

Abstract
In living systems, the chemical space and functional repertoire of proteins are dramatically expanded through the post-translational modification (PTM) of various amino acid residues. These modifications frequently trigger unique protein-protein interactions (PPIs) – for example with reader proteins that directly bind the modified amino acid residue – which leads to downstream functional outcomes. The modification of a protein can also perturb its PPI network indirectly, for example, through altering its conformation or subcellular localization. Uncovering the network of unique PTM-triggered PPIs is essential to fully understand the roles of an ever-expanding list of PTMs in our biology. In this review, we discuss established strategies and current challenges associated with this endeavor.
Keywords: post-translational modification, protein-protein interaction, genetic code expansion, expressed protein ligation, peptide probe, photo-affinity probe, Mass-spectrometry proteomics
Introduction
Although proteins are generally composed of only twenty canonical amino acids, the chemical space available to them is dramatically expanded in living systems by post-translational modifications (PTMs) of various amino acid side residues.[1] Such PTMs provide a powerful way to augment and regulate protein function.[1–4] The modified amino acid residue is often recognized by specific “reader” and “eraser” proteins (Figure 1), and these unique protein-protein interactions (PPIs) govern the complex regulation of the functional outcome associated with the PTM.[2,5] In addition, PTMs may also trigger unique PPIs indirectly; for example, through inducing dynamic structural changes or altered subcellular localization. Consequently, orchestrating new functionally important protein-protein interactions is a central mechanism underpinning the regulatory role played by many PTMs in our biology.
Figure 1.
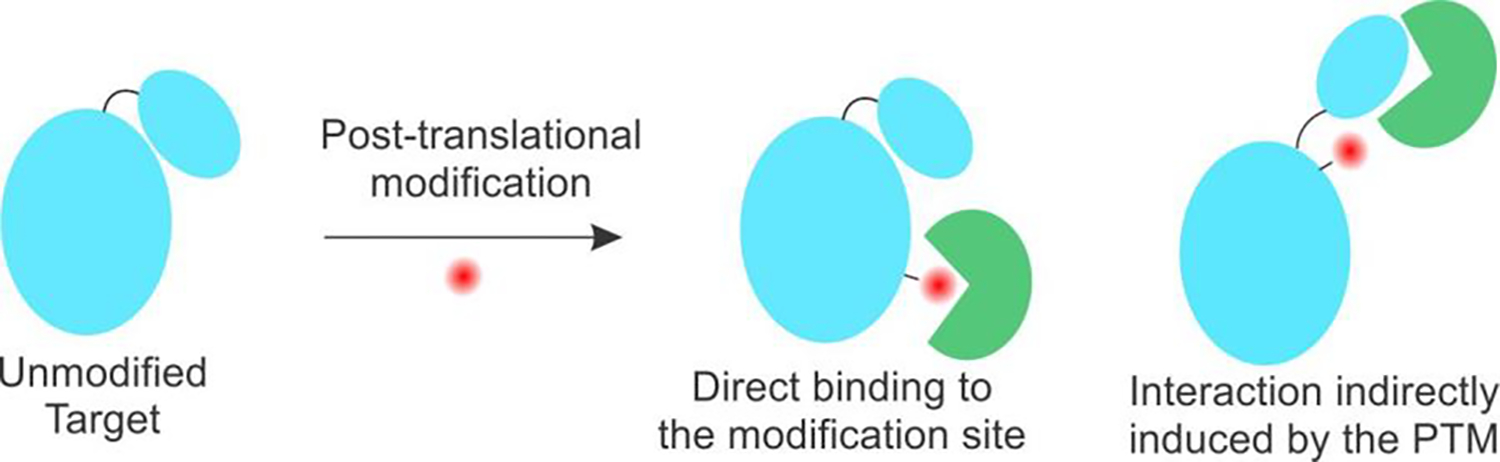
PTMs can lead to novel PPIs either directly or indirectly
Over the last two decades, rapid advances in proteomics have dramatically expanded the catalog of known PTMs and the sites in the proteome that are subjected to them,[6,7] highlighting a significantly larger role for these modifications in our biology. To fully understand their function, it is essential to systematically identify the unique set of PPIs that are triggered in response to a particular PTM. Doing so would depend on two key abilities: 1) to model the presence/absence of the PTM on a target protein, and 2) to reliably characterize the PPIs associated with each state. However, these were historically challenging to achieve for multiple reasons.[8] The biochemical origins of many PTMs are poorly understood. Even when the mechanism is known, reconstituting them cleanly to generate homogeneously PTM-labeled proteins is frequently not possible. This challenge makes it difficult to investigate the consequences of the PTM, including the identification of PTM-associated interaction partners. Furthermore, the strengths of different PTM-dependent PPIs span a wide range,[9] and many are insufficiently strong to allow the isolation of non-covalently bound interaction partners from the complex cellular milieu via pull-down or immunoprecipitation.[10,11] Here we discuss contemporary strategies that are being developed to overcome these limitations. We first focus on approaches to reliably model the presence or absence of a PTM on proteins, followed by strategies to identify PTM-specific interaction partners.
Strategies to model the presence or absence of PTMs on proteins
Some proteins are post-translationally modified at significant levels in cells under known conditions, such as upon the activation of a signal transduction pathway. In such cases, the naturally modified protein can be used as a probe to identify both general and PTM-specific interaction partners. To uniquely identify the latter group, a control protein-probe without a PTM can be used, which can be generated either by mutating the site of modification, or by knocking out/down the ‘writer’ protein that installs the PTM. This strategy was used to reveal that BARD1 binds p50 when the latter is phosphorylated at Ser337,[12] and to show that cereblon binds and degrades glutamine synthetase when the latter is acetylated at lysine 11 and 14.[13] However, this strategy is limited to modifications that occur naturally at significant levels.
