

Abstract
More than 800 million people suffer from kidney disease. Genetic studies, and follow-up animal model and cell biological experiments indicate the key role of proximal tubule metabolism. The kidney has one of the highest mitochondrial density. Mitochondrial biogenesis, mitochondrial fusion, fission, and mitochondrial recycling, such as mitophagy are critical for proper mitochondrial function. Mitochondrial dysfunction can lead to an energetic crisis, orchestrate different types of cell death (apoptosis, necroptosis, pyroptosis, ferroptosis), influence cellular calcium levels and redox status. Collectively, mitochondrial defect in renal tubules contributes to epithelial atrophy, inflammation, or cell death, orchestrating kidney disease development.
Keywords: Renal tubule cell, kidney disease, mitochondria, inflammation, cell death, mitophagy
The key contribution of proximal tubule metabolism to kidney disease development
The kidney maintains electrolyte, fluid balance and secretes hormones. More than 800 million people suffer from kidney disease. Kidney dysfunction will cause toxins, fluid, and electrolyte build-up. Without treatment, kidney disease can progress to end-stage kidney failure requiring life-sustaining renal replacement therapy. New drug development for kidney disease is limited by our poor mechanistic understanding of disease pathogenesis. Kidney function genetic studies highlighted important enrichment of disease-causing genes in the kidney proximal tubules. Follow-up animal model and cell biological experiments highlighted specific genes and pathways supporting the key role of metabolism and mitochondrial dysfunction in kidney disease development. Monogenic gene mutations of mitochondrial genes such as MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms), MERRF (myoclonus, epilepsy with ragged red fibers), and Leigh syndrome also present with renal phenotypes further supporting the role of mitochondria and metabolism in kidney disease [2].
The renal glomerulus has size and charge selectivity, so all metabolites, electrolytes, and proteins under 60kD are filtered into the tubules. The kidney proximal tubules are responsible for the reabsorption of all filtered nutrients, most electrolytes, and the secretion of some toxins. Electrolyte and nutrient reabsorption have a high-energy requirement; therefore the kidney tubules have one of the highest mitochondrial contents in the body[1].
Fatty acids are the preferred energy source for kidney tubules, which are then metabolized by fatty acid oxidation (FAO) and oxidative phosphorylation (OX-PHOS). Changes in metabolism and the potential build-up of intermediates could influence cellular functions[3]. Damaged mitochondria fail to supply sufficient ATP, causing an energetic deficit, resulting in atrophy or dedifferentiation of kidney tubule cells. Kidney tubule cell atrophy or dedifferentiation is defined by the loss of expression of markers of terminally differentiated tubule cells and sometimes loss of apical and basal polarity. A more severe hypoxic or toxic injury to kidney tubule cells will cause not only dedifferentiation but also epithelial cell death. Key molecules and interactions between cellular dysfunction, death pathways playing role in kidney disease are discussed in this review.
Inflammation is a key feature of acute and chronic kidney disease. Mitochondrial alterations in tubule cells are sufficient to induce sterile inflammation observed in acute and chronic kidney disease. Generation of mitochondrial reactive oxygen species play role in inflammasome activation [4]. Mitochondrial defect or improper clearing of damaged mitochondria can lead to the cytosolic release of mitochondrial DNA and activation of the cytosolic nucleotide sensors[5]. Inflammatory cell death mechanisms, such as pyroptosis and ferroptosis attract immune cells due to cytokine release[6]. Here we will review the multifaceted role of mitochondria and proximal tubule metabolism in kidney disease and dysfunction, including the contribution of energy deficit, dysfunctional metabolism, cell death, and sterile inflammation. Defining the molecular mechanism of kidney dysfunction would enable much-needed drug development and the application of precision medicine tools for kidney disease.
Energy balance
The primary function of the mitochondria is to generate ATP via a chain of biochemical reactions called the Krebs cycle [7]. Kidney tubules, especially proximal tubules transport kilograms of sodium chloride, other electrolytes, and nutrients daily. Kidney tubule cells preferentially oxidase fatty acids to generate energy (Fig.1). It has been known that kidney tubules can also burn ketones and lactate. While glucose utilization is almost undetectable in proximal renal tubules, it could be utilized in distal tubule segments [3]. Proximal kidney tubule cells can also generate glucose via a process called gluconeogenesis [8]. Gene expression analysis of diseased kidneys revealed lower expression of genes associated with fatty acid oxidation and oxidative phosphorylation [9].
Fig.1. Compromised fatty acid metabolism leads to kidney function impairment.
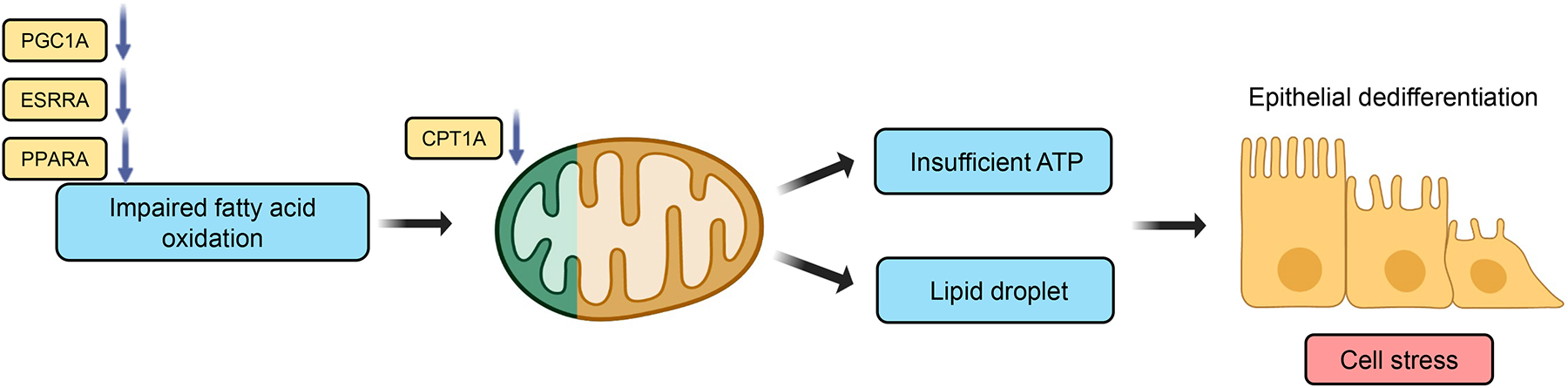
Fatty acids are the main source of energy for kidney tubule epithelial cells and are oxidized by fatty acid oxidation and mitochondrial oxidative phosphorylation. Several transcription factors such as Estrogen Related Receptor Alpha (ESRRA), Peroxisome Proliferator Activated Receptor Alpha (PPARA) play an important role in regulating fatty acid oxidation in kidney tubules, in addition to peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1a), which regulates mitochondrial biogenesis and metabolism. Lower expression of ESRRA, PGC1A, PPARA, and Carnitine Palmitoyltransferase 1A (CPT1A), was observed in acute and chronic kidney disease leading to impairment in fatty acid oxidation, subsequent reduction in ATP level, cellular lipid accumulation, contributing to functional impairment and dedifferentiation of tubule cells.
The rate-limiting enzyme in the mitochondrial oxidative phosphorylation is the carnitine palmitoyltransferase-1 (CPT1). Expression of CPT1A was lower in patients and animal models of kidney fibrosis [10]. Accumulation of short- and middle-chain acylcarnitine was observed in diseased human kidneys, likely reflecting impaired FAO [11]. Inhibition of CPT1 with etoxomir caused intracellular lipid deposition, ATP depletion, and epithelial dedifferentiation in mice, cellular changes observed in AKI and CKD [10]. Conversely, tubule-specific CPT1A transgenic mice had improved FAO and were protected from fibrosis and showed better kidney function following injury [11]. The dysfunctional mitochondria-induced epithelial impairment was largely attributed to the energetic deficit, however, the role of metabolic intermediate build-up cannot be excluded.
Peroxisome proliferator-activated receptor α (PPARA) is one of the key transcriptional regulators of lipid metabolism. PPARA is highly expressed by kidney proximal tubule cells. Genetic deletion of PPARA in mice caused increased lipid accumulation due to reduced FAO and exacerbated fibrosis in aging and diabetic kidneys [12]. The PPARA agonist, fenofibrate attenuated glomerular injury in obese animals. Fenofibrate also lowered tubulointerstitial injury in a mouse model of fatty acid-overload, folic acid injury nephropathy (FAN), unilateral ureteral obstruction (UUO) injury, and polycystic kidney disease [13, 14]. In patients, fenofibrate use was associated with reduced albuminuria and slower eGFR decline, irrespective of serum lipid levels. Unfortunately, fenofibrate leads to an acute rise of serum creatinine, likely due to directly interfering with tubule creatinine secretion [15].
