

Abstract
Early disease detection is a prerequisite for enacting effective interventions for disease control. Strains of the bacterial plant pathogen Xylella fastidiosa have recurrently spread to new crops in new countries causing devastating outbreaks. So far, investigation of outbreak strains and highly resolved phylogenetic reconstruction have required whole-genome sequencing of pure bacterial cultures, which are challenging to obtain due to the fastidious nature of X. fastidiosa . Here, we show that culture-independent metagenomic sequencing, using the Oxford Nanopore Technologies MinION long-read sequencer, can sensitively and specifically detect the causative agent of Pierce’s disease of grapevine, X. fastidiosa subspecies fastidiosa . Using a DNA sample from a grapevine in Virginia, USA, it was possible to obtain a metagenome-assembled genome (MAG) of sufficient quality for phylogenetic reconstruction with SNP resolution. The analysis placed the MAG in a clade with isolates from Georgia, USA, suggesting introduction of X. fastidiosa subspecies fastidiosa to Virginia from the south-eastern USA. This proof of concept study, thus, revealed that metagenomic sequencing can replace culture-dependent genome sequencing for reconstructing transmission routes of bacterial plant pathogens.
Keywords: metagenomics, nanopore sequencing, pandemic prevention, plant biosecurity, plant disease diagnostics
Data Summary
Metagenomic sequencing files have been submitted to the National Center for Biotechnology Information (NCBI) SRA (Sequence Read Archive) under BioProject PRJNA763353, with BioSample accession numbers SAMN21438496, SAMN21438497, SAMN21438498, SAMN21438499, SAMN21438500 and SAMN21438501. The metagenome-assembled genome was submitted to the NCBI Assembly database and assigned accession number GCA_020792795.1.
Impact Statement.
Disease outbreak investigation in humans, animals and plants has tremendously benefitted from the development of new sequencing technology. For example, food-borne bacterial disease outbreaks are now routinely investigated using phylogenetics based on whole-genome sequences obtained from pure cultures. However, time is critical when an outbreak occurs so that control measures can be enacted quickly. Therefore, metagenomic sequencing, which does not require culturing and can be performed on DNA directly extracted from a host sample, would be desirable. Here, we show that it is possible to assemble an entire genome sequence of a bacterial plant pathogen, Xylella fastidiosa , directly from metagenomic sequences obtained with the Oxford Nanopore Technologies MinION sequencer. Since X. fastidiosa grows slowly in culture, this represents significant time saving. The obtained genome sequence quality was better than many public genome sequences available for this pathogen. Thus, it was possible to reconstruct a highly resolved phylogenetic tree and infer the likely geographical origin of the investigated strain. In conclusion, this proof of concept study represents a significant step forward in replacing culture-dependent genome-based approaches with culture-independent metagenome-based approaches when investigating pathogen transmission chains.
Introduction
Pathogens are a constant threat to humans, animals and plants. In regard to plant diseases, international trade of plant materials (plant globalization) plays a major role in the dissemination of plant pathogens to new areas, where they may come in contact with new hosts and lead to emergence of new diseases. Fast, specific and sensitive detection and identification of pathogens, and tracing their transmission routes, thus, is needed to quickly contain emerging disease outbreaks to prevent them from turning into epidemics and pandemics [1].
Xylella fastidiosa is a prime example of a pathogen that has spread out of its geographical areas of origin in the Americas to countries around the world through movement of plant material, which has led to the emergence of disease outbreaks on several crops in many countries and, in some cases, significant economic damage [2, 3]. For example, based on phylogenetic reconstruction and circumstantial evidence, it is likely that a single strain of X. fastidiosa subspecies fastidiosa was introduced to California at the end of the nineteenth century leading to the emergence of Pierce’s disease of grapevine (Vitis vinifera) that has since spread throughout California, other USA wine growing regions, and countries in Europe and Asia [4, 5]. Citrus variegated chlorosis caused by ' X. fastidiosa subspecies pauca' emerged in Brazil in the 1980s, becoming one of the main citrus diseases in that country [6]. More recently, quick olive decline caused by another strain of ' X. fastidiosa subspecies pauca' has been introduced to Italy, threatening olive production in the Mediterranean basin [7]. X. fastidiosa is easily disseminated without being detected, because it can be latently present in asymptomatic shrubs and trees [8]. Once established in a new area, it can then spread quickly over long distances by insect transmission [9]. Moreover, the pathogen takes several days to up to 2 weeks to grow in culture, making culture-dependent identification impractical [5]. When symptoms do develop, they range from reduced yield and fruit size, wilting and dieback to decline and death. Unfortunately, there is no treatment for diseases caused by X. fastidiosa, and the main recourse for outbreak management is to reduce the insect vector population, destroy infected plants to prevent further spread of the pathogen, and/or replace infected plants with relatively more tolerant cultivars [10, 11].
Methods of detection for X. fastidiosa include microscopy, immunological assays and molecular methods [12], none of which require culturing. Also, identification of X. fastidiosa to the subspecies level by quantitative PCR (qPCR) and to the strain level using multilocus sequence typing (MLST) can be performed using DNA directly extracted from plants [13, 14]. However, to reconstruct transmission chains, whole-genome sequencing is necessary to identify a sufficient number of SNPs for phylogenetic analysis. This has only been possible from pure cultures [5]. Currently, more than 80 assembled genomes representing all X. fastidiosa subspecies are available in the National Center for Biotechnology Information (NCBI) Assembly database with genome sizes ranging from 2.4 to 2.7 Mb, a G+C content of 51–52 mol%, number of contigs ranging from 1 to over 400, ambiguous bases from 0 to over 300, and completeness generally over 99 %. There are approximately another 50 unassembled genomes in the NCBI Sequence Read Archive (SRA) database. Because of the slow growth of X. fastidiosa in culture, a culture-independent method would be desirable to more quickly investigate outbreaks. This is true not only for outbreaks caused by X. fastidiosa , but also for any other plant pathogen, in particular, for those that cannot be grown in pure culture at all, such as phytoplasmas [15].
Recently, culture-independent metagenomic sequencing (sequencing all the micro-organisms present in a sample simultaneously) has been explored for fast detection and identification of viral, fungal and bacterial plant pathogens [16], including X. fastidiosa [17, 18]. The Oxford Nanopore Technologies (ONT) MinION sequencer has attracted particular attention, because it requires minimal laboratory infrastructure for library preparation and sequencing, is of relatively low cost, and provides results almost in real-time [19]. Moreover, the long reads generated by the ONT MinION facilitate assembly of single-contig metagenome-assembled genomes (MAGs) as was shown, for example, for human stool samples [20]. We recently demonstrated the feasibility of metagenomic sequencing with the MinION for detection and identification of two tomato pathogens, Pseudomonas syringae pv. tomato (Pto) and Xanthomonas perforans [21]. Also, in this case, the long reads generated by MinION made it possible to obtain MAGs, which allowed identification to near strain-level resolution. However, it was not possible to reach sufficient resolution to identify transmission chains. This was probably due to the error rate of nanopore sequencing at the time, which is in a large part due to the challenge in basecalling, the process of translating the changes in electrical signal measured at each individual nanopore to a sequence of nucleotides [22]. This is in line with published MAGs of human pathogens obtained by nanopore sequencing, which have been used, for example, to characterize antibiotic resistance [23], but nanopore sequencing has only very recently been shown to provide SNP resolution suitable for phylogenetic reconstruction for outbreak investigations [24]. With respect to X. fastidiosa , identification has been limited to the subspecies level when employing the MinION for metagenomic sequencing [17].
