

Abstract
For decades, mitochondrial dysfunctions and the generation of reactive oxygen species have been proposed to promote the development and progression of the amyloid pathology in Alzheimer’s disease, but this association is still debated. It is unclear whether different mitochondrial dysfunctions, such as oxidative phosphorylation deficiency and oxidative stress, are triggers or rather consequences of the formation of amyloid aggregates. Likewise, the role of the different mitochondrial oxidative phosphorylation complexes in Alzheimer’s patients’ brain remains poorly understood.
Previous studies showed that genetic ablation of oxidative phosphorylation enzymes from early age decreased amyloid pathology, which were unexpected results. To better model oxidative phosphorylation defects in aging, we induced the ablation of mitochondrial Complex III (CIIIKO) in forebrain neurons of adult mice with amyloid pathology. We found that mitochondrial Complex III dysfunction in adult neurons induced mild oxidative stress but did not increase amyloid beta accumulation. On the contrary, CIIIKO-AD mice showed decreased plaque number, decreased Aβ42 toxic fragment and altered amyloid precursor protein clearance pathway. Our results support the hypothesis that mitochondrial dysfunctions alone, caused by oxidative phosphorylation deficiency, is not the cause of amyloid accumulation.
Keywords: Mitochondria dysfunctions, oxidative phosphorylation deficiency, Alzheimer’s disease, mouse model, Oxidative stress
2. Background
Alzheimer’s disease (AD) is the most common cause of dementia and decline in memory and cognition in the aging population. It is an age-related neurodegenerative disease characterized by the presence of extracellular amyloid plaques in cortex and hippocampus, mainly composed of amyloid beta (Aβ) fragment, and neurofibrillary tangles, caused by intracellular accumulation of hyperphosphorylated Tau protein [1]. Mitochondrial dysfunctions have been implicated in the development and progression of many neurodegenerative diseases, including AD, and a “mitochondrial hypothesis” has been proposed in which changes in mitochondrial function initiate the cascade that leads to the pathological hallmarks characteristic of AD, in particular to the formation of amyloid plaques [2]. The most relevant alterations include oxidative phosphorylation (OXPHOS) defects, elevated oxidative stress, perturbations in mitochondrial dynamics and mitophagy, alterations in mitochondrial transport, increased mitochondrial DNA (mtDNA) mutations, and defective calcium homeostasis [3]. Aging, the primary risk factor in AD [4], is also associated with decline of mitochondrial function in the central nervous system [5–9]. Accumulated mutations in mtDNA, which encodes proteins for several OXPHOS complexes, have been found in post mortem brains of AD patients [10–12] which also show decreased gene expression of all mtDNA-encoded genes and of the master mitochondrial biogenesis regulator, PGC-1α [13]. Early studies that report defects in OXPHOS in AD patients [14–17] describe, in particular, deficiency of cytochrome c oxidase (Complex IV of the electron transport chain), at least partially responsible for increase in reactive oxygen species (ROS) production, decrease in energy stores, and disruption of energy metabolism [18, 19].
Scarcer are data on the other complexes and in particular of Complex III. Decreased Complex III core protein 1 was observed in the temporal cortex of patients with AD [20], mitochondrial preparations of frontal cortex of AD patients show deficiencies of Complex II, III and IV but no differences in respiratory supercomplexes [21]. Other reports show how Complex III activity and Complex III/ Citrate Synthase (CIII/CS) ratio are higher in AD patients’ platelets [22].
Despite years of study, the role of the different mitochondrial OXPHOS complexes in the brain of AD patients remains poorly understood, especially regarding amyloid pathology. In particular, it is still unclear if different mitochondrial dysfunctions are a trigger or a consequence of plaque formation: ROS can affect amyloid pathology by enhancing beta-secretase and gamma-secretase activity and exacerbating amyloid beta fragment (Aβ) production and aggregation [23–26], but amyloid pathology, on the other side, can also negatively affect mitochondrial function. Moreover, Aβ directly associates with mitochondria, inhibits OXPHOS [15, 27–33], and can alter fission and fusion processes [34].
We previously reported that severe isolated Complex IV deficiency led to a reduction in plaque formation and Aβ steady-state levels in the APP/PS1 mouse model [35] and that depletion of mtDNA also led to a decrease in plaques in the same model [36].
These findings were surprising and raised the question that severe OXPHOS defects may not reflect age related OXPHOS decay, which are mild. To better model a timed OXPHOS defect, we crossed APP/PS1 transgenic mice with a RISP neuron specific conditional KO controlled by tamoxifen induction. We induced the Complex III dysfunction in 5 months old animals, and we analyzed the consequences on amyloid pathology after 3 months.
3. Methods
3.1. Mouse Procedures
The APP/PS1 transgenic mice carry a mutant APPSwe and a mutant PSEN1dE9 allele (The Jackson Laboratory, Stock #34832) [37], CaMKIIα-CreERT2 transgenic mice express a tamoxifen-inducible Cre-recombinase under the control of the mouse calcium/calmodulin-dependent protein kinase II alpha (CaMKIIα) promoter region [38] (The Jackson Laboratory, Stock #012362). Mice were crossed with RISPFF mice where exon 2 of the RISP gene is flanked by two loxP sites [39]. All animals used in this work had a C57Bl/6J background, backcrossed for 10 generations. Tamoxifen was prepared by first dissolving the powder in ethanol (20 mg per 100 μl) and mixing this solution with 900 μl corn oil for a final concentration of 20 mg/ml. 5-month-old AD-RISPKO mice (RISPFF- APP/PS1+/−-CaMKIIαCreERT2+/−) and control mice (RISPFF- APP/PS1+/−) were injected IP with 130 mg per kilogram body weight of tamoxifen once a day for 5 consecutive days. Mice were housed in a virus-antigen-free facility of the University of Miami, Division of Veterinary Resources in a 12-h light/dark cycle at room temperature and fed ad libitum. Sex differences in APP/PS1 mice are well known, with females accumulating amyloid at an earlier age than males and building up more amyloid deposits [40]. These differences are not limited to the amyloid pathology [41–44]. Therefore, for this study, we analyze only females. Control group indicated as “WT” is composed by 8 months old C57Bl/6J females.
3.2. Quantitative PCR
Total DNA was extracted from cortex using standard phenol/chloroform extraction. Quantitative PCR reactions done using SYBR chemistry (SsoAdvanced Universal Master Mix SYBR Green, Bio-Rad) were performed on a Bio-Rad CFX96/C1000 qPCR machine. The comparative ddCt method was used to determine the relative levels of undeleted and deleted Uqcrfs1 to a control genomic region [45]. To estimate the levels of “undeleted Uqcrfs1” we designed primers to amplify a region in Exon 2 of levels Uqcrfs1 (F:AACCAAGATGAGTACAGACA, R:AGAACCAAGAAGGAGATTGA); to estimate the levels of “deleted Uqcrfs1” we designed primers in the intron sequences flanking Exon 2 of Uqcrfs1 (F:TCATCCGAGACCCAGCAA, R:AGCACATAGCAGAGATACAA). Primers for beta-Actin were (F:GCGCAAGTACTCTGTGTGGA, R:CATCGTACTCCTGCTTGCTG).
3.3. Western Blots
Mice were perfused with phosphate-buffered saline, brains were isolated, and cortex was dissected and homogenized in PBS supplemented with cOmplete™ Protease Inhibitor Mixture (Roche) and Phosphatase inhibitor PhosSTOP™ (Roche) [12]. Protein concentrations were determined using the DC kit (Bio-Rad Laboratories). The homogenates were resolved on SDS-PAGE gels, transferred onto a polyvinylidene difluoride (PVDF) membrane, and hybridized with the antibodies raised against total OXPHOS rodent Cocktail, NDUFB8, NDUFA9, SDHA, UQCRFS1/RISP, VDAC1/Porin, CytC, COX1, ATP5a, UQCRC1/Core1, p62 (Abcam); GFAP, Synaptophysin, Nicastrin, Presenilin 1, LC3B, CHOP, Neurogranin (Cell Signaling); TUJ1 (Chemicon); Iba1 (Wako-016–20001); SOD2 (Upstate); APP C-Terminal Fragment, and Purified anti-β-Amyloid 1–16 Antibody clone 6E10 (Biolegend); 20s proteasome (Novus Biol). Bands were normalized for total protein loading (visualized by stain free technology, in the Chemidoc system, Biorad). As the same blots were used for hybridization of multiple antibodies, the following figures used the same loading control: Core1, VDAC1, Iba1 and p62; APP-fl, APP-Ct, and CHOP; synaptophysin and Rieske; 20s proteasome and SOD2; 6E10 and LC3b.
3.4. Enzymatic Activity Assays
Complex IV and citrate synthase activities were measured in tissue homogenates by spectrophotometric methods [46]. Briefly, homogenates from cortex were prepared using a hand-held rotor (VWR) to homogenize tissue in PBS plus Complete Protease Inhibitor Mixture (Roche) on ice. Cytochrome c (2mM) reduced with sodium dithionite was added to homogenates in 10mM KH2PO4, 1mg/ml BSA, and 120mM lauryl maltoside. Samples were read at 550–580nm with the slope reading taken for 2’ at 37°C. Potassium cyanide (240mM) was used to inhibit the reaction to ensure slope was specific to COX activity. Readings were normalized by protein concentration. Homogenates for citrate synthase were added to 50mM Tris- HCl, pH7.5, 20mM acetyl CoA, 10mM 5,5’-dithiobis (2-nitrobenzoic acid), and 0.2% Triton X-100 and performed at 412nm with 50mM oxaloacetate to start the reaction. Readings were obtained for 5’ at 30°C. Normalization was again to protein concentration.
3.5. Beta-Secretase Activity Assay
Beta-secretase activity was measured in cortical lysates using a FRET-based substrate, H-RE(EDANS)EVNLDAEFK(DABCYL)R-OH (Calbiochem), which contains a beta-secretase cleavage sequence with Swedish-type mutations. Homogenates lysed in PBS- 1% Triton-X100 and Complete Protease Inhibitor Mixture (Roche) were centrifuged at 10000g for 5’ at 4°C, and the supernatant was used for the analysis. The activity assay was conducted on 4μg of protein in a 100μl reaction volume that contained 12.5μM substrate and 20mM sodium acetate (pH4.4). The fluorescence from the released EDANS fluorophore was measured at 37°C with excitation and emission wavelengths of 355nm and 460nm respectively, using a Synergy H1 hybrid reader (Biotek). Background fluorescence from the substrate alone was subtracted from the readings. Fluorescence (arbitrary units) was recorded 30’ after the initiation of reaction.
