

Abstract
Introduction:
Identifying CSF-based biomarkers for the β-amyloidosis that initiates Alzheimer’s disease (AD) could provide inexpensive and dynamic tests to distinguish AD from normal aging and predict future cognitive decline.
Methods:
We developed immunoassays specifically detecting all C-terminal variants of secreted amyloid β-protein and identified a novel biomarker, the Aβ 37/42 ratio, that outperforms the canonical Aβ42/40 ratio as a means to evaluate the γ-secretase activity and brain Aβ accumulation.
Results:
We show that Aβ 37/42 can distinguish physiological and pathological status in 1) presenilin-1 mutant vs. wild-type cultured cells, 2) AD vs. control brain tissue, and 3) AD vs. cognitively normal (CN) subjects in CSF, where 37/42 (AUC 0.9622) outperformed 42/40 (AUC 0.8651) in distinguishing CN from AD.
Discussion:
We conclude that the Aβ 37/42 ratio sensitively detects presenilin/γ-secretase dysfunction and better distinguishes CN from AD than Aβ42/40 in CSF. Measuring this novel ratio alongside promising phospho-tau analytes may provide highly discriminatory fluid biomarkers for AD.
Keywords: Alzheimer’s disease, amyloid β-protein, CSF biomarkers
Introduction
Over the last three decades, many genetic, neuropathological, biochemical, and biomarker analyses have converged on the concept that accumulation of the 42-residue amyloid β-protein (Aβ 42) is an early and pathogenically critical event in both familial and ‘sporadic’ Alzheimer’s disease (AD). Sequential cleavages of the β-amyloid precursor protein (APP) by β- and γ-secretases yield a heterogeneous array of small peptides, the longer forms of which (Aβ 42 and Aβ 43) are highly prone to self-aggregation into initially soluble and then insoluble oligomers and fibrils, accompanied by progressive neuritic/synaptic, microglial, and astrocytic changes. Despite this emerging understanding of a key mechanistic role for Aβ 42, there remain questions about how well measurements of this and other related Aβ peptides in biological fluids, like cerebrospinal fluid (CSF), can accurately reflect alterations of APP processing and Aβ deposition in the patient’s brain and thus predict a clinical diagnosis of AD.
γ-Secretase, a unique intramembrane aspartyl protease, is a hetero-tetramer consisting of the proteins presenilin (PS), nicastrin (NCT), anterior pharynx defective-1 (APH-1) and presenilin enhancer-2 (PEN-2) [1–3]. The two presenilin homologs, PS1 and PS2, each have nine transmembrane domains (TMDs); following their auto-proteolytic cleavage, they become the catalytic subunit of γ-secretase that contains the active-site aspartates in TMD 6 and 7 [4]. γ-Secretase processes its substrates (>100 different type-I transmembrane proteins to date) [5] by a series of sequential hydrolytic cleavages within the TMDs, i.e., in the hydrophobic environment of the lipid bilayer. Following the initial shedding of the large ectodomain of a substrate by α-secretase (e.g., ADAM10 or ADAM17) or β-secretase (BACE1 or 2), the presenilin/γ-secretase complex then catalyzes multiple intramembrane cleavages. In the case of the substrate APP, this step releases the heterogeneous p3 and Aβ peptides. Recent evidence suggests that γ-secretase functions within a high molecular weight, multi-secretase complex that also contains either α- or β-secretase, thus allowing efficient sequential processing of substrates by α/γ or β/γ complexes to the final products [6, 7].
Following the α- or β-secretase cuts, γ-secretase processes the resultant membrane-anchored C-terminal fragments by an initial endo-proteolytic (ε) cleavage followed by a series of carboxypeptidase-like trimming steps (γ-cleavages). The ε-cleavage generates long peptides (Aβ 48 and Aβ 49) that remain membrane-anchored and undergo stepwise γ-cleavages every 3 amino acids until the respective Aβ 42 and Aβ 40 peptides are released. We have reported that this γ-secretase processing requires the stabilization of an enzyme-substrate scission complex that uses 3 amino acid binding pockets (S1’-S2’-S3’) in the presenilin active site. Substrate occupancy of these 3 pockets occurs after its initial binding (“docking”) but precedes catalysis, indicating that a conformational change (“unwinding”) in the bound helical substrate is needed to allow ε-cleavage [8]. It is now clear that secreted Aβ peptides are produced by γ-secretase along two proteolytic pathways: Aβ 49→46→43→40→37 and Aβ 48→45→42→39, 38 or 37 [9, 10]. The existence of the shortest Aβ peptides, Aβ 37, Aβ 38, and Aβ 39, has been demonstrated [11–17], but these have been little studied. Aβ 38 and 39 were found to result mainly from cleavage of Aβ 42 by removal of a tetra-peptide or tripeptide, respectively. Aβ 37 was described to result either from cleavage of Aβ 42 or from cleavage of Aβ 40 by removal of a pentapeptide or tripeptide, respectively.
Based on extensive biochemical, biophysical, and structural studies of γ-secretase, it has emerged that γ-secretase requires at least 6 steps to achieve the hydrolysis of a substrate: 1) initial activation of PS by an auto-proteolysis that generates the active PS heterodimer; 2) the docking of the substrate TMD to the γ-secretase complex; 3) substrate TMD unwinding and binding to the active site; 4) endopeptidase (ε) activity; 5) successive carboxypeptidase-like (γ) cleavages, and 6) release of the final products into the luminal and extracellular compartments. Although recent advanced cryo-EM analysis of γ-secretase has provided impressive structural information about the complex [18, 19], the still somewhat limited resolution of cryo-EM means that the fine details of PS1 residues and how they contribute to PS proteolytic function remain unclear. Through detailed measurement of all secreted Aβ products generated by wild-type or mutant PS1, we recently discovered that hydrophilic loop 1 of PS1 is required for proper sequential γ-secretase processivity by modulating the affinity between APP (substrate) and PS1 (enzyme) [20]. These findings should promote the rational design of the next generation of γ-secretase modulators to lower Aβ amyloidogenic peptides in the human brain without blocking substrate cleavage. However, further details of the critical carboxypeptidase-like (γ) proteolytic events and the availability of the resultant products in biological fluids as potential biomarkers remain to be clarified.
For many years, Aβ CSF and plasma biomarkers have focused on Aβ42 and Aβ40, with Aβ40 being the major secreted form and Aβ42 being a minor (~10%) secreted form that can under some circumstances give rise to Aβ42 oligomerization and amyloid plaque formation in the brain. Here, we take advantage of our newly developed sensitive immunoassays to detect all six secreted C-terminal variants of Aβ (Aβ43, Aβ42, Aβ40, Aβ39, Aβ38, and Aβ37), enabling us to study the carboxypeptidase-like (γ) products in detail. We asked whether a more comprehensive analysis of all Aβ peptides in human CSF would help us understand and better predict amyloid neuropathology and its clinical consequences, rather than relying solely on the canonical Aβ42/40 ratio.
Material and Methods
Participants for CSF analysis
The Mayo Clinic Study of Aging (MCSA) is a population-based, prospective study of residents living in Olmsted County, MN. Details of study design and participant recruitment are published [21, 22]. In 2004, the Rochester Epidemiology Project (REP) medical records linkage system was used to enumerate Olmsted County residents between the ages of 70 and 89, as described [23]. In 2012, the MCSA was extended to include those aged 50 and older. The present analyses included 79 participants (38 CN, 25 MCI, and 16 AD) in whom we measured CSF Aβ peptides. Subject enrollment, sample collection, and sharing of samples across sites were approved by the Mayo Clinic and the Olmsted Medical Center. Table 2 shows demographic characteristics and further details about the cohorts. The institutional review boards of the Mayo Clinic and the Olmsted Medical Center approved all study protocols and written informed consent was obtained from all participants.
Table 2.
Patient demographics in current study
| Brain samples |
LP-NCI
(n = 24) |
HP-NCI
(n = 18) |
AD
(n = 17) |
| Age of death (SD), y | 88.7 (5.1) | 92.2 (6.1) | 90.9 (6.4) |
| Years of education (SD), y | 17.3 (4.4) | 15.5 (2.8) | 15.9 (3.1) |
| Sex, female (%) | 11 (45.8%) | 17 (94.4%) | 12 (70.6%) |
| CSF samples |
CN
(n = 38) |
MCI
(n = 25) |
AD
(n = 16) |
| Age (SD), y | 74.9 (6.4) | 74.9 (8.3) | 75.2 (10.2) |
| Sex, female (%) | 12 (31.6%) | 11 (44%) | 5 (31.1%) |
Clinical categorization of participants
Subjects were designated as Alzheimer’s disease (AD), Mild cognitive impairment (MCI), or Cognitive normal (CN) based on a clinical and cognitive assessment every 15 months that includes separate assessments by a study coordinator, physician, and neuropsychologist. These visits included an interview by a study coordinator/nurse, a neurologic exam, and neuropsychological testing [22]. The neuropsychological battery included 9 tests covering 4 domains (memory, language, executive function, and visuospatial function), as described [22].
CSF analysis
CSF samples were obtained by lumbar puncture between the L3 and L4 intervertebral space. Lumbar puncture was performed early in the morning after fasting. CSF was collected and stored at −80°C in polypropylene tubes. All samples were thawed once before analysis. CSF samples were analyzed by sandwich immunoassays.
Generation of FAD PS1 expression vectors
A pcDNA3.1 vector harboring the WT human PS1 was used as the template to generate FAD PS1 expression vectors. To introduce mutations, the template was amplified by PCR into 2 DNA fragments with an overlapping sequence containing the mutated locus. Overlap PCR was done to generate the whole open-reading frame containing the mutation. PCR products were sub-cloned into the pcDNA3.1 vector for expression under a CMV promoter. Vectors were sequenced from both 5’ and 3’ ends to confirm successful mutagenesis.
Tissue culture and transfection of adherent cells
Adherent HEK cells were cultured in complete growth medium: Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 10 units/mL penicillin, and 10 mg/mL streptomycin (all from Life Technologies). For transfection, adherent HEK cells were seeded in 24-well dishes at a density of 5×105 cells per well. Transfection was carried out with PEI MAX reagent (Polysciences). Cells were incubated for 24 hours and media was changed for conditioning another 24 hours, at which time, media was harvested for ELISA, and cells were harvested for western blot.
NGN2-iN generation.
Induced neurons (iNs) were generated following as published [24] with minor modifications [25]. In brief, iPSCs were plated at a density of 95,000 cells/cm2 on a growth factor reduced matrigel (Corning #354230) coated plate and then were transduced with three lentiviruses: pTet-O-NGN2-puro (Addgene plasmid #52047, a gift from Marius Wernig), Tet-O-FUW-EGFP (Addgene plasmid #30130, a gift from Marius Wernig), and FUdeltaGW-rtTA (Addgene plasmid #19780, a gift from Konrad Hochedlinger). The cells were then dissociated with Accutase, plated at 200,000 cells/cm2 using Stemflex (Thermofisher) and ROCK inhibitor (10 μM) (D0). From D1 to D3, the media was gradually switched from KSR media [Knockout DMEM, 15% KOSR, 1x MEM-NEAA, 55 uM β-mercaptoethanol, and 1x GlutaMAX (Thermofisher)] to N2B media [DMEM/F12, 1x GlutaMAX, 1x N2 supplement B (Stemcell Technologies), 0.3% dextrose (Sigma)]. D1 media was 100% KSR, D2 was 50% KSR, 50% N2B, D3 was 100% N2B. Doxycycline (2 μg/ml, Sigma) was added from D1 to the end of the differentiation, and puromycin (5 μg/ml, Gibco) was added from D2 to the end of the differentiation. On D3, B27 supplement (1:100) (Life Technologies) was added. On D4, cells are replated at 50,000 cells/cm2 and from D4 to the end of differentiation D21, cells were cultured with NBM media (Neurobasal medium, 0.5x MEM-NEAA, 1x GlutaMAX, 0.3% dextrose) + 1:50 B27 + BDNF, GDNF, CNTF (10 ng/ml, Peprotech) and fed every 2–3 days.
