

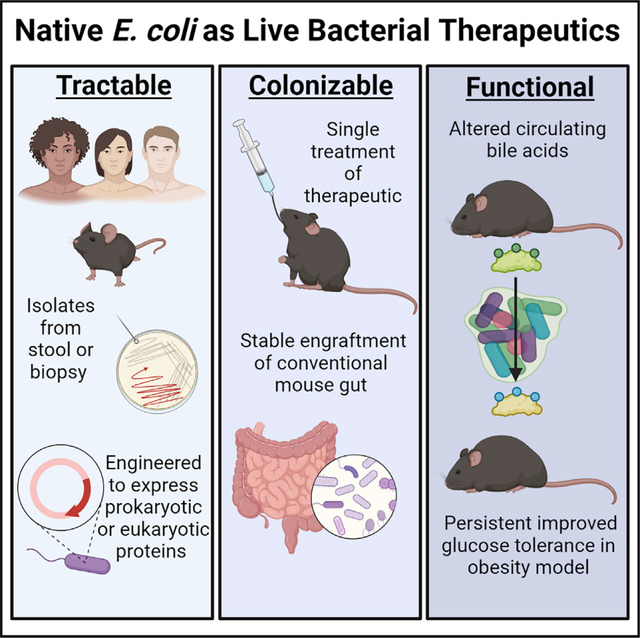
SUMMARY
Live bacterial therapeutics (LBTs) could reverse diseases by engrafting in the gut and providing persistent beneficial functions in the host. However, attempts to functionally manipulate the gut microbiome of conventionally raised (CR) hosts have been unsuccessful because engineered microbial organisms (i.e., chassis) have difficulty in colonizing the hostile luminal environment. In this proof-of-concept study, we use native bacteria as chassis for transgene delivery to impact CR host physiology. Native Escherichia coli bacteria isolated from the stool cultures of CR mice were modified to express functional genes. The reintroduction of these strains induces perpetual engraftment in the intestine. In addition, engineered native E. coli can induce functional changes that affect physiology of and reverse pathology in CR hosts months after administration. Thus, using native bacteria as chassis to “knock in” specific functions allows mechanistic studies of specific microbial activities in the microbiome of CR hosts and enables LBT with curative intent.
Graphical Abstract
In brief
Native E. coli strains isolated from mouse stool are genetically engineered for long-term engraftment in the conventional mouse gut and enable long-term systemic effects on the host, such as improvements in insulin sensitivity in mouse models of type 2 diabetes.
INTRODUCTION
Gut microbes sense and condition the luminal environment over time periods measured in years. Numerous chronic human diseases, including obesity (Muscogiuri et al., 2019; Zarrinpar et al., 2014), non-alcoholic fatty liver disease (Aron-Wisnewsky et al., 2020; Saran et al., 2020), type 2 diabetes (Gurung et al., 2020; Zarrinpar et al., 2014), atherosclerosis (Allaband et al., 2021; Jie et al., 2017; Xue et al., 2021), polycystic ovary syndrome (Rizk and Thackray, 2021), inflammatory bowel disease (Kostic et al., 2014), and cancer (Sepich-Poore et al., 2021), have protracted symptomology and have consequently been targets of potential engineered, live bacterial therapeutics (LBTs). LBTs can also be designed to express functions that address monogenic inborn errors of metabolism (e.g., phenylalanine metabolism in patients with phenylketonuria) and other orphan diseases (Isabella et al., 2018; Puurunen et al., 2021). Engineered bacteria also enable otherwise challenging mechanistic studies, allowing researchers to learn how individual bacterial functions or proteins in the microbiome contribute to host physiology, pathophysiology, or the microbiome community (Biteen et al., 2016). Although synthetic biologists have designed increasingly elaborate genetic circuits that in principle could risk-stratify patients and combat disease, these circuits have only worked in reduced-community models in vivo (i.e., gnotobiotic mice, antibiotic-treated mice) (Claesen and Fischbach, 2015; Mimee et al., 2016).
A key step in engineered LBTs is selection of a microbial host organism, or chassis, which would enable environment-sensing, regulated gene expression, and production of a therapeutic product. Current LBT chassis (e.g., Escherichia coli Nissle 1917, Bacteroides spp., Lactobacillus spp.) cannot engraft or even survive in the gut luminal environment. They require frequent re-administration and are difficult to localize to specific regions, such as the proximal gut, thereby resulting in unreliable delivery of function (Claesen and Fischbach, 2015; Isabella et al., 2018; Pedrolli et al., 2019). The lack of engraftment severely limits the use of LBTs for chronic conditions, for curative effect, or to help devise tools to study specific functions in the gut microbiome (Claesen and Fischbach, 2015; Pedrolli et al., 2019). Published human trials using engineered LBTs demonstrated safety but yielded disappointing physiological results (Braat et al., 2006; Puurunen et al., 2021). The authors attributed therapeutic failure to the inability of the chassis to survive long enough to express the transgene of interest. Hence, appropriate chassis selection remains a significant barrier to translating synthetic biology applications to human diseases.
LBTs face many challenges to survival in the luminal environment, both from the host (e.g., peristalsis, innate and adaptive immunity) and other native microorganisms (e.g., competition, niche availability) (Walker and Owen, 1990). Probiotic strains not adapted to the luminal environment cannot easily compete with microbes that have adapted to their specific luminal niche over thousands of generations (Huang et al., 2021). This phenomenon is apparent in most patients who have received fecal microbiota transplant, where the individual’s native microorganisms return and largely, or completely, displace the transplanted microbes (Li et al., 2016). However, early studies of native E. coli strains, such as MP-1 (Lasaro et al., 2014) and NGF-1 (Kotula et al., 2014; Riglar et al., 2017), demonstrate prolonged engraftment in the luminal environment of 70–200 days, respectively, in mice pre-treated with antibiotics and kept in specific pathogen-free (SPF) conditions. More recently, a wastewater Bacteroides ovatus strain that was engineered to be tunable to a prebiotic, porphyran, engrafted in the gut with 105–1012 colony-forming units (CFUs)/g in the feces of mice housed in gnotobiotic isolators for up to 25 days (Shepherd et al., 2018). However, whether native species, particularly a low-abundance organism such as native E. coli, can persistently engraft in non-antibiotic-treated, non-sterile, fully conventional conditions and be used to functionally change the luminal environment, induce physiological change, or treat disease, remains unclear.
In theory, native bacteria are already maximally adapted to the luminal environment of the host, thereby bypassing nearly all the barriers to engraftment, making them an ideal chassis for transgene delivery. The reluctance to use undomesticated native bacteria is driven by the reality that they are difficult to culture and modify, although recent studies demonstrate that they can be modified more consistently with novel methods (Brophy et al., 2018; Jin et al., 2022; Ronda et al., 2019; Rubin et al., 2022; Wiles et al., 2018). Studies in gnotobiotic mice show that a host monocolonized with a single species is resistant to colonization by the same, but not different, species (Lee et al., 2013), suggesting that if the niche of the engineered bacteria is already filled, engraftment would be more difficult. In addition, despite the early success with native strains (Lasaro et al., 2014; Riglar et al., 2017), many have assumed that E. coli are not good colonizers due to disappointment with their commensal lab strain or probiotic counterparts (e.g., E. coli Nissle 1917). This has led researchers to favor species that are far more abundant in the gut microbiome (e.g., Bacteroides spp., Lactobacillus spp.). However, these species are much more challenging to engineer. Hence, it may seem that the barriers to using native bacteria as chassis for transgene delivery, particularly for conventionally raised (CR) hosts, are high.
Here we present a proof-of-concept study that advances our ability to use engineered bacteria to effectively change physiology in CR wild-type (WT) hosts by demonstrating that native bacteria can serve as chassis for transgene delivery that modifies the host phenotype. This is accomplished by identifying a genetically tractable, native bacterial strain from a host (i.e., an undomesticated E. coli), modifying this strain to express a transgene of interest, and then reintroducing the engineered native bacteria to the host (Figure 1A). The transgenes of interest included bile salt hydrolase (BSH), a prokaryotic bile acid deconjugation enzyme (Figure S1A) that can potentially affect host metabolic homeostasis, and IL-10, a mammalian cytokine that can function as an anti-inflammatory agent. After a single treatment, engineered native bacteria engraft throughout the entire gut of CR hosts for the lifetime of the mouse, retain the function of their transgene, induce metabolomic changes, affect host physiology, and even ameliorate pathophysiological conditions. We evaluate the robustness of the approach by performing experiments in non-sterile facilities, testing biocontainment between co-housed littermates, and changing diets. We demonstrate the translational potential of this method by successfully and stably transforming native, undomesticated, human-derived E. coli that can be used for transgene delivery in humans to potentially treat disease. Although the individual steps of the proposed approach for engineering undomesticated native bacteria to express the desired function of interest are not particularly difficult, the combination is novel, and, put together, they clearly demonstrate that even resource-limited labs can accomplish what has yet to be achieved with other synthetic biology approaches: persistent functional manipulation of the luminal environment of CR hosts to study the physiological effects.
Figure 1. Gut native E. coli are genetically tractable and can serve as a chassis for transgene delivery.

(A) Experimental strategy of engineered native bacteria.
(B) Optimal characteristics of a chassis for transgene delivery for a potential LBT.
(C) Original isolate (EcAZ-1), GFP-producing strain (EcAZ-2), and GFP- and BSH-producing strain (EcAZ-2BSH+) plated on LB containing TDCA. Precipitate around EcAZ-2BSH+ is the result of TDCA deconjugated by the bacteria to DCA and qualitatively indicates enzyme functionality.
(D) Growth curve of EcAZ-2 compared with EcAZ-1, EcAZ-2BSH+ compared with EcAZ-1, and EcAZ-2IL10+ compared with EcAZ-2. The line represents an average of three measurements per strain.
(E) Proteomic analysis shows increased BSH protein expression in EcAZ-2BSH+ compared with EcAZ-1 and EcAZ-2 (n = 4).
(F) Log2 ratio of DCA to TDCA in a timed enzymatic assay (n = 3; see Figure S1G for raw values).
(G) IL-10 levels detected with ELISA from cell lysates (n = 6).
The marker covers some error bars in (F) and (G). Given the low number of samples for (E) and (F), we used a Student’s t test after the confirmation of normality with Q-Q plot. We used a Mann-Whitney U test for (G). (A) and (B) were created with BioRender.com.
See also Figure S1.
RESULTS
Gut native E. coli bacteria are genetically tractable
An ideal chassis is genetically tractable, can stably and persistently colonize the selected host, express the gene of interest persistently (and without much effect on bacterial fitness), and impose a functional change to alter the lumen and/or serum to affect host physiology (Figure 1B). Importantly, the strategy should be easily translated to humans, as this has been a particular challenge in adapting synthetic biology tools to clinical problems (Figure 1B). In our studies, we used an undomesticated, native E. coli, EcAZ-1, isolated from the stool of a CR-WT C57BL/6 male mouse acquired from Jackson Laboratory (Bar Harbor, ME). As measured using MUMmer4 alignments (Març ais et al., 2018), EcAZ-1 is a closely related strain to NGF-1 (Kotula et al., 2014; Riglar et al., 2017), with >99% identity across the full length of the main chromosome and 8-kb plasmid, and 88% in the 42-kb plasmid. E. coli MP-1 (Lasaro et al., 2014) is more distantly related, with ~97% identity across only 87% of the EcAZ-1 chromosome. The 8-kb plasmid of EcAZ-1 is present in MP-1 at >99% identity across its length.
