

Abstract
The microbial communities of humans are characteristic and complex mixtures of microorganisms that have co-evolved with their human hosts. The species that make up these communities vary between hosts as a result of restricted migration of microorganisms between hosts and strong ecological interactions within hosts, as well as host variability in terms of diet, genotype and colonization history. The shared evolutionary fate of humans and their symbiotic bacteria has selected for mutualistic interactions that are essential for human health, and ecological or genetic changes that uncouple this shared fate can result in disease. In this way, looking to ecological and evolutionary principles might provide new strategies for restoring and maintaining human health.
Nowhere in the study of human biology are basic concepts changing more rapidly than with respect to the human microbiota. Microorganisms were first shown to cause disease in humans in the 1800s, and after this finding, the popular and scientific views of the microbial world became dominated by the quest to understand, prevent and cure microbial disease. This led to millions of lives being saved through improved hygiene, vaccinations and antibiotics. However, most interactions between humans and microorganisms do not result in disease. Beneficial host–microbe interactions have been studied for more than a century, but it was not until the advent of molecular biology that the pathogen-dominated view of human-associated microorganisms began to change. Gene-sequence-based approaches have recently allowed complex microbial communities to be characterized more comprehensively and have removed the constraint of being able to identify only microorganisms that can be cultured, greatly increasing knowledge about commensal microorganisms and mutualistic microorganisms of humans1–12 (that is, organisms in a relationship in which one partner benefits and the other is unharmed, and organisms in a relationship in which both partners benefit, respectively), as well as human pathogens13–18. Researchers are now finding that host–microbe interactions are essential to many aspects of normal ‘mammalian’ physiology, ranging from metabolic activity to immune homeostasis19–25. With the availability of new tools to investigate complex microbial communities and the expanded appreciation for the importance of the human indigenous microbiota, this is an opportune time to apply ecological and evolutionary principles to improve the current understanding of both health and disease.
So far, the human microbiota has not been fully described, but it is clear that microorganisms are present in site-specific communities on the skin and mucosal surfaces and in the intestinal lumen. Each community contains microorganisms from certain families and genera that are found in the same habitat in many or most individuals, although at the species and strain levels the microbiota of an individual can be as unique as a fingerprint3,11,26. The microbial communities of other terrestrial vertebrates mainly contain lineages that are related to, but distinct from, those in humans27–31. These characteristics indicate that humans have co-evolved with their microbial partners. In this review, we examine evolutionary and ecological principles that are relevant to these relationships, and we consider microbial pathogenesis in this context.
Evolution of mutualism
In the 1960s, evolutionary biologists rejected the idea that natural selection would generally favour the good of the species (or any group), because individual types with the greatest reproductive success in a population increase in relative abundance regardless of the consequences for the population as a whole32. Since then, the evolution of traits that benefit individuals other than the trait bearer has been extensively researched, both theoretically and empirically. Although the field has been contentious at times, there is now general agreement about the conditions that promote cooperation, including mutualism between species32–34.
Organisms can have traits that contribute directly to their own fitness and also incidentally benefit members of another species. When such ‘by-product benefits’ occur in both directions, the result is a no-cost mutualism34. For example, plant polysaccharides that are not digestible by humans are the main substrates for microbial growth in the colon, whereas butyrate and other products of microbial fermentation are important energy sources for the host35,36. Intestinal symbionts are selected to be effective consumers of available resources through direct effects on their fitness, but this also benefits the host because resource competition provides an additional barrier to colonization by potential pathogens37–41.
If mutualistic by-product interactions such as the example above are possible, but not ecologically inevitable, then traits that improve the likelihood or stability of a relationship (for example, site-specific attachment molecules) might evolve in one or both partners. A species might also evolve to increase its own fitness by increasing the fitness of a mutualistic partner34. For example, microbial symbionts that secrete molecules that inhibit host pathogens (known as pathogen interference)38–40 or detoxify compounds that harm the host42 can augment the lifespan and reproductive capacity of the host, thereby giving the symbionts more opportunities to spread. Evolution to increase mutualistic benefits has been called ‘partner fidelity feedback’, and it is strengthened if the same lineages of partners interact across multiple generations33,34. Unlike traits that support mutualism incidentally, traits that evolve specifically to improve mutualism, such as the production of compounds dedicated to pathogen interference, can impose a direct fitness cost, although a net benefit would be expected in the context of the evolved mutualism34.
Mutualism-promoting traits with a direct cost for the bearer, however, create the potential for ‘cheating’. When organisms interact to create a shared benefit, cheaters are organisms that obtain the benefit without helping to create it. For example, a cheating microbial phenotype could result from a mutation that redirects resources towards faster growth of the microorganism itself instead of detoxification or pathogen interference. The cheater therefore increases its relative fitness in a host by avoiding costly contributions towards host fitness while benefiting from the improved host fitness that results from the mutualistic contributions of its competitors33,34. Various evolutionary outcomes are possible, including the absence of costly contributions to mutualism, contributions to mutualism that are below the level that would maximize the mutualistic benefit, and the coexistence of mutualists and cheaters in the community43. A possible example of this dynamic balance is that certain benefits attributed to probiotic bacterial species are characteristic of only a subset of the strains that make up the species38,40. For any mutualism that is not cost free, the partners can evolve mechanisms to protect their relationship from being exploited by cheaters32,34,44, and mutualism can be stronger and more stable where ecological features limit the potential for exploitation (discussed later).
The immune system is the most conspicuous set of anti-exploitation adaptations involved in human–microbial symbiosis. The gastrointestinal mucosa is intimately associated with the most abundant and diverse microbial communities in the human body, but the gut-associated immune system neither controls the composition of the gut microbiota nor remains ignorant of it. Instead, specialized tissues and cells actively sample the intestinal contents and initiate local immune responses that help to confine the microbiota to the gut, avoiding a damaging systemic inflammatory response to the microorganisms present in the healthy gut45. However, if host tissue is damaged46 or if microorganisms spread to normally sterile sites45, then there is a vigorous systemic response to clear the infection. Therefore, microorganisms are free to compete for resources in the gut, generating a robust and disease-resistant community37,41, but are prevented (usually) from exploiting the host to obtain additional resources. Recent work has also shown that the normal development and activity of the ‘host’ immune system is itself a result of mutualistic interactions20–22,24,25 (see page 819).
Humans and their collective microbiota are segmented into many local communities, each comprising an individual human with his or her symbionts. This ecological pattern, characterized by strong interactions within distinct local communities and limited interactions or migration between them, is described as a metacommunity. Another level of metacommunity organization exists because individual humans belong to social groups that tend to share a similar microbiota47,48. At both levels, the metacommunity structure allows selection to occur between the local units (that is, between individuals or between social groups), which promotes mutualism and restrains cheating within the human–microbe symbiosis32,49. Such selection occurs when a local symbiotic community succeeds or fails together, with more successful communities increasing in abundance or prevalence relative to less successful communities32. For example, a human individual or social group that carries a microbiota with strong defences against an abundant pathogen is likely to leave more progeny than those lacking such defences. If the progeny tend to carry the parental microbiota, then mutualistic microorganisms that make costly contributions to pathogen defence are favoured by selection between distinct local symbiotic communities. This community-level selection opposes the tendency for cheating non-defenders to increase in relative abundance within each local symbiotic community32. The greater similarity of the microbiota within a human family than between human families12 (and within, rather than between, chimpanzee social groups30) shows that there is, indeed, a shared evolutionary fate. The individualized microbiota of each person has a stake in his or her fitness.