3.6. Quantification of Aβ42 fragment
Cortices were homogenized in cold guanidine buffer (5M guanidine HCl, 50mM Tris-HCl pH8.0). Total Aβ42 content was quantified using a human Aβ42 colorimetric ELISA kit (Invitrogen) according to the manufacturer’s instruction with an incubation time of 16h at 4°C.
3.7. Immunostaining and Stereological Quantification of Amyloid Plaques
Anesthetized mice were perfused with ice-cold PBS, and 4% PFA. Brains were isolated, cut in half (coronal plane), further fixed in 4% PFA at 4°C ON, and cryoprotected by immersion in 30% sucrose solution for at least 24h (until they sink). The brains were then mounted with OCT and frozen in isopentane-liquid nitrogen. 20μm thick coronal sections were cut by cryostat (rostral to caudal direction) starting approximately at bregma −1.46.
Sections were incubated in 70% formic acid for 20’ and in 100% methanol + 0.3% H2O2 for 30’, blocked with 10% normal goat serum (NGS) for 30’ at RT and incubated with monoclonal anti-Aβ (1:500, 6E10 antibody, Chemicon) for 16h at 4°C. Sections were then incubated with secondary Antibody biotin-conjugated goat anti-mouse (KPL) for 1h at RT and Streptavidin-Peroxidase (KPL) for 30’ at RT. Staining was visualized using a solution of 0.05% 3,3’-diaminobenzidine (DAB), 50mM Tris-HCl pH7.2, 0.02% H2O2. Slides were then mounted with SurgiPath Sub-X mounting media (Leica). Images were captured with a Pathscan Enabler 5. As previously described [35] the total number of amyloid plaques per tissue area were counted from ten non-consecutive stained sections per animal (n=3–4 per group) scanning the whole area. Counting was performed independently by two blinded investigators. Fiji program was used for counting. Antibodies used for morphology analysis were: monoclonal anti-NeuN (1:500 Millipore), monoclonal anti-GFAP (1:500 Cell Signaling).
3.8. Nissl staining:
PFA-fixed frozen sections were dried for 3 hours, then incubated in 1:1 ethanol/chloroform overnight for de-fattening. Slides were then rehydrated through series of ethanol solutions, from 100% ethanol to distilled water. Staining was performed in warm (37°C) 0.1% cresyl violet solution (Abcam) for 5–10 minutes. Slides were then rinsed in running distilled water, dehydrated through series of ethanol solutions, from water to 100% ethanol, cleared in xylene, and mounted with a permanent mounting medium. Images were captured with an Pathscan Enabler 5.
3.9. Immunostaining of Oxidized Nucleic Acids
Histological sections were obtained as described above. Sections were microwave-heated twice for 4’ in 10mM sodium citrate buffer (pH6.0). Sections were then permeabilized in 0.2% Triton X-100 for 30’ and endogenous peroxidase activity was quenched by immersing section in 100% methanol containing 0.3% H2O2 for 30’. After blocking with 10% NGS for 1h at RT, sections were incubated with monoclonal antibody against 8-hydroxy-2’-deoxyguanosine (oh8dG) and 8-hydroxyguanosine (oh8G) (1:1000, QED Bioscience) for 16h at 4°C in 5% NGS. Sections were then incubated with secondary Antibody biotin-conjugated goat anti-mouse (KPL) for 1h at RT and Streptavidin-Peroxidase (KPL) for 30’ at RT. Staining was visualized using a solution of 0.05% 3,3’-diaminobenzidine (DAB), 50mM Tris-HCl pH7.2, 0.02% H2O2. For a negative control, sections were pre-incubated with 10μg/μl RNase (Qiagen) and 0.04U/μl DNase (Promega) in 50μl of 1ŔQ buffer (Promega) for 3 hours at 37°C prior to the primary antibody incubation. For a positive control, sections were pretreated with 30% H2O2 for 10 minutes before blocking with NGS. Counting was performed independently by two blinded investigators on 10 randomly chosen cortical regions per mouse. Fiji program was used for counting.
3.10. OxyBlot
Cortices were homogenized in cold PBS + Complete Protease Inhibitor Mixture (Roche). OxyBlot measures carbonyl groups introduced into proteins by oxidative reactions with ozone or oxides of nitrogen or by metal catalyzed oxidation. Carbonyl groups were quantified on 15mg of protein homogenates using S7150 Sigma-Aldrich OxyBlot Protein Oxidation Detection Kit.
3.11. Statistical Analysis
One-way ANOVA followed by Bonferroni post-test was used to evaluate the significance of the differences between three groups (WT, AD, CIIIKO-AD). Two-tailed, unpaired Student t test was used to determine the statistical significance between two different groups (AD, CIIIKO-AD). *** p<0.001, ** p< 0.01, *p< 0.05. Error bars represent standard error of the mean (SEM).
4. Results
4.1. Generation of CIIIKO-AD mice
To analyze the effects of an adult-onset Complex III defect on amyloid pathology, we developed and characterized a transgenic mouse carrying mutant APP and mutant presenilin 1 (APP/PS1) [37] with adult-onset Rieske Iron Sulfur Protein (RISP) defects. RISP is encoded by the Uqcrfs1 gene and is a catalytic subunit of mitochondrial OXPHOS Complex III. Because APP/PS1 mice develop amyloid deposits starting at 6–7 months of age [47], we wanted to induce a Complex III defect in adult mice. Therefore we crossed RISPFF mice (where exon 2 of the RISP gene is flanked by two loxP sites) [39] with a CaMKIIα-CreERT2 transgenic mouse, in which the expression of the Cre-recombinase is tamoxifen-inducible [38] (Fig. 1B). The experimental animals’ (CIIIKO-AD) genotype was RISPF/F-APP/PS1+/−-CaMKIIα-CreERT2+/−, whereas the control animals’ (AD) genotype was RISPF/F-APP/PS1+/− (Fig. 1A). CaMKIIα is expressed also in male germ cells [48], so in order to obtain the experimental animals, only females CaMKIIα-CreERT2+/− were used in the breeding scheme (Fig. 1A). We injected 5-month-old mice with tamoxifen, and we analyzed the amyloid pathology 3 months after inducing the Uqcrfs1 gene deletion, when the mice were 8 months old (5+3) (Fig. 1C).
Figure1: Generation of CIIIKO-AD mice.
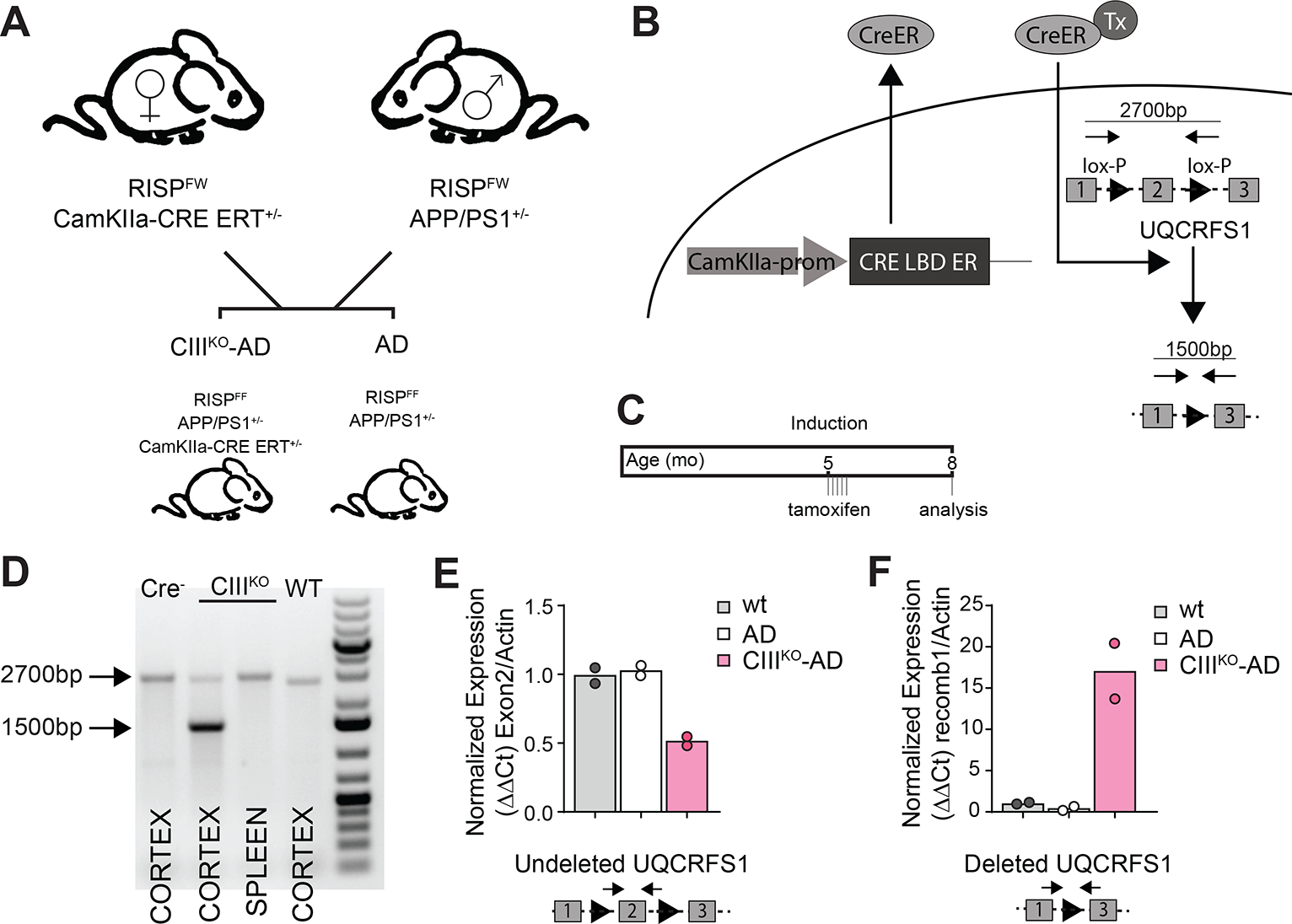
(A) breeding scheme to obtain CIIIKO-AD animals. (B) Cartoon depicting the induction of Cre-recombinase and the consequent excision of exon 2 of Uqcrfs1 gene encoding for RISP. (C) Cartoon depicting the induction schedule with tamoxifen injections. (D) PCR products showing the tissue-specific excision of exon 2 of Uqcrfs1 gene after tamoxifen induction. (E) qPCR amplifying exon 2 of Uqcrfs1 gene, showing the decrease of the WT allele in the genomic DNA from CIIIKO-AD animals’ cortex. (F) qPCR amplifying recombined Uqcrfs1 gene, showing the recombination of the excised gene in the genomic DNA from CIIIKO-AD animals’ cortex.