Nicotinamide adenine dinucleotide (NAD) is essential for mitochondrial respiration, as it is one of the key oxidizing agents in cells. It acts as a coenzyme in redox reactions, as a donor in ADP-ribosylation reactions, as a precursor of the second messenger for cyclic ADP-ribose, as well as a substrate for sirtuins that remove acetyl groups from proteins. Amongst the 7 mammalian sirtuins, SIRT3, SIRT4, and SIRT5 are located in the mitochondria [16]. SIRT3 interacts with enzymes responsible for energy generation and oxidative stress, including the long-chain acyl-coenzyme A dehydrogenase [17], pyruvate dehydrogenase [18], and MnSOD [19]. Sirt3 KO mice showed increased oxidative stress, apoptosis, resulting in an increase in susceptibility to ischemic, toxic [20], or obstructive (UUO) kidney injury [18]. The role of SIRT4, and 5 remains controversial. One study reported impaired energy metabolism and enhanced mitochondrial fragmentation in SIRT5 deficient human proximal tubular epithelial cells [21]. Another study showed improved kidney function and less tissue damage following either ischemia- or cisplatin-induced AKI in SIRT5 KO mice [22]. In addition, while SIRT1 is localized to the nucleus, it has been shown to regulate mitochondrial biogenesis and fatty acid oxidation by deacetylating the metabolic transcription factor PGC1a [23].
De novo biosynthesis is a key regulator of NAD levels. Impaired de novo NAD+ biosynthesis has been reported in acute kidney injury (AKI). Mice with heterozygous loss of quinolinate phosphoribosyltransferase (QPRT) had lower NAD+ levels and increased AKI susceptibility [24]. NAD supplementation was shown to be protective from kidney injury. The NAD precursor nicotinamide (NAM) restored solute uptake, oxygen consumption rate (OCR), and lipid metabolism in acute kidney injury models [25]. NAM treatment also reduced tubular atrophy, apoptosis, and inflammation in renal fibrosis induced by UUO [26]. Supplementation with nicotinamide mononucleotide (NMN), a NAD+ precursor, restored mitochondrial density, renal SIRT1 activity in aging mice, and protected mice from cisplatin-induced AKI [27]. These results indicate that NAD precursors could be promising for patients with kidney disease via improving mitochondrial dysfunction [28].
In summary, transcriptome profiling and animal model studies indicated an impaired FAO and OX-PHOS in acute and chronic kidney disease leading to an energy deficit and causing tubule dedifferentiation or atrophy. Increasing the activity of PPARA, CPT1, or supplementing mice with NAD or its precursors could improve FAO and OX-PHOS and kidney function, indicating the key role of tubule energy deficit in kidney disease.
Redox regulation and Oxidative stress
Defective mitochondria fail to maintain the proton gradient across the inner mitochondrial membrane and are the main source of reactive oxygen species (ROS) in most cells (Fig.2). Under physiological conditions, 0.2–2% of the electrons in the electron transport chain do not follow the normal transfer but directly leak out of the electron transport chain and interact with oxygen to produce superoxide or hydrogen peroxide. Complex I and complex III are considered to be the main sites for ROS production. In addition, NADPH oxidases located in the mitochondria or at the plasma membrane might also generate ROS. Kidney utilizes about 20% of the cardiac output, and are very susceptible to hypoxic injury [30]. Oxygen is required for the electron transport chain reaction, therefore, hypoxia leads to ROS production mainly at the complex III [29].
Fig.2. Increased mitochondrial reactive oxygen species (ROS) in kidney injury.
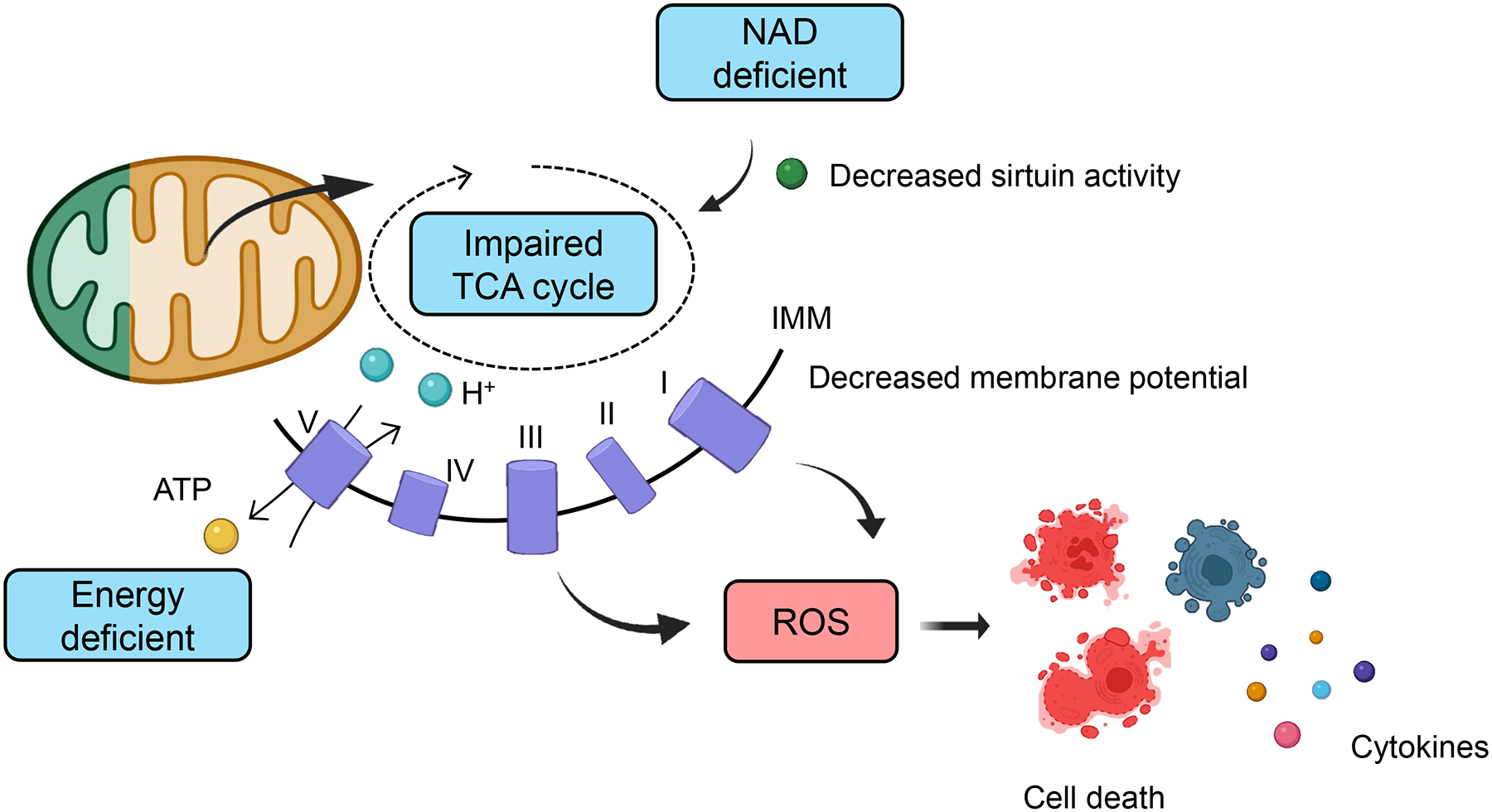
The citric acid cycle which takes place in the mitochondrial matrix generates cellular energy (ATP). The cycle consumes acetyl-CoA and water, reduces NAD+ to NADH, and releases carbon dioxide and water. Low NAD levels and NAD-dependent mitochondrial sirtuin activity in kidney disease are associated with impaired mitochondrial respiration, energy deficiency, and ROS accumulation in kidney tubule cells. The impaired Krebs cycle in damaged kidney tubule cells cause an energetic crisis. Cellular ATP in kidney tubule cells is critical for the re-absorptive capacity of proximal tubule cells. Increase cellular ROS can trigger cell death, specifically necroptosis, ferroptosis, and pyroptosis. Inflammatory cell death will cause the release of inflammatory cytokines.
