

Abstract
Conventional PD‐L1 immunohistochemical tissue biopsies only predict 20%–40% of non‐small cell lung cancer (NSCLC) patients that will respond positively to anti‐PD‐1/PD‐L1 immunotherapy. Herein, we present an immunogold biochip to quantify single extracellular vesicular RNA and protein (AuSERP) as a non‐invasive alternative. With only 20 μl of purified serum, PD‐1/PD‐L1 proteins on the surface of extracellular vesicles (EVs) and EV PD‐1/PD‐L1 messenger RNA (mRNA) cargo were detected at a single‐vesicle resolution and exceeded the sensitivities of their bulk‐analysis conventional counterparts, ELISA and qRT‐PCR, by 1000 times. By testing a cohort of 27 non‐responding and 27 responding NSCLC patients, AuSERP indicated that the single‐EV mRNA biomarkers surpass the single‐EV protein biomarkers in predicting patient responses to immunotherapy. Dual single‐EV PD‐1/PD‐L1 mRNA detection differentiated responders from non‐responders with an accuracy of 72.2% and achieved an NSCLC diagnosis accuracy of 93.2%, suggesting the potential for AuSERP to provide enhanced immunotherapy predictions and cancer diagnoses within the clinical setting.
1. INTRODUCTION
The immune system responds to cancer via a complex network of cellular interactions in which cytotoxic T cells, helper T cells, and natural killer cells are activated and work in concert against tumour cells (Verdegaal et al., 2016). However, many metastatic tumours have adopted methods to hijack immune checkpoints to evade immune recognition. The overexpression of programmed cell death ligand 1 (PD‐L1) on the surface of tumour cells, which binds to programmed cell death protein 1 (PD‐1) on T cells leading to a blockade of T cell activation, protects tumour cells from T cell‐mediated killing (Ribas, 2015). The manipulation of immune checkpoint pathways using immune checkpoint inhibitors (ICIs) has emerged as an effective form of immunotherapy, demonstrating positive and durable clinical outcomes (Gulley et al., 2017). For instance, patients with metastatic melanoma treated with concurrent ipilimumab (anti‐cytotoxic T lymphocyte‐associated molecule‐4 (anti‐CTLA‐4)) and nivolumab (anti‐PD‐1) achieved an overall survival rate of 79% in 2 years (Wolchok et al., 2013). However, a majority of cancer patients do not respond positively to immunotherapy. For example, the response rate to single‐agent PD‐1/PD‐L1 inhibition in patients with renal cell carcinoma is only 19% (Motzer et al., 2015). Hence, there is an urgency to determine which individual patients may benefit from PD‐1/PD‐L1 blockade and other immunotherapies.
Tumour PD‐L1 expression has been approved by the Food and Drug Administration (FDA) as a predictive biomarker for anti‐PD‐1/PD‐L1 immunotherapy, which is detected using immunohistochemistry (IHC) (Goodman et al., 2017). Four PD‐L1 IHC assays using four different anti‐PD‐L1 antibodies (22C3, 28–8, SP263, and SP142) on two different automated staining platforms (Dako and Ventana) have been registered with the FDA (Ancevski Hunter et al., 2018; Jotatsu et al., 2018). Patients with higher expression of PD‐L1 on their tissue biopsies are associated with improved response rates to PD‐1/PD‐L1 blockade (Garon et al., 2015). However, sampling at a single metastatic site may not represent the entire tumour burden in a highly heterogeneous cancer (Hong et al., 2018; Westphal & Lamszus, 2015). Hence, it is desirable to develop novel technologies to detect predictive biomarkers from bodily fluids in a non‐invasive manner. Such an approach can help integrate signals from all metastatic foci and can be repeated serially throughout immunotherapy with ease.
Circulating microRNAs (miRNAs) have been utilized as potential predictive biomarkers for anti‐PD‐1/PD‐L1 immunotherapy for non‐small cell lung cancer (NSCLC) (Fan et al., 2020; Shukuya et al., 2020). However, precise quantification of circulating miRNAs is highly challenging due to inconsistencies originating from pre‐ and post‐analytical variables and the inability to discriminate among closely related miRNAs (Saliminejad et al., 2019). In this regard, circulating messenger RNAs (mRNAs), which can be protected within extracellular vesicles (EVs) from in vivo degradation, are superior to circulating miRNAs as practical predictive biomarkers for clinical use (O'Brien et al., 2020).
EVs are lipid particles released from cells that vary from 30 nm to a few microns in diameters and are present in all biological fluids (e.g., blood, urine, and cerebral spinal fluid) (Mager, Breakefield & Wood, 2013). EVs can be classified by size as small EVs (sEVs; smaller than 200 nm) and medium/large EVs (m/lEVs; larger than 200 nm), by biochemical composition, or by the environmental conditions of their biogenesis in accordance with the Minimal Information for Studies of Extracellular Vesicles (MISEV2018) guideline (Thery et al., 2018). EVs contain different cargo, including proteins, RNA, DNA, and lipids that can be trafficked between cells and serve as mediators of intercellular communication (Xu et al., 2018). Due to the partial overlap in size of EVs and lipoprotein particles (LPPs) that are also present in bodily fluids, purification of EVs usually requires immunoaffinity‐based methods in addition to size‐based separation (e.g., filtration, size‐exclusion chromatography, and flow field‐flow fractionation) (Liangsupree et al., 2020; Zhang & Lyden, 2019).
