

Abstract
Astrocytes release functional mitochondria (Mt) that play regulatory and prosurvival functions on entering adjacent cells. We recently demonstrated that these released Mts could enter microglia to promote their reparative/prophagocytic phenotype that assists in hematoma cleanup and neurological recovery after intracerebral hemorrhage (ICH). However, the relevance of astrocytic Mt transfer into neurons in protecting brain after ICH is unclear. Here, we found that ICH causes a robust increase in superoxide generation and elevated oxidative damage that coincides with loss of the mitochondrial enzyme manganese superoxide dismutase (Mn-SOD). The damaging effect of ICH was reversed by intravenous transplantation of astrocytic Mt, which on entering the brain (and neurons), restored Mn-SOD levels and reduced neurological deficits in male mice subjected to ICH. Using an in vitro ICH-like injury model in cultured neurons, we established that astrocytic Mt on entering neurons prevented reactive oxygen species-induced oxidative stress and neuronal death by restoring neuronal Mn-SOD levels while at the same time promoted neurite extension and upregulation of synaptogenesis-related gene expression. Furthermore, we found that Mt genome-encoded small peptide humanin, which is normally abundant in Mt, could simulate Mt-transfer effect on neuronal Mn-SOD expression, oxidative stress, and neuroplasticity under ICH-like injury. This study demonstrates that adoptive astrocytic Mt transfer enhances neuronal Mn-SOD-mediated antioxidative defense and neuroplasticity in the brain, which potentiate functional recovery following ICH.
SIGNIFICANCE STATEMENT Mitochondrial dysfunction and antioxidant defense play essential roles in brain damage after ICH. Astrocytes release functional Mt that enters adjacent cells to help brain homeostatic function. Here, we show that systemic transplantation of astrocytic Mt restores ICH-impaired neuronal antioxidative defense, enhances neurite outgrowth, and improves stroke recovery after ICH. Our study suggests that systemic transplantation of astrocytic Mt could be considered as a novel and potentially promising strategy for ICH treatment.
Keywords: antioxidant defense, ICH, mitochondria, Mn-SOD, neuroplasticity, ROS
Introduction
Intracerebral hemorrhage (ICH) is one of the stroke subtypes with high mortality and morbidity and limited options for effective treatments (Keep et al., 2012; Hemorrhagic Stroke Academia Industry (HEADS) Roundtable Participants, Second HEADS Roundtable Participants, 2020). Soon after ICH, red blood cells (RBCs) within hematoma undergo progressive lysis, generating large amount of cytotoxic hemoglobin (Hb), heme, and iron, which robustly induce the generation of reactive oxygen species (ROS; Aronowski and Hall, 2005; Aronowski and Zhao, 2011; Keep et al., 2012; Hu et al., 2016). Under normal physiological states, various antioxidant systems, including superoxide dismutase (SOD), are activated to neutralize ROS; however, these systems are disrupted after hemorrhagic stroke (Wu et al., 2002; Aronowski and Hall, 2005; Aronowski and Zhao, 2011), and excessive accumulation of ROS causes irreversible cellular injuries through protein, lipid, or nucleic acid oxidation and mitochondrial dysfunction (Hall et al., 2000; Kim-Han et al., 2006; Lu et al., 2015; Hu et al., 2016).
Manganese superoxide dismutase (Mn-SOD; SOD2) is a mitochondria (Mt) localized antioxidant enzyme, which detoxifies superoxide anion (O2·−) in Mt (Slot et al., 1986) and is critical in protecting cells from oxidative damage after myriad cellular stress-relevant pathologies, including stroke (Murakami et al., 1998; Kim et al., 2002; Wu et al., 2002; Maier et al., 2006; Jung et al., 2009). Mn-SOD deficiency leads to ROS overproduction, neuronal death, hemorrhagic transformation, and exacerbation of infarction size and neurologic deficits after ischemic injury (Murakami et al., 1998; Fujimura et al., 1999; Kim et al., 2002; Maier et al., 2006). In neurons, signal transducer and activator of transcription 3 (STAT3) transcriptionally upregulates Mn-SOD expression on being phosphorylated at tyrosine (Y705) residue and binding to the Mn-SOD promoter (Jung et al., 2009). Phosphorylation of STAT3 is immediately decreased by ischemic injury in the mouse brain, leading to reduced Mn-SOD expression (Jung et al., 2009, 2011). The resulting Mn-SOD deficiency leads to superoxide accumulation in Mt causing oxidative damage and neuronal death after ischemic/reperfusion injuries (Kim et al., 2002; Jung et al., 2009; Sarafian et al., 2010), attesting to the unique importance of Mn-SOD in preventing damage to neurons under pathologic conditions.
Accumulating evidence reveals the importance of Mt transfer as a mechanism involved in cell-to-cell communication (Islam et al., 2012; Hayakawa et al., 2016, 2018; Jung et al., 2020; Nakamura et al., 2020). We and others have shown that astrocytes release functional Mts that play regulatory and prosurvival functions on entering adjacent cells in the brain (Hayakawa et al., 2016; Jung et al., 2020). Extracellularly released astrocytic Mts are incorporated into the neighboring neuron and promote neuronal survival and stroke recovery by increasing intracellular ATP levels in the mouse model of transient focal ischemia (Hayakawa et al., 2016). We also demonstrated that astrocyte-released Mt incorporates in the microglial cells to promote a reparative microglial phenotype with increased phagocytic capacity to promote hematoma clearance and functional recovery from ICH (Jung et al., 2020). This and present studies suggest that adoptive astrocytic Mt transfer could be considered as a potential approach to treat ICH. Because Mn-SOD is a mitochondrially localized antioxidative enzyme, there could be a beneficial effect of Mn-SOD when transplanted Mt enters targeted cells. Here, we propose that one of the therapeutic mechanisms of Mt transfer is to enhance the Mn-SOD-mediated antioxidant defense system in the recipient ICH brain.
Humanin (HN) is an Mt-derived small peptide transcribed from the Mt genome at the 16S ribosomal RNA locus that is either stored in Mt or secreted into cytosolic spaces following translational processing for autocrine or paracrine signaling (Lee et al., 2013). Our previous study demonstrated that HN is one of the key regulators of astrocytic Mt-mediated neurological recovery in a mouse ICH model by promoting microglia to a phagocytic/reparative and anti-inflammatory phenotype (Jung et al., 2020). HN inhibits ROS generation and protects cells from oxidative-stress-induced cell death, including increased phosphorylation of STAT3 (Hashimoto et al., 2005, 2009; Kim et al., 2016; Sreekumar et al., 2016).
In summary, we propose that transplantation of astrocytic Mt improves functional recovery from ICH, including enhancing the Mn-SOD-mediated neuronal antioxidant defense system and neuroplasticity in the brain.
Materials and Methods
Experimental animals
C57BL6/J male mice (3 months old) were purchased from The Jackson Laboratory and randomly assigned to their groups for each experiment. Female Sprague Dawley rats [embryonic day (E)17 timed pregnant] were purchased from Charles River Laboratories and used for primary cerebral cortical neuron and astrocyte cultures. Animals were maintained under a 12 h light/dark cycle and received a standard diet and ad libitum access to water. All procedures for animal experiments were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Animal Welfare Committee of the University of Texas Health Science Center at Houston.
Generation of Mn-SOD (SOD2)-deficient astrocytes in culture
To evaluate the role of Mn-SOD in astrocytic Mt, we generated hGFAPCreERT2:Mn-SODfl/fl mice by crossing GFAP-CreERT2 (hemizygotes) with Mn-SOD-floxed (Mn-SODfl/fl: homozygote) mice. Specifically, hGFAP-Cre/ERT2 [B6.Cg-Tg(GFAP-cre/ERT2)505Fmv/J, catalog #012849, The Jackson Laboratory] and C57BL6/SOD2LoxP mice were used. C57BL6/SOD2LoxP mice were generated using the cryorecovery service of The Jackson Laboratory with the approval of Ting-Ting Huang at Stanford University and agreement from Takahiko Shimizu in Japan, who originally possessed this mouse strain. To generate Mn-SOD knockout (Mn-SOD-CKO) astrocytes in cultured, postnatal day (P)0–2 mouse brains from hGFAPCreERT2:Mn-SODfl/fl mice were processed to generate primary astrocytic culture, as we described earlier (Zhao et al., 2017; Jung et al., 2020). To induce Mn-SOD-deficiency the astrocytes were treated with 1 µm 4-hydroxytamoxifen (4-OHT; Sigma-Aldrich) to induce Cre recombination. Genotyping was performed by using PCR and the following primer sets specific to the hGFAP-Cre transgene and the SOD2-flox allele; hGFAP-Cre transgene: 5′-CGAGGGGCATCTAGTGGAGAAG-3′, 5′-TTAGGGCTCAGGTTTGTCCAGAA-3′, 5′-AGCTTGGCTGGACGTAA-3′; SOD2-flox allele: 5′-GCCAGTCTAGCCCACTCCTT-3′, 5′-TCCCTGAACATGTCCATCAG-3′.
Tamoxifen-induced activation of cre recombination
Postnatal mouse brains (P0–2) from hGFAPCreERT2:Mn-SODfl/fl mice were used to generate Mn-SOD-deficient primary astrocytes. The Mn-SODfl/fl postnatal mouse brains were used as littermate controls. To induce Cre recombination, 1 µm 4-hydroxytamoxifen (4-OHT; Sigma-Aldrich) was added to astrocyte culture media for 48 h. After 48 h of 4-OHT treatment, the cells were washed with PBS, and fresh new medium was supplied to astrocyte cultures.
Astrocyte-conditioned medium and MitoTracker-labeled Mt
Astrocyte-conditioned medium (ACM), Mt-depleted ACM (mdACM), and ACM containing MitoTracker-labeled Mt were prepared following our and other's well-established protocols (Hayakawa et al., 2016; Jung et al., 2020). Briefly, to prepare ACM or mdACM, fresh culture media was supplied to astrocyte cultures (astrocytes plated on 25 cm2 flask in 3 ml of culture medium with 80–90% of cell confluency or at a density of 0.5–1 × 107 cells/flask). Twenty-four hours later, the culture media was harvested to collect extracellularly released Mt and was centrifuged at 1,000 rpm for 5 min to remove cell debris. The supernatant was carefully collected and used as ACM containing extracellular astrocytic Mt. mdACM was obtained by 0.22 µm filtration of ACM, as previously described (Hayakawa et al., 2016; Jung et al., 2020). To prepare the ACM containing labeled Mt, we used MitoTracker Red/CMXRos (catalog #M7512, Thermo Fisher Scientific), as previously described (Hayakawa et al., 2016; Jung et al., 2020). Briefly, the cultured astrocytes were treated with 200 nm MitoTracker Red/CMXRos (catalog #M7512, Thermo Fisher Scientific) and incubated for 30 min at 37°C. After washing with PBS three times, the astrocytes were supplied with fresh culture medium and incubated for 24 h to allow accumulation of secreted astrocytic Mt. The culture media was harvested and centrifuged at 1,000 rpm for 5 min to remove cell debris, and the supernatant was used for ACM-containing MitoTracker-labeled Mt. The extracellular Mt from cultured astrocytes was assessed using FACS analysis as previously described (Hayakawa et al., 2016; Jung et al., 2020). Briefly, MitoTracker Red/CMXRos-positive particles in ACM or mdACM were detected using a CytoFLEX S cytometer (Beckman Coulter).
ICH surgery
ICH mice models were induced by autologous blood injection, as described previously (Zhao et al., 2015; 2017; Jung et al., 2020). Briefly, C57BL/6 male mice (3 months old) were anesthetized with chloral hydrate (0.35g/kg, i.p.). An anesthetized mouse was placed in a stereotactic frame, and a burr hole in the skull was made at the coordination of 0.0 mm anterior and 3.0 mm lateral to the bregma. A 30 gauge needle was inserted into the left striatum 3.5 mm from the brain surface, and 18 µl of peripheral blood, harvested from the femoral vein of the mouse, was infused at 1 µl/min. Sham mice were operated on using the same procedure except for blood infusion. Core body temperature was monitored and maintained at 37.0 ± 0.5°C during the surgical procedure and 2 h following surgery. Next, 200 µl of ACM or mdACM (by 0.22 µm filtration of ACM to remove Mt particles) were injected intravenously at 1 h, 7 d, and 14 d post-ICH onset.
Evaluation of neurological deficit score
The corner test or foot fault test assessed the neurological deficit score (NDS), which was calculated following previously established protocols (Zhao et al., 2007, 2017; Jung et al., 2020). Tests were conducted pre-ICH and on days 7, 14, and 21 post-ICH. The behavioral examiner was blinded to the assignment of each animal group. A corner turn test was performed to identify sensorimotor deficits that reflect multiple asymmetries (Schaar et al., 2010). Briefly, the animal is placed facing the corner (formed by two Plexiglas boards connected at a 30° angle) and allowed to approach the corner (Schaar et al., 2010). After full turns back to the open edge, the numbers of right turns versus left turns of 10 trials were counted and the NDS was calculated as follows: NDS score = [absolute value of the difference between right turns and left turns] × 0.4, to make the maximum score 4.
The foot fault test was performed as we described previously (Zhao et al., 2007; 2017; Jung et al., 2020), with the following modification. Briefly, the animal is placed on the elevated horizontal ladder consisting of rungs (1.5 mm diameter and 5–20 mm randomly apart) and clear side walls (80 cm long and 15 cm high) and allowed to traverse the ladder. As the animal traverses the ladder, the video is recorded. Ten consecutive steps of ipsilateral forelimb per animal are evaluated for placing dysfunction. Depending on the quality of each step, the grading is the following: 0, balanced step on a full foot; 0.1, step on digits only; 0.2, step on waist only or a trial step/fixed; 0.3, step on one digit only or step-slip fixed or a trial step few times; and 0.4, slip, balance lost, or step missed (fault). Thus, the worse performance is scored as 4 (10 steps × 0.4 = 4).
In situ detection of O2·−production
O2·− production was evaluated by detecting oxidized dihydroethidium (HEt) in situ. HEt is a fluorescent probe oxidized to ethidium (Et) by superoxide anions (Murakami et al., 1998). Once HEt is incorporated into damaged cells and oxidized to Et by superoxide anion, the nucleus of the cells can be detected as red fluorescence (Murakami et al., 1998; Jung et al., 2009). In this study, the ROS production in ICH-operated mice was evaluated by in situ detection of oxidized HEt, as described previously (Murakami et al., 1998; Kim et al., 2002; Jung et al., 2009). Briefly, 1 mg HEt (catalog #D11347, Invitrogen) was dissolved in dimethyl sulfoxide and further diluted to 1 mg/ml in PBS following guidelines from the manufacturer. Mice were intravenously administered 200 µl 1 mg/ml HEt solution 15 min before ICH or sham operation. Twenty-four hours later, mice were anesthetized and subjected to cardiac perfusion with ice-cold PBS followed by 4% paraformaldehyde (PFA). Next, 20 µm sections were made using a microtome (catalog #HM450, Fisher Scientific). The Et fluorescence signal was detected by a Zeiss LSM 780 confocal laser scanning microscope using the following conditions: Excitation/Emission (Ex/Em) = 518 nm/606 nm.
