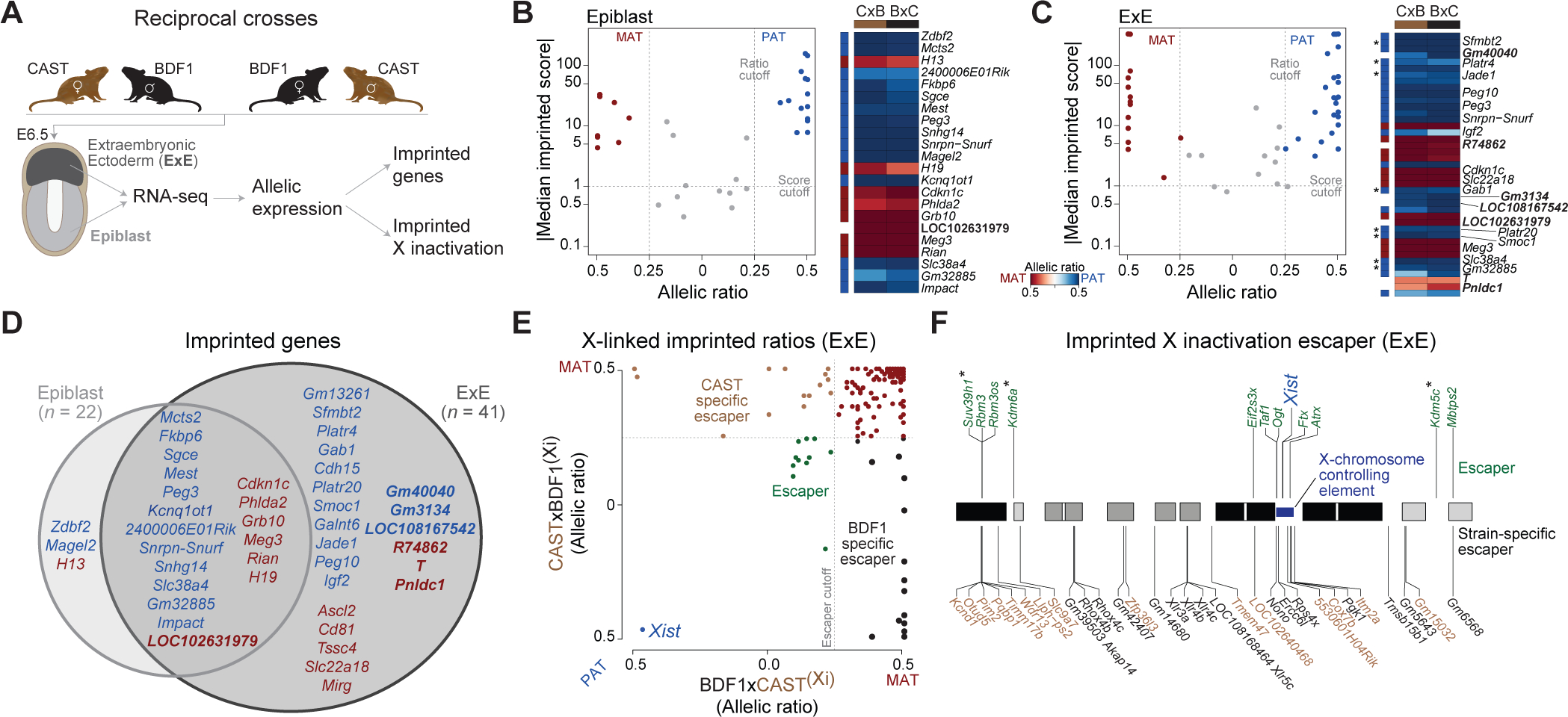
(A) Simplified schematic of our experimental system to obtain parent-specific expression landscapes (imprinting and X inactivation). E6.5 epiblast (light grey) and extraembryonic ectoderm (ExE, dark grey) are isolated from F1 reciprocal crosses (n = 11; 7 BDF1xCAST, 4m/3f and 4 CASTxBDF1, 1m/3f) and subjected to RNA-seq. Colors are used in figures throughout the manuscript to highlight the tissue of origin. (B-C) Imprinted genes identified in the E6.5 Epiblast and ExE (red: maternally expressed, blue: paternally expressed) using a median imprinted score cutoff and allelic ratios of 1 and 0.25, respectively (dashed lines). Corresponding heatmaps show the allelic ratios in the forward (BDF1xCAST) and reverse (CASTxBDF1) crosses. Allelic ratios are adjusted from an initial range of 0 to 1 such that 0 corresponds to equivalent expression between both alleles: 0 = Biallelic, 0.5 = 100% expressed from one allele (MAT (maternal), PAT (paternal), BDF1, or CAST). Previously uncharacterized imprinted genes are indicated in white, including maternal-specific expression of Brachyury (T) within the ExE. A more detailed explanation of the allelic ratio calculation is provided in the Methods. Asterisk indicates known non-canonical imprinting genes (see Table S1 sheet E)
(D) Overlap of imprinted genes between E6.5 Epiblast and ExE (red: maternally expressed, blue: paternal expressed).
(E) Scatter plot showing the allelic ratios for X-linked genes in ExE between forward (BDF1xCAST) and reverse (CASTxBDF1) crosses. Maternally expressed genes (red), XCI escaper genes (green), and strain-specific escape from the CAST (brown) and BDF1 (black) inactive X chromosome are indicated. The dashed line indicates the 0.25 allelic ratio threshold used to determine escaper genes.
(F) Chromosomal overview of genes that maintain biallelic expression on the Xi (“escapers”) including their shared or strain-specific status. Asterisk indicates escaper genes that function as chromatin modifiers.
Unsupervised clustering of gene expression confirmed epiblast and ExE lineage identity and our sample purity (Figure S1A). Overall, allelic ratios exhibited expected patterns: biallelic expression for autosomal genes, skewed XCI that favors expression of CAST alleles in Epiblast (Calaway et al., 2013), and imprinted XCI in ExE (Figure S1B). For autosomes, we identified 22 and 41 imprinted genes in the Epiblast and ExE, respectively, with 19 shared between lineages (Figure 1B–D, Table S1 sheet B-C). Allelic ratios of identified imprinted genes were not affected by embryo sex (Figure S1C). We also confirmed the eight known non-canonical imprinting genes (Sfmbt2, Platr4, Jade1, Gab1, Platr20, Smoc1, Slc38a4 and Gm32885) in ExE (Figure 1C, Asterisks), as well as seven putative, novel imprinted genes (Epiblast n = 1 and ExE n = 6). Our newly discovered imprinted loci are located in proximity to known imprinted regions and include six imprinted lncRNAs (LOC102631979, Gm40040, R74862, Gm3134, LOC108167542, and LOC102631979) as well as maternally-dominant expression of Brachyury (T) and Pnldc1.
To explore chromosome-level regulation, we next examined X-linked gene expression in female ExE samples, which undergo imprinted XCI (Figure 1E, Table S1 sheet D). Of 335 informative X-linked genes, 290 (86.5%) are maternally expressed, 11 (3.3%) escape inactivation (‘escaper,’ excluding Xist), and 33 (9.85%) escape X inactivation in a strain-specific manner (Cast n = 15, and BDF1 n = 18). Half of escaper genes cluster in proximity to the 1.85-Mb X-chromosome controlling element defined in Chadwick et al. (Chadwick et al., 2006), while others include notable epigenetic regulators (e.g., Suv39h1, Kdm6a, Kdm5c, Figure 1F). In summary, we mapped the parental-specific transcriptional landscape in the early embryonic and extraembryonic lineage for both males and females, allowing us to examine autosomal and X-chromosome-specific imprinting.
Zygotic perturbation of epigenetic pathways
Our comprehensive map of parent-specific expression allows us to systematically investigate the roles of key epigenetic pathways, which we accomplished via Cas9-based genetic disruption in F1 zygotes (Grosswendt et al., 2020; Smith et al., 2017) (Figure 2A, see STAR Methods). We disrupted the DNA methyltransferase Dnmt1, the histone 3 lysine 9 (H3K9) methyltransferases G9a and GLP (target genes Ehmt1 and Ehmt2, double KO), as well as the PRC1 and PRC2 complexes individually (target genes: Rnf2 and Eed, respectively) in BDF1xCAST zygotes. We collected Epiblast and ExE samples from a total of 30 knockout embryos at E6.5 and performed low input RNA-seq on matched pairs (Table S1 sheet E-F). Next, we validated effective gene disruption by comparing the global and target gene expression profiles between wildtype and KO embryos (Figure S2A–C). In addition, we verified gene disruption through targeted alignment of our RNA-seq data for the well-covered targets Eed and Rnf2 (Figure S2D, see STAR Methods) and further validated a representative subset for poorly covered targets using Sanger sequencing (3 of 3 ΔDnmt1 embryos and 3 of 9 ΔG9a-GLP embryos, Figure S2E). Finally, we confirmed that the overall lineage identity of KO samples was not impaired via unsupervised clustering of autosomal expression levels as well as of epiblast and ExE specific marker genes (Figure S2F–G). Within the Epiblast and ExE clusters, we found that ΔDnmt1 and ΔG9a-GLP embryos co-cluster, as do ΔRnf2 and ΔEed, in keeping with their more overlapping functions (Figure S2F) (Auclair et al., 2016; Jiang et al., 2020).
