

Abstract
From 2016 to 2020, high pathogenicity avian influenza (HPAI) H5 viruses circulated in Asia, Europe, and Africa, causing waves of infections and the deaths of millions of wild and domestic birds and presenting a zoonotic risk. In late 2021, H5N1 HPAI viruses were isolated from poultry in Canada and also retrospectively from a great black-backed gull (Larus marinus), raising concerns that the spread of these viruses to North America was mediated by migratory wild bird populations. In February and April 2022, H5N1 HPAI viruses were isolated from a bald eagle (Haliaeetus leucocephalus) and broiler chickens in British Columbia, Canada. Phylogenetic analysis showed that the virus from bald eagle was genetically related to H5N1 HPAI virus isolated in Hokkaido, Japan, in January 2022. The virus identified from broiler chickens was a reassortant H5N1 HPAI virus with unique constellation genome segments containing PB2 and NP from North American lineage LPAI viruses, and the remaining gene segments were genetically related to the original Newfoundland-like H5N1 HPAI viruses detected in November and December 2021 in Canada. This is the first report of H5 HPAI viruses’ introduction to North America from the Pacific and the North Atlantic-linked flyways and highlights the expanding risk of genetically distinct virus introductions from different geographical locations and the potential for local reassortment with both the American lineage LPAI viruses in wild birds and with both Asian-like and European-like H5 HPAI viruses. We also report the presence of some amino acid substitutions across each segment that might contribute to the replicative efficiency of these viruses in mammalian host, evade adaptive immunity, and pose a potential zoonotic risk.
Keywords: H5N1 clade 2.3.4.4, HPAI, reassortment, whole genome sequencing, Canada
1. Introduction
Wild aquatic birds are the natural reservoir host for low pathogenicity avian influenza (LPAI) viruses (Venkatesh et al. 2018). These viruses have been circulating within the wild bird populations, typically without causing overt clinical signs (Webster et al. 1992). Some H5 and H7 LPAI viruses can evolve to high pathogenicity avian influenza (HPAI) viruses when they infect domestic birds, primarily galliform species (Dietze et al. 2018). The wild bird–poultry interface is extremely important in the evolution of LPAI and HPAI viruses (La Sala et al. 2019; Verhagen, Fouchier, and Lewis 2021). Migratory aquatic birds as well as shorebirds can serve as transmission vectors for avian influenza viruses, introducing viruses to both wild birds and poultry in various geographic regions (Webster et al. 1992; Olsen et al. 2006). Viral amplification, through serial transmission in immunologically naive and partially immune wild birds, may significantly alter the genetic landscape and epidemiology of avian influenza viruses (Wikramaratna, Pybus, and Gupta 2014; Hill et al. 2016).
The HPAI viruses currently dominating many influenza outbreaks globally are derivatives of the goose-origin H5N1 HPAI virus (A/Goose/Guangdong/1/96) (Xu et al. 1999). Since 2014, genetically distinct HPAI H5Nx clade 2.3.4.4 viruses have caused considerable threats to poultry, remaining endemic in many parts of Eurasia and Africa and continuing to evolve into subclades that can cause epidemics globally (Lee et al. 2015; Fusaro et al. 2019; Poen et al. 2019; Bevins et al. 2022; King et al. 2022). In the last couple of years, the A/Goose/Guangdong/1/96 (GsGd) H5 clade 2.3.4.4 viruses have been frequently detected in domestic poultry and in a plethora of wild bird species in various European countries (Adlhoch et al. 2021a, 2021b; King et al. 2022). In North America, the first outbreak in poultry that was associated with a GsGd H5 clade 2.3.4.4 virus was in late 2014. A survey in wild birds identified an H5N8 subtype virus belonging to this clade in a gyrfalcon (Falco rusticolus) in the USA with all eight gene segments closely related to Eurasian GsGd viruses. Later in 2014 and 2015, a reassortant H5N2 virus, likely originated from H5N8, and local avian origin LPAI virus caused the first GsGd linked major outbreak in domestic poultry in several states in the USA and in British Columbia in Canada (Clement et al. 2015; Lee et al. 2015, 2016; Pasick et al. 2015). Concurrent with the active H5N2 outbreaks, both H5N8 and H5N2 viruses were confirmed in wild birds from the Pacific flyway of the USA (Ip et al. 2015; Bevins et al. 2016) and other major migratory bird flyways in the USA, except the Atlantic flyway (cited in Bevins et al. 2016). Additionally, an H5N1 HPAI virus with 19 amino acid deletions in the neuraminidase gene stalk region due to adaptation in the chickens, but harbouring a similar genetic constellation as the other HPAI viruses, was isolated from a backyard chicken and Cooper’s hawk in British Columbia (Berhane et al. 2016). In December 2021, H5N1 HPAI viruses from the 2.3.4.4b clade, genetically related to viruses circulating in Europe in early 2021, were isolated from poultry as well as a great black-backed gull (Larus marinus) in Newfoundland, Canada (Caliendo et al. 2022). In the USA, the H5N1 HPAI viruses were isolated from wild waterfowl from two Atlantic coastal states, coinciding with the movement of banded birds within the Atlantic flyway (Bevins et al. 2022). Wild bird migration that occurred via the Arctic may have carried these viruses from Europe across the Atlantic to Canada (Caliendo et al. 2022).
The incursions of these viruses in Newfoundland highlight the impact of migratory flyways in rapidly spreading novel viruses. In Japan, particularly in the Hokkaido region, in January 2022, the presence of an H5N1 HPAI virus was identified in a white-tailed eagle (Haliaeetus albicilla). In February 2022, Canada reported H5N1 viruses from a bald eagle (Haliaeetus leucocephalus) and, in April 2022, from a commercial chicken flock, both in British Columbia. The current study assessed the genetic and phylogenetic features of these recently identified viruses and analysed the likely routes of incursions.