Cells possess a robust ROS defense system. The mtROS (O2−) is first converted to H2O2 and further degraded to H2O by multiple antioxidant enzymes such as Cu/Zn SOD, MnSOD, catalase, thioredoxin peroxidase, or glutathione peroxidase. The role of these defense enzymes in kidney disease development has been demonstrated in multiple prior studies. Catalase deficient mice presented with enhanced mitochondrial oxidative stress when made diabetic [31]. Similarly, endothelial-specific thioredoxin reductase 2 knockout mice display renal abnormalities with greater oxidative stress [32]. Transgenic mice expressing glutathione peroxidase-1 had lower mitochondrial ROS and subsequently improved aging-associated fibrosis [33]. Uncoupling protein 2 (UCP2), a proton transporter lowers mtROS. Higher UCP2 expression was observed in UUO kidneys [34]. Deletion of UCP2 worsened ischemia [35] or bacterial lipopolysaccharide (LPS) injection-induced tubular injury [36].
Multiple antioxidants have been developed and marketed to prevent kidney disease in animal models. For example, the Nox1/4 inhibitor, GKT137831 reduced ROS production and subsequently lowered expression of proinflammatory and profibrotic markers in animal models of diabetic kidney disease [37]. A clinical trial with GKT137831 has been initiated in 2013, but its results still have not been made public (ClinicalTrials.gov Identifier: NCT02010242). Other anti-oxidants, for example, the citrate functionalized Mn3O4 nanoparticles (C-Mn3O4 NPs), biocompatible redox nanomedicine augmented the intracellular antioxidant defense system and protected from cisplatin-induced kidney tubule injury [38]. SS31 is a peptide-based antioxidant with mitochondrial targeting ability. SS31 treatment reduced mitochondrial damage, oxidative stress, inflammation, and apoptosis in streptozotocin-induced diabetic mice [39] or ischemia reperfusion-induced kidney injury [40]. MitoQ is yet another mitochondrial anti-oxidant with the potential to lower apoptosis and oxidative stress in human renal tubule cells exposed to high glucose [41].
The mechanism of increased ROS-induced tubule injury is not fully understood. Mitochondrial ROS can oxidize DNA, proteins, and lipids, and sensitize to inflammatory cell death pathways [4]. Mitochondrial ROS promote RIPK1 autophosphorylation, followed by recruitment of RIPK3 and MLKL to activate necroptosis [42]. Necroptosis plays an important role in kidney dysfunction [43]. RIPK3 deficient mice [44], catalytically inactive RIPK1 mice [45], MLKL knock-out mice [44], or mice treated with RIPK1 inhibitors; Necrostatin-1 [46], or Cpd-71 [47] had improved renal function and attenuated acute tubular injury. The role of necroptosis in kidney fibrosis and chronic kidney disease has also been demonstrated using the RIPK3 knock-out mice [48] and by Necrostatin-1 treatment [49]. Similarly, the ROS scavenger NAC (N-acetyl-L-cysteine) lowered necroptosis and alleviated kidney tubule injury after cisplatin injection [50].
Mitochondrial ROS can trigger the activation of the NLRP3 inflammasome, a key molecule in the pyroptosis pathway [51]. NLRP3 can activate caspase-1, and induce gasdermin D (GSDMD) cleavage, which is essential for pore formation and inflammatory cytokine release (IL-1B, IL-18). Caspase-1 KO mice showed protection from diabetic kidney disease and APOL1-associated glomerulosclerosis [52]. GSDMD knockout mice exhibited less tubular damage after toxic (cisplatin) or ischemic kidney injury [53]. Caspase-11 (human homolog caspase-4, -5) is also capable of cleaving GSDMD. Caspase-11 KO mice showed decreased tubule injury in contrast-induced acute kidney injury [54] and renal fibrosis in the UUO model of injury [55]. IL-1B blockade using a monoclonal antibody in diabetic db/db mice attenuated kidney dysfunction [56]. The effect of IL-18 in ischemic AKI has been demonstrated both in the IL-18 KO mice and via the therapeutic neutralization of IL-18 [57].
Ferroptosis is a recently identified cell death mechanism characterized by lipid peroxidation. ROS released by defective mitochondria can induce lipid peroxidation. The glutathione peroxidase 4 (GPX4) and the glutathione independent ferroptosis suppressor 1 (FSP1) are the key cellular defense systems against ferroptosis. GPX4 uses reduced glutathione (GSH) to detoxify lipid hydroperoxide, and FSP1 reduces ubiquinone (CoQ) to form ubiquinol (CoQH2). Increased tubule cell ferroptosis was reported both in acute and chronic kidney injury [58]. The main phenotype of the global GPX4 KO mice is tubule cell death and kidney injury, indicating the key role of GPX4 and ferroptosis in kidney tubule cells [59]. While GPX4 and FSP1 are mainly located in the plasma membrane, a recent paper identified dihydroorotate dehydrogenase (DHODH), as another ferroptosis defense system located in the mitochondrial membrane, suggesting the important role of mitochondria in ferroptosis [60]. DHODH catalyzes the conversion of dihydroorotate to orotate and generates ubiquinol to lower lipid peroxidation [60]. Ferrostatin-1 (Fer-1), a small-molecular inhibitor of lipid oxidation ameliorated both ischemic injury and diabetic kidney disease [61, 62]. Newly developed ferroptosis inhibitors, for example, XJB-5-131, showed a similar protective effect [63].
Important to note, that kidney function GWAS studies indicated the role of ferroptosis pathway genes in kidney disease. Functional annotation of an eGFR genetic locus prioritized Dipeptidase 1 (DPEP1) and Charged Multivesicular Body Protein 1 A (CHMP1A) as kidney disease risk genes [64]. Gene knock-out experiments confirmed the role of CHMP1A and DPEP1 in kidney disease. DPEP1 and CHMP1A seem to play role in iron import and export, respectively [65]. Higher iron concentration and increased ferroptosis were observed in CHMP1A haploinsufficient renal tubules. DPEP1 KO mice showed lower, while CHMP1A haploinsufficiency mice had more severe cisplatin or folic-acid injection-induced kidney injury [65].
In summary increased accumulation of reactive oxygen species in proximal tubules is a major contributor to pathology. Compounds that inhibit ROS generation or improve neutralization showed promise in animal models. Cellular ROS is an important trigger for inflammatory cell death pathways such as pyroptosis and ferroptosis.
Mitochondria Biogenesis
Mitochondrial numbers are a key determinant of mitochondrial function and it is the key mechanism to adapt to higher energy demand (Fig.2). Mitochondrial biogenesis is an important determinant of mitochondrial number. The peroxisome proliferator-activated receptor γ (PPARG) coactivator-1 of transcriptional coactivator α (PGC1a) is a master transcriptional regulator of mitochondrial biogenesis. Expression of PGC1a was lower in kidneys of patients with acute and chronic kidney disease [10]. Mice with genetic deletion of PCG1a appeared healthy at baseline but showed increased susceptibility to acute and chronic injury [28]. At the same time, tubule-specific transgenic expression PGC1a protected from acute kidney injury and fibrosis. PGC1a overexpression in tubule cells reversed the energy deficit via improving fatty acid oxidation [66]. In addition, PGC1a also increased levels of the de novo NAD biosynthetic enzymes [28]. PGC1a works in concert with other transcription factors such as nuclear factor erythroid 2-related factor 1 (NRF1) and NRF2. NRF1 and NRF2 regulate the expression of the electron transfer chain subunits encoded by the nuclear genome. NRF1 binds to the promoter and regulates the expression of TFAM (transcription factor A mitochondrial). NRF2 knockout mice exhibited increased tissue damage and fibrosis in disease models [67]. TFAM is essential for mitochondrial encoded gene transcription and replication [68]. Mice with tubule-specific deletion of TFAM showed abnormal OXPHOS, energy depletion (low ATP), cell death, and renal fibrosis [69]. The estrogen-related receptor alpha and gamma (ESRRA, ESRRRG) transcription factors are shown to play role in regulating mitochondrial biogenesis. Mice with ESRRA deletion appeared healthy at baseline but showed increased tubule injury in disease settings [70]. Mice with kidney tubule-specific ESRRG died of renal failure due to epithelial atrophy, tubule dilation, and cyst formation [71]. Pharmacological approaches have been developed for the restoration of mitochondrial biogenesis, for example via the beta 2-adrenergic receptor (β2AR) agonist, formoterol [72, 73], and a selective 5-HT1F receptor agonist, LY344864 [74], LY334370 [75, 76]. This approach showed therapeutic benefit in ischemic kidney injury.
