

Abstract
Human cytochrome P450 8B1 (CYP8B1) is involved in conversion of cholesterol to bile acids. It hydroxylates the steroid ring at C12 to ultimately produce the bile acid cholic acid. Studies implicated this enzyme as a good drug target for nonalcoholic fatty liver disease and type 2 diabetes, but there are no selective inhibitors known for this enzyme and no structures to guide inhibitor development. Herein, the human CYP8B1 protein was generated and used to identify and characterize interactions with a series of azole inhibitors, which tend to be poorly selective P450 inhibitors. Structurally related miconazole, econazole, and tioconazole bound with submicromolar dissociation constants and were effective inhibitors of the native reaction. CYP8B was cocrystallized with S-tioconazole to yield the first X-ray structure. This inhibitor bound in the active site with its azole nitrogen coordinating the heme iron, consistent with inhibitor binding and inhibition assay data. Additionally, the CYP8B1 active site was compared with similar P450 enzymes to identify features that may facilitate the design of more selective inhibitors. Selective inhibitors should promote a better understanding of the role of CYP8B1 inhibition in normal physiology and disease states and provide a possible treatment for nonalcoholic fatty liver disease and type 2 diabetes.
Keywords: cytochrome P450, membrane protein, CYP8B1, X-ray crystallography, NAFLD, type 2 diabetes mellitus
Abbreviations: cDNA, complementary DNA; CV, column volumes; CYP8B1, cytochrome P450 8B1; DMSO, dimethyl sulfoxide; FXR, farnesoid X nuclear receptor; LB, lysogeny broth; NAFLD, nonalcoholic fatty liver disease; PDB, Protein Data Bank; WT, wild type
Bile acids synthesized in the liver play important roles in normal human physiology and disease. The major human bile acids are cholic acid and chenodeoxycholic acid and their conjugates (1). All such bile acids are atypical nonplanar steroids and amphipathic, with their concave hydrophilic α-face dominated by hydroxyl groups and the convex hydrophobic β-face projecting methyl groups. Structural variations on this general scaffold result in various bile acids with distinct functionalities. Cholic acid differs from chenodeoxycholic acid in that cholic acid has an additional hydroxyl on the steroidal scaffold. In the classic bile acid synthesis pathway accounting for over 90% of human bile acids, cytochrome P450 8B1 (CYP8B1, sterol 12α-hydroxylase, E.C.1.14.14.139) installs this hydroxyl on carbon 12 of 7α-hydroxycholest-4-en-3-one to generate cholic acid (1). Thus, cholic acid is more polar, while chenodeoxycholic acid is more lipophilic. As a result, CYP8B1 activity controls the ratio of cholic acid to chenodeoxycholic acid and thus the overall hydrophobicity of the bile pool. This hydrophilic/lipophilic balance of bile acids has long been recognized as the major regulator of cholesterol homeostasis and intestinal fat absorption, but these bile acids also have important roles as signaling molecules (1). Bile acids specifically act on the nuclear farnesoid X receptor and the G protein–coupled bile acid receptor 1 (2). Through these pathways, bile acids regulate their own homeostasis, lipid metabolism, glucose homeostasis, and energy expenditure. These capabilities suggested modulation of the bile acid pool as a potential therapeutic strategy for nonalcoholic fatty liver disease (NAFLD) and type 2 diabetes mellitus (2).
NAFLD is a common disorder in which excess fat accumulates in hepatocytes of individuals who drink little or no alcohol (3). Associated with obesity, NAFLD can often progress to nonalcoholic steatohepatitis, fibrosis, and cirrhosis requiring liver transplant (3). In NAFLD patients, cholate-derived bile acids are much higher than chenodeoxycholate-derived bile acids (4). Cholic acid has the lowest critical micellar concentration (∼50 μM) of all bile acids and is highly efficient for mixed micelle formation with cholesterol and dietary cholesterol absorption (2). CYP8B1 inhibition would decrease that ratio of the 12α-hydroxylated or cholic acid bile acids to non-12α-hydroxylated or chenodeoxycholic acid bile acids. Therefore, a decreased level of cholic acid is expected to result in less intestinal cholesterol and fat absorption. CYP8B1 inhibition would also alter signaling through the farnesoid X nuclear receptor (FXR). Chenodeoxycholic acid is a strong FXR agonist, while cholic acid is weak (2). In NAFLD, increased chenodeoxycholic acid ratios activate FXR, upregulating genes important for glucose and triglyceride metabolism (5). Hepatic FXR activation represses gluconeogenesis and triglyceride synthesis and enhances the free fatty acid β-oxidation and triglyceride clearance (5). While a FXR agonist has reduced liver fibrosis (6) and is under phase III study in patients with nonalcoholic steatohepatitis (7), concerns have been raised about FXR effects in other tissues (8). Therefore, it seems likely that tissue-specific modulation of FXR would be desirable and CYP8B1 inhibition would accomplish this. Recent mouse Cyp8b1 knockdown studies also demonstrated significantly decreased steatosis and hepatic lipid content and a regression in hepatic steatosis (9). Taken together, CYP8B1 is a potential drug target for NAFLD.
CYP8B1 and type 2 diabetes are connected in 2 potential ways. First, one study showed that in healthy subjects and type 2 diabetic patients, increased ratio of 12α-hydroxylated bile acids over non-12α-hydroxylated bile acids is related to insulin resistance (10). Second, diabetic mice also exhibit elevated Cyp8b1 expression (11). To evaluate CYP8B1 as a potential therapeutic strategy for diabetes, knockdown studies were employed (12). In mice, Cyp8b1 knockdown resulted in increases in free fatty acids in the ileum lumen, increased release of plasma glucagon-like peptide 1 (GLP-1), and improved glucose tolerance, insulin sensitivity, and insulin secretion (12).
CYP8B1 knockdown using siRNA is a suboptimal therapeutic strategy with substantial delivery difficulties. A more viable strategy would be an orally active small molecule inhibitor selective for CYP8B1. However, no selective CYP8B1 inhibitors are currently known. Although Chung et al. recently designed and synthesized a CYP8B1 inhibitor (13), the effectiveness and selectivity of the inhibitor were not determined. Also, there are no CYP8B1 structures to guide inhibitor development and optimization, which hampers the definition of CYP8B1 roles in both normal physiology and disease states. Identification of a selective CYP8B1 inhibitor is likely to be challenging as one must consider other P450 enzymes, such as CYP8A1 and CYP7A1, with similar amino acid sequences and substrates. The work herein was initiated to support the discovery of selective CYP8B1 inhibitors. To our knowledge, this is the first report of generation of purified human CYP8B1 (14, 15, 16). This CYP8B1 bound and 12-hydroxylated its native substrate and was used to identify some tight-binding nonselective inhibitors and to determine the first X-ray structure of this critical human enzyme.