Human–microbe mutualism often involves more than two partners, although the same principles apply. For example, the colonic degradation of polysaccharides that provides butyrate for the host is a cooperative microbial process35,36. Extracellular enzymes from multiple species are required for complete hydrolysis of the polymers. In addition, some of the resultant sugars are consumed by strains that do not produce extracellular enzymes but provide growth factors to strains that do35. Some fermenters such as Bifidobacterium spp. release lactate as waste. Their fermentation efficiency is increased by lactate fermenters, such as Eubacterium hallii, that release butyrate as waste, and this butyrate is then used by the host36. Sugar-fermenting lactobacilli that produce neither hydrolytic enzymes nor growth factors could be considered cheaters from the perspective of polysaccharide degradation, but they could be considered mutualists of the entire symbiotic community if they interfere with the colonization of pathogens40. The butyrate-producing consortium as a whole is a mutualist of the host and would be favoured by community-level selection over consortia producing less desirable fermentation products36. However, selection for mutualistic functional traits such as butyrate production cannot entirely determine the composition of the microbiota, because communities of different composition can have similar functional characteristics in a given context. Not only selection on community-level traits but also competition within the community and chance colonization events affect the structure of the microbiota50.
Human microbial communities and health
The distribution of microorganisms in and on the human body reflects adaptations to life on land, which were made about 400 million years ago. Terrestrial vertebrates developed skin, lungs, internal fertilization, and protective membranes around the embryo. The skin became relatively impermeable, and mucous membranes were confined to protected sites. Because microorganisms generally thrive only in moist environments, these adaptations to a mostly dry environment have shaped the abundance, location and phenotypes of human-associated microorganisms and have limited the exchange of microorganisms between individuals.
The current understanding of the human microbiota relies heavily on cultivation-based approaches and therefore is biased and incomplete. Although imperfect, molecular approaches that identify microorganisms from small-subunit (16S) ribosomal RNA gene sequences offer advantages over cultivation. The 16S rRNA gene is typically chosen because it is present universally and can provide a taxonomic identification ranging from the domain and phylum level to approximately the species level. However, these methods have been used to study human microbial ecology for only a decade, and the available data are limited. There are few deep surveys of microbial-community membership in any human habitat and even fewer assessments of functional potential or activity. In general, 16S rRNA gene-sequence data have been collected from one site in a few humans at one time, representing a narrow range of health and disease states2,5–7, although there are studies that include several temporal or spatial samples per individual1,3,4,51. Sequence-dependent approaches that are less labour-intensive but yield lower-resolution data have been applied to a larger number of individuals, at various time points and under various conditions8,9,11,12,52. Even so, the microbial communities associated with only a small proportion of the diversity of human genotypes, lifestyles, diets and diseases have been investigated. One high-throughput method for obtaining information about bacterial communities is to use phylogenetic microarrays, which yield high-resolution data, but this method also depends on adequate 16S rRNA gene-sequence databases10. Like these microarray studies, metagenomic and proteomic analyses are just beginning to be published53,54. Technical and ethical constraints restrict sampling from humans; therefore, model systems will continue to be important, and examples of these are listed in Table 1.
Table 1 |.
Model systems for animal-microbe symbioses
| Type of symbiosis | Specific system (Host/symbiont species) | Host phylogenetic affiliation | Host tissue colonized | Reference |
|---|---|---|---|---|
| Highly complex consortia (102-103)* | Mus musculus (mouse) | Vertebrate chordate | Intestine | 19 |
| Danio rerio (zebrafish) | Vertebrate chordate | Intestine | 86 | |
| Microcerotermes spp. and Reticulitermes spp. (termites) | Insect arthropod | Hindgut | 87 | |
| Relatively simple consortia (~2–25)* | Hirudo medicinalis (leech) | Oligochaete annelid | Intestine | 88 |
| Lymantria dispar (gypsy moth) | Insect arthropod | Larval midgut | 89 | |
| Drosophila melanogaster (fruitfly) | Insect arthropod | Intestine | 90 | |
| Hydra oligactis and Hydra vulgaris | Hydrozoan cnidarian | Not determined | 91 | |
| Monospecific (1)* | Euprymna scolopes (sepiolid squid)/Vibrio fischeri | Cephalopod mollusc | Light organ | 92 |
| Eisenia fetida (earthworm)/Acidovorax spp. | Oligochaete annelid | Excretory tissues | 93 | |
| Steinernema spp./Xenorhabdus spp. and Heterorhabditis spp./Photorhabdus spp. | Entomopathogenic nematodes | Gut-associated vesicle or region | 94 |
Number of bacterial-symbiont phylotypes found reproducibly.
Despite the limited data available, analyses of the human microbiota have revealed intriguing features. Most of the phylogenetic diversity is found in shallow, wide radiations in a small subset of the known deep lineages26. Specifically, there are more than 50 bacterial phyla on Earth, but human-associated communities are dominated by only 4 phyla (Firmicutes, Bacteroidetes, Actinobacteria and Proteobacteria), with 9 other phyla (Chlamydiae, Cyanobacteria, Deferribacteres, Deinococcus–Thermus, Fusobacteria, Spirochaetes, Verrucomicrobia, and the candidate phyla TM7 and SR1) found in some sites and individuals (Fig. 1). In contrast to the paucity of phyla represented, the human microbiota contains an abundance of species and strains. Uniform probabilities of speciation and extinction over time would result in an exponential increase in the number of lineages throughout evolution. However, in humans, there is a marked excess of phylotype diversity at the species and strain level compared with the trends in more inclusive taxa (Fig. 2); there are similar patterns in other vertebrate hosts26. This finding might reflect a long history of stability in the types of microbial niche associated with terrestrial animals, together with factors (such as host heterogeneity and metacommunity structure) that promote diversification among organisms in similar niches. In contrast to the remarkable diversity of bacterial species, a striking but unexplained finding is that the only Archaea found frequently in humans are several species of methanogens. Methanobrevibacter smithii is abundant in the colon of some humans3,53. Also, Methanobrevibacter oralis and close relatives can be found within the subgingival crevice in the human mouth but only in the setting of moderate to severe disease55. Overall, the human microbiota is similar to that of other mammals at the phylum level, but most bacterial families and genera seem to be distinct (Fig. 3).
Figure 1 |. Site-specific distributions of bacterial phyla in healthy humans.