Mitochondria Dynamics, Shape, Size and Turn-over
Mitochondrial shape and size, which is controlled by fission and fusion, are yet another less well-understood determinant of function (Fig.2). Abnormalities in mitochondrial dynamics have been observed both in acute and chronic disease conditions [77]. DRP1 (dynamin-related protein 1) is the master regulator of mitochondrial fission. Proximal tubule-specific deletion of DRP1 or mdivi-1 treatment, a pharmacological inhibitor of DRP1, preserved mitochondrial structure, reduced oxidative stress, and protected against ischemic- or cisplatin-induced renal injury, inflammation, apoptosis [78, 79]. Mitofusin (MFN) 1 and 2 control mitochondrial fusion. Mice with proximal tubule-specific MFN2 deficiency showed accelerated kidney function recovery and enhanced survival after acute kidney injury [80].
Damaged or old mitochondria are degraded in the proteasome or via mitophagy. Mitophagy allows the selective degradation of damaged mitochondria by the autophagy machinery. Loss of mitochondrial membrane potential serves as a trigger for mitophagy (Fig.3). Mitophagy plays a role both in acute and chronic kidney disease. PINK1 and PARKIN are the key mitophagy inducers. PINK1 insertion into healthy mitochondria will induce PARL (rhomboid-like protein)-mediated PINK1 cleavage and rapid removal by the proteasome-dependent pathway. Under cellular stress, defective mitochondria with impaired membrane potential accumulate PINK1 in the outer membrane. PINK1 on the outer mitochondrial membrane recruit PARKIN for mitophagosome formation. PINK1 or PARKIN knockout mice have low mitophagy, develop severe mitochondrial damage, higher mtROS production, and increased cell death and kidney injury after injection of LPS [81], cisplatin [82], or UUO surgery [83]. The deficiency of the mitochondrial methylmalonyl-coenzyme A mutase (MMUT) is an inherited metabolic disorder associated with kidney injury. MMUT knock-out mice had blunted PINK1-directed mitophagy, leading to the accumulation of dysfunctional mitochondria, and kidney epithelial dysfunction [84].
Fig.3. Mitophagy plays key role in degrading impaired mitochondria.
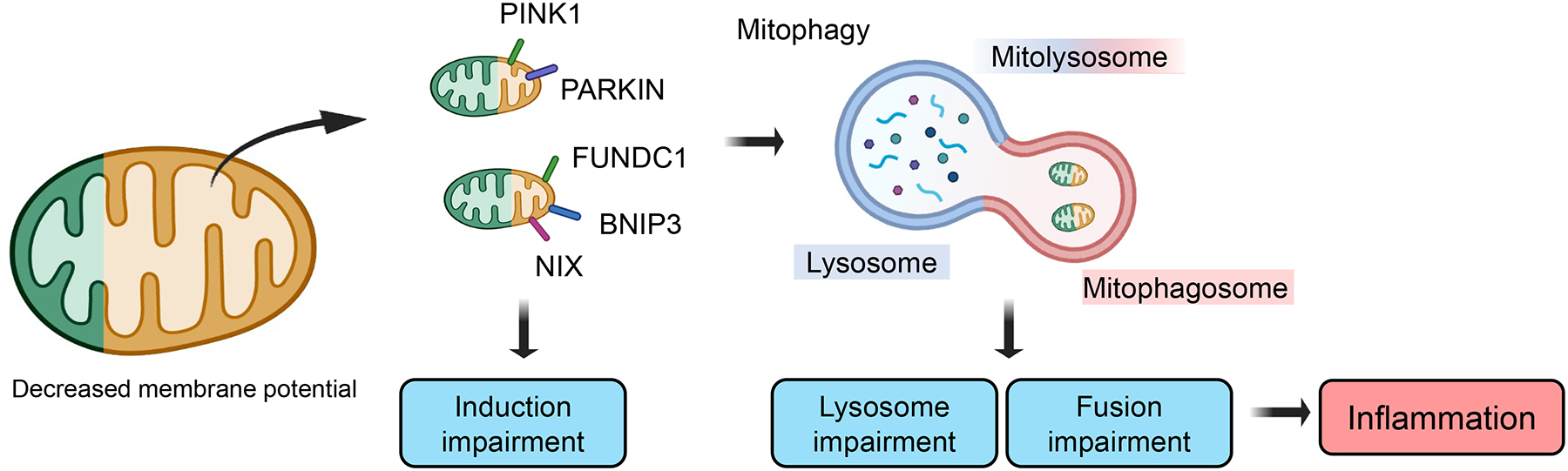
Mitophagy selectively eliminates damaged mitochondria. Mitochondrial injury can cause mitochondrial membrane depolarization and trigger mitophagy. Mitophagy receptors; PTEN Induced Kinase 1 (PINK1), PARKIN, FUN14 Domain Containing 1 (FUNDC1), BCL2 Interacting Protein 3 (BNIP3), NIX can initiate mitophagosome formation. Induction of mitophagy is followed by lysosome mitophagosome fusion, forming the mitolysosome. Impaired mitophagy can lead to the accumulation of defective mitochondria, inflammation, and worsening kidney injury.
Bcl2 interacting protein 3 (BNIP3), BNIP3L (NIX) and FUNDC1 are PINK1/Parkin independent mitophagy receptors. Mice with BNIP3 overexpression had lower apoptosis and dampened acute kidney injury [85]. BNIP3 is transcriptionally controlled by hypoxia-inducible factor 1-alpha (HIF1a). BNIP3 is an important mediator of the adaptive metabolic response by regulating mitochondrial ROS via mitophagy [86]. Overexpression of NIX by a recombinant adenoviral vector, reduced mitochondrial fragmentation and tubular cell apoptosis in high-protein diet-challenged mice [87]. FUNDC1 knock-out mice had higher ROS levels, enhanced inflammation, and impaired erythropoietin production [88]. A recent paper demonstrated that PHB2 (Prohibitin2) is another inner mitochondrial membrane mitophagy receptor [89]. PHB2 knockdown lowered mitophagy and augmented cell death while overexpression of PHB2 protected from inflammasome activation [90]. In addition to the genetic models, pharmacological enhancement of mitophagy has also been shown to be beneficial. MitoQ treatment (a mitochondria-targeted antioxidant) improved mitophagy and protected from diabetes-induced tubule injury [41]. Coenzyme Q10 (CoQ10) is an electron carrier in the mitochondrial respiration system. CoQ10 administration restored mitophagy in diabetic kidney disease and improved kidney function [91].
Mitochondrial dynamics and quality control is critical for kidney function, especially mitophagy plays an important role in the elimination and recycling of damaged mitochondria. Enhancing mitophagy is a potential strategy to improve mitochondrial health and showed benefit in animal models.
The role of mitochondria in controlling inflammation
Severe mitochondrial damage and defect in mitochondrial clearance can lead to a leakage of the mitochondrial DNA (mtDNA) into the cytosol. The exact mechanism of the mtDNA leakage is still not fully understood (Fig.4). BAK and BAX pores in the mitochondrial outer membrane can lead to inner mitochondrial membrane herniation and cytosolic release of mtDNA [6]. The presence of mtDNA in the cytosol is a sign of pathogenic infection and is recognized by the cytosolic nucleotide sensing pathways. The nucleic acid receptors can be divided into two main categories: immune sensing receptors, which include Toll-like receptor 3 (TLR3), TLR7, TLR8, TLR9, retinoic acid inducible gene I (RIG-I), melanoma differentiation associated gene 5 (MDA5), absent in melanoma 2 (AIM2) and cyclic GMP–AMP synthetase (cGAS); and nucleic acid receptors, including double-stranded RNA (dsRNA)-activated protein kinase R (PKR), IFN-induced protein with tetratricopeptide repeats 1 (IFIT1), 2′−5′-oligoadenylate synthetase 1 (OAS1) and ribonuclease L (RNase L), and adenosine deaminase acting on RNA 1 (ADAR1). Two teams independently reported the activation of cGAS-stimulator of interferon genes (STING) cytosolic DNA sensing pathway in diseased kidney tubule cells including the cisplatin-induced injury model [92] and tubule specific TFAM knock-out mice [69]. Pharmacological inhibition of STING, using C176, or genetic deletion of STING attenuated kidney injury both in acute and chronic kidney disease models [69]. A recent study, analyzing the risk variant APOL1 induced glomerular disease model indicated that kidney disease-associated APOL1 variant altered mitophagy, leading to cytosolic leakage of mtDNA and activation of cGAS and STING [93, 94]. STING activates downstream molecules, including TBK1, IRF3, and IRF7, and induces the expression of interferon-stimulated genes (ISGs). ISG15 is reported to be released to the extracellular space and regulate immune cells migration and activation [95]. Bst2, called Tetherin, promotes dendric cell activation [96] or T cell proliferation [97]. NFkB is also a downstream target of STING. The role of NFkB in kidney inflammation has been well described [98].
Fig.4. The key role of mitochondria in eliciting an inflammatory response.