Results
Generation of recombinant CYP8B1
To generate recombinant CYP8B1, a synthetic complementary DNA (cDNA) was designed. Most microsomal human cytochrome P450 enzymes have an N-terminal transmembrane helix, and hydropathy plot analysis indicated this was likely the case for CYP8B1. As truncation of this hydrophobic sequence typically increases expression and increases solubility favorable for crystallization, the N-terminal 25 amino acids were removed and replaced by a sequence coding for MAKKTSS. Additionally, a sequence coding for a six-residue histidine tag was appended to the C terminus. The entire gene was codon optimized and subcloned into the pCWori+ vector for Escherichia coli expression. As is typical for membrane P450 enzymes, the resulting CYP8B1 protein was membrane bound, likely via a typical secondary membrane-binding site. Thus, addition of detergent was used to solubilize CYP8B1. A three-column purification protocol was developed yielding highly purified protein, as indicated by SDS-PAGE (Fig. 1A), which eluted as a monomer on size-exclusion chromatography (data not shown). The resulting CYP8B1 protein demonstrated a heme Soret peak with a maximum absorbance at 420 nm (Fig. 1, B). This absorbance profile is consistent with water coordination of the heme iron in the active site. When the protein is reduced and exposed to carbon monoxide, the difference spectrum showed much greater absorbance at 420 nm compared to the absorbance at 450 nm (Fig. 1C). This was initially concerning given that the 450 nm absorbance is correlated with a catalytically active form of the enzyme, while 420 nm is sometimes correlated with an inactive form of cytochrome P450 proteins, though this is not consistent between P450 enzymes and can be reversible (17, 18, 19). However, when substrate was added to CYP8B1, the same reduced carbon monoxide difference spectrum displayed only absorbance at 450 nm (Fig. 1D). Thus, the presence of substrate appeared to alter the enzyme heme environment toward a state that is typically active. Nonetheless, because of this result, it was particularly important to determine if the enzyme could bind ligands and perform catalysis normally.
Figure 1.

Characterization of recombinant, purifiedwild typeCYP8B1. (A), SDS-PAGE, (B) UV-visible absorbance spectrum, indicating heme incorporation with a Soret λmax at 420 nm, (C) reduced carbon monoxide difference spectrum, and (D) reduced carbon monoxide difference spectrum in the presence of 7α-hydroxycholest-4-en-3-one. CYP8B1, cytochrome P450 8B1.
CYP8B1 substrate binding
As is typical for cytochrome P450 enzymes, the ability to bind ligands was evaluated by determining shifts of the heme Soret peak from the initial value. A shift to shorter wavelengths is typically observed when ligands bind in the active site such that they displace a water from the heme iron (type I binding). Conversely, a shift to longer wavelengths is typically observed when ligands not only displace this water but have a ligand nitrogen that coordinates the heme iron (type II binding). While the former is often observed for substrates, the latter typically is consistent with inhibition since it blocks O2 binding at this site required for catalysis.
Titrations of CYP8B1 with its native substrate 7α-hydroxycholest-4-en-3-one (Fig. 2A) did yield the expected blue shift in absorbance. The difference spectra indicated decreases in absorbance at 427 nm and increasing absorbance at 392 nm (Fig. S1), consistent with a type I interaction. Plotting the changes in absorbance versus ligand concentration revealed that the dissociation constant (KD) value for this substrate was 18.0 nM (95% CI: 14.9–21.8 nM) (Fig. 2B, blue). This value is consistent with high affinity of CYP8B1 for its native substrate.
Figure 2.

Comparisons ofwild typeCYP8B1 (blue) and W281F mutant(red)function. (A), structures and names for CYP8B1 substrate and product. 7α-hydroxycholest-4-en-3-one (B) binding and (C) metabolism to 7α,12α-dihydroxycholest-4-en-3-one. D, summary of the derived parameters for wild type and W281F CYP8B1 7α-hydroxycholest-4-en-3-one binding and metabolism. CYP8B1, cytochrome P450 8B1.
CYP8B1 catalysis
CYP8B1 hydroxylates C12 of 7α-hydroxy-4-en-3-one to form 7α,12α-dihydroxycholest-4-en-3-one (Fig. 2A). The recombinant CYP8B1 herein was able to perform this reaction (Fig. 2C, blue, Fig. S2) with a Km of 0.5 μM (95% confidence interval [CI] 0.4–0.6 μM) and a kcat of 7.4 min−1 (95% CI 6.8–8.0 min−1). The resulting CYP8B1 catalytic efficiency (kcat/Km) is 14.7 μM−1min−1.
Recombinant CYP8B1 W281F substrate binding and catalysis
Tryptophan 281 in CYP8B1 may be important for the proper protein function. In CYP7A1, when the corresponding conserved active site tryptophan (W284 in substrate recognition site 4 (20) or SRS-4 in the I helix) was mutated to phenylalanine, the spectral shift in response to cholesterol binding was abolished (21). To investigate the role of W281, CYP8B1 with a W281F mutation was recombinantly generated, expressed, and purified the same way as WT CYP8B1. This CYP8B1 W281F mutant still bound the native substrate 7α-hydroxy-4-en-3-one with the same spectral shift as the WT enzyme (Fig. S1). However, compared to WT protein, the W281F mutant demonstrated both lower affinity in the form of a 2-fold higher KD value (34.3 nM (95% CI 25.1–46.8 nM)) and a decreased population of CYP8B1-binding substrate at saturation in the form of an approximately 2-fold decrease in Amax value (Fig. 2B, red; Fig. 2D).
The W281F mutant, CYP8B1 was able to convert 7α-hydroxy-4-en-3-one to 7α,12α-dihydroxy-4-en-3-one (Fig. 2C, red; Fig. S2). The Km for this reaction was 0.54 μM (95% CI 0.39–0.75 μM) (Fig. 2D), which is similar with the Km of WT CYP8B1. However, the kcat value for W281F was 1.20 min−1, which is less than one-sixth of the WT protein. The catalytic efficiency was thus correspondingly reduced more than 6-fold to 2.2 μM−1min−1.
Altogether, these results confirm that W281 is important in substrate binding and catalysis.
CYP8B1 inhibitor binding
Since specific inhibitors of CYP8B1 have the potential to be valuable clinically but are largely unknown, an initial strategy was to evaluate the ability of CYP8B1 to bind several nonspecific P450 inhibitors containing azole groups, which can often coordinate the heme iron with high affinity. A number of these are commercially available and target other P450 enzymes as clinically useful antifungals (fluconazole, econazole, clotrimazole, miconazole, tioconazole, and ketoconazole) or retinoic acid metabolism blocking agents (liarozole). While addition of the triazole-containing fluconazole did not induce a shift of the CYP8B1 Soret peak and thus may not bind, all of the others drugs aforementioned contain imidazoles and shifted the Soret peak to longer wavelengths (424.5–425.5 nm, type II binding), consistent with an interaction between the CYP8B1 active site heme Fe and the ligand nitrogen. Titrations with each compound were consistent with single site binding with dissociation constants ranging from 60 to 210 nM for econazole, miconazole, and tioconazole up to 1.3 to 1.6 μM for clotrimazole, liarozole, and ketoconazole (Table 1, Fig. S3). These results not only indicated that CYP8B1 can interact typically with type II imidazole–containing ligands but that it can do so with relatively high affinities.
Table 1.
CYP8B1 binding and inhibition by azoles
| Compound | Structure | Binding |
Inhibition |
||
|---|---|---|---|---|---|
| Shift in Soret peak | KD (μM) | Turnover at 80 μM (%) | IC50 (μM) | ||
| Miconazole | 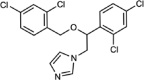 |
Type II | 0.098 (0.085–0.11) | 0.75 | 0.460 (0.315–0.629) |
| Econazole | 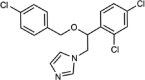 |
Type II | 0.057 (0.048–0.068) | 1.7 | 0.803 (0.663–0.963) |
| Tioconazole | 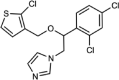 |
Type II | 0.21 (0.18–0.24) | 1.4 | 3.94 (3.13–5.06) |
| Clotrimazole | 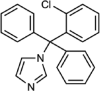 |
Type II | 1.6 (1.5–1.8) | 6.8 | 7.36 (4.83–14.7) |
| Liarozole | 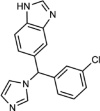 |
Type II | 1.3 (1.2–1.5) | 30 | 37.7 (25.5–61.8) |
| Ketoconazole | 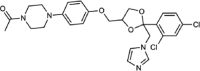 |
Type II | 1.3 (1.1–1.6) | 16 | N.D. |
| Fluconazole | 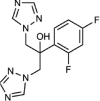 |
None | N.D | 95 | N.D. |
N.D., not determined.