Because of its high impact and the difficulty of growing it in culture, here, we used X. fastidiosa for a proof of concept study. We show how metagenomic sequencing using MinION in combination with improved basecalling, the genome-based identification platform LINbase [25] and phylogenetic reconstruction can go beyond sensitive and specific detection and allow phylogenetic reconstruction at SNP resolution. We describe how this approach could be integrated into a worldwide platform for easy and quick sharing of genomic and metagenomic sequencing results for early plant disease outbreak detection for fast and effective intervention to help prevent plant disease epidemics and pandemics.
Methods
Petiole collection and DNA extraction
Five symptomatic grapevine (V. vinifera) samples and one healthy negative control sample (see Table 1 for metadata) were collected in late summer or early fall (August or September). Each symptomatic sample consisted of at least six leaves (including petioles) with marginal leaf necrosis harvested from different portions of the canopy of a single vine. The negative control sample (sample 11) was collected from a geographical area where Pierce’s disease had not been reported (Winchester, VA, USA). Samples were placed in a plastic bag, sealed and transported to the laboratory in a cooler containing ice, then shipped overnight to Virginia Tech’s Plant Disease Clinic (Blacksburg, VA, USA). Six petioles were randomly selected, bundled and cut into pieces (~4 mm length), and 100 mg was transferred to a 2 ml screw-cap tube, which contained garnet matrix A (~180 mg) and a 6.35 mm ceramic sphere (MP BioMedicals). Tubes were frozen in liquid nitrogen and homogenized at a speed of 5 m s−1 for 60 s in a FastPrep-24 (MP BioMedicals). The Isolate II plant DNA kit (Bioline USA) was used to extract DNA according to the manufacturer's instructions, using the supplied CTAB lysis buffer. DNA was stored at −80 °C until use.
Table 1.
Sample metadata, qPCR results and summary of sequencing results
|
Characteristic |
11 |
368 |
669 |
228 |
255 |
382 |
|---|---|---|---|---|---|---|
|
Symptoms |
No |
Yes |
Yes |
Yes |
Yes |
Yes |
|
Grape cultivar |
Chardonnay |
Petit Verdot |
Cabernet Franc |
Viognier |
Vidal Blanc |
Cabernet Franc |
|
Location of collection |
Winchester, VA |
Middleburg, VA |
Smithfield, VA |
Charlottesville, VA |
Fairlawn, VA |
Faber, VA |
|
Year of collection |
2020 |
2013 |
2020 |
2013 |
2013 |
2013 |
|
DNA concentration (ng μl−1) |
13.9 |
22.7 |
24.7 |
22.8 |
23.9 |
28.7 |
|
DNA quality (260/280) |
1.56 |
1.74 |
1.74 |
1.72 |
1.69 |
1.73 |
|
qPCR C t [grape (RS1)] |
20.68 |
17.3 |
19.7 |
18.86 |
19.06 |
16.99 |
|
qPCR C t [Xf (rimM)] |
Undetermined |
17.94 |
20.4 |
34.03 |
35.87 |
18.73 |
|
Samples run per flow cell |
2 |
2 |
1* |
3 |
3 |
3 |
|
Total number of reads |
489 136 |
55 695 |
1 271 428 |
619 578 |
563 451 |
232 722 |
|
Total number of reads (ha1) |
518 818 |
59 693 |
1 305 206 |
735 768 |
649 927 |
271 524 |
|
Xf reads (wimp) |
0 |
640 |
26 643 |
17 |
8 |
2245 |
|
Xf percentage (wimp) |
0 |
1.208 |
2.327 |
0.003 |
0.002 |
1.024 |
|
Xf reads (wimp ha1) |
0 |
801 |
29 878 |
11 |
13 |
2906 |
|
Xf percentage (wimp ha1) |
0 |
1.342 |
2.289 |
0.001 |
0.002 |
1.070 |
|
Total length of reads (Mb) |
399.41 |
96.29 |
3962.02 |
1129.76 |
1247.70 |
520.69 |
|
Mean read length (bp) |
840 |
1728 |
3116 |
1823 |
2214 |
2237 |
|
Longest read length (bp) |
30 160 |
43 841 |
264 283 |
364 246 |
502 142 |
160 166 |
Xf, X. fastidiosa.
*After high-accuracy basecalling.
qPCR
X. fastidiosa was detected using a multiplex qPCR assay that targeted an endogenous grape gene, resveratrol synthase I mRNA (RS), and the X. fastidiosa 16S rRNA processing protein rimM gene. The use of an endogenous plant gene in a qPCR detection assay serves as an internal control in the amplification reaction, preventing false negatives that may occur due to operator error or inhibitors present in DNA extractions. The RS primers and TaqMan probe [26] and the rimM primers and TaqMan probe [27] were constructed with the JOE and FAM reporters, respectively, with Black Hole Quencher-1 (Integrated DNA Technologies) for use with the StepOnePlus (Life Technologies) real-time PCR system. Final reaction components consisted of 1× SensiFAST Probe Hi-ROX reaction (Bioline), 2 µl template DNA, 0.9 µM for each primer, 0.25 µM for each probe, and nuclease-free water to achieve a final volume of 20 µl. Final cycling conditions for the multiplex qPCR were: 5 min at 95 °C, followed by 40 cycles of 10 s at 95 °C and 30 s at 60 °C. Cycle threshold (C t) values were applied automatically by the StepOne software v. 2.1 in all qPCR assays. Sensitivity results are reported in Fig. S1 (available with the online version of this article).
Nanopore sequencing
Sequencing library preparation and sequencing
DNA quantity and quality were checked using the NanoDrop ONE (Thermo Scientific). Sequencing libraries were prepared following ONT protocols. First, DNA fragments were repaired using FFPE repair mix (New England Biolabs), the repaired samples were barcoded using the Native barcoding kit (ONT), the barcoded samples were mixed and ligated with the adapter, and the sequencing library was prepared using the ligation sequencing kit (ONT). The products from each of the steps were monitored by using NanoDrop ONE. The library was loaded on the MinION flow cell by using the flow cell priming kit (ONT) and was run using the MinKNOW software (version 4.2.8) for 72 h. Sample 669 was run on a single flow cell to maximize the number of reads. Samples 368 and 11 were run on a flow cell together with samples from other projects. Samples 228, 255 and 382 were run together on one flow cell in order to determine whether enough reads could be obtained when combining two to three samples on the same flow cell.
Basecalling
FAST5 files were used as input to the ONT Guppy CPU (version 5.0.7) basecalling tool with the fast-basecalling setting in MinKNOW software (version 21.02.1). Samples were re-basecalled before attempting assembly using the high-accuracy basecalling setting in ONT Guppy GPU (version 4.5.2) on the Virginia Tech high-performance computing cluster (ARC; Advanced Research Computing). All samples were basecalled with a minimum quality score of 7 and stored in fastq format files. All fastq files were deposited in the NCBI SRA with accession numbers SAMN21438496, SAMN21438497, SAMN21438498, SAMN21438499, SAMN21438500 and SAMN21438501.