The use of short peptides harboring the PTM in its native context is one of the most popular approaches for investigating PTM-triggered PPIs. Such modified peptides with defined sequences can be readily accessed through chemical synthesis. Proteins that bind post-translationally modified residues often retain significant affinity and selectivity for such minimal recognition motifs. Such peptides probes have been used to systematically screen for binding by suspected protein domains in many studies, such as for identifying YEATS and double PHD finger domains as readers of crotonyllysine,[14,15] and GAS41 as a reader for succinyllysine.[16] Conversely, modified peptide probes have also been useful for identifying the target sequence for a particular PTM-binding domain.[17,18] Peptide probes have also been valuable for evaluating crosstalk between two different PTMs within the same motif.[19,20] Moreover, modified peptide-probes have also been employed in a high-throughput manner using, for example, peptide microarrays,[21–23] or in combination with quantitative mass-spectrometry following a pull-down.[24] There are countless other examples where peptide probes have been useful to characterize PTM-dependent PPIs.[25] Although peptide based probes are popular due to their ease of access, these do not capture the context of the full protein, which may partially or completely compromise their ability to bind native interaction partners.[26,27] Furthermore, peptide probes are not suitable to investigate PTM-triggered PPIs that do not directly bind the modified residue. These limitations have inspired efforts at generating full-length proteins homogeneously labeled with PTMs at chosen sites.[8]
Using a natural amino acid that ‘mimics’ the chemical properties of the modified residue is another popular strategy to model PTMs. For example, negatively charged aspartic acid and glutamic acid residues are often used to mimic the effect of phosphorylation, charge-neutral glutamine has been used to model lysine acetylation, and positively charged arginine is used to mimic an unacylated lysine residue.[12,28–32] However, such substitutions may not always accurately reproduce the consequences triggered by the actual PTM because of significant structural differences. Furthermore, this approach cannot be used for the vast majority of PTMs that are not structurally similar to one of the canonical amino acids.
A PTM can also be introduced onto proteins using the natural ‘writer’ enzyme(s) responsible for its installation, provided the pathway is established and can be reconstituted.[33,34] Engineered variants of such ‘writer’ proteins, many of which modify numerous substrate proteins, can be developed to target specific proteins. For example, it was possible to achieve protein-selective O-GlcNAcylation by fusing O-GlcNAc transferase (OGT) to a nanobody that selectively binds a target protein.[35] However, biochemical origin of many PTMs are either unknown or hard to reconstitute to modify only the desired site(s), compromising the general utility of this strategy.
Protein semisynthesis using expressed protein ligation (EPL) enables the assembly of full-length proteins from synthetic peptides and recombinantly expressed protein fragments.[36,37] Using this approach, it has been possible to access proteins site-specifically labeled with many different PTMs. For example, this strategy has been used to generate site-specifically succinylated,[38] glutarylated,[39] and ubiquitilated[40] histones to evaluate how these PTMs affect their interaction with other chromatin components. In another example, protein semisynthesis was used to generate Ubc9, an E2-ligase, that is site-specifically modified with SUMO, and further equipped with a photo-affinity probe and a biotin group. This trifunctional protein probe was used to covalently capture the E3 ligase RanBP2 that associates with this complex.[41] Although protein semisynthesis has been widely used to study PTM biology, there are several challenges limiting its scope. This strategy is often restricted to proteins that are smaller in size, and those that can survive the demanding workflow. For example, EPL is typically performed under denaturing conditions, and proteins that are not easily refolded may not be amenable to this technology.[37] Finally, application of this technology is restricted largely to in vitro experiments, precluding the investigation of delicate PPIs that require the context of a living cell.
Genetic code expansion (GCE) technology has emerged as a powerful strategy for generating full-length proteins homogeneously incorporating PTMs at predefined sites.[42,43] This technology uses engineered nonsense-suppressing aminoacyl-tRNA synthetase (aaRS)/tRNA pairs to enable co-translational site-specific incorporation of non-canonical amino acids (ncAAs) into proteins in living cells. Engineered aaRS/tRNA pairs have been developed to genetically encode numerous PTMs of interest, including several lysine PTMs,[44–46] as well as PTMs of tyrosine,[47–51] serine,[52,53] threonine,[54] and arginine.[55] It has been also possible to genetically encode ncAAs that structurally mimic the PTM, but are resistant to removal by endogenous machinery.[49,53,56] A significant advantage of the GCE technology is the ability produce homogeneously modified proteins directly in living cells, which may enable capturing delicate PPIs that require the cellular context. However, only a subset of known PTMs can be currently incorporated using this technology. PTMs with large and complex structures remains particularly challenging to encode, as it has been difficult to engineer existing aaRSs to recognize such ncAAs. The unpredictable ncAA incorporation efficiency is another challenge commonly encountered with the GCE approach.
GCE and other site-specific protein modification approaches have also been used to introduce unique functionalities into proteins that can be subsequently leveraged to chemically/enzymatically install a PTM mimic. For example, taking advantage of its unique reactivity and relatively low abundance, engineered cysteine residues have been chemoselectively functionalized to incorporate mimics of lysine acetylation and methylation.[57,58] Chemoselective modification of dehydroalanine, which can be generated from cysteine or selenium-containing ncAAs, has also been used to introduce mimics of various PTMs onto proteins.[59] Moreover, similar post-translational selective functionalization approaches have facilitated the introduction of more complex modifications such as glycosylation, [60,61], SUMOylation, [62] and uniquitination.[63]
Approaches to identify PTM-associated PPIs
Potential interactors for a particular PTM can sometimes be predicted based on existing biochemical, structural or genetic information, or through homology analysis with established PPIs targeting structurally similar PTMs. In such cases, the suspected proteins can be screened to confirm binding using peptide or protein probes (Figure 2A). For example, reader domains for novel lysine PTMs crotonylation and succinylation were confirmed by screening predicted protein domains for binding PTM-labeled peptide probes in vitro.[14–16] The limitation of this approach is the requirement of preexisting insight.
Figure 2.