To verify that Cre-recombinase excised exon 2 of the Uqcrfs1 gene specifically in brain of CIIIKO-AD mice after tamoxifen induction, we compared DNA extracted from cortex and spleen of CIIIKO-AD and AD mice injected with tamoxifen. We could detect the gene deletion in cortex of CIIIKO-AD mice, but neither in spleen of CIIIKO-AD mice nor in cortex of control mice (Fig. 1D), indicating the specificity of the system. We estimated the levels of exon 2 excision in cortex by qPCR and found that ~50% of the Uqcrfs1 gene was deleted (Fig. 1E). Considering that the samples used to extract DNA contained not only CaMKIIα+ neurons but also glia and CaMKIIα− neurons, the excision of the Uqcrfs1 gene in the target neurons was actually higher than the 50% detected by this method.
4.2. Effects of ablation of RISP on OXPHOS complexes
To verify the levels of RISP protein in CaMKIIα+ neurons, we performed a western blot assay on cortical homogenates of WT, AD, and CIIIKO-AD mice (Fig. 2A). When quantifying a mitochondrial protein, it is critical that the amounts of the protein of interest is normalized to the amounts of mitochondria. This is important because the levels of mitochondria can change dramatically, particularly in tissues with oxidative phosphorylation defects. To analyze the levels of RISP in relation to the levels of mitochondria, we normalized it to mitochondrial membrane protein VDAC1. The level of RISP was reduced to ~39.2% compared to AD and to ~34.2% compared to WT animals (Fig. 2C). We also normalized RISP to steady-state level of UQCRC1/Core 1 (another Complex III subunit located in a different module of the fully assembled Complex III) and calculated that RISP was reduced to about ~39.2% compared to AD and to ~33.1% compared to WT animals (Fig. 2D). To determine whether the decrease of the Complex III subunits affected the other OXPHOS complexes, we analyzed the steady-state levels of representative proteins from each complex (Fig. 2F, 2G). We analyzed NDUFS3 and NDUFB8 (for Complex I), SDHA and SDHB (for Complex II), COXI and CYTC (for Complex IV), and ATP5a (for ATPase). When normalized to mitochondrial membrane protein VDAC1, no proteins belonging to the other complexes were significantly affected compared to AD mice (Fig. 2G). We measured the steady-state levels of non-OXPHOS mitochondrial membrane proteins, VDAC1 and TIM23. VDAC1 and TIM23 were unchanged in CIIIKO-AD compared to AD control. TIM23 was slightly decreased compared to WT animals (Fig. 2E).
Figure2: RISP and mitochondrial proteins expression in Cortex.
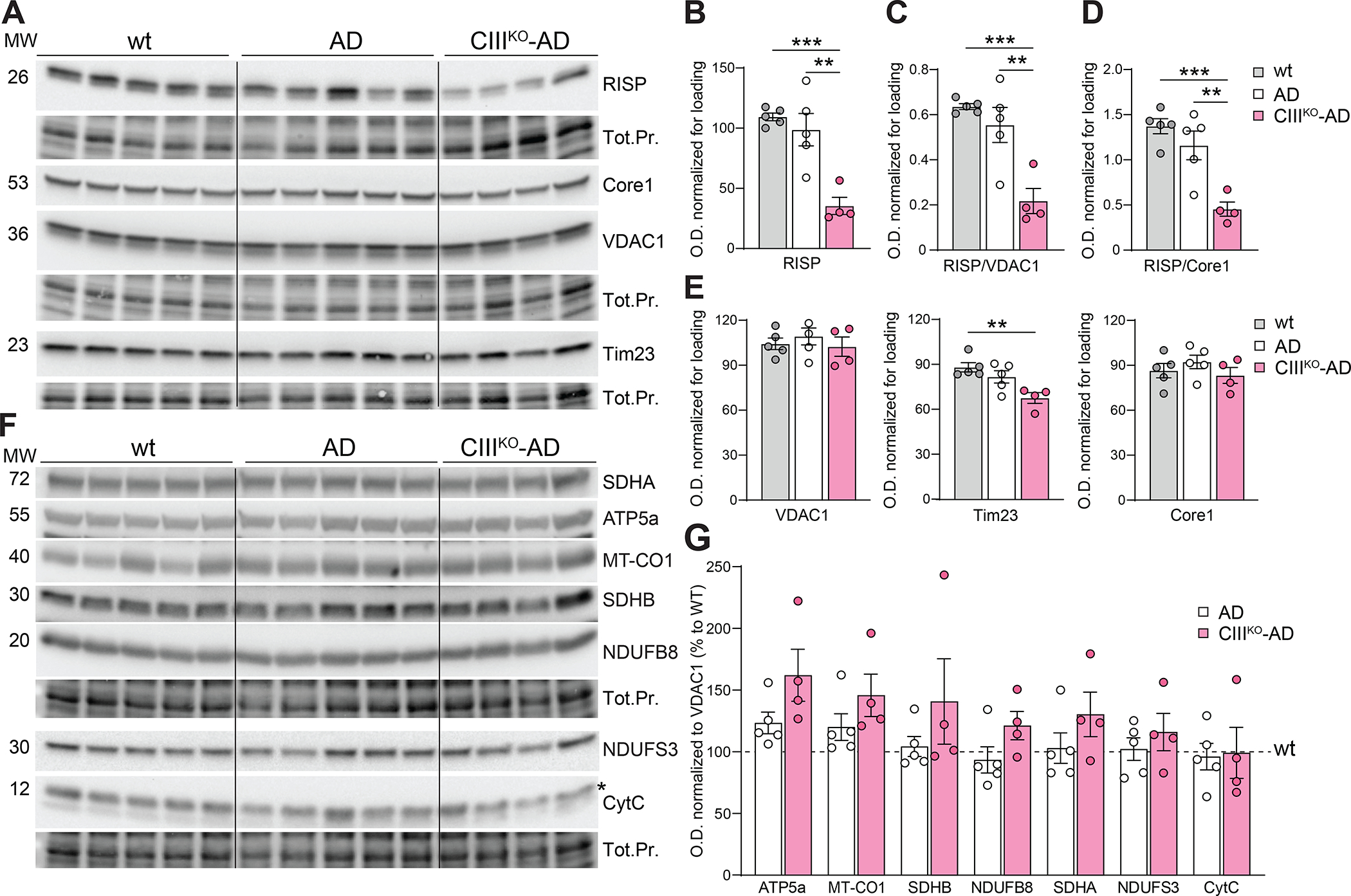
(A) Western blots of homogenates from cortex of 8-month-old females WT, AD, and CIIIKO-AD probing for RISP, Core1, VDAC1 and Tim23 and relative loading controls (Total Protein). (B-D) Quantification of steady state level of RISP normalized to total protein (Tot.Pr.) (B), to VDAC1 (C) and to Core1 (D) in cortex of 8-month-old females. (E) Quantification of steady state level of VDAC1, Tim23 and Core1 normalized to total protein. (F) Western blots of homogenates from cortex of 8-month-old females WT, AD and CIIIKO-AD probing for mitochondrial proteins NDUFS3 and NDUFB8 (Complex I), SDHA (Complex II), Cyt C and COXI (Complex IV), ATP5A (Complex V) and relative loading controls (Total Protein). (G) Relative quantifications of mitochondrial proteins normalized to VDAC1, expressed as a percentage of control WT animals. Bars represent means ±SEM of n=4–5 for each group, each point is represented by a circle.
To determine if the induced decrease of RISP was enough to cause a defect in Complex III activity, we measured OXPHOS complex enzymatic activities in cortical homogenates. Complex III activity normalized to citrate synthase (CS) was reduced to 65% (± 7) compared to control (Fig. 3A). These homogenates also contain glial and endothelial cells, in which the Uqcrfs1 gene has not been knocked out. Because of this, we have to consider an underestimation of the actual decrease in Complex III activity in affected neurons. Complex IV and citrate synthase activities were not changed in cortical homogenates (Fig. 3B, 3C).
Figure3: Induced Complex III deficiency.