Metagenomic analysis of unassembled reads
Basecalled fastq files were used as input to the cloud computing-based wimp (What’s In My Pot?) (v 2021.03.05) analysis tool (ONT) [28], through the platform epi2me for species-level identification of Xylella in all the samples. wimp uses Centrifuge [29] and NCBI’s RefSeq database to assign taxonomy to long reads rapidly. For the rarefaction analysis, reads identified as X. fastidiosa by wimp were randomly subsampled using the command fasta-subsample --seed<random number><fasta file><number of subsampled reads>from the MEME Suite (version 5.3.3) [30].
In parallel, fastq files were used as input to sourmash (version 3.5.0) [31]. Sourmash is a command-line tool, with multiple utilities, that uses hash sketches to rapidly compute sequence similarity between a query and a database of reference genomes. One of the utilities is sourmash gather, which uses a greedy algorithm to partition metagenomes into individual genomes to calculate the containment and average abundance values. Publicly available reads of 54 X. fastidiosa genomes in the NCBI SRA database were assembled in-house using the de novo assembler SPAdes [32]. These assemblies together with 80 NCBI Xylella genome assemblies were used to build a custom reference database after checking their quality with the checkM tool [33]. First, sketches of the custom reference database were created with the sourmash command sourmash compute --scaled 1000 k 31<fastq file>. Sketches of each nanopore fastq read file were created with the sourmash commands sourmash compute --scaled 1000 k 31<fastq file> --track-abundance to obtain the average abundance values. Then, sourmash gather was used with the following command: sourmash gather -k 31 and --threshold-bp 15000<query.sig><reference_db.sig> -o<output.csv>to obtain the values p_match, p_query and avg_abundance. The utility sourmash gather gives two values in relation to containment, p_match and p_query with p_match being the fraction and p_query being the fraction .
Metagenome assembly
After high-accuracy basecalling, reads were used as input to the de novo assembler Flye (version 2.8.3) [34] with the following command: flye --nano-raw<fastq file> --out-dir<directory name> --genome-size 2.7 m --meta -i 5. Other polishing parameters were explored, but there were no improvements in the assemblies beyond five iterations.
Metagenomic analysis of assembled reads
The longest scaffold (consisting of two contigs) from the high-accuracy basecalled 669 assembly (named MAG 669) was used as input to the webserver LINbase [25] to identify the closest related strain and the corresponding LIN group with the function ‘Identify using a genome sequence.’ MAG 669 was aligned with the X. fastidiosa subsp. fastidiosa Temecula 1 genome using the ANIm function in JSpeciesWS [35] using default parameters. A total of 67 publicly available X. fastidiosa subsp. fastidiosa genomes along with MAG 669 were used for the reconstruction of the phylogenetic trees. All 68 genome sequences were annotated using the prokka gene annotation tool (version 1.14.6) [36] with default settings. The annotated files were used as input for Roary (version 3.13.0) [37] to obtain a core-gene alignment. The following parameters were used: -e to obtain a multiFASTA core-gene alignment file as output, -n for fast core-gene alignment with the option MAFFT and -p for multithreading. A total of 1138 genes were used to construct the core genome. The core-gene alignment file was used as input for iq-tree (version 2.0.3) [38] using automated model-selection to obtain the final maximum-likelihood-based phylogenetic tree. The final phylogenetic tree was visualized using the ggtree package in R [39]. The same set of 68 genomes were used for Split-K-mer analysis (ska; version 1.0) [40]. All sequences were first converted into split k-mer sketches using the ska fasta subcommand with default k-mer length of 15 (sequences of 15 bp flanking a middle base i.e. split-base, on both ends). The resulting split k-mer sketches were merged into a single file using the ska merge subcommand. A reference-free alignment was performed on the merged file using the ska align subcommand. As for the core-genome tree, the alignment file was used as input for iq-tree to obtain the final maximum-likelihood-based phylogenetic tree, which was visualized using the ggtree package in R [39]. SNPs between selected genomes were counted with Snippy [41].
Results
Analysis flowchart
To determine detection sensitivity and specificity of metagenomic sequencing with the ONT MinION system and test its ability to provide sequencing data sufficient for phylogenetic reconstruction, we developed the workflow depicted in Fig. 1. In short, five symptomatic grapevine samples and one healthy control sample (from a geographical area with no reported Pierce’s disease) were collected in Virginia, USA (Table 1). DNA was extracted from grapevine petioles, which are known to carry a high concentration of X. fastidiosa DNA. DNA was analysed for quantity and quality, and the presence of X. fastidiosa was determined by qPCR (Table 1). One symptomatic sample (with ID 669) was sequenced on an individual MinION flow cell to obtain as many reads as possible, while the other symptomatic samples were combined in groups of two or three to determine whether enough reads could be obtained for detection while saving on flow cells. The negative control sample (sample 11) was sequenced on the same flow cell as an unrelated healthy plant sample from another project to avoid any possible cross-contamination from infected samples. After sequencing and basecalling, the ONT tool wimp [28] and the third-party tool sourmash [31, 42] were used to determine the presence and abundance of sequences belonging to X. fastidiosa and its subspecies. Results were compared with respect to sensitivity of detection with those obtained by qPCR. Finally, reads were assembled with the aim of obtaining MAGs to perform phylogenetic reconstruction for outbreak investigation.
Fig. 1.

Workflow for culture-independent detection and identification of X. fastidiosa . (a) DNA is extracted from grapevine petioles and used for DNA sequencing library preparation and, as a control, for qPCR. (b) The prepared DNA is then sequenced with the ONT MinION system and basecalled. Basecalled reads are either used directly for detection and identification of X. fastidiosa at the species and subspecies rank, or assembled for phylogenetic reconstruction for outbreak investigation. Created with BioRender.com.
Detection sensitivity and specificity
The DNA concentration of the samples was consistent, varying only slightly between 13.9 ng μl−1 for the healthy sample and 28.7 ng μl−1 for sample 382, which is in the range of the minimum concentration of 25 ng μl−1 recommended by ONT (Table 1). Also, nucleic acid quality varied little with the 260/280 ratio (nucleic acid/protein) ranging from 1.56 to 1.74, which is slightly lower than the 1.8–2.0 recommended by ONT. Values for C t (the PCR cycle after which a PCR product becomes detectable) obtained from the qPCR tests ranged from 17 to 21 for the grape gene resveratrol synthase 1 (RS1) used as an internal control, showing that the extracted DNA for all samples was amplifiable. C t values for the X. fastidiosa gene rimM, encoding a 16S rRNA processing protein, ranged from 18 to 36 (except for sample 11, the negative control, for which no rimM product was detected), revealing a wide range of pathogen abundance in the samples with sample 669 containing the highest pathogen abundance (C t of 17.94) and sample 255 containing the lowest pathogen abundance (C t of 35.87, at the limit of detection by qPCR).
The total number of reads obtained by MinION sequencing varied greatly between samples, ranging from 55 000 (sample 368) to almost 1.3 million (sample 669). Since the difference in the number of samples run per flow cell ranged only between one and three, and DNA concentration and quality were similar between samples, this was probably due to inconsistencies in sequencing library preparation (i.e. users making small changes inadvertently) and flow cell quality. Mean read length was more consistent between samples, ranging from 840 bp for the negative control, i.e. sample 11, to 3116 bp for sample 669. The longest overall read was obtained for sample 255, with a length of over 500 000 bp. Because of the differences in read number and length, the total length of the obtained metagenomes also varied greatly, from 96 Mb for sample 368 to almost 4.2 Gb) for sample 669.