There have been some efforts to characterize PD‐1/PD‐L1 proteins and PD‐L1 mRNA from EVs using western blot (Ricklefs et al., 2018), enzyme‐linked immunosorbent assay (ELISA) (Chen et al., 2018), flow cytometry (Theodoraki et al., 2018), quantitative reverse transcription polymerase chain reaction (qRT‐PCR) (Yu et al., 2019), and droplet digital PCR (ddPCR) (Del Re et al., 2018). However, these existing methods primarily focus on the bulk analysis of total proteins and RNA extracted from many EVs, lending averaged information, and limiting the assay's resolution and sensitivity (Kim et al., 2018). During the characterization process of bulk‐analysis methods, EVs are broken down to obtain their internal contents, leading to the loss of molecular information at individual EV levels (Wang et al., 2020), understating the impact of heterogeneity. Therefore, it is imperative to develop technologies that provide an accurate and efficient analysis of the molecular content at a single‐EV level. Several single‐EV analytical technologies have been proposed for EV molecular analysis, including analysis by fluorescence imaging of immobilized single EVs (Fraser et al., 2019; Lee et al., 2018; Liu et al., 2019), flow cytometry of single EVs via target‐initiated engineering (Shen et al., 2018), droplet‐based single‐exosome‐counting ELISA (droplet digital ExoELISA) (Liu et al., 2018), digital detection integrated with surface‐anchored nucleic acid amplification (Tian et al., 2018), proximity‐dependent barcoding assay (Wu et al., 2019), and immuno‐droplet digital PCR (iddPCR) (Ko et al., 2020). These methods successfully improved the limit of detection (LOD) and demonstrated heterogeneous protein profiles of single EVs unachievable by bulk‐analysis methods. Despite this progress, detecting mRNAs and low abundance proteins (such as PD‐1/PD‐L1) in single EVs remains challenging due to the inherent limitations of signal‐to‐background thresholding (Yekula et al., 2020).
Herein, we describe a novel technology that enables single‐EV capture and detection to quantify low abundance biomarkers for immunotherapy. We engineered a highly sensitive immunogold‐based biochip to co‐quantify PD‐1/PD‐L1 proteins and mRNAs in single EVs sorted from NSCLC patient serum. Multiplexed analyses enabled the simultaneous detection of multiple targets in a single assay, thereby promoting the discovery of novel biomarker combinations for an improved diagnosis, prognosis, and prediction. Our gold nanoparticle‐based single extracellular vesicular RNA and protein (AuSERP) biochip was formulated on a gold‐coated glass coverslip functionalized with polyethylene glycol (PEG), to prevent non‐specific binding, and gold spherical nanoparticles (NPs), to amplify the signal and improve single‐EV sensitivity. Different antibodies were tethered on the chip surface to capture and sort EVs into subpopulations based on their membrane protein compositions. Anti‐PD‐1/PD‐L1 antibodies and the tyramide signal amplification (TSA) method were used to quantify the corresponding membrane proteins on the captured single EVs. Molecular beacons (MBs) with target‐specific probes were also fused with the captured single EVs to identify and quantify PD‐1/PD‐L1 mRNAs. Molecular characterization at the single‐EV level was achieved by coupling AuSERP that maximizes the signal‐to‐noise ratio (SNR) with high‐resolution total internal reflection fluorescence (TIRF) microscopy. Serum samples from 27 NSCLC non‐responders and 27 NSCLC responders collected before immunotherapy were analysed for four single‐EV biomarkers (PD‐1/PD‐L1 proteins and mRNAs). We showed that AuSERP was highly sensitive to detecting single‐EV protein and mRNA. Compared to single‐EV protein biomarkers, single‐EV mRNA cargo excelled at diagnosing NSCLC and predicting patient responses to immunotherapy. By combining single‐EV PD‐1/PD‐L1 mRNA biomarkers, AuSERP diagnosed NSCLC patients and predicted patient responses to anti‐PD‐1/PD‐L1 immunotherapy with accuracies of 93.2% and 72.2%, respectively, suggesting the potential applications of AuSERP as a non‐invasive and competitive alternative to diagnose cancer patients and predict immunotherapy responses.
2. RESULTS
2.1. AuSERP maximizes the signal‐to‐noise ratio and offers high‐throughput analyses for single‐EV detection
High‐resolution TIRF microscopy has been widely used in cell biology to detect single molecules due to its high SNR (Ter‐Ovanesyan et al., 2017). This imaging technique restricts excitation to a precise focal plane near the coverslip and eliminates out‐of‐focus fluorescence, allowing single‐molecule detection (Kudalkar et al., 2016). High‐resolution TIRF microscopy has been applied to visualize EVs pre‐stained with a fluorescent membrane dye on a glass slide (Ter‐Ovanesyan et al., 2017). However, for protein‐specific on‐chip EV labelling, the non‐specific binding of labelling antibodies to the glass coverslip complicates differentiating true binding events from background noise. We designed a biochip with a PEG coating to prevent the non‐specific binding of biomolecules to the glass surface. We first deposited a thin gold coating (thickness ∼ 12 nm) onto a glass coverslip via titanium (thickness ∼ 2 nm), which serves as a ‘metal glue’, and subsequently coated it with PEG (2 kDa). AuSERP was then assembled by attaching the functionalized glass coverslip to a silicone gasket with 64 chambers (Figure 1a). This design is highly scalable, allowing for up to four AuSERP biochips to be placed into a tray for high‐throughput robotic processing of up to 256 samples. The treated glass surface was then functionalized with streptavidin‐conjugated gold NPs and a cocktail of biotinylated antibodies to capture specific EV subpopulations (Figure 1b). To further minimize non‐specific binding, the treated glass was blocked with 3% (w/v) bovine serum albumin (BSA) and 0.05% (v/v) Tween‐20 before and after EV capture. The thin gold coating was made to further improve the SNR of TIRF microscopy through the surface plasmon resonance (SPR) effect, which takes place when total internal reflection occurs at a metal film‐liquid interface (Fang, 2015; Sun et al., 2010). We previously showed that a biochip coated with a thin gold film and PEG could sensitively quantify target RNAs within EVs in bulk for an early non‐invasive cancer diagnosis (Hu et al., 2017). In that design, EVs were captured on the biochip using cationic lipoplex nanoparticles (CLNs) via electrostatic fusion, and EV RNA cargos were detected with MBs encapsulated within the CLNs. However, the immobilization of highly positively charged CLNs (zeta potential ∼ 30 mV) on the biochip surface caused significantly high background noise for RNA detection (Hu et al., 2017), which impairs the capability and sensitivity required for single‐EV analyses. To enable both protein and RNA detections at a single‐EV level, in the present study, we designed the novel AuSERP biochip in which single EVs were captured and sorted via antibodies tethered onto gold NPs.