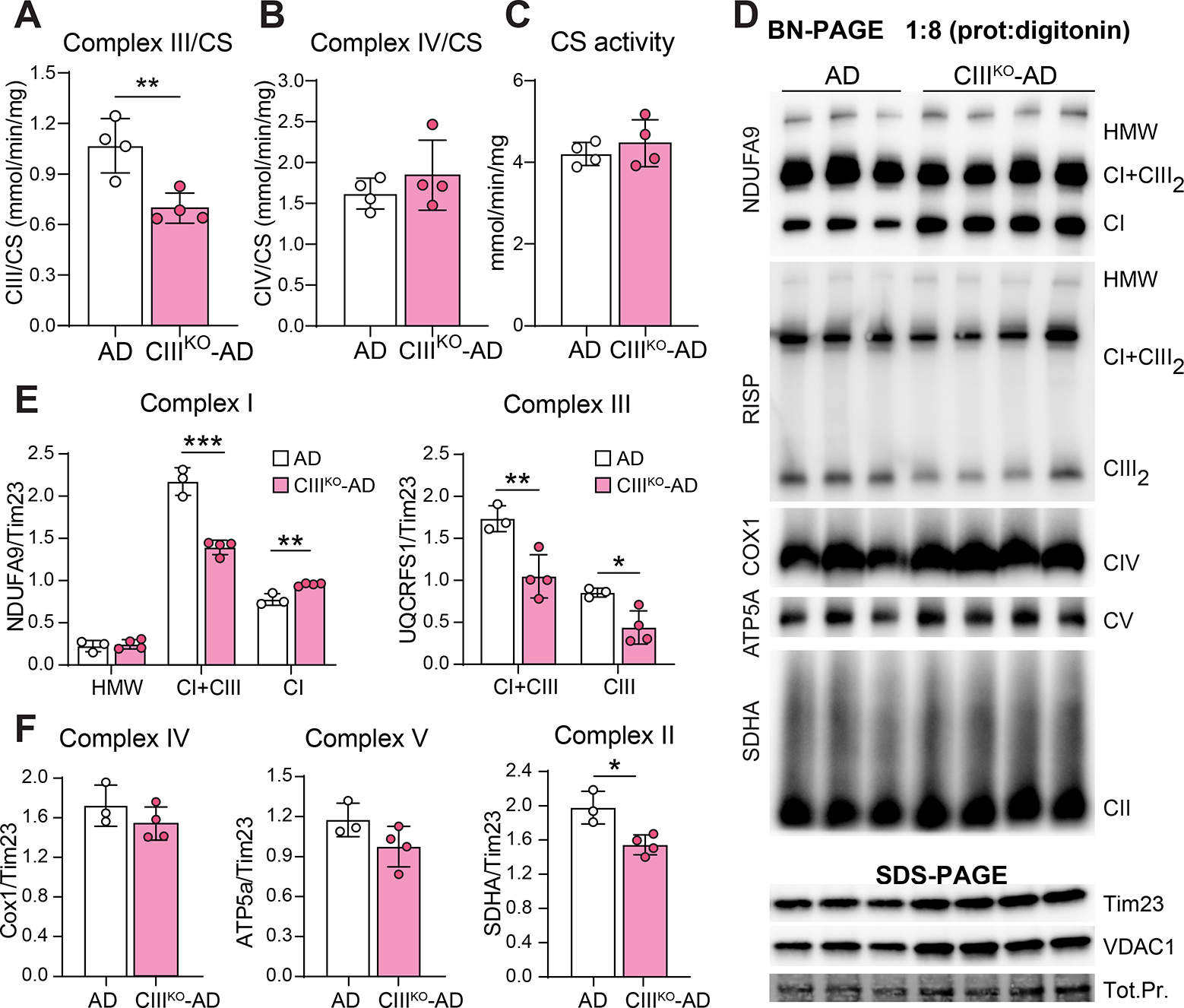
(A-C) Spectrophotometric Complex III/Citrate Synthase (CIII/CS) activity ratio (A), Complex IV/Citrate Synthase (CIV/CS) activity ratio (B), and Citrate Synthase (CS) activity measured in cortical homogenates of 8-month-old AD and CIIIKO-AD females. (D) Steady state levels of complexes and supercomplexes measured by BN-PAGE in homogenates from cortex of 8-month-old AD and CIIIKO-AD females. The antibodies used for the different complexes were: NDUFA9 (Complex I), RISP (Complex III), COX1 (Complex IV), ATP5a (Complex V), and SDHA (Complex II). Mitochondrial content was measured by western blot of the same homogenates using antibody against Tim23 and VDAC1. (E-F) Quantification of blue natives from cortical homogenates normalized for mitochondrial content (Tim23 normalized to total protein (Tot.Pr.)). Bars represent means ±SEM of n=3–4 for each group, each point is represented by a circle.
In order to analyze if the loss of RISP caused a reorganization of the respiratory complexes, mitochondrial proteins were extracted with digitonin and separated by blue native gel electrophoresis followed by western blot (Fig. 3D). Complex I, III and IV were analyzed using anti NDUFA9, RISP and COX1 antibodies, respectively. TIM23 and VDAC1 were used as mitochondrial loading control. Mitochondrial supercomplexes are indicated in the figure as HMW (High Molecular Weight) and CI+CIII2. Complex V and II were analyzed using ATP5a and SDHA antibodies, respectively. We detected a decrease of CI+CIII2 architectures in cortex of CIIIKO-AD mice, and an increase of free Complex I (Fig. 3E). Assembled Complexes IV, V and II did not show changes (Fig. 3F).
4.3. Neurodegeneration and neuroinflammation in CIIIKO-AD mice
The brains of 8-month-old CIIIKO-AD mice did not show significant differences in size or weight compared to AD controls. To detect if the defect in Complex III affected the neuroanatomy of the brain, we first analyzed the gross morphology of different brain regions by hematoxylin & eosin staining and we did not detect any gross nor massive anatomical alteration (Fig. 4A). We then performed Nissl staining (which is commonly used to stain nucleic acids in the nervous tissue) and immunohistochemistry staining for NeuN (marker of neuronal nuclei) on PFA-fixed frozen sections. We did not detect gross changes in the neuronal population in different areas analyzed (Fig. 4B–C). We then performed western blots analysis to measure the levels of neuronal marker TUJ1 on cortical homogenates of WT animals, AD, and CIIIKO-AD. We detected a decrease in TUJ1 in cortex from CIIIKO-AD females compared to both controls (Fig. 4D). We further measured the levels of Synaptophysin, which participates in different steps of synaptic biogenesis, vesicle protein sorting, vesicle priming, synapse formation, and exo- and endocytosis and is considered a marker for axonal damage. We did not detect significant changes in cortical homogenates (Fig. 4D). We also measured the levels of Neurogranin, a small synaptic protein whose expression is regulated by synaptic activity [49, 50]. We did not find significant changes (Fig. 4D), suggesting that the neuronal activity was not changed by the Complex III depletion.
Figure4: Neuronal content and Neuroinflammation:
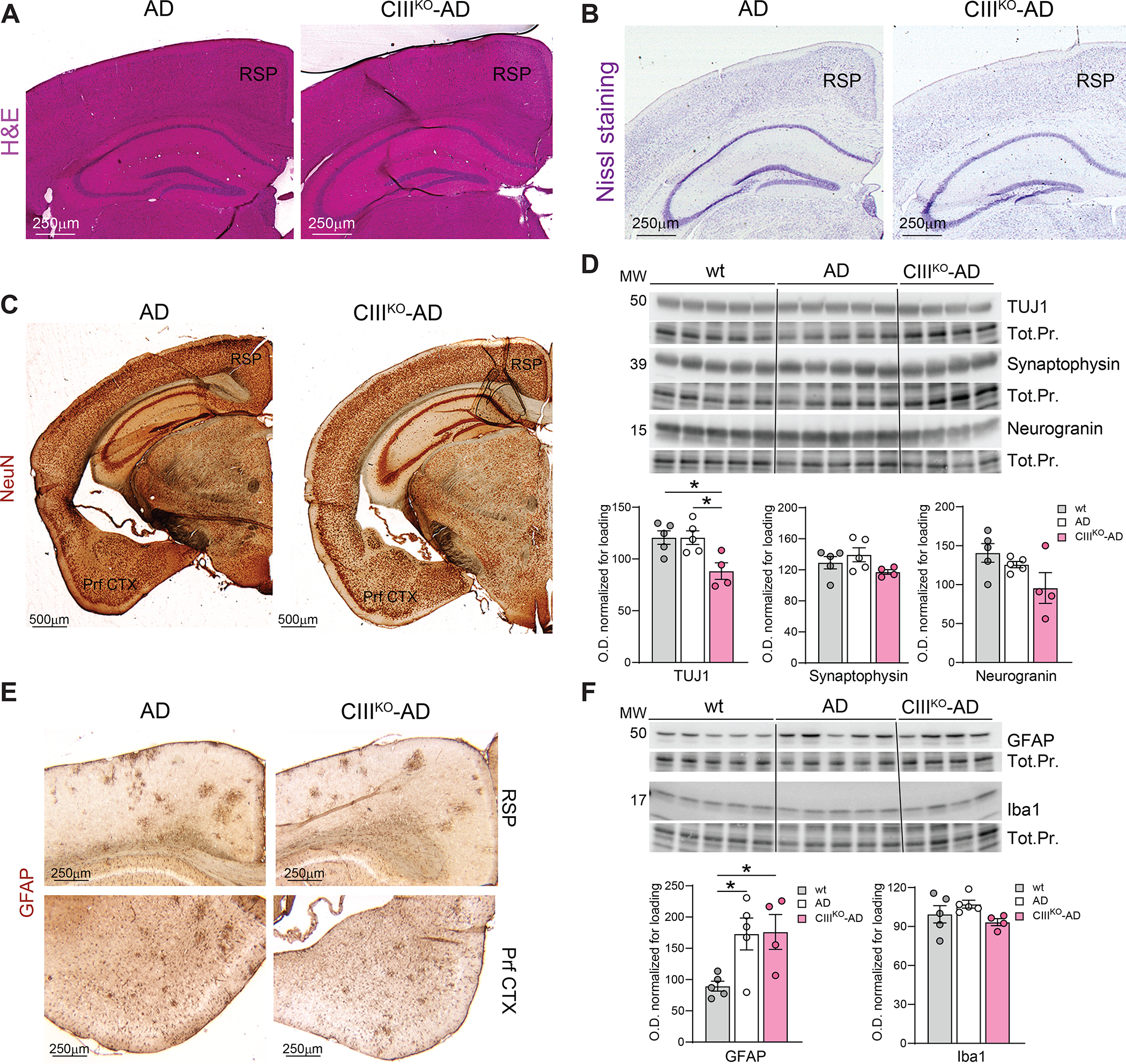
(A) Representative images of H&E staining on histological sections from AD and CIIIKO-AD mice. (B) Representative images of Nissl staining on histological sections from AD and CIIIKO-AD mice. (C) Representative images of IHC probing for neuronal marker NeuN staining on coronal sections of 8-month-old AD and CIIIKO-AD females. (D) Western blot probing for Class III β-Tubulin (TUJ1), Synaptophysin, and Neurogranin on cortical homogenates from 8-month-old WT, AD, and CIIIKO-AD females (n=4–5/group) and relative quantifications normalized to loading. (E) Representative images of IHC probing for glial marker GFAP on cortex of 8-month-old AD and CIIIKO-AD females. (F) Western blot probing for glial marker GFAP and microglial marker Iba1 on cortical homogenates from 8-month-old WT, AD, and CIIIKO-AD females (n=4–5/group) and relative quantifications normalized to loading. RSP: Retrosplenial area; Prf CTX: Piriform Cortex. Tot.Pr.: Total Protein.