To answer the most relevant question of how many of the obtained sequences were of X. fastidiosa , we first used the tool wimp [28], which is provided by ONT and uses NCBI RefSeq as a reference database. The number of reads assigned to X. fastidiosa ranged from 8 in sample 255 (0.002 % of total reads) to 26 643 in sample 669 (2.327 % of total reads) as shown in Table 1 (see Table S1 for full results). Therefore, metagenomic sequencing detected X. fastidiosa even in the sample with the lowest pathogen abundance. However, since samples 228, 255 and 382 were run on the same flow cell, we cannot exclude the possibility that the small number of X. fastidiosa reads in samples 228 and 255 were due to cross-contamination from sample 382. Not a single read was misidentified as X. fastidiosa among the 489 000 total reads in the negative control sample 11, revealing a high specificity of metagenomic sequencing using MinION. However, if a pathogen that is closely related to X. fastidiosa were present in the same sample, such as Xanthomonas citri pv. viticola, it is possible that some reads of this pathogen could be misidentified as X. fastidiosa .
Since the detection limit of metagenomic sequencing depends on the number of reads that are obtained, we wanted to determine the minimum number of reads that are necessary for consistent detection of X. fastidiosa . Thus, we performed a rarefaction analysis on samples 669, 382 and 368 by randomly subsampling reads ten times at different total read numbers and considered the number of reads sufficient for detection when all 10 subsamples contained at least five reads identified by wimp as X. fastidiosa (Fig. 2, Table S2). The minimum read number ranged from 500 for sample 669 to approximately 1500 for samples 368 and 382. Since samples 228 and 255 only contained 17 and 8 X . fastidiosa reads, respectively, out of approximately 500 000 total reads (Table 1), no rarefaction analysis was necessary to determine that at least 500 000 reads were necessary to detect X. fastidiosa in these samples. Therefore, the number of reads necessary to detect X. fastidiosa ranges from approximately 500 reads for heavily infected samples to approximately 500 000 reads for very mildly infected samples.
Fig. 2.

Detection sensitivity of metagenomic sequencing with the ONT MinION based on wimp [28] analysis for samples 368, 382 and 669. The x-axis shows the total number of reads that were randomly subsampled. The y-axis reports the corresponding mean number of X. fastidiosa (Xf) reads based on ten subsamples taken for each subsample size. The error bars show the standard deviation.
Resolution of identification using unassembled reads
So far, we have looked at simply detecting the presence of species X. fastidiosa . However, identification to subspecies level is important for making decisions about disease intervention to control an outbreak. Fig. 3(a) shows the results obtained by wimp for reads assigned at the subspecies rank as a percentage out of all reads identified as X. fastidiosa . Unfortunately, wimp only assigned a small number of reads to subspecies, while most reads were assigned only at the species rank to X. fastidiosa overall. Moreover, some reads in all samples were assigned to ' X. fastidiosa subspecies morus', a mulberry pathogen, which is not known to include grape as a host. Therefore, we decided to use a second taxonomic classification tool, sourmash [31], in combination with a custom reference database consisting of 134 publicly available X. fastidiosa genomes, which belong to all published subspecies (Table S2). sourmash does not analyse reads individually, but the sourmash command gather identifies the overlap between a k-mer sketch computed for the combined metagenomic sequences in a sample and k-mer sketches computed for each genome in the reference library (see Methods for details). Based on the average abundance of the set of overlapping k-mers in the metagenome, sourmash infers how many copies of each genome are present in the metagenome. Fig. 3(b) shows the results by subspecies as a percentage of subspecies genome copies out of all X. fastidiosa genome copies. For sample 669, the majority of X. fastidiosa sequences were assigned to subspecies fastidiosa (71.7%), strongly suggesting that this subspecies was the causative agent, as expected for a grapevine with symptoms of Pierce’s disease. For sample 382, sourmash only identified X. fastidiosa subspecies fastidiosa . For sample 368, instead, sourmash only identified X. fastidiosa subspecies multiplex . While this subspecies is not known to cause Pierce’s disease, we have determined its presence by MLST on symptomatic grapevines in Virginia in the past (data available on request). The very small number of X. fastidiosa reads in samples 228 and 255 made subspecies assignment in these samples impossible using sourmash.
Fig. 3.

Identification of X. fastidiosa at the subspecies rank using (a) wimp [28] and (b) sourmash [31, 42]. The x-axes represents the sample IDs (described in Table 1). The y-axes shows the relative abundance of subspecies as a percentage of all sequences assigned to X. fastidiosa at the species rank. Note that sample 11 was the negative control with no X. fastidiosa reads identified by either wimp or sourmash, and sourmash did not identify X. fastidiosa in samples 228 and 255. Xf, X. fastidiosa ; Xff, X. fastidiosa subsp. fastidiosa ; Xfm, X. fastidiosa subsp. multiplex ; Xfp, 'X. fastidiosa. subsp. pauca'; Xfs, 'X. fastidiosa subsp. sandyi'.
Since sourmash gather provides abundance values for each individual genome in the reference database (Table S4), it also allowed us to infer which reference genome had the highest similarity to the genomes present in the metagenomes. For sample 669, this was genome 16M3, suggesting that the X. fastidiosa subsp. fastidiosa strain that caused Pierce’s disease in sample 669 (isolated in Virginia in 2020) is closely related to 16M3 (isolated in Georgia in 2016). However, similarity alone is not enough to make any conclusions about phylogenetic relationship and epidemiological linkage between strains; in other words, this result was not sufficient to conclude that the X. fastidiosa subsp. fastidiosa strain present in sample 669 was derived from a strain introduced into Virginia from Georgia.
Assembly of a complete X. fastidiosa genome from metagenomic reads
Since using the unassembled reads did not allow resolution of phylogenetic relationships between the X. fastidiosa sequences in our samples and public X. fastidiosa genomes, we performed assembly with the goal of obtaining MAGs that could then be precisely identified. We had originally basecalled our samples with the fast, but low accuracy, basecalling option of ONT’s Guppy basecalling tool [43]. Using Guppy’s high-accuracy setting instead slightly increased the number of reads identified as X. fastidiosa (Table 1). We then assembled the high accuracy reads de novo using the tool Flye [44]. For samples 368 and 382, the longest contig length ranged from 90 670 to 493 966, revealing that we did not have enough reads for assembly of complete MAGs. However, for sample 669, the longest contig had a length of 2 523 846 bp, approximately as long as the best publicly available X. fastidiosa subsp. fastidiosa genome, Temecula1, with a length of 2 521 148 bp. To confirm that this contig presented a complete X. fastidiosa MAG, we compared it with the Temecula 1 genome using the ANIm function in JSpeciesWS [35] and found the two genomes to have an average nucleotide identity (ANI) of 99.78 % and to align over 99.03 % of their length (Fig. 4a). We then determined the assembly quality of the 669 MAG compared to the Temecula 1 reference genome (Table 2). It can be seen that the 669 MAG is of only slightly lower quality with a completeness percentage of 96.61 % and 100 ambiguous bases (for a comprehensive comparison of the 669 MAG quality with X. fastidiosa genome assemblies obtained from pure cultures, see Table S3).
Fig. 4.