Figure 2. Epigenetic regulation of autosomal and X chromosome-specific imprinting.
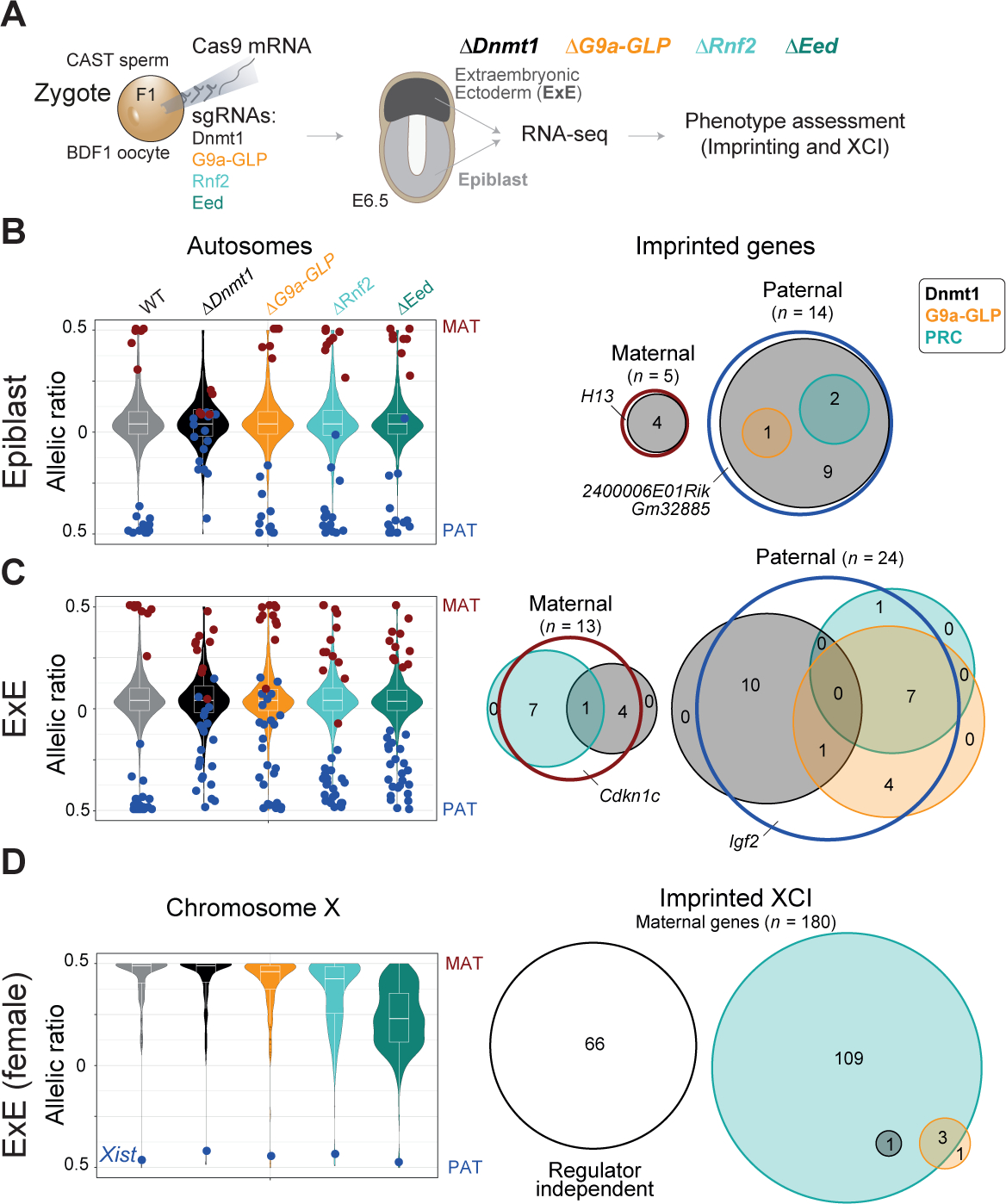
(A) Schematic overview for our strategy to assign roles for selected epigenetic regulators to parent-specific gene expression. Target epigenetic regulators are disrupted by injection of Cas9 and sgRNAs into hybrid F1 (BDF1xCAST) zygotes (ΔDnmt1 n = 3, ΔG9a-GLP n = 9, ΔRnf2 n = 8, ΔEed n = 10). E6.5 Epiblast (light grey) and extraembryonic ectoderm (ExE, dark grey) were collected from each selected embryo. Regulator colors are used throughout the rest of the manuscript: Dnmt1, black; G9a and GLP, orange, gene name Ehmt1 and Ehmt2; PRC1 member Rnf2, light turquoise; and PRC2 member Eed, turquoise.
(B) Left: Violin plots of the median allelic ratios of autosomal genes from WT and regulator mutants in Epiblast. Maternal and paternal expressed imprinted genes are indicated with red and blue dots, respectively. Right: Venn diagram shows the intersection of each epigenetic regulators’ contribution to imprint status. An epigenetic regulator was counted as relevant for silencing imprinted genes if the change in allelic ratio between WT and KO was ≥ 20%. PRC1 and PRC2 were summarized as PRC by using the higher delta. In Epiblast, DNA methylation-dependent imprinting is most frequent. Regulator independent imprinted genes have a delta allelic ratio < 20% in all disrupted regulators (see Figure S3B). Imprinted genes with less than two informative allelic ratio values in any regulator disruption data set are not shown.
(C) As in (B) for ExE. Extraembryonic imprints appear to depend on a more diverse set of regulators.
(D) Left: Violin plots displaying the median allelic ratio of X-linked genes from WT and regulator mutants in female ExE. The blue dot highlights the allelic ratio of the lncRNA Xist. Successful extraembryonic XCI depends on PRC2 and, to a lesser degree, PRC1. Notably, paternal Xist expression is largely stable in all regulator mutants examined (Figure S2H). Right: Venn diagram for imprinted XCI as shown for autosomal imprinting in (B) and (C). An epigenetic regulator was counted as relevant for silencing X-linked genes if the delta allelic ratio change between WT and KO for any regulator was ≥ 20%. Regulator independent X-linked genes have a delta allelic ratio < 20% in all disrupted regulators.
The extraembryonic lineage uses an expanded set of imprinting mechanisms
To test which epigenetic regulators are responsible for parent-specific regulation, we first examined the allelic landscape of autosomes. After excluding imprinted genes with less than two informative allelic ratios in any experimental cohort, we recovered 19 and 37 imprinted genes in Epiblast and ExE respectively (Figure S3A, bottom). We observed loss of imprinted silencing for most previously identified alleles within the ΔDnmt1 Epiblast, the majority of which are also deregulated in ExE (Figure 2B, Table S1 sheet G). In contrast, the ExE showed several other specific epigenetic dependencies on parent-of-origin expression. For example, G9a appears to repress a discrete set of maternal alleles, which also depend to a lesser degree on PRC regulators, whereas ΔRnf2 and ΔEed embryos preferentially influence the expression of paternal alleles independent of H3K9 or DNA methylation (Figure 2C, Table S1 sheet H). After examining our mutant data, only five imprinted genes (three in Epiblast and two in ExE) cannot be explained by any of these three pathways and remain imprinted across our mutant cohorts, although four of these are very near our cut-off for calling a gene as differentially regulated (Figure S3B, delta allelic ratio < 20%).
We also find that the PRCs stabilize imprinted X inactivation, with PRC2 disruption more severe than PRC1 (Figure 2D, Figure S2H). Loss of PRC2 regulation during imprinted XCI mirrors the response of PRC2-dependent imprints, with muted reactivation of the silenced locus (in this case the paternal X chromosome). Despite this chromosome-wide effect, we do not observe disrupted Xist imprinting or changes in allelic expression for ~36% of X-linked genes across all mutant regulators examined. Together, the preserved imprinting of Xist and the muted reactivation of paternal alleles suggest that PRC2 acts to stabilize subsets of Xist lncRNA target genes but does not substantially compromise the initial differentiation of this lineage, an observation we explore in greater detail below.
G9a-GLP control a discrete set of non-canonical imprinted genes
In Epiblast, the majority of imprinted genes appear to depend on DNA methylation (n = 14 out of 19 genes, a remaining 3 of which do not meet our criteria for differential allele-specific expression in any KO, Figure 3A left, Figure S3A–B). Zdbf2 and Slc38a4 are notable exceptions: Zdbf2 is reactivated in both ΔRnf2 and ΔEed embryos, whereas Slc38a4 primarily relies on G9a-GLP, in line with previous reports (Auclair et al., 2016; Greenberg et al., 2017). Notably, most epiblast-associated imprinted regions exhibit similar changes in the ExE, suggesting that they are constitutive imprints and do not depend on their respective lineage (Figure 3A right, Figure 3B, Table S1 sheet I). In contrast, 20 out of 37 informative ExE-imprinted genes are DNA methylation independent (Figure 3A right). Of these, seven maternally expressed loci predominantly rely on PRC1 and 2 to silence the paternal allele (Figure 3B top). All of these are either known targets or in proximity to the paternally expressed lncRNAs Kcnq1ot1 (Slc22a18, Ascl2, Tssc4, R74862, Cd81) and Airn (T, Phlda2). Notably, T expression has been previously observed within the early ExE prior to its canonical induction within the primitive streak of the epiblast (Rivera-Pérez and Magnuson, 2005). To contextualize our result, we reanalyzed previously published single-cell RNA-seq data of mouse gastrulation (Grosswendt et al., 2020) and find that extraembryonic T expression is transient, restricted to the earliest ExE progenitor state and downregulated during trophoblast differentiation (Figure S3C–D). The novel imprinting of T likely reflects its proximity to Airn, which is believed to recruit repressive complexes to paternal loci and expands to repress ~10 Mb of chr17 in mature placenta (Andergassen et al., 2017, 2019; Nagano et al., 2008; Schertzer et al., 2019; Terranova et al., 2008)
Figure 3. G9a controls non-canonical imprinting at endogenous retrovirus containing promoters.