Green fluorescent protein (GFP) linked with kanamycin resistance was introduced to EcAZ-1 via phage transduction to create a traceable version of the bacteria, EcAZ-2 (Figures 1C and S1A). EcAZ-2 was then engineered, separately, to form two different engineered native bacteria. The first, EcAZ-2BSH+, expresses BSH (Figures S1B and S1C), a bacterial gene that causes specific bile acid biotransformations demonstrated to affect the host metabolic phenotype in reduced-community microbiome studies (e.g., gnotobiotic mice, antibiotic-treated mice) (Jones et al., 2008; Joyce et al., 2014a; Yao et al., 2018). In vitro BSH activity was monitored with the aid of taurodeoxycholic acid (TDCA) plates; as EcAZ-2BSH+ deconjugates TDCA to deoxycholic acid (DCA), these plates form a white precipitate around the bacterial colony (Figure 1C). The second gene, IL-10 (to form EcAZ-2IL10+; Figure S1D), is a mammalian anti-inflammatory cytokine. Although continuous administration of IL-10-expressing probiotics has been successful in treating preclinical models of colitis (Steidler et al., 2000), this therapy has not successfully translated to humans, perhaps due the inability of chassis to survive in the luminal environment (Braat et al., 2006). The addition of gfp and kanamycin resistance, as well as bsh and Il-10, did not affect the in vitro growth rate of EcAZ-2, EcAZ-2BSH+, and EcAZ-2IL10+ (Figure 1D; Figure S1E). Sequencing of EcAZ-1 and EcAZ-2 demonstrates that these two bacteria remain almost identical. Other than the deliberate engineering, there are no other indels and only ~4,500 SNPs, which is in the range of the error rate of Illumina sequencers. The same is true for EcAZ-2BSH+, with fewer than ~10,000 SNPs versus EcAZ-1 and ~4,000 versus EcAZ-2, and no unexpected indels. Thus, native E. coli are genetically tractable, can express prokaryotic and eukaryotic transgenes, and can potentially serve as chassis for transgene delivery.
We performed additional assays to quantitatively assess protein expression in EcAZ-2BSH+ and EcAZ-2IL10+. Proteomic analysis demonstrates that BSH is one of the most highly expressed proteins in EcAZ-2BSH+, whereas it is not detectable in EcAZ-1 or EcAZ-2 (Figure 1E; Figure S1F). Constitutively expressed BSH produced by EcAZ-2BSH+ converts TDCA to DCA in a controlled in vitro assay rapidly, whereas no shift is seen with EcAZ-1 or EcAZ-2 (Figure 1F; Figure S1G). For EcAZ-2IL10+, IL-10 was detected in cell lysates but not the supernatant (Figure 1G). Thus, proteins produced by native E. coli are functional and highly expressed constitutively.
Engineered native E. coli can engraft in the gut lumen
Next, we assessed the candidate chassis’ ability to stably colonize the murine gut by gavaging 1010 CFUs of engineered bacteria with different E. coli chassis in CR-WT C57BL/6, non-antibiotic-treated mice. EcAZ-2 stably maintained colonization in 100% (n = 8) of mice for over 110 days in a non-sterile, low-barrier facility (Figures 2A and S2A). Domesticated lab bacteria that are often used as chassis for LBT applications (i.e., E. coli MG1655 and E. coli Nissle 1917), lost colonization over time (p = 0.02, log rank test, Figure S2A) and did not maintain colonization long-term (p < 0.0001, Fisher’s exact test, Figure 2B). Mice that had detectable non-native E. coli in their stool were nearly two orders of magnitude lower (as determined by CFU/g of stool) than those treated with EcAZ-2 (Figure 2A). Co-housing of mice colonized with EcAZ-2 with non-colonized mice did not lead to horizontal transmission between mice, despite the opportunity for coprophagia (Figure 2C). This is likely due to an insufficiently high enough dose of bacteria being transmitted through coprophagia, as gavage with 102 and 104 CFUs of EcAZ-2 in CR-WT C57BL/6, non-antibiotic-treated mice did not lead to engraftment in any mice in the non-sterile low-barrier facility, whereas 106 CFUs led to sporadic—and 108 CFUs more consistent—engraftment (Figure 2D). Mice gavaged with 108 CFUs were significantly more likely to be colonized than those gavaged with lower doses at every time point measured (Fisher’s exact test, Figure S2B).
Figure 2. Engineered native E. coli can engraft in the luminal environment.

(A) Long-term colonization after gavage with 1010 CFUs of E. coli MG1655 and E. coli Nissle 1917 (two lab-strain E. coli often used as chassis for function delivery), as well as EcAZ-2 in non-antibiotic-treated CR-WT mice housed in a low-barrier, non-sterile facility after a single gavage. Sample measures in the gray area denote mice where the bacteria were not detectable at a limit of detection of Log10 of 2.
(B) Colonization at 110 days (Fisher’s exact test; 4–8 mice/condition).
(C) Colonization in co-housed mice housed in a low-barrier, non-sterile facility after a single gavage, where one mouse received 1010 CFUs of EcAZ-2 and the other received vehicle via oral gavage. Arrow indicates change in stool collection protocol where mice were separated from their cage mates 24 h prior to their weekly stool collection. Mice were returned to their cage mates right after stool collection. Thus, although uncolonized mice are exposed to EcAZ-2 through coprophagia, the level is not sufficient to lead to engraftment.
(D) Colonization after gavage of different initial doses of EcAZ-2 in non-antibiotic-treated CR-WT mice housed in a low-barrier, non-sterile facility after a singlegavage (n = 5).
(E) Colonization after gavage of 1010 CFUs of EcAZ-2 in the stool of non-antibiotic-treated CR-WT mice in an SPF facility on a stable diet (10% fat) or on diets with different macronutrient profiles (non-fat diet [NFD] or high-fat diet [HFD]). Diet was changed 4 days post-gavage (4 mice/condition).
(F) Colonization after gavage of 1010 CFUs of EcAZ-2 or EcAZ-2BSH+ in non-antibiotic-treated CR-WT mice housed in an SPF facility (4–12 mice/condition).
(G) Colonization after gavage of 1010 CFUs of EcAZ-2 or EcAZ-2IL10+ in non-antibiotic-treated CR-WT mice housed in an SPF facility (4 mice/condition).
(H) Colonization of gastrointestinal tract in CR-WT mice 3 months after a single gavage in an SPF facility. Sample measures in the gray area denote mice where the bacteria were not detectable at a limit of detection of Log10 2. (4 mice/condition.)
(I) Colonization of EcAZ-2 and EcAZ-2IL10+ in the gastrointestinal tract of CR-WT mice housed in an SPF facility 79 days after gavage (same mice depicted in Figure 2E; 5/condition).
All error bars indicate standard error of the mean. Samples that were not detectable were excluded from mean and standard error of mean calculations. The marker covers some error bars in (E), (F), and (G).
See also Figure S2.
Colonization of EcAZ-2 was unaffected by changes to the diet macronutrient profile (Figure 2E). The addition of BSH or IL-10 activity does not significantly alter the colonization level of EcAZ-2; this native strain remains stably colonized in CR-WT mice for the life of the animal, in reproducible experiments (Figures 2F and S2C, for BSH; Figures 2G and S2D, for IL-10) in both SPF and non-sterile facilities. Sex of the CR-WT host did not affect how well EcAZ-2 colonized the host (Figure S2E).
To determine whether BSH function was retained during engraftment, bacterial isolates from mice treated with EcAZ-2BSH+ were grown on TDCA plates. Isolates from every region of the gastrointestinal (GI) tract of every mouse treated with EcAZ-2BSH+, across every experiment, retained BSH functionality, confirmed by deconjugation of TDCA to DCA (see Figure S2F). Likewise, cell lysates of bacterial isolates obtained from mice treated with EcAZ-2IL10+ at 89 days after gavage produced IL-10 at far higher levels than the negative control bacteria (i.e., EcAZ-2; Figure S2G).
We cultured gastrointestinal tissues (i.e., duodenum, jejunum, ileum, cecum, and rectum) to determine the extent of bacterial colonization in the various regions of the gut. In contrast to what has been reported for engineered non-native E. coli (Isabella et al., 2018), EcAZ-2 colonizes the proximal gut/small intestine regardless of whether it contains the bsh gene, Il-10 gene, or not, with the highest colonization in the cecum and ileum for the BSH strain and cecum and jejunum for the IL-10 strain (Figures 2H, 2I, and S2H). Colonization throughout the GI tract was consistent in fed and fasted mice (Figure S2H), and isolates from throughout the GI tract retained transgene activity (Figure S2I). These experiments demonstrate that EcAZ-2 can stably and persistently colonize CR-WT hosts for perpetuity, while retaining functionality of a gene of interest.
Engineered native E. coli can perform a luminal function
After demonstrating that native bacteria are genetically tractable and can persistently colonize the proximal gut and colon, we assessed whether the transgene delivered by the EcAZ-2 chassis can functionally manipulate the luminal environment of CR hosts. We used bsh as our transgene to evaluate the native bacterial chassis because its functions are well-described and it has been studied in reduced-community/gnotobiotic models (Jones et al., 2008, 2014a; Yao et al., 2018). Bile acid changes induced by BSH activity are presumed to affect multiple metabolic systems, including glucose, lipid, and bile acid homeostasis, as well as immunity/inflammation and thermogenesis (Joyce et al., 2014b; Wahlström et al., 2016). Hepatic primary conjugated bile acids are deconjugated by BSH (Figure 3A). Whereas the ileum has active transporters for the highly polar conjugated bile acids, deconjugated bile acids are less polar and are reabsorbed through passive diffusion. In the distal gut, deconjugated bile acids are substrates for luminal bacterial bile acid biotransformations that convert them to secondary bile acids. Secondary bile acids can be reabsorbed by passive diffusion, conjugated by the liver into conjugated secondary bile acids, and re-released into the lumen. Hence, increased BSH activity can affect all of these various bile acid pools.
Figure 3. Native E. coli can be used to change luminal metabolome without measurable effects in the microbiome.

(A) Summary diagram showing effects of BSH on fecal bile acid metabolism. The hypothesized effect of increased BSH on the different bile acid pools is shown with red arrows. A question mark denotes uncertainty.
(B–F) (B) Total fecal bile acids, (C) primary bile acids, (D) primary conjugated bile acids, (E) primary unconjugated bile acids, and (F) secondary bile acids in fecal samples collected from mice 12 weeks after a single gavage with vehicle, EcAZ-2, and EcAZ-2BSH+. Significant differences were determined by Kruskal-Wallis test with post hoc Dunn’s multiple comparison test comparing EcAZ-2 and EcAZ-2BSH+.
(G) Faith’s phylogenetic distance (a measure of α-diversity) from 16S rRNA gene sequencing performed on stool samples collected pre-treatment and at 2, 10, and 16 weeks post-gavage (left) and from the terminal ileum samples at the time of euthanasia (right).
(H) Relative abundance of E. coli as detected by 16S rRNA gene sequencing performed on stool samples collected pre-treatment and at 2, 10, and 16 weeks post-gavage and from the terminal ileum samples at the time of euthanasia.
(I) Principal coordinate analysis of weighted UniFrac β-diversity of the fecal samples collected pre-treatment and at 2, 10, and 16 weeks post-gavage.
(J) Principal coordinate analysis of weighted UniFrac β-diversity of the terminal ileum microbiome at the time of euthanasia.
(A) was created with BioRender.com.
See also Figure S3.
To confirm that BSH-expressing native E. coli are functional in vivo, we determined the effect of EcAZ-2BSH+ on the bile acids of mono-colonized gnotobiotic mice. Whereas mice mono-colonized with EcAZ-2 had only primary conjugated bile acids and no deconjugated bile acids (because they lack bacteria that can deconjugate), mice mono-colonized with EcAZ-2BSH+ had greatly reduced primary conjugated fecal bile acids (Figure S3A) and increased primary deconjugated fecal bile acids (Figure S3B), consistent with increased BSH activity. Quantitative analysis of the conjugated and unconjugated individual primary bile acids showed that EcAZ-2BSH+ significantly shifted the log ratio of cholic acid to taurocholic acid (CA:TCA), and β-muricholic acid to tauro-β-muricholic acid (bMCA:TbMCA), toward the deconjugated bile acids in mono-colonized mice (Figure S3C).
To determine whether engineered native bacteria expressing BSH can functionally manipulate the fully intact microbiome of CR mice, we gavaged EcAZ-2 or EcAZ-2BSH+ into 11-week-old CR-WT mice. 12 weeks after the single oral administration, stool samples were collected and analyzed for bile acids. CR mice colonized with EcAZ-2BSH+ had significantly higher total fecal bile acid levels than CR mice colonized with EcAZ-2 (Figure 3B). In addition, CR mice gavaged with EcAZ-2BSH+ had decreased primary fecal bile acids (Figure 3C) and decreased primary conjugated bile acids (Figure 3D) compared with mice treated with EcAZ-2. Surprisingly, primary unconjugated bile acids were also significantly decreased in EcAZ-2BSH+-treated mice compared with EcAZ-2 controls (Figure 3E). However, this could be due to increased conversion of deconjugated bile acids to secondary bile acids, which was significantly elevated in EcAZ-2BSH+ mice (Figure 3F). In fact, treatment with EcAZ-2BSH+ affected nearly every fecal bile acid measured, including TbMCA, a potent farnesoid X receptor (FXR) antagonist, and DCA, a potent G protein-coupled bile acid receptor 1 (TGR5) agonist (Figure S3D; Wahlström et al., 2016). These fecal bile acid data confirm that BSH is functional in vivo. Moreover, they demonstrate that native E. coli are valuable chassis that can express a function of interest intraluminally at least 12 weeks after a single gavage in CR-WT mice.