CYP8B1 inhibition
To validate that the binding observed for azoles aforementioned was inhibitory, CYP8B1 was first evaluated for its ability to generate the native 7α,12α-dihydroxycholest-4-en-3-one product in the presence of a single, high (80 μM) concentration of each azole inhibitor (Table 1). The remaining activity ranged from less than 1% for miconazole to 30% for liarozole. The only azole that did not significantly inhibit CYP8B1 at this concentration was fluconazole. Most azoles showing significant inhibition at this single high concentration were then evaluated at multiple inhibitor concentrations to determine the IC50 (Fig. S4). These IC50 values roughly correlated with the KD values determined before (Table 1). Miconazole and econazole are the strongest inhibitors with submicromolar IC50 values, followed by tioconazole and clotrimazole with IC50 values under 10 μM, and liarozole near 40 μM. These results demonstrate that a range of azole inhibitors act on CYP8B1. Identifying structural features of ligands beyond the azole moiety that are compatible with unique features of the rest of CYP8B1 active site should enable the design of more selective inhibitors that might have clinical utility.
Structure of CYP8B1 cocrystallized with the nonselective inhibitor (S)-tioconazole
No structures of human CYP8B1 or close orthologs in other species to guide more selective inhibitor development, but crystallization and structure determination of such membrane P450 enzymes are often facilitated by cocrystallization with high affinity ligands. Thus, structure determination was pursued with the high affinity azoles identified herein. Screening revealed that CYP8B1 cocrystallized with tioconazole to yield single, suitably sized oval crystals. X-ray data were collected from a single crystal, solved by molecular replacement employing the structure of human CYP8A1 (Protein Data Bank [PDB]: 2IAG), and refined (Table 2) to generate a 2.6 Å structure with one molecule in the asymmetric unit.
Table 2.
X-ray data collection, refinement, and validation statistics
| Statistic | CYP8B1/(S)-tioconazole |
|---|---|
| Data collection | |
| Space group | P 21 21 21 |
| Cell dimensions | |
| a, b, c (Å) | 58.28, 89.07, 107.85 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution (Å)a | 41.24–2.60 (2.64–2.60) |
| Redundancya | 5.9 (5.5) |
| Rpima | 0.053 (0.475) |
| Mn(I/sd)a | 15.8 (1.3) |
| CC ½a | 0.925 (0.585) |
| Completenessa (%) | 98.9 (98.9) |
| Total Reflectionsa | 104,655 |
| Unique Reflectionsa | 17,925 |
| Refinement | |
| Resolution (Å) | 41.24–2.60 |
| No. reflections | 397,249 |
| Rwork/Rfree | 0.21/0.27 |
| Molecules per asymmetric unit | 1 |
| Number of non-hydrogen atoms/B factor | |
| Protein | 7429/53.6 |
| Ligand | 36/82 |
| Glycerol | 14/70 |
| Heme | 73/51 |
| Water | 17/51 |
| RMS deviations | |
| Bond lengths (Å) | 0.005 |
| Bond angles (º) | 0.744 |
| Coordinate error (Maximum-likelihood) (Å) | 0.38 |
| Ramachandran plot: preferred/allowed/outliers (%) | 97.55/2.23/0.22 |
Highest resolution shell is shown in parenthesis.
The overall structure reveals a typical cytochrome P450 fold with helices D, E, I, and L surrounding the heme prosthetic group and helices F and G forming the roof of the catalytic domain (Fig. 3A). The active site adjacent to the heme is large (999 Å3) and relatively flat (due to spatial restriction from W281) but elongated (Fig. 3B). The active site further opens to the protein exterior and bulk solvent in 3 directions. A glycerol molecule from the protein buffer occupies part of channel 1 between the F helix and β1 (Fig. 3B). Two other channels extend between the B′ and C helices (channel 2) and between the I helix and β1-4 (channel 3).
Figure 3.

CYP8B1 structure. (A), overview of CYP8B1 (ribbons colored red N terminus to blue C terminus) in complex with tioconazole and glycerol (gray sticks). The heme is shown in black sticks with a red sphere for the iron. (B), active site cavity (gray mesh). CYP8B1, cytochrome P450 8B1.
Consistent with the type II spectral changes observed in the binding studies, (S)-tioconazole is clearly bound in the active site with its imidazole nitrogen forming a coordinate covalent bond to the CYP8B1 heme iron with a distance of 2.56 Å (Fig. 4A). CYP8B1 was cocrystallized with tioconazole whose stereochemistry was unknown; it was clear from the density that the S enantiomer was bound. While the electron density for the azole, phenyl, and thiophene rings are clear, the density is much poorer for the chloro substituents for each of the latter rings, which may suggest flexibility and/or multiple conformations.
Figure 4.

CYP8B1active site. (A) The tioconazole ligand with simulated annealing composite omit map and (B) residues within 5 Å of tioconazole (sticks). Coloring as in Figure 3. CYP8B1, cytochrome P450 8B1.
Regardless, the active site in the vicinity of the tioconazole ligand is dominated by 2 primary features (Fig. 4B). First, it is mainly lined by hydrophobic residues. Residues within 5 Å of tioconazole include F99, Y102, A103, L106, V107, F111, F213, V217, M278, M279, W281, A282, N286, P354, T355, L357, and F482 (Fig. 4B). These include residues of traditional substrate recognition sites (SRS) (20) SRS-1 in or near the B′ helix (99, 102, 103, 106, 107, 111), SRS-2 at the end of helix F (213, 217), SRS-4 in the I helix (278, 279, 286), SRS-5 (P354-L357), and SRS-6 or β4−1 (F482). This suggests that both hydrophobic and π-stacking and halogen-π interactions would be attractive features in a more selective inhibitor. Second, while the active site is elongated, its depth immediately over the heme is constrained due to the spatial hinderance of W281 and N286 of I helix on one side of the active site (Fig. 4B). To accommodate (S)-tioconazole, the carbon linker between its chiral center and the imidazole bend of almost 90° accommodates the dichlorophenyl and chlorothienyl rings. The indole moiety of W281 interacts with D210, which in turn interacts with R479 near the protein surface. This hydrogen bonding network may be essential for the proper function of CYP8B1, as W281 mutation significantly alters enzyme function (Fig. 2).
Docking of native substrate
While a cocrystal structure with the native substrate has not yet been achieved, the orientation of this 7α-hydroxycholest-4-en-3-one molecule is of significant interest. Thus, this substrate was docked into the current CYP8B1 structure. After tioconazole was removed, the native substrate was added and docked using induced-fit docking with Glide. One of the top-scoring poses oriented 7α-hydroxycholest-4-en-3-one in a manner consistent with the known C12-hydroxylation product (Fig. 5). This pose was the fourth unique pose and had a score of −8.6 kcal/mol. The distance between the heme iron and C12 was 5 Å. The long axis of the steroid nucleus was oriented along with the I helix but not parallel to it. The steroid α face was directed toward the I helix with the 7α-hydroxyl projecting toward W281 in such a way as to suggest this interaction might significantly impact ligand positioning. The steroid β face with its 2 projecting methyls was generally directed toward the B′ helix. Finally, the long C17 hydrocarbon side chain extended out of channel 2 between the B′ and I helices (Fig. 3B). Better scoring poses were in unproductive orientations, roughly orthogonal to the heme plane with the A ring ketone oriented toward the heme and the flexible C17 side chain pointed up into the area occupied by the glycerol.
Figure 5.

The native 7α-hydroxycholest-4-en-3-one substrate (gray sticks) computationally docked into the CYP8B1 structure (blue ribbons). Heme, black sticks.