(A) Allelic ratio of imprinted genes and the corresponding changes between wildtype (BDF1xCAST) and regulator disrupted embryos within the Epiblast (left) and ExE (right) lineage. Heatmap ranked by the allelic ratio change between WT and ΔDnmt1. For ExE, boxes highlight DNA methylation-independent, non-canonical expressed genes (Asterisk indicates previous description in the literature) and lncRNA directed PRC targets. Notably, ExE-specific imprinting is most apparent for a set of paternally-expressed, G9a-GLP controlled loci that only weakly depend on PRCs. Imprinted genes with less than two informative allelic ratio values in any experimental data set are not shown.
(B) Flow diagram outlining changes in the imprinted landscape between Epiblast and ExE for maternally (top) and paternally (bottom) expressed genes.
(C) Genome browser tracks for E6.5 ExE WGBS and RNA-seq data that cover two non-canonical G9a dependent imprinted clusters Jade1 (top) and Slc38a4 (bottom). Boxes highlight G9a-dependent hypomethylated DMRs (Overlapping ERV LTRs are indicated).
(D) Identified ExE DMRs using WT and ΔG9a WGBS data (1kb window, n = 3,691, |delta cutoff| ≥ 20%). Pie chart showing the proportion of hypo- and hypermethylated DMRs (top). Feature enrichment of the identified DMRs over background was calculated for intergenic, genic (±1kb of TSS), and different repeat classes using the Fisher’s exact test (bottom).
Notably, half of paternally expressed, ExE-specific imprinted genes appear to rely on G9a-GLP (n = 12 ΔG9a-GLP, n = 11 ΔDnmt1, n = 1 ΔPRC2), which include all previously defined H3K27me3-dependent non-canonical imprinted genes (Chen et al., 2019; Hanna et al., 2019; Inoue et al., 2017a) (Figure 3A right, Figure 3B bottom, Table S1 sheet J). These results suggest an interaction between H3K9 and H3K27 methylation where H3K9me may be the dominant modification for maintaining non-canonical imprinting long term. We also see some indication that these non-canonical imprints are distributed in clusters. For example, genes such as Platr4 and 2400006E01Rik are close to the known non-canonical imprinted gene Jade1, suggesting that this pathway may control a broader imprinted locus on chromosome 3 (Figure 3C top). G9a-GLP dependent cluster regulation was also observed for the non-canonical imprinted genes Slc38a4 and Gm32885 on chromosome 15 (Figure 3C bottom). Finally, we also found G9a-GLP dominant regulation of the paternally expressed Sfmbt2 gene, which spans one of the genome’s largest miRNA clusters (Inoue et al., 2020).
G9a recognizes ERV-driven promoter elements
A recent study reported that the long terminal repeats (LTR) of endogenous retroviruses (ERVs) can act as alternative promoters for non-canonical imprinted genes and found that maternally-inherited H3K27me3 transitions to DNA methylation-based silencing in extraembryonic tissues (Hanna et al., 2019). To examine the effects of G9a-GLP on these elements, we examined the DNA methylation dynamics of our G9a-sensitive genes using published whole-genome bisulfite sequencing (WGBS) data in regulator disrupted E6.5 ExE samples (ΔDnmt1, ΔRnf2, ΔEed and ΔG9a) (Grosswendt et al., 2020) as well as newly generated data for our G9a-GLP double KO (Figure 3C). Strikingly, all of our identified non-canonical imprinted genes show local loss of DNA methylation around their transcription start sites (TSSs) in ΔG9a WGBS data, but not for data generated from either PRC mutant (Figure 3C, Figure S3E). Furthermore, within these imprinted loci, every G9a-dependent differentially methylated region (DMR) overlaps an ERV LTR (Figure 3C, Figure S3E). This finding further strengthens our prior RNA-seq based observation that H3K9me appears crucial for non-canonical imprinting and suggests that G9a is necessary to recruit DNA methylation in this context. Next, we investigated whether G9a regulation of ERV-driven promoters is a general regulatory mechanism or specific for non-canonical imprinted regions. To address this question, we defined ExE DMRs using WT and ΔG9a WGBS data (see STAR Methods) and identified 3,392 hypomethylated (91.8%) and 299 hypermethylated (8.1%) DMRs (Figure 3D, Table S1 sheet K). The highest enrichment for hypomethylated DMRs over background was observed for promoters (P = 4×10−17, odds-ratio=1.5, +/− 1kb TSS) and LTRs (P = 7.9×10−50, odds-ratio=1.6). In summary, we find that G9a is the critical regulator for ERV-driven non-canonical imprinting and propose that this could represent the cooption of a more general regulatory mechanism.
Epigenetic regulation of the inactive X chromosome in females
We next leveraged our data to explore the architecture of X inactivation as it is regulated by distinct epigenetic pathways within the extraembryonic ectoderm. Out of 180 maternally expressed X-linked ExE genes (informative in all investigated ExE samples), 114 (63.4%) change their allelic expression in at least one epigenetic regulator mutant. These are almost entirely explained by PRC-based regulation, for which PRC2 is dominant: 74 X-linked genes pass our thresholds in ΔEed embryos (65%), while another 35 genes are shared between ΔRnf2 and ΔEed (30.7%) (Figure 4A). Regulator-dependent genes also show an increased expression in ΔEed embryos compared to wildtype, likely due to the activity of both alleles, whereas regulator independent genes maintain wildtype levels (Figure S4A). This synergy between PRC1 and PRC2, together with previous reports that maternal EED is sufficient to initiate and establish imprinted XCI (Harris et al., 2019), confirms that Polycomb-based repression is critical for propagating chromosome-wide epigenetic suppression.
Figure 4. Polycomb-based repression is critical to maintain the imprinted X chromosome.
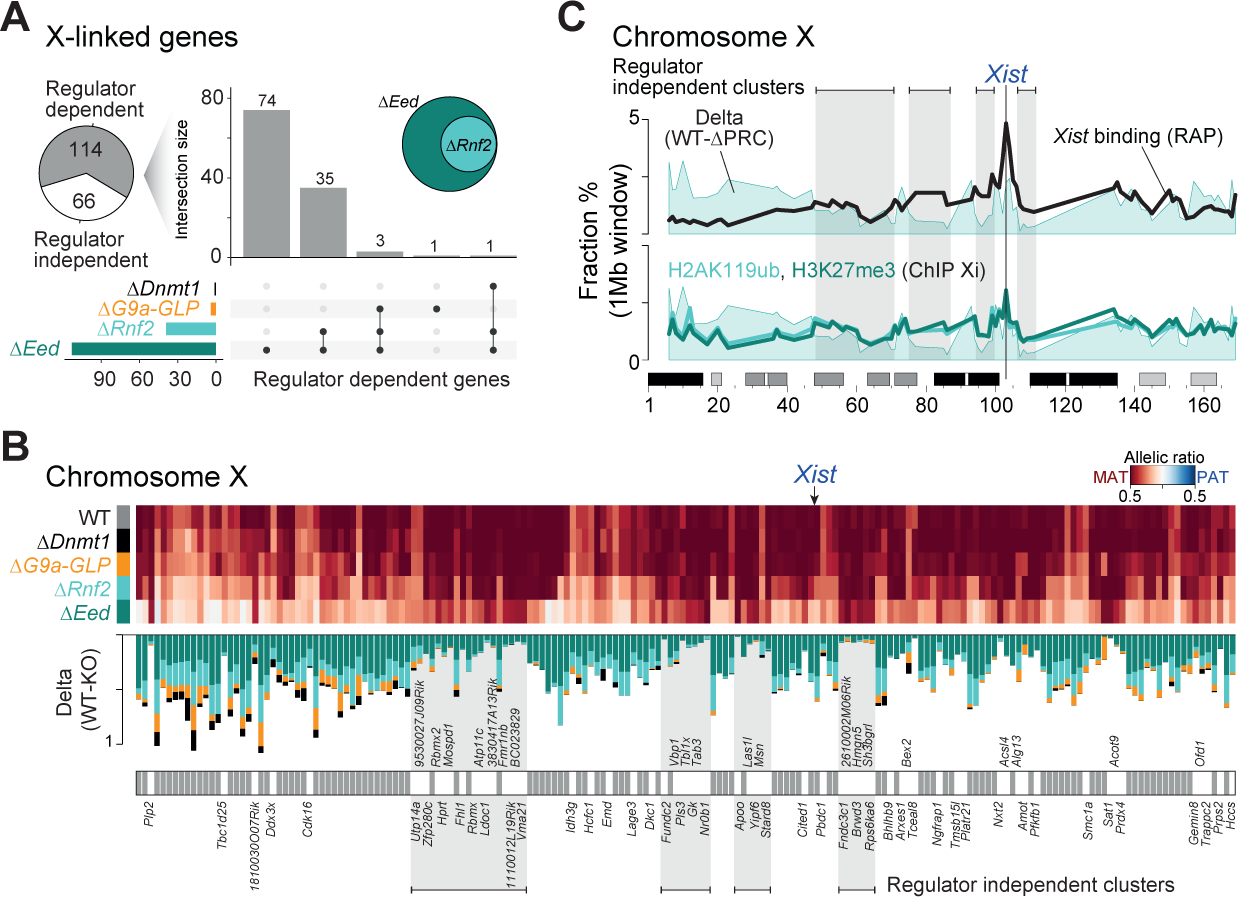
(A) Pie chart showing the proportion of regulator dependent or independent genes for imprinted XCI. A gene was called “regulator dependent” if the allelic ratio changes between WT and KO by ≥ 20% for any regulator. UpSet plot shows the intersection between the four disrupted regulator members: ΔDnmt1, ΔG9a-GLP, ΔRnf2, and ΔEed. The inset Venn diagram highlights the high overlap between ΔEed and ΔRnf2 dependent X-linked genes. Genes that escape imprinted XCI were excluded from this analysis.