2. Materials and methods
2.1. Clinical samples
2.1.1. Bald eagle
On 2 February 2022, a bald eagle, which was unable to fly, was found on the ground at the Stanley Park, Vancouver, British Columbia. It was admitted to a wildlife rehabilitation facility on 3 February 2022 and died the following day. A pool of oropharyngeal and cloacal swabs was collected on 10 February 2022 and submitted to the National Centre for Foreign Animal Disease (NCFAD), Winnipeg, Manitoba, for testing for the avian influenza virus.
2.1.2. Broiler chickens
A commercial poultry farm that housed 29,500 birds of 32-day-old broilers in the northern part of Okanagan Valley, British Columbia, experienced a sudden onset and unusually high mortality over a 4-day period starting from 12 April 2022. On the first and second day, 28 and 68 birds died, respectively. Two-hundred and ten birds died on the third day, and 308 birds died on the fourth day of the onset of the outbreak. Affected birds showed severe congestion of the head and conjunctiva. During necropsy on acutely dead birds, the findings were severe congestion of the skeletal muscles, trachea, lungs, and kidneys. On 13 April 2022, a pool of oropharyngeal swabs collected from dead chickens was received at NCFAD.
2.2. RNA extraction, virus detection, and isolation
Total RNA was extracted from both submissions using the MagMax 1835 Nucleic Acid Isolation Kit (ThermoFisher Scientific, Waltham, MA, USA) as per the manufacturer’s recommendation using the KingFisher Duo prime system platform (ThermoFisher Scientific, Waltham, MA, USA). The spiked Enteroviral armoured RNA (ARM-ENTERO; Asuragen) was used as an exogenous extraction and reaction control. For virus isolation, PCR positive samples from both submissions were inoculated into 9-day-old embryonated specific pathogen-free chicken eggs via the allantoic route. The samples were tested for influenza A using the matrix, H5 and H7 gene-specific real-time Reverse Transcription-Polymerase Chain Reaction (RT-PCR) assays as described previously (Spackman et al. 2002; Weingartl et al. 2010).
2.3. Nanopore sequencing and genome assembly
Eight genome segments of the viruses were amplified directly from the clinical specimens as described previously (Zhou et al. 2009). The amplified RT-PCR products were purified using QIAquick PCR purification kit (Qiagen, Hilden, Germany), and the concentration of the amplicon used for sequencing was determined with the Qubit® dsDNA BR Assay Kit (Invitrogen, Waltham, MA) using the Qubit® Fluorometer (ThermoFisher Scientific, Waltham, MA, USA). Nanopore sequencing was performed on an Oxford Nanopore GridION sequencer with an R9.4.1 flowcell after library construction using the rapid barcoding kit (SQK-RBK004). Base-called and demultiplexed sequence data were processed using an NCFAD-developed pipeline, nf-VirONTus (v1.1.0), to perform reference mapping to other avian influenza viruses, including sequences from the 2021 to 2022 outbreaks and genome assembly.
2.4. Phylogenetic data set
We searched Global Initiative on Sharing All Influenza Data (GISAID) and collected H5 sequences from Europe, Africa, and Asia that were detected from 01 January 2020 to 29 March 2022. To these HA gene sequences, we added one sequence from Hokkaido, Japan, (EPI_ISL_11330431) isolated from a white-tailed eagle and the two unpublished sequences from British Columbia, Canada, from a bald eagle and broiler chicken. We employed the Basic Alignment Search Tool from GISAID on the other genome segments of the Japanese and the Canadian viruses and collected the top 100 results of this search. Sequences were aligned using MAFFT v7.490 and trimmed to the starting ATG and ending STOP codons. Maximum likelihood trees were inferred using IQ-TREE 2.1.3. We used TreeTime, a Python-based framework, for phylogenetic analysis using an approximate maximum likelihood approach to reroot trees and estimate ancestral nodes whilst inferring molecular-clock phylogenies (Sagulenko, Puller, and Neher 2018). Clades not necessary for this analysis were collapsed to allow for easy visualisation. Times for the most recent common ancestor (TMRCA) were calculated in TreeTime. We generated geographical maps and gene cassettes for the viruses from the broiler chicken and bald eagle in British Columbia to illustrate the geographical origin of each of the gene segments. Geographical maps were generated in Mapchart.
Several studies have demonstrated the continuous movement and introduction of Eurasian lineage avian influenza viruses to North America (Winker et al. 2007; Dusek et al. 2014) due to the regular intercontinental movement of wild birds to and from breeding and non-breeding sites across the East Pacific, East Atlantic, and East Asian Flyways (reviewed in Lycett, Duchatel, and Digard 2019). Based on data presented in this study and from phylogenetic analysis, we highly predicted the H5N1 HPAI viruses have been introduced to Canada via the Pacific and Atlantic flyways.
Amino acid comparisons for the HA1 domain of HA gene of the isolates from British Columbia and Hokkaido were calculated using the custom python script Flutile (https://github.com/flu-crew/flutile). We compared these viruses with a candidate vaccine H5N8 clade 2.3.4.4 virus (candidate vaccine virus [CVV]) A/Gyrfalcon/Washington/41088-6/2014. Amino acid differences identified in putative antigenic sites have been annotated. The genome sequences of the bald eagle and broiler chicken viruses were screened with Fludb sequence feature identifier for genetic mutations that impact viral phenotypic characteristics of importance, which increase virulence, mammalian receptor adaptation, or antiviral resistance.