Sequential human brain extractions for Aβ analyses
The medial prefrontal cortex (BA9/46) (mPFC) was obtained for 59 participants of the Religious Order Study or Memory and Aging Project [26]. Individuals were selected based on clinical and pathological diagnoses of AD. LP-NCI (low pathology non-cognitively impaired) had neither a clinical nor pathological diagnosis of AD or other dementias. HP-NCI (high pathology NCI) had a pathological diagnosis of AD but not a clinical diagnosis. AD had both a clinical and pathological diagnosis of AD. No participants had a clinical diagnosis of dementia with Lewy bodies or a pathological diagnosis of hippocampal sclerosis. For analyses of Aβ levels in the mPFC, the grey matter was dissected and homogenized in Tris-buffered saline (TBS) and centrifuged at 100,000 g for 30 minutes. The supernatant was collected (“TBS-soluble”) and the pellet homogenized in 1% Triton X-100 in TBS and centrifuged at 100,000 g. The supernatant was collected (“Triton-soluble”) and the pellet homogenized in 6M guanidine hydrochloride (GuCl) using a sonicator and centrifuged at 100,000 g, with the supernatant collected (“GuCl-soluble”).
Aβ immunoassay (MSD)
Conditioned media from cultured cells, human brain extracts, and human CSF were diluted with 1% BSA in wash buffer (TBS supplemented with 0.05% Tween). For Aβ x-37/38/39/40/42/43 assays, each well of an uncoated 96-well multi-array plate (MesoScale Discovery, #L15XA-3) was coated with 30 μL of a PBS solution containing 3 μg/mL of 266 capture antibody to the mid-region of soluble Aβ (original gift of P. Seubert, Elan, plc) and incubated at room temperature overnight. Detection antibody solutions were prepared to contain biotinylated monoclonal antibodies specifically recognizing the C-terminal residue of each Aβ isoform, plus 100 ng/mL Streptavidin Sulfo-TAG (MesoScale Discovery, #R32AD-5), and 1% BSA diluted in wash buffer. Following overnight incubation for capture antibody coating, 25 μL/well of the sample, followed by 25 μL/well of detection antibody solution was incubated for 2 hr at RT with shaking at >300 rpm, and washing of wells with wash buffer (TBS supplemented with 0.05% Tween) between incubations. The plate was read and analyzed according to the MSD manufacturer’s manual.
Immunostaining of human brain sections
Autopsied brain sections were provided by the Neuropathology Division of the Department of Pathology, Brigham and Women’s Hospital. Formalin-fixed paraffin-embedded sections (5 μm) of temporal cortices from 3 AD patients were used in this study. Immunohistochemistry was carried out as described [27]. Briefly, the rehydrated sections were incubated with 88% formic acid for 20 minutes, and endogenous peroxidases were inactivated by treatment with 0.3% H2O2 in PBS containing 0.3% Triton X-100. The sections were incubated with primary antibody at 4°C overnight and then with biotinylated secondary antibodies for 1 hour. For visualization, the sections were treated with avidin-biotin-HRP complex (Vector) and then with DAB substrate (TCI) with or without intensification by nickel ammonium sulfide. Photomicrographs were taken with an ECLIPSE E600 microscope (Nikon) equipped with Mlchrome 5 Pro camera (Tucsen) and brightness/contrast were adjusted with ImageJ (NIH).
Quantification and statistical analysis
All statistical analyses were performed using GraphPad Prism 9 software. Statistical details of experiments are described in the text and/or figure legends.
Results
Aβ42/40 is insufficient in distinguishing some PS1 enzymatic changes caused by FAD mutations
To date, 341 distinct missense mutations in PS1 or PS2 have been found to cause early-onset familial AD (FAD), highlighting the critical role of PS1/2 structure-function relationships in AD pathogenesis. Each of the PS1 or PS2 mutations associated with clinical AD induces a conformational change of the protein within the γ-secretase complex that alters its proteolytic activity. As regards APP processing, these PS structural changes are reflected in quantitative analyses of the products of PS/γ-secretase: Aβ peptides of differing lengths and the complementary tri-, tetra-, and penta-peptides that arise from sequential hydrolyses from the ε- to the various γ-cleavage sites. The latter hydrophobic peptides are released into the cytosol or retained in the transmembrane and are difficult to isolate and quantify accurately, whereas Aβ monomers are aqueously soluble and measurable.
We have used CRISPR/Cas9 on human HEK-293 cells to generate PS1/2 dKO cells in order to eliminate endogenous PS activity. As previously characterized [20], the best 293 dKO line obtained, had no detectable PS1 and PS2 expression by immunoblot band, generated < 10% of the total Aβ levels made by wild type (WT) 293 cells. Next, we built a library of PS1 expression constructs for 131 distinct FAD mutations. Then, we overexpressed each of these FAD mutants together with WT APP-C99 (the immediate substrate of γ-secretase) in the dKO 293 cells and performed detailed measurements of all secreted C-terminal variants of Aβ. We built sensitive and specific sandwich immunoassays to detect Aβ43, Aβ42, Aβ40, Aβ39, Aβ38, and Aβ37, as shown in Fig. 1a, which is a schematic representation of the APP transmembrane protein sequence, the assay antibody epitopes, and the processive cleavage pathways. We further demonstrated the performance of each assay (including its LLoQ) on synthetic Aβ calibrators in Fig. S1a. Further, we validated the specificity of each assay by testing if an assay cross-reacted with the other 5 Aβ calibrators, as shown in Fig. S1b. We found that 1) Aβ x-37, 38, 39, 40, and 43 assays were very specifically recognizing only the cognate synthetic Aβ peptide they ought to recognize; 2) Aβ x-42 assay could recognize Aβ43 as well, but with much lower affinity, which is consistent with a previous report on the same antibody 21F12 [28].
Figure 1. Aβ 37/42 is an analyte more sensitive to PS1 enzymatic changes caused by FAD mutations.
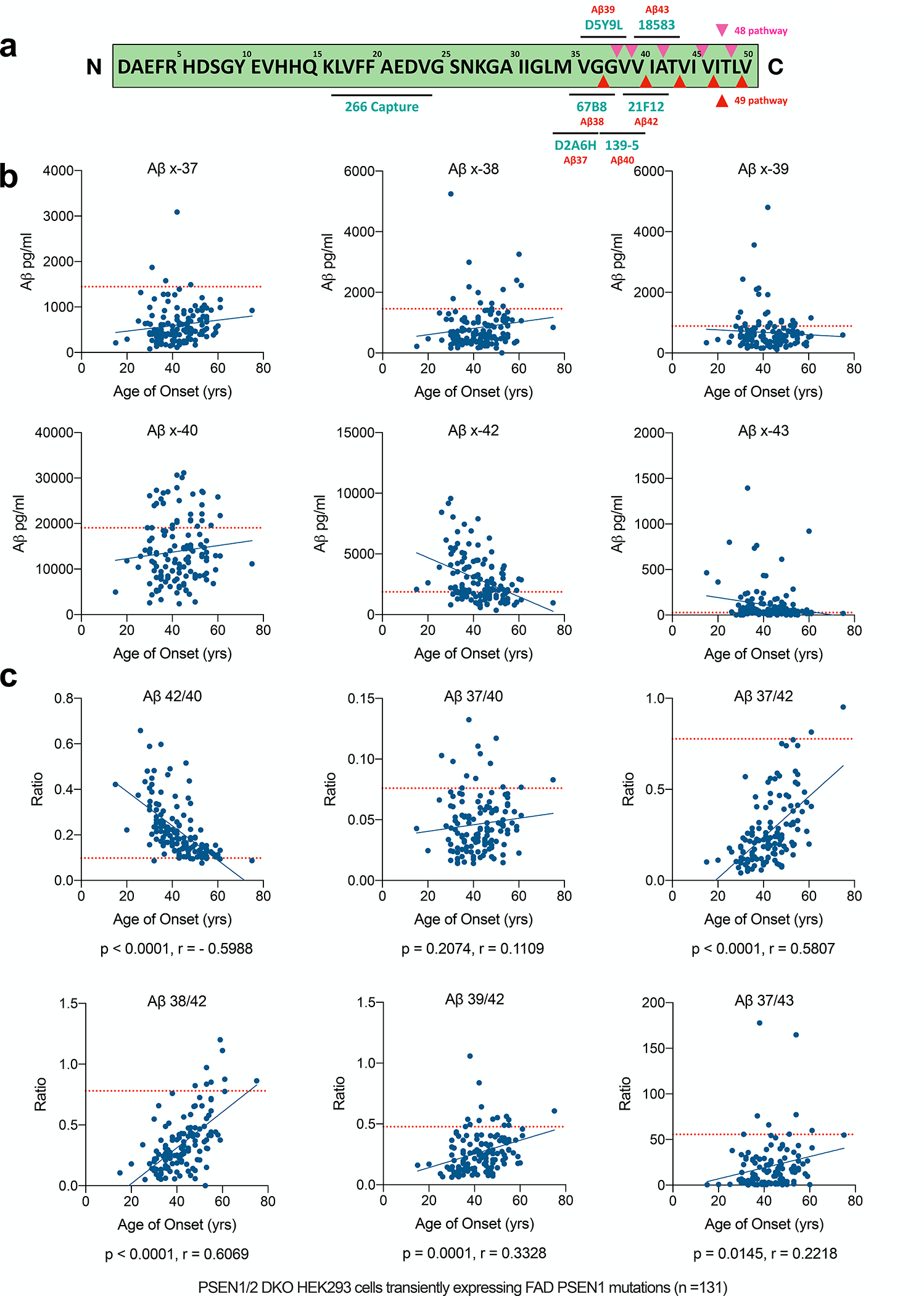
a) Schematic of APP transmembrane region protein sequence, assay antibody epitopes, and the Aβ49 (red arrows) and 48 (purple arrows) processive cleavage pathways. b-c) Measurements of concentrations and ratios of Aβs secreted from transfectants of 131 FAD mutants in PS1/2 dKO HEK293 cells, with the levels from WT-PS1 transfectants labeled as the red dotted line.
We measured all six secreted C-terminal variants from dKO cells expressing each of the 131 FAD PS1 mutations, for all of which the approximate age of onset (AOO) of their clinical AD symptoms is known. Compared to WT PS1 transfectants, the FAD PS1 transfectants generally secreted less short-Aβ peptides (x-37, x-38, x-39, x-40) and more long-Aβ peptides (x-42, x-43) (Fig. 1b and Table 1). Aβ x-42 levels themselves showed a significant correlation with AOO (p < 0.0001 by Pearson correlation). As the WT or mutant PS1 were transiently expressed in the cells, to normalize the amount of secreted Aβs from different transfectants, we further used the ratio between a specific secreted Aβ and the total secreted Aβ C-terminal variants (37+38+39+40+42+43), as shown in Fig. S2a. We found that, after this normalization, secreted Aβ x-37, 38, 40, 42, and 43 all significantly correlated with AOO, with the statistical analysis of the data in Fig. 1b and Fig. S2a compared in Fig. S2b.
Table 1.