(B) Regulator-independent X-linked genes are organized into distinct spatial clusters. Allelic ratios of maternally expressed X-linked genes and the corresponding changes between wildtype (BDF1xCAST) and regulator disrupted ExE lineages are shown (X-linked gene matrix ranked by the genomic position). Regulator-independent X-linked genes are indicated. A gene was called “regulator independent” if the allelic ratio changes between WT and KO is < 20% for every regulator.
(C) Regulator independent regions occur in domains with high Xist enrichment and repressive chromatin. 1Mb windows summarizing (average) paternal X expression changes between WT and PRC, for Xist enrichment over input (RAP) (Engreitz et al., 2013) and for H3K27me3 and H2AK119ub enrichment on the inactive X (Żylicz et al., 2019) across the entire X-chromosome.
To learn more about the remaining 66 regulator independent genes (36.6%), we plotted allelic ratios according to their genomic position across the X chromosome. PRC-independent genes exist in defined clusters and appear to be independent of their WT expression levels (Figure 4B, Figure S4B). We therefore hypothesized that these clusters might represent initial Xist target loci, for which imprinted Xist expression (and localization) is sufficient to maintain paternal silencing. Specifically, we cross-referenced our results against Xist RNA Antisense Purification (RAP) data, a method that maps lncRNA interactions with chromatin (Engreitz et al., 2013) as well as H3K27me3 and H2AK119ub ChIP-seq specific for the inactive X chromosome (Żylicz et al., 2019). PRC-independent loci exhibit strong enrichment for Xist binding as well as for H3K27me3 and H2AK119ub, whereas PRC2-dependent loci are comparatively Xist depleted (Figure 4C). The striking overlap between Xist and PRC-independent silencing suggest that the primary mechanisms of Xist recruitment persist in the absence of Polycomb, but chromosome-wide dispersal of this signal is compromised. Taken together, our findings highlight a central, albeit likely secondary, role for PRC1 and 2 in translating local Xist recruitment to stably silence the surrounding area.
Three distinct mechanisms for encoding parental-specific gene regulation
Our results enable an expansive inventory of genes that exhibit parent-specific expression in the early embryo and placenta according to their dependencies on key epigenetic pathways. From our perturbation data, we are able to assign the regulation of imprinted regions in Epiblast (n = 13) and ExE (n = 20) to one of three distinct epigenetic mechanisms (Figure 5A). First, known canonical ICRs are established and maintained by germline DMRs. Second, paternally unmethylated ICRs enable lncRNA transcription, which then recruits repressive machinery to distal target genes in cis. Third, a G9a-dominant mechanism controls ERV-driven, non-canonical imprinting in extraembryonic tissues at genes that are often clustered together. Of the thirteen regulator-dependent imprinted regions in the embryonic lineage, all are associated with gDMRs and primarily represent ICRs (Figure 5B): eleven regions are directly linked to gDMRs, while two regions, Zdbf2 and Slc38a4, translate gDMR information in cis via PRC2 with G9a acting as a secondary mechanism (Auclair et al., 2016; Greenberg et al., 2017). Lastly, the Kcnq1 region directs DNMTs via the lncRNA Kcnq1ot1 to silence the distant gene Phlda2, in line with a previous report (Mohammad et al., 2010).
Figure 5. Three distinct epigenetic mechanisms for controlling parent-specific gene regulation.

(A) Illustration of identified mechanisms for parental-specific gene regulation: Imprinted control region (ICR), ICR-directed lncRNA deployment, and non-canonical G9a dominant.
(B) Ideograms of mouse chromosomes show the position of epiblast-specific imprinted genes. Pie charts on the top highlight the proportion of the allelic ratio change between WT and ΔDnmt1, ΔG9a-GLP, and ΔPRC samples. The circle size denotes the combined delta change. The symbols below each imprinted region highlight the mechanisms defined in (A).
(C) Ideograms as in (B) for the ExE lineage. Question marks (?) highlight speculative mechanisms as informed by our data and the literature. One region includes the solo imprinted gene Glant6, which seems to depend equally on all three regulators. The second region harbors the two maternally expressed genes (T and Pnldc1) that are in proximity to the Igf2r region and thus likely targets of the paternally expressed lncRNA Airn.
(D) Summary of X-linked regulator dependent and independent genes, as well as genes that escape the process of imprinted XCI. The model on the right illustrates the mechanism for how the inactive X is maintained in a silent state by Xist and Polycomb.
The extraembryonic lineage shares regulation for the majority of these ICR-regulated regions (n = 11) but includes additional ICR-controlled lncRNA clusters, as well as seven non-canonical G9a-regulated imprinted regions (Figure 5C). Two lncRNA controlled clusters are regulated by Kcnq1ot1 and Airn and incorporate multiple secondary targets, all located in previously defined extraembryonic-specific silencing domains that largely depend on PRC2 and 1 to translate ICR status across expanded territories (Andergassen et al., 2017, 2019; Pandey et al., 2008; Schertzer et al., 2019; Terranova et al., 2008; Wagschal et al., 2008). Non-canonical G9a-dominant imprinted regions also tend to lie within defined clusters, such as the Smbt2, Jade1, and Slc38a4 imprinted regions and involve the regulation of ERV-driven gene loci (Figure 5C). Finally, our data provide an improved understanding of imprinted XCI, including Xist-based PRC2 recruitment that is most important for maintaining suppression beyond Xist’s primary chromosomal targets (Figure 5D).
DISCUSSION
In this study, we systematically perturbed multiple repressive pathways in highly polymorphic embryos to investigate the scope of epigenetically maintained parental imprinting. We find that most canonically imprinted genes are shared between the embryonic and extraembryonic lineages. In contrast, non-canonical imprinting is largely restricted to the placenta. For instance, we identify an H3K9 methylation-based mechanism that suppresses maternal ERV-driven promoters, which could also represent a more general strategy for gene regulation outside of imprinting. Previously, oocyte-specific H3K27me3 was described as a mechanism for non-canonical imprinting within preimplantation embryos (Inoue et al., 2017a). However, our investigation finds only a slight effect in zygotic PRC2 mutants on the allelic ratios of these targets, but complete loss of imprinting and associated reactivation in G9a mutants. These results suggest that H3K9me is critical for non-canonical imprinting, at least from implantation forward.
We also provide mechanistic insights of long-range silencing by imprinted lncRNAs in extraembryonic lineages. Previous reports observed that Kcnq1ot1 and Airn are expressed from the zygote stage and deployed to locally recruit PRCs (Andergassen et al., 2019; Schertzer et al., 2019; Terranova et al., 2008). Notably, we find that ΔDnmt1 embryos activate the silent Kcnq1ot1 allele but only show minor allelic expression changes of secondary PRC2-dependent targets. We do observe significant downregulation of several Kcnq1ot1 targets, suggesting that the reactivated lncRNA can suppress the targets prematurely and outside of their natural developmental window. Finally, Kcnq1ot1 targets in PRC mutants show biallelic expression, hinting that oocyte-specific PRC may be sufficient to initiate imprinted silencing, but zygotic PRC is required to maintain it. A similar mechanism may apply for our discovered imprinted maternal expression of Pnldc1 and T that both lie within Airn’s silencing domain, although Airn itself is only lowly expressed during this developmental period (Andergassen et al., 2017, 2019; Schertzer et al., 2019). The finding that T, a noted master regulator of the primitive streak, is transiently imprinted in placental progenitors also warrants deeper investigation, particularly given the extensive variability and incomplete penetrance of many T mutant alleles (Kispert and Herrmann, 1994).