Discussion
Recombinant CYP8B1 characterization
To our knowledge, human CYP8B1 has not previously been recombinantly expressed and purified. However, typical modifications for human membrane P450 engineering, including truncation of the single membrane–spanning N-terminal helix and addition of a C-terminal His tag, resulted in good yields of folded protein incorporating the heme prosthetic group that could be highly purified and characterized. Spectral characterization yielded the typical Soret peak for P450 enzymes with water coordinated to the active site heme iron. However, the reduced carbon monoxide difference spectra revealed some interesting observations. Substrate-bound CYP8B1 had the canonical difference peak at 450 nm reflective of normal cysteine thiolate interaction with the heme iron, consistent with catalytically active protein. However, in the absence of substrate, the reduced carbon monoxide difference spectrum for CYP8B1 has a peak primarily at 420 nm with only a small or no peak at 450 nm. Many have suggested that such “P420” protein is catalytically inactive and not reversible to P450 (19, 22). However, over the course of working with many different human P450 enzymes, we have observed a number of cases where the “P420” state reverts to a “P450” state in terms of its spectrum and catalytic activity but none so markedly as CYP8B1. In this case, the simple addition of substrate converts almost total “P420” to total “P450,” so it is clearly reversible spectrally. In catalytic assays where substrate would be present, the enzyme is catalytically active with its native substrate. The literature suggests 2 possible explanations for the “P420” state. One is that the proximal Cys thiolate becomes protonated and dissociates from the heme iron. The other is that this Cys-Fe interaction is substituted for a His-Fe interaction due to some conformational change of the proximal side of the heme (23). While some P450 enzymes that form the spectral “P420” form have histidines near the proximal cysteine, in CYP8B1 the most reasonably located histidines are all more than 8 Å away from Fe in either (1) the C helix (H124), which can be repositioned in some P450 structures depending on the state (24), or (2) His428, found in a flexible loop preceding the proximal cysteine. Regardless of the mechanism, in CYP8B1, substrate binding on the distal site of the heme alters the protein conformation on the proximal side, either via small changes in Cys-Fe interaction or via larger conformational changes to relocate a histidine hear the iron.
Further evidence that interactions on the distal side of the heme alter proximal arrangements is found in the W281F mutant. While most mutations are more likely to destabilize proteins, this mutation consistently demonstrated more P450 species and less P420 species in the reduced carbon monoxide difference spectra compared with WT CYP8B1 (Fig. S5), but at the same time it impaired substrate binding and significantly reduced catalytic efficiency (Fig. 2). It is possible that the conformational stability is a tradeoff for catalytic activity.
CYP8B1 function
Recombinant, purified CYP8B1 bound both substrate and inhibitors with the typical red-shift and blue-shift of the Soret peak and is consistent with their expected binding modes—water displacement versus water replacement. The enzyme bound its native substrate with a KD of 18 nM and catalyzed its 12-hydroxylation with a Km of 0.5 μM. Since these 2 values are only equivalent when the dissociation rate (koff) is large relative to kcat (7.4 min−1), the substrate dissociation rate constant must be quite slow. In many P450 enzyme structures, the substrate is observed in an enclosed active site with no channels sufficient for substrate or product passage, thus requiring protein conformational changes with each catalytic cycle. CYP8B1 does have channels between the active site and the exterior bulk solvent when cocrystallized with tioconazole. Thus, either the native substrate is tightly held within the active site despite the existence of these channels or the protein conformation closes further when substrate is bound to facilitate substrate retention.
The active site residue W281 has previously been suggested to be important in CYP8B1 and several related enzymes. Herein, the W281F mutant decreased affinity for its native substrate ∼2-fold but decreased turnover even more drastically, ∼6-fold. These data indicate that the major effect is on the catalytic step. The current structure demonstrates that this tryptophan is located directly over the heme iron (Fe-Trp 8.2 Å) and docking suggests it is near the 7αOH substituent of the substrate. Altogether, this information suggests that W281 is involved in substrate positioning for optimal catalysis. This tryptophan residue is conserved in several other related P450 enzymes, including the CYP8A1 and CYP7A1 active sites, and aligns with a tryptophan in CYP7B1 and CYP39A1 (25). In CYP7A1 and CYP8A1, this tryptophan serves as a “low ceiling” keeper (26) but also interacts with an asparagine in F-helix, which forms a salt bridge with arginine near the protein surface. In CYP8B1, this triad is conserved among W281, D210, and R479. In CYP7A1, the W283F mutation abolished the spectral response to its native substrate, cholesterol (21), and impaired catalysis. In CYP7B1, an R486C mutation in this triad correlates with neurodegenerative disease, which is possibly caused by aberrant oxysterol levels (27). Such information supports the hypothesis that this network is important for proper function in several human cytochrome P450 enzymes.
Toward CYP8B1 inhibitors
Little attention has been paid to the design and synthesis of selective CYP8B1 inhibitors (13), despite significant evidence that such molecules might be clinically useful in the treatment of NAFLD and type 2 diabetes (9, 12). Herein, CYP8B1 was demonstrated to bind a relatively broad range of azoles, all with interactions between the azole nitrogen and the heme iron. While such compounds are often poorly selective in discriminating among the 57 human cytochrome P450 enzymes, they often have intrinsically high affinities, modulated by the compatibility of the active site with the remainder of the ligand structure. Such is the case here.
The best azoles had affinities under 100 nM and submicromolar IC50 values. The 3 most effective inhibitors, miconazole, econazole, and tioconazole, are all very structurally similar, differing only in substitutions of a thiophene versus phenyl ring and the specific chlorine substitution pattern. Within this series, the KD values suggest that the slightly larger phenyl ring and parachlorination of econazole is favorable over the thiophene and orthochlorination of tioconazole, while additional chlorination at the meta position yields the most effective inhibitor miconazole. While specific halogen interactions are not apparent in the crystal structure with tioconazole, these data suggest that the chlorination substitution pattern should be considered in optimizing binding interactions. If econazole and miconazole bind in similar orientations as that observed for tioconazole, it is possible that their monochlorophenyl or dichlorophenyl rings interact with F482 via either π-π stacking or halogen-π interactions (Fig. 4B).
Clotrimazole and liarozole are less effective inhibitors. Their structures are more compact without a methylene bridge between the iron-coordinating imidazole and the rest of the structure. When the methylene is present between the imidazole and the sp3 carbon from which the other ring systems project, the ligands would be more flexible, which may facilitate conformations whereby the nonimidazole rings are more compatible with the available space in this “low ceiling” binding pocket.
A much larger inhibitor that still binds with a micromolar KD value is ketoconazole. While the iron-binding and dichlorophenyl arms of this inhibitor are identical to the tightest-binding compound miconazole, the third arm is significantly elongated. Inspection of this compound and the CYP8B1 active site suggests that the elongated arm might be accommodated by extending out of either of the 2 channels originating near the nonimidazole rings of tioconazole.
Finally, despite a core structure somewhat similar to other compounds and 2 potential iron-coordinating triazoles, fluconazole showed no binding and almost no inhibitory effects to CYP8B1. This could be due to steric hindrance from the central hydroxyl or due to the fact that triazoles tend to have weaker binding to cytochrome P450 hemes (28).
Overall, the identification of tight-binding ligands provides an excellent starting point for the design of CYP8B1 inhibitors. Additionally, the initial structure–activity relationships discussed herein suggest that affinity can be successfully optimized by modifications of the ligand structure apart from the iron-coordinating group. The next challenge is to identify inhibitor modifications that might provide good selectivity for CYP8B1 over other human cytochrome P450 enzymes.