We expected that changes in intraluminal bile acids would lead to global changes in gut microbiome composition. However, microbiome analysis showed that EcAZ-2 and EcAZ-2BSH+ had no detectable effect in any microbiome measures. Compared with mice treated with vehicle, we found no significant differences in Faith’s phylogenetic diversity in the fecal microbiome at 2, 10, and 16 weeks post-gavage or in the ileum after euthanasia (Figure 3G). Other measures of α-diversity such as richness (Figure S3E) and the Shannon index (Figure S3F) found a transient difference at week 2 between mice in the three conditions that disappeared by the later time points. Differential abundance analysis using ALDEx2 (Fernandes et al., 2014) found no compositional differences at significance (p ≤ 0.05), even before any type of multiple hypothesis correction between EcAZ-2 and vehicle, either with or without BSH, or any between EcAZ-2 and EcAZ-2BSH+, either across all samples or at any specific time point alone. Confirmatory analysis with ANCOM (Mandal et al., 2015) demonstrated no compositional difference between EcAZ-2 and EcAZ-2BSH+, and only two amplicon sequence variants (ASVs) between EcAZ-2 and vehicle (assigned as Lachnospiraceae and Bacteroidaceae), and one between EcAZ-2BSH+ and vehicle (assigned as Clostridiacae) that was inconsistent across time points. Hence, despite inducing changes in the luminal metabolome, engineered native bacteria in this case did not induce a compositional shift in the gut microbiome.
There were no E. coli detectable by ASV matching the 16S rRNA gene sequence of E. coli prior to gavage. Although E. coli could be identified in some of the samples from mice treated with EcAZ-2 or EcAZ-2BSH+, most had levels of E. coli in stool that were statistically indistinguishable as a group from the vehicle-treated controls (Figure 3H). Overall, based on 16S rRNA gene sequencing, E. coli comprised <0.1% of the fecal microbiome. Principal coordinate analysis of the UniFrac weighted β-diversity performed at 2, 10, and 16 weeks could not distinguish the three treatment groups (Figure 3I; pseudo-F = 1.304, p = 0.233). The lack of a difference is reflected in the ileum as well (Figure 3J; pseudo-F = 1.114, p = 0.339). This demonstrates that, even at low abundances, the native E. coli can be used to introduce a function of interest in CR hosts, and that they do so without significantly altering the microbial community already established in the host.
Engineered native E. coli bacteria affect host physiology
We next investigated whether transgene delivery and expression by the native E. coli chassis can affect host physiology by first determining the impact of colonization with EcAZ-2BSH+ on circulating bile acids. Mice colonized with EcAZ-2BSH+ had reduced total serum bile acids, increased primary bile acids, and decreased secondary bile acids, though these did not achieve significance (Figures 4A and S4A). There were larger changes in specific circulating bile acids, including known agonists and antagonists of FXR, such as increased TCA and TbMCA, respectively, compared with their deconjugated counterparts (Figures 4B, 4C, and S4B). We measured the impact of these differences in circulating bile acids on hepatic gene expression, focusing on genes important for bile acid metabolism. We found increased expression of Fxr, Shp, and Cyp27a1 (Figure 4D), consistent with increased bile acid biosynthesis through the alternative pathway, perhaps to alleviate the higher excretion of bile acids in the stool of mice colonized with EcAZ-2BSH+. This demonstrates that the addition of a single gene into the microbiome via engineered native bacteria can significantly alter host physiology.
Figure 4. Native E. coli can be used to physiologically change the host and treat pathophysiological conditions.

(A) Total, primary, and secondary serum bile acids from mice treated with a single gavage of vehicle, EcAZ-2, or EcAZ-2BSH+ 12 weeks prior.
(B) Serum levels of CA (top), TCA (middle), and the log2 ratio of CA to TCA in mice treated with vehicle, EcAZ-2, or EcAZ-2BSH+.
(C) Serum levels of bMCA (top), TbMCA (middle), and the log2 ratio of bMCA to TbMCA in mice treated with vehicle, EcAZ-2, or EcAZ-2BSH+.
(D) Hepatic gene expression of Fxr, Shp, Cyp7a1, and Cyp27a1, as determined by qRT-PCR. Significant differences were determined by a Student’s t test after normality verified through Q-Q plot.
(E) Mouse weights of CR-WT mice treated with a single gavage of vehicle, EcAZ-2, and EcAZ-2BSH+.
(F) Fasting (16 h) and postprandial (30 min) glucose and insulin levels. Significant differences were determined by Kruskal-Wallis test with post hoc Dunn’s multiple comparison test comparing all three conditions done separately for fasted and fed measures.
(G) Mouse weights of Ob/Ob mice treated with a single gavage of EcAZ-2 and EcAZ-2BSH+.
(H) Glucose tolerance test in Ob/Ob mice treated with a single gavage of EcAZ-2 and EcAZ-2BSH+ 15 weeks prior. Significance determined with Mann-Whitney U test corrected for multiple comparisons. Inset shows area under the curve. Significance determined by Mann-Whitney U test.
For (A), (B), and (C), significant differences were determined by a Kruskal-Wallis test with post hoc Dunn’s multiple comparison test comparing EcAZ-2 and EcAZ-2BSH+.
See also Figure S4.
To determine whether the transgene delivery with the native E. coli chassis can induce long-lasting change in host physiological phenotype, we metabolically characterized the cohort of mice that received vehicle, EcAZ-2, or EcAZ-2BSH+ approximately 12 weeks after gavage. Unlike what has been reported in gnotobiotic and reduced-community mice (Joyce et al., 2014a; Yao et al., 2018), increased BSH activity induced with engineered native bacteria did not affect the body weight of the host (Figure 4E). The addition of EcAZ-2 or EcAZ-2BSH+ did not affect food intake (Figure S4C). 12 weeks after a single gavage with engineered native bacteria, CR mice on a normal chow diet colonized with EcAZ-2BSH+ had reduced postprandial blood glucose and insulin levels compared with those colonized with EcAZ-2 after a mixed meal challenge (Figure 4F). We repeated the mixed meal challenge in a separate experiment using female mice with a shorter duration of colonization with engineered native bacteria (Figure S4D). As with the male mice, the female mice treated with EcAZ-2BSH+ had lower postprandial insulin levels (Figure S4E).
Decreased postprandial insulin induced by EcAZ-2BSH+ suggests improved insulin sensitivity and glucose tolerance. To determine whether transgene delivery with a native E. coli chassis could be used to ameliorate disease, we investigated the effect of EcAZ-2BSH+ in a genetic model of type 2 diabetes, the Ob/Ob mice. EcAZ-2 and EcAZ-2BSH+ stably colonized CR Ob/Ob mice (Figure S4F). Similar to what we had observed in CR-WT mice, EcAZ-2BSH+ did not induce a change in body weight (Figure 4G). Despite identical body weights, Ob/Ob mice colonized with EcAZ-2BSH+ had significant improvements in insulin sensitivity compared with the control cohort (Figure 4H) after receiving a single treatment of engineered native bacteria more than 3 months earlier. Thus, these experiments show that transgene delivery using native E. coli affects the serum metabolome, extra-intestinal (e.g., hepatic) gene expression, and host physiology, and ameliorates disease months after a single administration.
Humans have genetically tractable native E. coli
To determine whether the engineered native bacteria strategy can be translated to humans, we isolated E. coli strains from biopsies obtained from multiple regions of the gastrointestinal tracts of volunteers undergoing routine outpatient endoscopy. Nine isolated strains were assessed for antibiotic sensitivity, tractability via transduction, transformation with assorted sizes of plasmids, and conjugation (a subset shown in Table 1). Many isolates were sensitive to commonly used laboratory antibiotics, including kanamycin, chloramphenicol, and carbenicillin. All isolates evaluated were resistant to transduction via P1vir when lysates were generated from E. coli MG1655 or E. coli Nissle 1917. Strains were transformable with several differently sized plasmids. Conjugation using a modified F-plasmid (Figure S5) proved to be a universally successful method for manipulation for 10/10 human-derived E. coli, quantified for a subset of the strains shown in Table 1. To encourage plasmid retention, conjugation machinery was removed post-transfer. Thus, the first steps of the engineered native bacteria approach, identification/isolation of genetically tractable bacteria and modification to express a gene of interest, can be translated to humans. Further studies are necessary to determine whether the autologous transfer of engineered native bacteria can lead to long-lasting colonization of a human host and treatment of long-standing chronic conditions and genetic diseases.
Table 1.
Human-derived E. coli isolated from intestinal biopsies are genetically tractable
| Metadata |
Antibiotic MIC (mg/mL) |
Transformation (% efficiency) |
Conjugation |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Strain | Host species | Tissue | Cm | Kan | Carb | Small plasmid (4.4 kb) | Medium plasmid (35 kb) | Large plasmid (70 kb) | Efficiency (or limit of detection) |
|
| |||||||||
| EcAZ-1 | mouse (C57BL6) | feces | 5 | 6.25 | 25 | 4.25 | 4.51E–4 | 6.60E–5 | 5.98E–3 |
| AZ-61 | human-2 | rectum | 5 | 6.25 | 50 | 0.067 | 1.97E–4 | 1.99E–5 | 1.31E–5 |
| AZ-116 | human-4 | ileum | 10 | 6.25 | resistant | - | 2.58E–6 | 2.42E–5 | 2.58E–3 |
| AZ-120 | cecum | 5 | 6.25 | 25 | 0.23 | 1.18E–6 | 1.44E–5 | 5.13E–3 | |
Abbreviations: Cm, chloramphenicol; Kan, kanamycin; Carb, carbenicillin; see also Figure S5.
DISCUSSION
Most therapies that target microbiome composition do not have a detectable impact on the gut microbiome and are not robust to the interpersonal diversity and plasticity of the microbiome in human hosts (Kristensen et al., 2016; Mimee et al., 2016). In addition, many different configurations of the gut microbiota lead to the same functional result (Human Microbiome Project, 2012; Turnbaugh et al., 2009), suggesting that microbial function may be difficult to manipulate with therapies that target the composition of the microbiome alone. To develop a better mechanistic understanding of the microbe-host relationship and more effective microbiome-mediated therapies, approaches based on functional modulation of the gut microbiome are necessary. However, these approaches have been difficult to develop, and none have demonstrated long-term engraftment with a change in physiology and/or improvement of pathologic phenotype.
To address the inability of engineered bacteria to colonize the CR host, only three strategies have shown success (Bober et al., 2018; Claesen and Fischbach, 2015; Mimee et al., 2016; Riglar and Silver, 2018; Sheth et al., 2016): using native E. coli as chassis (Kotula et al., 2014; Lasaro et al., 2014; Riglar et al., 2017); creating exclusive metabolic niches for an engineered probiotic strain, which can then outcompete resident microorganisms (Shepherd et al., 2018); and engineering non-colonizing bacteria to distribute mobile genetic elements (e.g., plasmids) to native bacteria in vivo (Ronda et al., 2019). However, none of these approaches have been demonstrated to engraft in the proximal gut, achieve engraftment in non-sterile facilities, alter luminal or serum metabolome, affect host physiology, or demonstrably ameliorate disease, as shown here.
A promising approach has been to find genes that improve engraftment in commonly used chassis, such as E. coli Nissle 1917 (Crook et al., 2019). More commonly, investigators have been developing tools to manipulate families of bacteria that are found in large numbers in the gut microbiome to determine whether they can outcompete or coexist with native strains and engraft in the intestinal lumen (Bermúdez-Humarán et al., 2013; Guglielmetti et al., 2013; Lim et al., 2017; Whitaker et al., 2017). Of particular interest has been Bacteroides spp., including B. ovatus and B. thetaiotaomicron (Lim et al., 2017; Mimee et al., 2015; Shepherd et al., 2018). These bacteria and other engineered probiotics have been exclusively assessed under reduced-community (as opposed to CR) conditions (i.e., gnotobiotic mice, antibiotic-treated mice) and have not yet demonstrated whether they can be used to alter the metabolome and host physiology, and reverse pathology in fully conventional mice. Moreover, these efforts have advanced our understanding of bacterial engraftment, biogeography of microorganisms and luminal micro-niches, and the biological requirements for bacterial colonization and function delivery. However, they also indicate that the amount of bacterial engineering and experimentation necessary to develop chassis that can colonize the gut is daunting and out of the reach for resource-limited labs without dedicated synthetic biologists. Moreover, they are problematic for studying steady-state mechanisms in the gut, unlikely to work in animal models where diet and host genetics are manipulated, and limit feasibility of these techniques for adoption and interventions in humans.