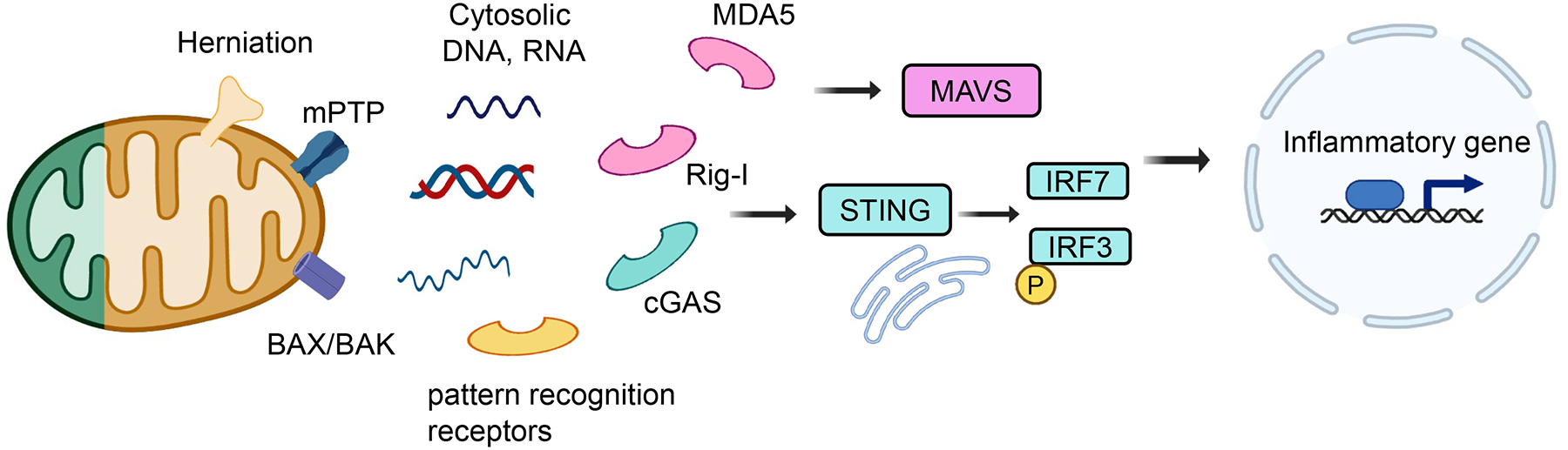
The mitochondria can serve as a signaling platform to control inflammation. Mitochondrial DNA (mtDNA) is released from damaged mitochondria into the cytosol through BCL2-associated X protein (BAX)/ BCL2-antagonist/killer (BAK), mitochondrial permeability transition pore (mPTP), or by mitochondria herniation. Cytosolic nucleotides are recognized by several nucleotide sensing pathways such as Cyclic GMP–AMP synthase (cGAS) - Stimulator of interferon genes (STING)- Interferon Regulatory Factor 3 (IRF3)/ IRF7, Retinoic acid-inducible gene I (RIG-I)/ Melanoma differentiation-associated protein 5 (MDA5)-Mitochondrial antiviral signaling protein (MAVS). These pathways activate pro-inflammatory genes including interferon-stimulated genes. Genetic deletion or pharmacological inhibitor of cGAS/STING or RIG-I attenuated kidney disease development.
The cytosolic mitochondrial DNA or necrotic cell DNA taken up by macrophages can also activate AIM2, another cytosolic nucleotide sensing pathway [99]. In one study, AIM2 KO mice showed reduced caspase-1 activation and IL-1B expression and were protected from kidney injury induced by UUO [100]. On the other hand, in a nephrotoxic serum (NTS)–induced glomerulonephritis model study, the AIM2 KO mice exhibited more severe glomerular crescent formation, tubular injury and inflammation [101], implying context-depend roles of AIM2. Mice with genetic deletion of RIG-I were also protected from tissue fibrosis and kidney disease [102].
Toll-like receptors (TLRs) expressed on endosomes recognize cytosolic nucleotides and trigger the innate immune response and inflammatory cascades. Cytosolic mtDNA has been shown to activate TLR9 [103], however, the role of TLR9 in kidney disease is not fully understood. Kidney injury was attenuated in the cecal ligation and puncture (CLP)-induced sepsis model in mice with global TLR9 loss [104]. Mice lacking TLR9 only in renal proximal tubules presented with a more severe AKI [105].
Apoptosis
The mitochondria play a key role in orchestrating a multitude of cell death mechanisms. Apoptosis is a non-inflammatory cell death mechanism occurring without the rupture of the cell membrane. The release of cytochrome c from the mitochondria to the cytosol through BAX/BAK pores activates subsequent caspases such as caspase-9, followed by the activation of the execution caspases such as caspase-3 (Fig.5). Apoptosis has been observed both in acute and chronic kidney disease and is likely a leading mechanism resulting in tubule cell and podocyte loss in acute and chronic kidney disease. The effect of pan-caspase inhibitors in acute kidney injury and kidney fibrosis remains inconclusive. An earlier study showed that z-VAD (pan-caspase inhibitor) inhibited apoptosis of kidney tubule cells, and prevented inflammation and tissue injury following ischemia [106]. In a later study, z-VAD treatment failed to ameliorate renal injury in the ischemia-reperfusion model [107]. In addition, the specificity of z-VAD has also been questioned, including its potential effect on autophagy [108]. Caspase-3 KO mice had less severe microvascular endothelial cell apoptosis and reduced renal fibrosis after ischemic injury [109]. Renal tubular cell apoptosis was also attenuated in BAX/BAK double KO mice with improved renal function after ischemic injury [110]. Kidney function genome-wide association study identified caspase-9 as a kidney disease risk gene. Lowering caspase-9 expression protected mice from kidney injury and fibrosis [111], indicating the causal role of apoptosis in disease development.
Fig.5. Apoptosis.
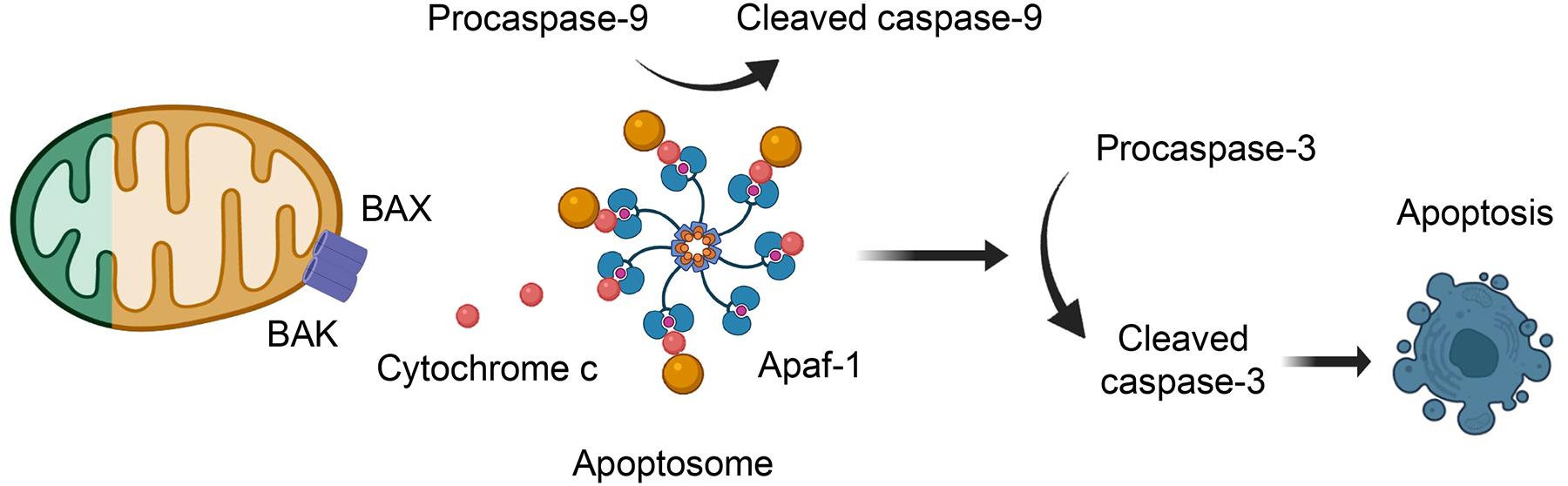
Proximal tubule apoptosis is a prominent feature of acute and chronic kidney disease. Intrinsic apoptosis is triggered by mitochondria damage. BCL2-associated X protein (BAX)/ BCL2-antagonist/killer (BAK) pores in the outer membrane of mitochondria will cause translocation of cytochrome c from the mitochondria to the cytosol to initiate the formation of the apoptosome consisting of caspase-9, Apaf1, and cytochrome-c. The apoptosome will activate executionary caspases such as caspase-3. Apoptosis will cause epithelial cell loss. Animal studies, analyzing BAX/BAK double KO, Caspase-9, and Caspase-3 KO mice or chemical caspase-inhibitors indicated the protective role of apoptosis from kidney disease development.