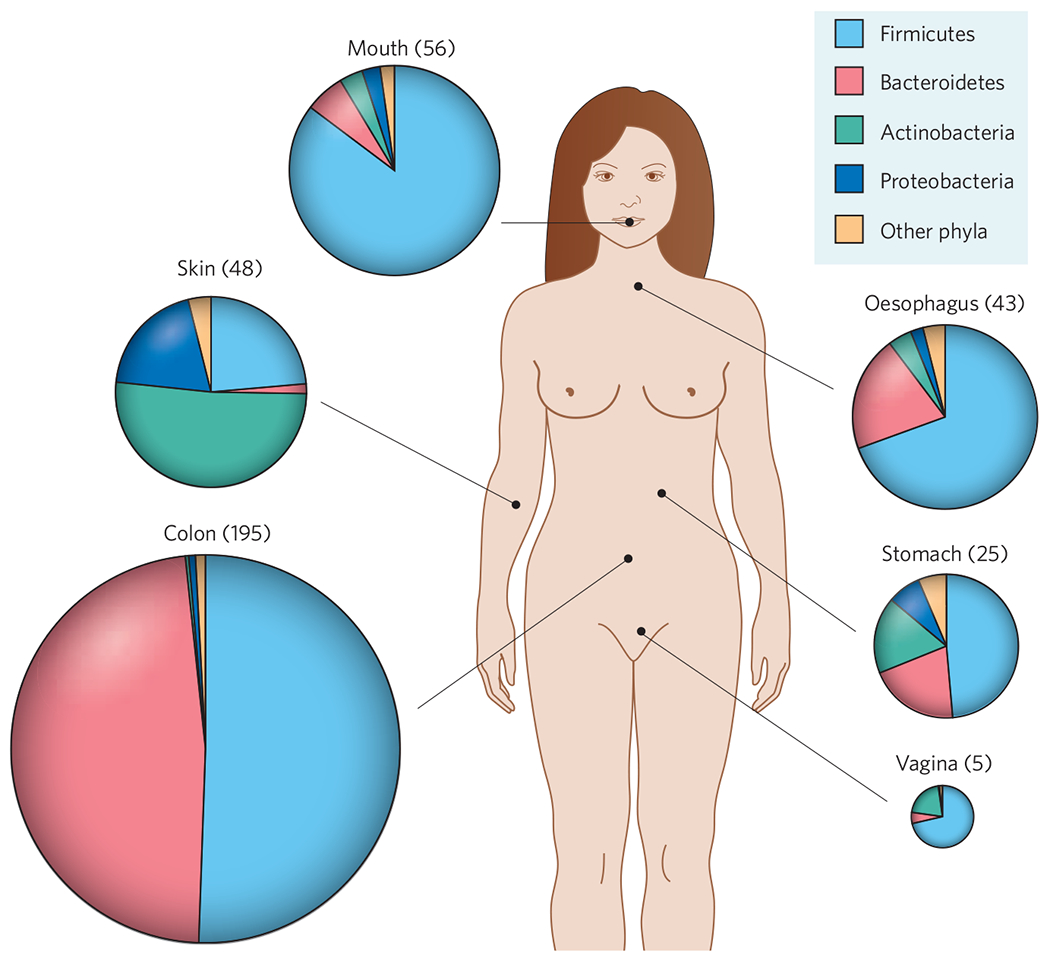
The area of the chart for each site represents the average number of distinct phylotypes (approximate species-level taxa, based on 16S rRNA gene-sequence analysis) per individual. (The mean number of phylotypes per individual is shown in parentheses; 3–11 individuals were studied per habitat.) The coloured wedges represent the proportion of phylotypes belonging to different phyla. More than 50 bacteria phyla exist, but human microbial communities are overwhelmingly dominated by the 4 that are shown. The relative abundance of these phyla at most sites tends to be consistent across individuals: for example, in almost all humans studied so far, Bacteroidetes and Firmicutes predominate in the colon. By contrast, the composition of the vaginal microbiota is more variable; most women have a preponderance of Firmicutes with few other representatives, whereas a minority of women have a preponderance of Actinobacteria with few other representatives. An estimated 20–80% of human-associated phylotypes (depending on habitat) are thought to have eluded cultivation so far. Data taken from refs 1–7.
Figure 2 |. Patterns of human-associated microbial diversity.
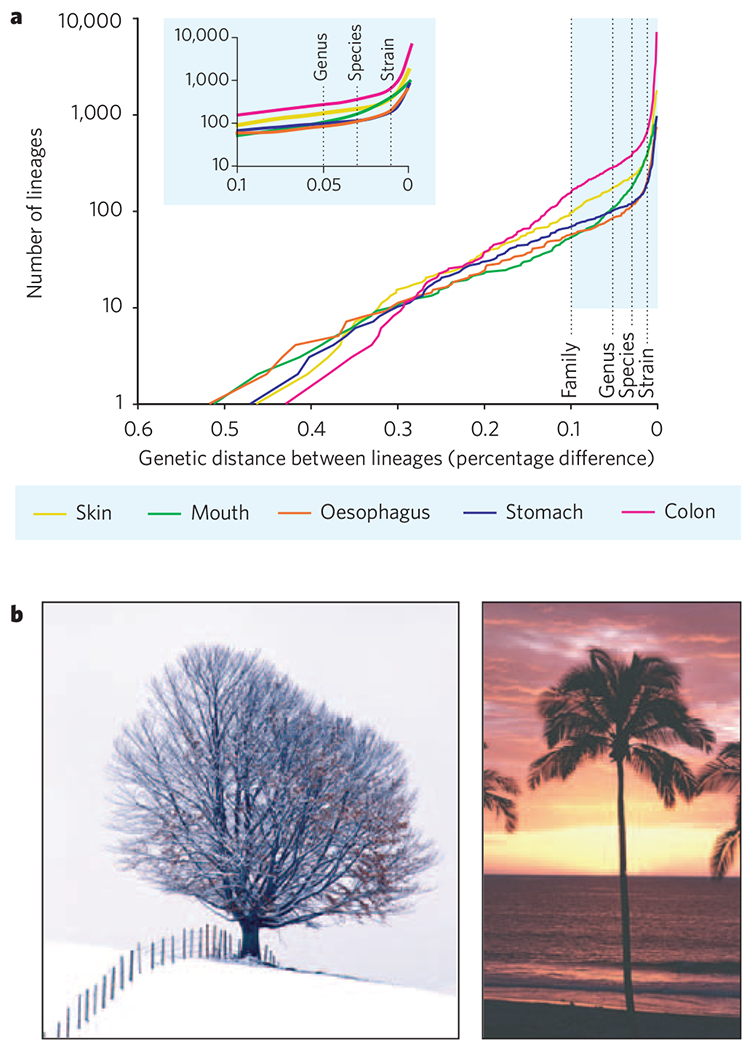
a, Lineage-by-distance analysis of 16S rRNA gene-sequence data from human microbial communities in specific habitats. The x axis shows the percentage difference threshold (Olsen correction), over 1,241 unambiguously aligned positions of near full-length 16S rRNA gene sequences, for delineating separate lineages. The y axis shows the number of distinct lineages that exist at the distance threshold. If speciation and extinction occur with constant probabilities as 16S rRNA gene sequences diverge, this would result in an exponentially increasing number of lineages with diminishing evolutionary distances between them (a straight line on a semi logarithmic plot). Such a pattern seems to hold from the phylum level (largest distances between lineages) to approximately the species level. However, relative to this trend, all sites have an excess of recently diverged lineages. The excess lineages accumulate in the range of 16S rRNA gene divergence that is typically associated with species and strains. The inset depicts a portion of the same data at a larger scale. Samples were taken from 3–11 individuals, depending on the site. Data taken from refs 1–5. b, When displayed as a dendrogram, 16S rRNA gene-based patterns of microbial diversity in soil and aquatic environments generally resemble the tree shape on the left, with new branches arising at all distances from the root. Patterns of diversity in vertebrate-associated communities resemble the tree shape on the right, with few branches arising close to the root and many branches arising close to the branch tips.