Here, we demonstrate a simple technique that advances our ability to use engineered bacteria to effectively change physiology in CR-WT hosts, using tools and approaches that can be adopted by nearly any lab. By combining existing techniques to native strains, we demonstrate that undomesticated E. coli derived from the host can be used as chassis to knockin specific functions into the gut microbiome, thus making mechanistic studies of the gut microbiome attainable by most labs without advanced synthetic biology experience. E. coli are easily culturable, common in the human gut (Bailey et al., 2010; Bok et al., 2018; Li et al., 2010; Najjuka et al., 2016; Stoppe et al., 2017), and tools to engineer them have existed for decades. Previous studies have also demonstrated that native bacteria are tractable and can engraft, although these studies were not focused on affecting host physiology or reversing pathology (Lasaro et al., 2014; Riglar et al., 2017). In this study, we show that by using native E. coli as our chassis, the resultant engineered native bacteria can: (1) stably colonize CR-WT hosts for months, possibly in perpetuity after a single gavage, without the need for pre-treatment for microbiome depletion; (2) engraft the proximal gut in addition to the colon; (3) demonstrate natural biocontainment between co-housed mice; and (4) alter specific luminal metabolites as intended, that leads to (5) a change in serum metabolites, which then (6) change host metabolic homeostasis and core physiological processes, and thus (7) prevent dysmetabolic phenotypes. Moreover, our preliminary data show that the initial steps of this process can be translated to humans, where we have found readily tractable, native E. coli that could be transformed to express a gene. Importantly, we show that identical cohorts that only differ in a single prokaryotic gene in a single low-abundance bacterial strain can have a dramatically different metabolic phenotype. Although this has been demonstrated with pathogenicity islands (e.g., Shiga toxin), we now demonstrate that a beneficial bacterial gene can also have systemic effects.
Earlier studies of an E. coli strain isolated from mouse fecal samples from a different facility (i.e., NGF-1) (Lasaro et al., 2014; Ziesack et al., 2018) likewise demonstrated long-term colonization (Riglar et al., 2017). However, it was not clear whether undomesticated E. coli strains could be used to study specific bacterial functions, affect the luminal environment, and shape host physiology. Given that E. coli abundance is <0.1% of the microbiome, others have focused on chassis that belong to more highly abundant strains. Interestingly, our study shows that colonization of the host luminal environment with our engineered E. coli did not affect the overall composition of the microbiome nor the relative abundance of E. coli, as detected by 16S rRNA gene sequencing analysis. This suggests several possibilities, including that (1) E. coli potentially occupies a particular niche that enables close interaction with the host without too much change in the overall composition of the microbiome, and that (2) E. coli is replacing or co-existing with its clonal relatives. Large microbiome resource studies, such as the Human Microbiome Project and American Gut Project (Lloyd-Price et al., 2019; McDonald et al., 2018), show limited detection of E. coli by 16S rRNA gene sequencing from fecal samples, whereas infants have higher levels (Chu et al., 2017), suggesting that E. coli abundance may be affected by niche availability. Moreover, low-abundance individual microbes can have large ecosystem effects because of the niche they occupy and the functions they perform (Banerjee et al., 2018); thus, a microbe need not be abundant to have a big impact. Overall, our ability to find genetically tractable E. coli in humans suggests that this approach can be translated to patients.
Our results clearly demonstrate that native E. coli are particularly adept at affecting host physiology and, considering the engineering toolbox that has been developed to genetically manipulate them, E. coli make an effective chassis. In addition, having chassis that are unable to engraft has been proposed as a mechanism for biocontainment as it would improve the safety profile by removing concerns about LBT proliferation, transmission to healthy cohabitants, and therapy delivery beyond the end of treatment (Claesen and Fischbach, 2015; Isabella et al., 2018). However, given the inability of current developed methods to effect change in CR hosts, including humans, the development of techniques to make transgene expression tunable (Lim et al., 2017; Whitaker et al., 2017), and our results showing biocontainment with co-housed mice, we suggest that transgene delivery with engineered native chassis will be a far more effective approach.
The technical capability to change microbiome function in CR mice is critical to understanding the role of the gut microbiome in various physiological and pathophysiological processes. Many animal models of disease cannot be recapitulated in reduced microbiome community conditions. For example, baseline metabolic activity in microbiome-depleted (e.g., gnotobiotic, antibiotic-treated) mice is vastly different from that in CR mice (Wichmann et al., 2013; Zarrinpar et al., 2018). This could be why the effects of BSH on host metabolic physiology have been difficult to understand, as prior studies using different reduced-community models have led to contradictory results (Joyce et al., 2014a; Yao et al., 2018). Though BSH is a prevalent function in the luminal environment, it has been identified as an important bacterial function in metabolic homeostasis and immunity (Dantas Machado et al., 2022; Joyce et al., 2014b; Wahlström et al., 2016), making it an ideal first bacterial function to evaluate. We selected our particular BSH (from the strain Lactobacillus salivarius) due to its promiscuity and effectiveness (Jones et al., 2008). However, we acknowledge that BSH from other strains could have no effect, or a different effect, on host physiology than what we have shown. Hence, functional manipulation of the microbiome should be paired with other omics analysis (e.g., metabolomics, host transcriptomics) to better understand the host-microbe relationship.
Because deconjugated bile acids do not have dedicated transporters and require reabsorption through passive diffusion, increased luminal BSH activity should lead to an increase in fecal excretion of bile acids, though this has not been reported in microbiome-depleted models (Joyce et al., 2014a). Moreover, they ignore the downstream compositional, metabolomic, and proteomic effects that can lead to changes to the luminal community that could amplify or dampen the effects of the knocked-in function. For example, though increased BSH activity is hypothesized to lead to an increase in primary unconjugated bile acids, we observed the opposite. Instead, because deconjugated bile acids are substrates that some bacteria can convert to secondary bile acids, we observed an increase in secondary bile acids. This observation is apparent in CR mice but not in mono-colonized or reduced-community mice. Although our present work focuses on providing a proof-of-concept study, the BSH results show something different from reduced-community studies—a decrease in serum insulin in the setting of unchanged body weight—which profoundly changes our understanding of how microbiome bile acid biotransformations affect host metabolism (Joyce et al., 2014b). It is unclear why bile acids are such powerful microbial signaling agents. Unlike amino acids and carbohydrates, which are shuttled to the liver through the portal circulation, medium- and long-chain fatty acids bypass the liver because they are absorbed through the lymphatic system (Ticho et al., 2019; Xiao et al., 2019). Hence, bacterially modified bile acids may serve as a nutrient signal that informs the liver about luminal fatty acid absorption. This hypothesis warrants further investigation with dedicated experiments, which can now be tested with engineered native bacteria.
Many investigators may favor using non-engrafting species, such as food-grade L. lactis and generally recognized as safe (GRAS) E. coli Nissle as chassis because there is natural biocontainment of the eventual LBTs (i.e., lack of survival). Moreover, the Food and Drug Administration (FDA) has already approved LBTs with these chassis for phase I trials (Braat et al., 2006; Puurunen et al., 2021). Because of the failure of these trials in demonstrating physiological change in human hosts, and that the FDA will not approve LBTs that are ineffective in treating disease, better chassis for functional manipulation of the luminal environment are needed. Moreover, patient advocacy groups such as the Juvenile Diabetes Research Foundation, the National Phenylketonuria Alliance, and the Crohn’s and Colitis Foundation, as well as economists who are concerned about rising healthcare costs (Holman, 2020; Martin et al., 2021), are encouraging scientists to investigate therapeutics with curative intent. In their guidance to industry, the FDA stated that LBTs “(M)ay be isolated from human hosts… (with the goal) to stimulate other potentially beneficial cellular processes as a result of transient persistence and/or long-term colonization with the microorganisms” (Food and Drug Administration, 2016). Hence, engineered native bacteria fit the FDA’s definition of LBTs. Recent therapies with CAR-T already demonstrate the FDA’s willingness to approve therapies where cells from individuals are modified and used, autologously, to treat chronic or debilitating diseases (June and Sadelain, 2018). Hence, the FDA already has language in their guidance regarding LBTs, as well as precedence in autologous cell-based therapeutics that support the potential use of engineered native bacteria for the treatment of chronic diseases. Nevertheless, biocontainment will be an important issue for engrafting LBTs such as engineered native bacteria. Though native chassis demonstrate natural biocontainment (as demonstrated in Figures 2C and 2D) additional kill-switch biological circuits (Stirling et al., 2017) may need to be employed prior to use in humans. Even if engineered native bacteria are not a clinically viable path, they have inherent value as a tool for understanding the microbe-host relationship and for performing more mechanistic studies to better understand how specific bacterial functions affect host physiological processes.
The central novelty of engineered native E. coli is that they demonstrate: (1) stable, long-term engraftment of low-abundance organisms in the absence of antibiotic pre-treatment or selection; (2) persistent metabolomic change from a single treatment; (3) persistent physiological change from a single treatment; and (4) reversal of the pathological phenotype after a single treatment. The strategy of using native bacteria as a chassis to introduce transgenes of interest can be used for different organ systems (e.g., skin, lungs, vagina), expanding our ability to perform mechanistic studies and providing a strategy for the creation of therapeutic engineered native bacteria in any microbiome- mediated/modulated disease. Moreover, personalized engineered native bacteria will help overcome the barriers that have limited the use of engineered bacteria in humans, including interpersonal differences and micro-niche changes induced by disease. Finally, life-long compliance with chronic therapies is a substantial burden in patients with chronic diseases, which contributes to their poor morbidity and mortality—and to rising healthcare costs. Some engineered probiotics in development for disease intervention require multiple doses (sometimes up to 3 times daily), which may add significant complications for feasibility, cost, and adherence. If the engineered native bacteria strategy is successfully developed and translated to humans, it has the potential to introduce novel, curative biotherapeutics that improve treatment of chronic diseases without relying on patient compliance.
Limitations of the study
Our analysis of isolated human native E. coli demonstrates that these bacteria are quite resistant to genetic manipulation. However, several recent publications report more effective methods of engineering native bacteria (Brophy et al., 2018; Jin et al., 2022; Rubin et al., 2022; Wiles et al., 2018). Any function engineered into native bacteria will likely reduce the fitness of bacteria unless there is selective pressure to maintain the transgene of interest. Moreover, some genes may be more burdensome than others. Nevertheless, in these proof-of-concept studies we have demonstrated that, even without selective pressure, native E. coli have the potential to maintain an engineered function for years. IL-10 and BSH production are likely not burdensome enough to affect chassis fitness and native E. coli are resilient enough to continue to constitutively express the transgenes of interest. However, whether this observation can be generalized to all other functions potentially introduced with native E. coli chassis is unlikely. Moreover, it is unclear whether other native chassis are as resilient, and thus would elicit the same physiological response, as the native E. coli described in this study.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Amir Zarrinpar (azarrinpar@ucsd.edu).
Materials availability
EcAZ-1, EcAZ-2, EcAZ-2BSH+, and EcAZ-2IL10+ will be made available subject to a materials transfer agreement with the University of California, San Diego.
Data and code availability
Data have been deposited at the European Nucleotide Archive under accession number ENA:PRJEB39003 and are publicly available as of the date of publication. Proteome data was uploaded to massive.ucsd.edu and can be accessed using the following identifiers: PXD032187.
All original code has been deposited at https://github.com/ZarrinparLab/2022_enb_paper and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Experiments
All animal experiments were conducted in accordance with the guidelines of the IACUC of the University of California, San Diego. C57BL/6 mice (Jackson Laboratories) or B6.Cg-Lepob/J (Jackson Laboratories) were housed in a specific pathogen free facility (irradiated chow and autoclaved bedding), or when specified, in a separate section of the vivarium that was a low barrier facility (non-irradiated chow and non-autoclaved bedding; e.g., Figures 1A and 1D). Mice were housed 4–5 per cage unless otherwise stated. They were fed a normal-chow diet (Diet 7912, Teklad Diets, Madion, WI). After an acclimation period, 12-week-old male mice were pseudorandomized into two groups. This pseudorandomization was based on the initial weight of the mice; in the end, the mean and standard deviation of the weight of the mice was the same between all groups. Mice were gavaged with 200 μL of either PBS or ~1×1010 cfu/mL of the corresponding bacterial strain unless otherwise stated. Body weight and food consumption were monitored every 1–2 weeks. In the experiment including mice on a 0% fat diet, weight was monitored more frequently, and the study was terminated due to poor health (e.g. low weight, instances of bone fractures) of the animals in accordance with our Animal Care Protocol.