Our approach simultaneously allowed us to shed light on XCI maintenance in vivo. We elucidated both PRCs as critical factors that maintain portions of the inactive X chromosome in a repressed state, while perturbing DNA methylation or H3K9me pathways had no impact. Finally, we identified PRC-independent gene clusters that resemble the early binding sites of Xist-mediated suppression. Different territories on ChrX appear to either depend on direct Xist binding or propagate this local cue to distal areas. Because H3K27me3 and H2AK119ub are distributed across both primary and secondary Xist targets, our data support the role of PRCs in translating primary Xist recruitment to chromosome-wide epigenetic silencing, as opposed to the establishment of these territories de novo. This model would be consistent with observations linking XCI to primary interactions between Xist, the repressor SPEN, and other major epigenetic suppressors beyond Polycomb (Chu et al., 2015; Dossin et al., 2020; McHugh et al., 2015; Minajigi et al., 2015; Monfort et al., 2015). However, it remains unclear if the independent nature of primary Xist targets would persist further into development or erode without auxiliary PRC activity.
Together, our data provide a comprehensive inventory of the epigenetic mechanisms of parent-specific imprinting, which is also fundamental for many X-linked diseases and imprinting disorders where unlocking the silent healthy allele presents an attractive therapeutic strategy. Moreover, our study provides a platform for future investigation into the molecular genetics of parental imprinting and X inactivation by combining zygotic genome perturbation with polymorphic strains. For example, we establish that non-canonical imprinting requires G9a to maintain maternal silencing of ERV promoter-containing genes but do not yet understand the nature of imprint establishment in the maternal germline. G9a’s global effect on genome regulation is more subtle than has been typically observed for other key factors associated with H3K9 methylation, such as TRIM28/KAP1 or SETDB1, which both exhibit peri-implantation knockout lethality and substantial deregulation of multiple ERV classes (Dodge et al., 2004; Karimi et al., 2011; Rowe et al., 2010; Shibata et al., 2011). Notably, these alternate K9-methylation-based regulators have also been associated with imprint maintenance of classical ICRs through Kruppel-Associated Box Zinc-Finger Proteins (KRAB-ZFPs) such as ZFP57 (Li et al., 2008; Messerschmidt et al., 2012). Our finding that G9a is alternatively associated with imprinting of several discrete ERV subfamilies also agrees with comparative analyses in mouse embryonic stem cells and early embryos that show a more muted response to G9a knockout compared to those of the DNMTs, TRIM28, or SETDB1 (Maksakova et al., 2013; Tachibana et al., 2002; Wagschal et al., 2008; Zylicz et al., 2015). Whether or not the ERV classes associated with G9a-based imprinting utilize a similar interface with either SETDB1, TRIM28, or novel KRAB-ZFPs remains to be explored.
Conditional knockouts in the female germline will further expand our understanding of the relationship between the epigenetic machinery that encodes these imprints and those that interpret them to facilitate maintenance. For example, recent investigations of non-canonical, histone-dependent imprinting have found that maternal H2AK119ub1 recruits zygotic H3K27me3 after fertilization, suggesting multiple routes to relay parent-specific epigenetic information into offspring (Chen et al., 2021; Mei et al., 2021). Similarly, our PRC mutants exhibit limited reactivation of autosomal imprints or of paternal X reactivation, with only some leaky expression, whereas Dnmt1 and G9a mutations have little or no effect. It is possible that these regulators provide additional levels of repression that can only be observed when PRCs are also absent.
Upon completion of this work, it appears that very few, if any, parent-specific allelic expression cannot be explained by one of the three reported mechanisms described above. However, little is known about how these non-canonical imprints are innovated over evolutionary time or the degree to which they are conserved across eutherian mammals in comparison to classic ICRs. Moreover, their striking enrichment in the placental lineage further highlights this tissue as a domain for expanded epigenetic innovation in mammals. Future studies in these areas will provide greater clarity for the roles these additional imprinting mechanisms play in supporting fetal development.
Limitations of the study
Our zygotic genome perturbation strategy allowed us to investigate and assign the mechanisms that maintain parent-specific imprinting genome-wide. However, a few points should be considered in this context. First, our ability to detect imprinted genes is based upon the prevalence and density of discrete polymorphisms that distinguish the CAST/EiJ father and BDF1 mother in our RNA-seq data, which only allowed us to examine an average of 9,271 and 9,226 out of 31,159 available genes for the Epiblast and ExE, respectively. Although the total number of genes available for SNP-based comparison in this cross is high (86.9% of 35,855 total gene annotations), our analysis was limited to genes with high expression in the earliest progenitors of the embryo and placenta. We cannot address the possibility that other imprinted genes and mechanisms may become more prominent later in development. Second, we also can not entirely rule out that some replicates may generates incomplete null alleles and hence create occasional hypomorphs. Our large replicate power should mitigate the risk of misinterpretation and we do observe that all our targets are significantly downregulated, implying substantial gene disruption and nonsense-mediated mRNA decay of residual mutant transcripts. Third, we are also unable to distinguish whether or not the differential effects in gene expression from similar regulator mutants, such as ΔRnf2 (PRC1) and ΔEed (PRC2), reflect differential retention of maternal factors into the blastocyst. It is possible that maternal factors might still compensate for the zygotic perturbation over a transient period. Future investigations will be required to determine the exact influence of oocyte inherited regulators in codifying parent-specific imprints to support embryonic development. Finally, we did not explore redundant or cooperative functions across multiple regulator classes and synergy between multiple epigenetic pathways may mask other forms of parental imprinting not captured here. Double or triple regulator mutant strategies would permit a quantitative investigation for each independent regulator’s contribution to maintaining parent-specific imprinting.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to by the Lead Contact, Alexander Meissner (meissner@molgen.mpg.de).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
All datasets have been deposited in the Gene Expression Omnibus (GEO) and are accessible under GSE171206. Previously published data used in this study include WGBS data and single-cell RNA sequencing data from GSE137337.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
We used 8 week-old male CAST/EiJ (CAST) as sperm donors for ICSI, 6–12 week old B6D2F1/J (BDF1) female mice were purchased from the Jackson Laboratory to provide oocytes. 22–24g female CD-1 mice were purchased from Charles River Laboratory and used as pseudopregnant donors, and Swiss-Webster vasectomized males (9 weeks old) were purchased from Taconic Biosciences for mating to CD-1 females. All procedures were performed in our specialized specific-pathogen-free facility, followed all relevant animal welfare guidelines and regulations, kept on a twelve hour light dark cycle from 6 am to 6 pm, and provided a standard diet. Embryos were collected without considering sex, but individual sexes were analyzed after sequencing and showed no effects on imprinting (Figure S1C). All animal experiments were approved by Harvard University IACUC protocol (28–21).
METHOD DETAILS
Disrupting epigenetic regulators in F1 background using zygotic CRISPR–Cas9 injection
Embryos were generated as previously described (Wang et al., 2013). Briefly, BDF1 strain female mice (age 6–8 weeks, Jackson Labs) were superovulated by serial Pregnant Mare Serum Gonadotropin (5 IU per mouse, Prospec Protein Specialists) and human chorionic gonadotropin (5 IU, Millipore) injections 46 h apart. The following day, MII stage oocytes were isolated in M2 media supplemented with hyaluronidase (Millipore) and stored in 25 μl drops of pre-gassed KSOM with half-strength concentration of amino acids (Millipore) under mineral oil (Irvine Scientific). Zygotes were generated by piezo-actuated intracytoplasmic sperm injection (ICSI, see (Grosswendt et al., 2020)) using thawed CAST strain sperm in batches of 30–50 oocytes and standard micromanipulation equipment, including a Hamilton Thorne XY Infrared laser, Eppendorf Transferman NK2 and Patchman NP2 micromanipulators, and a Nikon Ti-U inverted microscope. Zygotes for whole-genome bisulfite sequencing were generated by natural mating with BDF1 males. For the reciprocal cross, BDF1 males were naturally mated to CAST females and screened for copulation plugs, after which E6.5 stage embryos were isolated accordingly, with the date of the copulation plug score day E0.5.
For zygotic disruption, pronuclear stage 3 (PN3) zygotes were injected with 200 ng μl−1 Cas9 mRNA and a 100 ng μl−1 equimolar ratio of 3–4 sgRNAs targeting different exons of an epigenetic regulator gene locus (designed using ChopChop (Labun et al., 2016) and the IDT CRISPR–Cas9 guide RNA checker, as previously described in (Smith et al., 2017)). We designed the gRNA to avoid CAST SNPs to ensure that the gRNAs are equally effective between alleles. Injections utilized the same microinjection setup and Piezo-actuated injection of front-filled 6–7 μm injection needles. At around 84 h after fertilization, cavitated blastocysts were transferred into the uterine horns of pseudopregnant CD-1 strain females (25–35g, Charles River) generated by mating with vasectomized SW strain males (Taconic), which results in a 24 h offset in gestational time to accommodate implantation, after which animals were monitored for 5 days for embryo isolation at E6.5.
Embryo isolation and library preparation
At E6.5, animals were euthanized and the uterine horns removed. Purified epiblast and ExE were isolated according to (Chenoweth and Tesar, 2010) with a few modifications. Briefly, E6.5 embryos were removed from deciduae and transferred to independent 25 μL drops of M2 media. Using a glass flame-pulled capillary, Reichert’s membrane was removed, and the embryo carefully bisected along the epiblast, ExE boundary. Then, each Epiblast/ExE pair was transferred into an individual drop of dissociation medium containing 0.5% trypsin and 2.5% pancreatin in PBS (w/v, Sigma). Embryos were cultured with slow orbital rotation at 4°C for 15 minutes, after which they were transferred into new M2 drops. After ~5 minutes of resting, Epiblast and ExE were passed through a slightly narrower flame-pulled glass capillary to remove the visceral endoderm without disrupting the target tissue. Finally, each tissue was serially washed in 0.1% BSA in PBS prior to snap freezing in lysis buffer.