Aβs secretion from PS dKO 293 cells co-transfected with WT or mutant PSEN1 and APP-C99 (pg/ml)
| AOO (yrs) | Aβ x-37 | Aβ x-38 | Aβ x-39 | Aβ x-40 | Aβ x-42 | Aβ x-43 | |
|---|---|---|---|---|---|---|---|
| WT-PSEN1 | 1546.54 | 1478.58 | 947.71 | 19945.49 | 1832.06 | 28.05 | |
| WT-PSEN1 | 1424.56 | 1521.39 | 846.68 | 20513.23 | 2026.34 | 31.15 | |
| WT-PSEN1 | 1367.12 | 1368.03 | 863.40 | 16696.32 | 1745.90 | 20.95 | |
| D40del (delGAC) | 48.00 | 1492.97 | 1636.89 | 1072.35 | 20615.23 | 1988.16 | 34.76 |
| E69D | 55.00 | 976.90 | 1123.85 | 704.64 | 13673.39 | 1319.32 | 22.52 |
| A79V | 53.00 | 605.65 | 1543.53 | 610.41 | 13205.98 | 1589.18 | 35.90 |
| V82L | 53.00 | 398.56 | 639.00 | 740.00 | 27083.21 | 2073.23 | 284.99 |
| I83T | 55.00 | 672.28 | 907.30 | 593.69 | 16218.21 | 2051.43 | 40.15 |
| L85P | 20.00 | 290.93 | 468.71 | 444.23 | 11827.96 | 2620.52 | 363.14 |
| V89L (G>C) | 46.00 | 625.01 | 1407.48 | 644.92 | 14485.76 | 2403.17 | 43.10 |
| C92S | 53.50 | 794.28 | 1226.15 | 877.23 | 26838.88 | 4006.24 | 56.56 |
| V94M | 53.00 | 981.35 | 1062.39 | 621.72 | 12736.50 | 1270.96 | 27.14 |
| V96F | 52.50 | 711.19 | 0.00 | 683.74 | 15341.04 | 3403.84 | 48.53 |
| V97L | 36.50 | 922.72 | 1036.18 | 813.71 | 17445.85 | 2012.16 | 29.87 |
| T99A | 43.00 | 1392.16 | 1650.27 | 985.70 | 25047.81 | 2916.71 | 56.56 |
| F105I | 53.00 | 925.33 | 1131.90 | 1073.06 | 20655.24 | 2999.07 | 45.89 |
| F105V | 52.00 | 717.59 | 849.09 | 906.59 | 17628.17 | 2347.69 | 41.16 |
| L113P | 43.00 | 648.77 | 434.20 | 971.31 | 6215.50 | 1515.77 | 11.93 |
| Y115D | 29.00 | 642.87 | 1292.59 | 1171.87 | 19089.38 | 9167.92 | 109.59 |
| Y115H | 35.00 | 438.87 | 405.68 | 558.27 | 6179.35 | 1526.73 | 34.76 |
| T116N | 35.00 | 612.20 | 815.37 | 686.76 | 9435.60 | 2464.06 | 22.52 |
| T116I | 48.00 | 528.93 | 1395.08 | 605.99 | 9469.68 | 2122.29 | 11.93 |
| T116R | 35.00 | 317.01 | 318.65 | 412.02 | 8548.74 | 5111.09 | 178.09 |
| P117A | 35.00 | 177.26 | 393.95 | 246.03 | 3299.15 | 1278.66 | 22.52 |
| P117L | 30.25 | 622.35 | 1100.84 | 740.94 | 14392.16 | 4989.74 | 44.97 |
| E120D (A>T) | 48.00 | 920.11 | 1462.01 | 1015.48 | 22045.57 | 3963.11 | 51.06 |
| E120K | 39.00 | 394.08 | 389.60 | 548.73 | 14054.53 | 6895.07 | 127.82 |
| I83_M84del | 36.00 | 406.87 | 759.44 | 590.30 | 26730.13 | 5044.51 | 733.95 |
| E120G | 30.00 | 77.07 | 160.10 | 249.06 | 2583.38 | 801.98 | 11.93 |
| L134R | 53.00 | 1204.35 | 2090.92 | 927.85 | 24127.90 | 3780.80 | 58.05 |
| N135D | 35.00 | 297.76 | 407.35 | 504.90 | 8317.60 | 1951.06 | 44.05 |
| N135Y | 32.00 | 477.90 | 355.12 | 444.23 | 11388.10 | 5490.94 | 231.68 |
| N135S | 33.00 | 209.07 | 232.58 | 264.49 | 5361.16 | 1807.99 | 48.53 |
| M139T | 49.00 | 807.04 | 859.80 | 812.83 | 13961.76 | 2062.33 | 27.14 |
| M139I | 37.00 | 1577.28 | 1005.53 | 2096.57 | 18302.15 | 3898.40 | 20.78 |
| M139K | 37.00 | 396.88 | 481.47 | 298.63 | 10516.99 | 1754.42 | 58.79 |
| M139V | 41.00 | 1266.82 | 723.21 | 1289.09 | 15074.84 | 3165.84 | 32.40 |
| I143N | 47.50 | 432.78 | 1275.87 | 279.24 | 4485.32 | 1958.70 | 18.92 |
| I143M | 50.00 | 464.09 | 1087.71 | 384.61 | 8034.80 | 1499.32 | 11.93 |
| I143V | 55.00 | 789.77 | 1354.78 | 679.03 | 12497.59 | 1911.76 | 29.87 |
| I143T | 35.00 | 591.71 | 1094.57 | 441.38 | 7771.47 | 2244.30 | 20.78 |
| I143F | 55.00 | 349.38 | 893.97 | 355.55 | 9068.61 | 1247.87 | 18.92 |
| M146V | 40.00 | 583.19 | 985.19 | 563.38 | 17117.29 | 2609.66 | 34.76 |
| M146I | 45.00 | 492.76 | 967.76 | 512.13 | 14485.76 | 2040.52 | 34.76 |
| M146L | 43.00 | 392.49 | 604.90 | 377.11 | 9150.66 | 1670.19 | 14.58 |
| TI47I | 42.00 | 508.29 | 1089.76 | 1340.52 | 7827.06 | 2536.88 | 0.00 |
| T147P | 32.00 | 1179.10 | 1362.78 | 899.59 | 23960.35 | 2068.87 | 41.16 |
| L150P | 60.00 | 584.89 | 3256.36 | 524.70 | 25857.23 | 2927.55 | 921.04 |
| L153V | 36.00 | 608.59 | 1164.25 | 656.10 | 18447.80 | 4692.06 | 58.79 |
| Y154C | 41.00 | 255.54 | 384.73 | 365.07 | 7412.62 | 975.66 | 430.89 |
| Y154N | 40.00 | 265.76 | 581.63 | 423.46 | 18960.09 | 3110.63 | 434.89 |
| H163P | 33.00 | 237.08 | 405.68 | 422.48 | 13600.92 | 4220.69 | 91.26 |
| H163R | 45.00 | 251.50 | 524.26 | 379.27 | 10482.86 | 1736.93 | 24.14 |
| H163Y | 47.00 | 327.94 | 705.57 | 463.32 | 12615.87 | 1657.06 | 28.53 |
| A164V | 75.00 | 926.58 | 840.14 | 590.30 | 11165.17 | 973.45 | 16.87 |
| W165G | 36.00 | 1277.43 | 1037.67 | 3563.53 | 24429.23 | 7449.12 | 52.68 |
| W165C | 42.00 | 3089.53 | 1182.58 | 4801.76 | 27899.21 | 5727.84 | 46.78 |
| L166del | 38.00 | 411.36 | 753.14 | 932.06 | 8040.98 | 1879.00 | 20.78 |
| L166V | 42.00 | 112.13 | 172.27 | 242.97 | 4767.79 | 885.02 | 38.08 |
| L166H | 30.00 | 397.63 | 5249.21 | 589.92 | 16246.34 | 9568.57 | 59.51 |
| L166R | 37.00 | 361.47 | 366.21 | 345.24 | 13135.11 | 3096.55 | 763.49 |
| L166P | 15.00 | 212.08 | 219.58 | 335.86 | 4949.36 | 2087.41 | 464.24 |
| I167del | 38.00 | 1279.00 | 2180.40 | 1938.94 | 26983.90 | 5831.77 | 44.97 |
| S169del | 42.00 | 985.42 | 1991.05 | 1924.98 | 30664.73 | 7890.59 | 63.03 |
| S169L | 31.00 | 813.09 | 704.49 | 546.32 | 11730.80 | 5266.73 | 35.90 |
| S169P | 31.00 | 1871.47 | 1792.84 | 2431.81 | 19077.62 | 8039.78 | 33.60 |
| L171P | 38.00 | 642.68 | 368.70 | 2139.92 | 12098.54 | 2021.98 | 22.52 |
| L173F | 50.00 | 1028.02 | 294.91 | 568.07 | 8773.80 | 1389.61 | 0.00 |
| L173W | 26.00 | 1318.53 | 422.16 | 553.12 | 12811.22 | 8435.97 | 34.76 |
| L174R | 48.00 | 414.82 | 297.41 | 371.68 | 15294.97 | 5171.21 | 140.98 |
| L174M | 59.00 | 537.24 | 2396.63 | 351.57 | 12942.79 | 1995.80 | 55.04 |
| F175S | 61.00 | 1165.51 | 2226.42 | 1157.34 | 21737.70 | 2870.09 | 28.53 |
| F176L | 51.00 | 921.78 | 1313.38 | 726.70 | 19432.52 | 2001.19 | 16.49 |
| F177S | 30.00 | 990.85 | 690.97 | 1348.35 | 13573.62 | 5504.69 | 29.59 |
| E184D | 42.00 | 717.46 | 1011.29 | 850.15 | 20280.72 | 3278.52 | 32.50 |
| I202F | 54.00 | 1040.80 | 1006.18 | 974.83 | 14710.44 | 1736.55 | 6.32 |
| G206A | 55.00 | 523.71 | 608.72 | 484.22 | 7659.71 | 988.51 | 0.00 |
| G206D | 33.00 | 494.56 | 715.02 | 762.56 | 10576.88 | 1911.97 | 13.84 |
| G206V | 30.00 | 415.64 | 274.91 | 805.78 | 6768.73 | 2207.52 | 0.00 |
| G206S | 32.00 | 503.19 | 583.25 | 956.78 | 9633.85 | 2263.48 | 16.49 |
| G209R | 46.00 | 251.15 | 230.13 | 207.53 | 5293.68 | 2729.45 | 179.81 |
| G209V | 41.30 | 456.36 | 362.49 | 368.53 | 12096.31 | 4470.52 | 32.50 |
| G209A | 40.00 | 483.64 | 647.15 | 703.74 | 9594.65 | 2605.26 | 16.49 |
| G209E | 43.00 | 167.36 | 189.48 | 235.89 | 2397.42 | 575.80 | 0.00 |
| S212Y | 46.50 | 618.80 | 917.03 | 608.23 | 15112.27 | 2165.02 | 31.08 |
| I213F | 33.00 | 342.80 | 364.18 | 484.64 | 24415.83 | 6829.26 | 241.09 |
| I213T | 45.00 | 855.43 | 623.90 | 515.66 | 14708.16 | 1533.98 | 16.49 |
| H214N | 46.00 | 449.95 | 893.03 | 506.84 | 12122.80 | 1868.39 | 24.71 |
| H214Y | 41.00 | 360.04 | 622.25 | 425.59 | 11447.28 | 1480.97 | 16.49 |
| H214D | 55.00 | 733.53 | 520.54 | 468.51 | 11869.31 | 1260.43 | 22.89 |
| G217D | 36.00 | 776.16 | 1110.67 | 827.80 | 24482.49 | 3745.34 | 69.84 |
| Q222H | 35.00 | 500.79 | 721.01 | 762.56 | 25424.62 | 4361.24 | 117.52 |
| L226F | 33.00 | 136.43 | 174.33 | 164.86 | 5117.69 | 1294.78 | 28.05 |
| S230R | 58.00 | 385.87 | 332.75 | 292.65 | 6804.69 | 796.47 | 31.08 |
| A231P | 53.00 | 466.69 | 500.92 | 571.04 | 22119.75 | 3315.71 | 115.61 |
| A231T | 52.00 | 397.03 | 472.37 | 264.48 | 8509.63 | 968.69 | 0.00 |
| M233T | 38.00 | 1123.48 | 2991.78 | 1028.37 | 8485.56 | 3937.30 | 6.32 |
| M233I | 28.00 | 635.86 | 1110.79 | 884.00 | 14164.60 | 6142.56 | 0.00 |
| M233V | 31.00 | 263.95 | 478.70 | 336.58 | 4338.23 | 1683.59 | 0.00 |
| M223L | 46.00 | 219.33 | 350.14 | 114.42 | 5375.00 | 837.20 | 0.00 |
| L235P | 33.00 | 187.19 | 246.83 | 235.14 | 8136.71 | 1879.80 | 36.49 |
| L235R | 45.00 | 214.79 | 233.25 | 202.49 | 6802.44 | 1949.32 | 80.86 |
| L235V | 47.00 | 683.34 | 594.37 | 590.23 | 11412.50 | 1464.33 | 26.42 |
| F237I | 46.00 | 514.98 | 384.37 | 281.78 | 8452.47 | 874.79 | 0.00 |