Human volunteers
For human-derived strains, volunteers undergoing outpatient endoscopy were selected in accordance with VA San Diego IRB #H130266. Meta data including sex, gender identity, and age of the participants were not recorded as they did not pertain to the goal of the study.
METHOD DETAILS
Bacterial Isolation
For murine derived strains; stool was collected from a male C57BL/6 male mouse acquired from Jackson Laboratory (Bar Harbor, ME). Stool was suspended in sterile DI water and homogenized for 2 min. at 3500 rpm in Mini-Beadbeater-24 (Biospec, Bartlesville, OK). Homogenized stool was plated on MacConkey agar containing lactose. Potential E. coli colonies were selected based on colony shape and ability to ferment lactose. They were cultured for isolation twice. 16S rRNA genes were amplified using the primers described below (Illumina Part #15044223 Rev. B) and sequenced using Sanger sequencing (Eton Biosciences, La Jolla, CA) to confirm identity.
For human-derived strains, volunteers undergoing outpatient endoscopy were selected in accordance with VA San Diego IRB #H130266. Biopsies were collected from various regions of the GI tract adjacent to sites that were needed for clinical indications (e.g. polyps) and immediately transferred to sterile PBS. Tissue samples were homogenized, and bacterial growth, isolation, and identification was performed as described above. 16S rRNA gene amplicon PCR forward primer =
5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG;
16S amplicon PCR reverse primer =
5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC
Strain Development
The index native murine E. coli used for transformation was labeled as EcAZ-1. Genome assembly for this strain can be found at the European Nucleotide Archive under accession number ENA:PRJEB39003. Pacbio and Illumina reads were assembled using Unicycler (https://doi.org/10.1371/journal.pcbi.1005595) v.0.5.0 using default settings and annotated with Prokka (https://doi.org/10.1093/bioinformatics/btu153) v.1.14.6 with an assigned species of Escherichia coli and adding Swissprot proteins (downloaded 6/16/2021) as an additional annotation source. For EcAZ-2, ECAZ-1 was exposed to P1vir lysate trained on MG1655 with kanamycin resistance (aph) and fluorescence (GFP-mut2) cassette added at the attB site using recombination (pSIM18). Successful transductants were selected for using kanamycin resistance (aph) and fluorescence (EcAZ-2). EcAZ-1 was nearly 100-fold more resistant to phage transduction than E. coli Nissle. For the construction of EcAZ-2 containing bile salt hydrolase (BSH; EcAZ-2BSH+), EcAZ-1 was exposed to P1vir lysate trained on MG1655 containing chloramphenicol resistance (cat) and bsh cassette added at the yfgG site using recombination (pSIM18). Successful transductant was selected for functionality of BSH and chloramphenicol resistance. Verification of BSH functionality was verified by plating on lactose-containing LB plates supplemented with TDCA. Precipitation of DCA indicated functioning BSH. The successful transductant was then exposed to lysate with kanamycin resistance (aph) and fluorescence (GFP-mut2). Successful transductant was selected for gain of kanamycin resistance. The bsh gene used for this study naturally occurs in Lactobacillus salivarius JCM1046 and is effective at altering bile acids in germ free and antibiotic-treated, conventionalized mice when expressed by an E. coli Nissle chassis (https://doi.org/10.1073/pnas.1323599111). The first insertion event (for the cassette containing aph and gfp) occurred between chromosomal bases 3,267,361–3,274,418, which covers a Ribosomal RNA operon, murI (glutamate racemase) and btuB (vitamin B12 / E colicin / bacteriophage BF23 outer membrane porin BtuB). The second insertion event (for the cassette containing cat and bsh) occurred between chromosomal bases 1,886,038–1,887,917, which covers ydcM (putative transposase), ybhB (putative kinase inhibitor), and ybhC (outer membrane lipoprotein).
For the construction of EcAZ-2 containing IL-10 (EcAZ-2IL10+), EcAZ-1 was exposed to a lysate trained on MG1655 containing chloramphenicol resistance (cat) and human Il10. The successful transductants were selected for via IL-10 gene detection by PCR and chloramphenicol resistance. Insertion occurred in the yfgG (metal ion stress response protein) gene. Verification of IL-10 expression was performed by growing a overnight cultures of 6 isolates per mouse in Super Optimal Broth (SOB), 12.5 ug/mL of Kanamycin and 10 ug/mL of chloramphenicol. The OD600 of the cultures were read and the lowest value was used to normalize the amount of bacteria used on the ELISA. The normalized cultures were centrifuged and and resuspended in Assay diluent (10% FBS in 1× PBS). The samples underwent four freeze-thaw cycles and the cell lysates were tested on a human IL-10 detection ELISA (Catalog 430601, Biolegend). The supernatant that was drawn off each sample before normalization was also tested on a human IL-10 detection ELISA (Catalog 430601, Biolegend).
Growth Curves
Overnight cultures of SOB, supplemented with no antibiotic for EcAZ-1, 12.5 ug/mL of kanamycin for EcAZ-2, and 10 ug/mL of chloramphenicol for EcAZ-2BSH+ and EcAZ-2IL10+, were back diluted 1:100 in the same media described and loaded in triplicate in a 96-well plate. Blanks for each media were also loaded in triplicate. Incubation and measurements were done in an Infinite M Nano+ plate reader (Teacn, Morrisville, NC) at 37°C. OD600 measurements were performed every 10 minutes for 18 hours directly after 5 seconds of shaking. OD was determined by subtracting out the average blank for the media type for each time point.
Antibiotic Susceptibility Testing
Human bacterial isolates were assessed for susceptibility to commonly used laboratory antibiotics chloramphenicol, kanamycin, and carbenicillin. Overnight cultures of each strain were grown in LB broth. Antibiotics were diluted in Mueller Hinton broth to the following working concentrations: (1) 10 ug/mL, 5 ug/ml, and 2.5 ug/mL of chloramphenicol, (2) 25 ug/mL, 12.5 ug/mL, and 6.25 ug/mL of kanamycin, (3) 100 ug/mL, 50 ug/mL, and 25 ug/mL. Overnight cultures were back diluted 1:1000 into Mueller Hinton broth with antibiotics in a 96-well plate and incubated at 37°C for 18 hours. OD600 measurements were performed at the beginning and end of the 18 hour incubation directly after 5 seconds of shaking. OD was determined by subtracting out the average blank for the media type for each time point.
Transformation Efficiency
Human strains were tested for susceptibility to transformation via electroporation. Electrocompetent cells were prepared in 1mM MOPS buffer. Three plasmid sizes were assessed included pTrcHisC (4.4kb), a modified version of pNCM (pNCM[ΔccdB::aadA Δrep::[cat gfp]], roughly 70kb), and an additionally modified version of pNCM (pNCM[traMJ::[aph FRT] Δtra ΔfinO202::[N25-gfp]], roughly 35kb). All plasmid transformations were assessed at 1800 V, 200 ohms, and 25 uF. Strains were recovered in SOC for 1.5 hours and plated on selective and non-selective LB plates. After overnight incubation at 37°C, colonies were counted and CFU/mL was determined for successful transformants and viable cells present. Efficiency was determined as CFU/mL on selective plates divided by CFU/mL on non-selective plates. Due to complications with antibiotic selection for the modified version of pNCM, some transformation efficiencies could not be calculated. In these instances, the limit of detection is reported.
Conjugation Efficiency
Human bacterial isolates were assessed for susceptibility to conjugation using a 2,6-diaminopimelic acid dependent donor strain containing a modified F-plasmid, pNCM, containing a kanamycin resistance marker and green fluorescent protein with a deletion in finO to promote conjugation. The human and donor strains were grown to mid-log phase growth (OD600 0.4–0.6) in SOC. The human strains were each mixed 1:1 with the donor strain and incubated in SOC with no shaking for 4 hours. Successful conjugates were selected on LB plates for the ability to grow without DAP, and for a gain of GFP and kanamycin resistance. After overnight incubation at 37°C, colonies were counted and CFU/mL was determined for successful conjugants and viable cells present. Efficiency was determined as CFU/mL on selective plates divided by CFU/mL on non-selective plates.
Colonization Assessment
For stool, we collected 3–5 stool pellets from individual mice and weighed them. 1 mL of sterile deionized water was added to each sample along with one sterile chrome bead (Neta Scientific, Hainesport, NJ). Samples were homogenized for 2 min. at 3500 rpm in Mini-Beadbeater-24 (Biospec, Bartlesville, OK). For control samples, homogenized stool was plated on kanamycin and chloramphenicol to check for bacterial contamination. For EcAZ-2 colonized mice, samples were plated on chloramphenicol to check for unintended bacterial contamination and diluted and plated on kanamycin to calculate CFU/gram of stool. The EcAZ-2BSH+ and EcAZ-2IL10+ stool was diluted and plated on kanamycin and chloramphenicol.
Collected and tested tissues include duodenum defined as the first 10 cm of small intestine, jejunum defined as the next 10 cm of small intestine, Ileum defined as the next 5 cm of small intestine, cecum, and colon.
For tissues, mice were euthanized and tissue samples were harvested and immediately placed on ice. Dissection tools were disinfected between animals. Samples were processed and plated as described for stool. For each region of the GI tract, bacterial colonies were tested for enzyme functionality by patching from non-selective plates onto lactose and taurodeoxycholic acid (TDCA) containing plates. Up to 25 colonies for each region for each mouse were tested. In cases where 25 colonies did not grow, all colonies were tested.
For each region of the GI tract, verification of IL-10 expression was performed by growing an overnight culture of isolates in SOB, 12.5 ug/mL of Kanamycin and 10 ug/mL of chloramphenicol. In order to lyse the bacteria, the cultures were centrifuged, and the resulting pellet was resuspended in Assay diluent (10% FBS in 1× PBS). The samples were then flash frozen and defrosted for 4 rounds to lyse the cells and used on a IL-10 detection ELISA (Catalog 430601, Biolegend).
Co-Housing Experiment
Three pairs of 6-month-old male C57BL/6 mice were co-housed in a low barrier facility (where food and bedding were not autoclaved). From each cage, one mouse was gavaged with 200 μL of PBS while the other was gavaged with 200 μL ~1×1010 cfu/mL EcAZ-2. After gavage, mice were single housed for a week. Five days post-gavage, baseline stool was collected from each mouse. The pairs were then reunited with their cage mate. Initial stool collection demonstrated coprophagia but no persistent colonization. After 19 days, the protocol was modified so that mice were separated and individually housed one day prior to stool collection. After stool collection, each mouse was reunited with their cage mates until the next stool collection. Thus, with the revised protocol, coprophagic incidents would no longer appear and only incidents of horizontal transfer/colonization would be detectable.
Dosing experiment
20 mice were singly housed with 5 mice per condition. All mice were house in the low barrier facility. Mice were gavaged with 200 μL of either ~1×108 cfu/mL, 1×106 cfu/mL, 1×104 cfu/mL, or 1×102 cfu/mL of EcAZ-1. Stool collections were performed 2 days, 5 days, 1 week, 2 week, and 20 days after gavage as described above.
Gnotobiotic Mice
Germ-free C57BL/6 mice were bred and maintained in sterile flexible film isolators and screened for bacterial, viral, and fungal contamination as described (https://doi.org/10.30802/AALAS-JAALAS-18-000130). Mice were fed autoclaved chow (Envigo) and transferred into the Sentry SPP cage (Allentown). Mice were inoculated with corresponding bacteria oral gavage. Stool was collected for bile acid analysis 7 days later. Mono-colonization was confirmed upon euthanasia of the gnotobiotic mice.