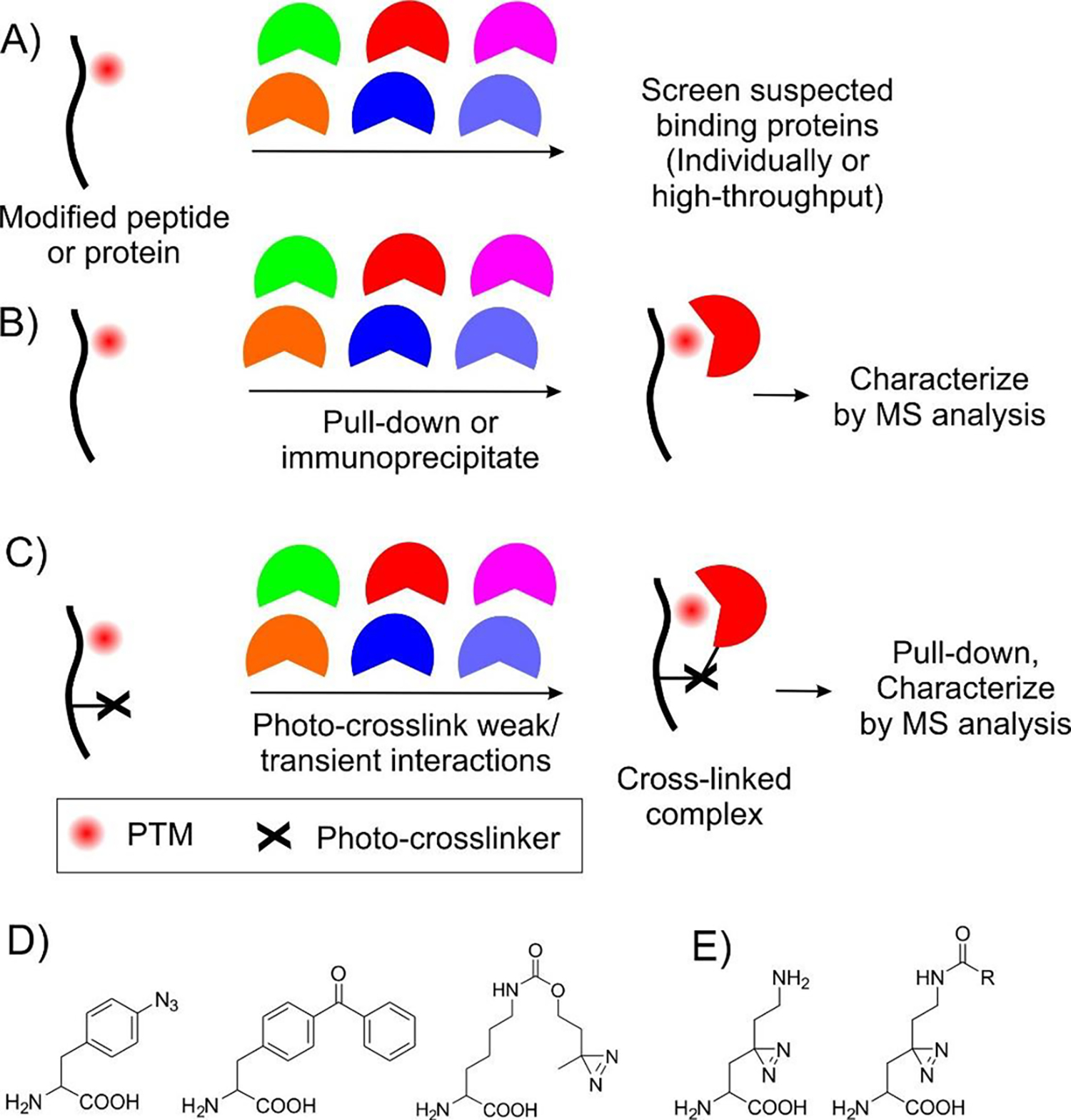
A-C show different approaches to identify novel PTM-associated PPIs. D) Structures of ncAAs with photo-crosslinkers frequently used to capture PPIs. E) Novel lysine-derived ncAAs, harboring a diazirine group, which can be incorporated into proteins and used to capture interactors that bind this residue
Performing binding screens at a much higher throughput can enable identification of binding partners in the absence of a preexisting insight. Using synthetic peptide microarrays, higher throughput screening can be performed in vitro.[21,23] The ability to genetically encode various PTMs has opened the door to adapting established high-throughput cell-based PPI assays [64] for identifying PTM-dependent PPIs. For example, it has been possible to express a library of thousands of predicted phosphoserine-containing peptides from the human proteome in E. coli, using co-translational incorporation of phosphoserine, and screen these for binding potential interactors using a high-throughput fluorescence-based two-hybrid screen.[65] Other examples include the identification of Nop56p and Nop58p as arginine-methylation-dependent interaction partner of Nop1p through two-hybrid assay,[19] and of a PPI between RhPIP2 and RhPTM that is regulated by phosphorylation through a split-ubiquitin membrane yeast two-hybrid (MYTH) assay.[66]
Using the modified peptide or protein as a bait to pull-down or immunoprecipitate non-covalently bound interaction partners from the complex cellular milieu, followed by their identification using immunostaining or mass-spectrometry, is a popular strategy to identify unknown PPIs (Figure 2B). From such experiments, PTM-specific interactors can be identified by their differential enrichment upon using protein/peptide probes either with or without the PTM. For example, this strategy was used to identify readers of trimethyl-lysine,[24] to confirm that phosphorylation of PKM2 by ERK2 at Ser 37 recruits PIN1,[67] that phosphorylation of OPTN by TBK1 enhances its binding to ubiquitin chains,[32] that Wnt5a activation of ROR1 induces binding to 14-3-3ζ,[68] and that lysine-acetylated glutamine synthetase is a substrate for cereblon.[13] A key limitation of this approach is that many PTM-specific PPIs are not strong enough to survive the isolation step.
An attractive way to overcome this limitation is the use of photo-affinity probes, which can enable the capture of proximal non-covalent interactors through the formation of a stable covalent linkage (Figure 2C). Established photo-affinity probes such as aromatic azides, benzophenone, and diazirines (Figure 2D) can be readily incorporated into chemically synthesized peptides. Peptide probes harboring the photo-crosslinking functionality, in the presence or absence of the PTM, have been used in conjunction with quantitative MS-proteomics to identify PTM-specific interaction partners.[69–75] Fundamental limitations of peptide probes, as described earlier, restricts the scope of this approach.