Figure 3 |. Relationships between bacterial 16S rRNA gene sequences from the intestinal microbiota of animals.
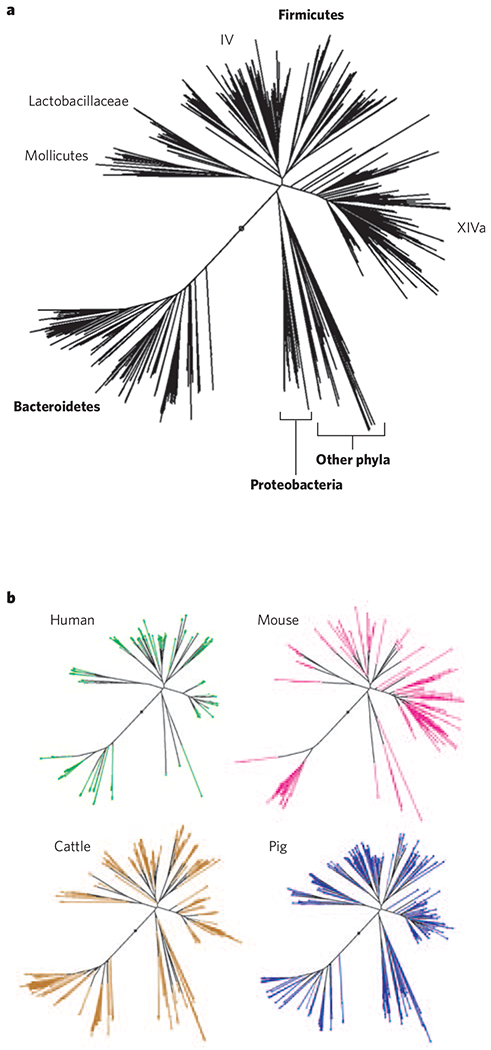
A set of aligned, high-quality, full-length sequences was obtained from Greengenes95. Sequences derived from one human stool sample and caecal samples from one mouse family were chosen to obtain approximately the same number of sequences as obtained from multiple studies of the bovine rumen and pig caecum and colon (range 617–748 sequences per host species). a, A neighbour-joining tree was created from 1,241 unambiguously aligned positions in all 2,735 sequences, with selected taxa indicated. Mollicutes, Lactobacillaceae and Clostridium clusters IV and XIVa are within the Firmicutes96. b, Host-specific trees were created with the same topology as the entire tree, shown in part a, but they depict only the sequences derived from the indicated host species. Branches shared with at least one other host species are shown in black, and branches specific to a single species are coloured. The same phyla and classes predominate in these animals (evident from the overlapping tree topologies and shared branches), although their relative abundances vary. By contrast, most genera and many families are specific to a single host species (coloured branches).
Multiple samples of the microbiota that are taken separated in time or space from a single body habitat within one individual are generally more similar to each other than they are to samples from the same habitat in another individual3,9,11, although temporal variation in the skin microbiota of an individual is as great as the variation between individuals4. In addition, the bacterial communities at a given site are more similar between human family members than between unrelated individuals12, but more studies are necessary to distinguish the effects of genetic relatedness and a shared early environment50. Antibiotics can markedly affect the composition of the microbiota in the short term, with most (but not all) families and genera of gut microorganisms returning to typical levels within weeks of exposure51,56. However, pathogens can exploit the reduced competitiveness of a community disturbed by antibiotics, thereby establishing themselves in the host39,57. The degree to which the unique bacterial communities of an individual are re-established after antibiotic treatment is unclear, but particular antibiotic-resistant strains that colonize or evolve during treatment can persist for years58,59.
A human infant acquires its microbiota from the environment. In humans, symbionts are not vertically transmitted (that is, transmitted through the germ line), as they are in some invertebrate animals. Colonization, succession and diversification occur within characteristic windows of time in the various microbial habitats in the body, ranging over the first weeks, months or years of life10,60,61. The composition of faecal communities in early infancy, for example, is dynamic and reflects opportunistic environmental exposures, especially to the mother. The introduction of solid foods then begins the transition to an individualized, adult-like microbiota10. The assembly of bacterial communities on tooth surfaces also follows a consistent pattern as teeth emerge62, as well as after the removal of pre-existing biomass (that is, plaque)52,60. The horizontal transfer of microorganisms to every human generation favours strains that are locally abundant at that time (for example, those present in parents and kin), but colonization remains somewhat stochastic50. The mixing of lineages from different sources that occurs during community assembly is analogous to the reassortment of parental alleles during sexual reproduction49, and it promotes the adaptation of community composition to local conditions and the rapid spread of beneficial strains. However, strains that become locally abundant by cheating can also spread.
Competition for niches within the human microbiota is ubiquitous and occurs together with the selective forces that promote mutualism in the community as a whole. Microorganisms can even compete and cooperate simultaneously. For example, Bacteroides thetaiotaomicron and M. smithii facilitate each other’s growth by complementary energy metabolism, while competing for nitrogen63. Cooperatively crosslinked biofilms containing multiple species promote the colonization of tooth surfaces, even while the constituent species compete with each other for individual binding sites64. Symbionts that are highly prevalent and abundant probably have effective mechanisms for competing for resources: for example, B. thetaiotaomicron65 and Bifidobacterium longum66 have a wide variety of inducible genes encoding factors involved in the binding, uptake and degradation of plant- and host-derived polysaccharides. Competition within the human microbiota involves not only resources but also interference; that is, the direct inhibition of one strain by another in a resource-independent manner. In some cases, the metabolic byproducts of one species (such as lactate or short-chain fatty acids) inhibit other microorganisms. In other cases, dedicated compounds are generated solely because of their inhibitory effect: for example, reactive oxygen species and the peptide antibiotics known as bacteriocins39,40. The immediate fitness costs and context-dependent benefits of dedicated interference compounds result in selection for diversity: for example, the capacity to produce and resist bacteriocins evolves rapidly among closely related strains67,68. Both resource competition and interference competition contribute to the resistance of the intact microbiota to colonization by pathogens37–40.
Microbial evolution and human disease
Microbial symbionts occupy a complex adaptive landscape. Many traits affect fitness, and many different trait combinations can generate a local optimum fitness (that is, a fitness peak). Natural selection generally acts on subtle phenotypic differences to move microorganisms ‘uphill’ towards a fitness peak, but larger changes can move an organism onto the slope of a different fitness peak (Fig. 4). The fitness of a symbiont depends on environmental features that can change, such as the coexisting microbiota, the diet of the host, and which species and even particular individual is the host. Thus, the adaptive landscape is dynamic.
Figure 4 |. Adaptive landscapes.