Immunohistochemistry
All animal were euthanized and procedures for immunohistochemistry were performed with minor modifications following our previously established protocol (Jung et al., 2020). Briefly, mice were anesthetized and subjected to cardiac perfusion with cold PBS. After cardiac perfusion, dissected brains were immediately transferred to 4% PFA for fixation, incubated for 24 h, and then moved into 30% sucrose. After 24 h, 20 µm sections were made using a microtome with freezing unit (catalog #HM450, Fisher Scientific), and the sections were preserved in an anti-freezing buffer (20 mm sodium phosphate, 20 mm sodium hydroxide, 30% glycerol, and 30% ethylene glycerol in distilled water) at −20°C until processed for immunostaining. Sections were permeabilized with 0.3% Triton-X in PBS for 30 min at room temperature and blocked in blocking buffer (5% goat serum, 3% bovine serum albumin, and 0.1% Triton X in PBS) for 1 h at room temperature. After washing with PBS, the sections were incubated with neuronal nuclei (NeuN) Alexa 488 conjugated antibody (1:200; catalog #MAB377X, Millipore), Synapsin 1/2 (1:200; catalog #106003, Synaptic Systems), MAP-2 (1:200; catalog #MA5-12826, Invitrogen), and Mn-SOD rabbit polyclonal antibody (1:200; catalog #24127-1-AP, Proteintech) for 24 h at 4°C. For secondary antibodies staining, samples were incubated with Alexa Flour 546 goat anti-rabbit antibody (1:500; catalog #A11035, Invitrogen) or Alexa Flour 488 goat anti-mouse antibody (1:500; catalog #A11029, Invitrogen) or Alexa Flour 488 goat anti-rabbit antibody (1:500; catalog #A11034, Invitrogen) for 2 h at room temperature. After nuclei counterstain with DAPI in PBS for 10 min at room temperature, cells were mounted with Prolong Diamond Antifade (Invitrogen). Images were captured by a Zeiss LSM 780 confocal laser scanning microscope.
Primary astrocyte cultures
Primary rat or mouse astrocyte cultures were prepared using E17-day-old Sprague Dawley rat brains or postnatal (P0-2) mouse brains, respectively, as described previously (Zhao et al., 2017; Jung et al., 2020). Briefly, the brain tissues were dissected for collecting cortices, and the meninges removed. The cortices were digested in 0.25% trypsin to make a single cell suspension and plated on poly-D-lysine-coated 75-cm2 flasks. The mixed glial cells were cultured in DMEM containing 10–20% fetal bovine serum (FBS), 50 U/ml penicillin, and 50 µg/ml streptomycin for 2 weeks. Once the mixed glial cells were fully confluent, the cells were trypsinized with 0.25% trypsin and replated onto new poly-D-lysine-coated 75-cm2 flasks. Several hours after plating, the flasks of cells were subjected to shaking at 220 rpm overnight at 37°C incubator with 5% CO2 to remove loosely attached microglia and oligodendrocyte precursor cells from the cell monolayer. The floating cells were discarded the following day, and fresh media was supplied to pure astrocyte cultures. Astrocytes were cultured in a humidified incubator at 37°C with 5% CO2.
Primary neuron cultures
Primary rat neurons were prepared from cerebral cortices using E17 Sprague Dawley rat brains with minor modifications from our previously established protocols (Jung et al., 2009, 2015). Briefly, the brain tissues were dissected for collecting cortices and the meninges removed. The cortices were digested in 0.25% trypsin to make a single-cell suspension. The cells were seeded on poly-D-lysine-coated culture plates in DMEM containing 10% FBS, 50 U/ml penicillin, and 50 µg/ml streptomycin. Twenty-four hours after seeding, the culture medium was replaced with Neurobasal medium with 2% B-27, 0.5 mmol/l glutamine, and 1% Antibiotic-Antimycotic in six-well or 24-well plates.
Real-time oxygen consumption
Real-time oxygen consumption of astrocytic Mt in ACM or mdACM was determined using an extracellular O2 consumption assay kit (catalog #ab197243, Abcam) according to instructions from the manufacturer. The astrocytes culture media, DMEM containing 10% FBS, was used as negative control. Briefly, 20 µl O2 consumption reagent was added to 300 µl ACM or mdACM. After gently mixing, the fluorescence was immediately measured (every 1.5 min up to 30 min) using the filter combination of Ex/Em = 380 nm/650 nm.
In vitro ICH-like injury
To mimic in vitro ICH-like injury, we used RBC lysates in the cell cultures. RBC isolation and lysis were prepared following our previously established protocols (Zhao et al., 2017; Jung et al., 2020). Briefly, the peripheral blood from a mouse or rat was obtained via cardiac puncture. RBCs were isolated using the Ficoll-Plaque density gradient. RBCs were lysed with distilled water to form the lysate. RBC lysates from 3 × 107 RBCs were added to each well (3 × 105 neurons/well) of the cell culture plate to mimic ICH-induced lethal injury. The RBCs lysates were further diluted 1:3 using distilled water to mimic ICH-induced sublethal injury (1 × 107 RBCs lysates onto 3 × 105 neurons/well of the cell culture plate) and salvageable tissue at the perihematoma area in the brain. This ICH-induced sublethal injury was used for neurite extension assessment and synaptogenesis-related gene expression.
Assessment of cell viability
In vitro neuronal viability was measured by lactate dehydrogenase (LDH) assay using a Cytotoxicity Detection Kit (Roche Applied Science) following guidelines from the manufacturer. LDH is an intracellular enzyme and is released rapidly on plasma membrane damage. Thus, the amount of LDH remained in the cell can be used to probe cell viability. Briefly, cultured neurons (3.0 × 105 neurons/well) were pretreated with mdACM, ACM, or 100 ng/ml recombinant HN protein (catalog #CSB-MP814209HU, Cusabio) for 24 h, followed by exposure to RBC lysates for 48 h. After washing with PBS three times, neurons were lysed with 0.1% Triton X for 15 min. After centrifugation at 14,000 rpm for 15 min, the cell extracts containing LDH were harvested. By adding LDH reaction buffer into cell lysates, absorbance was measured at 490 nm to detect the amount of remained LDH in cytosol, which corresponds to cell viability.
Measurement of ROS
Intracellular ROS was measured using MitoSOX Red mitochondrial superoxide indicator for live-cell imaging (catalog #M36008, Thermo Fisher Scientific) following the guidelines of the manufacturer. Cultured neurons were pretreated with mdACM, ACM, or 100 ng/ml recombinant HN protein for 24 h, followed by exposure to RBC lysates for 12 h. After washing the neurons with PBS three times, 5 µm MitoSOX reagent was added, incubated at 37°C for 10 min, and washed with PBS three times. Absorbance at 548 nm determined the neuronal ROS production.
Quantitative RT-PCR
For RNA isolation from the brain, mice were killed at 24 h or 21 d after ICH and perfused with ice-cold PBS. The brain tissues were removed and snap frozen. Tissue samples were homogenized (brain) and lysed (tissue culture) with TRIzol reagent. For RNA isolation from cultured neurons or astrocytes, the cells were washed with PBS three times and immediately frozen. Total RNA was extracted using RNA isolation kit (RNeasy Mini Kit, QIAGEN) and converted with amfiRivert Platinum One cDNA synthesis Platinum Master Mix (catalog #R6100, GenDEPOT) according to the protocols of the manufacturer. Real-time quantitative PCR (RT-qPCR) was performed using amfiSure qGreen Q-PCR Master Mix without ROX (catalog #Q5600, GenDEPOT). The following primers sets were used: Rat Sod2 Forward (Fw), 5′-AAGCGTGACTTTGGGTCTTT-3′; Rat Sod2 Reverse (Rv), 5′-ATCCCCAGCAGTGGAATAAG-3′; Rat Syn1 Fw, 5′- GTGTCAGGGAACTGGAAGACC-3′; Rat Syn1 Rv, 5′-AGGAGCCCACCACCTCAATA-3′; Rat Syp Fw, 5′-ACTACTCCTCGTCGGCTGAA-3′; Rat Syp Rv, 5′-ACAGGGTCCCTCAGTTCCTT-3′; Rat Dlg4, QuantiTect Primer Assay, Assay name, Rn_Dlg4_1_SG, QIAGEN; Rat Gapdh Fw, 5′-AGA CAG CCG CAT TTC TTG T-3'; Rat Gapdh Rv, 5′-CTT GCC GTG GTA GAG TCA T-3'; Mouse Sod2 Fw, 5′-ACA GCG CAT ACT CTG TGT GA-3'; Mouse Sod2 Rv, 5′-GGG GGA ACA ACT CAA CTT TT-3'; Mouse Gapdh Fw, 5′-TGT TCC TAC CCC CAA TGT GT-3'; Mouse Gapdh Rv, 5′-TGT GAG GGA GAT GCT CAG TG-3′.
Western blot analysis
Samples were lysed with RIPA buffer containing halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). The whole-cell lysates were centrifuged at 14,000 × g for 15 min at 4°C. Protein content was evaluated using a Bio-Rad Protein Assay Kit (catalog #500-0006, Bio-Rad). A total of 30 µg protein sample was boiled with Laemmli sample buffer (catalog #NP0007, Invitrogen) for 5 min at 90°C, followed by separation on 4–20% gradient Tris-Glycine polyacrylamide gels (Thermo Fisher Scientific). Proteins were blotted onto nitrocellulose membranes with the iBlot2 Dry Blotting System (Thermo Fisher Scientific). Membranes were blocked (catalog #927-60001, LI-COR) and incubated with primary antibodies against Synapsin 1/2 (catalog #106003, Synaptic Systems), PSD-95 (catalog #20665-1-AP, Proteintech), p-STAT3 (catalog #D3A7, Cell Signaling Technology), Mn-SOD (catalog #24127-1-AP, Proteintech), 3-nitrotyrosine (catalog #X1719M, Exalpha Biologicals), and β-actin (catalog #D6A8, Cell Signaling Technology) at 4°C overnight and 30 min incubation at room temperature. After washing three times with 0.5% Tween 20 Tris-buffered saline (TBS), membranes were incubated with IRDye 800CW donkey anti-rabbit IgG (catalog #926-33214, LI-COR) and IRDye 800CW donkey anti-mouse IgG (catalog #926-32212, LI-COR) secondary antibodies for 1 h and washed with 0.5% Tween 20 TBS. All primary and secondary antibodies were used at 1:1000 or 1:2000 dilution, respectively. Images were visualized using LI-COR Odyssey CLx, and signal intensity was measured with ImageJ software (National Institutes of Health).
Measurement of neurite length
Neurite outgrowth measurements in cultured neurons were performed as previously described (Pekcec et al., 2013). Rat neurons were seeded at 5 × 104 cells/well of poly-D-lysine-coated 12-well plates. At day in vitro (DIV)1, neurons were pretreated with ACM, mdACM, or 100 ng/ml recombinant HN protein for 24 h then exposed to 1.7 × 106 RBCs lysate for 96 h. Phase-contrast images were captured at DIV7 using an Olympus IX81 motorized inverted microscope. Length of individual neurites from each treatment group were manually measured using ImageJ software.
Immunocytochemistry
For immunocytochemistry, primary rat neurons were seeded (5 × 104 cells/well) onto poly-D-lysine-coated German glass in 24-well plates. At DIV2, neurons were pretreated with ACM or mdACM for 24 h, then exposed to RBCs lysate (1.7 × 106 RBCs/well) for 96 h. After washing with PBS, neurons were fixed with 4% PFA for 15 min, permeabilized with 0.3% Triton-X in PBS for 30 min at room temperature, and blocked in blocking buffer (10% goat serum, 3% BSA, and 0.1% Triton-X in PBS) for 3 h at room temperature. After washing with PBS, cells were incubated with primary antibodies against Synapsin 1/2 (1:200; catalog #106003, Synaptic Systems) and MAP-2 (1:200; catalog #MA5-12826, Invitrogen) overnight at 4°C. After washing with PBS three times, cells were incubated with appropriate Alexa 488 or Alexa 568 conjugated secondary antibodies at 1:500 dilution for 2 h at room temperature. Nuclei were stained with DAPI in PBS for 10 min at room temperature. Cells were mounted with ProLong Diamond Antifade (Invitrogen). Images were captured by Zeiss LSM 800 confocal laser scanning microscopy with 63× oil immersion objectives in 1024 × 1024 scanning format.
Knockdown using siRNA
Primary neurons or astrocytes were seeded in 12-well plates at 3.0 × 105 cells/well or 24-well plates at 5.0 × 104 cells/well, or 6-well plates at 7.5 × 105 cells/well, respectively. siRNA knockdown targeted rat Sod2 using the following sense: 5′-GAUACAUSGUUUGCAAGAATT-3′ and antisense 5′-UUCUUGCAAACUAUGUAUCTT-3′ sequences (catalog #s103032120, QIAGEN). Neurons and astrocytes were transfected with 10 nmol/l or 200 nmol/l siRNA, respectively. Scrambled siRNA (Scr si; catalog #SIC001, Sigma-Aldrich) acted as the negative control. Transfection used HiPerFect siRNA Transfection reagent (catalog #301704, QIAGEN) following the guidelines from the manufacturer.
Experimental design and statistical analysis
Experimental animals were randomly assigned to each group for in vivo experiments. The sample size was determined by our previous studies using in vivo and in vitro ICH models (Zhao et al., 2007, 2015, 2017; Jung et al., 2020) and power analysis using STATMATE 3 software (GraphPad) with the following parameters: estimated SD of each group at 0.1–0.2, a significant difference among group by power of 80–90%, and expected difference among groups at 20–30%. GraphPad Prism version 7.04 software was used for statistical analysis. One-way ANOVA followed by Fisher's least significant difference (LSD) was used to compare multiple groups. One caveat in using Fisher's LSD following ANOVA is that Fisher's LSD may not adequately correct for multiple comparisons and thus could increase false positives. To ensure the statistical conclusions, we confirmed the results by using other tests (eg., Tukey's method) following ANOVA, which may better correct for multiple comparisons (data not shown). Repeated measures one-way ANOVA with a multiple comparison test using Tukey's method was used to compare groups in continuously measured oxygen consumption assay. An unpaired t test assessed the difference between two groups. For in vivo behavioral test, repeated measures two-way ANOVA followed by Fisher's LSD was used between groups at different time points. All the statistical tests were two sided. Data are shown as mean ± SEM. Statistical significance was considered as follows: p < 0.05 (*p < 0.05, **p < 0.01). All the detailed information regarding statistical analysis in each experiment are addressed in the figure legends.