| F237L | 47.00 | 752.22 | 533.15 | 431.61 | 11610.62 | 1311.43 | 18.83 |
| Q223R | 33.00 | 401.42 | 553.44 | 619.65 | 27309.18 | 5861.76 | 1393.90 |
| L248P | 42.00 | 792.17 | 1158.82 | 877.00 | 20660.37 | 4487.02 | 35.20 |
| L248R | 54.00 | 488.43 | 376.11 | 311.66 | 8624.73 | 906.10 | 6.32 |
| A246P | 47.00 | 542.04 | 894.98 | 708.99 | 17439.46 | 6312.23 | 88.96 |
| Y256S | 25.00 | 692.23 | 1315.93 | 352.04 | 10432.68 | 3902.22 | 799.69 |
| P267A | 45.00 | 918.07 | 1402.97 | 867.08 | 31142.12 | 4543.71 | 91.33 |
| P267L | 59.00 | 438.28 | 384.17 | 237.39 | 9861.54 | 1021.88 | 16.49 |
| P267S | 35.00 | 220.75 | 270.81 | 225.98 | 8621.72 | 1860.09 | 69.84 |
| R278I | 50.00 | 164.81 | 164.07 | 193.86 | 2787.89 | 351.31 | 212.18 |
| R278S | 39.00 | 144.06 | 299.88 | 159.79 | 6151.93 | 1206.30 | 182.45 |
| R278T | 37.00 | 171.79 | 328.12 | 205.02 | 7183.98 | 1465.37 | 256.96 |
| R278K | 40.00 | 169.53 | 437.78 | 162.85 | 9649.68 | 1741.75 | 239.27 |
| E280A | 48.00 | 418.70 | 904.10 | 485.47 | 14424.95 | 2706.68 | 76.20 |
| E280K | 48.00 | 687.49 | 1402.97 | 604.03 | 20808.60 | 2943.58 | 99.25 |
| L282R | 43.00 | 209.49 | 303.56 | 240.36 | 11095.80 | 1089.63 | 133.66 |
| L282V | 44.30 | 619.28 | 739.30 | 708.69 | 30128.05 | 4166.33 | 62.94 |
| L282F | 49.00 | 474.06 | 539.66 | 517.65 | 26497.82 | 2517.26 | 118.47 |
| V272A | 31.00 | 402.85 | 315.84 | 301.35 | 16799.08 | 3407.66 | 80.21 |
| V272D | 43.00 | 249.44 | 207.12 | 196.48 | 5926.89 | 782.89 | 117.52 |
| F386S | 48.00 | 289.09 | 491.61 | 355.28 | 14768.92 | 3907.38 | 612.41 |
| F388L | 43.00 | 609.74 | 466.37 | 361.16 | 10166.28 | 1088.59 | 13.84 |
| F386I | 50.00 | 268.88 | 447.98 | 221.29 | 8340.43 | 1581.78 | 69.10 |
| G384A | 34.00 | 384.43 | 434.87 | 221.29 | 5354.11 | 1784.31 | 124.01 |
| L424F | 57.00 | 930.85 | 1089.01 | 646.99 | 19611.88 | 2465.48 | 40.14 |
| L424H | 40.00 | 386.82 | 489.22 | 477.47 | 19637.83 | 4384.95 | 121.72 |
| L424R | 33.00 | 240.06 | 363.55 | 232.88 | 13283.99 | 3529.54 | 154.05 |
| L424V | 30.00 | 434.27 | 587.81 | 515.66 | 26126.44 | 4094.13 | 195.17 |
| E318G | 61.00 | 988.89 | 1063.06 | 555.84 | 12872.48 | 1213.58 | 16.49 |
| E9 Del | 45.00 | 210.15 | 345.40 | 192.10 | 6476.67 | 1207.34 | 173.08 |
Next, we calculated the classic Aβ42/40 ratio shown by numerous groups including ours to be increased by FAD PS1 mutations (e.g., [7, 20, 29–33]). There was a strong inverse correlation between Aβ42/40 level in the conditioned medium (CM) and that mutant’s estimated clinical AOO (Fig.1c, 1st panel). Compared to WT PS1 (mean ratio=0.098), most FAD PS1 mutations increased the 42/40 ratio in CM (Fig. 1c), but 22 (~20%) of the 131 mutations had 42/40 ratios within ± 20% of WT PS1. Hence, Aβ42/40 is not sufficiently sensitive to distinguish PS1 enzymatic changes caused by some FAD mutations. To search for a more sensitive correlation, we hypothesized that Aβ37 as the final end-product of processive γ-secretase cleavages would be a better indicator of the loss of presenilin carboxypeptidase-like function from FAD mutations [34, 35]. In accord with this concept, we found that the Aβ37/42 ratio in the CM strongly differentiated WT from FAD mutant PS1 effects (Fig. 1c, right panel); > 75% of the PS1 mutations produced an Aβ37/42 ratio lower than 50% of the WT PS1 ratio. Moreover, only 6 of 131 mutants had a 37/42 ratio at or above the level of WT PS1 (Fig. 1c, right panel). Thus, Aβ37/42 was the best benchmark for differentiating FAD mutants from WT PS1, a result consistent with a much smaller collection of FAD mutations analyzed in our recent report [20].
Extracellular Aβ37/42 ratio as a novel biomarker for presenilin activity in iPSC-derived patient neurons bearing AD-causing PS1 mutations
Considering the limitations of the transient and non-neural HEK293 over-expression system, we next examined iPSC-derived human neurons from two independent FAD mutation carriers and their unaffected family members (from the DIAN study [36]) as a far more physiological system. These NGN2-induced neurons are highly useful tools to analyze PS1 proteolytic functions in culture, as they reflect what may happen in the patient’s brain, without artifacts from gene copy numbers or transfection efficiency and relying on entirely endogenous components [37]. We measured the concentrations of Aβx-37, Aβx-40, and Aβx-42 as well as the Aβ37/40, Aβ42/40 and Aβ37/40 ratios (Fig. 2a). The ratios of peptides secreted by the non-carrier (WT) neurons were similar, demonstrating the consistency of this analytical culture system. Aβ37/40, Aβ42/40, and Aβ37/42 ratios significantly distinguished carriers of PS1 mutations G217R and H163R from their WT relatives, with 37/42 showing the largest difference between WT and mutant, consistent with the HEK293 cell data.
Figure 2. Aβ 37 is an analyte more sensitive to chemical modulations of γ-secretase activity.
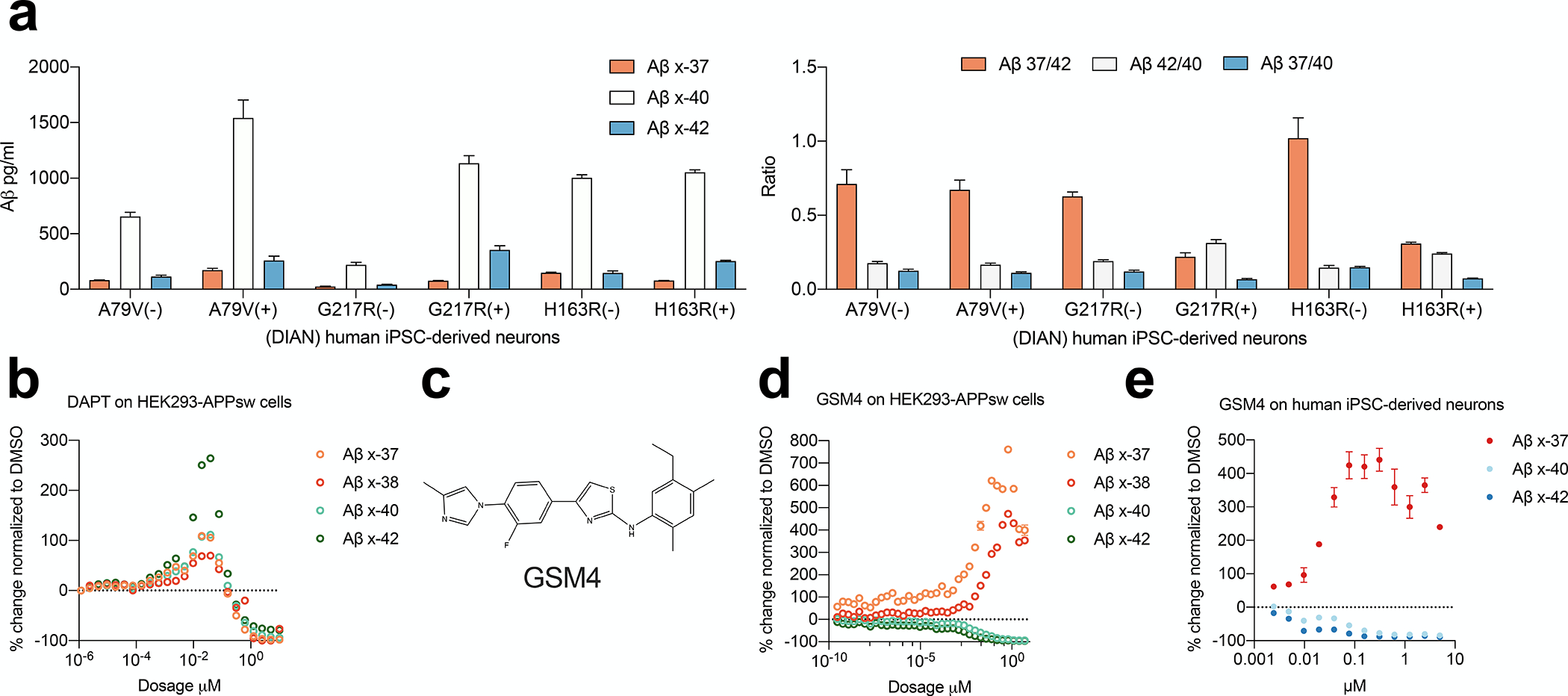
a) Indicated concentrations and ratios of Aβs secreted from iPSC-derived human neurons (means ± SD, n=6, Non-paired Student’s t-test: ***, p<0.001; ****, p<0.0001). b) Aβs secreted from HEK293-swAPP cells treated with DAPT from 10 μM down to 1.1 pM (n=2 technical replicates; means ± SEM). c) Molecular representation of the GSM4. d-e) Aβs secreted from HEK293-swAPP cells and on human iPSC-derived neurons treated with serial doses of GSM4 (n=2 technical replicates; means ± SEM).
Aβ37 production is more sensitive to pharmacological inhibition and modulation of γ-secretase
We next explored the γ-secretase proteolytic properties reflected by the novel Aβ37/42 ratio, which already serves as a highly sensitive read-out of PS dysfunction across our large, diverse panel of FAD mutations mentioned above. First, we used a well-characterized γ-secretase inhibitor (GSI), DAPT, to treat HEK293 cells expressing the APP KM670/671NL (“Swedish”) FAD mutation (HEK293-APPsw), which markedly enhances β-secretase cleavage and thus total Aβ production. We applied serially diluted DAPT doses ranging from 1.12 pM up to 10 μM. Secreted Aβx-37, x-38, x-40, and x-42 were quantified in the conditioned media (Fig. 2b). We observed the “paradoxical” increase of Aβ generation, especially x-42, in the 20 nM to 200 nM low dose range of DAPT, an effect reported multiple times with GSIs (e.g., [38]). Further analysis of the pattern of DAPT inhibition showed that Aβ37 was more suppressed by DAPT than Aβ40 or Aβ42, suggesting that Aβ37 production is somewhat more sensitive to the pharmacological action of a γ-secretase inhibitor. We, therefore, turned to γ-secretase modulators (GSMs), which appears as an attractive small-molecule therapeutic approach targeting the β-amyloidosis of AD, as they allow γ-cleavage of all substrates to occur but shift the specific cleavages in the APP TMD toward shorter Aβ peptides such as Aβ37 and 38, probably by enhancing the substrate and enzyme affinity [20]. This GSM approach has the twin advantages of lowering the strongly pro-aggregating Aβ42 and 43 peptides and raising the levels of the demonstrably anti-aggregating Aβ37 and 38 peptides [39]. We treated both the HEK293-APPsw and the iPSC-derived neurons with a very potent GSM, GSM4 (Fig. 2c), as reported [40]; this compound was resynthesized and characterized by us recently [41]. We observed the expected dose-dependent increase in short Aβs (37, 38) and decrease in long Aβs (40, 42) (Fig. 2d, e). In the HEK293-APPsw cells, GSM4 at as low as 0.29 pM could elevate Aβ37 by 118% but only decrease Aβ42 by 23% (Fig. 2d). This striking difference suggests Aβ37 production is more sensitive to the chemical modulation of γ-secretase, as it also is to the DAPT inhibitor (above). Based on these GSI and GSM sensitivities, we propose that Aβ37 production and the “short-to-long” Aβ37/42 ratio could have special utility in analyzing PS/γ-secretase dysfunction in biological samples, including in human extracellular fluids.