Comparison of the MAG 669 sequence with other X. fastidiosa subsp. fastidiosa genomes. (a) Alignment of the 669 MAG with the genome of X. fastidiosa subsp. fastidiosa Temecula 1 using ANIm implemented in JSpeciesWS [35] (99.78 % ANI, 99.03 % nucleotides aligned) and visualized using Ribbon [50]; (b) Result page of LINbase [25] when using MAG 699 as the query with the ‘identify using a genome sequence’ function. The most similar genome to the query (the ‘best match’) is reported together with its LIN. The ANI value between the query and the best match is shown above the LIN. Below the best match, the taxa to which the query belongs are listed as well.
Table 2.
Comparison of the sequence quality of MAG 669 with the X. fastidiosa subsp. fastidiosa reference genome
Temecula 1 and the genome of the closely related X. fastidiosa subsp. fastidiosa isolate XF51 CCPM1 were both obtained from pure cultures.
|
Characteristic |
MAG 669 |
Temecula 1 |
XF51 CCPM1 |
|---|---|---|---|
|
Length (Mb) |
2.52 |
2.52 |
2.8 |
|
Contigs |
2 |
2 |
115 |
|
Scaffolds |
1 |
2 |
115 |
|
N50 (contigs) (bp) |
2 522 580 |
2 519 802 |
68 803 |
|
N50 (scaffolds) (bp) |
2 523 846 |
2 519 802 |
68 803 |
|
Completeness |
96.61 |
99.64 |
97.76 |
|
Contamination |
0.12 |
0 |
6.51 |
|
Ambiguous bases |
100 |
0 |
0 |
With an assembled genome, we were able to take advantage of the genome-based classification platform LINbase [25], which assigns genomes to genera, species and within-species classes based on ANI. LINbase assigned the 669 MAG to X. fastidiosa subsp. fastidiosa, because its ANI to the most similar X. fastidiosa subsp. fastidiosa genome in LINbase, genome X. fastidiosa subsp. fastidiosa XF51 CCPM1 (see Table 2 for genome characteristics and quality), was over 99.899%, which is significantly higher than the ANI that distinguishes X. fastidiosa subsp. fastidiosa genomes from each other (98.5%) (Fig. 4b). X. fastidiosa subsp. fastidiosa XF51 CCPM1 was isolated in Georgia (USA), as was strain 16M3, which had been identified as the genome with the highest relative abundance by sourmash. This increased our suspicion that the Virginia strain was epidemiologically linked to Georgia, but was still insufficient evidence to conclude so.
Using a MAG for phylogenetic reconstruction at SNP resolution
Finally, the high quality 669 MAG made it possible to perform a phylogenetic analysis together with all other publicly available X. fastidiosa subsp. fastidiosa genomes, including genomes of three Georgia isolates, to further investigate the possibility that the X. fastidiosa subsp. fastidiosa strain in sample 669 was linked to Georgia. To do so, we reconstructed a core-genome tree based on the alignment of 1138 genes present one time in all included genomes, using the tool Roary [37], and an alignment-free whole-genome SNP tree based on a split k-mer analysis using the tool ska [40] (Fig. 5a, b). In both phylogenetic trees, MAG 669 grouped in a clade with the three Georgia strains with 100 % bootstrap support, revealing that MAG 669 shares a most recent common ancestor with strains from the south-eastern USA, but not strains from California. Also, the relatively small number of SNPs that distinguish MAG 669 from the Georgia strains XF51 and 16M3 (116 and 148, respectively) and the much higher number of SNPs that distinguish MAG 669 from the Californian Temecula 1 strain (796 SNPs) is in line with this result (Tables S4 and S5). Since the Georgia strains were isolated between 2009 and 2016, and sample 669 was collected in 2020, we hypothesize that they are epidemiologically linked and that the ancestor of X. fastidiosa subsp. fastidiosa in sample 669 may have been introduced to Virginia from the south-eastern USA by interstate movement of plant material or northward movement of infected X. fastidiosa subsp. fastidiosa sharpshooter insect vectors.
Fig. 5.
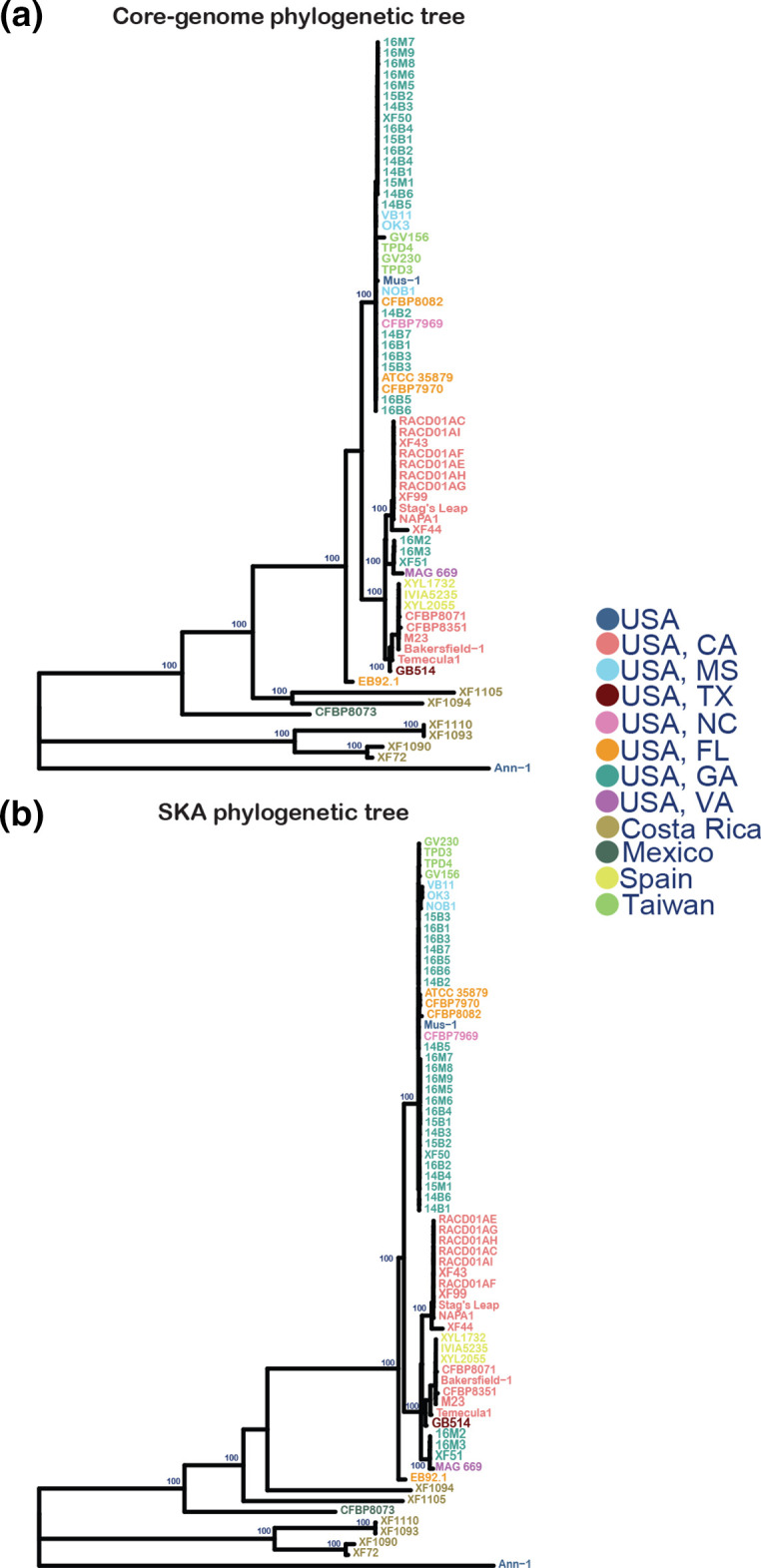
Phylogenetic trees of MAG 669, all high-quality publicly available genomes of X. fastidiosa subsp. fastidiosa isolates from the USA, a selection of international X. fastidiosa subsp. fastidiosa isolates and ' X. fastidiosa subsp. sandyi' Ann 1 as an outgroup. (a) Core-genome maximum-likelihood tree using 1138 core genes; (b) whole-genome maximum-likelihood SNP tree based on split k-mer analysis. Genomes are coloured according to isolation location. Percentage bootstrap support values >96 % are shown.