Quantitative Multiplex Proteomics
For sample preparation, cell pellets from vehicle, EcAZ-2, and EcAZ-2BSH+ E. coli were analyzed by quantitative multiplex proteomics at the UCSD Collaborative Center for Multiplexed Proteomics (https://doi.org/10.1016/j.cell.2020.07.040). EcAZ-1, EcAZ-2, and EcAZ-2BSH+ were grown overnight in LB with their respective antibiotics in replicate then back diluted 1:20 into 10mL LB broth and grown to mid-log phase (OD600 0.4–0.6). Samples were resuspended in equal volumes of lysis buffer (6M urea, 7% SDS, 50 mM TEAB, titrated to pH 8.1 with phosphoric acid) and sonicated. Disulfide bonds were reduced in 5 mM dithiothreitol (DTT) at 56 °C for 30 min and free cysteines were alkylated in 15 mM iodoacetamide (IAA) in a darkened environment for 20 min. The alkylation reaction was quenched for 15 min at room temperature through the addition of an equivalent volume of DTT as in the reduction reaction. Proteins were then trapped using ProtiFi S-Trap columns, digested with trypsin (Promega, V5113), and eluted according to manufacturing protocols. Eluents were desalted on C18 columns using instructions provided by the manufacturer (Waters). Desalted samples were dried under vacuum. Samples were subjected to peptide quantification using a Pierce Colorimetric Peptide Quantification Assay kit per the manufacturer’s instructions. 50 μg of each sample was separated for further processing, with four samples aliquoted twice to reach a total of 16 samples.
For sample labeling, samples were resuspended in a solution of 30% anhydrous acetonitrile (ACN) with 200 mM HEPES, pH = 8.5. Tandem mass tag (TMT) labels (Thermo Fisher Scientific; Catalog Number: A44520, Lot Number: WJ327115) were suspended in anhydrous ACN to a final concentration of 20 mg/mL, and 7 μL were added to each resuspended sample. Samples were randomized in the TMT 16plex. The labeling reaction was allowed to proceed for 1 h at room temperature, after which excess label was quenched through the addition of 8 μL of 5% hydroxylamine for 15 min. 50 μL of 1% trifluoroacetic acid (TFA) was added to each sample, and samples were combined and desalted on C18 columns (Waters) as above. The multiplexed samples were dried under vacuum.
For Fractionation, multiplexed samples were next subjected to fractionation using reverse phase high pH liquid chromatography. Briefly, samples were fractionated on an Ultimate 3000 high performance liquid chromatography system fitted with fraction collector, C18 column (4.6 × 250 mm), solvent degasser, and variable wavelength detector. Multiplexed samples were fractionated on a gradient ranging from 22% to 35% ACN with 10 mM ammonium bicarbonate (ABC) over 60 min. The resulting 96 fractions were concatenated using methods previously described (https://doi.org/10.1002/pmic.201000722). Briefly, alternating wells were combined within each column, resulting in 24 total fractions. Alternating concatenated fractions were used for proteomic analysis.
For mass spectrometry-based proteomic analysis, all proteome mass spectrometry data were collected on an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific) with an in line Easy-nLC. Previously described methods were utilized for data collection (https://doi.org/10.1038/s41380-021-01339-z). Briefly, proteome data was collected using a “low resolution” method.
For data processing and normalization, raw files were searched using the SEQUEST algorithm in Proteome Discoverer 2.5 against the reference proteome for E. coli strain NGF-1 with the BSH1 sequence added (Uniprot Accession Q1WR93). Data were searched using a precursor mass tolerance of 50 ppm and fragment mass tolerance of 0.6. Static modifications were specified as follows: TMTpro on lysines and N-termini and carbamidomethylation of cysteines. Dynamic modifications were specified to include oxidation of methionines. Resulting peptide spectral matches were filtered at a 0.01 FDR by the Percolator module against the decoy database. Peptide spectral matches were exported, summed to the protein level, and batch corrected as previously described (https://doi.org/10.1074/mcp.M116.066019). Following batch correction, each sample normalized using Box-Cox power transformation (MASS R package, version 7.3–55), abundances were scaled between 0–1 (reshape R package, version 0.8.8), and each sample was standardized by its mean (https://doi.org/10.1111/j.2517-6161.1964.tb00553.x). Sample duplicates were averaged at this step. Final values represent normalized relative protein abundances.
BSH Activity Assay
Overnight cultures of LB, supplemented with no antibiotic for EcAZ-1, 12.5 ug/mL of kanamycin for EcAZ-2, and 10 ug/mL of chloramphenicol for EcAZ-2BSH+ were back diluted 1:20 in 1mL LB with the appropriate antibiotics and grown to mid-log phase (OD600 0.4–0.6). Cultures were centrifuged and the cell pellet was resuspended in 1mL of 0.1M sodium phosphate buffer, pH 6.0. Bacteria underwent freeze-thaw lysis and spun to pellet cell debris. Supernatant was mixed with 50uM TDCA and aliquots were removed and quenched in equal volume of 0.5M HCl at each time point. Samples were stored at −80°C until mass spectroscopy analysis.
Stool and Serum Bile Acids
Bile acids were extracted from samples as described before (https://doi.org/10.1038/s41467-018-05336-9, https://doi.org/10.1007/s00216-016-0048-1). Briefly, stool samples were homogenized and extracted in methanol (10 mg of sample/100 μL) containing heavy internal standards. Serum samples (25 μL) were extracted with 75 μL of methanol containing heavy internal standards. After vortexing for 10 minutes and centrifuging (16,000 × g, 4 C, 10 min), supernatants were transferred to glass vials for injection. Bile acids were analyzed on a Dionex Ultimate 3000 LC system (Thermo) coupled to a TSQ Quantiva mass spectrometer (Thermo) fitted with a Kinetex C18 reversed phase column (2.6 μm, 150 × 2.1 mm i.d., Phenomenex). The following LC solvents were used: solution A, 0.1 % formic acid and 20 mM ammonium acetate in water, solution B, acetonitrile/methanol (3/1, v/v) containing 0.1 % formic acid and 20 mM ammonium acetate. The following reversed phase gradient was utilized: at a flow rate of 0.2 mL/min with a gradient consisting of 25–29 % B in 1 min, 29–33 % B in 14 min, 33–70 % B in 15 min, up to 100 % B in 1 min, 100 % B for 9 min and re-equilibrated to 25 % B for 10 min, for a total run time of 50 min. The injection volume for all samples was 10 μL, the column oven temperature was set to 50°C and the autosampler kept at 4°C. MS analyses were performed using electrospray ionization in positive and negative ion modes, with spray voltages of 3.5 and −3 kV, respectively, ion transfer tube temperature of 325°C, and vaporizer temperature of 275°C. Multiple reaction monitoring (MRM) was performed by using mass transitions between specific parent ions into corresponding fragment ions for each analyte. Results were quantified using isotopically labeled internal standards. Data were averaged across samples.
The bile acids measured using this technique include CA, TCA, bMCA, TbMCA, α-muricholic acid (aMCA), tauro-α-muricholic acid (TaMCA), chenodeoxycholic acid (CDCA), taurochenodeoxycholic acid (TCDCA), DCA, TDCA, hyocholic acid (HCA), taurohyocholic acid (THCA), ω-muricholic acid (oMCA), tauro-ω-muricholic acid (ToMCA), ursodeoxycholic acid (UDCA) and lithocholic acid (LCA). Glycine conjugated bile acids were not detected. Of note, no UDCA was detected in the stool. Total bile acid measurements was the sum of all bile acids measured using mass spectrometry. Primary bile acids included CA, TCA, aMCA, TaMCA, bMCA, TbMCA, CDCA, and TCDCA. Secondary bile acids included DCA, TDCA, oMCA, ToMCA, UDCA, and LCA.
16S rRNA Gene Sequence/Microbiome Data Processing and Analysis
16S rRNA gene amplicon sequences were processed using Qiime2 (version 2020.6) using default parameters (https://doi.org/10.1038/s41587-019-0209-9). ASVs were generated by the Deblur method, trimmed to 120bp (https://doi.org/10.1128/mSystems.00191-16). Taxonomy was assigned via the sklearn plugin (https://doi.org/10.1101/2020.10.05.326504; https://doi.org/10.1186/s40168-018-0470-z) against the Silva v123 model (https://doi.org/10.1093/nar/gks1219), and ASVs were filtered to remove Eukaryotic, Mitochondrial, Plastid, and unassigned taxa. The phylogenetic diversity metrics and PCoA were then calculated on samples rarefied to 4000 reads.
qPCR from Liver and Terminal Ileum
The livers and terminal ileum from control and EcAZ-2BSH+ mice were flash frozen in liquid nitrogen. The frozen tissues were homogenized and RNA was extracted using TriZol reagent according to manufacturer’s instructions. 1ug of RNA was reverse transcribed using quanta biosciences qScript cDNA supermix (Cat# 95048–025). The samples were diluted 1:50 and qRTPCR was run using FastStart Universal SYBR Green Master (Rox) (Cat# 4913850001) on an Applied Biosystems QuantStudio 5. The data were analyzed and gene expression was expressed relative to mrpl46.
| Primers | Sequence |
|---|---|
|
| |
| CYP7a1 Fwd | GGGATTGCTGTGGTAGTGAGC |
| CYP7a1 Rev | GGTATGGAATCAACCCGTTGTC |
| CYP27a1 Fwd | CCAGGCACAGGAGAGTACG |
| CYP27a1 Rev | GGGCAAGTGCAGCACATAG |
| Fxr Fwd | GCTTGATGTGCTACAAAAGCTG |
| Fxr Rev | CGTGGTGATGGTTGAATGTCC |
| Nr0b2 Fwd | TGGGTCCCAAGGAGTATGC |
| Nr0b2 Rev | GCTCCAAGACTTCACACAGTG |
Blood Collection and Serum Biomarker Measurements
Blood was collected from fasted (16 h) or re-fed animals (30 min). Re-feeding was performed by oral gavage of Ensure (Abbott, Columbus, Ohio). In male mice, serum insulin was performed using the Bio-Plex Pro Mouse Diabetes 8-Plex Assay (Bio-Rad, Hercules, CA) as per the manufacturer’s protocol.
Glucose Tolerance
Glucose tolerance assays were performed on fasted mice (16 h, Pur-O-Cel bedding) by monitoring glucose levels after a glucose bolus (10 uL of 20% glucose/kg of body weight (BW)) via oral gavage. Glucose concentration was measured from a tail snip using a OneTouch Ultra glucometer. Tail tips were anesthetized with a topical anesthetic (Actavis, Parsippany-Troy Hills, NJ) prior to snip.
Insulin Measurement
10-week old female mice were fasted (5 h, Pur-O-Cel bedding) and then bled from the tail vein to collect fasted plasma. Two days later, mice were again fasted (14–16 h, Pur-O-Cel bedding) and then orally gavaged with 0.5g glucose/kg body weight of Ensure (Abbott, Columbus, Ohio) to measure insulin response to a mixed meal. 30 minutes after the gavage, blood was collected through a submandibular bleed. Serum insulin from fasted and fed blood was measured using a mouse ELISA (Crystal Chem Ultra Sensitive Mouse ELISA Kit).
QUANTIFICATION AND STATISTICAL ANALYSIS
All comparisons of EcAZ-2 to EcAZ-2BSH+ were calculated using Mann-Whitney U test or Kruskal-Wallis test with post-hoc Dunn’s multiple comparison test as specified with the exception of Figures 1E, 1F, 4D, and S3A–S3C, which used a student’s t-test due to low sample size. When student’s t-test was used, normality of data was confirmed with a Q-Q plot prior to the test. For qualitative 2×2 comparisons (Figures 2B and S2B), a Fisher’s exact test was used to determine significance.