The Aβ 37/42 ratio correlates with neuropathology in ‘sporadic’ AD
Although autosomal dominant FAD represents < 0.1% of all AD cases, it may offer a unique opportunity to gain insights into the molecular mechanisms underlying cases of ‘sporadic’ Alzheimer’s disease (SAD), where PS and APP are both WT. To search for common mechanistic ground between the Aβ biochemistry of FAD and SAD, we asked whether γ-activity could be a driving factor in the latter cases, as well. Earlier studies have suggested that the process of aging in the brains of both lower primates and humans might alter PS/γ-secretase function, as observed in in vitro γ-secretase proteolytic assays from brain tissue extracts coupled with ELISAs of Aβ42/40, Aβ43/40, and Aβ38/42 [42–44]. We quantified Aβ37/42 together with the classic Aβ42/40 ratio in the postmortem brain tissue of 59 participants of the Religious Order Study (ROS) or the Rush Memory and Aging Project (MAP), two longitudinal studies of aging and dementia (demographics in Table 2). These 59 brains were categorized by ROS-MAP into 3 AD-relevant neuropathological groups: low-pathology with no cognitive impairment (LP-NCI), high-pathology with no cognitive impairment (HP-NCI), and Alzheimer’s disease (AD). We performed sequential extractions of frozen cerebral cortex into 3 fractions: TBS-soluble, 1% Triton-soluble, and 6 M guanidine HCl (GnCl)-soluble, and measured Aβx-37, x-40, and x-42 in each.
In the TBS-soluble extracts, HP-NCI and AD cortex had significantly more Aβx-37 than did LP-NCI, while Aβx-40 and x-42 did not significantly distinguish the groups (Fig. 3a). The canonical Aβ42/40 ratio was significantly lower overall in LP-NCI than in HP-NCI and AD, while Aβ 37/42 was significantly higher overall in LP-NCI than in HP-NCI and AD and showed greater differences between the groups; only a few LP-NCI subjects were down in the range of the HP-NCI and AD brains (Fig. 3a, far right). Next, in the Triton-soluble extracts (Fig. 3b), Aβ42/40 showed no significant differences between the LP-NCI and AD, whereas Aβ 37/42 was again very significantly higher in LP-NCI than both the HP-NCI and AD brains. In the GnCl soluble extracts, Aβ37/42 was again markedly (> 60-fold) and significantly higher in the LP-NCI brains than the HP-NCI and AD brain, whereas the 42/40 ratio showed smaller mean differences among the groups (Fig. 3c). Collectively, the data in Fig. 3 reveals that the strongly elevated Aβ37/42 ratio in all three sequential extracts significantly distinguishes LP-NCI from HP-NCI and AD brain tissue, especially in the TBS-soluble and Triton-soluble fractions. Importantly, the 37/42 ratio shows less overlap of values between brains with low and high AD-type pathology than does the classical 42/40 ratio (Fig. 3a–c).
Figure 3. Brain Aβ 37/42 correlated closely with AD-relevant brain pathology.
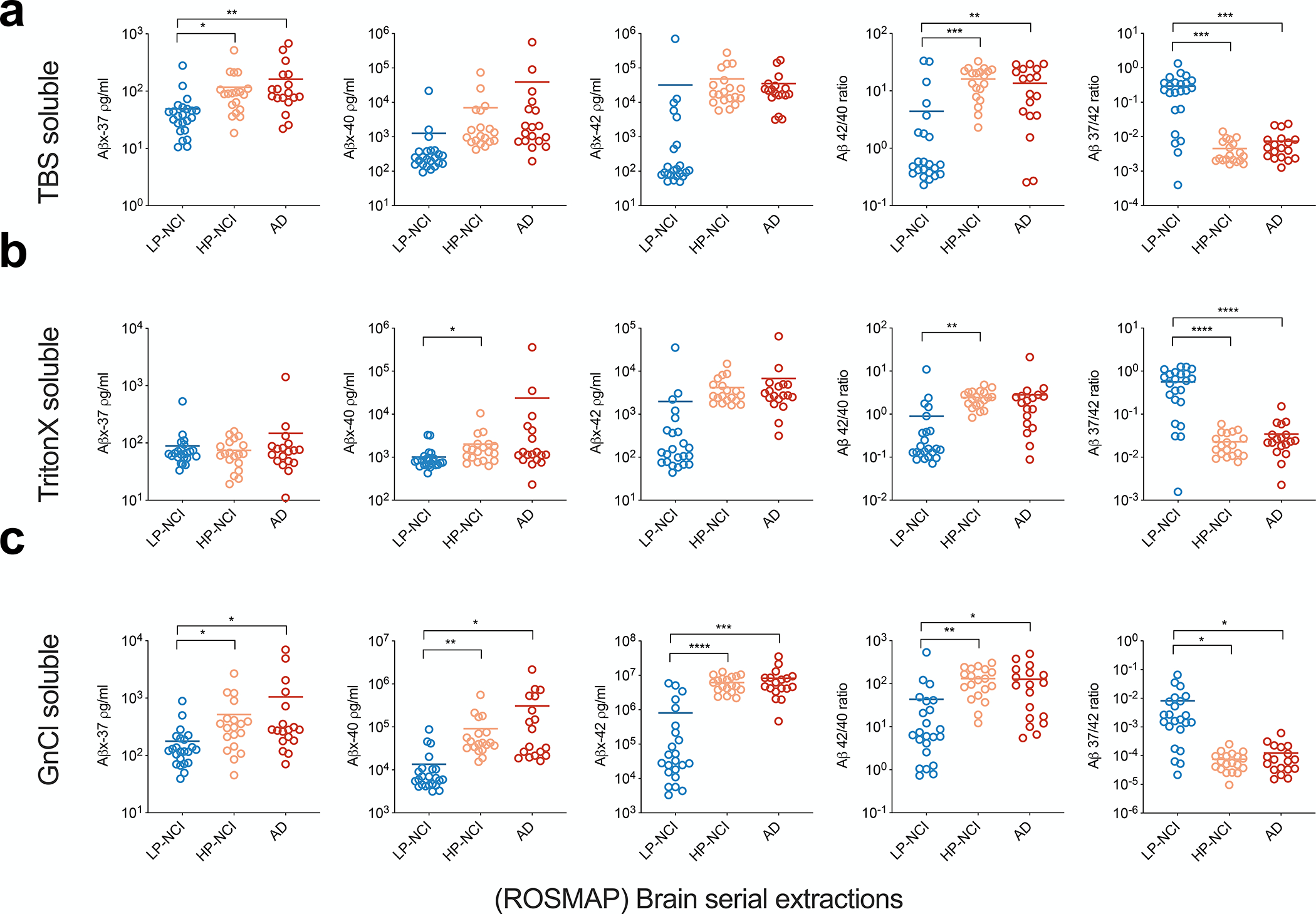
a) Aβ peptides measured in Tris-soluble fractions. b) Aβs measured in Triton-soluble fractions. c) Aβs measured in GnCl-soluble fractions. All graphs show means ± SDs; n=2 technical replicates. Non-paired Student’s t-test: * p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
We also found that there was little Aβ37 peptide aggregation into plaques in the AD brain. Immunohistochemistry for Aβ37 vs. Aβ42 (antibody 21F12) on formalin-fixed paraffin sections showed that Aβ37 was mainly found in cerebral blood vessels such as arterial walls, while Aβ42 was deposited in plaques (S3a-b). To further validate this finding, we also assessed Aβ37 (DAB+Ni; purple) and Aβ42 (DAB; brown) by double-labeling the same AD brain section (Fig. S3c–d). This finding is consistent with previous reports using MALDI imaging mass spectrometry [45], and immunohistochemistry [11]. Interestingly, we have recently obtained evidence that the ratio of secreted Aβ37/42 in the conditioned media of iPSC-derived human neurons generated from various ROS-MAP subjects correlates strongly with the subject’s AD-type neuropathology [37]. The cumulative findings from the ROS-MAP collection of rigorously phenotyped brains support our hypothesis that the Aβ37/42 ratio is strongly associated with SAD, as it is with FAD.
Aβ37/42 ratio as a biomarker for AD in CSF
In light of the above evidence that the Aβ37/42 ratio can distinguish AD brains from low-pathology control brains, we asked whether this ratio could also serve as a potential fluid biomarker for AD patients. We quantified all detectable soluble Aβ species (Aβx-37, x-38, x-39, x-40, x-42, and x-43) in 79 cerebrospinal fluid (CSF) samples collected from a cohort of clinically well-defined patients from the Mayo Clinic Alzheimer’s Disease Research Center (demographics in Table 2) divided into 3 groups: cognitively normal (CN), mild cognitive impairment (MCI) and Alzheimer’s disease (AD), as shown in Fig. 4a. First, we validated our Aβx-37 assay for measuring human CSF (Fig. S4a–b). We made serial dilutions of a pooled CSF sample from 2- to 32-fold and observed perfect linearity of signals so that the signal was still above LLoQ at 32-fold dilution. The recovery ratio was 100 ± 6%, indicating no significant matrix effects in CSF for this specific assay. Furthermore, we immunodepleted (ID) Aβ37 using anti-total Aβ (4G8) from a pooled CSF sample and found > 70% depletion of signals, with an anti-Aβ42 (21F12) antibody serving as a good negative control for the ID (Fig. S4b). These data indicate that the Aβx-37 assay is specific, linear, and reliable for complex human biological fluids such as CSF.
Figure 4. CSF Aβ 37/42 ratio distinguishes AD from both CN and MCI.
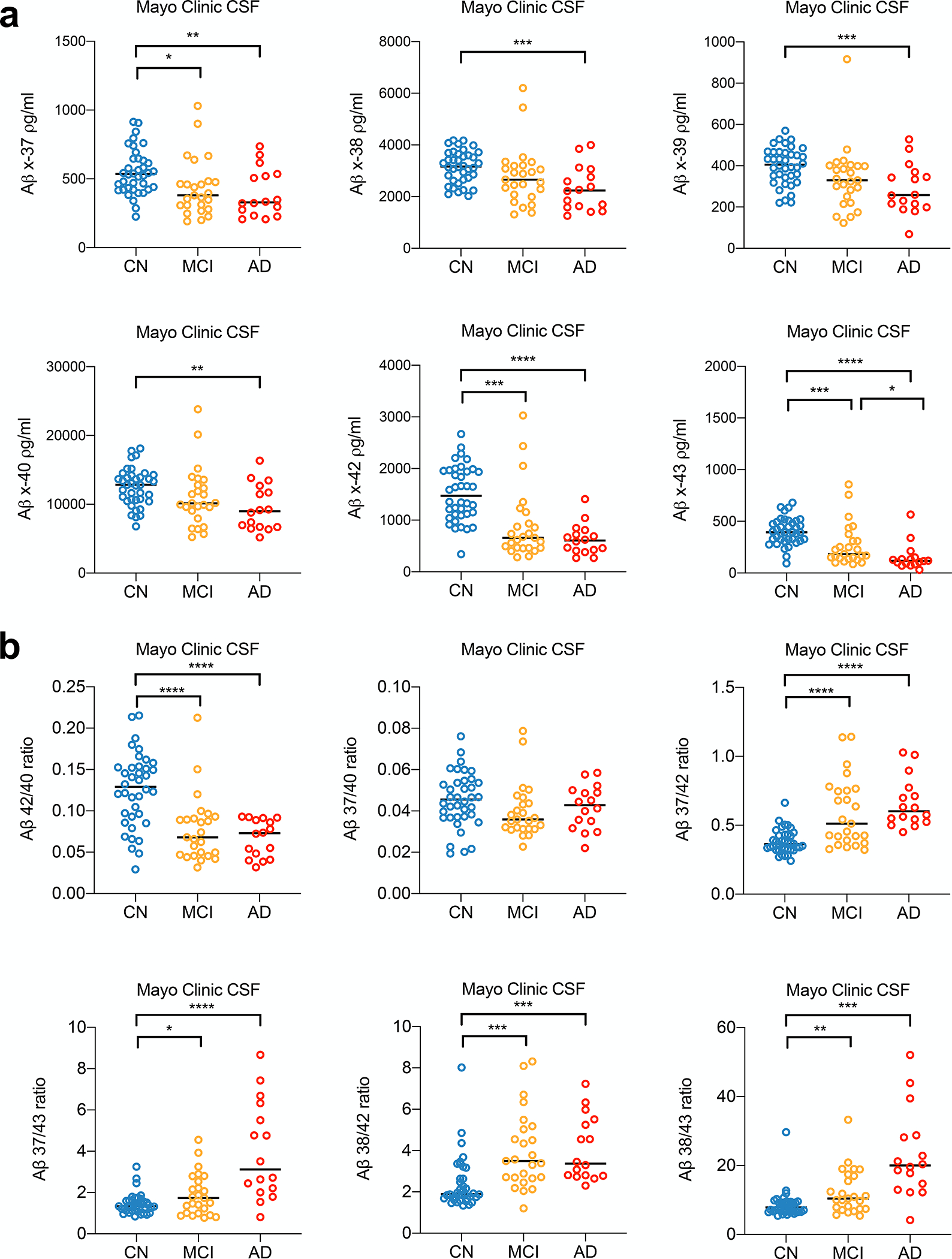
a-b) Aβs measured and ratios calculated in CSF samples from 3 clinical diagnoses (n=2 technical replicates, mean ± SD, Non-paired Student’s t-test: * p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001).