The GCE technology has enabled site-specific incorporation of ncAAs harboring photo-affinity probes into full-length proteins (Figure 2D), which has been adapted for interrogating PTM-associated PPIs. For example, a lysine analog containing the small diazirine photo-crosslinker group on the sidechain was incorporated across the proteome (Figure 2E).[76] Two variants of the same lysine-analog were also incorporated site-specifically, carrying either a photocage or the crotonyl modification on Nε (Figure 2E).[77] The small built-in diazirine group in both cases enabled capture of lysine-PTM-specific PPIs. Application of this approach is currently restricted to lysine PTMs. and it is conceivable that the built-in diazirine group may perturb the native binding interactions at the proximal Nε in some cases. Incorporation of two distinct ncAAs – one encoding the PTM of interest and the other harboring the photo-crosslinking probe – has been recently reported.[78] This strategy allows flexible placement of the photo-crosslinker relative to the PTM in a full-length protein to systematically optimize capture efficiency, and it can be extended to other genetically encoded PTMs. Furthermore, this technology is uniquely suited to capture PPIs indirectly triggered by PTMs. However, application of this technology can be somewhat limited by the low efficiency of incorporating two distinct ncAAs. For protein-based PTMs such as SUMO, the photo-crosslinker ncAA has been directly incorporated into it to identify interacting proteins.[79] Conversely, genetically encoded photo-crosslinkers have also been incorporated into the binding site of known PTM-reader proteins to capture previously unknown targets.[80,81] It should be noted that photo-crosslinking groups typically have a short capture radius, making their crosslinking efficiency highly sensitive to the site of incorporation. Screening multiple sites are sometimes necessary to achieve efficient capture of unknown targets.[78] Finally, in addition to photoaffinity probes, genetically encoded chemical crosslinkers have also been used to capture interaction partners. For example, an electrophilic tyrosine derivative was used to covalently capture phosphatases that erase phosphorylation of distinct tyrosine residues of the HER2 receptor.[82]
Conclusion
Even though significant progress has been made in the last few decades to define the dynamic PPI networks that are regulated by PTMs, it is evident that our current understanding is limited. This is especially true for the rapidly expanding catalog of newly discovered PTMs. It is challenging to develop a single approach to address this issue that would be suitable for all PTMs, given the remarkable diversity of their structure and properties. As this review highlights, many different creative approaches have been developed to explore PPIs associated with different PTMs. In addition, computational approaches are also being explored to predict PTM-associated PPIs.[83–85] Expanding the current scope of these diverse approaches would be critical to broaden our understanding of the vast network of PTM-associated PPIs that underlie all aspects of our biology. In particular, it will be important to install previously inaccessible PTMs, or their structural analogs, into full-length substrate proteins, both in vitro and in living cells. A combination of engineered PTM-writers, the ability to enhance the endogenous PTM levels, an expanded toolbox of GCE platforms, and cellular delivery of modified proteins synthesized in vitro would be beneficial to this end. It will be also important to expand our capability to capture PPIs associated with the modified form of the protein through improved covalent capture methods, as well as advanced proteomic technologies for characterizing proteome-wide protein-protein interactions.
Table 1.
Strategies to model the presence and absence of PTMs on proteins
| Strategy | Advantages | Limitations | Representative references |
|---|---|---|---|
| Proteins naturally modified in cells under known conditions | • Straightforward to access with few manipulations • The absence of PTM can be readily modeled by mutating the target site or knocking out/down the ‘writer’ protein |
• Conditions leading to many PTMs are not known • Degree of endogenous modification is often heterogeneous • Site-specific modification may be difficult to achieve |
[12,13] |
| Use of small synthetic peptides harboring the PTM in its native context | • Readily generated through chemical synthesis • Wide variety of modifications can be accessed • Possible to model multiple PTMs within the same peptide • Can be equipped with photo-crosslinkers • Can be used in higher throughput format such as peptide microarray |
• Does not capture the context of the full-length folded protein, which may partially or completely abolish interaction with partners • Cannot be used to capture PTM-triggered PPIs that do not directly involve the modified residue |
[14–25] |
| Use of natural amino acids that mimic a PTM | • Easy to implement through straightforward mutagenesis • Introduces modification at specific site(s) |
• Significant structural differences often result in imperfect mimicry • Most PTMs cannot be modeled using a natural amino acid |
[12,28–32] |
| Using the endogenous ‘writer’ protein(s) to install a PTM | • Can be relatively simple to generate using recombinant writers • Engineered writers can be generated to target specific substrate proteins |
• Biochemical origin of many PTMs are unknown or hard to reconstitute • Can be difficult to homogeneously modify specific site(s) • Some PTMs are not enzymatically installed |
[33–35] |
| Protein semi-synthesis through expressed protein ligation (EPL) | • Homogeneously modified full-length proteins can be generated • A wide variety of natural/synthetic modifications can be installed • Multiple, different modifications can be installed |
• Technically demanding • EPL is performed under denaturing conditions; refolding the resulting protein may be challenging • Typically precludes experiments in living cells • Internal modifications on larger proteins are challenging to access |
[36–41] |
| Genetic code expansion (GCE) | Homogeneously modified full-length proteins can be generated Close structural mimics of PTMs can be incorporated that are resistant to removal Modified proteins can be expressed in living cells The modified residue can be theoretically incorporated into any site of any protein that can be recombinantly expressed |
A limited number of PTMs have been genetically encoded Efficiency of incorporation is site-dependent Incorporation of multiple modifications into one proteins can be challenging |
[42,43,45–50,52–56] |
Table 2.