3. Results
Phylogenetic trees inferred from the HA gene sequences showed that white-tailed eagle (H. albicilla) sampled in Hokkaido in January 2022 and the bald eagle (H. leucocephalus) sampled in British Columbia a month later were very similar to each other genetically and more distantly to viruses identified in November 2021, mainly from wild birds in a number of countries in Europe. Across the gene segments, the genetic clustering was similar relative to the HA gene sequence patterns, with the exception of the NS gene segment, where the white-tailed eagle (H. albicilla) and the bald eagle virus are genetically distinct from each other. However, both these NS segments were phylogenetically related to other NS genes from recent European H5 2.3.4.4b viruses (Fig. 1).
Figure 1.
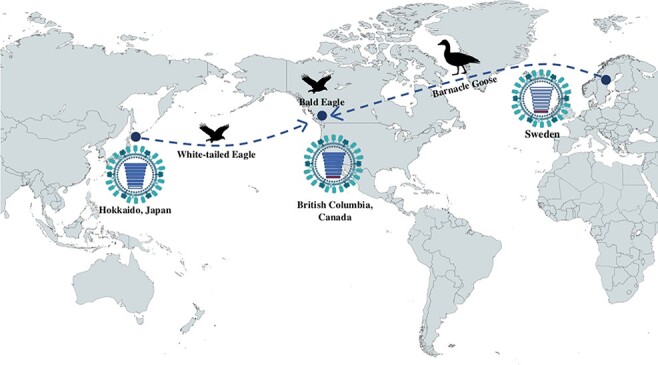
Putative geographic origin of the genomic composition of the virus isolated from a bald eagle in British Columbia, Canada. The NS gene segment from the bald eagle virus is genetically closely related to a virus isolated from a Barnacle Goose in Sweden, whilst the other gene segments are most similar to a virus isolated from a white-tailed eagle in Hokkaido, Japan. Geographic maps have been generated in Mapchart.
Using time-resolved phylogenetic trees for each gene segment (Supplementary Data S1 and S2), the TMRCA of all the gene segments for the bald eagle virus ranged from August 2021 to January 2022 (Table 1).
Table 1.
TMRCA of all gene segments for the Canadian bald eagle and the chicken isolates. The dates have been given with lower and higher confidence interval bounds.
| Bald eagle | Chicken | |||||
|---|---|---|---|---|---|---|
| Segment | Date | Lower bound | Higher bound | Date | Lower bound | Higher bound |
| HA | 18 November 2021 | 02 April 2021 | 03 January 2022 | 16 November 2020 | 30 September 2020 | 15 December 2020 |
| MP | 08 September 2021 | 05 June 2021 | 04 December 2021 | 03 March 2021 | 07 November 2020 | 03 March 2021 |
| NA | 03 January 2022 | 01 January 2022 | 03 January 2022 | 28 September 2021 | 27 September 2021 | 29 September 2021 |
| NP | 31 August 2021 | 12 December 2020 | 29 December 2021 | 23 March 2017 | 16 February 2015 | 18 October 2018 |
| NS | 02 October 2021 | 25 August 2021 | 02 November 2021 | 11 May 2021 | 03 March 2021 | 11 May 2021 |
| PA | 26 September 2021 | 23 May 2021 | 14 December 2021 | 11 May 2021 | 02 April 2021 | 11 May 2021 |
| PB1 | 14 September 2021 | 24 May 2021 | 05 December 2021 | 16 December 2020 | 07 November 2020 | 16 December 2020 |
| PB2 | 20 August 2021 | 03 January 2021 | 16 December 2021 | 08 April 2014 | 17 August 2012 | 31 December 2015 |
For the HA gene sequences, the virus from a broiler chicken in British Columbia sampled in April 2022 was genetically closely related to viruses identified in wild birds in South Carolina in 2021 and also clusters within the Newfoundland group from late 2021. However, the other gene segments have a more mixed genetic relationship. PA and NS cluster with Eurasian H5 HPAI strains from spring 2021, and PB1 with strains from late 2020 and early 2021, again from Europe, whereas PB2 and NP locate on long branches and are genetically closely related to viruses from the North American lineage: NP genetically closer to a virus isolated from a ruddy turnstone (Arenaria interpres) in Delaware in 2020 and PB2 to a virus detected in a northern shoveler (Spatula clypeata) in Utah in 2017. The broiler chicken virus presented long branch lengths within the MP gene, and it clustered together with strains from West Africa detected in early 2021, whilst the NA segment was genetically related to a Japanese virus from late 2021 isolated from a chicken (Fig. 2).
Figure 2.
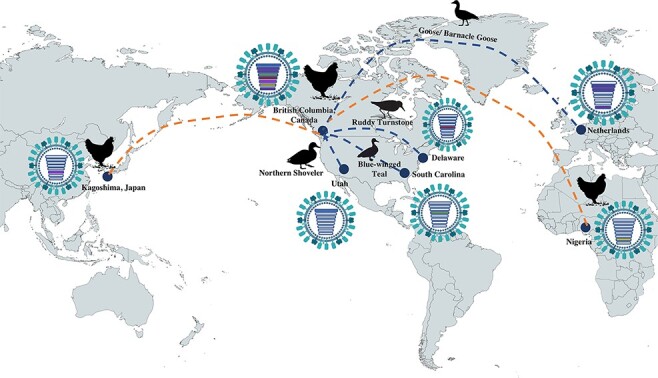
Putative geographic origin of the genomic composition of the virus isolated from chickens in British Columbia, Canada. The chicken genomic cassette is composed of viral segments (PB2, HA, and NP) genetically closely related to viruses from the North American lineage. The PB1, PA, and NS are closely related to viruses from the Netherlands and Denmark. The European viruses have spread to Canada potentially by Geese or Barnacle Geese. The NA is genetically related to a chicken virus detected in Japan in late 2021, whilst the MP gene was closely related to a chicken virus detected in West Africa. Geographic maps have been generated in Mapchart.