FIGURE 1.
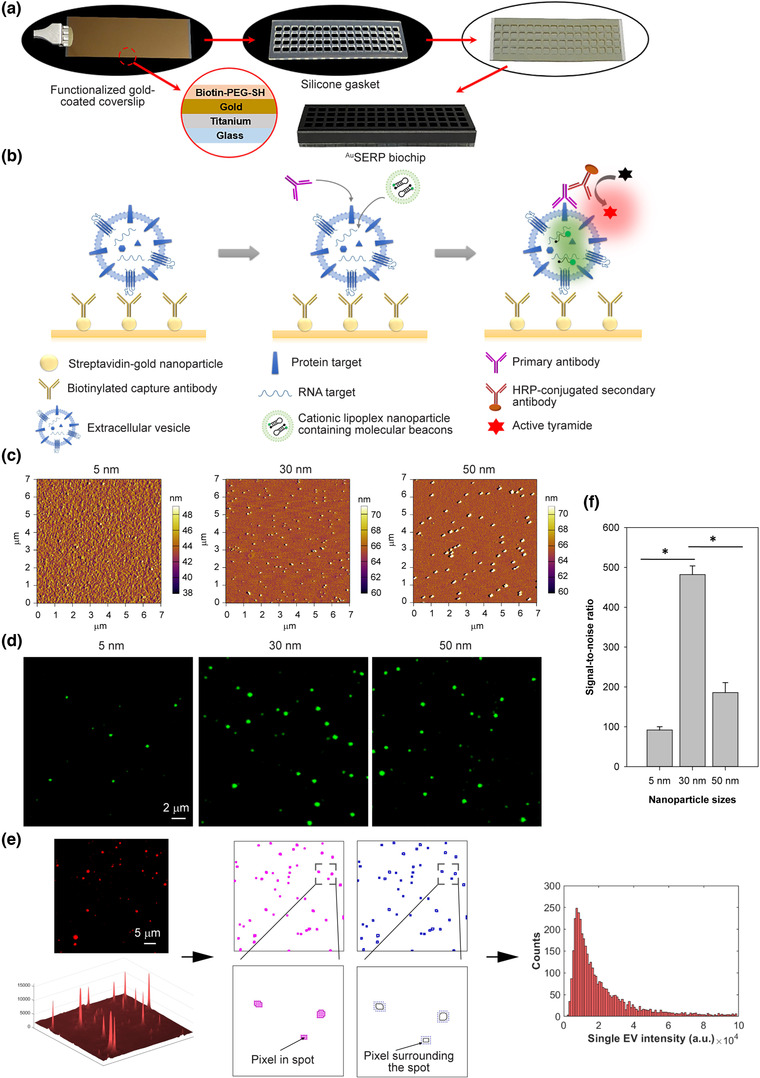
Design, characterization, and optimization of AuSERP. (a) AuSERP assembly. A functionalized gold‐coated coverslip was attached to a silicone gasket with 64 chambers for high‐throughput analysis of single‐EV biomarkers. (b) A schematic representation of the mechanism of detection for protein and mRNA biomarkers present in single extracellular vesicles (EVs) using AuSERP. A gold‐coated coverslip with PEG‐tethered gold nanoparticles (NPs) conjugated to capture antibodies was used to immobilize single EVs. Proteins on the surface of the single EVs were detected using the corresponding primary antibody and a tyramide signal amplification (TSA) method, resulting in fluorescent signals. mRNA cargo was identified using target‐specific molecular beacons (MBs) encapsulated in cationic lipoplex nanoparticles (CLNs), resulting in fluorescent signals. (c) Atomic force microscopy (AFM) images of AuSERP coated with different‐sized gold NPs (5, 30, and 50 nm). (d) Representative total internal reflection fluorescence (TIRF) microscopy images of CD63 protein expression on the surface of H1568 single EVs captured with AuSERP with different NP sizes. The images were cropped and enlarged from their original images, which are provided in Figure S1(b). (e) Image processing workflow. Background noise signals surrounding bright spots were first removed using the Wavelet denoising method. All the pixels in each spot were then identified and subtracted by the mean intensity of pixels surrounding the spot. The noise‐subtracted intensities of each pixel within the spot were summed into a net intensity of the single EV. Net intensities of all single EVs in 100 TIRF microscopy images were then collected to generate a histogram of net fluorescence intensity. (e) A signal‐to‐noise ratio (SNR) comparison of AuSERP with different sized gold NPs for single‐EV capture. The signal was calculated from the total fluorescence intensity of the CD63 surface protein levels on every single EV (individual fluorescence spot). The data were expressed as mean ± SD; n = 3; *P < 0.0001, Student's t‐test. a.u., arbitrary units
We first investigated the effect of streptavidin‐conjugated gold NP size (5, 30, and 50 nm) on the SNR of AuSERP as metal NPs exhibit localized SPR that can be observed as a strong UV‐Vis absorption band absent from bulk metal (Haes & Van Duyne, 2004) whereby gold NPs better enhance fluorescent signals via higher refractive index (RI) changes and resonance angle shifts (Uludag & Tothill, 2012). Atomic force microscopy (AFM) images showed that NPs of all sizes were uniformly dispersed on the surface (Figure 1c and Figure S1a). The dispersion of NPs on AuSERP promoted a single‐EV resolution. With the same concentration loaded into each well (0.005% (w/v) based on gold), the number of NPs coated on the surface decreased as particle size increased. EVs from H1568 cells, an NSCLC cell line, were used to evaluate AuSERP. As previously described, EVs were first isolated from cell culture supernatant using tangential flow filtration (TFF) to remove residual proteins and small‐molecules impurities (Zhang et al., 2021). The purified EVs were then captured on the chip using a cocktail of anti‐CD9 and anti‐CD63 biotinylated antibodies. CD63 and CD9 are abundant tetraspanin surface antigens on EVs. This immunoaffinity‐based approach was designed to separate EVs from remaining LPPs present in bodily fluids after TFF. CD63 staining of the captured EVs was used to evaluate the effect of NP sizes on the fluorescence signal intensity and background noise. TIRF microcopy images and histograms of net fluorescence intensity revealed that surfaces coated with 30‐nm gold NPs had the highest signal and the lowest background, while both signal and background were lower for those coated with 5‐nm NPs and higher for those coated with 50‐nm NPs (Figure 1d and Figure S1b‐c). Fluorescence intensities of biomarker signals from TIRF microscopy images were quantified using a custom‐built MATLAB algorithm, as shown