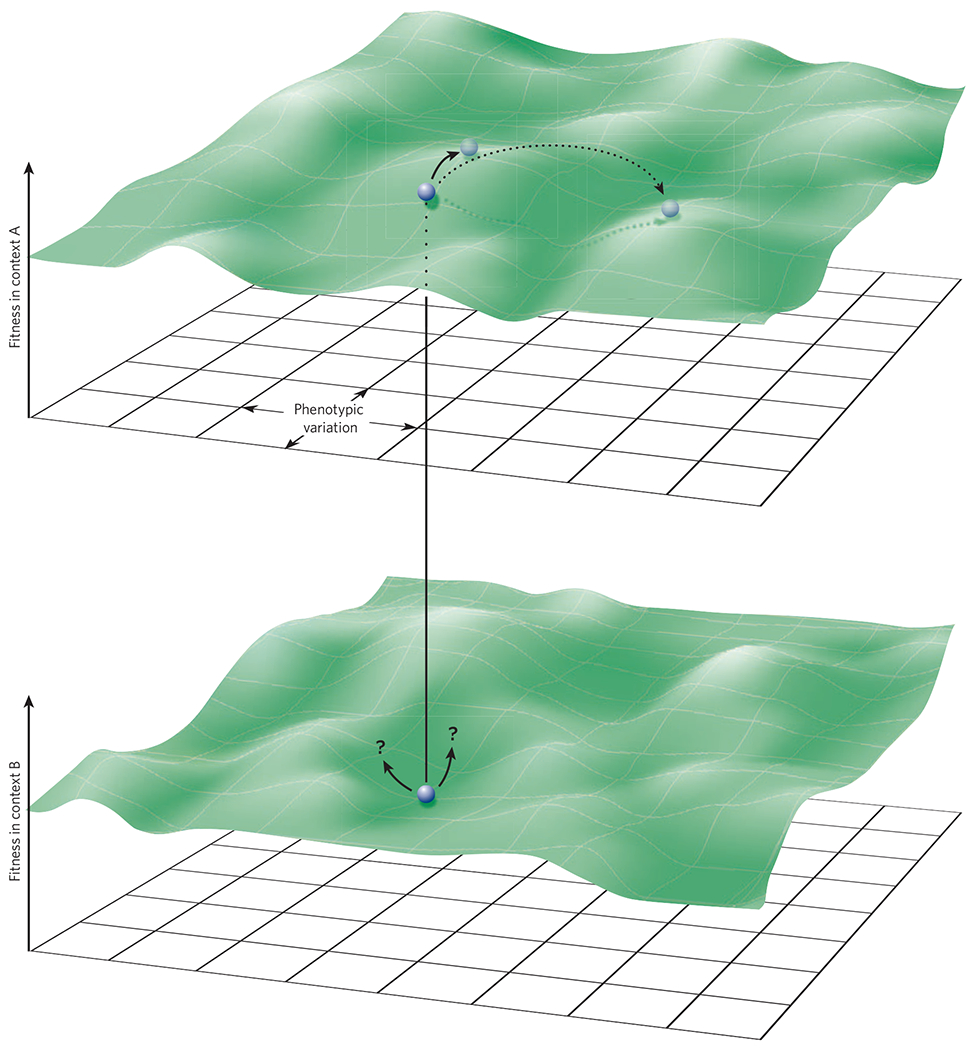
The plane is a conceptual representation of the multidimensional phenotypes that are available to a microorganism. The height of the surface above the plane represents the fitness of the corresponding phenotypes in a given ecological context, including biotic and abiotic components of the environment. In a given environment (context A, upper panel), for mutations that have a small effect, a phenotype (circle) under natural selection will tend to evolve along the steepest path uphill towards higher fitness (solid arrow), eventually moving the mean phenotype of a population to a local fitness maximum. Mutations that have a large effect, such as horizontal gene transfer, can shift a phenotype to the slope of a different fitness peak (dashed arrow). This can markedly alter the outcome for the host; for example, it can result in pathogenesis instead of mutualism. The valley separating the peaks represents phenotypes of low fitness, such as those that are likely to elicit an immune response but lack the adaptations necessary to survive it. For a given phenotype, a change in context (for example, a change in host diet, alterations in coexisting microbial populations, or transfer to a different host or host species; context B, lower panel) can have subtle or marked effects on fitness. A phenotype near a fitness peak in context A might be in a valley of low fitness in context B. If the microorganism survives, the subsequent course of evolution might depend on the direction of phenotypic change caused by the next mutation.
Changes in the genotype or environment of a non-pathogenic symbiont can result in the invasion of host tissue. The usual outcome is then an immune response that eliminates the infection. This high rate of microbial mortality imposes a strong selective pressure: the rare changes that enable a symbiont to survive such a challenge would involve avoiding immune recognition or circumventing immune control, at least until some progeny have been transmitted to a new host. Alternatively, changes that increase the opportunities for a non-pathogen to be transmitted to a new host reduce the dependence of the microorganism on the fate of the current host. In this case, the selective pressures on the fitness of the symbiont are less constrained by the need to preserve host fitness as well. In either case, the microorganism can begin adapting towards a fitness peak as a pathogen.
All human microbial symbionts must be able to establish themselves in new hosts. The adaptations of mutualistic or commensal microorganisms towards this end can facilitate a pathogenic lifestyle as well. For example, the biochemical mechanisms for sensing host environments, interacting with host surfaces and even communicating with the host are often the same in human pathogens, commensal microorganisms and mutualistic microorganisms15,69–71. Not surprisingly, many common human pathogens are closely related to non-pathogenic symbionts: examples are found in the genera Staphylococcus, Streptococcus, Neisseria and Enterococcus, and in the family Enterobacteriaceae13–18. It is not coincidental that these taxa tolerate the aerobic environment between hosts, whereas the more abundant, but less aerotolerant, taxa of the colon have fewer known pathogens as close relatives. The greater ability of aerotolerant taxa to be transmitted to a new host weakens the selection for mutualism in the current host33,34. In general, the pathogenic phenotypes in taxa that contain abundant non-pathogenic symbionts have multiple evolutionary origins13–18, emphasizing that pathogenicity is not necessarily a considerable evolutionary barrier for microorganisms. By contrast, other pathogens have originated only once72,73, but the continued emergence of new diseases is a reminder that there might be many unoccupied pathogen fitness peaks at present.
The evolution of pathogen virulence has received considerable attention, largely centred on the paradox that pathogens both harm and depend on their hosts74. The view that highly virulent pathogens originated recently, with selection inevitably reducing virulence over time, has been supplanted by the realization that there is an optimal level of virulence (for the pathogen) that depends on the biology of its host interactions74,75. For example, if pathogen transmission is inherently damaging to the host (as occurs with Salmonella enterica serovar Typhimurium76), then selective pressure on the pathogen balances the benefit of higher transmission against the loss of host viability as a result of higher virulence. By contrast, pathogens with environmental reservoirs (for example, Vibrio cholerae), transmission vectors (for example, Plasmodium falciparum) or environmentally resistant propagules (such as spores; for example, Clostridium tetani) might be able to afford a higher level of virulence than those that depend on direct transmission77. For pathogens that depend on normal host activity for transmission, such as sexually transmitted pathogens (for example, Chlamydia trachomatis and Treponema pallidum), low virulence and/or long latency can promote the spread of pathogen. In host populations with a reduced potential for pathogens to encounter new hosts, the optimal virulence is reduced to allow the host to survive long enough to ensure pathogen transmission78.