We compared the HA1 domain of the HA amino acid sequence of the broiler chicken and bald eagle viruses with A/Gyrfalcon/Washington/41088-6/2014—a zoonotic CVV that was recommended after the major outbreak in poultry in the USA in 2015—to assess if this CVV was likely antigenically similar to these new 2022 emerging strains (Table 2), were it to be used in any poultry vaccine. We identified amino acid substitutions in putative antigenic sites in all the three isolates. The bald eagle and the broiler chicken viruses had substitutions in antigenic site B (A185E, D195T, and V198I) and antigenic site C (E268G). The broiler chicken virus also had an amino acid substitution in antigenic site D (V210A). The Hokkaido virus had the same amino acid substitutions as the bald eagle, but the substitutions differ from the CVV.
Table 2.
Comparative amino acid substitutions of the isolates from Canada and Japan to H5N8 A/Gyrfalcon/Washington/41,088-6/2014. Putative antigenic positions were annotated based on H5 numbering. Amino acid residues 185, 195, and 198 form antigenic site B; amino acids 210 and 268 form antigenic sites D and C, respectively.
| Amino acid residues | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Viruses | 14 | 94 | 104 | 114 | 119 | 141 | 185 | 195 | 198 | 210 | 223 | 236 | 268 | 269 | 282 | 325 |
| A/Gyrfalcon/Washington/41,088-6/2014_H5N8 | K | T | L | T | R | S | A | D | V | V | S | N | E | M | I | R |
| A/Bald_Eagle/Canada/OTH-33-36/2022_H5N1 | E | S | I | K | P | E | T | I | R | D | G | V | V | |||
| A/white-tailed_eagle/Hokkaido/R22/2022_H5N1 | E | S | I | K | P | E | T | I | R | D | G | V | V | K | ||
| A/Chicken/BC/FAV-0228/2022_H5N1 | E | S | M | I | K | P | E | T | I | A | R | D | G | V | V | K |
We have documented amino acid residues present in the polymerase and nucleoprotein segments of the H5N1 HPAI viruses obtained from the bald eagle and broiler chicken known to influence influenza A virus phenotypes. Some of the amino acid residues in influenza A viruses were previously confirmed to enhance virulence in mice and increase the polymerase activity and replication in mammalian cells. A summary of these amino acid residues is presented in Table 3. The adaptive mutations observed may play roles in cross-species barrier transmission with subsequent pandemic potential.
Table 3.
Multiple host-adaptive mutations in viral polymerases and nucleoprotein of GsGd lineage H5N1 HPAI virus isolated from bald eagle and chicken (+ = present; − = absent). Relevant references are provided for influenza A viruses.
| Species involved | |||||
|---|---|---|---|---|---|
| Proteins | Amino acid mutation | Bald eagle | Chickens | Predicted activity | References |
| PB2 | K389R | + | + | ↑Polymerase activity in mammalian cells | Yamayoshi et al. (2014); Yamayoshi et al. (2018) |
| K482R | + | + | ↑Polymerase activity in mammalian cells | Yamayoshi et al. (2014); Yamayoshi et al. (2018) | |
| V598T | + | + | ↑polymerase activity & ↑replication in mammalian and avian cells ↑Virulence in mice |
Xiao et al. (2016) | |
| PB1 | D3V | + | + | ↑Polymerase activity & ↑replication in mammalian and avian cells | Elgendy et al. (2017) |
| D622G | + | + | ↑Polymerase activity in mammalian cells ↑Virulence in mice |
Feng et al. (2016) | |
| PA | S37A | + | + | ↑Polymerase activity mammalian cells | Yamayoshi et al. (2014) |
| P190S | + | + | ↓Virulence in mice | DesRochers et al. (2016) | |
| N383D | + | + | |||
| N409S | + | + | ↑Polymerase activity & ↑replication in mammalian cells | Yamayoshi et al. (2014) | |
| NP | M105V | + | − | ↑Virulence in chickens | Tada et al. (2011a, 2011b) |
| A184K | + | + | ↑Replication in avian cells ↑Virulence in chickens ↑IFN response |
Wasilenko, Sarmento, and Pantin-Jackwood (2009) | |
| N319K | + | − | ↑Polymerase activity and replication in mammalian cells | Gabriel et al. (2005, 2008) | |
4. Discussion
Understanding intra- and intercontinental dispersal of H5N1 HPAI viruses in avian hosts is imperative to reduce the global impact of the viruses, both on food security and in zoonotic risk. In 2014 and 2015, multiple outbreaks linked to the GsGd lineage H5N2 HPAI subtype did affect many aquatic wild bird species, birds of prey and domestic poultry in the vast areas of the USA and in British Columbia, Canada (Pasick et al. 2015; Lee et al. 2016; Ramey et al. 2018, 2022). The outbreaks were contained successfully in poultry by depopulating, movement control, prompt diagnosis, and enhancing biosecurity. The shorter duration of circulation of the H5N2 HPAI virus in wild birds and poultry probably suppressed an acquired adaptive mutation from taking place that gradually prevented the maintenance of the virus in the local wetland birds of North America (DeJesus et al. 2016; Lee et al. 2017). There was no indication of GsGd lineage viruses from the domestic and local wild birds in all North American bird flyways since 2016 and until the new cases were found in Newfoundland, Canada, in December 2021.
In the current study, we demonstrate the presence of two ancestrally and genetically different GsGd lineage H5N1 HPAI clade 2.3.4.4b viruses isolated from domestic chickens and bald eagle in British Columbia in 2022. This work revealed that the current outbreak viruses in British Columbia were linked with viruses circulating in Asia, Europe, and West Africa and the ongoing expansion of H5N1 HPAI viruses from parts of Canada and the USA. The two viruses, although identified from the same province, are distinct based on phylogenetic reconstruction, suggesting they might have different incursion routes.