Neurodegeneration and metabolic defects are often accompanied by neuroinflammation, therefore we examined the extent of gliosis in these animals. We performed immunohistochemistry and western blot analysis with antibodies against GFAP (marker of glial cells) and Iba1 (marker of microglia). We did not detect significant changes between CIIIKO-AD and AD controls, neither in the morphology of GFAP and Iba1 positive cells (Fig. 4E), nor in the steady-state levels of both markers (Fig. 4F).
4.4. CIIIKO-AD mice show fewer amyloid plaques and fewer Aβ42 fragment
Starting at 6 months of age, APP/PS1 mice accumulate amyloid plaques, with most of them present in the cerebral cortex [36]. By 8–9 months, the plaque number reaches 1.5–2 plaques per mm2 [35, 36]. To analyze if the partial defect of Complex III affected the formation of the amyloid plaques, we performed a stereological count on 8-month-old AD and CIIIKO-AD mice. We visualized the plaques by immunohistochemistry on serial coronal sections with an anti-human Aβ antibody (6E10) that recognizes the amyloidogenic portion of human APP. Cortical plaques from CIIIKO-AD mice were significantly reduced in number compared to AD brains (Fig. 5A–B).
Figure5: Amyloid plaques and Aβ content.
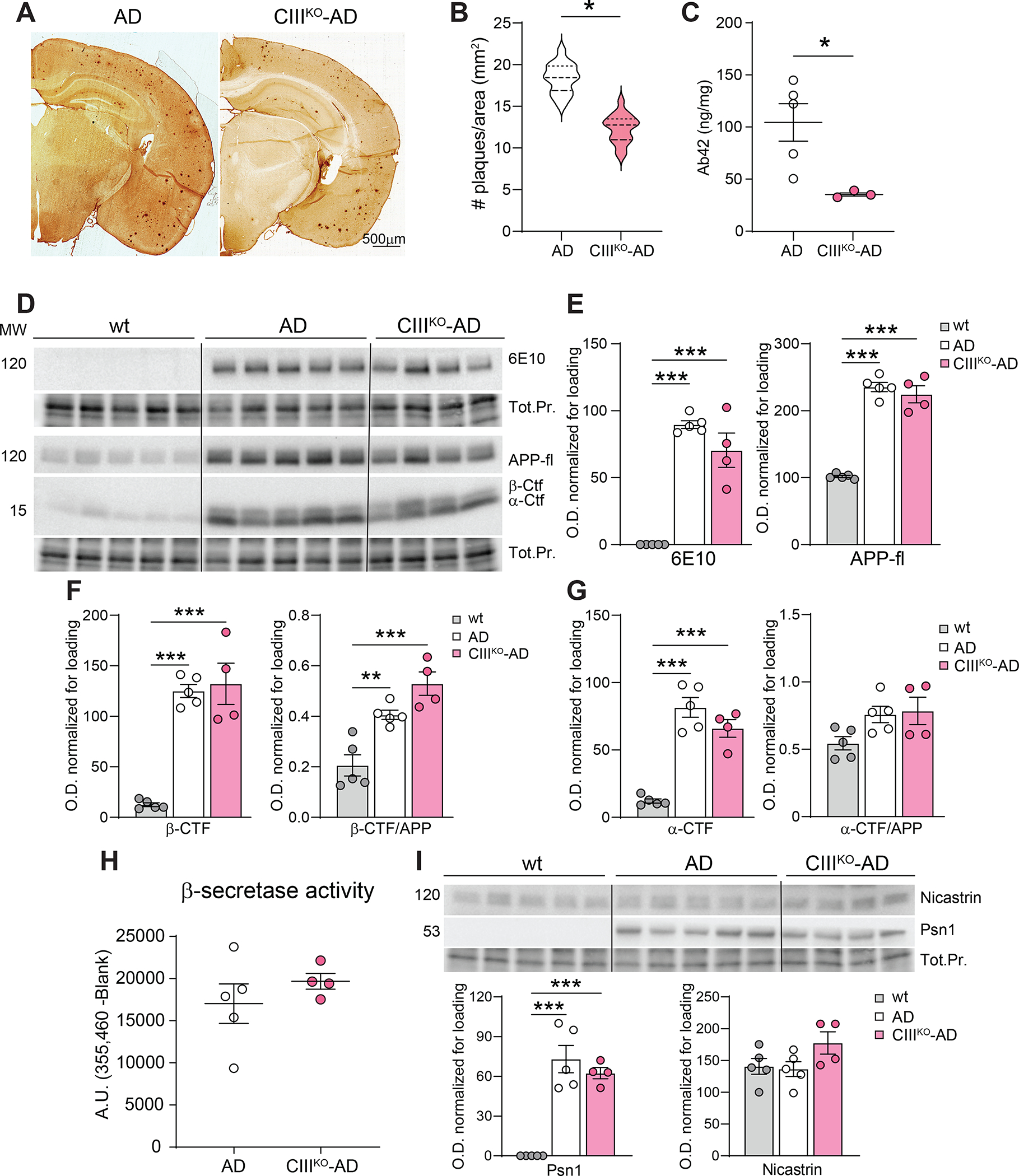
(A) Representative images of coronal sections of AD and CIIIKO-AD females’ brains, immunostained with an anti-Aβ antibody (6E10) to detect amyloid plaques. (B) Violin plot visualizing the quantification of cortical amyloid plaques in AD and CIIIKO-AD females (n=3/group). (C) Content of Aβ42 fragments in cortical and hippocampal homogenates from AD and CIIIKO-AD females (n 3–5/group). (D) Western blot analysis detecting the presence of total APP (6E10, APP-fl) and carboxy-terminal fragments (β-Ctf and α-Ctf) in cortical homogenates from WT, AD, and CIIIKO-AD females. (E) Quantification of optical density of APP-fl with two different antibodies. (F) Quantification of optical density signal from Western blot analysis of β-Ctf normalized to total protein (Tot.Pr.) and normalized to APP. (G) Quantification of optical density signal from Western blot analysis of α-Ctf normalized to total protein (Tot.Pr.) and normalized to APP. (H) Enzymatic activity of beta-secretase in cortical homogenates of AD and CIIIKO-AD females (n=4–5/group). (I) Western blot analysis and relative quantification probing for Nicastrin and Presenilin 1 (Psn1) in cortical homogenates from WT, AD, and CIIIKO-AD females (n=4–5/group).
The plaque number is correlated with Aβ42 content and to secretase activity [51, 52]. To determine whether the diminished number of plaques in CIIIKO-AD mice correlated with the amount of Aβ42 peptide, we performed an ELISA on protein homogenates from cortex of AD and CIIIKO-AD mice. Lysates from CIIIKO-AD mice’s brains showed a reduction of Aβ42 of ~66% (Fig. 5C).
We then analyzed the full-length APP content in the different genotypes by western blot. We used two antibodies, the anti-β-Amyloid 1–16 antibody (6E10 clone) which recognizes the amino acid residue 1–16 of human APP, therefore it is only present in the transgenic mice, and the APP-Ct which recognizes both human and mouse C-terminal fragment of APP, therefore recognizes both the human transgenic APP and the endogenous APP.
Quantifications of both antibodies showed no significant differences between AD and CIIIKO-AD mice (Fig. 5E), meaning that the total APP burden was not affected by the CIII deficiency.
We consequently analyzed APP processing by performing western blot analyses of the different fragments derived from the APP sequential cleavage. Using the APP-Ct antibody we could analyze in the same gel the un-cleaved APP (APP-fl at ~100kd) and the C-terminal fragments (Ctf) formed after the first secretase cleavage. The β-Ctf (~15kd) derives from the beta-secretase cleavage that occurs in the amyloidogenic processing of APP, while the α-Ctf (~12kd) derives from the alpha-secretase cleavage in the non-amyloidogenic processing of APP. We analyzed the steady-state level of β-Ctf and the ratio between β-Ctf /APP (Fig. 5F) and detected no significant changes between AD and CIIIKO-AD mice. No changes were also detected in the levels of α-Ctf and α-Ctf /APP between AD and CIIIKO-AD mice (Fig. 5G). One of the crucial events triggering the formation of amyloid fragments from APP is the cleavage by beta-secretase. We measured beta-secretase activity with a FRET-based assay in cortical lysates from AD and CIIIKO-AD mice brains, but we did not detect significant differences between the 2 groups (Fig. 5H). The steady state levels of the major gamma-secretases involved in the formation of Aβ42 (Nicastrin and Presenilin 1) in cortical homogenates were also not altered (Fig. 5I).
4.5. CIIIKO-AD mice show increased 20s proteasome and increased oxidative stress
To investigate the possibility that the decrease in Aβ42 fragments occurred because of an increase in its degradation, we analyzed the levels of two proteins involved in general cellular pathways for protein degradation: p62 and LC3B (autophagy pathway), and 20S proteasome (ubiquitin proteasome system). We did not detect changes in the steady state levels of p62 and LC3bII/I ratio between AD and CIIIKO-AD mice, even though LC3bII/I was increased in the CIIIKO-AD mice if compared to WT (Fig. 6A–B). When we measured the levels of 20s proteasome, we detected a significant increase in CIIIKO-AD mice compared to both controls, suggesting an alteration of the proteasome system (Fig. 6C–D). The levels of CHOP, a transcription factor that plays a key role in the cellular Integrated Stress Response [53], was not altered (Fig. 6C–D).
Figure6: Protein degradation and oxidative stress.