Approaches to identify PTM-associated PPIs
| Strategy | Advantages | Limitations | Representative references |
|---|---|---|---|
| Screening potential interaction partners for binding modified peptide/protein probe | • Straightforward workflow • Can be employed at a higher throughput using strategies like peptide microarray, or two-hybrid analyses |
• Requires existing insight into possible potential interactors | [14–16,21–23,64] |
| Immunoprecipitation or pull-down of non-covalently bound interactors using modified peptide or protein probe | • Established workflow; widely used • Can be used in complex milieu, such as cell-free extract, to isolate native complexes • Can be coupled with quantitative MS experiments to characterize binding partners with high confidence • Enables the identification of unanticipated interactors |
• Interactions that are weak or transient are challenging to detect, as these do not survive the isolation step | [13,24,32,67,68] |
| Small peptide probes harboring the PTM and a photo-crosslinker group | Almost any short sequence, with a wide variety of modifications, can be readily generated through synthesisEnables the identification of weaker binding interactionsCan be used in complex milieu such as cell-free extract to isolate interaction partnersCan be coupled with quantitative MS experiments to characterize binding partners with high confidence• Enables the identification of unknown interactors | • The photo-affinity probe may perturb the binding interaction • Photo-affinity probes have a short capture radius, making the cross-linking efficiency site-dependent. • Limitations intrinsic to small peptide probes still apply |
[69–75] |
| Full-length proteins harboring genetically encoded photo-affinity probes: The same ncAA encodes both the PTM and photo-crosslinker | • Homogeneously modified full-length proteins can be generated in living cells • The proximity of the photo-crosslinker group to proteins directly binding the PTM is favorable for crosslink formation • Incorporation of a single ncAA introduces both the PTM and the photo-affinity probe |
• Currently restricted to lysine PTMs • The photo-crosslinker group may perturb interaction with binding partners • Cannot be used to capture PTM-triggered PPIs that do not directly involve the modified residue |
[76,77] |
| Full-length proteins harboring genetically encoded photo-affinity probes: Two different ncAAs encode the PTM and the photo-crosslinker group, respectively | • Homogeneously modified full-length proteins can be generated in living cells • Flexibility to optimally position the photo-crosslinker relative to the PTM for high capture efficiency • Enables the capture of PPIs indirectly triggered by the PTM |
• Efficiency of incorporating two distinct ncAAs into one protein can be low • Can only be used for PTMs that have been already genetically encoded |
[78] |
Acknowledgement
AC acknowledges support from NIGMS (R35GM136437)
Footnotes
Declaration of Interest: Authors declare no conflict of interest
References [*of special interest; **of outstanding interest]
- 1.Walsh CT, Garneau-Tsodikova S, Gatto GJ Jr: Protein posttranslational modifications: the chemistry of proteome diversifications. Angewandte Chemie International Edition 2005, 44:7342–7372. [DOI] [PubMed] [Google Scholar]
- 2.Seet BT, Dikic I, Zhou M-M, Pawson T: Reading protein modifications with interaction domains. Nature reviews Molecular cell biology 2006, 7:473–483. [DOI] [PubMed] [Google Scholar]
- 3.Sims RJ, Reinberg D: Is there a code embedded in proteins that is based on post-translational modifications? Nature reviews Molecular cell biology 2008, 9:815–820. [DOI] [PubMed] [Google Scholar]
- 4.Deribe YL, Pawson T, Dikic I: Post-translational modifications in signal integration. Nature structural & molecular biology 2010, 17:666–672. [DOI] [PubMed] [Google Scholar]
- 5.Patel DJ, Wang Z: Readout of epigenetic modifications. Annual review of biochemistry 2013, 82:81–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olsen JV, Mann M: Status of large-scale analysis of post-translational modifications by mass spectrometry. Molecular & cellular proteomics 2013, 12:3444–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani D, Burgess MW, Gillette MA, Jaffe JD, Carr SA: Integrated proteomic analysis of post-translational modifications by serial enrichment. Nature methods 2013, 10:634–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Conibear AC: Deciphering protein post-translational modifications using chemical biology tools. Nature Reviews Chemistry 2020, 4:674–695. * Review article describing contemporary chemical biology strategies used to study protein post-translational modifications
- 9.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert J-P, Barsyte-Lovejoy D, Felletar I, Volkmer R, Müller S, Pawson T, et al. : Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149:214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perkins JR, Diboun I, Dessailly BH, Lees JG, Orengo C: Transient protein-protein interactions: structural, functional, and network properties. Structure 2010, 18:1233–1243. [DOI] [PubMed] [Google Scholar]
- 11.Acuner Ozbabacan SE, Engin HB, Gursoy A, Keskin O: Transient protein–protein interactions. Protein engineering, design and selection 2011, 24:635–648. [DOI] [PubMed] [Google Scholar]
- 12.Wu L, Crawley CD, Garofalo A, Nichols JW, Campbell P-A, Khramtsova GF, Olopade OI, Weichselbaum RR, Yamini B: p50 mono-ubiquitination and interaction with BARD1 regulates cell cycle progression and maintains genome stability. Nature communications 2020, 11:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Nguyen T, Lee JE, Sweredoski MJ, Yang S-J, Jeon S-J, Harrison JS, Yim J-H, Lee SG, Handa H, Kuhlman B, et al. : Glutamine triggers acetylation-dependent degradation of glutamine synthetase via the thalidomide receptor cereblon. Molecular cell 2016, 61:809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiong X, Panchenko T, Yang S, Zhao S, Yan P, Zhang W, Xie W, Li Y, Zhao Y, Allis CD, et al. : Selective recognition of histone crotonylation by double PHD fingers of MOZ and DPF2. Nature chemical biology 2016, 12:1111–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Sabari BR, Panchenko T, Wen H, Zhao D, Guan H, Wan L, Huang H, Tang Z, Zhao Y, et al. : Molecular coupling of histone crotonylation and active transcription by AF9 YEATS domain. Molecular cell 2016, 62:181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang Y, Jin J, Chung MWH, Feng L, Sun H, Hao Q: Identification of the YEATS domain of GAS41 as a pH-dependent reader of histone succinylation. Proceedings of the National Academy of Sciences 2018, 115:2365–2370. * Biochemical and structural insights led to the identification of GAS41 as a potential reader for succinyl-lysine. This hypothesis was confirmed using in vitro binding, pull-down, and structural analyses