We observed significant reductions of all 6 Aβ species in MCI and AD CSF compared to the CN group (Fig. 4a and Table 3). It should be pointed out that the biological mechanisms may differ among the peptides: reduced long Aβ species such as Aβ40, 42, and 43 may reflect their aggregation in MCI and AD brain tissue, whereas reduced short Aβ species such as Aβ37, 38, and 39 may reflect the relative loss of production due to impaired γ-processivity that drives final cleavage towards Aβ 37. Next, we calculated peptide ratios, as we had for conditioned media and human brain extracts (Fig. 4b). Compared to the Aβ42/40 ratio, we again found the “short:long” Aβ ratio better distinguished CN and AD or CN and MCI, as demonstrated by analysis of receiver operating characteristic (ROC) curves (Fig. 5a). The short:long Aβ ratios outperformed the Aβ42/40 ratio, with Aβ37/42 ratio as the best short:long ratio, having an impressive ROC AUC of 0.9622 in distinguishing between CN and AD. Further analysis of this dataset revealed that 1) two short:long Aβ ratios (37/42 and 38/43) strongly correlate with each other in CSF (Fig. 5b); and 2) both the 37/42 and 38/43 ratios correlate significantly with the Mini-Mental State Examination (MMSE) scores measured when the CSF samples were drawn (Fig. 5b), while 42/40 ratio also correlates with MMSE scores, but less significantly (r = 0.3532, p = 0.0016).
Table 3.
Concentration of Aβs in CSF samples (pg/ml)
| Diagnosis group | Aβ x-37 | Aβ x-38 | Aβ x-39 | Aβ x-40 | Aβ x-42 | Aβ x-43 |
|---|---|---|---|---|---|---|
| CN | 417.18 | 2247.26 | 311.51 | 9792.00 | 1151.73 | 280.28 |
| 532.15 | 3424.53 | 492.67 | 14263.50 | 1658.32 | 519.83 | |
| 522.50 | 3596.65 | 442.86 | 15162.53 | 1042.51 | 392.01 | |
| 542.80 | 3191.45 | 426.05 | 13675.80 | 1662.17 | 310.26 | |
| 423.73 | 2373.97 | 281.08 | 9069.03 | 1292.72 | 250.18 | |
| 740.41 | 3296.89 | 450.68 | 13659.09 | 2203.85 | 276.66 | |
| 632.63 | 3518.88 | 466.32 | 14508.52 | 1989.48 | 493.77 | |
| 471.16 | 2854.43 | 389.79 | 12847.65 | 1954.71 | 445.23 | |
| 914.71 | 4043.02 | 528.91 | 15174.66 | 1870.35 | 603.55 | |
| 422.18 | 3134.90 | 400.24 | 11390.10 | 976.41 | 329.76 | |
| 480.07 | 3734.71 | 410.67 | 13069.90 | 1111.41 | 394.09 | |
| 843.47 | 4177.56 | 453.54 | 18120.41 | 2182.51 | 632.28 | |
| 398.67 | 2317.82 | 318.03 | 10411.02 | 1090.00 | 330.07 | |
| 554.95 | 2658.80 | 342.79 | 9269.79 | 1997.16 | 443.33 | |
| 648.22 | 3397.28 | 351.94 | 10707.56 | 2288.72 | 446.79 | |
| 543.50 | 3112.73 | 319.71 | 13331.40 | 1941.29 | 480.36 | |
| 563.96 | 2945.23 | 393.68 | 12081.20 | 1122.82 | 323.53 | |
| 467.26 | 3548.06 | 461.69 | 13850.32 | 1346.44 | 503.41 | |
| 287.46 | 2816.36 | 386.86 | 12704.69 | 838.43 | 305.52 | |
| 647.25 | 3718.82 | 466.91 | 14520.59 | 2408.23 | 637.12 | |
| 581.44 | 3414.57 | 485.08 | 13919.62 | 1839.99 | 520.50 | |
| 434.32 | 2167.86 | 220.43 | 8106.18 | 1237.42 | 233.73 | |
| 795.66 | 4076.53 | 569.91 | 17106.30 | 1649.12 | 507.98 | |
| 440.39 | 2618.42 | 359.09 | 10458.33 | 826.45 | 456.83 | |
| 906.59 | 3689.51 | 510.72 | 14202.41 | 2668.89 | 681.50 | |
| 757.54 | 3288.65 | 431.33 | 11084.78 | 1935.45 | 474.37 | |
| 614.00 | 3072.17 | 392.40 | 11641.10 | 1638.55 | 380.55 | |
| 693.06 | 3818.41 | 468.86 | 13629.38 | 2034.54 | 525.71 | |
| 421.99 | 2235.97 | 254.33 | 8376.03 | 1323.46 | 292.89 | |
| 760.99 | 3650.27 | 432.92 | 12823.03 | 1969.20 | 424.82 | |
| 226.00 | 2732.93 | 359.48 | 11654.32 | 340.71 | 92.13 | |
| 516.26 | 2022.45 | 231.94 | 6788.11 | 1221.29 | 158.87 | |
| 383.29 | 4169.24 | 527.16 | 17758.74 | 861.35 | 392.60 | |
| 576.77 | 3075.22 | 351.98 | 10758.54 | 1354.18 | 352.51 | |
| 461.62 | 2099.05 | 221.76 | 8327.96 | 1210.59 | 326.21 | |
| 396.74 | 3140.49 | 431.71 | 13424.11 | 853.92 | 327.45 | |
| 340.15 | 3982.95 | 510.81 | 16829.75 | 914.40 | 403.06 | |
| 538.88 | 2512.88 | 304.09 | 10619.54 | 1587.37 | 384.16 | |
| MCI | 380.18 | 2655.13 | 366.04 | 11898.55 | 742.68 | 376.01 |
| 476.32 | 2952.76 | 424.94 | 15198.18 | 1351.95 | 544.10 | |
| 463.58 | 3192.76 | 398.04 | 13992.01 | 616.71 | 183.21 | |
| 458.78 | 3351.87 | 417.03 | 12780.46 | 403.16 | 100.75 | |
| 1030.20 | 6206.62 | 916.21 | 20144.54 | 3029.10 | 756.75 | |
| 439.19 | 2559.47 | 323.42 | 10195.33 | 467.01 | 135.00 | |
| 308.25 | 3085.19 | 401.43 | 13571.47 | 678.10 | 309.25 | |
| 348.60 | 2344.63 | 294.42 | 9919.30 | 466.41 | 125.83 | |
| 309.54 | 1986.79 | 220.29 | 7943.94 | 736.66 | 251.99 | |
| 395.77 | 3337.77 | 398.37 | 12331.13 | 1231.21 | 451.67 | |
| 666.62 | 2882.59 | 337.23 | 9059.61 | 875.12 | 169.90 | |
| 248.06 | 1791.05 | 123.02 | 5677.27 | 282.24 | 86.05 | |
| 639.70 | 3526.33 | 479.08 | 13742.62 | 934.28 | 228.33 | |
| 366.13 | 2478.88 | 329.18 | 9597.11 | 859.03 | 248.68 | |
| 901.08 | 2932.80 | 379.73 | 11450.60 | 2433.89 | 435.99 | |
| 200.30 | 1375.39 | 174.80 | 6398.26 | 560.59 | 141.75 | |
| 356.12 | 3047.69 | 384.22 | 11409.94 | 456.19 | 160.43 | |
| 191.91 | 1309.30 | 152.49 | 5238.66 | 301.40 | 110.72 | |
| 483.65 | 2453.00 | 300.97 | 9610.41 | 1153.73 | 225.07 | |
| 313.45 | 2078.56 | 251.04 | 10146.69 | 461.52 | 167.30 | |
| 279.87 | 2290.59 | 309.68 | 9966.47 | 657.02 | 308.64 | |
| 227.17 | 1568.49 | 153.18 | 6456.95 | 584.55 | 151.07 | |
| 267.76 | 1672.94 | 214.33 | 6670.59 | 493.89 | 149.64 | |
| 670.85 | 5446.76 | 403.78 | 23809.43 | 2052.33 | 856.88 | |
| 459.45 | 3210.46 | 282.53 | 9676.74 | 405.72 | 101.33 | |
| AD | 737.04 | 3851.94 | 481.96 | 16339.13 | 1407.18 | 566.24 |
| 226.63 | 1586.25 | 188.55 | 6351.89 | 459.73 | 133.95 | |
| 207.19 | 1257.66 | 68.91 | 5178.71 | 381.52 | 30.82 | |
| 207.45 | 1699.71 | 198.47 | 6959.08 | 268.48 | 102.98 | |
| 323.20 | 1632.90 | 215.27 | 6453.81 | 589.69 | 115.46 | |
| 506.41 | 2260.49 | 334.82 | 8787.39 | 806.86 | 92.03 | |
| 233.45 | 1848.20 | 230.08 | 7388.39 | 407.44 | 106.45 | |
| 535.53 | 2382.84 | 259.58 | 9163.66 | 847.38 | 151.40 | |
| 334.11 | 2586.74 | 345.17 | 11468.01 | 469.09 | 84.56 | |
| 296.91 | 3065.58 | 407.81 | 13454.80 | 426.31 | 126.88 | |
| 616.37 | 3124.12 | 369.84 | 12677.18 | 687.38 | 134.47 | |
| 348.52 | 1432.58 | 179.34 | 6677.23 | 620.90 | 71.21 | |
| 275.97 | 1406.93 | 216.17 | 6703.41 | 269.28 | 73.01 | |
| 324.45 | 2219.50 | 256.61 | 9234.34 | 720.00 | 338.64 | |
| 674.76 | 3993.89 | 528.15 | 13804.31 | 667.63 | 119.22 | |
| 520.55 | 2757.47 | 342.98 | 11708.41 | 1040.77 | 212.80 |
Figure 5. CSF Aβ 37/42 ratio distinguishes AD from CN and correlates with cognitive scores.