Discussion
In this work, we used the causative agent of Pierce’s disease of grapevine, X. fastidiosa subsp. fastidiosa , for a proof of concept study to investigate the potential of metagenomic sequencing with the ONT MinION system for plant pathogen detection and phylogenetic reconstruction for outbreak investigation. We found that this approach is comparable to the detection sensitivity of qPCR and can be used in place of whole-genome sequencing of cultured isolates in whole-genome phylogenetic tree reconstruction. This approach is similar to recent reports using metagenomics for the investigation of food-borne outbreaks caused by Salmonella using Illumina sequencing [37] or nanopore sequencing [38].
Metagenomic sequencing of plant samples with the MinION sequencer or Illumina sequencing platforms had been used previously to detect viral, fungal and bacterial plant pathogens [16], including X. fastidiosa [17, 45, 46]. However, when the MinION was used previously to detect X. fastidiosa , the obtained sequencing reads were simply aligned to reference genomes to find the genome to which the highest number of reads mapped without further phylogenetic analysis [17]; identification was limited to subspecies, or sequencing was performed only after PCR of MLST loci making whole-genome-based phylogenetic analysis impossible [45]. More recently, the Illumina iSeq and MiSeq were used for detection and identification of X. fastidiosa as well [18]. Both Illumina platforms detected X. fastidiosa with a similar sensitivity as qPCR and identified sequences with subspecies resolution, even in mixed infections. However, phylogenetic reconstruction was not possible. Even the MLST loci, usually used for X. fastidiosa strain typing [47], were only partially represented in the obtained metagenomic sequences, and the short reads limited the possibility of assembling MAGs.
Similar to the Illumina results [40], we observed that metagenomic sequencing with the MinION was as sensitive as qPCR in samples for which we obtained approximately 500 000 reads. Therefore, when using an entire flow cell, with which more than one million reads can be obtained, the detection sensitivity may be even higher than qPCR. Importantly, not a single read was misidentified as X. fastidiosa in our negative control sample, demonstrating the high specificity of long-read metagenomic sequencing (although we should note that based on our results there is a very low abundance of other bacteria in the grapevine petiole, reducing the risk of misidentifying other bacteria as X. fastidiosa ). Therefore, metagenomic sequencing with the MinION is an excellent approach to determine whether X. fastidiosa is present in a plant sample when it is suspected as the causative agent of an emerging disease outbreak on a new crop in an area where it was not previously present. However, because of the shorter protocols and lower cost of qPCR [estimated at $4 (£3.25) per qPCR reaction; £1=$1.23] compared to the cost of metagenomic sequencing [almost $1000 (£812.50) when using one sample per flow cell], qPCR will remain the detection method of choice when a high volume of samples needs to be processed in an area where X. fastidiosa is already known to be endemic. Sequencing multiple samples on one MinION flow cell may be an option in some situations as well, balancing cost with read number. However, the highly variable number of reads we obtained per flow cell makes it difficult to decide the number of samples to combine in a single run.
One challenge when using unassembled metagenomic reads for identification is that it is difficult to conclude which X. fastidiosa subspecies is present. Here, as well as in other studies [46], metagenomic reads mapped to more than one subspecies for all samples when using wimp and for sample 669 when using sourmash. This may be due to the high rate of recombination in X. fastidiosa , which can lead to horizontal gene transfer between strains of different subspecies [48]. Therefore, reads of some genes in a metagenome may align to X. fastidiosa genomes of subspecies that are different from the subspecies present in the sequenced sample. Obtaining enough reads of sufficient length to assemble a MAG, as we did in this study for sample 669 using an entire flow cell, thus, is important for precise identification. In fact, once the 669 MAG was assembled, its identification as X. fastidiosa subsp. fastidiosa was highly reliable using the LINbase platform, since its ANI to the most similar X. fastidiosa subsp. fastidiosa genome (99.89%) was much higher than the minimal pair-wise ANI found between X. fastidiosa subsp. fastidiosa genomes (98.5%).
Besides identifying the 669 MAG at the subspecies rank, LINbase also grouped the 669 MAG with genome X. fastidiosa subsp. fastidiosa XF51 CCPM1 from a Georgia isolate at an ANI threshold of 99.899 %. If additional high-quality genomes of Georgia X. fastidiosa subsp. fastidiosa isolates (or isolates from other states in the south-eastern USA) had been included in LINbase, it would have been possible to assign the 669 MAG to an ANI cluster (which is called a ‘LINgroup’ in LINbase) [25], corresponding to isolates from the south-eastern USA. This would have strongly suggested that the Virginia isolate was epidemiologically linked to the Georgia isolates even without phylogenetic analysis. However, the other public genome sequences of Georgia isolates were of insufficient quality to be added to LINbase (they were fragmented into more than 500 contigs, making accurate ANI computations impossible). Since genome quality is of a lower concern for phylogenetic reconstruction (sequencing errors increase the ANI of a MAG to other bacterial genomes but because sequencing errors are not shared with other genomes they do not change the phylogenetic position in a tree), it was possible to include these genomes in the alignment-based core-genome tree and in the split k-mer-based SNP tree. Both trees allowed us to assign the 669 MAG with high confidence (100 % bootstrap support) to the same clade as the three Georgia isolates, suggesting that X. fastidiosa subsp. fastidiosa was not introduced to Virginia on grapevine nursery stock purchased from California but from a sharpshooter insect vector moving north from the south-eastern USA or via transport of X. fastidiosa subsp. fastidiosa infected plant material from the south-eastern USA to Virginia. While this finding needs to be confirmed with additional X. fastidiosa subsp. fastidiosa samples, such information is important for disease control and prevention, since it informs growers of the high risk of future introductions of X. fastidiosa subsp. fastidiosa to Virginia from southern locations. More broadly, this result indicates that metagenomic sequencing with the MinION can be used to reconstruct pathogen transmission chains with accuracy similar to using genome sequences of cultured isolates, as has been the state of the art [5].