- 17.Munier CC, De Maria L, Edman K, Gunnarsson A, Longo M, MacKintosh C, Patel S, Snijder A, Wissler L, Brunsveld L, et al. : Glucocorticoid receptor Thr524 phosphorylation by MINK1 induces interactions with 14-3-3 protein regulators. Journal of Biological Chemistry 2021, 296:100551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharma P, Lioutas A, Fernandez-Fuentes N, Quilez J, Carbonell-Caballero J, Wright RH, Di Vona C, Le Dily F, Schüller R, Eick D, et al. : Arginine citrullination at the C-terminal domain controls RNA polymerase II transcription. Molecular cell 2019, 73:84–96. e87. [DOI] [PubMed] [Google Scholar]
- 19.Smith D-L, Erce MA, Lai Y-W, Tomasetig F, Hart-Smith G, Hamey JJ, Wilkins MR: Crosstalk of phosphorylation and arginine methylation in disordered SRGG repeats of Saccharomyces cerevisiae fibrillarin and its association with nucleolar localization. Journal of molecular biology 2020, 432:448–466. [DOI] [PubMed] [Google Scholar]
- 20.Chen P, Zhuo Y, Tian S, Zhang T, Zhai G, Fan E, Ma Z, Zhang Y, Zhang K: An Integrated Approach for Combinatorial Readout of Dual Histone Modifications by Epigenetic Tandem Domains. Analytical chemistry 2020, 92:6218–6223. [DOI] [PubMed] [Google Scholar]
- 21.Hernandez DP, Dittmar G: Peptide array–based interactomics. Analytical and Bioanalytical Chemistry 2021, 413:5561–5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramberger E, Suarez-Artiles L, Perez-Hernandez D, Haji M, Popp O, Reimer U, Leutz A, Dittmar G, Mertins P: A Universal Peptide Matrix Interactomics Approach to Disclose Motif-Dependent Protein Binding. Molecular & cellular proteomics 2021, 20:100135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shanle EK, Shinsky SA, Bridgers JB, Bae N, Sagum C, Krajewski K, Rothbart SB, Bedford MT, Strahl BD: Histone peptide microarray screen of chromo and Tudor domains defines new histone lysine methylation interactions. Epigenetics & chromatin 2017, 10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, et al. : Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 2010, 142:967–980. [DOI] [PubMed] [Google Scholar]
- 25.Lin J, Li XD: Peptide-based approaches to identify and characterize proteins that recognize histone post-translational modifications. Chinese Chemical Letters 2018, 29:1051–1057. [Google Scholar]
- 26.Hsu WW, Wu B, Liu WR: Sirtuins 1 and 2 are universal histone deacetylases. ACS chemical biology 2016, 11:792–799. [DOI] [PubMed] [Google Scholar]
- 27.Tsai W-W, Wang Z, Yiu TT, Akdemir KC, Xia W, Winter S, Tsai C-Y, Shi X, Schwarzer D, Plunkett W, et al. : TRIM24 links a non-canonical histone signature to breast cancer. Nature 2010, 468:927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W, Feng Y, Guo Q, Guo W, Xu H, Li X, Yi F, Guan Y, Geng N, Wang P: SIRT1 modulates cell cycle progression by regulating CHK2 acetylation− phosphorylation. Cell Death & Differentiation 2020, 27:482–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui W, Shen X, Agbas E, Tompkins B, Cameron-Carter H, Staudinger JL: Phosphorylation Modulates the Coregulatory Protein Exchange of the Nuclear Receptor Pregnane X Receptor. Journal of Pharmacology and Experimental Therapeutics 2020, 373:370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wiese M, Bannister AJ, Basu S, Boucher W, Wohlfahrt K, Christophorou MA, Nielsen ML, Klenerman D, Laue ED, Kouzarides T: Citrullination of HP1γ chromodomain affects association with chromatin. Epigenetics & chromatin 2019, 12:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Erber L, Luo A, Chen Y: Targeted and Interactome Proteomics Revealed the Role of PHD2 in Regulating BRD4 Proline Hydroxylation*[S]. Molecular & Cellular Proteomics 2019, 18:1772–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, et al. : Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proceedings of the National Academy of Sciences 2016, 113:4039–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gorelik A, Bartual SG, Borodkin VS, Varghese J, Ferenbach AT, van Aalten DM: Genetic recoding to dissect the roles of site-specific protein O-GlcNAcylation. Nature structural & molecular biology 2019, 26:1071–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vinkemeier U, Cohen S, Moarefi I, Chait B, Kuriyan J, Darnell J Jr: DNA binding of in vitro activated Stat1 alpha, Stat1 beta and truncated Stat1: interaction between NH2-terminal domains stabilizes binding of two dimers to tandem DNA sites. The EMBO journal 1996, 15:5616–5626. [PMC free article] [PubMed] [Google Scholar]
- 35.Ramirez DH, Aonbangkhen C, Wu H-Y, Naftaly JA, Tang S, O’Meara TR, Woo CM: Engineering a proximity-directed O-GlcNAc transferase for selective protein O-GlcNAcylation in cells. ACS chemical biology 2020, 15:1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thompson RE, Muir TW: Chemoenzymatic semisynthesis of proteins. Chemical reviews 2019, 120:3051–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agouridas V, El Mahdi O, Diemer V, Cargoët M, Monbaliu J-CM, Melnyk O: Native chemical ligation and extended methods: mechanisms, catalysis, scope, and limitations. Chemical reviews 2019, 119:7328–7443. [DOI] [PubMed] [Google Scholar]
- 38.Jing Y, Ding D, Tian G, Kwan KCJ, Liu Z, Ishibashi T, Li XD: Semisynthesis of site-specifically succinylated histone reveals that succinylation regulates nucleosome unwrapping rate and DNA accessibility. Nucleic acids research 2020, 48:9538–9549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bao X, Liu Z, Zhang W, Gladysz K, Fung YME, Tian G, Xiong Y, Wong JWH, Yuen KWY, Li XD: Glutarylation of histone H4 Lysine 91 regulates chromatin dynamics. Molecular cell 2019, 76:660–675. e669. * Protein semi-synthesis was used to generate site-specifically glutarylated histones. Using this approach it was shown that glutarylation at histone H4K91 destabilizes nucleosome in vitro.