The observed level of virulence for a pathogen, however, does not necessarily correspond to its evolutionary optimum. Many pathogens are zoonotic (that is, transmitted from animals to humans)79 and can be adapted to a low-virulence niche in their primary host; an example is enterohaemorrhagic Escherichia coli in cattle80. Unless transmission by humans contributes to the evolutionary success of the pathogen, excessive (or suboptimal) virulence in humans exerts no selective pressure on the microorganism. Competition between different strains of a pathogen (as a result of co-infection, as occurs with Plasmodium spp., or in-host evolution, as occurs with human immunodeficiency virus) can affect virulence, because an optimally virulent pathogen (as measured by transmission success) might not be the best competitor during mixed infections in a single host81. A rapidly replicating, excessively virulent strain might kill the host or provoke a successful immune response before transmission of a co-infecting, less virulent strain, even if the latter strain is optimally virulent when infecting a host alone81. Competition between pathogens can also decrease virulence. The production of extracellular iron-scavenging molecules (known as siderophores) contributes to the virulence of many bacterial pathogens, but cheating lineages that consume siderophores without producing them reduce virulence, thereby benefiting the host82. The diverse biology of host–pathogen and pathogen–pathogen interactions precludes simple predictions about the effect of interpathogen competition on virulence81,83.
The importance of opportunity for the origin of pathogens is emphasized by a recent analysis of the 25 infectious diseases that cause the most human death and disability84. The preferred host of a pathogen is thought to change most easily to a species closely related to the current host85. Indeed, although primates constitute only a small proportion of all animal species on Earth, they are the origin of a large proportion of these serious human diseases. However, an even larger proportion of these diseases originated from domestic animals, reflecting greater opportunities for the symbionts of domesticated species to be transmitted to humans84. With the advent of agriculture, changes in human populations simultaneously created a new niche for deadly pathogens. Ten of these 25 major infectious diseases could have arisen only after urbanization, because they depend on human–human transmission and quickly kill infected individuals or leave them with lifelong immunity84. Such ‘crowd’ diseases could not have survived in the small dispersed human societies present before agriculture. Common pathogens derived from human mutualistic microorganisms have also exploited these changes in the human population, with many clonal lineages being disseminated globally13–18.
Urbanization and global travel have eroded some of the barriers to microbial transmission between social groups that contribute to the metacommunity structure of the human–microbe symbiosis. The diminished fidelity of host and symbiont lineages to each other (both within and between generations) and reduced opportunities for community-level selection between human social groups have reduced the strength of selection for mutualism. Microbial cheaters that allocate resources to their own growth and dissemination instead of pathogen interference or other costly contributions to host fitness can now spread globally, instead of merely within a tribe. Symbionts that colonize an infant who resides in an urban area include many microorganisms that are not derived from the infant’s relatives, much less from an extended kin group with a consistent lifestyle and geographic range over generations. This disruption of co-evolved mutualism between humans and human microbiota, as a result of changes in human ecology, contributes to the increasing prevalence of chronic and degenerative disease in industrialized countries21,47.
Paths forward
Researchers have only just begun to describe the microbial communities that are associated with humans and the extent of the interactions between host and microbiota. Understanding this symbiotic ‘landscape’ will require research that spans the biological hierarchy from molecules to communities and is informed by ecological and evolutionary theory. Only with an integrated approach will it be possible to comprehend the complex ecology of human health and the many ways in which interactions between humans and microorganisms can go awry.
The first step in improving our understanding is to describe the composition of microbial communities in each habitat of the human body and how this varies over time, among individuals and with respect to variables such as diet, host genotype and health status. This project is now in its early stages, with the first successful forays having laid the groundwork for more ambitious studies, such as the Human Microbiome Project (see page 804).
Several recent studies highlight remarkable examples of how a co-evolved microbiota can markedly affect host biology at the molecular level19–25, and these findings call for a complete re-examination of human physiology and immunology44. Attributes that were assumed to be human traits have been shown to result from human–microbe interactions.
Although human studies are essential, the technical and ethical limitations of carrying out experiments and obtaining samples from humans mean that experimental model systems also need to be used. These two approaches offer complementary information. The relevance of human studies is clear. But experimental model systems have two main advantages: they highlight evolutionary conserved features that are likely to be crucial for function, and they show diversity (how a single ‘goal’ is accomplished differently), thereby exposing the essence of a characteristic. Models for the study of symbioses range from binary relationships between an invertebrate and one microbial species to complex vertebrate systems involving consortia of microorganisms (Table 1). For models with complex consortia, gnotobiotic techniques are used to manipulate the symbiosis experimentally. By contrast, using simpler consortia facilitates the molecular dissection of interactions in the intact natural setting. The genetic tools available for some model hosts allow the identification of genes and proteins that control host responses and manage the consortia.
From the microbial perspective, the host is a simply a complex environment — the distinction between human health and disease is important only as far as it affects microbial fitness. To think that we can intervene effectively in human–microbe relationships without considering microbial ecology and evolution is folly, as demonstrated by the spread of antibiotic-resistant microorganisms13,14,16,17,58,59 and by the connections between some modern diseases and alterations in the human microbiota21,47. The principles and mechanisms that underlie microbial community structure and host–symbiont relationships must become incorporated into our definitions of human health. It will be crucial to consider the role of microbial communities, and not just individual species, as pathogens and mutualists55. Moreover, one of the goals of medical intervention during disease should be minimizing damage to the health-associated homeostasis between humans and their microbiota. Medical and general educational curricula will need to be modified accordingly.
Acknowledgements
Research in the laboratory of D.A.R. is supported by funds from the Doris Duke Charitable Foundation, the Horn Foundation, the Office of Naval Research and the National Institutes of Health (NIH). Research in the laboratory of M.M.-N. is supported by the NIH and the National Science Foundation. D.A.R. is a recipient of an NIH Director’s Pioneer Award and a Doris Duke Distinguished Clinical Scientist Award.