Results
ICH disrupts the mitochondrial antioxidative defense system and induces oxidative damage in the affected brain hemisphere
Hematoma degradation products trigger massive ROS generation, oxidative stress, and disruption of antioxidant defense systems (Wu et al., 2002; Aronowski and Hall, 2005; Aronowski and Zhao, 2011). This imbalance between oxidative stress and antioxidant defense results in ROS-mediated cellular stress and Mt dysfunction, contributing to brain injuries after ICH (Kim-Han et al., 2006; Lu et al., 2015). Thus, first we examined the effect of ICH on expression of Mn-SOD, an antioxidant enzyme specifically localized in Mt that plays an essential role in protecting cells from oxidative stress. In mice subjected to ICH, the Mn-SOD expression was significantly decreased in the ipsilateral hemisphere compared with the contralateral hemisphere 24 h after stroke (Fig. 1A). The significant reduction of the Mn-SOD level in the ipsilateral hemisphere after ICH with immunohistochemistry was confirmed by Western blot analysis (Fig. 1B). We also found a significant reduction of Mn-SOD mRNA levels in ipsilateral hemisphere compared with contralateral hemisphere (Fig. 1C). We next examined the O2·− production in ICH-affected hemispheres using in situ O2·− generation probe HEt. Animals were injected with HEt right before ICH, and 24 h later the oxidized HEt signal (red fluorescence) was assessed. We found a robust increase in O2·− formation in the perihematoma regions (Fig. 1D). Superoxide radicals themselves produce cellular injury, but also by reacting with nitric oxide, they induce peroxynitrite formation that is cytotoxic through nitration of tyrosine residue on many vital proteins (Ding et al., 2014; Duan et al., 2016). Thus, in our next experiment, we measured 3-nitrotyrosine (3-NT) formation after ICH. As expected, we found a significant increase in the 3-NT formation in the ipsilateral hemispheres of ICH-affected brains (Fig. 1E), confirming the overall increase in superoxide availability. These data demonstrate that ICH disrupts mitochondrial-dependent superoxide generation coinciding with reduced Mn-SOD expression in the ICH-affected brain.
Figure 1.
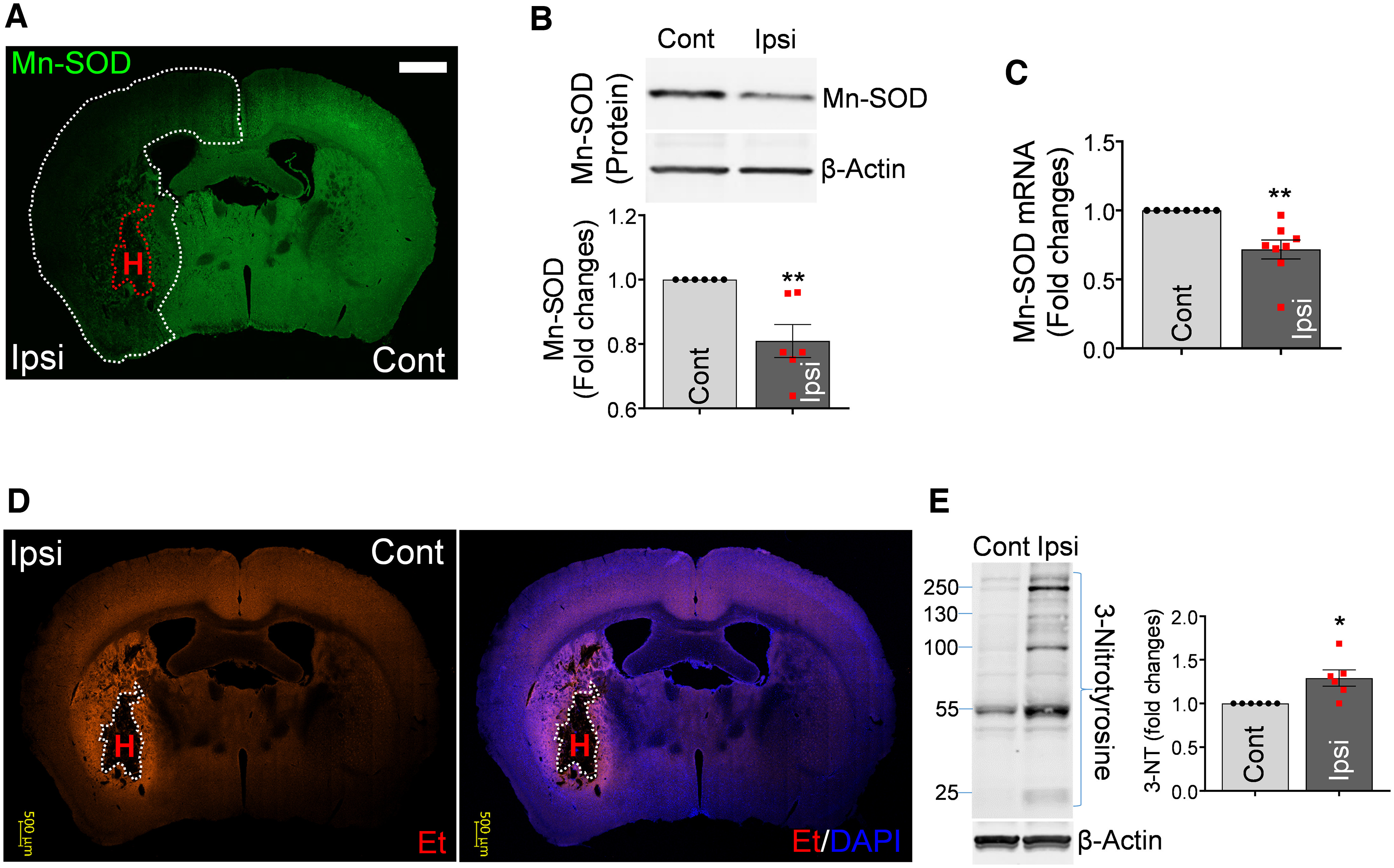
ICH disrupts mitochondrial antioxidative defense and induces oxidative/nitrosative damage in the affected brain hemisphere. A, Immunohistochemistry; immunoreactivity for Mn-SOD (green) in the coronal section of mouse brain at 24 h after ICH. Red dotted line delineates hematoma location (H), and white dotted line delineates ICH-affected area with reduced Mn-SOD expression in the ipsilateral hemisphere. Scale bar, 1000 µm. Cont, Contralateral; Ipsi, ipsilateral. B, Representative Western blot image and illustrating bar graph showing Mn-SOD protein quantitation in the Cont and Ipsi hemispheres at 24 h after ICH. The significance of Mn-SOD/β-actin protein levels was assessed by two-tailed unpaired t test (n = 6 per group), **p < 0.01 (p = 0.0041, Cont vs Ipsi), t value (t = 3.706). C, Mn-SOD mRNA levels in the Cont and Ipsi hemispheres at 24 h after ICH, evaluated by RT-qPCR. Values represent fold change of mRNA levels in ipsilateral hemispheres compared with contralateral hemispheres. Data are shown as mean ± SEM. The significance of Mn-SOD mRNA levels was assessed by two-tailed unpaired t test (n = 8 per group), **p < 0.01 (p = 0.0012, Cont vs Ipsi), t value (t = 4.051). D, In situ detection of intracellular O2·− in mice at 24 h after ICH that were intravenously injected with 200 µl 1 mg/ml HEt (superoxide indicator) solution 15 min before ICH onset. HEt is incorporated into damaged cells, and on oxidation by superoxide anion is converted to Et, which generates red fluorescence (Murakami et al., 1998; Jung et al., 2009). White dotted line delineates hematoma area (H). Nuclei were counterstained with DAPI (blue). Scale bar, 500 µm. E, Representative Western blot image and quantitative bar graph showing 3-NT protein levels in Cont and Ipsi hemispheres at 3 h after ICH-inducing surgery. The significance of 3-NT/β-actin protein levels was assessed by two-tailed unpaired t test (n = 6 per group), *p < 0.05 (p = 0.0110, Cont vs Ipsi), t value (t = 3.114).
Astrocytic Mt improves neurological outcome after ICH model in mice
Our studies (Jung et al., 2020) and others' (Hayakawa et al., 2016) demonstrated that astrocytes secrete functional Mt, which can enter the neighboring cells in the brain. Hayakawa et al. (2016) demonstrated that the astrocytic Mts were found within the adjacent neurons in ischemic mouse brains and that they amplified cell survival signals and enhanced ATP generation under ischemic injury in vitro (Hayakawa et al., 2016). Although this seminal study coined the concept of Mt-based therapy for stroke-induced brain damage, it is clear there are still many pending questions regarding the mechanism by which transferred Mt mediates neuroprotection. ROS overproduction after ICH causes cellular/mitochondrial damage leading to progression of brain injury (Aronowski and Hall, 2005; Aronowski and Zhao, 2011; Hu et al., 2016; Hemorrhagic Stroke Academia Industry (HEADS) Roundtable Participants, Second HEADS Roundtable Participants, 2020). Thus, the objective of next experiment was to test whether mitigating ROS by replacement of damaged Mt through systemic administration of functional Mt, which is the source of the antioxidant enzyme Mn-SOD, can protect neurons and improve recovery after ICH.
To test the hypothesis, we first examined that systemically administered astrocytic Mt indeed enters the ICH-affected brain. The Mts of astrocytes in culture were fluorescently labeled with MitoTracker-Red/CMXRos, and the extracellularly released labeled Mts from astrocytes were collected and then via the femoral vein injected into mice at 24 h after ICH onset. Twenty-four hours after the injection, these labeled Mts were detected in the perihematoma tissue (Fig. 2A). We calculated, using five independent brains, that 57.7–69.7% of neurons (NeuN-positive cells) in the ipsilateral hemisphere of ICH-injured brains contained injected labeled astrocytic Mt (Fig. 2B,C). These data indicate that systemically transplanted astrocytic Mt can enter the brain and be taken up by the neurons in ICH-affected brain tissue.
Figure 2.

Astrocytic Mt improves neurologic outcomes in ICH mice and restores Mn-SOD level in neurons in the ICH-affected hemispheres. A, B, The ACM containing MitoTracker-Red/CMXRos-labeled Mt (red) was intravenously injected at 24 h after ICH onset, and the animals were killed 24 h after Mt injection and processed for immunohistochemistry. A, Representative confocal microscopy image of the perihematomal area of the brain of mouse, which was injected with ACM containing MitoTracker-Red/CMXRos-labeled Mt (red). Nuclei were counterstained with DAPI (blue). Scale bar, 50 µm. B, Representative confocal microscopy image of neurons in the ICH-affected hemisphere (brains 1 and 2), which were injected with ACM containing MitoTracker-Red/CMXRos-labeled Mt (red). Arrows indicate Mt incorporated into neurons. Neurons were labeled with NeuN (green) and nuclei with DAPI (blue). Scale bar, 5 µm. C, The graph shows the percentage of neurons (NeuN+ cells) that are positive for astrocytic Mt in the ICH-affected hemisphere (collected from 2–5 fields per brain) as assessed for five independent mice brains (1–5). D, NDS established by corner turn test in ICH-injured mice that were intravenously injected with ACM or mdACM at 1 h, 7 d, and 14 d after ICH onset. The significance of NDS was assessed by repeated measures two-way ANOVA with multiple comparison and Fisher's LSD test (n = 8–9 per group), *p < 0.05 (p = 0.0431, ACM-treated mice vs mdACM-treated mice at 21 d), t value (t = 2.067). E, Representative images of immunofluorescent staining of Mn-SOD in neurons (NeuN-positive cells) in the ICH-affected area of the brain at 21 d after ICH in mice that intravenously received ACM or mdACM at 1 h, 7 d, and 14 d after ICH. White arrows indicate the cells showing high Mn-SOD (red) in the NeuN (green)-positive cells as result of treatment with ACM. Nuclei were counterstained with DAPI (blue). Scale bar, 40 µm. F, The dot graph illustrating the levels of Mn-SOD immunofluorescence signal intensity (measured in brain sections; 9 randomly selected fields per each brain) in the ipsilateral hemisphere of ACM-treated (red squares) versus mdACM-treated mice (black circles), as described in E; n = 5 animals per treatment group. The significance was assessed by two-tailed unpaired t test (n = 5 mice per group), *p < 0.05 (p = 0.0425, ACM-treated mice vs mdACM-treated mice), t value (t = 2.41). All data are shown as mean ± SEM. To test whether Mn-SOD level in ACM may affect protective effect of ACM, we also used ACMs from two populations of astrocytes that were exposed to Mn-SOD siRNA to deplete Mn-SOD or to scrambled siRNA (control). NDS was evaluated by the foot fault test in ICH-injured mice, which were treated with ACMs obtained from these astrocytes (Extended Data Fig. 2-1).
Next, we examined whether administration of astrocytic Mt can improve outcomes after ICH. Our and others' earlier studies demonstrated that Mts are secreted by cultured primary astrocytes in large amounts, they range from 300 to 1100 nm in size, and thus are effectively removed from ACM by 0.22 µm filtration (Hayakawa et al., 2016; Jung et al., 2020). We previously confirmed that 78% of Mt particles from ACM were removed through a 0.22 µm filter (Jung et al., 2020). We subjected animals to ICH and intravenously injected ACM or mdACM (by filtration) once a week, starting 1 h after ICH, for 3 weeks (three injections total) and measured the NDS of ICH mice with the corner turn test. The NDS score was significantly reduced in ACM-treated mice at day 21 (Fig. 2D). Our data suggest that systemically administered astrocytic Mts enter brain, are incorporated into the neurons in the perihematoma area, and improve neurological outcome following ICH. When analyzing brains of these mice after completing the behavioral test (21 d), we found that ACM-receiving animals, compared with animals receiving mdACM, showed significant restoration of Mn-SOD in the neurons throughout perihematomal brain areas (Fig. 2E,F). To examine whether Mn-SOD is important for ACM-mediated stroke recovery, by using Mn-SOD siRNA (vs scrambled siRNA), we generated Mn-SOD-deficient astrocytes. The Mn-SOD level in ACMs from astrocytes treated with Mn-SOD siRNA versus scrambled siRNA was reduced by 23% based on Western blot analysis for Mn-SOD protein levels in ACM. We used ACMs from these two populations of astrocytes to test whether Mn-SOD level in ACM may have an impact on the protective effect of ACM. Thus, we subjected mice to ICH and intravenously injected ACM from Mn-SOD knockdown astrocytes (Mn-SOD-KD-ACM) or ACM from astrocytes treated with scrambled siRNA (scr-ACM). ACM was delivered once a week, starting 1 h after ICH, for 3 weeks (three injections total), and neurological deficit was determined on days 3, 7, 14, and 21. Mice receiving Mn-SOD-KD-ACM showed significantly worse NDS at day 3 after ICH compared with the mice receiving the scr-ACM and no difference in behavior during consecutive (days 7–21) testing (Extended Data Fig. 2-1). This result may suggest that the higher level of Mn-SOD may benefit therapeutic capacity of transplanted Mt.