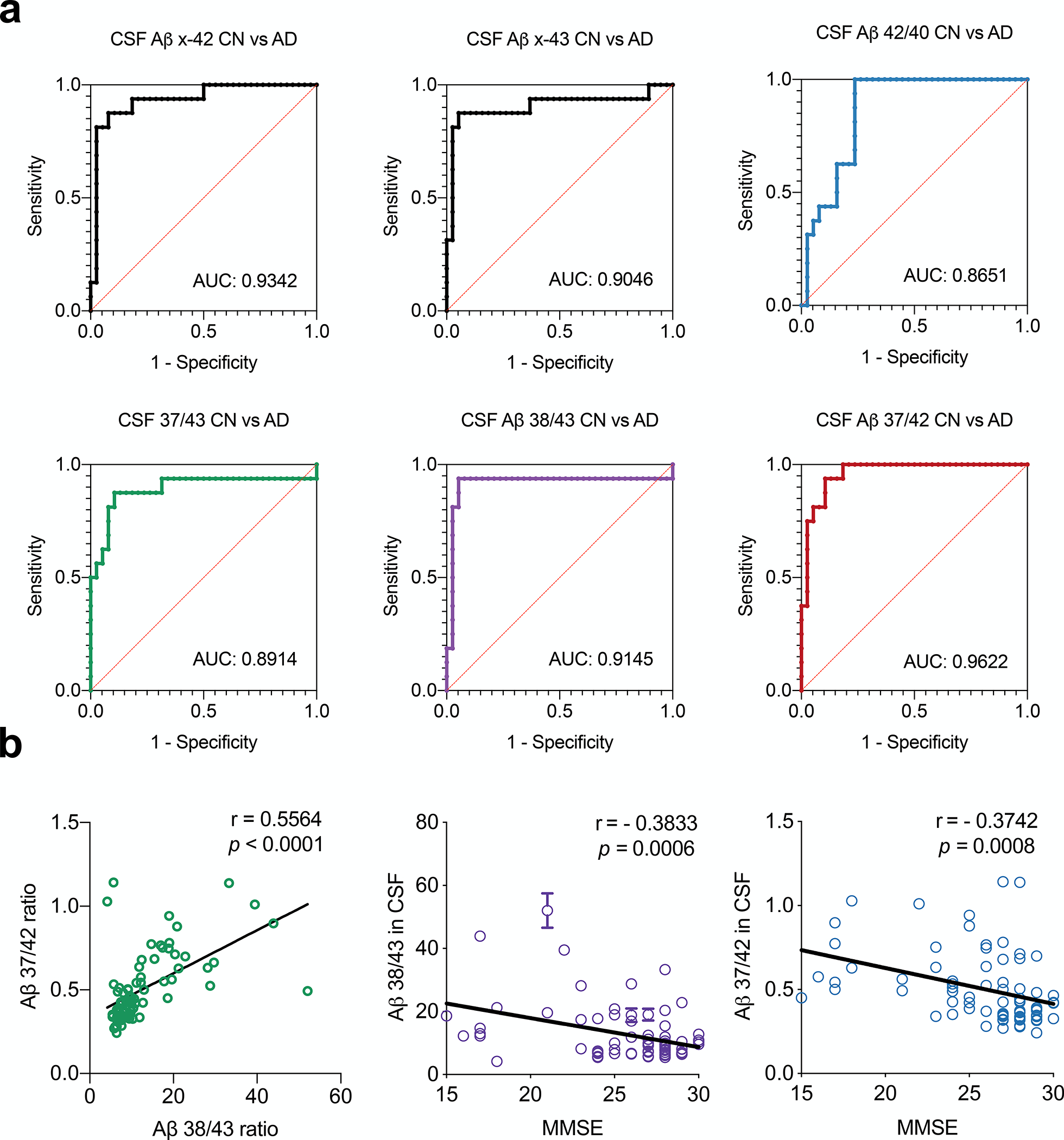
a) Area under the curve (AUC) was calculated by plotting a Receptor Operator Curve (ROC) between CN and AD subjects for Aβ ratios. b) Plot of CSF Aβ 37/42 ratio vs. Aβ 38/43, left. Plots of CSF Aβ 37/42 and Aβ 38/43 vs. the Mini-Mental State Exam (MMSE) scores of patients at the time of CSF collection (p value analyzed by Pearson correlation coefficient; r analyzed by linear regression.
Discussion
Our data from cultured cells, human brain and CSF all support the hypothesis that the Aβ37/42 ratio, validated here for the first time, is a novel metric for the common pathogenic features of APP processing in FAD and SAD. From recent SAD clinical trials of four Aβ antibodies that each lower brain levels of Aβ (aducanumab; gantenerumab; lecanemab; donanemab), we have learned that factors associated with positive biomarker signals and some apparent clinical benefit include high antibody exposure over prolonged intervals and early initiation of treatment, preferably at the early symptomatic or even pre-symptomatic phases. Given the genetic, neuropathological and biomarker evidence that Aβ accumulation begins long before symptoms, an inexpensive set of fluid biomarkers that can help predict future cognitive dysfunction at early stages of Aβ deposition could be highly advantageous for AD therapeutic development and management. To address this goal, we need a deeper understanding of APP processing and Aβ metabolism in normal and AD subjects. In this study, we provide data supporting a new and improved biomarker, Aβx-37, and specifically its ratio to Aβx-42, as a compelling means to evaluate γ-secretase processivity and brain and CSF Aβ accumulation. When ratioed to Aβ42 in the same sample, Aβ37 was able to sensitively distinguish physiological from pathological status in 1) PS1 mutant vs. WT cultured cells; 2) sporadic AD vs. control brain tissue; and 3) AD vs. normal CSF.
Practical fluid biomarkers for quantifying Aβ accumulation were heretofore described exclusively as Aβ42 and Aβ40. Recent studies suggest that the Aβ42/40 ratio in plasma is decreased by 10–20% in AD patients vs. age-matched controls [46–48]. This is a small difference, considerably less than the >30% fall in this ratio in AD CSF [47, 49–51]. Here, we propose a novel ratio between the final (shortest) product of processive γ-secretase cleavage, Aβ 37, and the longer, more hydrophobic and self-aggregating pathogenic species, Aβ42. We document an increase of the mean Aβ37/42 ratio by ~70% (0.3816 to 0.6610) in AD CSF (Fig. 4b). Moreover, the Aβ 37/42 ratios from CSF correlated significantly with MMSE, a widely used basic measure of cognitive function that has responded to Aβ antibody treatment in some clinical trials, making 37/42 a unique biomarker, since the 42/40 ratio in plasma has been quantitatively correlated with Aβ pathology (amyloid-PET positivity) [46, 47] but not level of cognitive function. Previous studies reported impaired γ-secretase activity with age by analyzing non-human primate brains aged 4–36 years [43] as well as in aged human brains that correlated with the level of Aβ pathology [42]. Impaired γ-secretase processivity could also be implicated in diverse cellular functions, as its hundreds of substrates are involved in many different cellular events. Regarding Aβ, we find that the Aβ 37/42 ratio correlates with 1) age of onset of clinical symptoms in FAD PS1 mutant carriers; 2) brain pathology of SAD patients; and 3) cognitive consequences of AD pathology.
An important advantage of our Aβ37 analyte is that its immunoassay is highly practical and reproducible, with low CVs. It can be performed on a standard immunoassay platform, in our case the MesoScale Discovery instrument, without the need for an expensive ultrasensitive (e.g., bead-based) platform. This technical advantage will help ensure that Aβ37 is measured accurately and reproducibly by clinical labs in the future. Moreover, a conceptual advantage of the assay is that it creates a ratio of the shortest product of presenilin-mediated processive processing of APP to the longest such product (Aβ42; Aβ43 is often undetectable in human biofluids), thereby maximizing the ability to detect a delta in γ-secretase trimming efficiency. Taking advantage of this highly sensitive and specific assay (Fig. 5a), we now plan to analyze larger longitudinal cohorts of CSF and also translate this assay into plasma, in order to quantify Aβ dyshomeostasis in brain and biofluids at earlier stages of AD pathogenesis.
In conclusion, the comprehensive immunochemical quantification of all Aβ C-terminal variants in cultured cells, brain tissue and CSF has led to the identification and validation of the novel Aβ37/42 ratio that holds promise as a more sensitive and specific CSF biomarker for clinical diagnosis in this head-to-head comparison (Fig. 5a) with the Aβ42/40 ratio, an already compelling and increasingly used biomarker of β-amyloidosis.
Supplementary Material
Supplementary Figure 1. Performance of novel Aβ assays on the MSD platform. a) Standard curves of new Aβ assays using synthetic peptide calibrators, Lower Limit of Quantification (LLOQ) was calculated by the lowest standard >2 times background. b) Test of specificity of each immunoassay using all 6 distinct Aβ calibrator peptides.
Supplementary Figure 4. Assay validation for Aβ x-37 in CSF. a) Measurement of Aβ x-37 from pooled human CSF after serial dilutions (n=2 technical replicates). b) Measurement of Aβ x-37 from pooled CSF before vs. after immunodepleting with anti-Aβ antibody 4G8 or control IgG.
Supplementary Figure 2. Normalized Aβs secreted from transfectants of 131 FAD mutants in PS dKO 293 cells. a) Each secreted Aβ is normalized to the total of secreted Aβs (Aβ37+38+39+40+42+43) and presented as a ratio, with the levels from WT-PS1 transfectants labeled as the red dotted line. b) Statistical analysis of data from Fig. 1b and Fig S1a (p value analyzed by Pearson correlation coefficient; r analyzed by linear regression.
Supplementary Figure 3. Immunohistochemistry of Aβ37 and Aβ42 in human (AD) brain. a-b) Immunostaining of Aβ37 and 42 on serial paraffin sections. c-d) Double labeling of Aβ37 (DAB+Ni; purple) and Aβ42 (DAB; brown) on the serial paraffin section. Scale bar = 200 μm for a-c) and 100 μm for d).
RESEARCH IN CONTEXT.
Systematic review: To better understand the dynamic of Aβ in the pathogenesis of AD, rigorous characterization and quantification of all secreted Aβ from human brains and biofluids are critical and essential. To date, there have been no study fully characterized all six secreted C-terminal variants of Aβ in brain and CSF systematically.
Interpretation: Through a systematic analysis using culture cells including human iPSC derived neurons, human brain tissues and CSF, we provide an improved biomarker, Aβ 37/42 ratio. It sensitively detects presenilin/γ-secretase dysfunction; correlates with brain amyloid deposition and better distinguishes CN from AD than Aβ42/40 in CSF.
Future directions: These findings support and encourage further research in the development of plasma Aβ37/42 ratios in combination with other surrogate biomarkers of cortical Aβ deposition to facilitate the development and clinical implementation of novel drug candidates against AD.
Acknowledgments
We are grateful to members of the Selkoe laboratory for helpful discussions. We thank Dr. R. Petersen and colleagues, Mayo Clinic, Rochester, MN for generously providing CSF samples.
Data collection and sharing for this project was supported by The Dominantly Inherited Alzheimer Network (DIAN, U19AG032438) funded by the National Institute on Aging (NIA), the Alzheimer’s Association (SG-20-690363-DIAN), the German Center for Neurodegenerative Diseases (DZNE), Raul Carrea Institute for Neurological Research (FLENI), Partial support by the Research and Development Grants for Dementia from Japan Agency for Medical Research and Development, AMED, and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), Spanish Institute of Health Carlos III (ISCIII), Canadian Institutes of Health Research (CIHR), Canadian Consortium of Neurodegeneration and Aging, Brain Canada Foundation, and Fonds de Recherche du Québec – Santé. This manuscript has been reviewed by DIAN Study investigators for scientific content and consistency of data interpretation with previous DIAN Study publications. We acknowledge the altruism of the participants and their families and contributions of the DIAN research and support staff at each of the participating sites for their contributions to this study.
This work was funded by the following NIH grants: R01 AG06173 (DJS), R01 AG071865 (DJS and LL), PPG AG015379 (DJS), R01 AG055909 (TYP) and R03 AG063046 (LL).
The brain tissue results published here are in whole or in part based on data obtained from the AMP-AD Knowledge Portal (https://adknowledgeportal.synapse.org/). Study data were provided by the Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago. ROS-MAP data collection was supported through funding by NIA grants P30AG10161, R01AG15819, R01AG17917, R01AG30146, R01AG36836, U01AG32984, U01AG46152, the Illinois Department of Public Health, and the Translational Genomics Research Institute.
Funding:
This work was funded by National Institutes of Health grants R01 AG006173 (DJS), R01 AG071865 (DJS and LL) and P01 AG015379 (DJS), R01 AG055909 (TYP), and the Davis APP program at BWH (DJS). LL was supported by the National Institutes of Health grant R03AG063046, a MADRC development project grant, and a BWH Program for Interdisciplinary Neuroscience pilot grant. The funders had no role in data collection, analysis, or decision to publish.
Abbreviations:
- AD
Alzheimer’s disease
- oAβ
oligomeric amyloid β-protein
- LLoQ
Lower Limit of Quantification
- MSD
Meso Scale Discovery Assay
- CLIA
Clinical Laboratory Improvement Amendments
- CN
cognitively normal
Footnotes
Declaration of Interests
DJS is a director and consultant of Prothena Biosciences. The other authors declare no competing financial interests.