in Figure 1(e), which allows for a rapid and automated single‐EV image analysis. Briefly, our algorithm recognized a collective of spots with distinguished edges and removed background noise signals surrounding the bright spots using the Wavelet denoising method. To ensure all analyses are conducted at the single‐vesicle level, all collectives of spots falling within a threshold of 3–8 pixels, set through a user interface, were identified as biomarker signals from single EVs (all other signals were discarded as EV aggregates or noise). The size‐based threshold was included as EV aggregates produce larger fluorescent spots than single EVs in TIRF microscopy images (Figure S2). Although EVs with very low copy numbers may fall below this threshold, appropriate exclusion criteria are necessary to differentiate signals derived from single EVs versus noise from controls. It is worth mentioning that other technologies used for the analysis of single EVs, such as single‐molecule localization microscopy and nanoflow cytometry, have also established similar thresholding techniques with respect to a control sample (Lennon et al., 2019; Silva et al., 2021; Tian et al., 2020). All the pixels within each spot were then identified and subtracted by the mean intensity of pixels surrounding the spot. The noise‐subtracted intensities of each pixel within the spot were then summed into a net intensity of the single EV. Net intensities of all single EVs in 100 TIRF microscopy images were then collected to generate a histogram of net fluorescence intensity and calculate the total fluorescence intensity (TFI), where the TFI is equal to the area under the histogram. Consequently, the SNR of AuSERP was defined as
| (1) |
where TFIEV is the TFI of signals obtained from EV samples, TFIneg is the mean TFI of signals obtained from blanks (negative control – phosphate‐buffered saline (PBS)), and SDneg (or noise) is the standard deviation of signals obtained from blanks (by three independent experiments). Among the three sizes, 30 nm was shown to produce the highest SNR (P < 0.0001, Figure 1f). Therefore, 30‐nm gold NPs were chosen for our subsequent experiments. Our results were consistent with the observation that the size of gold NPs significantly affects the level of signal enhancement due to a critical balance between surface coverage, distance from the coverslip surface, and the size‐dependent RI shifts (Lyon et al., 1999).
2.2. AuSERP enables single‐EV PD‐L1 protein detection with ∼ 1000 times more sensitivity than conventional ELISA
We next evaluated AuSERP coated with 30‐nm gold NPs for PD‐1/PD‐L1 protein and mRNA characterization of single EVs derived from in vitro cell model systems. The schematic representation of single‐EV PD‐1/PD‐L1 protein and mRNA detection on AuSERP is shown in Figure 1(b). A TSA method was employed to boost PD‐1/PD‐L1 protein signals on single‐EV surfaces. This method uses the enzyme horseradish peroxidase (HRP) – a secondary antibody – to convert labelled tyramide molecules at the epitope detection site into a highly reactive oxidized intermediate, which binds rapidly and covalently to electron‐rich tyrosine residues present in proteins near the epitope (Faget & Hnasko, 2015). Therefore, the TSA method can generate high‐density labels of a target protein, making it 10–200 times more sensitive than conventional immunostaining methods. PD‐1/PD‐L1 mRNAs inside the single EVs were detected using CLNs encapsulating MBs (CLN‐MBs) to provide a strong signal of target RNAs due to increased delivery efficiency of MBs into EVs compared to free MBs. As positively charged lipid nanoparticles, CLNs fuse with negatively charged EVs and deliver many MBs into the EVs (Wu et al., 2013). In contrast, the delivery of free MBs into EV lipid bilayers without any assistance is restricted to Brownian motion. Thus, our technology allows for the co‐quantification of PD‐1/PD‐L1 molecular contents inside and on the surface of single EVs, thereby providing more comprehensive information on single EVs from NSCLC patient serum.