The virus from the bald eagle identified in British Columbia in February 2022 has no evidence of epidemiological connection with the virus identified in chickens from the same province in April 2022. Time-resolved maximum likelihood phylogenetic reconstruction of the HA gene sequences placed the H5N1 HPAI virus isolated from the bald eagle with a virus isolated from a white-tailed eagle from Japan in 2022 and, importantly, distinct from the Newfoundland H5N1 HPAI viruses. As with the original viruses isolated from chickens and a great black-backed gull in Newfoundland in December 2021, the H5N1 HPAI HA gene sequence obtained from a chicken in British Columbia in April 2022 was clustered with the HA gene sequences of European H5 HPAI viruses circulating in spring 2021 (Caliendo et al. 2022).
Gene segment analyses of the H5N1 HPAI virus obtained from chickens in British Columbia showed that the Eurasian HPAI virus upon introduction into North American wild birds has undergone reassortment with local LPAI viruses. The emergence of reassortant viruses with novel genome constellations is not unexpected as similar reassortments have also occurred during the 2014/2015 introduction of GsGd lineage H5Nx HPAI virus into the North America (Lee et al. 2016) and recently in Eurasia after the emergence of the HPAI viruses in 2020 in the Russian Federation (Lewis et al. 2021). The continuation of virus evolution through genetic reassortment may lead to the co-circulation of viruses carrying complex patterns of constellation of internal gene segments. A longitudinal survey is required in the North American wild birds to identify the host niche in which these H5 HPAI viruses are being maintained, expanded, and transmitted. However, as the North American lineage viruses in wild birds have been spatially segregated for longer periods, it is interesting to monitor the relative fitness of the HPAI reassortant viruses, which acquired internal genes from North American lineage viruses to hold footage in the dynamic wild bird population in North America.
The genetic footprints of all segments of the viruses, integrating migratory wild bird flyways and our phylogenetic data, provide evidence that the H5N1 HPAI viruses have been introduced to British Columbia likely through two prominent flyways—the East Asia-Australasia/Pacific and the Atlantic flyways. The presumption of H5N1 HPAI virus dispersions from more Nordic regions to British Columbia is supported by transcontinental but interhemispheric migratory birds. The timing of detections of the genetically distinct H5N1 in bald eagle in British Columbia and white-tailed eagle in Japan presumably is informative of the new introduction of the virus from East Asia. The flight ranges of the white-tailed eagles are suggested to stretch as far as the USA islands in the North Bering Sea and occasionally to the Alaska’s western shores (www.birds-of-north-america.net/White-tailed_Eagle.html). In 2014 and 2015 in the USA and Canada, H5N8 HPAI clade 2.3.4.4 virus was introduced from East Asia into North America through the Beringian Crucible via intercontinental associations of waterfowl that finally dispersed the virus into the Pacific flyway (Lee et al. 2015). The interspecies transmission of the virus, however, was enhanced through genetic reassortment of H5N8 with a North American avian origin LPAI virus resulting in the generation of H5N2 HPAI virus responsible for the outbreaks in Canada and the USA (Lee et al. 2016). However, we cannot rule out the possibility of introduction from the most northern Nordic regions over Greenland and Iceland. The close genetic relationship between the chicken viruses in British Columbia and Nigeria, West Africa, highlights how the intercontinental movement of these viruses is being facilitated by the overlapping of bird flyways. The GsGd lineage viruses were introduced into West Africa in 2005 (Cattoli et al. 2009) and since then remained as a threat to poultry populations in the region ever since. West Africa is located at the crossroads of two migratory flyways—the Black Sea/Mediterranean and the East Atlantic—and Nigeria has been previously shown to facilitate the spread of the virus to other countries in the region, mainly due to a socioeconomic situation that prevents efficient disease control (Ekong et al. 2012).
The correlation between the spread of avian influenza virus and seasonal bird flyways can be depicted by actively recovering ringed birds and from available larger data sets. Ring-recovery data showed that banded dabbling ducks in the Atlantic flyway are frequently encountered within the Atlantic and to the inland flyways in the USA (Bevins et al. 2022). The Atlantic flyway analysis through ringed-recovery and clustering of the chicken H5N1 HPAI virus from British Columbia with European H5N1 viruses favoured the incursion of GsGd H5N1 viruses to British Columbia via the Atlantic flyway. We hypothesise that a direct interhemispheric migratory water bird including geese could possibly have dispersed the virus to chickens in British Columbia. In a recent study, we identified the Atlantic flyway as the likely route of the initial incursion of GsGd H5N1 viruses to East Canada and east Coast USA states in 2021 from Western Europe (Caliendo et al. 2022). The increased number of HPAI genotypes in Western Europe in almost all seasons implies strong dissemination and maintenance of the virus in wild birds. The continued continental expansion of introduced H5N1 viruses on the East Coast of the USA and the movement of birds through the North American flyways have likely contributed to the dispersion of the virus to British Columbia. The dispersions of avian influenza viruses were found several times higher within-flyway than the between-flyway, even though flyway intersects are expected, implying that the viruses could reach British Columbia by either of the North American flyways (Fourment, Darling, and Holmes 2017).
The detection of H5N1 viruses in long-distance migratory dabbling ducks, such as the American wigeon (Mareca americana), indicates that multiple wild bird species serve as carriers and potentially as amplifiers of the virus. Avian species such as migrating ruddy turnstone (A. interpres) can serve as a transmission vector for HPAI virus from wintering grounds to Delaware Bay, a critical stopover site for shorebirds during spring migration. These species may serve in connecting intercontinental associations of waterfowl and virus incursions. The same species have been found to carry the highest number and subtypes of influenza virus in the Delaware Bay ecosystem (Poulson et al. 2020).