Concluding remarks
The kidney tubules have one of the highest mitochondrial density to generate energy for the transport of large amounts of sodium and other solutes. Defect in mitochondrial biogenesis, dynamics and mitophagy contribute to kidney disease development by the cells failing to meet the cellular energetic requirement. Mitochondrial damage has been widely recognized in acute or chronic kidney injury. Mitochondrial injury triggers multiple cell death mechanisms (apoptosis, necroptosis, pyroptosis, ferroptosis) contributing to epithelial cell loss, inflammation, and kidney disease. Defective mitochondria release ROS and further mitochondrial damage can cause the cytosolic leakage of mitochondrial DNA, activate cytosolic nucleotide sensors, induce inflammation, and enhance kidney disease development (Fig.6). Future studies shall aim to understand the relationship and hierarchy between the different mitochondrial alterations observed in diseased states. Furthermore, as the mitochondria interact with other organelles such as the nucleus, endoplasmic reticulum, and peroxisome, future work should aim to better dissect mitochondrial dysfunction and organelle interaction in disease. It would also be desirable to develop non-invasive methods to monitor mitochondria function in patients. Drugs, that target mitochondrial dysfunction could be promising for the treatment and prevention of renal disease. Some of these drugs including those that target oxidative stress, mitochondria biogenesis, and cell death show promising effects in animal models, however, their effectiveness in patients with kidney disease has not been established.
Fig.6. The key role of mitochondria in kidney disease development.
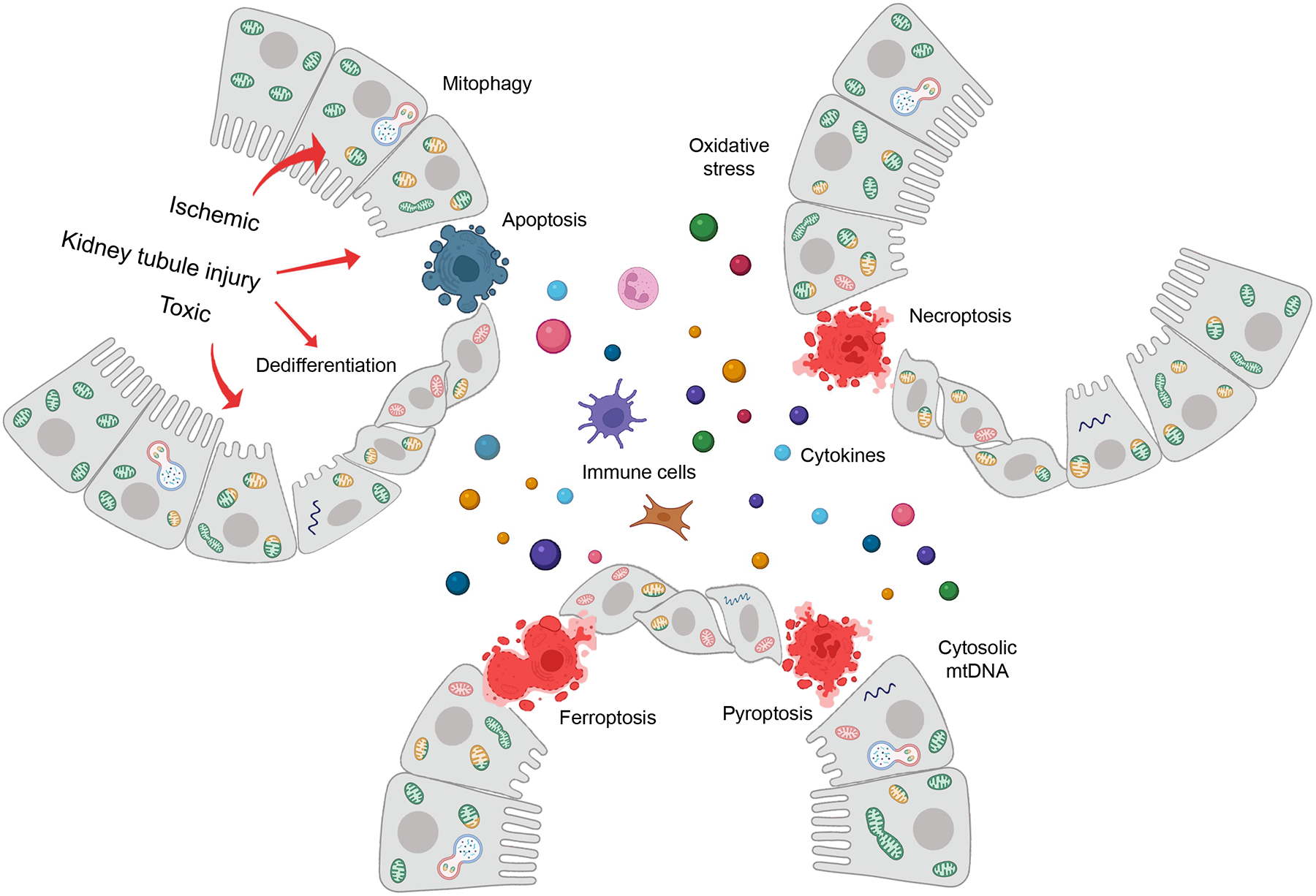
The figure illustrates the overall biological changes in response to mitochondria damage in kidney tubules following toxic or ischemic injury. Mitochondrial injury in kidney tubule cells will lead to energy deficiency, increase reactive oxygen species (ROS) generation, the cytosolic release of mitochondrial DNA. Mitochondria biogenesis, change in shape, size, and turn-over play role in kidney tubule function. Severe injury will lead to cell death including apoptosis, necroptosis, pyroptosis, ferroptosis, and enhance inflammation and fibrosis by releasing pro-inflammatory cytokines, attracting immune cells, or activating fibroblasts.
Outstanding question box.
Fatty acid oxidation and mitochondrial oxidative phosphorylation play key roles in maintaining kidney function. What are the specific enzymatic steps, genes impaired in the pathway? How does altered fatty acid metabolism lead to kidney dysfunction. Is kidney dysfunction caused by impaired energy production? Can we observe the accumulation of metabolic intermediates? Do metabolic intermediates contribute to kidney dysfunction?
What are the key pathways for mitochondrial quality control? How can we improve mitochondrial quality control in kidney tubules? Can we enhance mitophagy to improve kidney function?
What is the contribution of genetics? Which genes are affected and how do these genes influence kidney tubule metabolism?
Mitochondria play a central role in multiple cell death mechanisms (apoptosis, necroptosis, pyroptosis, ferroptosis). What is the relationship between different cell death mechanisms? Multiple different cell death mechanisms have been observed in acute and chronic kidney disease. How do different cell death mechanisms lead to adaptive or maladaptive kidney repair?
Highlight.
Kidney function genetic studies and follow-up omics functional analysis highlighted the key role of kidney proximal tubules and metabolisms in kidney disease development.
The kidney has the second-highest mitochondrial density in the body to enable the active reabsorption of nutrients and electrolytes.
Altered redox balance, impaired cellular energetics, increased cell death and inflammation observed as a consequence of mitochondrial dysfunction causing kidney disease development.
Acknowledgements
This work has been supported in the Susztak lab by the National Institute of Health NIH R01 DK087635, DK076077.