An in vitro PD‐L1 model was developed by stimulating H1568 cells with interferon‐gamma (IFN‐γ), a cytokine secreted by activated effector T cells. IFN‐γ is critical for innate and adaptive immunity and is known to upregulate PD‐L1 expression on tumour cells (Abiko et al., 2015; Kil et al., 2017). In the present study, IFN‐γ significantly increased the PD‐L1 protein expression on H1568 cells as shown by IHC (Figure 2a) and ELISA (P < 0.001, Figure 2b). IHC was performed using an FDA‐approved diagnostic assay, Dako PD‐L1 IHC 22C3 pharmDx (Jotatsu et al., 2018). An anti‐PD‐L1 antibody (Cell Signalling Technology (CST)) was also used to conduct PD‐L1 immunofluorescence on the cells (Figure S3a). The result was comparable to the PD‐L1 clinical assay, indicating the CST anti‐PD‐L1 antibody's efficiency in binding to PD‐L1 antigens. This antibody was, therefore, selected for further PD‐L1 protein characterization on single EVs with AuSERP.
FIGURE 2.
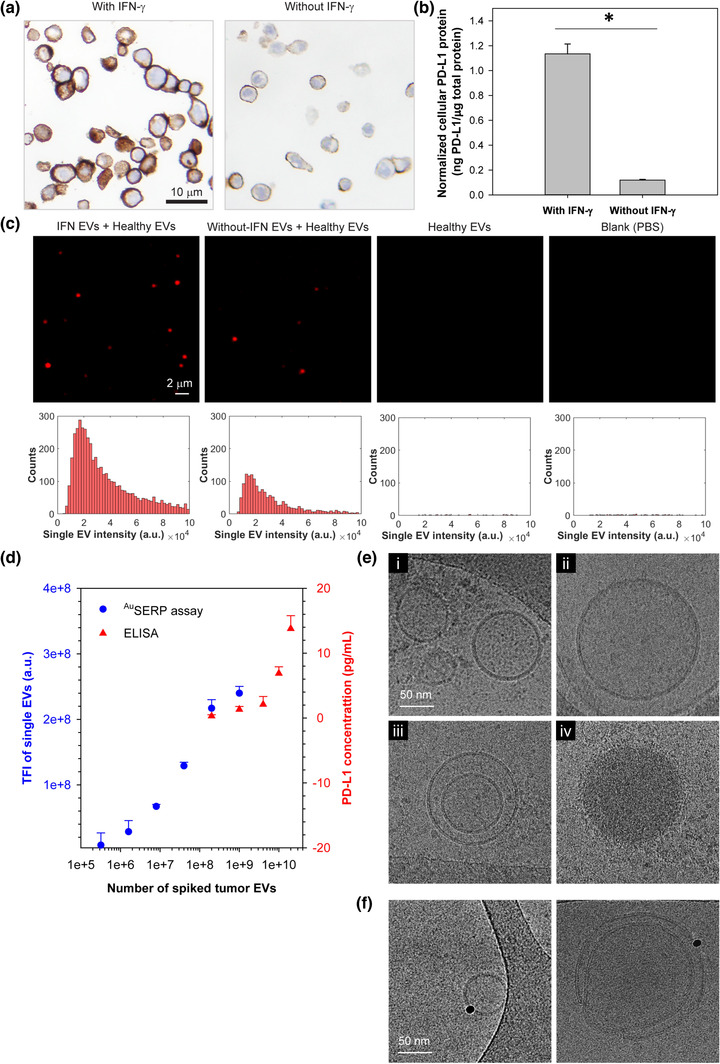
In vitro model and characterization of cellular and single‐EV PD‐L1 protein. (a) Immunohistochemistry (IHC) of PD‐L1 protein in H1568 cells with/without interferon‐gamma (IFN‐γ) stimulation. Cell nuclei and PD‐L1 protein were stained blue (by haematoxylin) and brown (by Dako PD‐L1 IHC 22C3 pharmDx), respectively. (b) Quantification of PD‐L1 protein levels in H1568 cells measured by ELISA and normalized by the total protein expressed by the cells measured with a BCA Protein Assay. The data were expressed as mean ± SD; n = 3; *P < 0.001, Student's t‐test. (c) Representative TIRF microscopy images and their corresponding histograms of PD‐L1 protein expression on the surface of single EVs derived from H1568 cells with/without IFN‐γ stimulation in comparison to healthy donor EVs and PBS as controls. The single‐EV PD‐L1 protein signals were characterized with AuSERP using anti‐PD‐L1 antibodies and the TSA method. H1568 EVs were spiked in healthy donor EVs at a 1:1 ratio with 5 × 1010 particles/ml each. Healthy donor EVs were purified from healthy donor serum and then diluted in PBS to reach the target concentration. The images were cropped and enlarged from their original images, which are provided in Figure S3(b). (d) A performance evaluation of the AuSERP for PD‐L1 protein detection in comparison to ELISA (the average values are included in Table S3). EVs derived from IFN‐γ‐stimulated H1568 cells were spiked in healthy donor EVs at different concentrations ranging from 0 to 5 × 1010 particles/ml. The healthy donor EV concentration was kept constant at 5 × 1010 EVs/ml for all samples. The limit of detection (LOD) of AuSERP for PD‐L1 protein was ∼ 106 spiked tumour EVs, ∼ 1000 times lower than ELISA. The data were expressed as mean ± SD; n = 3. TFI, total fluorescence intensity; a.u., arbitrary units. (e) Cryogenic transmission electron microscopy (cryo‐TEM) images of EVs produced by IFN‐γ‐stimulated H1568 cells. (f) Cryo‐TEM images of immunogold labelled PD‐L1 protein on the EV surface
PD‐L1 protein on the surface of single EVs produced by H1568 cells with/without IFN‐γ stimulation was characterized using AuSERP with the CST anti‐PD‐L1 antibody. EVs from H1568 cells were spiked in healthy donor EVs at a 1:1 ratio (H1568 EVs: healthy donor EVs) with 5 × 1010 particles/ml each to simulate NSCLC clinical samples and characterize PD‐L1 expression on single EVs. Healthy donor EVs at 5 × 1010 particles/ml and PBS blank samples were also examined as healthy donor controls and negative controls, respectively. EV staining procedures were performed without a permeabilization buffer to preserve the PD‐L1 protein on the single‐EV membrane surface. TIRF microscopy images and their corresponding histograms showed that PD‐L1 protein expression on EVs from H1568 cells with/without IFN‐γ stimulation were successfully detected with AuSERP and were significantly greater than the healthy donor control and the negative control (P < 0.001, Figure 2c and Figure S3b). The minimal signal detected in the blank control (PBS) may be derived from the substrate autofluorescence and/or non‐specific binding of detection antibodies, which were insignificant compared to the true signal.