Analysis of opportunities for selectively targeting CYP8B1
The greatest challenge in clinical CYP8B1 inhibitor design is likely selectivity compared to CYP8A1 and CYP7A1. CYP8A1 (prostaglandin I2 synthase) (29) has the highest amino acid sequence identity (41%), but its prostaglandin H2 substrate is fairly distinct from the CYP8B1 steroidal substrate. CYP7A1 is less identical in terms of amino acid sequence (36%) but acts on the similar steroidal substrate cholesterol. Although CYP7A1 hydroxylates the steroid nucleus at C7 and CYP8B1 hydroxylates C12, there are potential similarities of the active sites. While inhibition of CYP8B1 is desirable for treatment of NAFLD and type 2 diabetes, inhibition of CYP8A1 and CYP7A1 are undesirable as they result in essential hypertension (30) and hypercholesterolemia (31), respectively. Thus, the first CYP8B1 structure herein will be compared to structures of both CYP8A1 and CYP7A1, which have been determined previously (25, 29).
CYP8A1 converts prostaglandin H2 to prostacyclin without the typical requirement for cytochrome P450 reductase or NADPH as an electron donor (29). Despite their different substrates and distinctive catalytic reactions of all the human cytochrome P450 enzymes, CYP8A1 and CYP8B1 have the most similar overall and active site amino acid sequences. Two human CYP8A1 structures are available, one with the inhibitor minoxidil (PDB 3B6H) (29) and one which is ligand free (PDB 1IAG) (29). These structures have an overall Cα RMSD 0.1 Å with most distinctions occurring on the proximal side of the heme rather than the distal active site cavity (29). The ligand-free CYP8A1 structure (PDB 2IAG) aligns with the CYP8B1/tioconazole structure with a Cα RMSD of 1.54 Å (Fig. 6A). Overlay of the active sites (Fig. 6B) illustrates the strong conservation of actives site residues and orientations, including the tryptophan serving as a “low ceiling” keeper (26) (W281 in CYP8B1), the asparagine protruding into the active site (N286 in CYP8B1), and the noted interactions between the I helix tryptophan, F helix aspartic acid (D210 in CYP8B1), and surface arginine (R479 in CYP8B1). There are notable differences in the topology of the CYP8B1 and CYP8A1 active sites, however. These are primarily related to differences in the heme porphyrin plane and the positioning of the propionates. Largely because of these differences, the volume of the CYP8A1 active site is slightly larger (1237 Å3, Fig. 7A) with 2 major differences. First, the CYP8A1 active site extends toward the distal side of the heme below the 2 propionates (Fig. 7A, circle 1). Second, additional active site volume is also observed in CYP8A1 between the I and F helices next to W282 (Fig. 7A, circle 2), which is thought to accommodate the long side chain of prostaglandin H2, based on a substrate analog structure of zebrafish CYP8A1 (29). Conversely, unique active site cavity volume is observed above the heme plane in CYP8B1 next to channel 3 (Fig. 3B). Thus, inhibitor designs with larger sizes directly over the heme could be exploited to design inhibitors that are more selective for CYP8B1 than for CYP8A1.
Figure 6.

Structural comparisons of CYP8B1 (pale cyan) with CYP8A1 (raspberry) and CYP7A1 (orange). Comparisons of CYP8B1 with CYP8A1 (A) overall and (B) active sites. Comparisons of CYP8B1 with CYP7A1 (C) overall and (D) active sites.
Figure 7.

Comparison of active site cavities. Active site cavities (gray mesh) for (A) CYP8A1 (raspberry) and (B) CYP7A1(orange) for comparison with Figure 3B. Heme is shown as black sticks with red sphere for iron.
CYP7A1 is the other human P450 enzyme that should probably be a first-line counter target. Although it has lower amino acid sequence identity with CYP8B1 (36%), their steroidal substrates are very similar. CYP7A1 or cholesterol 7α-hydroxylase catalyzes the first and rate-limiting step in the classic bile acid synthetic pathway (25). In addition, CYP7A1 can also hydroxylate cholest-4-en-3-one to form the CYP8B1 substrate 7α-hydroxy-cholest-4-en-3-one (25), suggesting the possibility that high similarities will be observed in their active site architectures. A structure of CYP7A1 with the artificial T104L mutation has been generated in complex with this cholest-4-en-3-one substrate (PDB 3SN5) (25). Alignment of the CYP7A1/cholest-4-en-3-one structure with the CYP8B1/tioconazole structure herein similarly yields a low Cα RMSD of 1.35 Å, consistent with relatively close overlay of the overall folds (Fig. 6C). The most significant differences are observed for the F and G helices and the B′ helix. Although active site residue identities are again highly conserved, there are more substantial differences in their placements between CYP7A1 and CYP8B1 (Fig. 6D) than were observed between CYP8A1 and CYP8B1 (Fig. 6B), even though the heme plane and propionates are more similar between the former 2 enzymes. The volume of the CYP7A1 active site is 1035 Å3 (Fig. 7B), which is more similar with the CYP8B1 active site volume than CYP8A1. Compared with CYP8B1, the CYP7A1 active site extends further to the roof of the active site (Fig. 7B, circle 1) and between helices C and I (Fig. 7B, circle 2), the latter of which accommodates the long aliphatic chain of its cholest-4-en-3-one substrate. More cavity volume is observed between B′ helix and the heme propionate due to the position of B′ helix. These differences might be usefully exploited in designing more selective inhibitors.
Conclusions
CYP8B1 appears to be a promising drug target for the treatment of NAFLD and type 2 diabetes. However, the roles of CYP8B1 in both normal physiology and disease states are still poorly understood, and no selective inhibitors have yet been developed as either tool or clinical compounds. Currently, the only study of CYP8B1 depletion was done in mice using cyp8b1 siRNA, which is a suboptimal therapeutic strategy with substantial delivery difficulties. A selective CYP8B1 inhibitor would be a more desirable probe of the physiology and as a potential drug. The generation, functional characterization, and determination of the first CYP8B1 structure herein should significantly advance this effort. Specific details of the CYP8B1 active site architecture, particularly compared to CYP7A1 and CYP8A1, should facilitate structure-based design of selective CYP8B1 inhibitors. This in turn should promote a better understanding of the role of CYP8B1 inhibition in normal physiology and disease states and potentially provide a possible treatment for NAFLD and type 2 diabetes.
Experimental procedures
Expression and purification of CYP8B1 and W281F mutant
The human CYP8B1 cDNA was synthesized with codon optimization for E. coli expression (Genscript). Nucleotides coding for WT amino acids 1 to 25 composing the transmembrane helix were truncated and replaced with nucleotides coding for the sequence MAKKTSS. Nucleotides were also added at the C terminus to code for a six-residue histidine tag just prior to the stop codon. The resulting sequence was cloned into the pCWori+ vector. For the mutant, an extra mutation W281F was made.