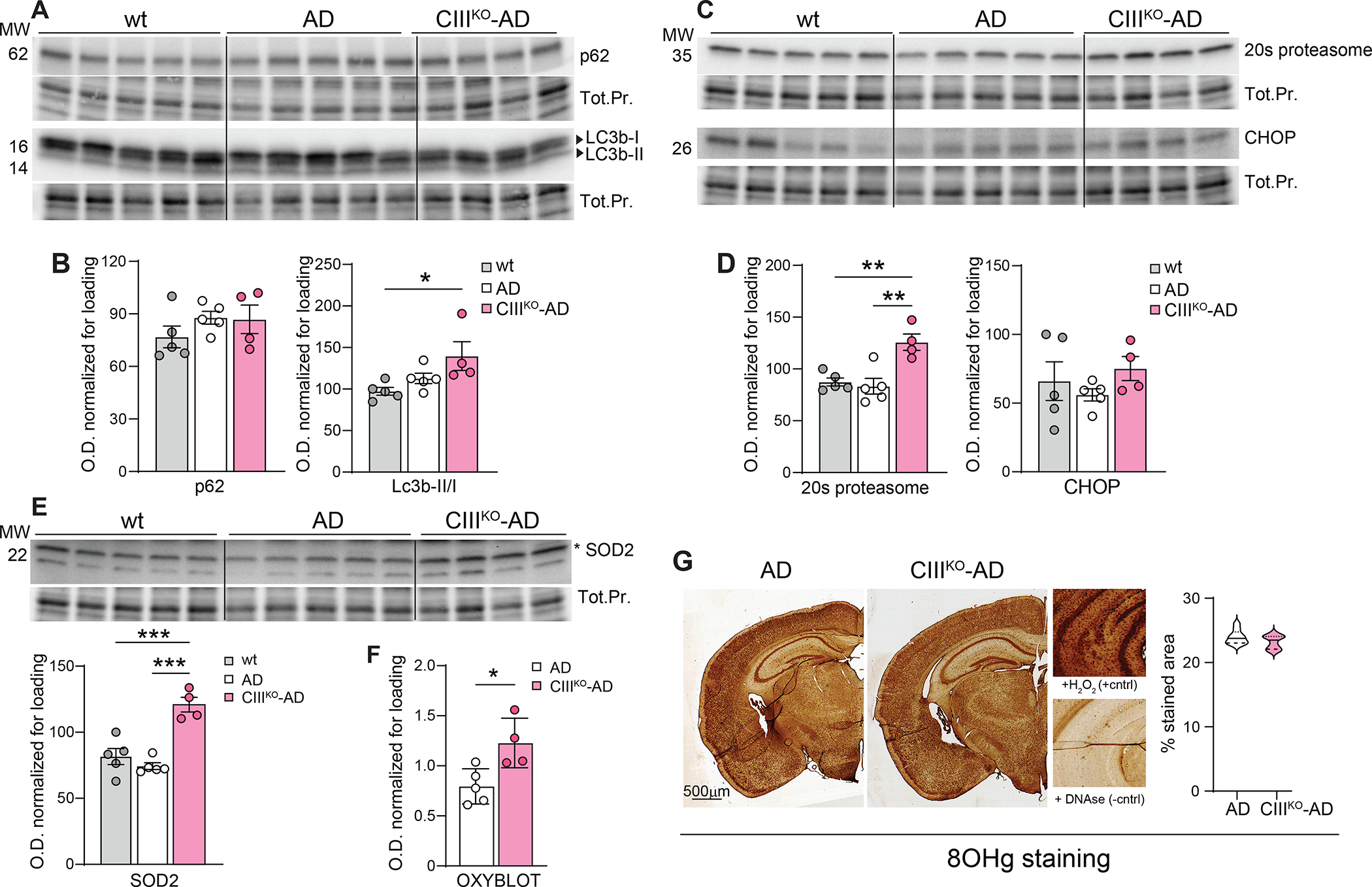
(A) Western blot analysis probing for p62 and LC3B (autophagy pathway), in cortical homogenates from 8-month-old WT, AD, and CIIIKO-AD females (n=4–5/group). (B) relative quantification of p62/protein loading and Lc3bII/Lc3bI. (C) Western blot analysis probing for 20S proteasome (ubiquitin proteasome system) and CHOP (Integrated Stress Response) in cortical homogenates from 8-month-old WT, AD, and CIIIKO-AD females (n=4–5/group) and (D) relative quantifications normalized to total protein loading. (E) Western blot analysis probing for SOD2 and relative quantification normalized to total protein, in cortical homogenates from WT, AD and CIIIKO-AD females (n=4–5/group). (F) Quantification of carbonyl groups added to protein side chains as consequence of oxidative stress (Oxyblot) in cortical homogenates from AD and CIIIKO-AD females (n=4–5/group). (G) Representative images of coronal sections of AD and CIIIKO-AD females’ brains, immunostained with an anti-8OHg antibody to detect DNA/RNA oxidation and relative quantification. In the close-up: positive control (H2O2 treatment) and negative control (DNAse treatment).
Plaque formation, OXPHOS defects, and proteasome system have been both correlated with oxidative stress. AD patients’ brains show increased levels of lipid peroxidation, DNA strand breaks, and oxidized DNA bases [54]. To investigate for signs of oxidative stress, we measured the steady-state level of SOD2, a protein that is typically increased as a consequence of oxidative stress. We detected increased levels of SOD2 in cortex of CIIIKO-AD mice (Fig. 6E). Oxidative stress also causes oxidative modification to proteins with consequent addition of carbonyl groups to protein side chains. To provide a more direct indication of the oxidative state of the different brain regions, we performed an OxyBlot (Sigma), that identifies these carbonyl groups. Cortical homogenates showed proteins with more carbonylated groups, indicating a higher status of oxidative stress (Fig. 6F).
Previously we showed that the cRISPKO animals displayed oxidative stress in the cortex, particularly in the piriform region [55]. To analyze if in our model the oxidative stress was also localized in specific regions of the brain, we performed an 8-hydroxy-deoxy-guanosine (8-OHdG)/8-hydroxy-guanosine (8-OHG) staining. 8-OHdG and 8-OHG are a product of DNA and RNA oxidative damage respectively, therefore an indirect result of the presence of ROS. We did not detect changes between AD and CIIIKO-AD mice (Fig. 6F), showing that the CIII defect did not exacerbate oxidative damage to nucleic acids.
5. Discussion
Although the primary cause of Alzheimer’s disease (AD) is still unknown, mitochondrial OXPHOS dysfunction has been implicated in the development and progression of AD. Several studies reported OXPHOS defects in postmortem AD brains [14, 16, 17, 56] and mutations in mitochondrial DNA (mtDNA), which encodes proteins for several OXPHOS complexes [10–12]. Moreover, it has been shown recently that mitochondrial Complex I abnormalities are associated with tau load in AD patients [57]. By better understanding the metabolic consequences of OXPHOS defects in the CNS and in AD pathology, novel therapeutic approaches, potentially applicable to AD, could be developed.
Different OXPHOS deficiencies influence development and brain pathology in distinctive ways. We have previously compared the phenotypes of mice in which Complex I (NDUFS3) [58], Complex III (RISP) or Complex IV (COX10) [55] have been knocked out in CamKIIα+ neuronal cells. These models showed some interesting differences. The Complex I neuronal KO (Ndufs3 nKO) mice died around 5 months with a fatal encephalopathy, ataxia, neuronal cell death and massive gliosis [58]. The Complex III deficient mice (Uqcrfs1 nKO) were all dead by 5 months and showed degeneration of the piriform cortex with elevated levels of oxidative damage [55]. The Complex IV deficient mice (Cox10 nKO) survived much longer with a progressive CNS defect and showed a neuronal loss localized to the hippocampus [55]. These findings are also different from our observations with a model of neuronal mtDNA damage, which showed prominent degeneration of the striatum prior to the degeneration of the cortex and hippocampus [59].
Therefore, different OXPHOS deficiencies could influence the development of neurodegeneration, oxidative stress, neuroinflammation and amyloid pathology in distinctive ways. Surprisingly, inhibition of different mitochondrial complexes resulted in a decrease of plaque burden and Aβ accumulation. Zhang et al. [60] reported that a mild inhibition of Complex I reduced levels of Aβ and phospho-Tau in three animal models. In these animals, there was no sign of oxidative damage or inflammation, and mitochondrial bioenergetics was increased. AMP-activated protein kinase was upregulated, GSK3β activity was reduced, and restoration of axonal trafficking resulted in elevated levels of neurotrophic factors and synaptic proteins [60]. Our lab previously generated a neuron-specific Complex IV conditional KO mouse (knocking out Cox10, subunit of CIV) crossed with the APP/PS1 mouse (COXd-AD) [35]. The plaque burden in cortex and hippocampus was significantly decreased in COXd-AD mice compared to APP/PS1 controls. This reduction was accompanied by a reduction of total Aβ42 without a significant alteration in the level of APP. In that model, beta-secretase activity was lower and oxidative stress was reduced. The Aβ pathology in this model, however, was studied at 4 months, because the early ablation of COX10 caused a massive neurodegeneration.
In a different model (AD-mito-PstI), we induced mtDNA damage in cortex and hippocampus of APP/PS1 mouse for 2 months (6 to 8 months of age) [36]. Also, in this model, the OXPHOS deficiency caused by mtDNA depletion had a negative effect on Aβ42 content and, consequently, on plaque burden. This effect was associated with an alteration in APP processing, independent from beta-secretase activity and from oxidative stress. On the other hand, in the mutator mouse that show accumulation of mtDNA mutations and premature aging, Aβ42 levels and plaque density were increased. The increased amyloid pathology was not caused by an elevated Aβ production but by a decreased clearance by the insulin degrading enzyme (IDE) [61].
A frequent criticism, leveled by proponents of OXPHOS defects causing plaque and Aβ accumulation, is that the OXPHOS defect in these different models was too severe and already present from birth, not mimicking OXPHOS defects associated with normal aging. To address this criticism, we analyzed the consequences of a partial, and adult-onset Complex III deficiency on the amyloid pathology. We chose Complex III, because defects in this complex have been associated with increased ROS [55]. The lack of Complex III activity in neurons causes premature death by 5 months of age [55]. To avoid such a strong phenotype, we used an inducible model in which the expression of cre-recombinase is activated by tamoxifen injections at 5-months. This caused the excision of exon 2 of the Uqcrfs1 gene and a depletion of protein Rieske iron–sulfur protein (RISP), catalytic subunit of Complex III, with a consequent Complex III deficiency in CamKIIα+ neurons. This late CIII deficiency allowed us to avoid a massive neurodegeneration (we detected only a mild decrease of TUJ1 in cortex) and to analyze the amyloid pathology 3 months later, when the mice were 8 months old and showed a peak in the amyloid pathology.