Additionally, the PD‐L1 protein expression on single EVs from IFN‐γ‐stimulated H1568 cells was considerably higher than on single EVs derived from cells without stimulation (P < 0.005, Figure 2c and Figure S3b). Similar to cells, previous studies revealed that the levels of PD‐L1 protein on tumour‐derived EVs were also upregulated following IFN‐γ stimulation (Chen et al., 2018; Ricklefs et al., 2018). Thus, our platform was sensitive enough to differentiate PD‐L1 protein levels on single EVs under these different conditions. To indicate the robustness of AuSERP for single‐EV characterization, we tested an Abcam anti‐PD‐L1 antibody (clone 28‐8) and compared it with the CST anti‐PD‐L1 antibody. Total fluorescence intensities of PD‐L1 protein signals provided by both antibodies were comparable (P > 0.05, Figure S3c). However, there was a slight qualitative difference, where larger spot sizes and fewer spots were detected by the Abcam antibody (Figure S3d).
We then compared the LOD of AuSERP with the most sensitive commercial ELISA kit, which has a LOD of 0.6 pg/ml to demonstrate the enhanced sensitivity provided by diverging from bulk‐analysis methods and detecting PD‐L1 on the surface of non‐lysed EVs at a single‐vesicle resolution. EVs produced from IFN‐γ‐stimulated H1568 cells were spiked into healthy donor EVs at different concentrations ranging from 0 to 5 × 1010 particles/ml and quantified for PD‐L1 protein expression using AuSERP and ELISA. The healthy donor EV concentration was kept constant at 5 × 1010 EVs/ml for all samples. While the minimum sensitivity of ELISA was ∼ 109 spiked H1568 EVs, our system could detect as low as ∼ 106 spiked H1568 EVs (Figure 2d). As such, our platform outperformed ELISA in analytical sensitivity by ∼ 1000 times. The LOD of our system was determined using the SNR method, in which an SNR of three is generally accepted for estimating the LOD (Shrivastava & Gupta, 2011; Uludag & Tothill, 2012).
We also characterized the size, concentration, morphology, and structure of EVs from H1568 cells to understand their physical properties. Size distributions of EVs with/without IFN‐γ stimulation measured by tunable resistive pulse sensing (TRPS) are shown in Figure S4(a‐b). In both cases, most EVs were sEVs. There was also no significant difference in the number of EVs produced by the cells with/without IFN‐γ stimulation (P > 0.05, Figure S4c). As such, IFN‐γ did not increase the number of EVs secreted by the cells, consistent with a previous report (Poggio et al., 2019), but upregulated the level of PD‐L1 protein on EVs (Figure 2c and Figure 3b). EVs isolated from H1568 cells with IFN‐γ stimulation were also observed using cryogenic transmission electron microscopy (cryo‐TEM). Most of the EVs were intact and had a round shape with a clear lipid bilayer/membrane and a translucent internal structure, indicating little material within the lumen (Figure 2e/i‐ii and Figure S4d/i). However, some particles had a round shape with electron‐dense cargo and no visible lipid membrane (Figure 2e/iv). Single, double, and multilayer vesicles with different sizes were also visualized (Figure 2e/i‐iii and Figure S4d). These observations demonstrate the vast heterogeneity of EVs, as mentioned in previous studies (Emelyanov et al., 2020; Yuana et al., 2013). The presence of PD‐L1 protein on the surface membrane of single EVs was also confirmed by immunogold labelling with cryo‐TEM imaging, where single gold NPs bound to the membranes of single EVs via PD‐L1 (Figure 2f).