Neuronally incorporated astrocytic Mt moves along the axon of neurites of immature neurons in culture
Hayakawa et al. (2016) demonstrated the incorporation of transferred astrocytic Mts into primary neurons in culture. Here, we expanded their finding by asking how long the transferred astrocytic Mts can be retained inside the neurons and whether they are transported along neurites as the neurites grow. First, we asked whether the astrocyte-released Mts are indeed functional or not. To answer this, we measured mitochondrial oxygen consumption from ACM or mdACM (filtered ACM) preparation. We found by measuring Mt oxygen consumption, that the astrocyte-released Mts are fully functional (Fig. 3A). The oxygen consumption levels in ACM were significantly higher than in mdACM (Fig. 3A). In addition, the oxygen consumption levels in mdACM were very similar to the levels in negative control (nonconditioned fresh culture media), suggesting again that the filtration method was indeed effective in removal of Mts from ACM (Fig. 3A).
Figure 3.

Mt functionality and neuronally incorporated astrocytic Mt moves along the axonal growth. A, Real-time oxygen consumption in ACM (red circles), mdACM (blue squares), and astrocyte culture media alone (DMEM with 10% FBS, as a negative control, green triangles); 20 µl O2 consumption reagent was added to 300 µl ACM, mdACM, or media in each well. The fluorescence was immediately measured every 1.5 min up to 30 min. Data are shown as mean ± SEM. Repeated measures one-way ANOVA assessed the significant changes in oxygen consumption with a multiple comparison test using Tukey's method (n = 3 per group), *p < 0.01 (ACM vs mdACM, q = 28.17). B, Schematic diagram of Mt transfer from astrocytes to neurons in vitro. Rat cortical astrocytes in culture were stained with 200 nm MitoTracker-Red/CMXRos to label astrocytic Mt, followed by washing and incubation in fresh media for 24 h. The medium was collected, half of the media was filtered to generate mdACM, and then ACM or mdACM was transferred to neurons in culture. Scale bar, 20 µm. C, Representative phase-contrast images of two rat cultured primary neurons, which were treated with ACM or mdACM. Images were captured at 0, 4, 24, 72, 120, and 168 h after treatment of ACM or mdACM. Yellow arrowheads point to MitoTracker-Red/CMXRos-labeled Mt incorporated into the neurons. Scale bar, 10 µm. Neuronally incorporated astrocytic Mt redistributes along the axonal growth (Extended Data Fig. 3-1).
Next, we labeled the Mt in astrocytes using MitoTracker-Red/CMXRos, collected labeled ACM, prepared mdACM, then transferred ACM/mdACM onto cultured immature neurons (day 1 after seeding; Fig. 3B), and live imaged the gradual incorporation of MitoTracker-Red/CMXROS-labeled astrocytic Mt into cultured neurons, over 168 h (Fig. 3C). The neuronally incorporated red labeled Mts appeared 4 h after ACM transfer and were found there for at least 168 h (Fig. 3C). As the neurites extended, these astrocytic Mts were found along the neurite (primary axon) at 120 h or 168 h (Fig. 3C, Extended Data Fig. 3-1, yellow arrowheads), indicating that the incorporated Mts gradually migrate from soma to neurite as the neurites grow. The neurite outgrowth, including axon elongation, requires a consistent supply of energy with proper Mt transport from the soma to distal areas (Morris and Hollenbeck, 1993; Han et al., 2016). Our data suggest that the astrocyte-released Mts have physiological oxygen consumption levels (Fig. 3A), and they are transported along the axon during neuronal growth (Fig. 3C, Extended Data Fig. 3-1, yellow arrowheads).
Astrocytic Mt protects neurons against ICH-like injury via inhibition of ROS overproduction
We found that Mn-SOD levels were decreased in ICH-affected mice brains (Fig. 1A–C). We also found that administered astrocytic Mts entered the NeuN-positive cells in ICH brains that coincide with significant restoration of Mn-SOD levels and improved neurologic deficits (Fig. 2). Because Mn-SOD is a mitochondrial localized protein, we now examined whether the protective effect of astrocytic Mt against ICH is because of the restoration of neuronal Mn-SOD by astrocytic Mt uptake/transfer.
To test this as a possibility, we used the validated in vitro ICH-like injury model (Zhao et al., 2017) based on exposure of cultured neurons to RBCs (main components of hematoma). Hb, heme, and iron in RBC robustly promote ROS generation, leading to neuronal death and brain tissue damage (Regan and Panter, 1996; Felberg et al., 2002; Wagner et al., 2003; Nakamura et al., 2004; Aronowski and Hall, 2005). We exposed neurons in culture to lysed RBCs and established that lysed RBCs resulted in an approximate eight- to nine-fold increase in ROS production by the injured neurons, compared with vehicle control (Fig. 4A). This ROS production in neurons was effectively prevented by ACM but not by mdACM (Fig. 4A). Next, we tested whether the astrocytic Mt transfer can protect neurons from ICH-like injury. We found that the neuronal viability was effectively restored by ACM but not by mdACM (Fig. 4B). Moreover, we found that the resulting ICH-like injury-induced ROS generation in neurons was effectively prevented by ACM and that ACM collected from astrocytes after longer incubation time offered more robust benefits in reducing ROS (Extended Data Fig. 4-1A,B). Specifically, cultured neurons under ICH-like injury pretreated with ACMs that were collected from astrocytic culture at 48 h of incubation showed 34% more reduction in ROS production, compared with ACM that was collected at 24 h (Extended Data Fig. 4-1A).
Figure 4.

Astrocytic Mt protects neurons from ICH-like injury by upregulating Mn-SOD and inhibiting ROS overproduction in culture. A, ROS generation, assessed with MitoSOX Red mitochondrial superoxide indicator, by neurons in culture (3.0 × 105 neurons/well) pretreated with ACM or mdACM for 24 h before ICH-like injury for 12 h in vitro (RBC lysates, 3.0 × 107 RBCs/well). The significant changes in ROS generation were assessed by one-way ANOVA/Fisher's LSD test (n = 4 per group), **p < 0.01 (p = 0.0004, CON vs VEH plus ICH-like injury], t value (t = 4.841); **p < 0.01 (p = 0.0011, VEH plus ICH-like injury vs ACM plus ICH-like injury), t value (t = 4.271); **p < 0.01 (p = 0.0002, ACM plus ICH-like injury vs mdACM plus ICH-like injury), t value (t = 5.41). CON, Control; VEH, vehicle. ACM collected from astrocytes on longer incubation is more effective in preventing ROS generation from neurons under ICH-like injury (Extended Data Fig. 4-1). B, Viability of neurons under ICH-like injury conditions and treatment protocol as described for A. Neuronal viability was measured by LDH assay. Values represent fold change in viabilities versus CON. The significance in neuronal viability was assessed by one-way ANOVA/Fisher's LSD test (n = 4 per group), **p < 0.01 (p < 0.0001, CON vs VEH plus ICH-like injury), t value (t = 5.943); **p < 0.01 (p < 0.0001, VEH plus ICH-like injury vs ACM plus ICH-like injury), t value (t = 9.686); **p < 0.01 (p < 0.0001, ACM plus ICH-like injury vs mdACM plus ICH-like injury), t value (t = 8.784). C, Viability of neurons (3.0 × 105 neurons/well) that were transfected with 10 nm Mn-SOD-siRNA (SOD2 si) or Scr si for 24 h before exposure to ACM or control media for 24 h and then exposed to ICH-like injury in vitro (RBC lysates, 3.0 × 107 RBCs/well) for 48 h. Values represent fold change of viabilities compared with Scr si group. The significance in neuronal viability was assessed by one-way ANOVA/Fisher's LSD test (n = 18 in Scr si, n = 15 in Scr si plus ICH-like injury, 7 in Scr si plus ACM plus ICH-like injury, 25 in SOD2 si plus ICH-like injury, and 11 in SOD2 si plus ACM plus ICH-like injury), **p < 0.01 (p < 0.0001, Scr si vs Scr si plus ICH-like injury), t value (t = 5.354); *p < 0.05 (p = 0.0232, Scr si plus ICH-like injury vs SOD2 si plus ICH-like injury), t value (t = 2.32); **p < 0.01 (p < 0.0001, Scr si plus ICH-like injury vs Scr si plus ACM plus ICH-like injury), t value (t = 5.963); **p < 0.01 (p = 0.0023, Scr si plus ACM plus ICH-like injury vs SOD2 si plus ACM plus ICH-like injury), t value (t = 3.156); **p < 0.01 (p < 0.0001, SOD2 si plus ICH-like injury vs SOD2 si plus ACM plus ICH-like injury), t value (t = 5.42). D, Representative Western blot image and quantitating bar graph showing Mn-SOD protein levels in cultured neurons treated with ACM or mdACM under ICH-like injury in vitro. The significant changes in Mn-SOD/β-actin protein level were assessed by one-way ANOVA/Fisher's LSD test (n = 5 per group), **p < 0.01 (p = 0.0060, CON vs ICH-like injury), t value (t = 3.166); **p < 0.01 (p = 0.0009, ICH-like injury vs ACM plus ICH-like injury), t value (t = 4.041); **p < 0.01 (p = 0.0008, ACM plus ICH-like injury vs mdACM plus ICH-like injury), t value (t = 4.113). Mn-SOD is present in ACM but not in mdACM, and Mn-SOD level is higher in astrocytes than in neurons, in vitro (Extended Data Fig. 4-2). E, Mn-SOD mRNA levels in cultured neurons treated with ACM or SI plus ACM under ICH-like injury in vitro. SI, STAT3 inhibitor. Neurons were pretreated with ACM or 2 μm SI plus ACM for 24 h, followed by exposure to ICH-like injury for 48 h. Values represent fold change of Mn-SOD mRNA levels compared with CON group. The significant changes in Mn-SOD mRNA level were assessed by one-way ANOVA/Fisher's LSD test (n = 8 per group), **p < 0.01 (p = 0.0032, CON vs ICH-like injury), t value (t = 3.223); **p < 0.01 (p < 0.0001, ICH-like injury vs ACM plus ICH-like injury), t value (t = 12.19); **p < 0.01 (p = 0.0025, ACM plus ICH-like injury vs SI plus ACM plus ICH-like injury), t value (t = 3.316). All data are shown as mean ± SEM.
To understand whether restoration of viability by ACM was dependent on the presence of neuronal Mn-SOD, we used Mn-SOD-specific siRNA transfection to knock down Mn-SOD in cultured neurons before ICH-like injury, with or without ACM treatment. Scrambled siRNA was used as control. The efficacy of the knockdown of Mn-SOD in neurons with siRNA was 43% based on qRT-PCR analysis for Mn-SOD mRNA level. As anticipated, Mn-SOD-deficient neurons showed statistically lower viability than control neurons when subjected to ICH-like injury (Fig. 4C, second vs fourth bar). The neuronal viability was improved by astrocytic Mt (ACM) in control neurons (Fig. 4C, second vs third bar) and in Mn-SOD-deficient neurons (Fig. 4C, fourth vs fifth bar). The restoration of viability by ACM in control neurons was significantly higher than in Mn-SOD-deficient neurons (Fig. 4C, third vs fifth bar). These data demonstrate that (1) the naive neuronal Mn-SOD is important in protecting neurons from ICH-like injury, and (2) astrocytic Mts, which carry Mn-SOD, can enhance resistance of both control and Mn-SOD-deficient neurons.
Astrocytic Mt upregulates Mn-SOD expression in neurons by modulating STAT3 under ICH-like injury
Under ICH-like injury, we found that Mn-SOD protein levels were increased in the neurons treated with ACM but not with mdACM (Fig. 4D). Also, we found that only ACM and not mdACM contains Mn-SOD (Extended Data Fig. 4-2A). Along with the findings that astrocytes have higher levels of Mn-SOD than neurons (Extended Data Fig. 4-2B,C), these data may suggest that astrocytic Mt has the unique ability to transfer Mn-SOD from astrocytes to neurons.
Our earlier study showed that STAT3 is a master transcription factor regulating Mn-SOD expression in neurons (Jung et al., 2009). To test whether the increase in Mn-SOD in neurons in response to astrocytic Mt could (in addition to direct transfer through intra-Mt Mn-SOD content) involve STAT3, we added STAT3 inhibitor Stattic (SI; catalog #sc-202818, Santa Cruz Biotechnology) to neurons in culture under ICH-like injury, before adding ACM. Then we compared the Mn-SOD levels between vehicle-treated and STAT3 inhibitor-treated neurons. We found that STAT3 inhibitor significantly reduced the induction of Mn-SOD mRNA in response to Mt transfer (Fig. 4E). Overall, these results suggest that astrocytic Mt increases neuronal Mn-SOD through carrying over the Mn-SOD during the Mt transfer into the neurons and also by upregulating neuronal Mn-SOD expression through STAT3 activity.
Mn-SOD-deficient astrocytic Mt fails to inhibit ROS overproduction and protects neurons against ICH-like injury
To document the Mn-SOD role in neuroprotection mediated by astrocytic Mt transfer, we generated astrocyte-specific conditional Mn-SOD knockout (Mn-SOD-CKO) mice using the tamoxifen-induced Cre-loxP system. Mn-SODfl/fl mice were used as littermate controls. Specifically, we cultured primary astrocytes from GFAPCreERT2:Mn-SODfl/fl mice versus Mn-SODfl/fl mice brains and treated them with 4-OHT to generate Mn-SOD-CKO astrocytes. Using mRNA-level and Western blot analyses, we confirmed the partial deficiency of Mn-SOD in Mn-SOD-CKO astrocytes (Extended Data Fig. 5-1).
Ultimately, we collected the astrocytic Mt (ACM) from these Mn-SOD-deficient (Mn-SOD-CKO) or Mn-SOD-proficient (Mn-SODfl/fl) astrocytes, and transferred them onto neurons in culture (Fig. 5A). Twenty-four hours later, we examined the neuronal Mn-SOD level, under ICH-like injury. As expected, control astrocytic Mt robustly increased neuronal Mn-SOD level; however, the level of Mn-SOD in neurons receiving Mt from Mn-SOD-deficient astrocytes was significantly lower (Fig. 5B). In agreement with the essential role of Mn-SOD in ROS neutralization, we determined that ACM from Mn-SOD-CKO astrocytes, compared with control ACM, are not effective in preventing ROS overproduction in neurons subjected to ICH-like injury (Fig. 5C, second vs third or fourth bar in the graph). In agreement with the above results, the astrocytic Mts collected from Mn-SOD-deficient astrocytes were less effective in protecting neurons from ICH-like injury (Fig. 5D). Although the astrocytic Mts from control astrocytes significantly restored the viability of the neurons (Fig. 5D, first vs second bar on the graph), the restoration of viability by the Mn-SOD-deficient Mt was significantly less pronounced (Fig. 5D, second vs third bar). These data suggest that Mn-SOD carried by astrocytic Mt plays a neuroprotective role in ICH-like injury.