WT and W281F expression plasmids were transformed into E. coli DH5α cells already containing the pGro7 plasmid for expressing the GroES–GroEL chaperone complex (Takara Bio) and plated on a lysogeny broth (LB) agar plate containing 100 μg/ml carbenicillin for pCWori+ selection and 20 μg/ml chloramphenicol for pGro7 selection and grown at 37 °C overnight. A single colony was then inoculated into 5 ml LB media with the same antibiotics and grown for 6-8 h at 37 °C with shaking. A 50 μl sample of this culture was then introduced into 200 ml LB media and grown overnight with the same antibiotics, with shaking at 37 °C. For expression, 900 ml of terrific broth with 2 × potassium phosphate buffer and the same antibiotics in a 2.8 L Fernbach flask were inoculated with 20 ml of the overnight culture. This culture was incubated at 37 °C and 220 rpm until an absorbance at 600 nm (A600) of 0.4 to 0.6 was reached. At that point, 1 mM δ-aminolevulinic acid and 4 mg/ml arabinose were added as a heme precursor and to induce expression of the groES-groEL genes, respectively. When an A600 of 0.8 was reached, CYP8B1 expression was induced with 1 mM IPTG. The temperature and shaking were reduced to 25 °C and 190 rpm, respectively, for 48 to 66 h, after which bacterial cells were pelleted by centrifugation at 6930 × g at 4 °C for 10 min. The cell pellet was resuspended in 10% of the culture volume of resuspension buffer (500 mM potassium phosphate buffer, pH 7.4, 20% (v/v) glycerol, 500 mM sodium chloride) containing two tablets of 0.8 × SigmaFast protease inhibitor (Millipore Sigma). The resuspended sample was homogenized using a Dounce homogenizer with pestle A followed by pestle B. Lysozyme was added to 0.3 mg/ml. Cells were then disrupted by ultrasonication (Fisher Scientific Sonic Dismembrator Model 100) 10 times for 30 s with 30 s interval between every 2 sonication periods. CYP8B1 was extracted with 14 mM CHAPS (GoldBio) for 1 h at 4 °C. Cell debris and membranes were removed by ultracentrifugation at 12,000 × g for 50 min at 4 °C. The resulting supernatant was loaded on a 30 ml Ni-NTA column (Ni/NTA Superflow, Qiagen) equilibrated with 2 column volumes (CV) of loading buffer (100 mM potassium phosphate buffer, pH 7.4, 500 mM NaCl, 20% (v/v) glycerol, 14 mM CHAPS). After loading, the column was first washed with 5 CV loading buffer, followed by 5 CV Ni-NTA wash buffer (100 mM potassium phosphate buffer, pH 7.4, 500 mM NaCl, 20% (v/v) glycerol, 14 mM CHAPS, 5.6 mM histidine) to remove weakly bound contaminants. CYP8B1 was then eluted with 6 CV Ni-NTA elution buffer (10 mM potassium phosphate, pH 7.4, 100 mM NaCl, 20% (v/v) glycerol, 10 mM CHAPS, 80 mM histidine). The eluted CYP8B1 fractions typically had a ratio of absorbance at 420 to 280 of 0.7, which were pooled and concentrated to 20 to 50 ml using an Amicon ultra 50 kDa filter and then diluted 6-fold with CM wash buffer (5 mM potassium phosphate, pH 7.4, 20% (v/v) glycerol, 1 mM EDTA) containing 4 mM CHAPS. Two 5 ml HiTrap CM FF column (GE Healthcare) were connected in series for cation-exchange chromatography (total CV = 10 ml). Columns were equilibrated with 5 CV CM wash buffer. After loading, these columns were washed with 10 CV CM wash buffer and CYP8B1 subsequently eluted with a gradient from 0 to 500 mM NaCl formed by the CM wash buffer and CM elution buffer (50 mM potassium phosphate, pH 7.4, 500 mM NaCl, 20% (v/v) glycerol, 1 mM EDTA). Fractions with at least equal absorbance at 280 and 420 nm were pooled and concentrated to 1-2 ml using an Amicon ultra 50 kDa filter before injecting onto a size-exclusion column (GE Healthcare HiLoad 16/60 Superdex 200 pg). This column was equilibrated and run with CM elution buffer. Fractions with at least equal absorbance at 280 and 420 nm were pooled. The purity of the protein was examined by measuring the ratio of the absorbance of Soret peak to the absorbance at 280 nm (typically 1.0) and by SDS-PAGE. Purified CYP8B1 was also characterized using UV-visible absorbance spectrum and reduced carbon monoxide difference spectrum.
Expression and purification of human cytochrome P450 reductase
The human full-length cytochrome P450 reductase with K59Q mutation was synthesized with codon optimization for E. coli expression (Genscript). A six-residue histidine tag was added at the C terminus just prior to the stop codon. This modified cDNA was cloned into pET29a(+) vector and transformed into E. coli C41(DE3)/pGro7 cells. The protein was expressed and purified as described (32) except that cells were lysed by a homogenizer (Avestin, Inc EmulsiFlex-C3 High Pressure Homogenizer) instead of a French press.
Crystallization and structural determination
CYP8B1 (10 mg/ml) in CM elution buffer (50 mM potassium phosphate, pH 7.4, 500 mM NaCl, 20% (v/v) glycerol, 1 mM EDTA) was saturated with 30 μM tioconazole as evidenced by a shift of the Soret peak to 425 nm. CYP8B1 crystals were grown using sitting-drop vapor diffusion equilibration. The CYP8B1–tioconazole complex was equilibrated against 5% 2-methyl-2,4-pentanediol, 0.1 Hepes pH 7.5, 10% PEG 10,000 at 25 °C. Crystals formed within 24 h. Crystals were cryoprotected with mother liquor plus 30% glycerol and flash cooled in liquid nitrogen. Diffraction data were collected on beamline 21-ID-G of the advanced photon source and processed using HKL 2000 (33) The CYP8B1 structure was solved by molecular replacement using Phaser (34) with a CYP8A1 search model (PDB entry 2IAG). Model building and refinement were performed iteratively with Coot (35) and Phenix.refine (https://phenix-online.org/documentation/reference/refinement.html) (36), respectively. The ligand (S)-tioconazole was generated using elBOW (37) in Phenix. Active site void volumes were calculated using VOIDOO (38), with probe radius = 1.4 Å and grid spacing of 1.0. Figures were generated with PyMOL (39).
Ligand-binding assay
Ligand-binding affinities were determined by observing shifts in the CYP8B1 (in 50 mM potassium phosphate, 20% glycerol) absorbance spectrum as described previously (28). Clotrimazole, ketoconazole, and liarozole (dissolved in dimethyl sulfoxide [DMSO]) were titrated into 1 μM CYP8B1 in a cuvette with 1 cm path length. Because of their higher affinities, econazole, tioconazole, and miconazole (dissolved in DMSO) were titrated into 0.2 μM CYP8B1, and the 7α-hydroxy-4-cholesten-3-one substrate (dissolved in ethanol) was titrated into 0.1 μM CYP8B1 or CYP8B1 W281F in a cuvette with 5 cm path length to increase signal to noise. Changes in absorbance (local absorbance maximum minus minimum) were plotted against ligand concentration and analyzed using the single site–specific binding equation using GraphPad Prism version 8.0.0 for Mac OS X (GraphPad Software). All titration experiments were repeated in triplicate.
Determination of kinetic parameters
To determine the steady state kinetic parameters of CYP8B1 and CYP8B1 W281F for 12α-hydroxylation of its native substrate 7α-hydroxycholest-4-en-3-one, in vitro activity assays were conducted. A total reaction volume of 1 ml (for WT CYP8B1) or 0.5 ml (for CYP8B1 W281F) contained 0.01 μM CYP8B1 (WT and mutant) and 0.06 μM human cytochrome P450 reductase in 50 mM potassium phosphate buffer, pH 7.4 containing 20% glycerol. CYP8B1 and reductase were mixed and preincubated for 10 min at room temperature before adding 7α-hydroxycholest-4-en-3-one (0.01–10 μM, dissolved in ethanol; total of 0.2% ethanol). Reactions were initiated by the addition of 200 μM NADPH and allowed to proceed at 37 °C for 1.5 min (for WT) or 2 min (for CYP8B1 W281F) and terminated with the addition of 1 ml chloroform. For all reactions, substrate utilization was less than 10%. The internal standard of 40 μM 11-deoxycortisol (dissolved in DMSO) was added to all reactions. Sterols were extracted twice with 1 ml (for the WT) or 0.5 ml (for the mutant) chloroform, dried under the hood, resuspended in 100 μl 40% acetonitrile, and injected (60 μl for WT, 30 μl for the mutant) onto a reverse-phase HPLC column (Phenomenex, Luna, 5 μm, C18, 150 × 4.6 mm). Chromatography was performed at 40 °C with a flow rate of 0.8 ml/min using an acetonitrile-water gradient (phase A: 10% acetonitrile, phase B: 100% acetonitrile). The following method was used: 0 to 15 min 40% B and 15 to 45 min 80% B. Sterols were detected by UV absorbance at 240 nm. Retention times of the internal standard, product, and substrate were as follows: 11-deoxycortisol (internal standard) at 9.9 min, 7α,12α-dihydroxycholest-4-en-3-one at 24.5 min, and 7α-hydroxycholest-4-en-3-one at 39.5 min. Quantification of the product 7α,12α-dihydroxycholest-4-en-3-one was performed by determining the ratio of the peak area for the product over the peak area of the internal standard using a calibration curve. Initial reaction velocities were plotted against the respective 7α,12α-dihydroxycholest-4-en-3-one concentration and fit to the Michaelis–Menten equation using GraphPad Prism version 8.0.0 for Mac OS X (GraphPad Software). These experiments were performed in triplicate.