References
- 1.Aas JA, Paster BJ, Stokes LN, Olsen I & Dewhirst FE Defining the normal bacterial flora of the oral cavity. J. Clin. Microbiol 43, 5721–5732 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bik EM et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc. Natl Acad. Sci. USA 103, 732–737 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eckburg PB et al. Diversity of the human intestinal microbial flora. Science 308, 1635–1638 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao Z, Tseng CH, Pei Z & Blaser MJ Molecular analysis of human forearm superficial skin bacterial biota. Proc. Natl Acad. Sci. USA 104, 2927–2932 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pei Z et al. Bacterial biota in the human distal esophagus. Proc. Natl Acad. Sci. USA 101, 4250–4255 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verhelst R et al. Cloning of 16S rRNA genes amplified from normal and disturbed vaginal microflora suggests a strong association between Atopobium vaginae, Gardnerella vaginalis and bacterial vaginosis. BMC Microbiol. 4, 16 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou X et al. Characterization of vaginal microbial communities in adult healthy women using cultivation-independent methods. Microbiology 150, 2565–2573 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Lay C et al. Colonic microbiota signatures across five northern European countries. Appl. Environ. Microbiol 71, 4153–4155 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsuki T, Watanabe K, Fujimoto J, Takada T & Tanaka R Use of 16S rRNA gene-targeted group-specific primers for real-time PCR analysis of predominant bacteria in human feces. Appl. Environ. Microbiol 70, 7220–7228 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palmer C, Bik EM, DiGiulio DB, Relman DA & Brown PO Development of the human infant intestinal microbiota. PLoS Biol. 5, e177 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vanhoutte T, Huys G, De Brandt E & Swings J Temporal stability analysis of the microbiota in human feces by denaturing gradient gel electrophoresis using universal and group-specific 16S rRNA gene primers. FEMS Microbiol. Ecol 48, 437–446 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Zoetendal EG, Akkermans ADL, Akkermans-van Vliet WM, de Visser JAGM & de Vos WM The host genotype affects the bacterial community in the human gastrointestinal tract. Microb. Ecol. Health Dis 13, 129–134 (2001). [Google Scholar]
- 13.Leavis HL, Bonten MJ & Willems RJ Identification of high-risk enterococcal clonal complexes: global dispersion and antibiotic resistance. Curr. Opin. Microbiol 9, 454–460 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Miragaia M, Thomas JC, Couto I, Enright MC & de Lencastre H Inferring a population structure for Staphylococcus epidermidis from multilocus sequence typing data. J. Bacteriol 189, 2540–2552 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Callaghan MJ, Jolley KA & Maiden MC Opacity-associated adhesin repertoire in hyperinvasive Neisseria meningitidis. Infect. Immun 74, 5085–5094 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson DA & Enright MC Multilocus sequence typing and the evolution of methicillin-resistant Staphylococcus aureus. Clin. Microbiol. Infect 10, 92–97 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Robinson DA, Sutcliffe JA, Tewodros W, Manoharan A & Bessen DE Evolution and global dissemination of macrolide-resistant group A streptococci. Antimicrob. Agents Chemother 50, 2903–2911 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wirth T et al. Sex and virulence in Escherichia coli: an evolutionary perspective. Mol. Microbiol 60, 1136–1151 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bäckhed F et al. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl Acad. Sci. USA 101, 15718–15723 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cash HL, Whitham CV, Behrendt CL & Hooper LV Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 313, 1126–1130 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guarner F et al. Mechanisms of disease: the hygiene hypothesis revisited. Nature Clin. Pract. Gastroenterol. Hepatol 3, 275–284 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Kelly D et al. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-γ and RelA. Nature Immunol 5, 104–112 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Martin FP et al. A top-down systems biology view of microbiome-mammalian metabolic interactions in a mouse model. Mol. Syst. Biol 3, 112 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mazmanian SK, Liu CH, Tzianabos AO & Kasper DL An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122, 107–118 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S & Medzhitov R Recognition of commensal microflora by Toll-like receptors is required for intestinal homeostasis. Cell 118, 229–241 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Ley RE, Peterson DA & Gordon JI Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124, 837–848 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Gong J et al. 16S rRNA gene-based analysis of mucosa-associated bacterial community and phylogeny in the chicken gastrointestinal tracts: from crops to ceca. FEMS Microbiol. Ecol 59, 147–157 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Mackie RI, Rycyk M, Ruemmler RL, Aminov RI & Wikelski M Biochemical and microbiological evidence for fermentative digestion in free-living land iguanas (Conolophus pallidus) and marine iguanas (Amblyrhynchus cristatus) on the Galapagos archipelago. Physiol. Biochem. Zool 77, 127–138 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Nelson KE et al. Phylogenetic analysis of the microbial populations in the wild herbivore gastrointestinal tract: insights into an unexplored niche. Environ. Microbiol 5, 1212–1220 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Uenishi G et al. Molecular analyses of the intestinal microbiota of chimpanzees in the wild and in captivity. Am. J. Primatol 69, 367–376 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Wilson KH, Brown RS, Andersen GL, Tsang J & Sartor B Comparison of fecal biota from specific pathogen free and feral mice. Anaerobe 12, 249–253 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Wilson DS Biological communities as functionally organized units. Ecology 78, 2018–2024 (1997). [Google Scholar]
- 33.Foster KR & Wenseleers T A general model for the evolution of mutualisms. J. Evol. Biol 19, 1283–1293 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Sachs JL, Mueller UG, Wilcox TP & Bull JJ The evolution of cooperation. Q. Rev. Biol 79, 135–160 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Flint HJ Polysaccharide breakdown by anaerobic microorganisms inhabiting the mammalian gut. Adv. Appl. Microbiol 56, 89–120 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Flint HJ, Duncan SH, Scott KP & Louis P Interactions and competition within the microbial community of the human colon: links between diet and health. Environ. Microbiol 9, 1101–1111 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Fons M, Gomez A & Karjalainen T Mechanisms of colonisation and colonisation resistance of the digestive tract. Part 2: bacteria/bacteria interactions. Microb. Ecol. Health Dis 12, 240–246 (2000). [Google Scholar]
- 38.Reid G & Bruce AW Probiotics to prevent urinary tract infections: the rationale and evidence. World J. Urol 24, 28–32 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Brook I The role of bacterial interference in otitis, sinusitis and tonsillitis. Otolaryngol. Head Neck Surg 133, 139–146 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Servin AL Antagonistic activities of lactobacilli and bifidobacteria against microbial pathogens. FEMS Microbiol. Rev 28, 405–440 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Tilman D Niche tradeoffs, neutrality, and community structure: a stochastic theory of resource competition, invasion, and community assembly. Proc. Natl Acad. Sci. USA 101, 10854–10861 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pool-Zobel B, Veeriah S & Bohmer FD Modulation of xenobiotic metabolising enzymes by anticarcinogens — focus on glutathione S-transferases and their role as targets of dietary chemoprevention in colorectal carcinogenesis. Mutat. Res 591, 74–92 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Doebeli M, Hauert C & Killingback T The evolutionary origin of cooperators and defectors. Science 306, 859–862 (2004). [DOI] [PubMed] [Google Scholar]
- 44.McFall-Ngai M Adaptive immunity: care for the community. Nature 445, 153 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Macpherson AJ, Geuking MB & McCoy KD Immune responses that adapt the intestinal mucosa to commensal intestinal bacteria. Immunology 115, 153–162 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matzinger P The danger model: a renewed sense of self. Science 296, 301–305 (2002). [DOI] [PubMed] [Google Scholar]
- 47.O’Keefe SJ et al. Why do African Americans get more colon cancer than Native Africans? J. Nutr 137, 175S–182S (2007). [DOI] [PubMed] [Google Scholar]