References:
- [1].Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of γ-secretase activity. Nature cell biology 2003; 5: 486–488. [DOI] [PubMed] [Google Scholar]
- [2].Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc Natl Acad Sci U S A 2003; 100: 6382–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y et al. The role of presenilin cofactors in the γ-secretase complex. Nature 2003; 422: 438–441. [DOI] [PubMed] [Google Scholar]
- [4].Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 1999; 398: 513–517. [DOI] [PubMed] [Google Scholar]
- [5].Haapasalo A, Kovacs DM. The many substrates of presenilin/γ-secretase. J Alzheimers Dis 2011; 25: 3–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chen AC, Kim S, Shepardson N, Patel S, Hong S, Selkoe DJ. Physical and functional interaction between the α- and γ-secretases: A new model of regulated intramembrane proteolysis. J Cell Biol 2015; 211: 1157–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Liu L, Ding L, Rovere M, Wolfe MS, Selkoe DJ. A cellular complex of BACE1 and γ-secretase sequentially generates Aβ from its full-length precursor. Journal of Cell Biology 2019; 218: 644–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bolduc DM, Montagna DR, Seghers MC, Wolfe MS, Selkoe DJ. The amyloid-beta forming tripeptide cleavage mechanism of γ-secretase. Elife 2016; 5: e17578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kakuda N, Shoji M, Arai H, Furukawa K, Ikeuchi T, Akazawa K et al. Altered γ-secretase activity in mild cognitive impairment and Alzheimer’s disease. EMBO molecular medicine 2012; 4: 344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Takami M, Nagashima Y, Sano Y, Ishihara S, Morishima-Kawashima M, Funamoto S et al. γ-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl terminal fragment. Journal of Neuroscience 2009; 29: 13042–13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Reinert J, Richard BC, Klafki HW, Friedrich B, Bayer TA, Wiltfang J et al. Deposition of C-terminally truncated Aβ species Aβ37 and Aβ39 in Alzheimer’s disease and transgenic mouse models. Acta Neuropathologica Communications 2016; 4: [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wiltfang J, Esselmann H, Bibl M, Smirnov A, Otto M, Paul S et al. Highly conserved and disease-specific patterns of carboxyterminally truncated Abeta peptides 1–37/38/39 in addition to 1–40/42 in Alzheimer’s disease and in patients with chronic neuroinflammation. J Neurochem 2002; 81: 481–496. [DOI] [PubMed] [Google Scholar]
- [13].Bibl M, Esselmann H, Mollenhauer B, Weniger G, Welge V, Liess M et al. Blood-based neurochemical diagnosis of vascular dementia: A pilot study. Journal of neurochemistry 2007; 103: 467–474. [DOI] [PubMed] [Google Scholar]
- [14].Höglund K, Hansson O, Buchhave P, Zetterberg H, Lewczuk P, Londos E et al. Prediction of Alzheimer’s disease using a cerebrospinal fluid pattern of C-terminally truncated β-amyloid peptides. Neurodegenerative diseases 2008; 5: 268–276. [DOI] [PubMed] [Google Scholar]
- [15].Lewczuk P, Esselmann H, Bibl M, Paul S, Svitek J, Miertschischk J et al. Electrophoretic separation of amyloid β peptides in plasma. Electrophoresis 2004; 25: 3336–3343. [DOI] [PubMed] [Google Scholar]
- [16].Pannee J, Törnqvist U, Westerlund A, Ingelsson M, Lannfelt L, Brinkmalm G et al. The amyloid-β degradation pattern in plasma—a possible tool for clinical trials in Alzheimer’s disease. Neuroscience letters 2014; 573: 7–12. [DOI] [PubMed] [Google Scholar]
- [17].Portelius E, Andreasson U, Ringman JM, Buerger K, Daborg J, Buchhave P et al. Distinct cerebrospinal fluid amyloid β peptide signatures in sporadic and PSEN1 A431E-associated familial Alzheimer’s disease. Molecular Neurodegeneration 2010; 5: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bai X-c, Yan C, Yang G, Lu P, Ma D, Sun L et al. An atomic structure of human γ-secretase. Nature 2015; 525: 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhou R, Yang G, Guo X, Zhou Q, Lei J, Shi Y. Recognition of the amyloid precursor protein by human γ-secretase. Science 2019; 363: [DOI] [PubMed] [Google Scholar]
- [20].Liu L, Lauro BM, Wolfe MS, Selkoe DJ. Hydrophilic loop 1 of Presenilin-1 and the APP GxxxG transmembrane motif regulate γ-secretase function in generating Alzheimer-causing Aβ peptides. J Biol Chem 2021; 296: 100393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Petersen RC, Roberts RO, Knopman DS, Geda YE, Cha RH, Pankratz VS et al. Prevalence of mild cognitive impairment is higher in men: The Mayo Clinic Study of Aging. Neurology 2010; 75: 889–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Roberts RO, Geda YE, Knopman DS, Cha RH, Pankratz VS, Boeve BF et al. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology 2008; 30: 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].St Sauver JL, Grossardt BR, Yawn BP, Melton III LJ, Pankratz JJ, Brue SM et al. Data resource profile: the Rochester Epidemiology Project (REP) medical records-linkage system. International journal of epidemiology 2012; 41: 1614–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013; 78: 785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Muratore CR, Zhou C, Liao M, Fernandez MA, Taylor WM, Lagomarsino VN et al. Cell-type Dependent Alzheimer’s Disease Phenotypes: Probing the Biology of Selective Neuronal Vulnerability. Stem Cell Reports 2017; 9: 1868–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. Religious orders study and rush memory and aging project. Journal of Alzheimer’s Disease 2018; 64: S161–S189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kitamoto TKJS, Ogomori K, Tateishi J, Prusiner SB. Formic acid pretreatment enhances immunostaining of cerebral and systemic amyloids. Laboratory investigation; a journal of technical methods and pathology 1987; 57: 230–236. [PubMed] [Google Scholar]
- [28].Jäkel L, Boche D, Nicoll JAR, Verbeek MM. Aβ43 in human Alzheimer’s disease: effects of active Aβ42 immunization. Acta Neuropathol Commun 2019; 7: 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T et al. Familial Alzheimer’s disease–linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron 1996; 17: 1005–1013. [DOI] [PubMed] [Google Scholar]
- [30].Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G et al. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nature medicine 1997; 3: 67–72. [DOI] [PubMed] [Google Scholar]
- [31].Citron M, Eckman CB, Diehl TS, Corcoran C, Ostaszewski BL, Xia W et al. Additive effects of PS1 and APP mutations on secretion of the 42-residue amyloid β-protein. Neurobiology of disease 1998; 5: 107–116. [DOI] [PubMed] [Google Scholar]
- [32].Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N et al. Secreted amyloid β–protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nature medicine 1996; 2: 864–870. [DOI] [PubMed] [Google Scholar]
- [33].Sun L, Zhou R, Yang G, Shi Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase. Proceedings of the National Academy of Sciences 2017; 114: E476–E485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Okochi M, Tagami S, Yanagida K, Takami M, Kodama TS, Mori K et al. γ-secretase modulators and presenilin 1 mutants act differently on presenilin/γ-secretase function to cleave Aβ42 and Aβ43. Cell Rep 2013; 3: 42–51. [DOI] [PubMed] [Google Scholar]
- [35].Chávez-Gutiérrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M et al. The mechanism of γ-secretase dysfunction in familial Alzheimer disease. The EMBO journal 2012; 31: 2261–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Karch CM, Hernández D, Wang J-C, Marsh J, Hewitt AW, Hsu S et al. Human fibroblast and stem cell resource from the Dominantly Inherited Alzheimer Network. Alzheimer’s research & therapy 2018; 10: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lagomarsino VN, Pearse II RV, Liu L, Hsieh Y-C, Fernandez MA, Vinton EA et al. Stem cell-derived neurons reflect features of protein networks, neuropathology, and cognitive outcome of their aged human donors. Neuron 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Agholme L, Clarin M, Gkanatsiou E, Kettunen P, Chebli J, Brinkmalm G et al. Low-dose γ-secretase inhibition increases secretion of Aβ peptides and intracellular oligomeric Aβ. Molecular and Cellular Neuroscience 2017; 85: 211–219. [DOI] [PubMed] [Google Scholar]
- [39].Moore BD, Martin J, de Mena L, Sanchez J, Cruz PE, Ceballos-Diaz C et al. Short Aβ peptides attenuate Aβ42 toxicity in vivo. Journal of Experimental Medicine 2018; 215: 283–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kounnas MZ, Danks AM, Cheng S, Tyree C, Ackerman E, Zhang X et al. Modulation of γ-secretase reduces β-amyloid deposition in a transgenic mouse model of Alzheimer’s disease. Neuron 2010; 67: 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bhattarai S, Liu L, Wolfe MS. Discovery of aryl aminothiazole γ-secretase modulators with novel effects on amyloid β-peptide production. bioRxiv 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kakuda N, Yamaguchi H, Akazawa K, Hata S, Suzuki T, Hatsuta H et al. γ-Secretase activity is associated with Braak Senile Plaque stages. Am J Pathol 2020; [DOI] [PubMed] [Google Scholar]
- [43].Nishimura M, Nakamura S-i, Kimura N, Liu L, Suzuki T, Tooyama I. Age-related modulation of γ-secretase activity in non-human primate brains. Journal of Neurochemistry 2012; 123: 21–28. [DOI] [PubMed] [Google Scholar]
- [44].Szaruga M, Veugelen S, Benurwar M, Lismont S, Sepulveda-Falla D, Lleo A et al. Qualitative changes in human γ-secretase underlie familial Alzheimer’s disease. Journal of Experimental Medicine 2015; 212: 2003–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kakuda N, Miyasaka T, Iwasaki N, Nirasawa T, Wada-Kakuda S, Takahashi-Fujigasaki J et al. Distinct deposition of amyloid-β species in brains with Alzheimer’s disease pathology visualized with MALDI imaging mass spectrometry. Acta neuropathologica communications 2017; 5: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Palmqvist S, Janelidze S, Stomrud E, Zetterberg H, Karl J, Zink K et al. Performance of Fully Automated Plasma Assays as Screening Tests for Alzheimer Disease-Related β-Amyloid Status. JAMA Neurol 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Schindler SE, Bollinger JG, Ovod V, Mawuenyega KG, Li Y, Gordon BA et al. High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology 2019; 10.1212/WNL.0000000000008081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].West T, Kirmess KM, Meyer MR, Holubasch MS, Knapik SS, Hu Y et al. A blood-based diagnostic test incorporating plasma Aβ42/40 ratio, ApoE proteotype, and age accurately identifies brain amyloid status: findings from a multi cohort validity analysis. Molecular neurodegeneration 2021; 16: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Janelidze S, Pannee J, Mikulskis A, Chiao P, Zetterberg H, Blennow K et al. Concordance Between Different Amyloid Immunoassays and Visual Amyloid Positron Emission Tomographic Assessment. JAMA Neurol 2017; 74: 1492–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Janelidze S, Zetterberg H, Mattsson N, Palmqvist S, Vanderstichele H, Lindberg O et al. CSF Aβ42/Aβ40 and Aβ42/Aβ38 ratios: better diagnostic markers of Alzheimer disease. Annals of clinical and translational neurology 2016; 3: 154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Palmqvist S, Mattsson N, Hansson O, Initiative ADN. Cerebrospinal fluid analysis detects cerebral amyloid-β accumulation earlier than positron emission tomography. Brain 2016; 139: 1226–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Performance of novel Aβ assays on the MSD platform. a) Standard curves of new Aβ assays using synthetic peptide calibrators, Lower Limit of Quantification (LLOQ) was calculated by the lowest standard >2 times background. b) Test of specificity of each immunoassay using all 6 distinct Aβ calibrator peptides.
Supplementary Figure 4. Assay validation for Aβ x-37 in CSF. a) Measurement of Aβ x-37 from pooled human CSF after serial dilutions (n=2 technical replicates). b) Measurement of Aβ x-37 from pooled CSF before vs. after immunodepleting with anti-Aβ antibody 4G8 or control IgG.
Supplementary Figure 2. Normalized Aβs secreted from transfectants of 131 FAD mutants in PS dKO 293 cells. a) Each secreted Aβ is normalized to the total of secreted Aβs (Aβ37+38+39+40+42+43) and presented as a ratio, with the levels from WT-PS1 transfectants labeled as the red dotted line. b) Statistical analysis of data from Fig. 1b and Fig S1a (p value analyzed by Pearson correlation coefficient; r analyzed by linear regression.
Supplementary Figure 3. Immunohistochemistry of Aβ37 and Aβ42 in human (AD) brain. a-b) Immunostaining of Aβ37 and 42 on serial paraffin sections. c-d) Double labeling of Aβ37 (DAB+Ni; purple) and Aβ42 (DAB; brown) on the serial paraffin section. Scale bar = 200 μm for a-c) and 100 μm for d).