Approximately 300–600 cells were collected from E6.5 embryos and directly transferred to 2.6 μl of Lysis Buffer (Takara Bio USA, Inc.) followed by snap-freezing at −80°C in preparation for cDNA synthesis using the SMART-Seq v4 assay. Full-length cDNA was prepared using the SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing and sequencing libraries prepared using the Nextera XT DNA library preparation kit (Illumina). The resulting libraries were evaluated using a 4200 TapeStation (Agilent Technologies) and quantified by qPCR. Libraries were pooled and sequenced on an Illumina NovaSeq SP or S1 flow cell using paired-end, 50 bp reads.
RNA-seq and analysis
The RNA-seq data were aligned to the mm10 reference genome using the STAR aligner (Dobin et al., 2013) (STAR version 2.5.0 c: –outFilterMultimapNmax 1). The read counts for every RefSeq isoform (RefSeq gene annotation (downloaded February 2018) were determined using the htseq-count python script (Anders et al., 2015) (version 0.6.1) and further normalized in R.
Differential expression analysis was performed with raw counts as input using the R package DESeq2 (version 1.28.1). Genes were called significant if their FDR-adjusted p-values were smaller or equal than 0.1 (Love et al., 2014).
The Allelome.PRO approach was used to calculate allele-specific expression from the RNA-seq data as outlined in (Andergassen et al., 2015). Briefly, the Allelome.PRO pipeline uses strain-specific single-nucleotide polymorphisms (SNPs) to assign RNA-seq reads to the corresponding allele in F1 crosses. To obtain allele-specific ratios and scores from a single replicate, we slightly modified the Allelome.PRO pipeline and uploaded the modified version on SourceForge (https://sourceforge.net/projects/allelomepro/). The Allelome.PRO approach requires a gene and SNP annotation:
For the gene annotation, we used RefSeq, including 35,856 genes. To avoid unreliable allele-specific calls, we modified the annotation for known overlapping imprinted genes: first, we removed all the isoforms of H13, Kcnq1, and Copg2 overlapping the genes Mcts2, Kcnq1ot1, and Mest2, respectively. Second, we removed the two genes Peg3os and Gm33149, which overlap the known imprinted genes Peg3 and Gm32885, respectively. Third, we truncated the gene Tsix to avoid overlap with Xist and Gm32061 to prevent overlap with the two imprinted genes Ndn and Magel2. Finally, we assigned the gene name Snrpn-Snurf to the Snrpn and Snurf isoforms since both belong to the same gene.
To generate the SNP annotation for our F1 crosses between CAST and BDF1 (BDF1: F1 cross between C57BL/6J female x DBA/2J), we first derived 20,606,390 high confidence SNPs between CAST and C57BL/6J and 20,507,026 between CAST and DBA/2J (Keane et al., 2011). From the two SNP annotation files, we only used SNPs where the C57BL/6J allele was shared between DBA/2J (shared SNP nr: 17,967,587). Finally, we used only exonic SNPs, resulting in a final number of 1,513,184 SNPs. The Allelome.PRO “minread” parameter was set to 1 to include SNPs covered by one read. Genes with less than 10 read overlapping SNPs were assigned as non-informative.
In our setup, the Allelome.PRO pipeline provides an allelic ratio that ranges between 0 and 1 (0 = 100% expression from BDF1 allele, 1 = 100% expression from CAST allele, 0.5 = Biallelic). We deducted 1 from the allelic ratio to center biallelic expression to 0, resulting in an allelic ratio range of −0.5 (100% BDF1) and 0.5 (100% CAST). For simplification, we provided the absolute allelic ratios in the figures and added the information on whether 0.5 means BDF1, CAST, MAT (maternal), or PAT (paternal).
To capture the imprinted landscape expression in the early embryo and placenta, we used an allelic ratio cutoff of 0.25 and an imprinted score cutoff of 1 based on the Allelome.PRO parameters that we established in (Andergassen et al., 2015). This study showed that most known imprinted genes had an allelic ratio cutoff above 75%, which in our allelic ratio range represents a cutoff of 0.25 and an imprinted score above 1 (calculated based on a mock analysis and an FDR of 1%)
T expression in WT (E6.5 - E8.5)
We used our previously published WT mouse gastrulation reference single-cell RNAseq data (Grosswendt et al., 2020) to analyze the expression of T (Brachyury) across time within the embryonic tissues and the extraembryonic ectoderm. UMAPs for cell state distribution and stage information are the same as for our previous publication. For T specific visualization, the Seurat function ‘FeaturePlot’ was used with parameters ‘order=T’ and ‘min.cutoff=0’ to create the T expression UMAP. To obtain the percent T positive cells per embryo we set the expression levels of T to 1 if expressed at all and used the Seurat function ‘AverageExpression’ to calculate the percent positive cells per embryo (across all cells that were assigned to embryonic or extraembryonic ectoderm cell states). The ggplot package was used for visualization.
Cutsite analysis
Cutsite analysis was done as previously published in (Grosswendt et al., 2020). Briefly, single reads covering the targeted genes were extracted from the initial alignment and were realigned against the intron-free cDNA sequence of the respective gene using STAR with default settings and ‘--alignEndsType EndToEnd --outSAMattributes NH HI NM MD’. The aligned reads were next classified with respect to the target site of the sgRNA as: (1) ‘spliced/deleted’ if they did not match any nucleotide but were spanning across the entire target site, (2) ‘mismatched’ if any of the nucleotides were aligned as a mismatch/deletion/insertion to the reference, (3) ‘complete’ if all nucleotides matched the target site. Reads that did not span the full target site were scored as uninformative and removed from the analysis.
Whole-genome bisulfite sequencing (WGBS) analysis
The E6.5 ExE ΔG9a-GLP sample was isolated and processed into WGBS libraries using the Accel-NGS Methyl-seq kit as described in (Grosswendt et al., 2020). We utilized our previously generated ExE WT (n = 2), ΔG9a (n = 1) samples (Grosswendt et al., 2020) together with the generated ΔG9a-GLP sample to define G9a specific differentially methylated regions (DMRs) in E6.5 ExE. CpGs with less than ten reads were removed for the downstream analysis. CpGs were binned over a 1kb window, filtering out all windows with less than 10 CpG. Window methylation levels were combined using the average. Next, we averaged WT and G9a knockout (ΔG9a and ΔG9a-GLP) methylation levels and calculated the delta (WT-KO). Differentially methylated regions between WT and KO samples were defined by having a minimum difference of 0.2 (|delta cutoff| ≥ 20%). Feature enrichment of the identified DMRs over background was calculated for intergenic DMRs, genic (±1kb of TSS, RefSeq annotation), and different repeat classes (RepeatMasker tracks, UCSC) using the Fisher’s exact test.
QUANTIFICATION AND STATISTICAL ANALYSIS
In the manuscript, n represents the number of independent biological replicates, defined as tissue derived from different individuals, as detailed in the main text, figures, figure legends, and methods. Statistical analysis was performed using R Statistical Software (R version 4.0.3). Feature enrichment analysis of the identified DMRs over background was calculated using Fisher’s exact test. All statistical details of experiments are described in the figure legends and the METHOD DETAILS section.
Supplementary Material
Table S1. RNA and whole-genome bisulfite sequencing analysis, Table S1 sheet A-D is related to Figure 1, and Table S1 sheet E-K is related to Figure 2–3.