For proteomics data, statistical tests assuming normalized data with equal means and unequal variances were used. To compare all three groups, Brown-Forsythe and Welch one-way ANOVA was used. A p-value of < 0.05 was considered statistically significant. Protein abundances were graphed using GraphPad Prism software (version 9.3.1).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Escherichia coli MG1655 | Coli Genetic Stock Center | Cat#: 6300 |
| Escherichia coli Nissle 1917 | Provided by Suckjoon Jun | N/A |
| Escherichia coli EcAZ-1 | This manuscript | N/A |
| Escherichia coli EcAZ-2 | This manuscript | N/A |
| Escherichia coli EcAZ-2BSH | This manuscript | N/A |
| Escherichia coli EcAZ-2IL10 | This manuscript | N/A |
| Escherichia coli AZ-61 | This manuscript | N/A |
| Escherichia coli AZ-116 | This manuscript | N/A |
| Escherichia coli AZ-120 | This manuscript | N/A |
| Escherichia coli AZ-122 | This manuscript | N/A |
| P1 vir | Coli Genetic Stock Center | Cat#: 12133 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Sodium taurodeoxycholate hydrate (TDCA) | Millipore Sigma | Cat#:T0875 |
| 2,6-diaminopimelic acid | Life Technologies | Cat#:B22391.03 |
| Tandem mass tag labels | Thermo Fisher Scientific | Cat#: A44520 |
| qScript cDNA supermix | Quantabio | Cat:# 95048-025 |
| FastStart Universal SYBR Green Master | Sigma-Aldrich | Cat#: 4913850001 |
|
| ||
| Critical commercial assays | ||
|
| ||
| IL-10 ELISA | Biolegend | Cat#: 430610 |
| Bio-Plex Pro Mouse Diabetes 8-Plex Assay | Bio-Rad | Cat#: 171F7001M |
| Ultra Sensitive Mouse ELISA Kit | Crystal Chem | Cat#: 90080 |
|
| ||
| Deposited data | ||
|
| ||
| Proteomics data | This manuscript: massive.ucsd.edu | PXD032187 |
| 16S sequences and genome assembly | ENA project | PRJEB39003 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6J mice | The Jackson Laboratory | Strain #000664 |
| Ob/Ob mice B6.Cg-Lepob/J | The Jackson Laboratory | Strain #000632 |
|
| ||
| Oligonucleotides | ||
|
| ||
| 16S rRNA gene amplicon PCR forward primer: 5’TCG TCGGCAGCGTCAGATGT GTATAAGAGACAGCCTAC GGGNGGCWGCAG | Klindworth et al., 2013 | https://doi.org/10.1093/nar/gks808 |
| 16S amplicon PCR reverse primer: 5’GTCTCGTGGGC TCGGAGATGTGTATAAGAGACAGGACTACHVGGGT ATCTAATCC | Klindworth et al., 2013 | https://doi.org/10.1093/nar/gks808 |
| CYP7a1 Fwd: GGGATTGCTGTGGTAGTGAGC | This manuscript | N/A |
| CYP7a1 Rev: GGTATGGAATCAACCCGTTGTC | This manuscript | N/A |
| CYP27a1 Fwd: CCAGGCACAGGAGAGTACG | This manuscript | N/A |
| CYP27a1 Rev: GGGCAAGTGCAGCACATAG | This manuscript | N/A |
| Fxr Fwd: GCTTGATGTGCTACAAAAGCTG | This manuscript | N/A |
| Fxr Rev: CGTGGTGATGGTTGAATGTCC | This manuscript | N/A |
| Nr0b2 Fwd: TGGGTCCCAAGGAGTATGC | This manuscript | N/A |
| Nr0b2 Rev: GCTCCAAGACTTCACACAGTG | This manuscript | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pTrcHisC | Invivogen | Cat#:1324578 |
| pNCM | GeneBank | Acc#: CP011496 |
| Modified pNCM: pNCM[traMJ::[aph FRT] ΔfinO102::[cat N25-gfp]] | This manuscript | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| MASS R package version 7.3-55 | https://cran.r-project.org/web/packages/MASS/index.html | N/A |
| reshape R package version 0.8.8 | http://had.co.nz/reshape | N/A |
| Qiime2 version 2020.6 | https://doi.org/10.1038/s41587-019-0209-9 | N/A |
| Prism | GraphPad | v9.3.1 |
|
| ||
| Other | ||
|
| ||
| Orbitrap Fusion mass spectrometer | Thermo Fisher Scientific | Cat#:IQLAAEGAAPFADBMBCX |
| Dionex Ultimate 3000 LC | Thermo Fisher Scientific | LC/MS-5TF-0330 |
| TSQ Quantiva mass spectrometer | Thermo Fisher Scientific | LC/MS-5TF-0330 |
Highlights.
Native E. coli from the murine and human gut can be engineered for transgene delivery
Engineered native E. coli (EcAZ) engrafts in conventional mice in non-sterile conditions
Single treatments with EcAZ can be used to induce persistent physiological change
Single treatments with EcAZ can persistently ameliorate chronic pathological states
ACKNOWLEDGMENTS
We would like to express our appreciation to Dr. Kim Barrett and Dr. David Brenner, who provided resources to initiate this project during its conceptualization. S.D.B. was supported by NIH F32 DK113721. N.S. is a Biolegend Fellow. A.R.S. is supported by the Glenn Foundation for Medical Research Postdoctoral Fellowships in Aging Research. I.M. was supported in part by a UCSD Eureka Foundation Grant. E.S.M. is supported by NIH F31 HD106762. A.C.D.M. is supported by NIH R01 HL148801–02S1. C.S. is supported in part by NIH T32 AR064194. L.-A.R. is supported in part by NIH T32 GM007752. D.J.G. is supported by the UCSD Collaborative Center of Multiplexed Proteomics. A.Z. is supported by AFAR Research Grant for Junior Faculty, National Phenylketonuria Alliance, American Heart Association Beginning Grant-in-Aid (16BGIA27760160), Kavli Institute for Brain and Mind at UC San Diego, Jon I. Isenberg Endowed Fellowship, AASLD Liver Scholar Award, AGA Microbiome Junior Investigator Award, VA Merit BLR&D Award I01 BX005707, and NIH K08 DK102902, R03 DK114536, R21 MH117780, R01 HL148801, R01 EB030134, R01 HL157445, and U01 CA265719. All authors receive institutional support from NIH P30 DK120515, P30 DK063491, P30 CA014195, P50 AA011999, and UL1 TR001442. This material is the result of work supported with resources and the use of facilities at the VA San Diego Healthcare Center. The funders had no role in study design, data collection, and interpretation, or the decision to submit the work for publication. The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the United States government.
Footnotes
DECLARATION OF INTERESTS
A.Z. and S.D.B. are co-founders and equity-holders in Endure Biotherapeutics. They have filed a provisional patent based on the work described here (US Provisional Patent No. 16/604,138).
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2022.06.050.
REFERENCES
- Allaband C, Lingaraju A, Martino C, Russell B, Tripathi A, Poulsen O, Dantas Machado AC, Zhou D, Xue J, Elijah E, et al. (2021). Intermittent hypoxia and hypercapnia alter diurnal rhythms of luminal gut microbiome and metabolome. mSystems, e0011621. 10.1128/mSystems.00116-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aron-Wisnewsky J, Vigliotti C, Witjes J, Le P, Holleboom AG, Verheij J, Nieuwdorp M, and Clément K (2020). Gut microbiota and human NAFLD: disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 17, 279–297. 10.1038/s41575-020-0269-9. [DOI] [PubMed] [Google Scholar]
- Bailey JK, Pinyon JL, Anantham S, and Hall RM (2010). Commensal Escherichia coli of healthy humans: a reservoir for antibiotic-resistance determinants. J. Med. Microbiol. 59, 1331–1339. 10.1099/jmm.0.022475-0. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Schlaeppi K, and van der Heijden MGA (2018). Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 16, 567–576. 10.1038/s41579-018-0024-1. [DOI] [PubMed] [Google Scholar]
- Bermúdez-Humarán LG, Aubry C, Motta JP, Deraison C, Steidler L, Vergnolle N, Chatel JM, and Langella P (2013). Engineering lactococci and lactobacilli for human health. Curr. Opin. Microbiol. 16, 278–283. 10.1016/j.mib.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Biteen JS, Blainey PC, Cardon ZG, Chun M, Church GM, Dorrestein PC, Fraser SE, Gilbert JA, Jansson JK, Knight R, et al. (2016). Tools for the microbiome: nano and beyond. ACS Nano 10, 6–37. 10.1021/acsnano.5b07826. [DOI] [PubMed] [Google Scholar]
- Bober JR, Beisel CL, and Nair NU (2018). Synthetic biology approaches to engineer probiotics and members of the human microbiota for biomedical applications. Annu. Rev. Biomed. Eng. 20, 277–300. 10.1146/annurev-bioeng-062117-121019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bok E, Mazurek J, Myc A, Stosik M, Wojciech M, and Baldy-Chudzik K (2018). Comparison of commensal Escherichia coli isolates from adults and young children in Lubuskie province, Poland: Virulence Potential, Phylogeny and Antimicrobial Resistance. Int. J. Environ. Res. Public Health 15, 617. 10.3390/ijerph15040617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braat H, Rottiers P, Hommes DW, Huyghebaert N, Remaut E, Remon JP, van Deventer SJ, Neirynck S, Peppelenbosch MP, and Steidler L (2006). A phase I trial with transgenic bacteria expressing interleukin-10 in Crohn’s disease. Clin. Gastroenterol. Hepatol. 4, 754–759. 10.1016/j.cgh.2006.03.028. [DOI] [PubMed] [Google Scholar]
- Brophy JAN, Triassi AJ, Adams BL, Renberg RL, Stratis-Cullum DN, Grossman AD, and Voigt CA (2018). Engineered integrative and conjugative elements for efficient and inducible DNA transfer to undomesticated bacteria. Nat. Microbiol. 3, 1043–1053. 10.1038/s41564-018-0216-5. [DOI] [PubMed] [Google Scholar]
- Chu DM, Ma J, Prince AL, Antony KM, Seferovic MD, and Aagaard KM (2017). Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat. Med. 23, 314–326. 10.1038/nm.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claesen J, and Fischbach MA (2015). Synthetic microbes as drug delivery systems. ACS Synth. Biol. 4, 358–364. 10.1021/sb500258b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook N, Ferreiro A, Gasparrini AJ, Pesesky MW, Gibson MK, Wang B, Sun X, Condiotte Z, Dobrowolski S, Peterson D, et al. (2019). Adaptive strategies of the candidate probiotic E. coli Nissle in the mammalian gut. Cell Host Microbe 25, 499–512.e8. 10.1016/j.chom.2019.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantas Machado AC, Brown SD, Lingaraju A, Sivaganesh V, Martino C, Chaix A, Zhao P, Pinto AFM, Chang MW, Richter RA, et al. (2022). Diet and feeding pattern modulate diurnal dynamics of the ileal microbiome and transcriptome. Cell Rep. 40, 111008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes AD, Reid JN, Macklaim JM, McMurrough TA, Edgell DR, and Gloor GB (2014). Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2, 15. 10.1186/2049-2618-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Food and Drug Administration (2016). Early clinical trials with live biotherapeutic products: chemistry, manufacturing, and control information – guidance for industry. https://www.fda.gov/files/vaccines,%20blood%20&%20biologics/published/Early-Clinical-Trials-With-Live-Biotherapeutic-Products–Chemistry–Manufacturing–and-Control-Information–Guidance-for-Industry.pdf.