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- 1.Bhargava P and Schnellmann RG (2017) Mitochondrial energetics in the kidney. Nat Rev Nephrol 13 (10), 629–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Toole JF (2014) Renal manifestations of genetic mitochondrial disease. Int J Nephrol Renovasc Dis 7, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tian Z and Liang M (2021) Renal metabolism and hypertension. Nat Commun 12 (1), 963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Linkermann A et al. (2014) Regulated cell death in AKI. J Am Soc Nephrol 25 (12), 2689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhong F et al. (2019) Emerging Role of Mitochondrial DNA as a Major Driver of Inflammation and Disease Progression. Trends Immunol 40 (12), 1120–1133. [DOI] [PubMed] [Google Scholar]
- 6.McArthur K et al. (2018) BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359 (6378). [DOI] [PubMed] [Google Scholar]
- 7.Mishra P and Chan DC (2016) Metabolic regulation of mitochondrial dynamics. J Cell Biol 212 (4), 379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hui S et al. (2020) Quantitative Fluxomics of Circulating Metabolites. Cell Metab 32 (4), 676–688.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hallan S et al. (2017) Metabolomics and Gene Expression Analysis Reveal Down-regulation of the Citric Acid (TCA) Cycle in Non-diabetic CKD Patients. EBioMedicine 26, 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang HM et al. (2015) Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med 21 (1), 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miguel V et al. (2021) Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J Clin Invest 131 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chung KW et al. (2018) Impairment of PPARα and the Fatty Acid Oxidation Pathway Aggravates Renal Fibrosis during Aging. J Am Soc Nephrol 29 (4), 1223–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka Y et al. (2011) Fenofibrate, a PPARα agonist, has renoprotective effects in mice by enhancing renal lipolysis. Kidney Int 79 (8), 871–82. [DOI] [PubMed] [Google Scholar]
- 14.Lakhia R et al. (2018) PPARα agonist fenofibrate enhances fatty acid β-oxidation and attenuates polycystic kidney and liver disease in mice. Am J Physiol Renal Physiol 314 (1), F122–f131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Attridge RL et al. (2013) Fenofibrate-associated nephrotoxicity: a review of current evidence. Am J Health Syst Pharm 70 (14), 1219–25. [DOI] [PubMed] [Google Scholar]
- 16.Yang W et al. (2016) Mitochondrial Sirtuin Network Reveals Dynamic SIRT3-Dependent Deacetylation in Response to Membrane Depolarization. Cell 167 (4), 985–1000.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirschey MD et al. (2010) SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464 (7285), 121–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y et al. (2021) Sirtuin 3 regulates mitochondrial protein acetylation and metabolism in tubular epithelial cells during renal fibrosis. Cell Death Dis 12 (9), 847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tao R et al. (2014) Regulation of MnSOD enzymatic activity by Sirt3 connects the mitochondrial acetylome signaling networks to aging and carcinogenesis. Antioxid Redox Signal 20 (10), 1646–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morigi M et al. (2015) Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Invest 125 (2), 715–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haschler TN et al. (2021) Sirtuin 5 depletion impairs mitochondrial function in human proximal tubular epithelial cells. Sci Rep 11 (1), 15510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiba T et al. (2019) Sirtuin 5 Regulates Proximal Tubule Fatty Acid Oxidation to Protect against AKI. J Am Soc Nephrol 30 (12), 2384–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weinberg JM (2011) Mitochondrial biogenesis in kidney disease. J Am Soc Nephrol 22 (3), 431–6. [DOI] [PubMed] [Google Scholar]
- 24.Poyan Mehr A et al. (2018) De novo NAD(+) biosynthetic impairment in acute kidney injury in humans. Nat Med 24 (9), 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bugarski M et al. (2021) Changes in NAD and Lipid Metabolism Drive Acidosis-Induced Acute Kidney Injury. J Am Soc Nephrol 32 (2), 342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng M et al. (2019) Nicotinamide reduces renal interstitial fibrosis by suppressing tubular injury and inflammation. J Cell Mol Med 23 (6), 3995–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guan Y et al. (2017) Nicotinamide Mononucleotide, an NAD(+) Precursor, Rescues Age-Associated Susceptibility to AKI in a Sirtuin 1-Dependent Manner. J Am Soc Nephrol 28 (8), 2337–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tran MT et al. (2016) PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531 (7595), 528–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamanaka RB and Chandel NS (2009) Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr Opin Cell Biol 21 (6), 894–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haase VH (2013) Mechanisms of hypoxia responses in renal tissue. J Am Soc Nephrol 24 (4), 537–41. [DOI] [PubMed] [Google Scholar]
- 31.Hwang I et al. (2012) Catalase deficiency accelerates diabetic renal injury through peroxisomal dysfunction. Diabetes 61 (3), 728–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kameritsch P et al. (2021) The mitochondrial thioredoxin reductase system (TrxR2) in vascular endothelium controls peroxynitrite levels and tissue integrity. Proc Natl Acad Sci U S A 118 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chu Y et al. (2020) Glutathione peroxidase-1 overexpression reduces oxidative stress, and improves pathology and proteome remodeling in the kidneys of old mice. Aging Cell 19 (6), e13154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang L et al. (2013) A microRNA-30e/mitochondrial uncoupling protein 2 axis mediates TGF-β1-induced tubular epithelial cell extracellular matrix production and kidney fibrosis. Kidney Int 84 (2), 285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou Y et al. (2017) UCP2 attenuates apoptosis of tubular epithelial cells in renal ischemia-reperfusion injury. Am J Physiol Renal Physiol 313 (4), F926–f937. [DOI] [PubMed] [Google Scholar]
- 36.Zhong X et al. (2019) UCP2 alleviates tubular epithelial cell apoptosis in lipopolysaccharide-induced acute kidney injury by decreasing ROS production. Biomed Pharmacother 115, 108914. [DOI] [PubMed] [Google Scholar]
- 37.Jha JC et al. (2014) Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J Am Soc Nephrol 25 (6), 1237–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adhikari A et al. (2021) Redox nanomedicine ameliorates chronic kidney disease (CKD) by mitochondrial reconditioning in mice. Commun Biol 4 (1), 1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang SK et al. (2019) Mitochondria-Targeted Peptide SS31 Attenuates Renal Tubulointerstitial Injury via Inhibiting Mitochondrial Fission in Diabetic Mice. Oxid Med Cell Longev 2019, 2346580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu D et al. (2020) ROS-responsive chitosan-SS31 prodrug for AKI therapy via rapid distribution in the kidney and long-term retention in the renal tubule. Sci Adv 6 (41). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiao L et al. (2017) The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol 11, 297–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y et al. (2017) RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun 8, 14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Belavgeni A et al. (2020) Ferroptosis and Necroptosis in the Kidney. Cell Chem Biol 27 (4), 448–462. [DOI] [PubMed] [Google Scholar]
- 44.Chen H et al. (2018) RIPK3-MLKL-mediated necroinflammation contributes to AKI progression to CKD. Cell Death Dis 9 (9), 878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Newton K et al. (2016) RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ 23 (9), 1565–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Linkermann A et al. (2013) The RIP1-kinase inhibitor necrostatin-1 prevents osmotic nephrosis and contrast-induced AKI in mice. J Am Soc Nephrol 24 (10), 1545–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang JN et al. (2019) RIPK1 inhibitor Cpd-71 attenuates renal dysfunction in cisplatin-treated mice via attenuating necroptosis, inflammation and oxidative stress. Clin Sci (Lond) 133 (14), 1609–1627. [DOI] [PubMed] [Google Scholar]
- 48.Shi Y et al. (2020) RIPK3 blockade attenuates kidney fibrosis in a folic acid model of renal injury. Faseb j 34 (8), 10286–10298. [DOI] [PubMed] [Google Scholar]
- 49.Xiao X et al. (2017) Inhibition of Necroptosis Attenuates Kidney Inflammation and Interstitial Fibrosis Induced By Unilateral Ureteral Obstruction. Am J Nephrol 46 (2), 131–138. [DOI] [PubMed] [Google Scholar]
- 50.Meng XM et al. (2018) NADPH oxidase 4 promotes cisplatin-induced acute kidney injury via ROS-mediated programmed cell death and inflammation. Lab Invest 98 (1), 63–78. [DOI] [PubMed] [Google Scholar]
- 51.Abais JM et al. (2015) Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal 22 (13), 1111–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shahzad K et al. (2016) Caspase-1, but Not Caspase-3, Promotes Diabetic Nephropathy. J Am Soc Nephrol 27 (8), 2270–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miao N et al. (2019) The cleavage of gasdermin D by caspase-11 promotes tubular epithelial cell pyroptosis and urinary IL-18 excretion in acute kidney injury. Kidney Int 96 (5), 1105–1120. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Z et al. (2018) Caspase-11-mediated tubular epithelial pyroptosis underlies contrast-induced acute kidney injury. Cell Death Dis 9 (10), 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miao NJ et al. (2019) Caspase-11 promotes renal fibrosis by stimulating IL-1β maturation via activating caspase-1. Acta Pharmacol Sin 40 (6), 790–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lei Y et al. (2019) Interleukin-1β Inhibition for Chronic Kidney Disease in Obese Mice With Type 2 Diabetes. Front Immunol 10, 1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu H et al. (2008) IL-18 contributes to renal damage after ischemia-reperfusion. J Am Soc Nephrol 19 (12), 2331–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deng F et al. (2019) Myo-inositol oxygenase expression profile modulates pathogenic ferroptosis in the renal proximal tubule. J Clin Invest 129 (11), 5033–5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Friedmann Angeli JP et al. (2014) Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16 (12), 1180–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mao C et al. (2021) Author Correction: DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 596 (7873), E13. [DOI] [PubMed] [Google Scholar]