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| M2 medium | Millipore Sigma | Cat. # MR-015-D |
| EmbryoMax® KSOM Medium (1X) w/1/2 Amino Acids | Millipore Sigma | Cat. # MR-106-D |
| Trypsin from bovine pancreas | Sigma Aldrich | Cat. # T9935-50MG |
| Mineral Oil for Embryo Culture | Irvine Scientific | Cat. # 9305-500 mL |
| Pancreatin from porcine pancreas | Sigma Aldrich | Cat. # P3292-25G |
| Bovine Serum Albumin | Sigma Aldrich | Cat. # B6917-100MG |
| Gibco® PBS, pH 7.4 | Thermo Fisher | Cat. #: 10010049 |
| Pregnant Mare Serum Gonadotropin (1000 IU) | Prospec Protein Specialists | Cat. # HOR-272 |
| Human Chorionic Gonadotropin | Millipore Sigma | Cat. # C1063-1VL |
| mMESSAGE mMACHINE® T7 ULTRA Kit | Thermo Fisher | Cat. # AM1345 |
| MEGAshortscript™ T7 Transcription Kit | Thermo Fisher | Cat. # AM1354 |
| RNA Clean & Concentrator™Kit - 25 | Zymo Research | Cat. # R1017 |
| Critical commercial assays | ||
| SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing | Takara Bio USA, Inc. | Cat. # 634890 |
| Nextera XT DNA | Illumina | Cat. # FC-131-1024 |
| Accel-NGS Methyl-seq kit | Swift Biosciences | Cat. # 30096 |
| Deposited data | ||
| RNA-seq | This paper | GEO: GSE171206 |
| WGBS data and single-cell RNA sequencing data | Grosswendt et al., 2020 | GEO: GSE137337 |
| Experimental models: Organisms/strains | ||
| B6D2F1/J | The Jackson Laboratory | Strain Code: 100006 |
| CAST/EiJ | The Jackson Laboratory | Strain Code: 000928 |
| Crl:CD1(ICR) | Charles River | Strain Code: 022 |
| Vasectomized Swiss-Webster Male Mice | Taconic | Strain Code: SW-M-Vasec |
| Oligonucleotides | ||
| T7_Cas9_F AGTCAGTTAATACGACTCACTATAGCCACCATGGACTATAAGGACCAC |
Smith et al., 2017 | Genewiz |
| T7_Cas9_R GAGGCTGATCAGCGAGCTCTAGGAATTC |
Smith et al., 2017 | Genewiz |
| T7_sgRNA_F AGTCAGTTAATACGACTCACTATAGN20GTTTTAGAGCTAGAAATAGCAAG |
Smith et al., 2017 | Genewiz |
| T7_sgRNA_R AAAAAAAGCACCGACTCGGTGCCAC |
Smith et al., 2017 | Genewiz |
| gRNAs See Table S1 sheet E for protospacer sequences |
Smith et al., 2017, Grosswendt et al., 2020 | Genewiz |
| Recombinant DNA | ||
| pX330-U6-Chimeric_BB-CBh-hSpCas9 | Addgene | Cat. # 44230 |
| Software and algorithms | ||
| Allelome.PRO | Andergassen et al., 2015 | https://sourceforge.net/projects/allelomepro/ |
| STAR aligner | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Integrative Genome Viewer | Robinson et al., 2011 | https://software.broadinstitute.org/software/igv/ |
| htseq-count | Anders et al., 2015 | https://htseq.readthedocs.io/en/release_0.11.1/count.html |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| ChopChop | Labun et al., 2016 | https://chopchop.cbu.uib.no/ |
| BioRender | BioRender | BioRender.com |
Highlights.
Comprehensive inventory of epigenetic mechanisms that regulate parent-specific imprints
Extraembryonic ectoderm uses broader array of imprinting mechanisms than the epiblast
G9a-mediated pathway controls ERV-driven promoters of non-canonical imprinted genes
Polycomb silences secondary X chromosome targets in the extraembryonic ectoderm
Andergassen, Smith et al. generate zygotic mutants to investigate epigenetic regulation of parent-specific imprinting in early mouse embryos. The study assigns imprinted genes to three key mechanisms, two of which are more abundant in the placenta. Furthermore, it sheds light on how imprinted X chromosome inactivation is coordinated in vivo.
ACKNOWLEDGEMENTS
RNA sequencing and library preparation were performed at the Bauer Core Facility at Harvard University. AM was supported by NIH grants (DP3K111898 and P01GM099117) and the Max Planck Society.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Almeida M, Bowness JS, and Brockdorff N (2020). The many faces of Polycomb regulation by RNA. Curr. Opin. Genet. Dev. 61, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andergassen D, Dotter CP, Kulinski TM, Guenzl PM, Bammer PC, Barlow DP, Pauler FM, and Hudson QJ (2015). Allelome.PRO, a pipeline to define allele-specific genomic features from high-throughput sequencing data. Nucleic Acids Res. 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andergassen D, Dotter CP, Wenzel D, Sigl V, Bammer PC, Muckenhuber M, Mayer D, Kulinski TM, Theussl HC, Penninger JM, et al. (2017). Mapping the mouse Allelome reveals tissue-specific regulation of allelic expression. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andergassen D, Muckenhuber M, Bammer PC, Kulinski TM, Theussl HC, Shimizu T, Penninger JM, Pauler FM, and Hudson QJ (2019). The Airn lncRNA does not require any DNA elements within its locus to silence distant imprinted genes. PLoS Genet. 15, e1008268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auclair G, Borgel J, Sanz LA, Vallet J, Guibert S, Dumas M, Cavelier P, Girardot M, Forné T, Feil R, et al. (2016). EHMT2 directs DNA methylation for efficient gene silencing in mouse embryos. Genome Res. 26, 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow DP (2011). Genomic Imprinting: A Mammalian Epigenetic Discovery Model. 10.1146/Annurev-Genet-110410-132459 45, 379–403. [DOI] [PubMed] [Google Scholar]
- Barlow DP, and Bartolomei MS (2014). Genomic imprinting in mammals. Cold Spring Harb. Perspect. Biol. 6, a018382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow DP, Stöger R, Herrmann BG, Saito K, and Schweifer N (1991). The mouse insulin-like growth factor type-2 receptor is imprinted and closely linked to the Tme locus. Nature 349, 84–87. [DOI] [PubMed] [Google Scholar]
- Bartolomei MS, Zemel S, and Tilghman SM (1991). Parental imprinting of the mouse H19 gene. Nat. 1991 3516322 351, 153–155. [DOI] [PubMed] [Google Scholar]
- Calaway JD, Lenarcic AB, Didion JP, Wang JR, Searle JB, McMillan L, Valdar W, and Pardo-Manuel de Villena F (2013). Genetic Architecture of Skewed X Inactivation in the Laboratory Mouse. PLoS Genet. 9, e1003853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattanach BM, and Kirk M (1985). Differential activity of maternally and paternally derived chromosome regions in mice. Nat. 1985 3156019 315, 496–498. [DOI] [PubMed] [Google Scholar]
- Cerase A, Pintacuda G, Tattermusch A, and Avner P (2015). Xist localization and function: new insights from multiple levels. Genome Biol. 2015 161 16, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick LH, Pertz LM, Broman KW, Bartolomei MS, and Willard HF (2006). Genetic control of X chromosome inactivation in mice: Definition of the Xce candidate interval. Genetics 173, 2103–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Yin Q, Inoue A, Zhang C, and Zhang Y (2019). Allelic H3K27me3 to allelic DNA methylation switch maintains noncanonical imprinting in extraembryonic cells. Sci. Adv. 5, eaay7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Djekidel MN, and Zhang Y (2021). Distinct dynamics and functions of H2AK119ub1 and H3K27me3 in mouse preimplantation embryos. Nat. Genet. 2021 534 53, 551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenoweth JG, and Tesar PJ (2010). Isolation and maintenance of mouse epiblast stem cells. Methods Mol. Biol. 636, 25–44. [DOI] [PubMed] [Google Scholar]
- Chu C, Zhang QC, da Rocha ST, Flynn RA, Bharadwaj M, Calabrese JM, Magnuson T, Heard E, and Chang HY (2015). Systematic discovery of Xist RNA binding proteins. Cell 161, 404–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeChiara TM, Robertson EJ, and Efstratiadis A (1991). Parental imprinting of the mouse insulin-like growth factor II gene. Cell 64, 849–859. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge JE, Kang Y-K, Beppu H, Lei H, and Li E (2004). Histone H3-K9 Methyltransferase ESET Is Essential for Early Development. Mol. Cell. Biol. 24, 2478–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dossin F, Pinheiro I, Żylicz JJ, Roensch J, Collombet S, Saux A. Le, Chelmicki T, Attia M, Kapoor V, Zhan Y, et al. (2020). SPEN integrates transcriptional and epigenetic control of X-inactivation. Nat. 2020 5787795 578, 455–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Pandya-Jones A, McDonel P, Shishkin A, Sirokman K, Surka C, Kadri S, Xing J, Goren A, Lander ES, et al. (2013). The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 341, 1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson-Smith AC, Cattanach BM, Barton SC, Beechey CV, and Surani MA (1991). Embryological and molecular investigations of parental imprinting on mouse chromosome 7. Nat. 1991 3516328 351, 667–670. [DOI] [PubMed] [Google Scholar]
- Greenberg MVC, Glaser J, Borsos M, Marjou F. El, Walter M, Teissandier A, and Bourc’his D (2017). Transient transcription in the early embryo sets an epigenetic state that programs postnatal growth. Nat. Genet. 49, 110–118. [DOI] [PubMed] [Google Scholar]