Figure 5.
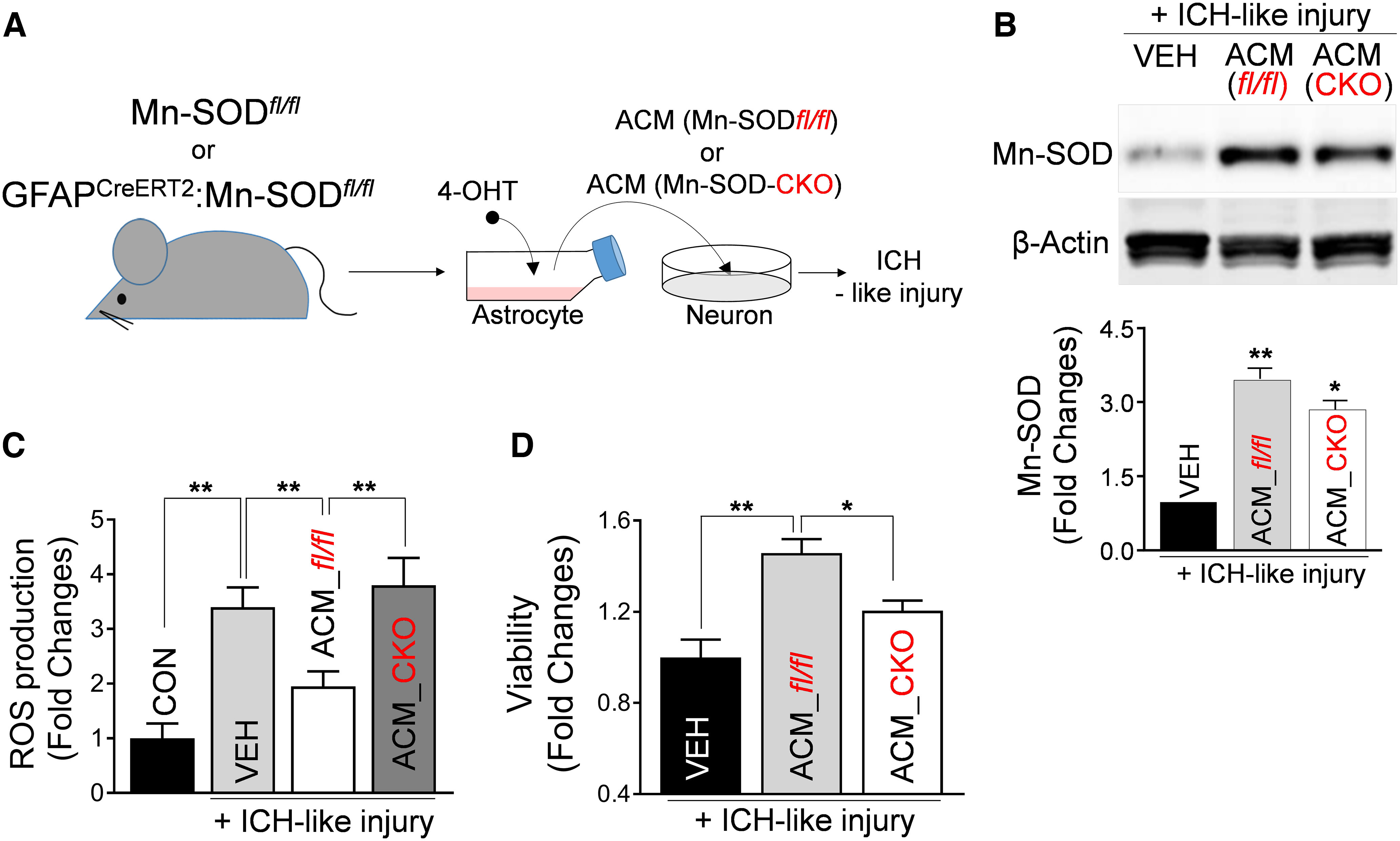
Mn-SOD-deficient astrocytic Mt fails to inhibit ROS overproduction and protects neurons against ICH-like injury. A, Schematic diagram of Mt transfer from Mn-SOD-deficient astrocytes to neurons in vitro. Astrocytes in culture generated from Mn-SODfl/fl mice or Mn-SODfl/fl: hGFAPCreERT2 mice were treated with 1 μm 4-OHT for 48 h to promote recombination. Knockdown of Mn-SOD in the cultured astrocytes from Mn-SODfl/fl: hGFAPCreERT2 mice was confirmed by Western blot analysis, as well as qRT-PCR (Extended Data Fig. 5-1). After washing the cells, fresh medium was supplied to astrocyte cultures. Twenty-four hours later, ACM from cultured astrocytes from Mn-SODfl/fl mice (ACM_Mn-SODfl/fl) or Mn-SODfl/fl: hGFAPCreERT2 mice (ACM_Mn-SOD-CKO) were collected and transferred onto cultured neurons. After a 24 h incubation with ACM from Mn-SODfl/fl or Mn-SOD-CKO astrocytes, the neurons were exposed to ICH-like injury for 48 h (for cell viability) or 12 h (for ROS generation). B, Representative Western blot image and illustrating graph showing Mn-SOD protein levels in cultured neurons that were pretreated with control culture media (VEH), ACM_Mn-SODfl/fl or ACM_Mn-SOD-CKO for 24 h, followed by exposure to ICH-like injury for 48 h. VEH, Vehicle. The significant changes in Mn-SOD/β-actin levels were assessed by one-way ANOVA/Fisher's LSD test (n = 6 per group), **p < 0.01 (p < 0.0001, VEH plus ICH-like injury vs ACM_Mn-SODfl/fl plus ICH-like injury), t value (t = 10.99); *p < 0.05 (p = 0.0173, ACM_Mn-SODfl/fl plus ICH-like injury vs ACM_Mn-SOD-CKO plus ICH-like injury), t value (t = 2.676). C, ROS generation in rat cortical neurons pretreated with ACM from Mn-SODfl/fl or Mn-SOD-CKO astrocytes for 24 h, followed by ICH-like injury for 12 h. The significant changes in ROS generation were assessed by one-way ANOVA/Fisher's LSD test (n = 13 per group), **p < 0.01 (p < 0.0001, CON vs ICH-like injury), t value (t = 4.668); **p < 0.01 (p = 0.0070, VEH plus ICH-like injury vs ACM_Mn-SODfl/fl plus ICH-like injury), t value (t = 2.82); **p < 0.01 (p = 0.0008, ACM_Mn-SODfl/fl plus ICH-like injury vs ACM_Mn-SOD-CKO plus ICH-like injury), t value (t = 3.598). CON. Control. D, Viability of rat cortical neurons treated with ACM_Mn-SODfl/fl or ACM_Mn-SOD-CKO for 24 h, followed by ICH-like injury for 48 h. Viabilities were measured by LDH assay. Values represent fold change of viabilities compared with VEH plus ICH-like injury group. The significance in cell viability was assessed by one-way ANOVA/Fisher's LSD test (n = 6 per group), **p < 0.01 (p = 0.0001, VEH plus ICH-like injury vs ACM_Mn-SODfl/fl plus ICH-like injury), t value (t = 5.159); *p < 0.05 (p = 0.0121, ACM_Mn-SODfl/fl plus ICH-like injury vs ACM_Mn-SOD-CKO plus ICH-like injury), t value (t = 2.854). Data are shown as mean ± SEM. ACM_fl/fl, ACM_Mn-SODfl/fl.
In neurons in culture, astrocytic Mt upregulates the expression of neuroplasticity-related genes and promote neurite outgrowth under ICH-like injury
Therapeutic approaches to treat ICH not only tackle rescuing neurons from acute injury but also include approaches to promote recovery and repair that require synaptic rearrangement. Mts are essential for neurite plasticity, including outgrowth and synaptogenesis during brain development (Morris and Hollenbeck, 1993; Mattson et al., 2008; Cheng et al., 2010; Han et al., 2016). In this study, we established that astrocytic Mts not only protected neurons from the oxidative stress caused by ICH but also improved synaptic plasticity as measured on day 21 after stroke. We found that the levels of synapsin 1/2 in the perihematomal area of mice that received ACM were higher compared with mdACM-receiving mice (Extended Data Fig. 6-1A,B). Astrocytic Mt transfer enhances dendritic extension in cultured neurons under oxidative stress such as oxygen-glucose deprivation/reoxygenation (Hayakawa et al., 2016). However, the mechanism behind astrocytic Mt-mediated neuroplasticity remains unclear.
Knowing that astrocytic Mt can improve neuronal survival under an ICH-like environment, we now used this model to gain more understanding of whether Mt transferred into neurons under ICH-like conditions affect neuronal propensity for repair and improved connectivity. In our ICH-like injury, we lowered the RBC number in RBC lysates (1.0 × 107 RBCs/3.0 × 105 neurons) to eliminate direct neuronal loss to simulate an insult level normally present in a more distal perihematomal brain, the area where most neuroplasticity events are anticipated. Using this model, we established that even with the retention of neuronal viability, this mild injury significantly reduced the expression of mRNA for neuroplasticity-related genes such as synapsin 1, synaptophysin, and PSD-95 (Fig. 6A). Next, we tested whether astrocytic Mt could reverse this process. Indeed, we found that treatment of neurons with ACM, but not with mdACM, reversed the reduction in mRNA of these neuroplasticity-related genes (Fig. 6B–D). To gain more insight into phenotypic changes associated with astrocytic Mt treatment, we examined whether Mt promotes neurite extension under mild ICH-like injury. We found that under mild ICH-like injury the neurite length at 4 d of incubation was significantly decreased, compared with control noninjured neurons (Fig. 6E). As expected, treatment of neurons with ACM significantly restored neurite length, whereas mdACM had no effect (Fig. 6E). In agreement with mRNA data (Fig. 6B,D), we also found that the levels of synapsin 1/2 and PSD-95 protein were significantly increased in ACM-treated but not in mdMCA-treated neurons (Fig. 6F). The findings of the enhanced neurite length, arborization, and synaptic protein expression in response to ACM treatment were further confirmed using immunochemistry for MAP2 to demarcate neuronal processes (dendrites primarily) and for synapsin 1/2 to map potential synapses throughout the processes (Fig. 6G). Altogether, our results demonstrate that under ICH-like injury, the astrocytic Mts transferred to neurons promote neuroplasticity.
Figure 6.
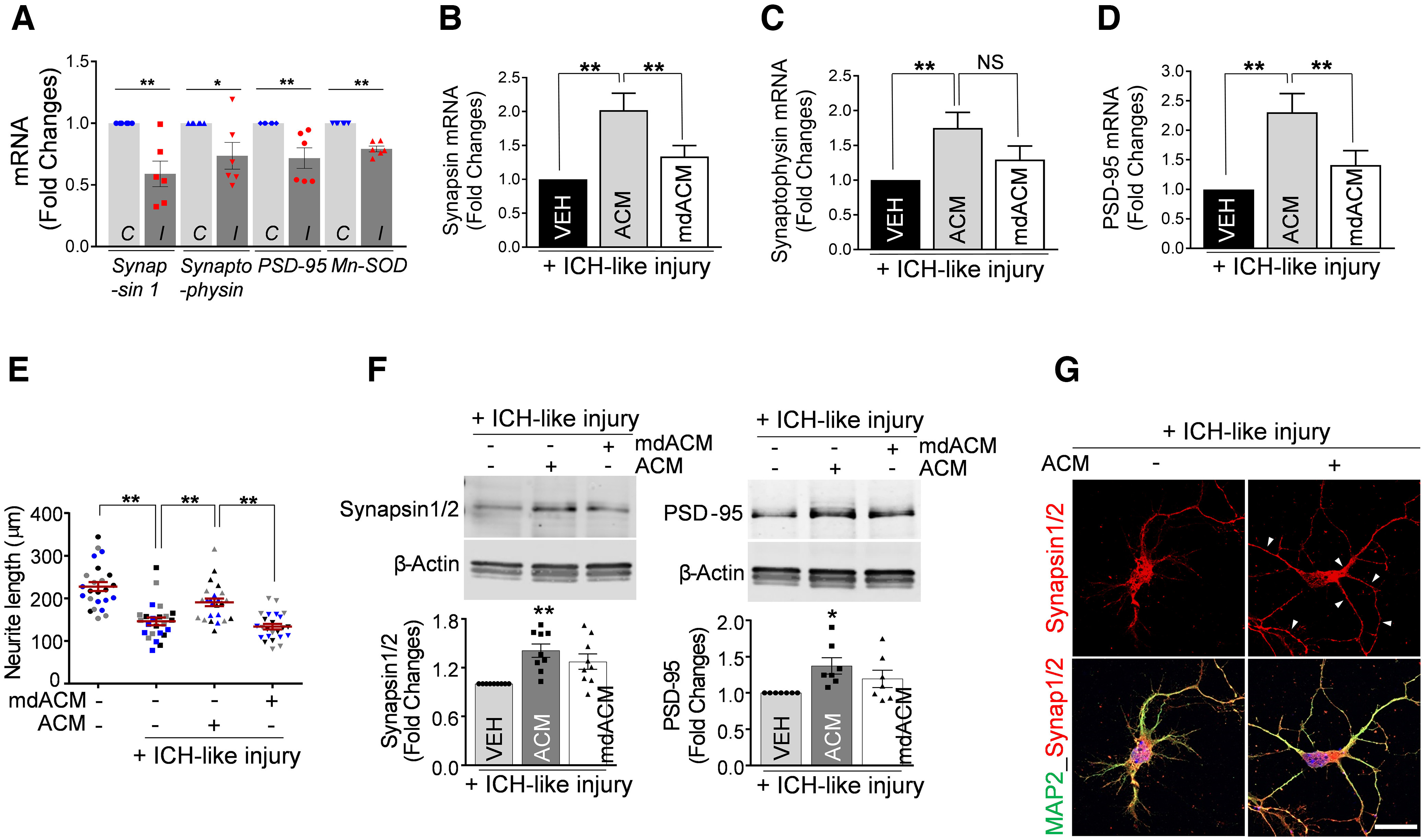
Astrocytic Mt upregulates the expression of synaptic plasticity-related genes and promotes neurite outgrowth under ICH-like injury. A, mRNA expression analysis for synaptic plasticity-related genes by qRT-PCR in cultured neurons (3.0 × 105 neurons/well) subjected to ICH-like injury (I) in vitro (RBC lysates, 1.0 × 107 RBCs/well) for 48 h versus untreated control (C). B–D, Cultured neurons were pretreated with ACM or mdACM for 24 h, followed by exposure to ICH-like injury in vitro (RBC lysates, 1.0 × 107 RBCs/well) for 48 h. A, The significant loss of synapsin 1, synaptophysin, PSD-95, and Mn-SOD mRNA was calculated using two-tailed unpaired t test (n = 6 per group), synapsin 1, **p < 0.01 (p = 0.0028, C vs I), t value (t = 3.944); synaptophysin, *p < 0.05 (p = 0.0355, C vs I), t value (t = 2.43); PSD-95, **p < 0.01 (p = 0.0068, C vs I), t value (t = 3.4); Mn-SOD, **p < 0.0001 (C vs I), t value (t = 8.995). B, Values represent fold change of synapsin 1 mRNA levels compared with VEH plus ICH-like injury group. The significance in mRNA levels changes was assessed by one-way ANOVA/Fisher's LSD test (n = 9 per group), **p < 0.01 (p = 0.0003, VEH plus ICH-like injury vs ACM plus ICH-like injury), t value (t = 4.196); **p < 0.01 (p = 0.0098, ACM plus ICH-like injury vs mdACM plus ICH-like injury), t value (t = 2.807). C, Values represent fold change of synaptophysin mRNA levels compared with VEH plus ICH-like injury group. The significance in mRNA levels changes was assessed by one-way ANOVA/Fisher's LSD test (n = 13 per group), **p < 0.01 (p = 0.0039, VEH plus ICH-like injury versus ACM plus ICH-like injury), t value (t = 3.084), NS; Non Significant. D, Values represent fold change of PSD-95 mRNA levels compared with VEH plus ICH-like injury group. The significance in mRNA levels changes was assessed by one-way ANOVA/Fisher's LSD test (n = 12 per group), **p < 0.01 (p = 0.0003, VEH plus ICH-like injury vs ACM plus ICH-like injury), t value (t = 3.99); *p < 0.01 (p = 0.0099, ACM plus ICH-like injury vs mdACM plus ICH-like injury), t value (t = 2.737). E, Neurite outgrowth quantification in rat cortical neurons (5.0 × 104 neurons/well) pretreated with ACM or mdACM, followed by exposure to ICH-like injury in vitro (RBC lysates, 1.7 × 106 RBCs/well) for 96 h. Eight individual neurites in culture from each treatment group (3 independent replicates for each treatment condition) were manually traced, and the length of each neurite was measured using ImageJ software. The significance in neurite length changes was assessed by one-way ANOVA/Fisher's LSD test (3 different colors represent 1 of 3 independent experiments-biological replicates, n = 3). Data are shown as mean ± SEM. The mean was calculated by averaging three values for each experiment; **p < 0.01 (p < 0.0001, VEH vs VEH plus ICH-like injury), t value (t = 8.413); **p < 0.01 (p = 0.0018, VEH plus ICH-like injury vs ACM plus ICH-like injury), t value (t = 4.574); **p < 0.01 (p = 0.0004, ACM plus ICH-like injury vs mdACM plus ICH-like injury), t value (t = 5.87). F, Representative Western blot images and quantitating bar graphs showing synapsin 1/2 and PSD-95 protein levels in cultured neurons pretreated with ACM or mdACM for 24 h, followed by exposure to ICH-like injury in vitro for 48 h. The significant changes in synapsin 1/2/β-actin and PSD-95/β-actin protein levels were assessed by one-way ANOVA/Fisher's LSD test (n = 9 in synapsin 1/2 and 7 in PSD-95), **p < 0.01 (p = 0.0005, VEH plus ICH-like injury vs ACM plus ICH-like injury in synapsin 1/2/β-actin), t value (t = 4.023); *p < 0.05 (p = 0.0121, VEH plus ICH-like injury vs ACM plus ICH-like injury in PSD-95/β-actin), t value (t = 2.789). G, Representative confocal microscopy images of cultured neurons pretreated with ACM for 24 h, followed by exposure to ICH-like injury in vitro for 48 h. Synapsin 1 and 2 are shown in red. Neurons were stained with MAP2 (green). Nuclei were counterstained with DAPI (blue). Scale bars: 20 µm. Data are shown as mean ± SEM. Synap1/2, Synapsin 1/2. Synapsin 1/2 is increased in perihematoma area of the brain after ICH in mice receiving astrocytic Mt in vivo (Extended Data Fig. 6-1).