To perform such outbreak investigations as part of routine diagnostics by plant disease clinics and plant protection agencies, two steps are required: (i) bioinformatic analysis of metagenomic sequencing needs to become accessible to non-bioinformaticians; and (ii) all plant pathogen MAGs obtained by metagenomic sequencing need to be uploaded to a worldwide database of plant pathogen genomes and MAGs. If these objectives were reached, each time that a disease outbreak with symptoms indicative of a possibly emerging disease occurred, metagenomic sequencing could be performed and MAGs could be assembled and uploaded to the worldwide database where they could be immediately compared with all other genomes and MAGs in the database. If two or more MAGs were highly similar to each other, an alert could be triggered and it might be early enough to stop the outbreak from turning into an epidemic or a pandemic. To make this vision a reality in epidemic and pandemic plant disease prevention, we are currently developing user-friendly pipelines for metagenomic sequence analysis by non-bioinformaticians. Additionally, we are improving and expanding the LINbase web server [25] to transform it into the genomeRxiv platform, which will make sharing sequencing results as easy as submitting a manuscript to a preprint server such as bioRxiv [49].
In conclusion, here, we have demonstrated that culture-independent metagenomic sequencing permits high-quality genome assembly of bacterial plant pathogens suitable for highly resolved phylogeny-based outbreak investigation. If combined with user-friendly sequence analysis tools and a platform for quick and easy sharing of sequence information, metagenomics could be adopted by plant disease clinics in routine diagnostics. This could lead to early detection of emerging plant disease outbreaks and provide the basis for effective interventions to prevent outbreaks from turning into epidemics and pandemics.
Supplementary Data
Funding information
This study was supported by the US Department of Agriculture Animal and Plant Health Inspection Service (contract AP20PPQS and T00C055). Funding to B.A.V., S.L. and M.N. was also provided in part by the Virginia Agricultural Experiment Station and the Hatch Program of the National Institute of Food and Agriculture, US Department of Agriculture.
Acknowledgements
We would like to thank Ms Kathryn Liu for help with DNA extraction.
Author contributions
Conceptualization: B.A.V., S.L., L.S.H. Methodology: B.A.V., S.L., L.S.H., E.B., S.Y. Software: M.A.J., P.S., R.M. Validation: M.A.J., H.L., E.B., S.Y. Formal analysis: M.A.J., H.L., P.S., R.M., E.B. Investigation: M.A.J., H.L., E.B., P.S., R.M. Resources: E.B., M.N. Data curation: M.A.J., P.S., R.M., E.B. Writing – original draft preparation: M.A.J., P.S., B.A.V. Writing – review and editing: E.B., L.S.H., S.L. Visualization: M.A.J., P.S. Supervision: B.A.V., S.L. Project administration: B.A.V., S.L. Funding: B.A.V., S.L., M.N., E.B.
Conflicts of interest
Life Identification Number and LIN are registered trademarks of This Genomic Life, Inc. Lenwood S. Heath and Boris A. Vinatzer report in accordance with Virginia Tech policies and procedures, and their ethical obligation as researchers, that they have a financial interest in the company This Genomic Life, Inc., that may be affected by the research reported in this paper. They have disclosed those interests fully to Virginia Tech, and they have in place an approved plan for managing any potential conflicts arising from this relationship.
Footnotes
Abbreviations: ANI, average nucleotide identity; MAG, metagenome-assembled genome; MLST, multilocus sequence typing; NCBI, National Center for Biotechnology Information; ONT, Oxford Nanopore Technologies; qPCR, quantitative PCR.
All supporting data, code and protocols have been provided within the article or through supplementary data files. One supplementary figure and five supplementary tables are available with the online version of this article.
References
- 1.Ristaino JB, Anderson PK, Bebber DP, Brauman KA, Cunniffe NJ, et al. The persistent threat of emerging plant disease pandemics to global food security. Proc Natl Acad Sci USA. 2021;118:e2022239118. doi: 10.1073/pnas.2022239118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schneider K, Mourits M, van der Werf W, Lansink AO. On consumer impact from Xylella fastidiosa subspecies pauca . Ecol Econ. 2021;185:107024. doi: 10.1016/j.ecolecon.2021.107024. [DOI] [Google Scholar]
- 3.European Food Safety Authority (EFSA) Update of the Xylella spp. host plant database. EFSA J. 2018;16:e05408. doi: 10.2903/j.efsa.2018.5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nunney L, Yuan X, Bromley R, Hartung J, Montero-Astúa M, et al. Population genomic analysis of a bacterial plant pathogen: novel insight into the origin of Pierce’s disease of grapevine in the U.S. PLoS One. 2010;5:e15488. doi: 10.1371/journal.pone.0015488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castillo AI, Bojanini I, Chen H, Kandel PP, De La Fuente L, et al. Allopatric plant pathogen population divergence following disease emergence. Appl Environ Microbiol. 2021;87:e02095-20. doi: 10.1128/AEM.02095-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coletta-Filho HD, Castillo AI, Laranjeira FF, de Andrade EC, Silva NT, et al. Citrus variegated chlorosis: an overview of 30 years of research and disease management. Trop Plant Pathol. 2020;45:175–191. doi: 10.1007/s40858-020-00358-5. [DOI] [Google Scholar]
- 7.Schneider K, van der Werf W, Cendoya M, Mourits M, Navas-Cortés JA, et al. Impact of Xylella fastidiosa subspecies pauca in European olives. Proc Natl Acad Sci U SA. 2020;117:9250–9259. doi: 10.1073/pnas.1912206117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Purcell AH, Hopkins DL. Fastidious xylem-limited bacterial plant pathogens. Annu Rev Phytopathol. 1996;34:131–151. doi: 10.1146/annurev.phyto.34.1.131. [DOI] [PubMed] [Google Scholar]
- 9.Chatterjee S, Almeida RPP, Lindow S. Living in two worlds: the plant and insect lifestyles of Xylella fastidiosa . Annu Rev Phytopathol. 2008;46:243–271. doi: 10.1146/annurev.phyto.45.062806.094342. [DOI] [PubMed] [Google Scholar]
- 10.Sicard A, Zeilinger AR, Vanhove M, Schartel TE, Beal DJ, et al. Xylella fastidiosa: insights into an emerging plant pathogen. Annu Rev Phytopathol. 2018;56:181–202. doi: 10.1146/annurev-phyto-080417-045849. [DOI] [PubMed] [Google Scholar]
- 11.Kyrkou I, Pusa T, Ellegaard-Jensen L, Sagot M-F, Hansen LH. Pierce’s disease of grapevines: a review of control strategies and an outline of an epidemiological model. Front Microbiol. 2018;9:2141. doi: 10.3389/fmicb.2018.02141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baldi P, La Porta N. Xylella fastidiosa: host range and advance in molecular identification techniques. Front Plant Sci. 2017;8:944. doi: 10.3389/fpls.2017.00944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dupas E, Briand M, Jacques M-A, Cesbron S. Novel tetraplex quantitative PCR assays for simultaneous detection and identification of Xylella fastidiosa subspecies in plant tissues. Front Plant Sci. 2019;10:1732. doi: 10.3389/fpls.2019.01732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cesbron S, Dupas E, Beaurepère Q, Briand M, Montes-Borrego M, et al. Development of a nested-multilocus sequence typing approach for a highly sensitive and specific identification of Xylella fastidiosa subspecies directly from plant samples. Agronomy. 2020;10:1099. doi: 10.3390/agronomy10081099. [DOI] [Google Scholar]