- Grosswendt S, Kretzmer H, Smith ZD, Kumar AS, Hetzel S, Wittler L, Klages S, Timmermann B, Mukherji S, and Meissner A (2020). Epigenetic regulator function through mouse gastrulation. Nature 584, 102–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna CW, and Kelsey G (2021). Features and mechanisms of canonical and noncanonical genomic imprinting. Genes Dev. 35, 821–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna CW, Pérez-Palacios R, Gahurova L, Schubert M, Krueger F, Biggins L, Andrews S, Colomé-Tatché M, Bourc’His D, Dean W, et al. (2019). Endogenous retroviral insertions drive non-canonical imprinting in extra-embryonic tissues. Genome Biol. 20, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris C, Cloutier M, Trotter M, Hinten M, Gayen S, Du Z, Xie W, and Kalantry S (2019). Conversion of random x-inactivation to imprinted x-inactivation by maternal prc2. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Jiang L, Lu F, Suzuki T, and Zhang Y (2017a). Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Jiang L, Lu F, and Zhang Y (2017b). Genomic imprinting of Xist by maternal H3K27me3. Genes Dev. 31, 1927–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Ogonuki N, Kamimura S, Inoue H, Matoba S, Hirose M, Honda A, Miura K, Hada M, Hasegawa A, et al. (2020). Loss of H3K27me3 imprinting in the Sfmbt2 miRNA cluster causes enlargement of cloned mouse placentas. Nat. Commun. 2020 111 11, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Ang JYJ, Lee AY, Cao Q, Li KY, Yip KY, and Leung DCY (2020). G9a Plays Distinct Roles in Maintaining DNA Methylation, Retrotransposon Silencing, and Chromatin Looping. Cell Rep. 33, 108315. [DOI] [PubMed] [Google Scholar]
- Karimi MM, Goyal P, Maksakova IA, Bilenky M, Leung D, Tang JX, Shinkai Y, Mager DL, Jones S, Hirst M, et al. (2011). DNA Methylation and SETDB1/H3K9me3 Regulate Predominantly Distinct Sets of Genes, Retroelements, and Chimeric Transcripts in mESCs. Cell Stem Cell 8, 676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, et al. (2011). Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kispert A, and Herrmann BG (1994). Immunohistochemical Analysis of the Brachyury Protein in Wild-Type and Mutant Mouse Embryos. Dev. Biol. 161, 179–193. [DOI] [PubMed] [Google Scholar]
- Labun K, Montague TG, Gagnon JA, Thyme SB, and Valen E (2016). CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 44, W272–W276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Ito M, Zhou F, Youngson N, Zuo X, Leder P, and Ferguson-Smith AC (2008). A Maternal-Zygotic Effect Gene, Zfp57, Maintains Both Maternal and Paternal Imprints. Dev. Cell 15, 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksakova IA, Thompson PJ, Goyal P, Jones SJ, Singh PB, Karimi MM, and Lorincz MC (2013). Distinct roles of KAP1, HP1 and G9a/GLP in silencing of the two-cell-specific retrotransposon MERVL in mouse ES cells. Epigenetics Chromatin 2013 61 6, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini-DiNardo D, Steele SJS, Levorse JM, Ingram RS, and Tilghman SM (2006). Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 20, 1268–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marahrens Y, Panning B, Dausman J, Strauss W, and Jaenisch R (1997). Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes Dev. 11, 156–166. [DOI] [PubMed] [Google Scholar]
- Matoba S, Wang H, Jiang L, Lu F, Iwabuchi KA, Wu X, Inoue K, Yang L, Press W, Lee JT, et al. (2018). Loss of H3K27me3 Imprinting in Somatic Cell Nuclear Transfer Embryos Disrupts Post-Implantation Development. Cell Stem Cell 23, 343–354.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, and Solter D (1984). Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 37, 179–183. [DOI] [PubMed] [Google Scholar]
- McHugh CA, Chen C-K, Chow A, Surka CF, Tran C, McDonel P, Pandya-Jones A, Blanco M, Burghard C, Moradian A, et al. (2015). The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 521, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei H, Kozuka C, Hayashi R, Kumon M, Koseki H, and Inoue A (2021). H2AK119ub1 guides maternal inheritance and zygotic deposition of H3K27me3 in mouse embryos. Nat. Genet. 2021 534 53, 539–550. [DOI] [PubMed] [Google Scholar]
- Messerschmidt DM, Vries W. de, Ito M, Solter D, Ferguson-Smith A, and Knowles BB (2012). Trim28 Is Required for Epigenetic Stability During Mouse Oocyte to Embryo Transition. Science (80-. ). 335, 1499–1502. [DOI] [PubMed] [Google Scholar]
- Minajigi A, Froberg J, Wei C, Sunwoo H, Kesner B, Colognori D, Lessing D, Payer B, Boukhali M, Haas W, et al. (2015). Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad F, Mondal T, Guseva N, Pandey GK, and Kanduri C (2010). Kcnq1ot1 noncoding RNA mediates transcriptional gene silencing by interacting with Dnmt1. Development 137, 2493–2499. [DOI] [PubMed] [Google Scholar]
- Monfort A, Di Minin G, Postlmayr A, Freimann R, Arieti F, Thore S, and Wutz A (2015). Identification of Spen as a Crucial Factor for Xist Function through Forward Genetic Screening in Haploid Embryonic Stem Cells. Cell Rep. 12, 554–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk D, Mackay DJG, Eggermann T, Maher ER, and Riccio A (2019). Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat. Rev. Genet. 20, 235–248. [DOI] [PubMed] [Google Scholar]
- Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, and Fraser P (2008). The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science (80-. ). 322, 1717–1720. [DOI] [PubMed] [Google Scholar]
- Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, Nagano T, Mancini-DiNardo D, and Kanduri C (2008). Kcnq1ot1 Antisense Noncoding RNA Mediates Lineage-Specific Transcriptional Silencing through Chromatin-Level Regulation. Mol. Cell 32, 232–246. [DOI] [PubMed] [Google Scholar]
- Peters J (2014). The role of genomic imprinting in biology and disease: an expanding view. Nat. Rev. Genet. 2014 158 15, 517–530. [DOI] [PubMed] [Google Scholar]
- Rivera-Pérez JA, and Magnuson T (2005). Primitive streak formation in mice is preceded by localized activation of Brachyury and Wnt3. Dev. Biol. 288, 363–371. [DOI] [PubMed] [Google Scholar]
- Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, Maillard PV, Layard-Liesching H, Verp S, Marquis J, et al. (2010). KAP1 controls endogenous retroviruses in embryonic stem cells. Nat. 2010 4637278 463, 237–240. [DOI] [PubMed] [Google Scholar]
- Schertzer MD, Braceros KCA, Starmer J, Cherney RE, Lee DM, Salazar G, Justice M, Bischoff SR, Cowley DO, Ariel P, et al. (2019). lncRNA-Induced Spread of Polycomb Controlled by Genome Architecture, RNA Abundance, and CpG Island DNA. Mol. Cell 75, 523–537.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata M, Blauvelt KE, Liem KF, and García-García MJ (2011). TRIM28 is required by the mouse KRAB domain protein ZFP568 to control convergent extension and morphogenesis of extra-embryonic tissues. Development 138, 5333–5343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleutels F, Zwart R, and Barlow DP (2002). The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 415, 810–813. [DOI] [PubMed] [Google Scholar]
- Smith ZD, Shi J, Gu H, Donaghey J, Clement K, Cacchiarelli D, Gnirke A, Michor F, and Meissner A (2017). Epigenetic restriction of extraembryonic lineages mirrors the somatic transition to cancer. Nature 549, 543–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surani MAH, Barton SC, and Norris ML (1984). Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nat. 1984 3085959 308, 548–550. [DOI] [PubMed] [Google Scholar]
- Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H, et al. (2002). G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 16, 1779–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terranova R, Yokobayashi S, Stadler MB, Otte AP, van Lohuizen M, Orkin SH, and Peters AHFM (2008). Polycomb Group Proteins Ezh2 and Rnf2 Direct Genomic Contraction and Imprinted Repression in Early Mouse Embryos. Dev. Cell 15, 668–679. [DOI] [PubMed] [Google Scholar]
- Tucci V, Isles AR, Kelsey G, Ferguson-Smith AC, Bartolomei MS, Benvenisty N, Bourc’his D, Charalambous M, Dulac C, Feil R, et al. (2019). Genomic Imprinting and Physiological Processes in Mammals. Cell 176, 952–965. [DOI] [PubMed] [Google Scholar]
- Wagschal A, Sutherland HG, Woodfine K, Henckel A, Chebli K, Schulz R, Oakey RJ, Bickmore WA, and Feil R (2008). G9a Histone Methyltransferase Contributes to Imprinting in the Mouse Placenta. Mol. Cell. Biol. 28, 1104–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, and Jaenisch R (2013). One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Cell 153, 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson CM, Ball ST, Dawson C, Mehta S, Beechey CV, Fray M, Teboul L, Dear TN, Kelsey G, and Peters J (2011). Uncoupling Antisense-Mediated Silencing and DNA Methylation in the Imprinted Gnas Cluster. PLOS Genet. 7, e1001347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylicz JJ, Dietmann S, Günesdogan U, Hackett JA, Cougot D, Lee C, and Surani MA (2015). Chromatin dynamics and the role of G9a in gene regulation and enhancer silencing during early mouse development. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Żylicz JJ, and Heard E (2020). Molecular Mechanisms of Facultative Heterochromatin Formation: An X-Chromosome Perspective. Annu. Rev. Biochem. 89, 255–282. [DOI] [PubMed] [Google Scholar]
- Żylicz JJ, Bousard A, Žumer K, Dossin F, Mohammad E, da Rocha ST, Schwalb B, Syx L, Dingli F, Loew D, et al. (2019). The Implication of Early Chromatin Changes in X Chromosome Inactivation. Cell 176, 182–197.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. RNA and whole-genome bisulfite sequencing analysis, Table S1 sheet A-D is related to Figure 1, and Table S1 sheet E-K is related to Figure 2–3.
Data Availability Statement
All datasets have been deposited in the Gene Expression Omnibus (GEO) and are accessible under GSE171206. Previously published data used in this study include WGBS data and single-cell RNA sequencing data from GSE137337.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.