CIIIKO-AD mice showed a decrease of plaque burden caused by a lower amount of Aβ42 fragment. In cortex, this was not accompanied by an alteration in the APP processing, nor by a difference in BACE1 activity or secretases steady-state levels.
Even if still controversial [62], a reduction in amyloid could be expected with a reduced neuronal activity as there seems to be a link between activity-dependent release of amyloid-β (Aβ) from neurons and formation of amyloid plaques. We therefore analyzed the levels of Neurogranin, a small synaptic protein whose expression is regulated by synaptic activity [49, 50] but we did not find significant changes, suggesting that the neuronal activity was not changed by the Complex III depletion.
Oxidative stress might affect the plaque formation by enhancing the expression and activity of BACE1 [63, 64] and by enhancing the secretion of soluble APPβ. In vitro and in vivo studies showed how Complex I and III- derived ROS led to elevated levels of Aβ [24]. Soluble APPα (sAPPα), which is the soluble moiety of APP released by secretases, has been shown to antagonize some Aβ-dependent toxicity. The functional target of sAPPα protection is not well defined, and its effect on OXPHOS function remains to be determined [65].
Recent studies have also reported that the treatment of different mouse models of amyloid pathology with mitochondrial-targeted antioxidants have protective effects that includes decrease in Aβ production and plaque accumulation [66, 67]. Therefore, the working hypothesis in the previous models was that a lower level of oxidative stress might have been the cause of a decrease in plaque number [35].
The fact that in our model we detected increased oxidative stress, led us to the conclusion that the mechanism by which interfering with OXPHOS complexes activity leads to a decrease of plaques in these animal models is independent from ROS formation. The observation that in the models in which mtDNA and Complex I were impaired, the decreased number of plaques was accompanied by no change in ROS, also supports our conclusion. Although ROS can exacerbate amyloid pathology, it is not clear if it has a primary role in the development of plaques.
ROS and mitochondrial dysfunctions are also deeply connected with the proteasome system: mitochondrial dysfunctions and impairment of the ubiquitin proteasome system (UPS) are two hallmarks of the ageing process, and both systems are implicated in the pathogenesis of many age-related diseases, including AD [68]. Even though extensive literature reports how acute mitochondrial dysfunctions, induced by altering elements of the electron transport chain, lead to an inhibition of the UPS [68–70], very few reports describe the 20s proteasome role after mild mitochondria dysfunction. 20s proteasome subunit recognizes and removes damaged proteins, and since oxidative stress promotes protein unfolding, proteasome-mediated degradation is enhanced upon exposure to H2O2 or O2. Following oxidative stress, the 26S proteasome disassembles in order to retrieve intact 20S and 19S particles [71]. Moreover, 20S proteasomes have been shown to be more resistant to oxidative stress in contrast to the 26S that is highly affected [72]. In our model we saw an increase of 20s proteasome subunit that would be consistent with an increased clearance of Ab42 fragments. If this increase is directly or indirectly due to the CIII dysfunction requires further investigation.
The combined data from this and other studies suggest that mitochondria dysfunction is not the prime contributor to the amyloid plaque accumulation, but rather a consequence of Aβ toxicity. The Aβ fragment’s typical localization is extracellular, but different studies showed that it can be found in other organelles, including mitochondria, in both human AD brain and mouse models where it causes impairment of electron transport chain (ETC) complexes and increased ROS production [32, 73]. Aβ can actively be imported into mitochondria through cellular trafficking systems and colocalize with the ETC inhibiting Complex IV [29, 74]. Aβ may also contribute to the disruption of mitochondrial fusion and fission processes, leading to mitochondrial fragmentation [34].
Any extrapolation to sporadic AD, however, needs to content with the limitations of the model, including the Complex III defect being restricted to neurons and plaques being formed by overexpression of hAPP and PSEN1.
Nonetheless, our results, together with previous reports, support a model in which oxidative phosphorylation deficiency in mouse models of amyloid pathology is not the cause of Aβ oligomer accumulation.
6. Conclusions
We reported that an adult-onset mitochondrial Complex III dysfunction in neurons of a mouse model of amyloid pathology induced mild oxidative stress but did not increase amyloid beta accumulation. CIIIKO-AD mice showed decreased plaque number, decreased Aβ42 toxic fragment and altered Ubiquitin Proteasome System. Our results support a model in which oxidative phosphorylation deficiency alone does not exacerbate amyloid pathology.
Funding
This work was supported primarily by the Florida Biomedical Foundation Ed and Ethel Moore Alzheimer’s Disease Research Program grant 5AZ06 (CTM), the National Institute of Health Grants K01AG057815 (MP), and 1R01NS079965 (CTM). The following grants also helped support this work: NIH grants 5R01EY010804, 1R01AG036871, R33ES025673 and the Muscular Dystrophy Association.
7. List of abbreviations
- Aβ
Amyloid beta
- AD
Alzheimer’s disease
- APP/PS1
amyloid precursor protein (Mo/HuAPP695swe), presenilin 1 (PS1-dE9)
- ATP5a
ATP synthase alpha-subunit gene
- BACE1
Beta-secretase 1, beta-site APP cleaving enzyme 1
- BN-PAGE
Blue native polyacrylamide gel electrophoresis
- CaMKIIα
calcium/calmodulin-dependent protein kinase II alpha
- COX1
Cyclooxygenase 1 (Cytochrome c oxidase)
- CS
Citrate Synthase
- CTF
APP C-Terminal Fragment
- CytC
Cytochrome C
- GFAP
Glial fibrillary acidic protein
- Iba1
Ionized calcium binding adaptor molecule 1
- LC3B
Autophagy marker Light Chain 3
- mtDNA
mitochondrial DNA
- NDUFB8
NADH:Ubiquinone Oxidoreductase Subunit B8
- NDUFA9
NADH:Ubiquinone Oxidoreductase Subunit A9
- OXPHOS
Oxidative Phosphorylation
- PBS
Phosphate-buffered saline
- PFA
Paraformaldehyde
- PGC-1α
peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- PVDF
polyvinylidene difluoride
- RISP
Rieske Iron Sulfur Protein
- ROS
reactive oxygen species
- RT
room temperature
- SDHA
Succinate Dehydrogenase Complex Flavoprotein Subunit A
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SOD2
Superoxide dismutase 2
- TUJ1
Neuron-specific class III beta-tubulin
- UQCRC1/Core1
Ubiquinol-Cytochrome C Reductase Core Protein 1
- UQCRFS1
Ubiquinol-Cytochrome C Reductase, Rieske Iron-Sulfur Polypeptide 1
- VDAC1/Porin
voltage-dependent anion channel
Footnotes
Competing interests
The authors have no relevant financial or non-financial interests to disclose
12. Statement and Declarations
Availability of supporting data
The complete datasets used and/or analyzed during the current study are available from the corresponding author on request.
Ethics approval
All experiments and animal husbandry were performed according to a protocol approved by the University of Miami Institutional Animal Care and Use Committee.