CYP8B1 inhibition assays
Potential inhibitors were initially screened by adding a fixed concentration of 80 μM to the assays described previously and determining the percentage activity remaining compared to a control reaction without inhibitor. These experiments were performed in duplicate or triplicate.
Subsequently, the concentration of azoles leading to 50% inhibition (IC50) of 7α-hydroxycholest-4-en-3-one metabolism by CYP8B1 was determined. These assays were conducted as described previously with the following modifications: (1) the concentration of the substrate 7α-hydroxycholest-4-en-3-one was held constant at the Km of 0.5 μM, (2) inhibitors (dissolved in DMSO) were added at increasing concentrations, (3) the reaction volume was decreased to 500 μl, and (4) the reaction time was increased to 2 min. Quantification of sterol products was also conducted as described in kinetic assay section except that (1) 30 μl of each sample was injected onto the column and (2) the methods/mobile phases had to be optimized to improve separation of inhibitors other than liarozole. The method for tioconazole was as follows: 0 to 12 min 40% B, 12 to 42 min 40% to 80% B, 42 to 70 min 80% B, and 70 to 75 min 40% B. For econazole and clotrimazole, the method was as follows: 0 to 12 min 40% B, 12 to 52 min 40% to 80% B, 52 to 75 min 80% B, 75 to 80 min 40% B. For miconazole, the same method was used but 10% acetonitrile with 0.03% acetic acid was substituted for 10% acetonitrile for A. The amount of the 7α,12α-dihydroxycholest-4-en-3-one product produced with no inhibitor present was set as 100% maximal activity. The amounts of the product produced in the inhibitor-containing samples were scaled to this value and reported as percentages of the maximal activity. These percentages were then plotted against the log inhibitor concentration in GraphPad Prism and fit to the log[inhibitor] versus response, four parameter variable slope equation. These experiments were repeated in triplicate.
Docking 7α-hydroxycholest-4-en-3-one into the CYP8B1 active site
Maestro V.2021-3 and the OPLS4 force field (40) were used to execute system preparation and the docking protocol. The dielectric constant was set to 1.0. The native substrate 7α-hydroxycholest-4-en-3-one was built and minimized, then prepared with the LigPrep module. The complex of CYP8B1 with S-tioconazole bound was stripped of waters and the glycerol molecule. The complex was set up using the Protein Preparation Workflow with the choice to cap all residues at sequence gaps. All hydrogens were minimized while the other atoms were held fixed. The system was then minimized with the ligand and all residues within 5 Å being free to move. The free residues were surrounded by a 5 Å shell of residues that were constrained with a force constant of 200 kcal/mol·Å2. The rest of the protein was held fixed. After this minimization, a 10 Å docking box was centered on S-tioconazole. A receptor grid was generated from this structure. The induced-fit docking area was defined by the central atom of S-tioconazole. The ligand was then removed and induced-fit docking (41) was executed with the Glide XP (42) protocol using default parameters. Residues within 5 Å were refined during the docking of 7α-hydroxycholest-4-en-3-one. A pose with C12 oriented toward the heme group was the fourth unique pose and had a score of −8.6 kcal/mol. Better scored poses were in unproductive orientations with the A ring’s ketone toward the heme and the flexible C17 tail pointed up filling the area occupied by the removed glycerol additive.
Data availability
All data is included in this article or the supplemental information statement, with the exception of the structure coordinates and structure factors. This latter information has been deposited with the PDB under accession code 7LYX.
Supporting information
This article contains supporting information.
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgments
Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. LS-CAT membership is partially supported by NIH grant P30 CA046592. Research reported in this publication was supported by the University of Michigan Center for Structural Biology (CSB). The CSB is grateful for support from the U-M Life Sciences Institute, the U-M Rogel Cancer Center, the U-M Medical School Endowment for Basic Sciences, and grants from the National Institute of Health.
Author contributions
J. L. and E. E. S. conceptualization; J. L. methodology; J. L., H. A. C., and E. E. S. investigation; J. L. and E. E. S. writing–original draft; J. L., H. A. C., and E. E. S. writing–review & editing; J. L. and E. E. S. visualization; E. E. S. supervision; E. E. S. funding acquisition.
Funding and additional information
The research was supported by NIH R37 GM076343 (to E. E. S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Biography

Jinghan Liu is a PhD candidate in the Department of Medicinal Chemistry at the University of Michigan. She studies the structure and function of human bile acid biosynthetic and xenobiotic cytochrome P450 enzymes. She uses X-ray crystallography and different protein assays to understand the P450 interaction with potential drugs, laying the groundwork for drug design for major human diseases and drug metabolism and toxicity predictions in neonates. linkedin.com/in/jinghan-liu-853241237
Edited by Joseph Jez
Supporting information
Supplemental Figures S1–S5
References
- 1.Staels B., Fonseca V.A. Bile acids and metabolic regulation: Mechanisms and clinical responses to bile acid sequestration. Diabetes Care. 2009;32(Suppl 2):S237–S245. doi: 10.2337/dc09-S355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiang J.Y. Recent advances in understanding bile acid homeostasis. F1000Res. 2017;6:2029. doi: 10.12688/f1000research.12449.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manne V., Handa P., Kowdley K.V. Pathophysiology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Clin. Liver Dis. 2018;22:23–37. doi: 10.1016/j.cld.2017.08.007. [DOI] [PubMed] [Google Scholar]
- 4.Puri P., Daita K., Joyce A., Mirshahi F., Santhekadur P.K., Cazanave S., et al. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology. 2018;67:534–548. doi: 10.1002/hep.29359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiao Y., Lu Y., Li X.Y. Farnesoid X receptor: a master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacol. Sin. 2015;36:44–50. doi: 10.1038/aps.2014.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han C.Y. Update on FXR Biology: promising therapeutic target? Int. J. Mol. Sci. 2018;19:2069. doi: 10.3390/ijms19072069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hindson J. Obeticholic acid for the treatment of NASH. Nat. Rev. Gastroenterol. Hepatol. 2020;17:66. doi: 10.1038/s41575-020-0264-1. [DOI] [PubMed] [Google Scholar]
- 8.Arab J.P., Karpen S.J., Dawson P.A., Arrese M., Trauner M. Bile acids and nonalcoholic fatty liver disease: molecular insights and therapeutic perspectives. Hepatology. 2017;65:350–362. doi: 10.1002/hep.28709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chevre R., Trigueros-Motos L., Castano D., Chua T., Corliano M., Patankar J.V., et al. Therapeutic modulation of the bile acid pool by Cyp8b1 knockdown protects against nonalcoholic fatty liver disease in mice. FASEB J. 2018;32:3792–3802. doi: 10.1096/fj.201701084RR. [DOI] [PubMed] [Google Scholar]