- Guglielmetti S, Mayo B, and Álvarez-Martín P (2013). Mobilome and genetic modification of bifidobacteria. Benef. Microbes 4, 143–166. 10.3920/BM2012.0031. [DOI] [PubMed] [Google Scholar]
- Gurung M, Li Z, You H, Rodrigues R, Jump DB, Morgun A, and Shulzhenko N (2020). Role of gut microbiota in type 2 diabetes pathophysiology. EBiomedicine 51, 102590. 10.1016/j.ebiom.2019.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holman HR (2020). The relation of the chronic disease epidemic to the health care crisis. ACR Open Rheumatol. 2, 167–173. 10.1002/acr2.11114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Jiang S, Huo D, Allaband C, Estaki M, Cantu V, Belda-Ferre P, Vá zquez-Baeza Y, Zhu Q, Ma C, et al. (2021). Candidate probiotic Lactiplantibacillus plantarum HNU082 rapidly and convergently evolves within human, mice, and zebrafish gut but differentially influences the resident microbiome. Microbiome 9, 151. 10.1186/s40168-021-01102-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isabella VM, Ha BN, Castillo MJ, Lubkowicz DJ, Rowe SE, Millet YA, Anderson CL, Li N, Fisher AB, West KA, et al. (2018). Development of a synthetic live bacterial therapeutic for the human metabolic disease phenylketonuria. Nat. Biotechnol. 36, 857–864. 10.1038/nbt.4222. [DOI] [PubMed] [Google Scholar]
- Jie Z, Xia H, Zhong SL, Feng Q, Li S, Liang S, Zhong H, Liu Z, Gao Y, Zhao H, et al. (2017). The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 8, 845. 10.1038/s41467-017-00900-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin WB, Li TT, Huo D, Qu S, Li XV, Arifuzzaman M, Lima SF, Shi HQ, Wang A, Putzel GG, et al. (2022). Genetic manipulation of gut microbes enables single-gene interrogation in a complex microbiome. Cell 185, 547–562.e22. 10.1016/j.cell.2021.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BV, Begley M, Hill C, Gahan CG, and Marchesi JR (2008). Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Natl. Acad. Sci. USA 105, 13580–13585. 10.1073/pnas.0804437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce SA, Macsharry J, Casey PG, Kinsella M, Murphy EF, Shanahan F, Hill C, and Gahan CG (2014a). Regulation of host weight gain and lipid metabolism by bacterial bile acid modification in the gut. Proc. Natl. Acad. Sci. USA 111, 7421–7426. 10.1073/pnas.1323599111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce SA, Shanahan F, Hill C, and Gahan CG (2014b). Bacterial bile salt hydrolase in host metabolism: potential for influencing gastrointestinal microbe-host crosstalk. Gut Microbes 5, 669–674. 10.4161/19490976.2014.969986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June CH, and Sadelain M (2018). Chimeric antigen receptor therapy. N. Engl. J. Med. 379, 64–73. 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, and Glöckner FO (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1. 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic AD, Xavier RJ, and Gevers D (2014). The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology 146, 1489–1499. 10.1053/j.gastro.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotula JW, Kerns SJ, Shaket LA, Siraj L, Collins JJ, Way JC, and Silver PA (2014). Programmable bacteria detect and record an environmental signal in the mammalian gut. Proc. Natl. Acad. Sci. USA 111, 4838–4843. 10.1073/pnas.1321321111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen NB, Bryrup T, Allin KH, Nielsen T, Hansen TH, and Pedersen O (2016). Alterations in fecal microbiota composition by probiotic supplementation in healthy adults: a systematic review of randomized controlled trials. Genome Med. 8, 52. 10.1186/s13073-016-0300-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasaro M, Liu Z, Bishar R, Kelly K, Chattopadhyay S, Paul S, Sokurenko E, Zhu J, and Goulian M (2014). Escherichia coli isolate for studying colonization of the mouse intestine and its application to two-component signaling knockouts. J. Bacteriol. 196, 1723–1732. 10.1128/JB.01296-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SM, Donaldson GP, Mikulski Z, Boyajian S, Ley K, and Mazmanian SK (2013). Bacterial colonization factors control specificity and stability of the gut microbiota. Nature 501, 426–429. 10.1038/nature12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Sun JY, Han LZ, Huang XH, Fu Q, and Ni YX (2010). Phylogenetic groups and pathogenicity island markers in fecal Escherichia coli isolates from asymptomatic humans in China. Appl. Environ. Microbiol. 76, 6698–6700. 10.1128/AEM.00707-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SS, Zhu A, Benes V, Costea PI, Hercog R, Hildebrand F, Huerta-Cepas J, Nieuwdorp M, Salojärvi J, Voigt AY, et al. (2016). Durable coexistence of donor and recipient strains after fecal microbiota transplantation. Science 352, 586–589. 10.1126/science.aad8852. [DOI] [PubMed] [Google Scholar]
- Lim B, Zimmermann M, Barry NA, and Goodman AL (2017). Engineered regulatory systems modulate gene expression of human commensals in the gut. Cell 169, 547–558.e15. 10.1016/j.cell.2017.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, Andrews E, Ajami NJ, Bonham KS, Brislawn CJ, et al. (2019). Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662. 10.1038/s41586-019-1237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal S, Van Treuren W, White RA, Eggesbø M, Knight R, and Peddada SD (2015). Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb. Ecol. Health Dis. 26, 27663. 10.3402/mehd.v26.27663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Març ais G, Delcher AL, Phillippy AM, Coston R, Salzberg SL, and Zimin A. (2018). MUMmer4: a fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944. 10.1371/journal.pcbi.1005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AB, Hartman M, Lassman D, and Catlin A; National Health Expenditure Accounts Team (2021). National Health Care Spending In 2019: steady growth for the fourth consecutive year. Health Aff. 40, 14–24. 10.1377/hlthaff.2020.02022. [DOI] [PubMed] [Google Scholar]
- McDonald D, Hyde E, Debelius JW, Morton JT, Gonzalez A, Ackermann G, Aksenov AA, Behsaz B, Brennan C, Chen Y, et al. (2018). American gut: an open platform for citizen science microbiome research. mSystems 3. e00031–e00018. 10.1128/mSystems.00031-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimee M, Citorik RJ, and Lu TK (2016). Microbiome therapeutics - Advances and challenges. Adv. Drug Deliv. Rev. 105, 44–54. 10.1016/j.addr.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimee M, Tucker AC, Voigt CA, and Lu TK (2015). Programming a human commensal Bacterium, Bacteroides thetaiotaomicron, to sense and respond to stimuli in the murine gut microbiota. Cell Syst. 1, 62–71. 10.1016/j.cels.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscogiuri G, Cantone E, Cassarano S, Tuccinardi D, Barrea L, Savastano S, and Colao A; on behalf of the Obesity Programs of nutrition, Education, Research and Assessment (OPERA) group (2019). Gut microbiota: a new path to treat obesity. Int. J. Obes. Suppl. 9, 10–19. 10.1038/s41367-019-0011-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najjuka CF, Kateete DP, Kajumbula HM, Joloba ML, and Essack SY (2016). Antimicrobial susceptibility profiles of Escherichia coli and Klebsiella pneumoniae isolated from outpatients in urban and rural districts of Uganda. BMC Res. Notes 9, 235. 10.1186/s13104-016-2049-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrolli DB, Ribeiro NV, Squizato PN, de Jesus VN, and Cozetto DA; Team AQA Unesp at iGEM 2017 (2019). Engineering microbial living therapeutics: the synthetic biology toolbox. Trends Biotechnol. 37, 100–115. 10.1016/j.tibtech.2018.09.005. [DOI] [PubMed] [Google Scholar]
- Puurunen MK, Vockley J, Searle SL, Sacharow SJ, Phillips JA 3rd, Denney WS, Goodlett BD, Wagner DA, Blankstein L, Castillo MJ, et al. (2021). Safety and pharmacodynamics of an engineered E. coli Nissle for the treatment of phenylketonuria: a first-in-human phase 1/2a study. Nat. Metab. 3, 1125–1132. 10.1038/s42255-021-00430-7. [DOI] [PubMed] [Google Scholar]
- Riglar DT, Giessen TW, Baym M, Kerns SJ, Niederhuber MJ, Bronson RT, Kotula JW, Gerber GK, Way JC, and Silver PA (2017). Engineered bacteria can function in the mammalian gut long-term as live diagnostics of inflammation. Nat. Biotechnol. 35, 653–658. 10.1038/nbt.3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riglar DT, and Silver PA (2018). Engineering bacteria for diagnostic and therapeutic applications. Nat. Rev. Microbiol. 16, 214–225. 10.1038/nrmicro.2017.172. [DOI] [PubMed] [Google Scholar]
- Rizk MG, and Thackray VG (2021). Intersection of polycystic ovary syndrome and the gut microbiome. J. Endocr. Soc. 5, bvaa177. 10.1210/jendso/bvaa177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronda C, Chen SP, Cabral V, Yaung SJ, and Wang HH (2019). Metagenomic engineering of the mammalian gut microbiome in situ. Nat. Methods 16, 167–170. 10.1038/s41592-018-0301-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin BE, Diamond S, Cress BF, Crits-Christoph A, Lou YC, Borges AL, Shivram H, He C, Xu M, Zhou Z, et al. (2022). Species- and site-specific genome editing in complex bacterial communities. Nat. Microbiol. 7, 34–47. 10.1038/s41564-021-01014-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saran AR, Dave S, and Zarrinpar A (2020). Circadian rhythms in the pathogenesis and treatment of fatty liver disease. Gastroenterology 158, 1948–1966.e1. 10.1053/j.gastro.2020.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepich-Poore GD, Zitvogel L, Straussman R, Hasty J, Wargo JA, and Knight R (2021). The microbiome and human cancer. Science 371. 10.1126/science.abc4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd ES, DeLoache WC, Pruss KM, Whitaker WR, and Sonnenburg JL (2018). An exclusive metabolic niche enables strain engraftment in the gut microbiota. Nature 557, 434–438. 10.1038/s41586-018-0092-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth RU, Cabral V, Chen SP, and Wang HH (2016). Manipulating bacterial Communities by in situ microbiome Engineering. Trends Genet. 32, 189–200. 10.1016/j.tig.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steidler L, Hans W, Schotte L, Neirynck S, Obermeier F, Falk W, Fiers W, and Remaut E (2000). Treatment of murine colitis by Lactococcus lactis secreting interleukin-10. Science 289, 1352–1355. [DOI] [PubMed] [Google Scholar]
- Stirling F, Bitzan L, O’Keefe S, Redfield E, Oliver JWK, Way J, and Silver PA (2017). Rational design of evolutionarily stable microbial kill switches. Mol. Cell 68, 686–697.e3. 10.1016/j.molcel.2017.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppe NC, Silva JS, Carlos C, Sato MIZ, Saraiva AM, Ottoboni LMM, and Torres TT (2017). Worldwide phylogenetic group patterns of Escherichia coli from commensal human and wastewater treatment plant isolates. Front. Microbiol. 8, 2512. 10.3389/fmicb.2017.02512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ticho AL, Malhotra P, Dudeja PK, Gill RK, and Alrefai WA (2019). Intestinal absorption of bile acids in health and disease. Compr. Physiol. 10, 21–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484. 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlström A, Sayin SI, Marschall HU, and Bäckhed F (2016). Intestinal crosstalk between bile acids and microbiota and its impact on Host Metabolism. Cell Metab. 24, 41–50. 10.1016/j.cmet.2016.05.005. [DOI] [PubMed] [Google Scholar]
- Walker RI, and Owen RL (1990). Intestinal barriers to bacteria and their toxins. Annu. Rev. Med. 41, 393–400. 10.1146/annurev.me.41.020190.002141. [DOI] [PubMed] [Google Scholar]
- Whitaker WR, Shepherd ES, and Sonnenburg JL (2017). Tunable expression tools enable single-cell strain distinction in the gut microbiome. Cell 169, 538–546.e12. 10.1016/j.cell.2017.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann A, Allahyar A, Greiner TU, Plovier H, Lundén GÖ, Larsson T, Drucker DJ, Delzenne NM, Cani PD, and Bäckhed F (2013). Microbial modulation of energy availability in the colon regulates intestinal transit. Cell Host Microbe 14, 582–590. 10.1016/j.chom.2013.09.012. [DOI] [PubMed] [Google Scholar]
- Wiles TJ, Wall ES, Schlomann BH, Hay EA, Parthasarathy R, and Guillemin K (2018). Modernized tools for streamlined genetic manipulation and comparative study of wild and diverse proteobacterial lineages. mBio 9. e01877–e01818. 10.1128/mBio.01877-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Stahel P, and Lewis GF (2019). Regulation of chylomicron secretion: focus on post-assembly mechanisms. Cell. Mol. Gastroenterol. Hepatol. 7, 487–501. 10.1016/j.jcmgh.2018.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Allaband C, Zhou D, Poulsen O, Martino C, Jiang L, Tripathi A, Elijah E, Dorrestein PC, Knight R, et al. (2021). Influence of intermittent hypoxia/hypercapnia on atherosclerosis, gut microbiome, and metabolome. Front. Physiol. 12, 663950. 10.3389/fphys.2021.663950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L, Seaton SC, Ndousse-Fetter S, Adhikari AA, DiBenedetto N, Mina AI, Banks AS, Bry L, and Devlin AS (2018). A selective gut bacterial bile salt hydrolase alters host metabolism. eLife 7, e37182. 10.7554/eLife.37182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrinpar A, Chaix A, Xu ZZ, Chang MW, Marotz CA, Saghatelian A, Knight R, and Panda S (2018). Antibiotic-induced microbiome depletion alters metabolic homeostasis by affecting gut signaling and colonic metabolism. Nat. Commun. 9, 2872. 10.1038/s41467-018-05336-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrinpar A, Chaix A, Yooseph S, and Panda S (2014). Diet and feeding pattern affect the diurnal dynamics of the gut microbiome. Cell Metab. 20, 1006–1017. 10.1016/j.cmet.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziesack M, Karrenbelt MAP, Bues J, Schaefer E, Silver P, and Way J (2018). Escherichia coli NGF-1, a genetically tractable, efficiently colonizing murine gut isolate. Microbiol. Resour. Announc. 7, e01416–e01418. 10.1128/MRA.01416-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data have been deposited at the European Nucleotide Archive under accession number ENA:PRJEB39003 and are publicly available as of the date of publication. Proteome data was uploaded to massive.ucsd.edu and can be accessed using the following identifiers: PXD032187.
All original code has been deposited at https://github.com/ZarrinparLab/2022_enb_paper and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.