8. Consent to participate: not applicable
9. Consent for publication: all authors approved the publication
11. References
- 1.Polanco JC, et al. , Amyloid-beta and tau complexity - towards improved biomarkers and targeted therapies. Nat Rev Neurol, 2018. 14(1): p. 22–39. [DOI] [PubMed] [Google Scholar]
- 2.Swerdlow RH, Burns JM, and Khan SM, The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta, 2014. 1842(8): p. 1219–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tapias V, Editorial: Mitochondrial Dysfunction and Neurodegeneration. Front Neurosci, 2019. 13: p. 1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hou Y, et al. , Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol, 2019. 15(10): p. 565–581. [DOI] [PubMed] [Google Scholar]
- 5.Bowling AC, et al. , Age-dependent impairment of mitochondrial function in primate brain. J Neurochem, 1993. 60(5): p. 1964–7. [DOI] [PubMed] [Google Scholar]
- 6.Hauptmann S, et al. , Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging, 2009. 30(10): p. 1574–86. [DOI] [PubMed] [Google Scholar]
- 7.Hong MG, et al. , Transcriptome-wide assessment of human brain and lymphocyte senescence. PLoS One, 2008. 3(8): p. e3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ross JM, et al. , High brain lactate is a hallmark of aging and caused by a shift in the lactate dehydrogenase A/B ratio. Proc Natl Acad Sci U S A, 2010. 107(46): p. 20087–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yao J, et al. , Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A, 2009. 106(34): p. 14670–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coskun PE, Beal MF, and Wallace DC, Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci U S A, 2004. 101(29): p. 10726–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krishnan KJ, et al. , Mitochondrial DNA deletions cause the biochemical defect observed in Alzheimer’s disease. Neurobiol Aging, 2012. 33(9): p. 2210–4. [DOI] [PubMed] [Google Scholar]
- 12.Lin MT, et al. , High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer’s disease brain. Hum Mol Genet, 2002. 11(2): p. 133–45. [DOI] [PubMed] [Google Scholar]
- 13.Bennett JP Jr. and Keeney PM, Alzheimer’s and Parkinson’s brain tissues have reduced expression of genes for mtDNA OXPHOS Proteins, mitobiogenesis regulator PGC-1alpha protein and mtRNA stabilizing protein LRPPRC (LRP130). Mitochondrion, 2020. 53: p. 154–157. [DOI] [PubMed] [Google Scholar]
- 14.Chagnon P, et al. , Distribution of brain cytochrome oxidase activity in various neurodegenerative diseases. Neuroreport, 1995. 6(5): p. 711–5. [DOI] [PubMed] [Google Scholar]
- 15.Chen JX and Yan SS, Role of mitochondrial amyloid-beta in Alzheimer’s disease. J Alzheimers Dis, 2010. 20 Suppl 2: p. S569–78. [DOI] [PubMed] [Google Scholar]
- 16.Long J, et al. , New evidence of mitochondria dysfunction in the female Alzheimer’s disease brain: deficiency of estrogen receptor-beta. J Alzheimers Dis, 2012. 30(3): p. 545–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mutisya EM, Bowling AC, and Beal MF, Cortical cytochrome oxidase activity is reduced in Alzheimer’s disease. J Neurochem, 1994. 63(6): p. 2179–84. [DOI] [PubMed] [Google Scholar]
- 18.Holper L, Ben-Shachar D, and Mann JJ, Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease. Neuropsychopharmacology, 2019. 44(5): p. 837–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Onyango IG, Khan SM, and Bennett JP Jr., Mitochondria in the pathophysiology of Alzheimer’s and Parkinson’s diseases. Front Biosci (Landmark Ed), 2017. 22: p. 854–872. [DOI] [PubMed] [Google Scholar]
- 20.Kim SH, et al. , Decreased levels of complex III core protein 1 and complex V beta chain in brains from patients with Alzheimer’s disease and Down syndrome. Cell Mol Life Sci, 2000. 57(12): p. 1810–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kenney PM and Bennett JP Jr., Alzheimer’s Disease Frontal Cortex Mitochondria Show a Loss of Individual Respiratory Proteins but Preservation of Respiratory Supercomplexes. Int J Alzheimers Dis, 2019. 2019: p. 4814783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fisar Z, et al. , Activities of mitochondrial respiratory chain complexes in platelets of patients with Alzheimer’s disease and depressive disorder. Mitochondrion, 2019. 48: p. 67–77. [DOI] [PubMed] [Google Scholar]
- 23.Guglielmotto M, et al. , The up-regulation of BACE1 mediated by hypoxia and ischemic injury: role of oxidative stress and HIF1alpha. J Neurochem, 2009. 108(4): p. 1045–56. [DOI] [PubMed] [Google Scholar]
- 24.Leuner K, et al. , Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid Redox Signal, 2012. 16(12): p. 1421–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paola D, et al. , Oxidative stress induces increase in intracellular amyloid beta-protein production and selective activation of betaI and betaII PKCs in NT2 cells. Biochem Biophys Res Commun, 2000. 268(2): p. 642–6. [DOI] [PubMed] [Google Scholar]
- 26.Tamagno E, et al. , Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis, 2002. 10(3): p. 279–88. [DOI] [PubMed] [Google Scholar]
- 27.Anandatheerthavarada HK, et al. , Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol, 2003. 161(1): p. 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calkins MJ, et al. , Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum Mol Genet, 2011. 20(23): p. 4515–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casley CS, et al. , Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem, 2002. 80(1): p. 91–100. [DOI] [PubMed] [Google Scholar]
- 30.Devi L, et al. , Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci, 2006. 26(35): p. 9057–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hansson Petersen CA, et al. , The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A, 2008. 105(35): p. 13145–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manczak M, et al. , Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet, 2006. 15(9): p. 1437–49. [DOI] [PubMed] [Google Scholar]
- 33.Reddy PH and Beal MF, Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol Med, 2008. 14(2): p. 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, et al. , Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A, 2008. 105(49): p. 19318–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fukui H, et al. , Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A, 2007. 104(35): p. 14163–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pinto M, et al. , Mitochondrial DNA damage in a mouse model of Alzheimer’s disease decreases amyloid beta plaque formation. Neurobiol Aging, 2013. 34(10): p. 2399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jankowsky JL, et al. , Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng, 2001. 17(6): p. 157–65. [DOI] [PubMed] [Google Scholar]
- 38.Madisen L, et al. , A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci, 2010. 13(1): p. 133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waypa GB, et al. , Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am J Respir Crit Care Med, 2013. 187(4): p. 424–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang J, et al. , Gender differences in the amount and deposition of amyloidbeta in APPswe and PS1 double transgenic mice. Neurobiol Dis, 2003. 14(3): p. 318–27. [DOI] [PubMed] [Google Scholar]
- 41.Zhou CN, et al. , Sex Differences in the White Matter and Myelinated Fibers of APP/PS1 Mice and the Effects of Running Exercise on the Sex Differences of AD Mice. Front Aging Neurosci, 2018. 10: p. 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mifflin MA, et al. , Sex differences in the IntelliCage and the Morris water maze in the APP/PS1 mouse model of amyloidosis. Neurobiol Aging, 2021. 101: p. 130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dominguez S, et al. , Sex Differences in the Phosphoproteomic Profiles of APP/PS1 Mice after Chronic Unpredictable Mild Stress. J Alzheimers Dis, 2020. 74(4): p. 1131–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li X, et al. , Sex differences between APPswePS1dE9 mice in A-beta accumulation and pancreatic islet function during the development of Alzheimer’s disease. Lab Anim, 2016. 50(4): p. 275–85. [DOI] [PubMed] [Google Scholar]
- 45.Schmittgen TD and Livak KJ, Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc, 2008. 3(6): p. 1101–8. [DOI] [PubMed] [Google Scholar]
- 46.Barrientos A, Fontanesi F, and Diaz F, Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr Protoc Hum Genet, 2009. Chapter 19: p. Unit19 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jankowsky JL, et al. , Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet, 2004. 13(2): p. 159–70. [DOI] [PubMed] [Google Scholar]
- 48.Choi CI, et al. , Simultaneous deletion of floxed genes mediated by CaMKIIalpha-Cre in the brain and in male germ cells: application to conditional and conventional disruption of Goalpha. Exp Mol Med, 2014. 46: p. e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones KJ, et al. , Rapid, experience-dependent translation of neurogranin enables memory encoding. Proc Natl Acad Sci U S A, 2018. 115(25): p. E5805–E5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garrido-Garcia A, et al. , Neurogranin Expression Is Regulated by Synaptic Activity and Promotes Synaptogenesis in Cultured Hippocampal Neurons. Mol Neurobiol, 2019. 56(11): p. 7321–7337. [DOI] [PubMed] [Google Scholar]
- 51.Mohajeri MH, Saini KD, and Nitsch RM, Transgenic BACE expression in mouse neurons accelerates amyloid plaque pathology. J Neural Transm (Vienna), 2004. 111(3): p. 413–25. [DOI] [PubMed] [Google Scholar]
- 52.Li R, et al. , Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer’s disease patients. Proc Natl Acad Sci U S A, 2004. 101(10): p. 3632–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaspar S, et al. , Adaptation to mitochondrial stress requires CHOP-directed tuning of ISR. Sci Adv, 2021. 7(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Misrani A, Tabassum S, and Yang L, Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front Aging Neurosci, 2021. 13: p. 617588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Diaz F, et al. , A defect in the mitochondrial complex III, but not complex IV, triggers early ROS-dependent damage in defined brain regions. Hum Mol Genet, 2012. 21(23): p. 5066–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sheng B, et al. , Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J Neurochem, 2012. 120(3): p. 419–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Terada T, et al. , Mitochondrial complex I abnormalities is associated with tau and clinical symptoms in mild Alzheimer’s disease. Molecular Neurodegeneration, 2021. 16(1): p. 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peralta S, et al. , Metformin delays neurological symptom onset in a mouse model of neuronal complex I deficiency. JCI Insight, 2020. 5(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pickrell AM, et al. , The striatum is highly susceptible to mitochondrial oxidative phosphorylation dysfunctions. J Neurosci, 2011. 31(27): p. 9895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang L, et al. , Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer’s Disease. EBioMedicine, 2015. 2(4): p. 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kukreja L, et al. , Increased mtDNA mutations with aging promotes amyloid accumulation and brain atrophy in the APP/Ld transgenic mouse model of Alzheimer’s disease. Mol Neurodegener, 2014. 9: p. 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Virtuoso A, et al. , The Spatiotemporal Coupling: Regional Energy Failure and Aberrant Proteins in Neurodegenerative Diseases. Int J Mol Sci, 2021. 22(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mouton-Liger F, et al. , Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2alpha pathway. Biochim Biophys Acta, 2012. 1822(6): p. 885–96. [DOI] [PubMed] [Google Scholar]
- 64.Tamagno E, et al. , Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J Neurochem, 2008. 104(3): p. 683–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lopez Sanchez MIG, et al. , Amyloid precursor protein drives down-regulation of mitochondrial oxidative phosphorylation independent of amyloid beta. Sci Rep, 2017. 7(1): p. 9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mao P, et al. , Mitochondria-targeted catalase reduces abnormal APP processing, amyloid beta production and BACE1 in a mouse model of Alzheimer’s disease: implications for neuroprotection and lifespan extension. Hum Mol Genet, 2012. 21(13): p. 2973–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McManus MJ, Murphy MP, and Franklin JL, The Mitochondria-Targeted Antioxidant MitoQ Prevents Loss of Spatial Memory Retention and Early Neuropathology in a Transgenic Mouse Model of Alzheimer’s Disease. Journal of Neuroscience, 2011. 31(44): p. 15703–15715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ross JM, Olson L, and Coppotelli G, Mitochondrial and Ubiquitin Proteasome System Dysfunction in Ageing and Disease: Two Sides of the Same Coin? Int J Mol Sci, 2015. 16(8): p. 19458–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang Q, et al. , Negative regulation of 26S proteasome stability via calpain-mediated cleavage of Rpn10 subunit upon mitochondrial dysfunction in neurons. J Biol Chem, 2013. 288(17): p. 12161–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meul T, et al. , Mitochondrial Regulation of the 26S Proteasome. Cell Rep, 2020. 32(8): p. 108059. [DOI] [PubMed] [Google Scholar]
- 71.Livnat-Levanon N, et al. , Reversible 26S proteasome disassembly upon mitochondrial stress. Cell Rep, 2014. 7(5): p. 1371–1380. [DOI] [PubMed] [Google Scholar]
- 72.Hohn A, Konig J, and Grune T, Protein oxidation in aging and the removal of oxidized proteins. J Proteomics, 2013. 92: p. 132–59. [DOI] [PubMed] [Google Scholar]
- 73.Adav SS, Park JE, and Sze SK, Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Mol Brain, 2019. 12(1): p. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Crouch PJ, et al. , Copper-dependent inhibition of cytochrome c oxidase by Abeta(1–42) requires reduced methionine at residue 35 of the Abeta peptide. J Neurochem, 2006. 99(1): p. 226–36. [DOI] [PubMed] [Google Scholar]