- 61.Martin-Sanchez D et al. (2017) Ferroptosis, but Not Necroptosis, Is Important in Nephrotoxic Folic Acid-Induced AKI. J Am Soc Nephrol 28 (1), 218–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim S et al. (2021) Correction: Characterization of ferroptosis in kidney tubular cell death under diabetic conditions. Cell Death Dis 12 (4), 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao Z et al. (2020) XJB-5–131 inhibited ferroptosis in tubular epithelial cells after ischemia-reperfusion injury. Cell Death Dis 11 (8), 629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Doke T et al. (2021) Transcriptome-wide association analysis identifies DACH1 as a kidney disease risk gene that contributes to fibrosis. J Clin Invest 131 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guan Y et al. (2021) A single genetic locus controls both expression of DPEP1/CHMP1A and kidney disease development via ferroptosis. Nat Commun 12 (1), 5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Han SH et al. (2017) PGC-1α Protects from Notch-Induced Kidney Fibrosis Development. J Am Soc Nephrol 28 (11), 3312–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nezu M and Suzuki N (2020) Roles of Nrf2 in Protecting the Kidney from Oxidative Damage. Int J Mol Sci 21 (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Picca A and Lezza AM (2015) Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions: Useful insights from aging and calorie restriction studies. Mitochondrion 25, 67–75. [DOI] [PubMed] [Google Scholar]
- 69.Chung KW et al. (2019) Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab 30 (4), 784–799.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsushida K et al. (2018) Estrogen-related receptor α is essential for maintaining mitochondrial integrity in cisplatin-induced acute kidney injury. Biochem Biophys Res Commun 498 (4), 918–924. [DOI] [PubMed] [Google Scholar]
- 71.Zhao J et al. (2018) Genomic integration of ERRγ-HNF1β regulates renal bioenergetics and prevents chronic kidney disease. Proc Natl Acad Sci U S A 115 (21), E4910–e4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cameron RB et al. (2019) Proximal Tubule β (2)-Adrenergic Receptor Mediates Formoterol-Induced Recovery of Mitochondrial and Renal Function after Ischemia-Reperfusion Injury. J Pharmacol Exp Ther 369 (1), 173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jesinkey SR et al. (2014) Formoterol restores mitochondrial and renal function after ischemia-reperfusion injury. J Am Soc Nephrol 25 (6), 1157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gibbs WS et al. (2018) Identification of dual mechanisms mediating 5-hydroxytryptamine receptor 1F-induced mitochondrial biogenesis. Am J Physiol Renal Physiol 314 (2), F260–f268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Garrett SM et al. (2014) Agonism of the 5-hydroxytryptamine 1F receptor promotes mitochondrial biogenesis and recovery from acute kidney injury. J Pharmacol Exp Ther 350 (2), 257–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gibbs WS et al. (2018) 5-HT(1F) receptor regulates mitochondrial homeostasis and its loss potentiates acute kidney injury and impairs renal recovery. Am J Physiol Renal Physiol 315 (4), F1119–f1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhan M et al. (2013) Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int 83 (4), 568–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Perry HM et al. (2018) Dynamin-Related Protein 1 Deficiency Promotes Recovery from AKI. J Am Soc Nephrol 29 (1), 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brooks C et al. (2009) Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest 119 (5), 1275–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gall JM et al. (2015) Conditional knockout of proximal tubule mitofusin 2 accelerates recovery and improves survival after renal ischemia. J Am Soc Nephrol 26 (5), 1092–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang Y et al. (2021) The PINK1/PARK2/optineurin pathway of mitophagy is activated for protection in septic acute kidney injury. Redox Biol 38, 101767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang Y et al. (2018) PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis 9 (11), 1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li S et al. (2020) Drp1-regulated PARK2-dependent mitophagy protects against renal fibrosis in unilateral ureteral obstruction. Free Radic Biol Med 152, 632–649. [DOI] [PubMed] [Google Scholar]
- 84.Luciani A et al. (2020) Impaired mitophagy links mitochondrial disease to epithelial stress in methylmalonyl-CoA mutase deficiency. Nat Commun 11 (1), 970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fu ZJ et al. (2020) HIF-1α-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol 36, 101671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang H et al. (2008) Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 283 (16), 10892–903. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 87.Xu D et al. (2019) NIX-mediated mitophagy protects against proteinuria-induced tubular cell apoptosis and renal injury. Am J Physiol Renal Physiol 316 (2), F382–f395. [DOI] [PubMed] [Google Scholar]
- 88.Geng G et al. (2021) Receptor-mediated mitophagy regulates EPO production and protects against renal anemia. Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wei Y et al. (2017) Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 168 (1–2), 224–238.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu Y et al. (2019) Prohibitin 2-mediated mitophagy attenuates renal tubular epithelial cells injury by regulating mitochondrial dysfunction and NLRP3 inflammasome activation. Am J Physiol Renal Physiol 316 (2), F396–f407. [DOI] [PubMed] [Google Scholar]
- 91.Sun J et al. (2019) CoQ10 ameliorates mitochondrial dysfunction in diabetic nephropathy through mitophagy. J Endocrinol. [DOI] [PubMed] [Google Scholar]
- 92.Maekawa H et al. (2019) Mitochondrial Damage Causes Inflammation via cGAS-STING Signaling in Acute Kidney Injury. Cell Rep 29 (5), 1261–1273.e6. [DOI] [PubMed] [Google Scholar]
- 93.Wu J et al. (2021) The key role of NLRP3 and STING in APOL1-associated podocytopathy. J Clin Invest 131 (20). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu J et al. (2021) APOL1 risk variants in individuals of African genetic ancestry drive endothelial cell defects that exacerbate sepsis. Immunity 54 (11), 2632–2649.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Perng YC and Lenschow DJ (2018) ISG15 in antiviral immunity and beyond. Nat Rev Microbiol 16 (7), 423–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li SX et al. (2016) Tetherin/BST-2 promotes dendritic cell activation and function during acute retrovirus infection. Sci Rep 6, 20425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Urata S et al. (2018) BST-2 controls T cell proliferation and exhaustion by shaping the early distribution of a persistent viral infection. PLoS Pathog 14 (7), e1007172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sanz AB et al. (2010) NF-kappaB in renal inflammation. J Am Soc Nephrol 21 (8), 1254–62. [DOI] [PubMed] [Google Scholar]
- 99.Zhong Z et al. (2018) New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560 (7717), 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Komada T et al. (2018) Macrophage Uptake of Necrotic Cell DNA Activates the AIM2 Inflammasome to Regulate a Proinflammatory Phenotype in CKD. J Am Soc Nephrol 29 (4), 1165–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chung H et al. (2021) AIM2 Suppresses Inflammation and Epithelial Cell Proliferation during Glomerulonephritis. J Immunol 207 (11), 2799–2812. [DOI] [PubMed] [Google Scholar]
- 102.Zhou Z et al. (2020) RIG-I aggravates interstitial fibrosis via c-Myc-mediated fibroblast activation in UUO mice. J Mol Med (Berl) 98 (4), 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang Q et al. (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464 (7285), 104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Naito Y et al. (2020) IL-17A activated by Toll-like receptor 9 contributes to the development of septic acute kidney injury. Am J Physiol Renal Physiol 318 (1), F238–f247. [DOI] [PubMed] [Google Scholar]
- 105.Han SJ et al. (2018) Kidney Proximal Tubular TLR9 Exacerbates Ischemic Acute Kidney Injury. J Immunol 201 (3), 1073–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Daemen MA et al. (1999) Inhibition of apoptosis induced by ischemia-reperfusion prevents inflammation. J Clin Invest 104 (5), 541–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Linkermann A et al. (2012) Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int 81 (8), 751–61. [DOI] [PubMed] [Google Scholar]
- 108.Herzog C et al. (2012) zVAD-fmk prevents cisplatin-induced cleavage of autophagy proteins but impairs autophagic flux and worsens renal function. Am J Physiol Renal Physiol 303 (8), F1239–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yang B et al. (2018) Caspase-3 Is a Pivotal Regulator of Microvascular Rarefaction and Renal Fibrosis after Ischemia-Reperfusion Injury. J Am Soc Nephrol 29 (7), 1900–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wei Q et al. (2013) Bax and Bak have critical roles in ischemic acute kidney injury in global and proximal tubule-specific knockout mouse models. Kidney Int 84 (1), 138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Doke T et al. (2021) Genome-wide association studies identify the role of caspase-9 in kidney disease. Sci Adv 7 (45), eabi8051. [DOI] [PMC free article] [PubMed] [Google Scholar]