Mitigating risk to poultry remains a priority during these widely spread H5 HPAI outbreaks in the world. Based on the assumption that the last incursion strain would be the closest prototype match in any emergency poultry vaccine bank in North America, we assessed changes in the critical antigenic sites of the HA protein in the two viruses that might impact emergency poultry vaccine use. The amino acid substitutions in key antigenic sites (e.g. site B) for both viruses may likely result in a reduction in phenotypic strain match of the hemagglutinin protein (Koel et al. 2014).
Virulence of influenza A viruses is often determined by the complex interactions between viral proteins and host factors, and as such, these interactions can enhance the polymerase activity and hence virus replication and pathogenicity in mammals (Gabriel, Herwig, and Klenk 2008; Yamayoshi et al. 2014; DesRochers et al. 2016). During sequence analysis, we did find some critical mutations of amino acid in PB2, PB1, PA, and NP gene segments that were previously associated with increased polymerase activity and replication in mammalian cells. Determining the significance of these critical amino acids present in the internal gene segments in virulence is beyond the scope of this paper; we recommend risk assessments to be conducted in various animal models using the new reassortant viruses.
Supplementary Material
Acknowledgements
The authors would like to thank the Canadian Food Inspection Agency field staff that were dealing with the ongoing avian influenza outbreaks. The authors also extend their appreciation to the Environment Climate Change Canada and Canadian Wildlife Health Cooperative field staff that were involved in Canada-wide avian influenza surveillance in wild birds. Drs John Pasick and Kathleen Hooper-McGrevy thank you for critically reviewing the manuscript.
Contributor Information
Tamiru N Alkie, National Centre for Foreign Animal Disease, 1015 Arlington Street, Winnipeg, Manitoba R3E 3M4, Canada.
Sara Lopes, Department of Pathobiology and Population Sciences, Hawkshead Campus, The Royal Veterinary College Hawkshead Lane, North Mymms, Hatfield, Hertfordshire AL9 7TA, UK.
Tamiko Hisanaga, National Centre for Foreign Animal Disease, 1015 Arlington Street, Winnipeg, Manitoba R3E 3M4, Canada.
Wanhong Xu, National Centre for Foreign Animal Disease, 1015 Arlington Street, Winnipeg, Manitoba R3E 3M4, Canada.
Matthew Suderman, National Centre for Foreign Animal Disease, 1015 Arlington Street, Winnipeg, Manitoba R3E 3M4, Canada.
Janice Koziuk, National Centre for Foreign Animal Disease, 1015 Arlington Street, Winnipeg, Manitoba R3E 3M4, Canada.
Mathew Fisher, National Centre for Foreign Animal Disease, 1015 Arlington Street, Winnipeg, Manitoba R3E 3M4, Canada.
Tony Redford, Animal Health Centre, BC Ministry of Agriculture and Food, 1767 Angus Campbell Road, Abbotsford, British Columbia V3G 2M3, Canada.
Oliver Lung, National Centre for Foreign Animal Disease, 1015 Arlington Street, Winnipeg, Manitoba R3E 3M4, Canada; Department of Biological Sciences, University of Manitoba, 50 Sifton Rd., Winnipeg, Manitoba R3T 2M5, Canada.
Tomy Joseph, Animal Health Centre, BC Ministry of Agriculture and Food, 1767 Angus Campbell Road, Abbotsford, British Columbia V3G 2M3, Canada.
Chelsea G Himsworth, Animal Health Centre, BC Ministry of Agriculture and Food, 1767 Angus Campbell Road, Abbotsford, British Columbia V3G 2M3, Canada; Canadian Wildlife Health Cooperative British Columbia, 1767 Angus Campbell Road, Abbotsford, British Columbia V3G 2M3, Canada; School of Population and Public Health, University of British Columbia, 2206 E Mall, Vancouver, British Columbia V6T 1Z3, Canada.
Ian H Brown, International Reference Laboratory for AI, Animal and Plant Health Agency-Weybridge, Woodham Lane, New Haw, Addlestone, Surrey KT15 3NB, UK.
Victoria Bowes, Animal Health Centre, BC Ministry of Agriculture and Food, 1767 Angus Campbell Road, Abbotsford, British Columbia V3G 2M3, Canada.
Nicola S Lewis, Department of Pathobiology and Population Sciences, Hawkshead Campus, The Royal Veterinary College Hawkshead Lane, North Mymms, Hatfield, Hertfordshire AL9 7TA, UK; International Reference Laboratory for AI, Animal and Plant Health Agency-Weybridge, Woodham Lane, New Haw, Addlestone, Surrey KT15 3NB, UK.
Yohannes Berhane, National Centre for Foreign Animal Disease, 1015 Arlington Street, Winnipeg, Manitoba R3E 3M4, Canada; Department of Animal Science, University of Manitoba, Chancellors Cir, Winnipeg, Manitoba R3T 2N2, Canada; Department of Veterinary Pathology, Western College of Veterinary Medicine, University of Saskatchewan, 52 Campus Dr., Saskatoon, Saskatchewan S7N 5B4, Canada.
Data availability
The sequences generated from the current study will be deposited in the Global Initiative on Sharing All Influenza Data EpiFlu™ database.
Supplementary data
Supplementary data are available at Virus Evolution online.
Funding
Costs associated with the work were covered under the Canadian Food Inspection Agency emergency funding for the 2022 outbreak (Y.B.). This project has been funded in whole or in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. 75N93021C00015 (N.S.L. and S.L.).
Conflict of interest:
The authors declare no conflict of interest.
Role of funding source
The project funders had no role in the project design, data generation or analysis or interpretation. The funders only covered the costs of laboratory reagents used in the study.