Mt-encoded HN may act as a factor mediating Mt-induced Mn-SOD upregulation, oxidative stress reduction, and promotion of neuroplasticity under ICH-like injury
Our recent study suggests that Mt genome-encoded small peptide HN is an important factor contributing to astrocytic Mt-based therapy for ICH by inducing microglial phagocytic/reparative phenotype (Jung et al., 2020). Thus, the objective of the next experiment was to establish whether HN, which is normally abundant in Mt and could be shuttles within Mt to neurons, could be a part of the Mt-transfer-induced effect on neuronal Mn-SOD expression, oxidative stress, and neuroplasticity.
STAT3 that controls Mn-SOD transcription in neurons (Jung et al., 2009) was earlier shown to be activated by HN (Hashimoto et al., 2005; Matsuoka and Hashimoto, 2010; Kim et al., 2016). In agreement with these reports, we now found that treatment of neurons in culture with recombinant HN significantly increased phosphorylation of STAT3 (p-STAT3 at Y705; Fig. 7A). Most important, in the context of ICH-like injury, HN also increased the neuronal levels of Mn-SOD protein (Fig. 7B) and effectively inhibited ROS overproduction induced by the injury (Fig. 7C). Furthermore, similar to Mt transfer, HN under ICH-like injury in vitro induced the neurite extension (Fig. 7D) and increased the expression of synapsin and PSD-95 mRNA (data not shown) and protein (Fig. 7E). These results suggest that Mt-derived HN could be an effector of astrocytic Mt transfer-mediated modulation of neuronal responses to ICH-like injury.
Figure 7.

Mt-encoded peptide HN promotes antioxidative mechanisms, upregulates synaptogenesis-related genes, and stimulates neuronal growth under ICH-like stress. A, Representative Western blot image and quantitating bar graph showing phosphorylation of STAT3 at Y705 in cultured neurons pretreated with 100 ng/ml recombinant HN protein for 24 h. The significant changes in p-STAT3/β-actin protein levels were assessed by two-tailed unpaired t test (n = 7 per group), **p < 0.01 (p < 0.0001, CON vs HN), t value (t = 7.561). CON, Control. B, Representative Western blot image and quantitating bar graph showing Mn-SOD protein levels in cultured neurons pretreated with 100 ng/ml recombinant HN protein for 24 h, followed by exposure to ICH-like injury for 48 h. The significant changes in Mn-SOD/β-actin protein levels were assessed by one-way ANOVA/Fisher's LSD test (n = 9 per group), **p < 0.01 (p < 0.0001, CON vs ICH-like injury), t value (t = 4.993); **p < 0.01 (p = 0.0050, ICH-like injury vs HN plus ICH-like injury), t value (t = 3.092). C, ROS generation in cultured neurons pretreated with 100 ng/ml recombinant HN protein for 24 h, followed by exposure to ICH-like injury for 12 h. The significant changes in ROS generation were assessed by one-way ANOVA/Fisher's LSD test (n = 16 per group), **p < 0.01 (p < 0.0001, CON vs ICH-like injury), t value (t = 4.535); *p < 0.05 (p = 0.0140, ICH-like injury vs HN plus ICH-like injury), t value (t = 2.556). D, Neurite outgrowth quantification in cultured neurons (5.0 × 104 neurons/well) pretreated with 100 ng/ml recombinant HN protein for 24 h, followed by exposure to ICH-like injury (RBC lysates, 1.7 × 106 RBCs/well) for 96 h. The six individual neurites in culture from each treatment group (3 independent replicates for each treatment condition) were manually traced, and the length of each neurite was measured using ImageJ software. The significance in the neurite length changes was assessed by one-way ANOVA/Fisher's LSD test (3 different colors represent 1 of 3 independent experiments-biological replicates, n = 3). Data are shown as mean ± SEM. The mean was calculated by averaging three values for each experiment; **p < 0.01 (p = 0.0023, CON vs ICH-like injury), t value (t = 5.073); *p < 0.05 (p = 0.0146, ICH-like injury vs HN plus ICH-like injury), t value (t = 3.394). E, Representative Western blot images and quantitating bar graphs showing synapsin 1/2 and PSD-95 protein levels in neurons pretreated with 100 ng/ml recombinant HN for 24 h, followed by ICH-like injury for 48 h. The significant changes in synapsin 1/2/β-actin and PSD-95/β-actin protein levels were assessed by one-way ANOVA/Fisher's LSD test (n = 10 in synapsin 1/2 and 9 in PSD-95), **p < 0.01 (p = 0.0087, CON vs ICH-like injury in synapsin 1/2/β-actin), t value (t = 2.828); **p < 0.01 (p = 0.0058, ICH-like injury vs HN plus ICH-like injury in synapsin 1/2/β-actin), t value (t = 2.996); **p < 0.01 (p < 0.0001, CON vs ICH-like injury in PSD-95/β-actin), t value (t = 5.361); **p < 0.01 (p < 0.0001, ICH-like injury vs HN plus ICH-like injury in PSD-95/β-actin), t value (t = 5.031). Data are shown as mean ± SEM.
Discussion
Mounting evidence suggests that Mt transfer between cells has signaling and beneficial effects, including supporting the energy in the recipient cell (Spees et al., 2006; Plotnikov et al., 2010; Islam et al., 2012; Pasquier et al., 2013; Liu et al., 2014; Wang and Gerdes, 2015; Hayakawa et al., 2016; 2018). We and others demonstrated Mt transfer in the CNS (Hayakawa et al., 2016; Chou et al., 2017; Hayakawa et al., 2018; Jung et al., 2020). Hayakawa et al. (2016) demonstrated that Mt transfer from astrocytes to neurons in the ischemic brain avert injury by increasing the ATP supply to neurons. Also, several animal studies and human trials have documented the benefit of autologous functional Mt transplantation into an injured organ to increase ATP in the injured tissue (McCully et al., 2009; Masuzawa et al., 2013; Cowan et al., 2016; McCully et al., 2016; Su et al., 2016; Kaza et al., 2017). The beneficial effects of Mt transfer are well accepted; however, the specific molecular mechanisms of action beyond the increase of ATP remain to be explored.
ROS accumulation generated by the blood degraded products and primarily iron in the ICH-affected brain induces Mt dysfunction (Wagner et al., 2003; Hu et al., 2016; Qu et al., 2016), and Mt in the perihematomal tissues from ICH patients have impaired respiratory function (Kim-Han et al., 2006; Lu et al., 2015). ICH-induced Mt dysfunction leads to enhanced ROS production and neuroinflammation, which further potentiate brain injuries (Lu et al., 2015; Zhou et al., 2017; Zheng et al., 2018; Chen et al., 2020). Therefore, restoration of Mt biological function by replacing ICH-affected Mt with a new healthy Mt could have a therapeutic potential in treating ICH. Here, we propose that transplantation of astrocytic Mt can enhance neuronal antioxidant defense through direct delivery of Mn-SOD to neurons within Mt, and by upregulating STAT3-mediated Mn-SOD transcription in the neurons, including by Mt peptide humanin, for better resistance to oxidative damage and promoting neuro recovery after ICH.
In this study, we investigated the potential mechanism behind the astrocytic Mt transfer-mediated neuroprotection, using in vivo ICH mouse model and in vitro ICH-like injury to neurons in culture. First, we showed that systemically administered astrocytic Mt entered the brain, including neurons, restored Mn-SOD levels, and improved functional recovery in the mouse ICH model. The ICH injury affected the mitochondrial free radical scavenging system, as shown by the robust reduction of Mn-SOD, ROS accumulation, and protein nitrosation in the brain (Fig. 1). Also, ICH-like injury in cultured neurons robustly increased neuronal ROS levels, lowered Mn-SOD level, and reduced neuronal survival, processes that were reversed by astrocytic Mt transfer to neurons (Fig. 4A,B,D,E). This study also implicates the STAT3/Mn-SOD pathway as a contributor to neuronal antioxidant system regulation through astrocytic Mt transfer. Astrocytic Mt-mediated increase in neuronal Mn-SOD may involve STAT3 activity to transcriptionally restore the reduced Mn-SOD levels caused by ICH (Fig. 4E). Our data suggest the importance of astrocytic Mt transplantation-induced Mn-SOD restoration in neurons to more effectively combat oxidative damage. Certainly, we are not excluding the possibility that other than neurons, brain cells also receive Mt, which may also contribute to functional recovery after ICH. Our previous study found that astrocytic Mts enter microglia where they enhance the PPARγ transcriptional pathway, which is known to promote reparative microglial phenotype and control phagocytosis-mediated hematoma clearance to enhance recovery after ICH (Jung et al., 2020). Thus, our overall findings suggest that it is the harmonious interplay between neurons and microglia (and potentially other cells) that on adapting astrocytic Mt helps in alleviating damage and promoting recovery after ICH.
In addition, this study demonstrates that astrocytic Mt may promote neuroplasticity during ICH recovery by upregulating synaptogenesis-related gene expression and neurite outgrowth. Mitochondrial biogenesis is essential for neuronal survival, growth, synaptogenesis, homeostasis, and recovery after injury (MacAskill and Kittler, 2010; Devine and Kittler, 2018). During the ICH recovery phase, neuronal growth to reconstruct the damaged brain circuitry is critical and requires a supply of functional Mt (Morris and Hollenbeck, 1993; Han et al., 2016). In our ongoing study, we found that extracellular Mt from astrocytes subjected to ICH-like injury in vitro had lower functionality (measured Mt membrane potential by JC-1 staining) compared with Mt from the control group (data not shown). This suggests that astrocytes following brain injury such as ICH may release subfunctional or nonfunctional Mt, which physiologically may not be capable of providing a benefit to the adjacent neurons and other cells on transfer. In this study, we observed the incorporation of functional astrocytic Mt into neurons that persisted for at least 7 d after transfer, moved along elongating neurites (Fig. 3C, Extended Data Fig. 3-1), and promoted neurite elongation during recovery post-ICH-like injury in vitro (Fig. 6E). Moreover, we observed that the transplantation of astrocytic Mt into ICH-injured mice significantly increased the level of synapsin 1/2 in the perihematomal area of the ICH-affected brain on day 21 after stroke in vivo (Extended Data Fig. 6-1A,B).