- 40. Debelouchina GT, Gerecht K, Muir TW: Ubiquitin utilizes an acidic surface patch to alter chromatin structure. Nature chemical biology 2017, 13:105–110. ** Homogeneously ubiquitylated histone H2B was generated using protein semi-synthesis. This was used to show that an acidic patch on ubiquitin binds to nucleosomes, preventing their tight packing and resulting in decompaction
- 41.Zhang Y, Hirota T, Kuwata K, Oishi S, Gramani SG, Bode JW: Chemical synthesis of atomically tailored SUMO E2 conjugating enzymes for the formation of covalently linked SUMO–E2–E3 ligase ternary complexes. Journal of the American Chemical Society 2019, 141:14742–14751. [DOI] [PubMed] [Google Scholar]
- 42.Chin JW: Expanding and reprogramming the genetic code. Nature 2017, 550:53. [DOI] [PubMed] [Google Scholar]
- 43.Young DD, Schultz PG: Playing with the molecules of life. ACS chemical biology 2018, 13:854–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim CH, Kang M, Kim HJ, Chatterjee A, Schultz PG: Site-specific incorporation of epsilon-N-crotonyllysine into histones. Angew Chem Int Ed Engl 2012, 51:7246–7249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neumann H, Peak-Chew SY, Chin JW: Genetically encoding N(epsilon)-acetyllysine in recombinant proteins. Nat Chem Biol 2008, 4:232–234. [DOI] [PubMed] [Google Scholar]
- 46.Xiao H, Xuan W, Shao S, Liu T, Schultz PG: Genetic incorporation of ε-N-2-hydroxyisobutyryl-lysine into recombinant histones. ACS chemical biology 2015, 10:1599–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Italia JS, Peeler JC, Hillenbrand CM, Latour C, Weerapana E, Chatterjee A: Genetically encoded protein sulfation in mammalian cells. Nature chemical biology 2020, 16:379–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neumann H, Hazen JL, Weinstein J, Mehl RA, Chin JW: Genetically encoding protein oxidative damage. J Am Chem Soc 2008, 130:4028–4033. [DOI] [PubMed] [Google Scholar]
- 49.Luo X, Fu G, Wang RE, Zhu X, Zambaldo C, Liu R, Liu T, Lyu X, Du J, Xuan W, et al. : Genetically encoding phosphotyrosine and its nonhydrolyzable analog in bacteria. Nat Chem Biol 2017, 13:845–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu CC, Schultz PG: Recombinant expression of selectively sulfated proteins in Escherichia coli. Nature biotechnology 2006, 24:1436–1440. [DOI] [PubMed] [Google Scholar]
- 51.Hoppmann C, Wong A, Yang B, Li S, Hunter T, Shokat KM, Wang L: Site-specific incorporation of phosphotyrosine using an expanded genetic code. Nat Chem Biol 2017, 13:842–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park HS, Hohn MJ, Umehara T, Guo LT, Osborne EM, Benner J, Noren CJ, Rinehart J, Soll D: Expanding the genetic code of Escherichia coli with phosphoserine. Science 2011, 333:1151–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rogerson DT, Sachdeva A, Wang K, Haq T, Kazlauskaite A, Hancock SM, Huguenin-Dezot N, Muqit MM, Fry AM, Bayliss R, et al. : Efficient genetic encoding of phosphoserine and its nonhydrolyzable analog. Nat Chem Biol 2015, 11:496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang MS, Brunner SF, Huguenin-Dezot N, Liang AD, Schmied WH, Rogerson DT, Chin JW: Biosynthesis and genetic encoding of phosphothreonine through parallel selection and deep sequencing. Nat Methods 2017, 14:729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mondal S, Wang S, Zheng Y, Sen S, Chatterjee A, Thompson PR: Site-specific incorporation of citrulline into proteins in mammalian cells. Nature Communications 2021, 12:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xie J, Supekova L, Schultz PG: A genetically encoded metabolically stable analogue of phosphotyrosine in Escherichia coli. ACS Chem Biol 2007, 2:474–478. [DOI] [PubMed] [Google Scholar]
- 57.Huang R, Holbert MA, Tarrant MK, Curtet S, Colquhoun DR, Dancy BM, Dancy BC, Hwang Y, Tang Y, Meeth K, et al. : Site-specific introduction of an acetyl-lysine mimic into peptides and proteins by cysteine alkylation. J Am Chem Soc 2010, 132:9986–9987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Simon MD, Chu F, Racki LR, de la Cruz CC, Burlingame AL, Panning B, Narlikar GJ, Shokat KM: The site-specific installation of methyl-lysine analogs into recombinant histones. Cell 2007, 128:1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dadová J, Galan SR, Davis BG: Synthesis of modified proteins via functionalization of dehydroalanine. Curr Opin Chem Biol 2018, 46:71–81. [DOI] [PubMed] [Google Scholar]
- 60.Chalker JM, Bernardes GJ, Davis BG: A “tag-and-modify” approach to site-selective protein modification. Accounts of chemical research 2011, 44:730–741. [DOI] [PubMed] [Google Scholar]
- 61.Hudak JE, Yu HH, Bertozzi CR: Protein glycoengineering enabled by the versatile synthesis of aminooxy glycans and the genetically encoded aldehyde tag. Journal of the American Chemical Society 2011, 133:16127–16135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fottner M, Brunner A-D, Bittl V, Horn-Ghetko D, Jussupow A, Kaila VR, Bremm A, Lang K: Site-specific ubiquitylation and SUMOylation using genetic-code expansion and sortase. Nature chemical biology 2019, 15:276–284. * A unique noncanonical amino acid was genetically encoded, which could be recognized by an engineered sortase to site-specifically install SUMOylation onto proteins
- 63.Fottner M, Weyh M, Gaussmann S, Schwarz D, Sattler M, Lang K: A modular toolbox to generate complex polymeric ubiquitin architectures using orthogonal sortase enzymes. Nat Commun 2021, 12:6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yakubu RR, Nieves E, Weiss LM: The methods employed in mass spectrometric analysis of posttranslational modifications (PTMs) and protein–protein interactions (PPIs). Advancements of mass spectrometry in biomedical research 2019, 1140:169–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Barber KW, Muir P, Szeligowski RV, Rogulina S, Gerstein M, Sampson JR, Isaacs FJ, Rinehart J: Encoding human serine phosphopeptides in bacteria for proteome-wide identification of phosphorylation-dependent interactions. Nature biotechnology 2018, 36:638–644. ** Genetic code expansion was used to generate a library of thousands of human-proteome-derived phosphoserine-containing peptides in E. coli. A FACS-based two-hybrid screen was also developed to screen various proteins for their ability bind specific phosphopeptides in this library in a high-throughput manner