- 48.Moore WE & Moore LH Intestinal floras of populations that have a high risk of colon cancer. Appl. Environ. Microbiol 61, 3202–3207 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Swenson W, Wilson DS & Elias R Artificial ecosystem selection. Proc. Natl Acad. Sci. USA 97, 9110–9114 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dethlefsen L, Eckburg PB, Bik EM & Relman DA Assembly of the human intestinal microbiota. Trends Ecol. Evol 21, 517–523 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Young VB & Schmidt TM Antibiotic-associated diarrhea accompanied by large-scale alterations in the composition of the fecal microbiota. J. Clin. Microbiol 42, 1203–1206 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li J et al. Identification of early microbial colonizers in human dental biofilm. J. Appl. Microbiol 97, 1311–1318 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Gill SR et al. Metagenomic analysis of the human distal gut microbiome. Science 312, 1355–1359 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klaassens ES, de Vos WM & Vaughan EE Metaproteomics approach to study the functionality of the microbiota in the human infant gastrointestinal tract. Appl. Environ. Microbiol 73, 1388–1392 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lepp PW et al. Methanogenic Archaea and human periodontal disease. Proc. Natl Acad. Sci. USA 101, 6176–6181 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jernberg C, Sullivan A, Edlund C & Jansson JK Monitoring of antibiotic-induced alterations in the human intestinal microflora and detection of probiotic strains by use of terminal restriction fragment length polymorphism. Appl. Environ. Microbiol 71, 501–506 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pepin J et al. Emergence of fluoroquinolones as the predominant risk factor for Clostridium difficile-associated diarrhea: a cohort study during an epidemic in Quebec. Clin. Infect. Dis 41, 1254–1260 (2005). [DOI] [PubMed] [Google Scholar]
- 58.Lofmark S, Jernberg C, Jansson JK & Edlund C Clindamycin-induced enrichment and long-term persistence of resistant Bacteroides spp. and resistance genes. J. Antimicrob. Chemother 58, 1160–1167 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Sjolund M, Tano E, Blaser MJ, Andersson DI & Engstrand L Persistence of resistant Staphylococcus epidermidis after single course of clarithromycin. Emerg. Infect. Dis 11, 1389–1393 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kolenbrander PE et al. Bacterial interactions and successions during plaque development. Periodontol. 2000 42, 47–79 (2006). [DOI] [PubMed] [Google Scholar]
- 61.Savage DC in Mucosal Immunology (eds Mestecky J et al. ) 19–34 (Elsevier, Boston, 2005). [Google Scholar]
- 62.Caufield PW et al. Natural history of Streptococcus sanguinis in the oral cavity of infants: evidence for a discrete window of infectivity. Infect. Immun 68, 4018–4023 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Samuel BS & Gordon JI A humanized gnotobiotic mouse model of host-Archaeal-bacterial mutualism. Proc. Natl Acad. Sci. USA 103, 10011–10016 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kolenbrander PE et al. Communication among oral bacteria. Microbiol. Mol. Biol. Rev 66, 486–505 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu J et al. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science 299, 2074–2076 (2003). [DOI] [PubMed] [Google Scholar]
- 66.Schell MA et al. The genome sequence of Bifidobacterium longum reflects its adaptation to the human gastrointestinal tract. Proc. Natl Acad. Sci. USA 99, 14422–14427 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Czárán TL, Hoekstra RF & Pagie L Chemical warfare between microbes promotes biodiversity. Proc. Natl Acad. Sci. USA 99, 786–790 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gordon DM, Riley MA & Pinou T Temporal changes in the frequency of colicinogeny in Escherichia coli from house mice. Microbiology 144, 2233–2240 (1998). [DOI] [PubMed] [Google Scholar]
- 69.Sperandio V, Torres AG, Jarvis B, Nataro JP & Kaper JB Bacteria-host communication: the language of hormones. Proc. Natl Acad. Sci. USA 100, 8951–8956 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shiner EK, Rumbaugh KP & Williams SC Inter-kingdom signaling: deciphering the language of acyl homoserine lactones. FEMS Microbiol. Rev 29, 935–947 (2005). [DOI] [PubMed] [Google Scholar]
- 71.Rendon MA et al. Commensal and pathogenic Escherichia coli use a common pilus adherence factor for epithelial cell colonization. Proc. Natl Acad. Sci. USA 104, 10637–10642 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wren BW The yersiniae — a model genus to study the rapid evolution of bacterial pathogens. Nature Rev. Microbiol 1, 55–64 (2003). [DOI] [PubMed] [Google Scholar]
- 73.Monot M et al. On the origin of leprosy. Science 308, 1040–1042 (2005). [DOI] [PubMed] [Google Scholar]
- 74.Brown NF, Wickham ME, Coombes BK & Finlay BB Crossing the line: selection and evolution of virulence traits. PLoS Pathog. 2, e42 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Woolhouse ME, Webster JP, Domingo E, Charlesworth B & Levin BR Biological and biomedical implications of the co-evolution of pathogens and their hosts. Nature Genet. 32, 569–577 (2002). [DOI] [PubMed] [Google Scholar]
- 76.Wickham ME, Brown NF, Boyle EC, Coombes BK & Finlay BB Virulence is positively selected by transmission success between mammalian hosts. Curr. Biol 17, 783–788 (2007). [DOI] [PubMed] [Google Scholar]
- 77.Walther BA & Ewald PW Pathogen survival in the external environment and the evolution of virulence. Biol. Rev. Camb. Philos. Soc 79, 849–869 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boots M & Mealor M Local interactions select for lower pathogen infectivity. Science 315, 1284–1286 (2007). [DOI] [PubMed] [Google Scholar]
- 79.Taylor LH, Latham SM & Woolhouse ME Risk factors for human disease emergence. Phil. Trans. R. Soc. Lond. B 356, 983–989 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Naylor SW, Gally DL & Low JC Enterohaemorrhagic E. coli in veterinary medicine. Int. J. Med. Microbiol 295, 419–441 (2005). [DOI] [PubMed] [Google Scholar]
- 81.Read AF & Taylor LH The ecology of genetically diverse infections. Science 292, 1099–1102 (2001). [DOI] [PubMed] [Google Scholar]
- 82.West SA & Buckling A Cooperation, virulence and siderophore production in bacterial parasites. Proc. R. Soc. Lond B 270, 37–44 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gardner A, West SA & Buckling A Bacteriocins, spite and virulence. Proc. R. Soc. Lond B 271, 1529–1535 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wolfe ND, Dunavan CP & Diamond J Origins of major human infectious diseases. Nature 447, 279–283 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Woolhouse ME, Taylor LH & Haydon DT Population biology of multihost pathogens. Science 292, 1109–1112 (2001). [DOI] [PubMed] [Google Scholar]
- 86.Cheesman SE & Guillemin K We know you are in there: conversing with the indigenous gut microbiota. Res. Microbiol 158, 2–9 (2007). [DOI] [PubMed] [Google Scholar]
- 87.Hongoh Y. et al. Intra- and interspecific comparisons of bacterial diversity and community structure support coevolution of gut microbiota and termite host. Appl. Environ. Microbiol 71, 6590–6599 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kikuchi Y & Graf J Spatial and temporal population dynamics of a naturally occurring two-species microbial community inside the digestive tract of the medicinal leech. Appl. Environ. Microbiol 73, 1984–1991 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Broderick NA, Raffa KF & Handelsman J Midgut bacteria required for Bacillus thuringiensis insecticidal activity. Proc. Natl Acad. Sci. USA 103, 15196–15199 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cox CR & Gilmore MS Native microbial colonization of Drosophila melanogaster and its use as a model of Enterococcus faecalis pathogenesis. Infect. Immun 75, 1565–1576 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fraune I & Bosch T Long-term maintenance of species-specific bacterial microbiota in the basal metazoan Hydra. Proc. Natl Acad. Sci. USA 104, 13146–13151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nyholm SV & McFall-Ngai MJ The winnowing: establishing the squid-Vibrio symbiosis. Nature Rev. Microbiol 2, 632–642 (2004). [DOI] [PubMed] [Google Scholar]
- 93.Davidson SK & Stahl DA Transmission of nephridial bacteria of the earthworm Eisenia fetida. Appl. Environ Microbiol 72, 769–775 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Goodrich-Blair H & Clarke DJ Mutualism and pathogenesis in Xenorhabdus and Photorhabdus: two roads to the same destination. Mol. Microbiol 64, 260–268 (2007). [DOI] [PubMed] [Google Scholar]
- 95.DeSantis TZ et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol 72, 5069–5072 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Collins MD et al. The phylogeny of the genus Clostridium: proposal of five new genera and eleven new species combinations. Int. J. Syst. Bacteriol 44, 812–826 (1994). [DOI] [PubMed] [Google Scholar]