The Mt-derived small peptide HN is also one of the key effectors in astrocytic Mt transfer-mediated neuroprotection in the ICH brain. We earlier demonstrated that astrocytic Mt-derived HN played a critical role in astrocytic Mt-mediated recovery by promoting a reparative microglia phenotype through phagocytic activity in a mouse ICH model (Jung et al., 2020). In the current study, we found that HN also participates in astrocytic Mt-enhanced neuronal antioxidation and neuroplasticity under ICH-like injury in vitro (Fig. 7). HN inhibits ROS generation and protects cells from oxidative stress-induced cell death by activation of STAT3 (Hashimoto et al., 2005, 2009; Kim et al., 2016; Sreekumar et al., 2016). Indeed, HN upregulated phosphorylation of STAT3 at Y705 (Fig. 7A) and increased Mn-SOD expression in neurons under ICH-like injury (Fig. 7B) and prevented ROS overproduction (Fig. 7C). HN released by astrocytes prevented synapse loss in hippocampal neurons under glutamate-induced dendritic atrophy (Zárate et al., 2019). Here, we also found that HN restored neurite extension inhibited by ICH injury in cultured neurons through upregulation of synapsin 1 and PSD-95, synaptogenesis-related genes (Fig. 7D,E). Although we have not provided direct evidence for the causal role of HN in Mt-mediated effect, as HN is enriched in Mt, it is highly likely that HN is at least in part involved in Mt-mediated effect.
In conclusion, our study reveals that systemic transplantation of astrocytic Mt in ICH promotes antioxidative protection and assists in functional recovery by enhancing Mn-SOD-mediated neuronal antioxidant defense and neuroplasticity in the brain.
Synapsin 1/2 is increased in perihematoma area of the brain after ICH in mice receiving astrocytic Mt in vivo. A, Representative immunofluorescence confocal images of Synapsin 1/2 (red) and MAP-2 (green) staining in ICH-affected perihematoma areas at 21 d after the injury in mice that were injected with ACM or mdACM at 1h, 7 d, and 14 d after ICH. Images show increased Synapsin 1/2 (red) in response to ACM versus mdACM treatment. Nuclei were counterstained with DAPI (blue). Scale bar, 40 µm. B, The bar graph showing difference in Synapsin 1/2 immunofluorescence signal intensity; n = 4 animals per treatment group. The significance was assessed by two-tailed unpaired t test (n = 4 per group), *p < 0.05 (p = 0.0139, ACM-treated mice vs mdACM-treated mice), t value (t = 3.435). All data are shown as mean ± SEM. Download Figure 6-1, TIF file (7.3MB, tif) .
Knockdown of Mn-SOD in the cultured astrocytes from Mn-SODfl/fl; hGFAPCreERT2 mice. A, Mn-SOD mRNA expression in cultured astrocytes (2.0 × 104 cells) generated from brains of Mn-SODfl/fl; hGFAPCreERT2 (Mn-SOD-CKO) mice or Mn-SODfl/fl mice were treated with 1 µm 4-OHT for 48 h. Mn-SODfl/fl as used as control. Data are shown as mean ± SEM. The significant changes in Mn-SOD mRNA levels were assessed by two-tailed unpaired t test (n = 10 pups, each pup was used to establish separate cultures), **p < 0.01 (p = 0.0046, Mn-SOD-CKO vs Mn-SODfl/fl), t value (t = 3.237). B, Representative Western blot image showing Mn-SOD protein levels in cultured astrocytes from Mn-SOD-CKO mice or Mn-SODfl/f mice, which were treated with 1 µm 4-OHT for 48 h. Mn-SODfl/fl was used as the control. Download Figure 5-1, TIF file (319KB, tif) .
Mn-SOD is present in ACM but not in mdACM (Mt deficient ACM-filtered ACM), and Mn-SOD level is higher in astrocytes than in neurons, in vitro. A, Western blot image shows the presence of Mn-SOD protein in ACM versus absence of Mn-SOD protein in mdACM. From each 200 µl of ACMs or mdACMs, 60 µl of each sample were loaded for Western blot analysis. For negative control, 60 µl DMEM only was used. The level of Mn-SOD in ACM on longer incubation is higher with 48 h versus 24 h incubation (lane 1 vs 3). Further prolongation of the incubation to 72 h did not result in further increase of Mn-SOD content in ACM (lane 3 vs 5). All the mdACM fractions showed no Mn-SOD (same as DMEM only; vehicle control; lanes 2, 4, 6, and). B, Western blot image shows the different level of Mn-SOD protein between the cultured primary neurons and cultured astrocytes (both generated from rat embryonic day 17 brains). The quantitative bar graph demonstrates the difference between Mn-SOD levels in neurons and astrocytes, assessed with ImageJ software. Data are shown as mean ± SEM. The significant changes in Mn-SOD levels were assessed by two-tailed unpaired t test (n = 4 per group), **p < 0.01 (p = 0.0022), t value (t = 5.096). N, Neurons; A, astrocytes. C, To ensure accuracy of the comparison, we loaded an equal amount of protein (30 µg) from neurons and astroglia lysates. The example of protein load is indicated by ruby gel staining. Download Figure 4-2, TIF file (3.5MB, tif) .
ACM collected from astrocytes upon longer incubation is more effective in protecting neurons from ICH-like injury. A, ROS generation, assessed with MitoSOX Red mitochondrial superoxide indicator, by neurons in culture (3.0 × 105 neurons/well) under 12 h of ICH-like injury conditions (RBC lysates, 3.0 × 107 RBCs/well) and pretreated (24 h before the insult) with ACMs that were collected from astrocytes at 24 h or 48 h of incubation. Five to 10 images of cultured neurons were captured per each culture well, for four independent culture wells per treatment condition, and the pixel numbers of MitoSOX Red+ particles were measured using ImageJ software. The significant differences in ROS production were assessed by one-way ANOVA/Fisher's LSD test (n = 4 per group; 4 biological replicates, *p < 0.05 (p = 0.0214, VEH plus ICH-like injury vs ACM 48 h plus ICH-like injury), t value (t = 4.425); *p < 0.05 (p = 0.0453, ACM 24 h plus ICH-like injury vs ACM 48 h plus ICH-like injury), t value (t = 3.313). B, Representative phase contrast images of neurons in culture with MitoSOX Red+ staining comparing VEH- vs ACM-pretreated condition both under ICH-like injury. Scale bar, 20 µm. Red fluorescent indicates the Mt superoxide, which was produced by neurons under ICH-like injury. Download Figure 4-1, TIF file (5.6MB, tif) .
Neuronally incorporated astrocytic Mts redistribute along the axonal growth. Rat cortical astrocytes were stained with 200 nm MitoTracker-Red/CMXRos to label astrocytic Mt for 30 min. Following three times of rigorous washing with PBS and incubation in fresh media for 24 h, the ACM was collected and then transferred to neurons in culture. Representative phase-contrast images of neurons with superimposed fluorescence (red) to show location of astrocytic Mt at day 5 and day 7 of coincubation are presented. Yellow arrowheads point to MitoTracker-Red/CMXRos-labeled Mt incorporated into the neurons. Scale bar, 20 µm. Download Figure 3-1, TIF file (13.5MB, tif) .
Treatment with Mn-SOD-deficient ACM is less effective in reducing neurological deficit at early post-ICH time compared with ACM from Mn-SOD-proficient astrocytes. NDS evaluated by foot fault test in ICH-injured mice, which were treated with ACM obtained from astrocytes that were exposed to Mn-SOD siRNA to deplete Mn-SOD (Mn-SOD-KD-ACM) or to scrambled siRNA (control; Scr-ACM) for 2 d. Mice received intravenous Scr-ACM or Mn-SOD-KD-ACM at 1 h, 7 d, and 14 d after ICH onset. The significance of NDS was assessed by repeated measures two-way ANOVA with multiple comparison and Fisher's LSD test (n = 9–10 mice per group), *p < 0.05 (p = 0.0290, Scr-ACM vs Mn-SOD-KD-ACM-treated mice at 3 d), t value (t = 2.221); p = 0.1567, Scr-ACM vs Mn-SOD-KD-ACM at 21 d), t value (t = 1.429). Download Figure 2-1, TIF file (255.6KB, tif) .
Footnotes
This work was supported by National Institutes of Health Grant R01 NS111590.
The authors declare no competing financial interests.
Reference
- Aronowski J, Hall CE (2005) New horizons for primary intracerebral hemorrhage treatment: experience from preclinical studies. Neurol Res 27:268–279. 10.1179/016164105X25225 [DOI] [PubMed] [Google Scholar]
- Aronowski J, Zhao X (2011) Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke 42:1781–1786. 10.1161/STROKEAHA.110.596718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Guo C, Feng H, Chen Y (2020) Mitochondria: novel mechanisms and therapeutic targets for secondary brain injury after intracerebral hemorrhage. Front Aging Neurosci 12:615451. 10.3389/fnagi.2020.615451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Hou Y, Mattson MP (2010) Mitochondria and neuroplasticity. ASN Neuro 2:e00045. 10.1042/AN20100019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou SH, Lan J, Esposito E, Ning M, Balaj L, Ji X, Lo EH, Hayakawa K (2017) Extracellular mitochondria in cerebrospinal fluid and neurological recovery after subarachnoid hemorrhage. Stroke 48:2231–2237. 10.1161/STROKEAHA.117.017758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan DB, Yao R, Akurathi V, Snay ER, Thedsanamoorthy JK, Zurakowski D, Ericsson M, Friehs I, Wu Y, Levitsky S, Del Nido PJ, Packard AB, McCully JD (2016) Intracoronary delivery of mitochondria to the ischemic heart for cardioprotection. PLoS One 11:e0160889. 10.1371/journal.pone.0160889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine MJ, Kittler JT (2018) Mitochondria at the neuronal presynapse in health and disease. Nat Rev Neurosci 19:63–80. 10.1038/nrn.2017.170 [DOI] [PubMed] [Google Scholar]
- Ding R, Chen Y, Yang S, Deng X, Fu Z, Feng L, Cai Y, Du M, Zhou Y, Tang Y (2014) Blood-brain barrier disruption induced by hemoglobin in vivo: involvement of up-regulation of nitric oxide synthase and peroxynitrite formation. Brain Res 1571:25–38. 10.1016/j.brainres.2014.04.042 [DOI] [PubMed] [Google Scholar]
- Duan X, Wen Z, Shen H, Shen M, Chen G (2016) Intracerebral hemorrhage, oxidative stress, and antioxidant therapy. Oxid Med Cell Longev 2016:1203285. 10.1155/2016/1203285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felberg RA, Grotta JC, Shirzadi AL, Strong R, Narayana P, Hill-Felberg SJ, Aronowski J (2002) Cell death in experimental intracerebral hemorrhage: the “black hole” model of hemorrhagic damage. Ann Neurol 51:517–524. 10.1002/ana.10160 [DOI] [PubMed] [Google Scholar]
- Fujimura M, Morita-Fujimura Y, Kawase M, Copin JC, Calagui B, Epstein CJ, Chan PH (1999) Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome C and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. J Neurosci 19:3414–3422. 10.1523/JNEUROSCI.19-09-03414.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall NC, Packard BA, Hall CL, de Courten-Myers G, Wagner KR (2000) Protein oxidation and enzyme susceptibility in white and gray matter with in vitro oxidative stress: relevance to brain injury from intracerebral hemorrhage. Cell Mol Biol (Noisy-le-grand) 46:673–683. [PubMed] [Google Scholar]
- Han SM, Baig HS, Hammarlund M (2016) Mitochondria localize to injured axons to support regeneration. Neuron 92:1308–1323. 10.1016/j.neuron.2016.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Suzuki H, Aiso S, Niikura T, Nishimoto I, Matsuoka M (2005) Involvement of tyrosine kinases and STAT3 in humanin-mediated neuroprotection. Life Sci 77:3092–3104. 10.1016/j.lfs.2005.03.031 [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Kurita M, Aiso S, Nishimoto I, Matsuoka M (2009) Humanin inhibits neuronal cell death by interacting with a cytokine receptor complex or complexes involving CNTF receptor alpha/WSX-1/gp130. Mol Biol Cell 20:2864–2873. 10.1091/mbc.e09-02-0168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH (2016) Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535:551–555. 10.1038/nature18928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Chan SJ, Mandeville ET, Park JH, Bruzzese M, Montaner J, Arai K, Rosell A, Lo EH (2018) Protective effects of endothelial progenitor cell-derived extracellular mitochondria in brain endothelium. Stem Cells 36:1404–1410. 10.1002/stem.2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemorrhagic Stroke Academia Industry (HEADS) Roundtable Participants, Second HEADS Roundtable Participants (2020) Recommendations for clinical trials in ICH: the Second Hemorrhagic Stroke Academia Industry Roundtable. Stroke 51:1333–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Tao C, Gan Q, Zheng J, Li H, You C (2016) Oxidative stress in intracerebral hemorrhage: sources, mechanisms, and therapeutic targets. Oxid Med Cell Longev 2016:3215391. 10.1155/2016/3215391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S, Bhattacharya J (2012) Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med 18:759–765. 10.1038/nm.2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung JE, Kim GS, Narasimhan P, Song YS, Chan PH (2009) Regulation of Mn-superoxide dismutase activity and neuroprotection by STAT3 in mice after cerebral ischemia. J Neurosci 29:7003–7014. 10.1523/JNEUROSCI.1110-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung JE, Kim GS, Chan PH (2011) Neuroprotection by interleukin-6 is mediated by signal transducer and activator of transcription 3 and antioxidative signaling in ischemic stroke. Stroke 42:3574–3579. 10.1161/STROKEAHA.111.626648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung JE, Karatas H, Liu Y, Yalcin A, Montaner J, Lo EH, van Leyen K (2015) STAT-dependent upregulation of 12/15-lipoxygenase contributes to neuronal injury after stroke. J Cereb Blood Flow Metab 35:2043–2051. 10.1038/jcbfm.2015.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung JE, Sun G, Bautista Garrido J, Obertas L, Mobley AS, Ting SM, Zhao X, Aronowski J (2020) The mitochondria-derived peptide humanin improves recovery from intracerebral hemorrhage: implication of mitochondria transfer and microglia phenotype change. J Neurosci 40:2154–2165. 10.1523/JNEUROSCI.2212-19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaza AK, Wamala I, Friehs I, Kuebler JD, Rathod RH, Berra I, Ericsson M, Yao R, Thedsanamoorthy JK, Zurakowski D, Levitsky S, Del Nido PJ, Cowan DB, McCully JD (2017) Myocardial rescue with autologous mitochondrial transplantation in a porcine model of ischemia/reperfusion. J Thorac Cardiovasc Surg 153:934–943. 10.1016/j.jtcvs.2016.10.077 [DOI] [PubMed] [Google Scholar]
- Keep RF, Hua Y, Xi G (2012) Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol 11:720–731. 10.1016/S1474-4422(12)70104-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GW, Kondo T, Noshita N, Chan PH (2002) Manganese superoxide dismutase deficiency exacerbates cerebral infarction after focal cerebral ischemia/reperfusion in mice: implications for the production and role of superoxide radicals. Stroke 33:809–815. 10.1161/hs0302.103745 [DOI] [PubMed] [Google Scholar]