2.3. AuSERP enables single‐EV PD‐L1 mRNA detection with ∼ 1000 times more sensitivity than conventional qRT‐PCR
The presence of PD‐L1 mRNA in plasma‐derived EVs of NSCLC patients was previously demonstrated using ddPCR (Del Re et al., 2018). Therefore, in addition to protein characterization, we aimed at detecting mRNAs inside the single EVs with AuSERP. We first designed a PD‐L1 MB and examined its fluorescent in situ hybridization capabilities with PD‐L1 mRNA in H1568 cells. The PD‐L1 MBs successfully localized PD‐L1 mRNAs in the cytoplasm, with stronger signals in the IFN‐γ‐stimulated cells compared to the cells without IFN‐γ stimulation (Figure 3a). This result was consistent with PD‐L1 mRNA quantification by qRT‐PCR in which cellular PD‐L1 mRNA was significantly increased with IFN‐γ stimulation (P < 0.0001, Figure 3b).
FIGURE 3.
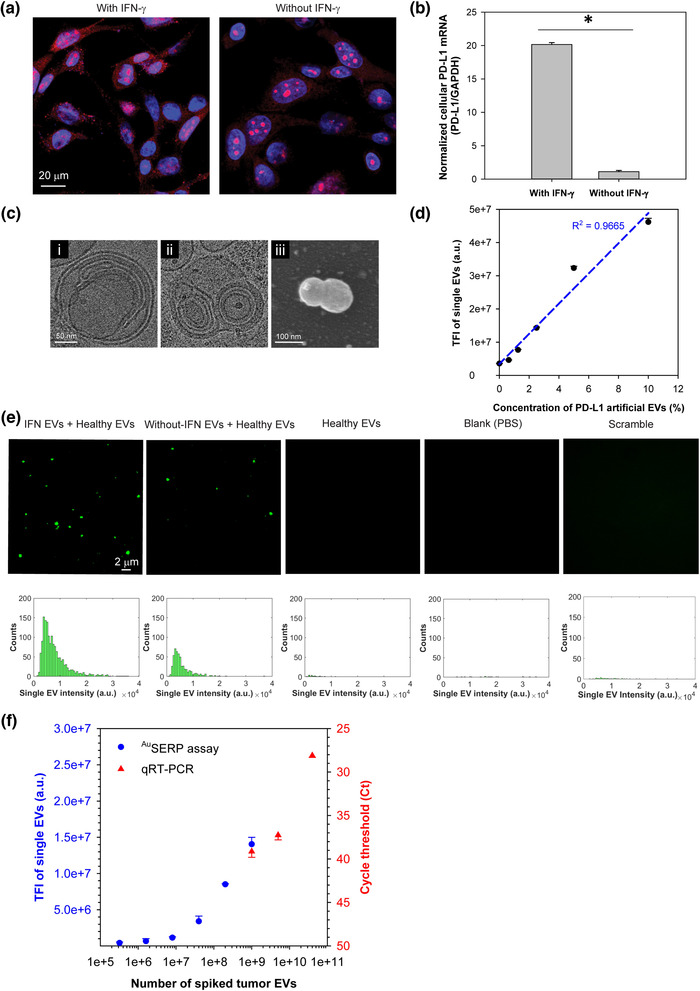
In vitro model and characterization of cellular and single‐EV PD‐L1 mRNA. (a) Confocal fluorescence microscopy images of PD‐L1 mRNA in H1568 cells with/without IFN‐γ stimulation. Cell nuclei and PD‐L1 mRNA were stained blue (by DAPI) and red (by PD‐L1 MBs via fluorescent in situ hybridization), respectively. (b) qRT‐PCR analysis of PD‐L1 mRNA levels in H1568 cells with/without IFN‐γ stimulation. The data were expressed as mean ± SD; n = 3; *P < 0.0001, Student's t‐test. (c) Cryo‐TEM images of a CLN‐MB with a typical “onion‐like” structure (i), the fusion of EVs produced by IFN‐γ‐stimulated H1568 cells with CLN‐MBs targeting PD‐L1 mRNA (ii), and a scanning electron microscopy (SEM) image of the fusion of EVs derived from IFN‐γ‐stimulated H1568 cells with CLN‐MBs targeting PD‐L1 mRNA (iii). (d) Calibration of PD‐L1‐targeting CLN‐MBs using artificial EVs made of liposomes encapsulating PD‐L1 single‐stranded DNA (ssDNA) oligos. There is a strong linear relationship between the resulting fluorescence signal and the concentration of artificial EVs (R2 = 0.9665, P < 0.0005, ANOVA). The data were expressed as mean ± SD; n = 3. (e) Representative TIRF microscopy images and their corresponding histograms of PD‐L1 mRNA expression in single EVs derived from H1568 cells with/without IFN‐γ stimulation in comparison to healthy donor EVs, PBS, and CLN‐scramble‐MBs as controls. The single‐EV PD‐L1 mRNA signals were characterized with AuSERP using PD‐L1‐targeting CLN‐MBs. H1568 EVs were spiked in healthy donor EVs at a 1:1 ratio with 5 × 1010 particles/ml each. The images were cropped and enlarged from their original images, which are provided in Figure S6(a). (f) A performance evaluation of AuSERP for PD‐L1 mRNA detection in comparison to qRT‐PCR (the average values are included in Table S4). EVs derived from IFN‐γ‐stimulated H1568 cells were spiked in healthy donor EVs at different concentrations ranging from 0 to 5 × 1010 particles/ml. The healthy donor EV concentration was kept constant at 5 × 1010 EVs/ml for all samples. The LOD of AuSERP for PD‐L1 mRNA detection was ∼ 106 spiked tumour EVs, ∼ 1000 times lower than qRT‐PCR (Ct values are presented in reverse order). The data were expressed as mean ± SD; n = 3. TFI, total fluorescence intensity; a.u., arbitrary units
To efficiently deliver MBs into EVs for RNA hybridization, we next encapsulated the designed MBs into CLNs. We previously reported that EV‐encapsulated cargo RNAs could be detected using CLNs containing target‐specific MBs (Hu et al., 2017). CLN‐MBs revealed an “onion‐like” structure with multiple wrapped lipid‐MB‐lipid layers (Figure 3c/i), as previously described (Weisman et al., 2004; Wu et al., 2013). The negatively charged MBs act as a bridge between the two positively charged liposomes. This process was repeated to form many layers in a single particle. Due to their positive charge, CLNs could fuse with negatively charged EVs via electrostatic interactions to form larger complexes, as shown in our cryo‐TEM image (Figure 3c/ii) and scanning electron microscopy (SEM) image (Figure 3c/iii). While the fusion of CLNs and single EVs is intrinsically efficient due to their electrostatic attraction, reducing the stochasticity of collisions by limiting the Brownian motion of the EVs via immobilization ensures a high yield of fusion events (Figure S5). This fusion allowed facile delivery of MBs into the EVs, leading to the binding of MBs to the target RNAs within the complexes' nanoscale confinement. The specificity of PD‐L1 CLN‐MBs was examined using artificial EVs containing PD‐L1 single‐stranded DNA (ssDNA) oligos. The resulting fluorescence signal showed a strong linear relationship with the concentration of artificial EVs, spiked in healthy donor EVs ranging from 0% to 10% (R2 = 0.9665, P < 0.0005, Figure 3d), indicating the high specificity of PD‐L1 CLN‐MBs to hybridize with PD‐L1 mRNA cargo.