- 66.Zhang S, Feng M, Chen W, Zhou X, Lu J, Wang Y, Li Y, Jiang C-Z, Gan S-S, Ma N: In rose, transcription factor PTM balances growth and drought survival via PIP2; 1 aquaporin. Nature plants 2019, 5:290–299. [DOI] [PubMed] [Google Scholar]
- 67.Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, Lyssiotis CA, Aldape K, Cantley LC, Lu Z: ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nature cell biology 2012, 14:1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu J, Chen L, Chen Y, Hasan MK, Ghia EM, Zhang L, Wu R, Rassenti LZ, Widhopf GF, Shen Z, et al. : Wnt5a induces ROR1 to associate with 14-3-3ζ for enhanced chemotaxis and proliferation of chronic lymphocytic leukemia cells. Leukemia 2017, 31:2608–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li X, Li XD: Chemical proteomics approaches to examine novel histone posttranslational modifications. Current opinion in chemical biology 2015, 24:80–90. [DOI] [PubMed] [Google Scholar]
- 70.Li X, Foley EA, Kawashima SA, Molloy KR, Li Y, Chait BT, Kapoor TM: Examining post-translational modification-mediated protein–protein interactions using a chemical proteomics approach. Protein Science 2013, 22:287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li X, Foley EA, Molloy KR, Li Y, Chait BT, Kapoor TM: Quantitative chemical proteomics approach to identify post-translational modification-mediated protein–protein interactions. Journal of the American Chemical Society 2012, 134:1982–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Xie Y, Chen L, Wang R, Wang J, Li J, Xu W, Li Y, Yao SQ, Zhang L, Hao Q: Chemical Probes Reveal Sirt2’s New Function as a Robust “Eraser” of Lysine Lipoylation. Journal of the American Chemical Society 2019, 141:18428–18436. * Peptide probes containing lipoylated lysine and a diazirine photo-crosslinker was used to identify Sirt2 as the eraser of lysine lipoylation
- 73.Liu Z, Yang T, Li X, Peng T, Hang HC, Li XD: Integrative chemical biology approaches for identification and characterization of “erasers” for fatty-acid-acylated lysine residues within proteins. Angewandte Chemie 2015, 127:1165–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang T, Liu Z, Li XD: Developing diazirine-based chemical probes to identify histone modification ‘readers’ and ‘erasers’. Chemical science 2015, 6:1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lin J, Bao X, Li XD: A tri-functional amino acid enables mapping of binding sites for posttranslational-modification-mediated protein-protein interactions. Molecular Cell 2021, 81:2669–2681. * A trifunctional amino acid was designed, containing a diazirine photo-crosslinker, an alkyne handle for purification, and a cleavable disulfide linker. When incorporated into peptide probes, it enables mapping of the binding site of PTM readers, and facilitated the identification of ClQBP as a histone chaperone
- 76.Yang T, Li X-M, Bao X, Fung YME, Li XD: Photo-lysine captures proteins that bind lysine post-translational modifications. Nature chemical biology 2016, 12:70–72. [DOI] [PubMed] [Google Scholar]
- 77. Xie X, Li X-M, Qin F, Lin J, Zhang G, Zhao J, Bao X, Zhu R, Song H, Li XD, et al. : Genetically encoded photoaffinity histone marks. Journal of the American Chemical Society 2017, 139:6522–6525. * Using genetic code expansion, lysine-derived ncAAs harboring a diazirine handle, and either a photo-labile protection group or a crotonyl group on the Nε, were site-specifically incorporated into proteins. The diazirine photo-crosslinker enables covalent capture of proteins that interact with this residue.
- 78. Zheng Y, Gilgenast MJ, Hauc S, Chatterjee A: Capturing post-translational modification-triggered protein–protein interactions using dual noncanonical amino acid mutagenesis. ACS chemical biology 2018, 13:1137–1141. ** Using genetic code expansion, N-acetyllysine and a benzophenone crosslinker were incorporated at two distinct sites of full-length histone H3. The benzophenone crosslinker enabled the covalent capture of the reader protein TRIM24.
- 79.Bruninghoff K, Aust A, Taupitz KF, Wulff S, Dörner W, Mootz HD: Identification of SUMO binding proteins enriched after covalent photo-cross-linking. ACS Chemical Biology 2020, 15:2406–2414. [DOI] [PubMed] [Google Scholar]
- 80. Sudhamalla B, Dey D, Breski M, Nguyen T, Islam K: Site-specific azide-acetyllysine photochemistry on epigenetic readers for interactome profiling. Chemical science 2017, 8:4250–4256. * A photo-crosslinker group (aromatic azide) was site-specifically incorporated into an N-acetyllysine reader domain to covalently capture its native substrates
- 81.Roy A, Barman S, Padhan J, Sudhamalla B: Engineering an acetyllysine reader with photocrosslinking amino acid for interactome profiling. Chemical Communications 2021, 57:9866–9869. [DOI] [PubMed] [Google Scholar]
- 82.Tang H, Dai Z, Qin X, Cai W, Hu L, Huang Y, Cao W, Yang F, Wang C, Liu T: Proteomic Identification of Protein Tyrosine Phosphatase and Substrate Interactions in Living Mammalian Cells by Genetic Encoding of Irreversible Enzyme Inhibitors. J Am Chem Soc 2018, 140:13253–13259. [DOI] [PubMed] [Google Scholar]
- 83.Linding R, Jensen LJ, Ostheimer GJ, van Vugt MA, Jørgensen C, Miron IM, Diella F, Colwill K, Taylor L, Elder K, et al. : Systematic discovery of in vivo phosphorylation networks. Cell 2007, 129:1415–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Joughin BA, Tidor B, Yaffe MB: A computational method for the analysis and prediction of protein: phosphopeptide-binding sites. Protein science 2005, 14:131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Humphreys IR, Pei J, Baek M, Krishnakumar A, Anishchenko I, Ovchinnikov S, Zhang J, Ness TJ, Banjade S, Bagde SR, et al. : Computed structures of core eukaryotic protein complexes. Science 2021, 374:eabm4805. * A promising computational approach to model protein-protein interactions