- Kim SJ, Guerrero N, Wassef G, Xiao J, Mehta HH, Cohen P, Yen K (2016) The mitochondrial-derived peptide humanin activates the ERK1/2, AKT, and STAT3 signaling pathways and has age-dependent signaling differences in the hippocampus. Oncotarget 7:46899–46912. 10.18632/oncotarget.10380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim-Han JS, Kopp SJ, Dugan LL, Diringer MN (2006) Perihematomal mitochondrial dysfunction after intracerebral hemorrhage. Stroke 37:2457–2462. 10.1161/01.STR.0000240674.99945.4e [DOI] [PubMed] [Google Scholar]
- Lee C, Yen K, Cohen P (2013) Humanin: a harbinger of mitochondrial-derived peptides? Trends Endocrinol Metab 24:222–228. 10.1016/j.tem.2013.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Ji K, Guo L, Wu W, Lu H, Shan P, Yan C (2014) Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc Res 92:10–18. 10.1016/j.mvr.2014.01.008 [DOI] [PubMed] [Google Scholar]
- Lu H, Jiang M, Lu L, Zheng G, Dong Q (2015) Ultrastructural mitochondria changes in perihematomal brain and neuroprotective effects of Huperzine A after acute intracerebral hemorrhage. Neuropsychiatr Dis Treat 11:2649–2657. 10.2147/NDT.S92158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill AF, Kittler JT (2010) Control of mitochondrial transport and localization in neurons. Trends Cell Biol 20:102–112. 10.1016/j.tcb.2009.11.002 [DOI] [PubMed] [Google Scholar]
- Maier CM, Hsieh L, Crandall T, Narasimhan P, Chan PH (2006) Evaluating therapeutic targets for reperfusion-related brain hemorrhage. Ann Neurol 59:929–938. 10.1002/ana.20850 [DOI] [PubMed] [Google Scholar]
- Masuzawa A, Black KM, Pacak CA, Ericsson M, Barnett RJ, Drumm C, Seth P, Bloch DB, Levitsky S, Cowan DB, McCully JD (2013) Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 304:H966–H982. 10.1152/ajpheart.00883.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka M, Hashimoto Y (2010) Humanin and the receptors for humanin. Mol Neurobiol 41:22–28. 10.1007/s12035-009-8090-z [DOI] [PubMed] [Google Scholar]
- Mattson MP, Gleichmann M, Cheng A (2008) Mitochondria in neuroplasticity and neurological disorders. Neuron 60:748–766. 10.1016/j.neuron.2008.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCully JD, Cowan DB, Pacak CA, Toumpoulis IK, Dayalan H, Levitsky S (2009) Injection of isolated mitochondria during early reperfusion for cardioprotection. Am J Physiol Heart Circ Physiol 296:H94–H105. 10.1152/ajpheart.00567.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCully JD, Levitsky S, Del Nido PJ, Cowan DB (2016) Mitochondrial transplantation for therapeutic use. Clin Transl Med 5:16. 10.1186/s40169-016-0095-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ (1993) The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci 104:917–927. 10.1242/jcs.104.3.917 [DOI] [PubMed] [Google Scholar]
- Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen SF, Chan PH (1998) Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci 18:205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Keep RF, Hua Y, Schallert T, Hoff JT, Xi G (2004) Deferoxamine-induced attenuation of brain edema and neurological deficits in a rat model of intracerebral hemorrhage. J Neurosurg 100:672–678. 10.3171/jns.2004.100.4.0672 [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Lo EH, Hayakawa K (2020) Placental mitochondria therapy for cerebral ischemia-reperfusion injury in mice. Stroke 51:3142–3146. 10.1161/STROKEAHA.120.030152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquier J, Guerrouahen BS, Al Thawadi H, Ghiabi P, Maleki M, Abu-Kaoud N, Jacob A, Mirshahi M, Galas L, Rafii S, Le Foll F, Rafii A (2013) Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J Transl Med 11:94. 10.1186/1479-5876-11-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekcec A, Yigitkanli K, Jung JE, Pallast S, Xing C, Antipenko A, Minchenko M, Nikolov DB, Holman TR, Lo EH, van Leyen K (2013) Following experimental stroke, the recovering brain is vulnerable to lipoxygenase-dependent semaphorin signaling. FASEB J 27:437–445. 10.1096/fj.12-206896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikov EY, Khryapenkova TG, Galkina SI, Sukhikh GT, Zorov DB (2010) Cytoplasm and organelle transfer between mesenchymal multipotent stromal cells and renal tubular cells in co-culture. Exp Cell Res 316:2447–2455. 10.1016/j.yexcr.2010.06.009 [DOI] [PubMed] [Google Scholar]
- Qu J, Chen W, Hu R, Feng H (2016) The injury and therapy of reactive oxygen species in intracerebral hemorrhage looking at mitochondria. Oxid Med Cell Longev 2016:2592935. 10.1155/2016/2592935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan RF, Panter SS (1996) Hemoglobin potentiates excitotoxic injury in cortical cell culture. J Neurotrauma 13:223–231. 10.1089/neu.1996.13.223 [DOI] [PubMed] [Google Scholar]
- Sarafian TA, Montes C, Imura T, Qi J, Coppola G, Geschwind DH, Sofroniew MV (2010) Disruption of astrocyte STAT3 signaling decreases mitochondrial function and increases oxidative stress in vitro. PLoS One 5:e9532. 10.1371/journal.pone.0009532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaar KL, Brenneman MM, Savitz SI (2010) Functional assessments in the rodent stroke model. Exp Transl Stroke Med 2:13. 10.1186/2040-7378-2-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot JW, Geuze HJ, Freeman BA, Crapo JD (1986) Intracellular localization of the copper-zinc and manganese superoxide dismutases in rat liver parenchymal cells. Lab Invest 55:363–371. [PubMed] [Google Scholar]
- Spees JL, Olson SD, Whitney MJ, Prockop DJ (2006) Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci U S A 103:1283–1288. 10.1073/pnas.0510511103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreekumar PG, Ishikawa K, Spee C, Mehta HH, Wan J, Yen K, Cohen P, Kannan R, Hinton DR (2016) The mitochondrial-derived peptide humanin protects RPE cells from oxidative stress, senescence, and mitochondrial dysfunction. Invest Ophthalmol Vis Sci 57:1238–1253. 10.1167/iovs.15-17053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Zhu L, Yu X, Cai L, Lu Y, Zhang J, Li T, Li J, Xia J, Xu F, Hu Q (2016) Mitochondrial transplantation attenuates airway hyperresponsiveness by inhibition of cholinergic hyperactivity. Theranostics 6:1244–1260. 10.7150/thno.13804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF (2003) Heme and iron metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab 23:629–652. 10.1097/01.WCB.0000073905.87928.6D [DOI] [PubMed] [Google Scholar]
- Wang X, Gerdes HH (2015) Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ 22:1181–1191. 10.1038/cdd.2014.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Hua Y, Keep RF, Schallert T, Hoff JT, Xi G (2002) Oxidative brain injury from extravasated erythrocytes after intracerebral hemorrhage. Brain Res 953:45–52. 10.1016/s0006-8993(02)03268-7 [DOI] [PubMed] [Google Scholar]
- Zárate SC, Traetta ME, Codagnone MG, Seilicovich A, Reines AG (2019) Humanin, a mitochondrial-derived peptide released by astrocytes, prevents synapse loss in hippocampal neurons. Front Aging Neurosci 11:123. 10.3389/fnagi.2019.00123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N, Grotta JC, Aronowski J (2007) Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann Neurol 61:352–362. 10.1002/ana.21097 [DOI] [PubMed] [Google Scholar]
- Zhao X, Sun G, Zhang J, Ting SM, Gonzales N, Aronowski J (2015) Dimethyl fumarate protects brain from damage produced by intracerebral hemorrhage by mechanism involving Nrf2. Stroke 46:1923–1928. 10.1161/STROKEAHA.115.009398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Ting SM, Liu CH, Sun G, Kruzel M, Roy-O'Reilly M, Aronowski J (2017) Neutrophil polarization by IL-27 as a therapeutic target for intracerebral hemorrhage. Nat Commun 8:602. 10.1038/s41467-017-00770-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Shi L, Liang F, Xu W, Li T, Gao L, Sun Z, Yu J, Zhang J (2018) Sirt3 ameliorates oxidative stress and mitochondrial dysfunction after intracerebral hemorrhage in diabetic rats. Front Neurosci 12:414. 10.3389/fnins.2018.00414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang S, Li Y, Yu S, Zhao Y (2017) SIRT1/PGC-1α signaling promotes mitochondrial functional recovery and reduces apoptosis after intracerebral hemorrhage in rats. Front Mol Neurosci 10:443. 10.3389/fnmol.2017.00443 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Synapsin 1/2 is increased in perihematoma area of the brain after ICH in mice receiving astrocytic Mt in vivo. A, Representative immunofluorescence confocal images of Synapsin 1/2 (red) and MAP-2 (green) staining in ICH-affected perihematoma areas at 21 d after the injury in mice that were injected with ACM or mdACM at 1h, 7 d, and 14 d after ICH. Images show increased Synapsin 1/2 (red) in response to ACM versus mdACM treatment. Nuclei were counterstained with DAPI (blue). Scale bar, 40 µm. B, The bar graph showing difference in Synapsin 1/2 immunofluorescence signal intensity; n = 4 animals per treatment group. The significance was assessed by two-tailed unpaired t test (n = 4 per group), *p < 0.05 (p = 0.0139, ACM-treated mice vs mdACM-treated mice), t value (t = 3.435). All data are shown as mean ± SEM. Download Figure 6-1, TIF file (7.3MB, tif) .
Knockdown of Mn-SOD in the cultured astrocytes from Mn-SODfl/fl; hGFAPCreERT2 mice. A, Mn-SOD mRNA expression in cultured astrocytes (2.0 × 104 cells) generated from brains of Mn-SODfl/fl; hGFAPCreERT2 (Mn-SOD-CKO) mice or Mn-SODfl/fl mice were treated with 1 µm 4-OHT for 48 h. Mn-SODfl/fl as used as control. Data are shown as mean ± SEM. The significant changes in Mn-SOD mRNA levels were assessed by two-tailed unpaired t test (n = 10 pups, each pup was used to establish separate cultures), **p < 0.01 (p = 0.0046, Mn-SOD-CKO vs Mn-SODfl/fl), t value (t = 3.237). B, Representative Western blot image showing Mn-SOD protein levels in cultured astrocytes from Mn-SOD-CKO mice or Mn-SODfl/f mice, which were treated with 1 µm 4-OHT for 48 h. Mn-SODfl/fl was used as the control. Download Figure 5-1, TIF file (319KB, tif) .
Mn-SOD is present in ACM but not in mdACM (Mt deficient ACM-filtered ACM), and Mn-SOD level is higher in astrocytes than in neurons, in vitro. A, Western blot image shows the presence of Mn-SOD protein in ACM versus absence of Mn-SOD protein in mdACM. From each 200 µl of ACMs or mdACMs, 60 µl of each sample were loaded for Western blot analysis. For negative control, 60 µl DMEM only was used. The level of Mn-SOD in ACM on longer incubation is higher with 48 h versus 24 h incubation (lane 1 vs 3). Further prolongation of the incubation to 72 h did not result in further increase of Mn-SOD content in ACM (lane 3 vs 5). All the mdACM fractions showed no Mn-SOD (same as DMEM only; vehicle control; lanes 2, 4, 6, and). B, Western blot image shows the different level of Mn-SOD protein between the cultured primary neurons and cultured astrocytes (both generated from rat embryonic day 17 brains). The quantitative bar graph demonstrates the difference between Mn-SOD levels in neurons and astrocytes, assessed with ImageJ software. Data are shown as mean ± SEM. The significant changes in Mn-SOD levels were assessed by two-tailed unpaired t test (n = 4 per group), **p < 0.01 (p = 0.0022), t value (t = 5.096). N, Neurons; A, astrocytes. C, To ensure accuracy of the comparison, we loaded an equal amount of protein (30 µg) from neurons and astroglia lysates. The example of protein load is indicated by ruby gel staining. Download Figure 4-2, TIF file (3.5MB, tif) .
ACM collected from astrocytes upon longer incubation is more effective in protecting neurons from ICH-like injury. A, ROS generation, assessed with MitoSOX Red mitochondrial superoxide indicator, by neurons in culture (3.0 × 105 neurons/well) under 12 h of ICH-like injury conditions (RBC lysates, 3.0 × 107 RBCs/well) and pretreated (24 h before the insult) with ACMs that were collected from astrocytes at 24 h or 48 h of incubation. Five to 10 images of cultured neurons were captured per each culture well, for four independent culture wells per treatment condition, and the pixel numbers of MitoSOX Red+ particles were measured using ImageJ software. The significant differences in ROS production were assessed by one-way ANOVA/Fisher's LSD test (n = 4 per group; 4 biological replicates, *p < 0.05 (p = 0.0214, VEH plus ICH-like injury vs ACM 48 h plus ICH-like injury), t value (t = 4.425); *p < 0.05 (p = 0.0453, ACM 24 h plus ICH-like injury vs ACM 48 h plus ICH-like injury), t value (t = 3.313). B, Representative phase contrast images of neurons in culture with MitoSOX Red+ staining comparing VEH- vs ACM-pretreated condition both under ICH-like injury. Scale bar, 20 µm. Red fluorescent indicates the Mt superoxide, which was produced by neurons under ICH-like injury. Download Figure 4-1, TIF file (5.6MB, tif) .
Neuronally incorporated astrocytic Mts redistribute along the axonal growth. Rat cortical astrocytes were stained with 200 nm MitoTracker-Red/CMXRos to label astrocytic Mt for 30 min. Following three times of rigorous washing with PBS and incubation in fresh media for 24 h, the ACM was collected and then transferred to neurons in culture. Representative phase-contrast images of neurons with superimposed fluorescence (red) to show location of astrocytic Mt at day 5 and day 7 of coincubation are presented. Yellow arrowheads point to MitoTracker-Red/CMXRos-labeled Mt incorporated into the neurons. Scale bar, 20 µm. Download Figure 3-1, TIF file (13.5MB, tif) .
Treatment with Mn-SOD-deficient ACM is less effective in reducing neurological deficit at early post-ICH time compared with ACM from Mn-SOD-proficient astrocytes. NDS evaluated by foot fault test in ICH-injured mice, which were treated with ACM obtained from astrocytes that were exposed to Mn-SOD siRNA to deplete Mn-SOD (Mn-SOD-KD-ACM) or to scrambled siRNA (control; Scr-ACM) for 2 d. Mice received intravenous Scr-ACM or Mn-SOD-KD-ACM at 1 h, 7 d, and 14 d after ICH onset. The significance of NDS was assessed by repeated measures two-way ANOVA with multiple comparison and Fisher's LSD test (n = 9–10 mice per group), *p < 0.05 (p = 0.0290, Scr-ACM vs Mn-SOD-KD-ACM-treated mice at 3 d), t value (t = 2.221); p = 0.1567, Scr-ACM vs Mn-SOD-KD-ACM at 21 d), t value (t = 1.429). Download Figure 2-1, TIF file (255.6KB, tif) .