- 15.Hogenhout SA, Oshima K, Ammar E-D, Kakizawa S, Kingdom HN, et al. Phytoplasmas: bacteria that manipulate plants and insects. Mol Plant Pathol. 2008;9:403–423. doi: 10.1111/j.1364-3703.2008.00472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Piombo E, Abdelfattah A, Droby S, Wisniewski M, Spadaro D, et al. Metagenomics approaches for the detection and surveillance of emerging and recurrent plant pathogens. Microorganisms. 2021;9:188. doi: 10.3390/microorganisms9010188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonants P, Griekspoor Y, Houwers I, Krijger M, van der Zouwen P, et al. Development and evaluation of a triplex taqman assay and next-generation sequence analysis for improved detection of Xylella in plant material. Plant Dis. 2019;103:645–655. doi: 10.1094/PDIS-08-18-1433-RE. [DOI] [PubMed] [Google Scholar]
- 18.Román-Reyna V, Dupas E, Cesbron S, Marchi G, Campigli S, et al. Metagenomic sequencing for identification of Xylella fastidiosa from leaf samples. mSystems. 2021;6:e0059121. doi: 10.1128/mSystems.00591-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jain M, Olsen HE, Paten B, Akeson M. The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol. 2016;17:239. doi: 10.1186/s13059-016-1103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moss EL, Maghini DG, Bhatt AS. Complete, closed bacterial genomes from microbiomes using nanopore sequencing. Nat Biotechnol. 2020;38:701–707. doi: 10.1038/s41587-020-0422-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mechan Llontop ME, Sharma P, Aguilera Flores M, Yang S, Pollok J, et al. Strain-level identification of bacterial tomato pathogens directly from metagenomic sequences. Phytopathology. 2020;110:768–779. doi: 10.1094/PHYTO-09-19-0351-R. [DOI] [PubMed] [Google Scholar]
- 22.Delahaye C, Nicolas J. Sequencing DNA with nanopores: troubles and biases. PLoS One. 2021;16:e0257521. doi: 10.1371/journal.pone.0257521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ciuffreda L, Rodríguez-Pérez H, Flores C. Nanopore sequencing and its application to the study of microbial communities. Comput Struct Biotechnol J. 2021;19:1497–1511. doi: 10.1016/j.csbj.2021.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buytaers FE, Saltykova A, Denayer S, Verhaegen B, Vanneste K, et al. Towards real-time and affordable strain-level metagenomics-based foodborne outbreak investigations using Oxford nanopore sequencing technologies. Front Microbiol. 2021;12:738284. doi: 10.3389/fmicb.2021.738284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tian L, Huang C, Mazloom R, Heath LS, Vinatzer BA. LINbase: a web server for genome-based identification of prokaryotes as members of crowdsourced taxa. Nucleic Acids Res. 2020;48:W529–W537. doi: 10.1093/nar/gkaa190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valsesia G, Gobbin D, Patocchi A, Vecchione A, Pertot I, et al. Development of a high-throughput method for quantification of Plasmopara viticola DNA in grapevine leaves by means of quantitative real-time polymerase chain reaction. Phytopathology. 2005;95:672–678. doi: 10.1094/PHYTO-95-0672. [DOI] [PubMed] [Google Scholar]
- 27.Harper SJ, Ward LI, Clover GRG. Development of LAMP and real-time PCR methods for the rapid detection of Xylella fastidiosa for quarantine and field applications. Phytopathology. 2010;100:1282–1288. doi: 10.1094/PHYTO-06-10-0168. [DOI] [PubMed] [Google Scholar]
- 28.What’s in my Pot? (WIMP), a quantitative analysis tool for real-time species identification. 2021. [ August 26; 2017 ]. https://nanoporetech.com/resource-centre/whats-my-pot-wimp-quantitative-analysis-tool-real-time-species-identification accessed.
- 29.Kim D, Song L, Breitwieser FP, Salzberg SL. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 2016;26:1721–1729. doi: 10.1101/gr.210641.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bailey TL, Johnson J, Grant CE, Noble WS. The MEME Suite. Nucleic Acids Res. 2015;43:W39–W49. doi: 10.1093/nar/gkv416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pierce NT, Irber L, Reiter T, Brooks P, Brown CT. Large-scale sequence comparisons with sourmash. F1000Res. 2019;8:1006. doi: 10.12688/f1000research.19675.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prjibelski A, Antipov D, Meleshko D, Lapidus A, Korobeynikov A. Using SPAdes de novo assembler. Curr Protoc Bioinformatics. 2020;70:e102. doi: 10.1002/cpbi.102. [DOI] [PubMed] [Google Scholar]
- 33.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 2019;37:540–546. doi: 10.1038/s41587-019-0072-8. [DOI] [PubMed] [Google Scholar]
- 35.Richter M, Rosselló-Móra R, Oliver Glöckner F, Peplies J. JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics. 2016;32:929–931. doi: 10.1093/bioinformatics/btv681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 37.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3693. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37:1530–1534. doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu G, Smith DK, Zhu H, Guan Y, Lam T-Y. Ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. 2017;8:28–36. [Google Scholar]
- 40.Harris SR. SKA: split kmer analysis toolkit for bacterial genomic epidemiology. bioRxiv. 2018:453142. doi: 10.1101/453142. [DOI] [Google Scholar]
- 41.Seemann T. Snippy: Rapid Haploid Variant Calling and Core Genome Alignment. 2015. https://github.com/tseemann/snippy
- 42.Titus Brown C, Irber L. sourmash: a library for MinHash sketching of DNA. JOSS. 2016;1:27. doi: 10.21105/joss.00027. [DOI] [Google Scholar]
- 43.Analysis solutions for nanopore sequencing data. 2021. https://nanoporetech.com/nanopore-sequencing-data-analysis
- 44.Kolmogorov M, Bickhart DM, Behsaz B, Gurevich A, Rayko M, et al. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat Methods. 2020;17:1103–1110. doi: 10.1038/s41592-020-00971-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Faino L, Scala V, Albanese A, Modesti V, Grottoli A, et al. Nanopore sequencing for the detection and identification of Xylella fastidiosa subspecies and sequence types from naturally infected plant material. Plant Pathol. 2021;70:1860–1870. doi: 10.1111/ppa.13416. [DOI] [Google Scholar]
- 46.Román-Reyna V, Dupas E, Cesbron S, Marchi G, Campigli S, et al. Metagenomic sequencing for identification of Xylella fastidiosa from leaf samples. mSystems. 2021;6:e0059121. doi: 10.1128/mSystems.00591-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuan X, Morano L, Bromley R, Spring-Pearson S, Stouthamer R, et al. Multilocus sequence typing of Xylella fastidiosa causing Pierce’s disease and oleander leaf scorch in the United States. Phytopathology. 2010;100:601–611. doi: 10.1094/PHYTO-100-6-0601. [DOI] [PubMed] [Google Scholar]
- 48.Potnis N, Kandel PP, Merfa MV, Retchless AC, Parker JK, et al. Patterns of inter- and intrasubspecific homologous recombination inform eco-evolutionary dynamics of Xylella fastidiosa . ISME J. 2019;13:2319–2333. doi: 10.1038/s41396-019-0423-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.bioRxiv 2021. https://www.biorxiv.org/
- 50.Nattestad M, Aboukhalil R, Chin C-S, Schatz MC. Ribbon: intuitive visualization for complex genomic variation. Bioinformatics. 2021;37:413–415. doi: 10.1093/bioinformatics/btaa680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.