- 10.Haeusler R.A., Astiarraga B., Camastra S., Accili D., Ferrannini E. Human insulin resistance is associated with increased plasma levels of 12α-hydroxylated bile acids. Diabetes. 2013;62:4184–4191. doi: 10.2337/db13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pathak P., Chiang J.Y.L. Sterol 12α-hydroxylase aggravates dyslipidemia by activating the ceramide/mTORC1/SREBP-1C pathway via FGF21 and FGF15. Gene Expr. 2019;19:161–173. doi: 10.3727/105221619X15529371970455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaur A., Patankar J.V., de Haan W., Ruddle P., Wijesekara N., Groen A.K., et al. Loss of Cyp8b1 improves glucose homeostasis by increasing GLP-1. Diabetes. 2015;64:1168–1179. doi: 10.2337/db14-0716. [DOI] [PubMed] [Google Scholar]
- 13.Chung E., Offei S.D., Aondo Jia U.T., Estevez J., Perez Y., Arman H.D., et al. A synthesis of a rationally designed inhibitor of cytochrome P450 8B1, a therapeutic target to treat obesity. Steroids. 2022;178:108952. doi: 10.1016/j.steroids.2021.108952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishida H., Noshiro M., Okuda K., Coon M.J. Purification and characterization of 7 alpha-hydroxy-4-cholesten-3-one 12 alpha-hydroxylase. J. Biol. Chem. 1992;267:21319–21323. [PubMed] [Google Scholar]
- 15.Eggertsen G., Olin M., Andersson U., Ishida H., Kubota S., Hellman U., et al. Molecular cloning and expression of rabbit sterol 12alpha-hydroxylase. J. Biol. Chem. 1996;271:32269–32275. doi: 10.1074/jbc.271.50.32269. [DOI] [PubMed] [Google Scholar]
- 16.Fan L., Joseph J.F., Durairaj P., Parr M.K., Bureik M. Conversion of chenodeoxycholic acid to cholic acid by human CYP8B1. Biol. Chem. 2019;400:625–628. doi: 10.1515/hsz-2018-0379. [DOI] [PubMed] [Google Scholar]
- 17.Omura T., Sato R. The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J. Biol. Chem. 1964;239:2370–2378. [PubMed] [Google Scholar]
- 18.Omura T., Sato R. The carbon monoxide-binding pigment of liver microsomes. II. Solubilization, purification, and properties. J. Biol. Chem. 1964;239:2379–2385. [PubMed] [Google Scholar]
- 19.Manna S.K., Mazumdar S. Reversible inactivation of cytochrome P450 by alkaline earth metal ions: auxiliary metal ion induced conformation change and formation of inactive P420 species in CYP101. J. Inorg. Biochem. 2008;102:1312–1321. doi: 10.1016/j.jinorgbio.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 20.Gotoh O. Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J. Biol. Chem. 1992;267:83–90. [PubMed] [Google Scholar]
- 21.Mast N., Graham S.E., Andersson U., Bjorkhem I., Hill C., Peterson J., et al. Cholesterol binding to cytochrome P450 7A1, a key enzyme in bile acid biosynthesis. Biochemistry. 2005;44:3259–3271. doi: 10.1021/bi047566a. [DOI] [PubMed] [Google Scholar]
- 22.McLean K.J., Warman A.J., Seward H.E., Marshall K.R., Girvan H.M., Cheesman M.R., et al. Biophysical characterization of the sterol demethylase P450 from Mycobacterium tuberculosis, its cognate ferredoxin, and their interactions. Biochemistry. 2006;45:8427–8443. doi: 10.1021/bi0601609. [DOI] [PubMed] [Google Scholar]
- 23.Sun Y., Zeng W., Benabbas A., Ye X., Denisov I., Sligar S.G., et al. Investigations of heme ligation and ligand switching in cytochromes P450 and P420. Biochemistry. 2013;52:5941–5951. doi: 10.1021/bi400541v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muralidhara B.K., Negi S., Chin C.C., Braun W., Halpert J.R. Conformational flexibility of mammalian cytochrome P450 2B4 in binding imidazole inhibitors with different ring chemistry and side chains. Solution thermodynamics and molecular modeling. J. Biol. Chem. 2006;281:8051–8061. doi: 10.1074/jbc.M509696200. [DOI] [PubMed] [Google Scholar]
- 25.Tempel W., Grabovec I., MacKenzie F., Dichenko Y.V., Usanov S.A., Gilep A.A., et al. Structural characterization of human cholesterol 7alpha-hydroxylase. J. Lipid Res. 2014;55:1925–1932. doi: 10.1194/jlr.M050765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ullrich V., Brugger R. Prostacyclin and thromboxane synthase: new aspects of hemethiolate catalysis. Angew. Chem. Int. Ed. Engl. 1994;33:1911–1919. [Google Scholar]
- 27.Goizet C., Boukhris A., Durr A., Beetz C., Truchetto J., Tesson C., et al. CYP7B1 mutations in pure and complex forms of hereditary spastic paraplegia type 5. Brain. 2009;132:1589–1600. doi: 10.1093/brain/awp073. [DOI] [PubMed] [Google Scholar]
- 28.Godamudunage M.P., Grech A.M., Scott E.E. Comparison of antifungal azole interactions with adult cytochrome P450 3A4 versus neonatal cytochrome P450 3A7. Drug Metab. Dispos. 2018;46:1329–1337. doi: 10.1124/dmd.118.082032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y.C., Chiang C.W., Yeh H.C., Hsu P.Y., Whitby F.G., Wang L.H., et al. Structures of prostacyclin synthase and its complexes with substrate analog and inhibitor reveal a ligand-specific heme conformation change. J. Biol. Chem. 2008;283:2917–2926. doi: 10.1074/jbc.M707470200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tuder R.M., Cool C.D., Geraci M.W., Wang J., Abman S.H., Wright L., et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1999;159:1925–1932. doi: 10.1164/ajrccm.159.6.9804054. [DOI] [PubMed] [Google Scholar]
- 31.Pullinger C.R., Eng C., Salen G., Shefer S., Batta A.K., Erickson S.K., et al. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J. Clin. Invest. 2002;110:109–117. doi: 10.1172/JCI15387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bart A.G., Scott E.E. Structural and functional effects of cytochrome b5 interactions with human cytochrome P450 enzymes. J. Biol. Chem. 2017;292:20818–20833. doi: 10.1074/jbc.RA117.000220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 34.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., et al. Phenix: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moriarty N.W., Grosse-Kunstleve R.W., Adams P.D. Electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D. Biol. Crystallogr. 2009;65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kleywegt G.J., Jones T.A. Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr. D. 1994;50:178–185. doi: 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]
- 39.Mura C., McCrimmon C.M., Vertrees J., Sawaya M.R. An introduction to biomolecular graphics. PLoS Comput. Biol. 2010;6 doi: 10.1371/journal.pcbi.1000918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu C., Wu C., Ghoreishi D., Chen W., Wang L., Damm W., et al. OPLS4: improving force field accuracy on challenging regimes of chemical space. J. Chem. Theor. Comput. 2021;17:4291–4300. doi: 10.1021/acs.jctc.1c00302. [DOI] [PubMed] [Google Scholar]
- 41.Sherman W., Day T., Jacobson M.P., Friesner R.A., Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 42.Friesner R.A., Murphy R.B., Repasky M.P., Frye L.L., Greenwood J.R., Halgren T.A., et al. Extra precision Glide: docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figures S1–S5
Data Availability Statement
All data is included in this article or the supplemental information statement, with the exception of the structure coordinates and structure factors. This latter information has been deposited with the PDB under accession code 7LYX.