Author contributions
T.H, M.S., J.K., W.X. M.F., and O.L. were involved in processing the laboratory samples and generation of the data. T.N.A., S.L., I.H.B., N.S.L., and Y.B were involved in the data analysis, interpretation of results, and design and drafting of the manuscript. T.R., C.G.H., and V.B. were involved in collecting epidemiological data. T.R. collected the bald eagle sample and associated epidemiological information. All authors revised and edited the manuscript’s preceding drafts and approved the final manuscript.
References
- Adlhoch C. et al. (2021a) ‘Avian Influenza Overview February – May 2021’, European Food Safety Authority Journal, 19: 6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2021b) ‘Avian Influenza Overview December 2020 – February 2021’, European Food Safety Authority Journal, 19: 6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berhane Y. et al. (2016) ‘Pathobiological Characterization of a Novel Reassortant Highly Pathogenic H5N1 Virus Isolated in British Columbia, Canada, 2015’, Scientific Reports, 6: 23380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevins S. N. et al. (2016) ‘Widespread Detection of Highly Pathogenic H5 Influenza Viruses in Wild Birds from the Pacific Flyway of the United States’, Scientific Reports, 6: 28980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2022) ‘Intercontinental Movement of Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4 Virus to the United States, 2021’, Emerging Infectious Diseases, 28: 1006–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caliendo V. et al. (2022) ‘Transatlantic Spread of Highly Pathogenic Avian Influenza H5N1 by Wild Birds from Europe to North America in 2021’, Scientific Reports, 12: 11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattoli G. et al. (2009) ‘Highly Pathogenic Avian Influenza Virus Subtype H5N1 in Africa: A Comprehensive Phylogenetic Analysis and Molecular Characterization of Isolates’, PLoS One, 4: e4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement T. et al. (2015) ‘Complete Genome Sequence of a Highly Pathogenic Avian Influenza Virus (H5N2) Associated with an Outbreak in Commercial Chickens, Iowa, USA, 2015’, Genome Announcements, 3: e00613–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus E. et al. (2016) ‘Changes in Adaptation of H5N2 Highly Pathogenic Avian Influenza H5 Clade 2.3.4.4 Viruses in Chickens and Mallards’, Virology, 499: 52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DesRochers B. L. et al. (2016) ‘Residues in the PB2 and PA Genes Contribute to the Pathogenicity of Avian H7N3 Influenza A Virus in DBA/2 Mice’, Virology, 494: 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietze K. et al. (2018) ‘From Low to High Pathogenicity - Characterization of H7N7 Avian Influenza Viruses in Two Epidemiologically Linked Outbreaks’, Transboundary and Emerging Diseases, 65: 1576–87. [DOI] [PubMed] [Google Scholar]
- Dusek R. J. et al. (2014) ‘North Atlantic Migratory Bird Flyways Provide Routes for Intercontinental Movement of Avian Influenza Viruses’, PloS One, 9: e92075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekong P. S. et al. (2012) ‘Spatio-temporal Epidemiology of Highly Pathogenic Avian Influenza (H5N1) Outbreaks in Nigeria, 2006-2008’, Preventive Veterinary Medicine, 103: 170–7. [DOI] [PubMed] [Google Scholar]
- Elgendy E. M. et al. (2017) ‘Identification of Polymerase Gene Mutations that Affect Viral Replication in H5N1 Influenza Viruses Isolated from Pigeons’, Journal of General Virology, 98: 6–17. [DOI] [PubMed] [Google Scholar]
- Feng X. et al. (2016) ‘Glycine at Position 622 in PB1 Contributes to the Virulence of H5N1 Avian Influenza Virus in Mice’, Journal of Virology, 90: 1872–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourment M., Darling A. E., and Holmes E. C. (2017) ‘The Impact of Migratory Flyways on the Spread of Avian Influenza Virus in North America’, BMC Evolutionary Biology, 17: 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusaro A. et al. (2019) ‘Disentangling the Role of Africa in the Global Spread of H5 Highly Pathogenic Avian Influenza’, Nature Communications, 10: 5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel G. et al. (2005) ‘The Viral Polymerase Mediates Adaptation of an Avian Influenza Virus to a Mammalian Host’, Proceedings of the National Academy of Sciences, 102: 18590–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel G., Herwig A., and Klenk H.-D. (2008) ‘Interaction of Polymerase Subunit PB2 and NP with Importin Alpha 1 Is a Determinant of Host Range of Influenza A Virus’, PLoS Pathogens, 4: e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill S. C. et al. (2016) ‘Antibody Responses to Avian Influenza Viruses in Wild Birds Broaden with Age’, Proceedings of the Royal Society B: Biological Sciences, 283: 20162159. https://www.birds-of-north-america.net/White-tailed_Eagle.html Classic Collection of North American Birds. Accessed July 05, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip H. S. et al. (2015) ‘Novel Eurasian Highly Pathogenic Avian Influenza A H5 Viruses in Wild Birds, Washington, USA, 2014’, Emerging Infectious Diseases, 21: 886–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King J. et al. (2022) ‘Highly Pathogenic Avian Influenza Virus Incursions of Subtype H5N8, H5N5, H5N1, H5N4, and H5N3 in Germany during 2020-21’, Virus Evolution, 8: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koel B. F. et al. (2014) ‘Antigenic Variation of Clade 2.1 H5N1 Virus Is Determined by a Few Amino Acid Substitutions Immediately Adjacent to the Receptor Binding Site’, mBio, 5: e01070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Sala L. F. et al. (2019) ‘Spatial Modelling for Low Pathogenicity Avian Influenza Virus at the Interface of Wild Birds and Backyard Poultry’, Transboundary and Emerging Diseases, 66: 1493–505. [DOI] [PubMed] [Google Scholar]
- Lee D.-H. et al. (2016) ‘Highly Pathogenic Avian Influenza Viruses and Generation of Novel Reassortants, United States, 2014–2015’, Emerging Infectious Diseases, 22: 1283–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2015) ‘Intercontinental Spread of Asian-Origin H5N8 to North America through Beringia by Migratory Birds’, Journal of Virology, 89: 6521–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2017) ‘Reoccurrence of Avian Influenza A (H5N2) Virus Clade 2.3.4.4 in Wild Birds, Alaska, USA, 2016’, Emerging Infectious Diseases, 23: 365–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis N. S. et al. (2021) ‘Emergence and Spread of Novel H5N8, H5N5 and H5N1 Clade 2.3.4.4 Highly Pathogenic Avian Influenza in 2020’, Emerging Microbes and Infections, 10: 148–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lycett S. J., Duchatel F., and Digard P. (2019) ‘A Brief History of Bird Flu’, Philosophical Transactions of the Royal Society B, 374: 20180257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen B. et al. (2006) ‘Global Patterns of Influenza A Virus in Wild Birds’, Science, 312: 384–8. [DOI] [PubMed] [Google Scholar]
- Pasick J. et al. (2015) ‘Reassortant Highly Pathogenic Influenza A H5N2 Virus Containing Gene Segments Related to Eurasian H5N8 in British Columbia, Canada, 2014’, Scientific Reports, 5: 9484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poen M. J. et al. (2019) ‘Co-circulation of Genetically Distinct Highly Pathogenic Avian Influenza A Clade 2.3.4.4 (H5N6) Viruses in Wild Waterfowl and Poultry in Europe and East Asia, 2017–18’, Virus Evolution, 5: vez004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulson R. et al. (2020) ‘Influenza A Viruses in Ruddy Turnstones (Arenaria interpres); Connecting Wintering and Migratory Sites with an Ecological Hotspot at Delaware Bay’, Viruses, 12: 1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramey A. M. et al. (2018) ‘Lessons Learned from Research and Surveillance Directed at Highly Pathogenic Influenza A Viruses in Wild Birds Inhabiting North America’, Virology, 518: 55–63. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2022) ‘Highly Pathogenic Avian Influenza Is an Emerging Disease Threat to Wild Birds in North America’, The Journal of Wildlife Management, 86: e22171. [Google Scholar]
- Sagulenko P., Puller V., and Neher R. A. (2018) ‘TreeTime: Maximum-Likelihood Phylodynamic Analysis’, Virus Evolution, 4: vex042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spackman E. et al. (2002) ‘Development of a Real-Time Reverse Transcriptase PCR Assay for Type A Influenza Virus and the Avian H5 and H7 Hemagglutinin Subtypes’, Journal of Clinical Microbiology, 40: 3256–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada T. et al. (2011a) ‘Emergence of Avian Influenza Viruses with Enhanced Transcription Activity by a Single Amino Acid Substitution in the Nucleoprotein during Replication in Chicken Brains’, Journal of Virology, 85: 10354–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2011b) ‘NP Body Domain and PB2 Contribute to Increased Virulence of H5N1 Highly Pathogenic Avian Influenza Viruses in Chickens’, Journal of Virology, 85: 1834–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh D. et al. (2018) ‘Avian Influenza Viruses in Wild Birds: Virus Evolution in a Multihost Ecosystem’, Journal of Virology, 92: e00433–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhagen J. H., Fouchier R. A. M., and Lewis N. (2021) ‘Highly Pathogenic Avian Influenza Viruses at the Wild–Domestic Bird Interface in Europe: Future Directions for Research and Surveillance’, Viruses, 13: 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasilenko J. L., Sarmento L., and Pantin-Jackwood M. J. (2009) ‘A Single Substitution in Amino Acid 184 of the NP Protein Alters the Replication and Pathogenicity of H5N1 Avian Influenza Viruses in Chickens’, Archives of Virology, 154: 969–79. [DOI] [PubMed] [Google Scholar]
- Webster R. G. et al. (1992) ‘Evolution and Ecology of Influenza A Viruses’, Microbiological Reviews, 56: 152–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingartl H. M. et al. (2010) ‘Genetic and Pathobiologic Characterization of Pandemic H1N1 2009 Influenza Viruses from a Naturally Infected Swine Herd’, Journal of Virology, 84: 2245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikramaratna P. S., Pybus O. G., and Gupta S. (2014) ‘Contact between Bird Species of Different Lifespans Can Promote the Emergence of Highly Pathogenic Avian Influenza Strains’, Proceedings of the National Academy of Sciences, 111: 10767–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winker K. et al. (2007) ‘Movements of Birds and Avian Influenza from Asia into Alaska’, Emerging Infectious Diseases, 13: 547–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C. et al. (2016) ‘PB2-588 V Promotes the Mammalian Adaptation of H10N8, H7N9 and H9N2 Avian Influenza Viruses’, Scientific Reports, 6: 19474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X. et al. (1999) ‘Genetic Characterization of the Pathogenic Influenza A/Goose/Guangdong/1/96 (H5N1) Virus: Similarity of Its Hemagglutinin Gene to Those of H5N1 Viruses from the 1997 Outbreaks in Hong Kong’, Virology, 261: 15–9. [DOI] [PubMed] [Google Scholar]
- Yamayoshi S. et al. (2018) ‘Enhanced Replication of Highly Pathogenic Influenza A (H7N9) Virus in Humans’, Emerging Infectious Diseases, 24: 746–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2014) ‘Virulence-Affecting Amino Acid Changes in the PA Protein of H7N9 Influenza A Viruses’, Journal of Virology, 88: 3127–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B. et al. (2009) ‘Single-Reaction Genomic Amplification Accelerates Sequencing and Vaccine Production for Classical and Swine Origin Human Influenza A Viruses’, Journal of Virology, 83: 10309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequences generated from the current study will be deposited in the Global Initiative on Sharing All Influenza Data EpiFlu™ database.