We further employed PD‐L1 CLN‐MBs for single‐EV PD‐L1 mRNA characterization with AuSERP. Consistent with the single‐EV PD‐L1 protein characterization, AuSERP successfully detected PD‐L1 mRNA in the single EVs derived from H1568 cells and quantitatively differentiated between the single EVs originating from cells with and without IFN‐γ stimulation (P < 0.005, Figure 3e and Figure S6a). Furthermore, the lack of fluorescence by CLNs containing scramble MBs indicates the specificity of the PD‐L1 MBs to PD‐L1 mRNA (Figure 3e). EVs produced from IFN‐γ‐stimulated H1568 cells were also spiked into healthy donor EVs to determine the LOD of our platform for single‐EV mRNA characterization compared to conventional qRT‐PCR. AuSERP could detect as low as ∼ 106 spiked H1568 EVs, which exceeded the sensitivity afforded by qRT‐PCR by ∼ 1000 times (Figure 3f). Altogether, AuSERP exhibited a linear range for the single‐vesicle detection of both PD‐L1 protein and mRNA at ∼ 107–109 single EVs (R2 = 0.9587, P < 0.05 for protein; R2 = 0.9699, P < 0.05 for mRNA). Higher concentrations of EVs resulted in the saturation of the surface and the assembly of EV aggregates, which could no longer be considered for single‐vesicle analyses (Figure S7a). Within the linear range, the dilution of EVs from IFN‐γ‐stimulated H1568 cells had no significant effect on the average intensity of the single EVs (P > 0.05 for protein, P > 0.05 for mRNA). The dilution‐independent intensity of the histograms within the linear range, further indicates that the fluorescent signal on TIRF microscopy images is derived from single EVs and not EV clusters (Figure S7b). Moreover, our system enabled the multiplexed detection of PD‐L1 protein and mRNA biomarkers simultaneously (Figure S6b). The increased sensitivity in comparison to bulk‐analysis methods and the ability to multiplex protein and mRNA to demonstrate EV heterogeneity highlight the importance of evaluating non‐lysed EVs at a single‐vesicle resolution. However, for the remainder of the experiments, single biomarker expression was performed to decrease the assay time.
2.4. AuSERP enables sensitive single‐EV PD‐1 protein and mRNA detection
T cells activated with anti‐CD3/CD28 antibodies and interleukin 2 (IL‐2) have been known to stimulate PD‐1 expression in the cells (Meng et al., 2018). EVs collected from activated T cells were purified and examined for PD‐1 protein and mRNA expression with AuSERP. The size distribution of the purified EVs is shown in Figure S8(a). T cell EVs were spiked into healthy donor EVs at different concentrations ranging from 0 to 5 × 1010 particles/ml. The healthy donor EVs were kept constant at 5 × 1010 particles/ml. Single EVs with detectable PD‐1 protein levels on their surface appeared as bright spots in TIRF microscopy images (Figure S8b). When the spiked EVs exceeded 106, the PD‐1 protein fluorescence intensities increased proportionally to the spiked EV concentration and were significantly higher than intensities from the healthy donor control and the blank control (P < 0.05, Figure S8c). Therefore, our assay allowed comparative and quantitative evaluations of PD‐1 protein expression on single EVs. PD‐1 mRNA in the single EVs was successfully detected and quantified with AuSERP (Figure S8d). To ensure that the probes utilized for AuSERP were specific to PD‐L1+/PD‐1+ protein and mRNA in EVs derived from H1568 cells and T cells, respectively, non‐human EVs were harvested from mouse embryonic fibroblasts (MEF). PD‐L1 protein and mRNA signals were absent in MEF EVs (P > 0.05 for protein, P > 0.05 for mRNA) as well as PD‐1 protein and mRNA (P > 0.05 for protein, P > 0.05 for mRNA), whereas CD63 was positive (P < 0.0001, Figure S9a), indicating that the probes for PD‐L1/PD‐1 protein and mRNA were specific to EVs derived from H1568 cells and T cells. These trends were also observed with high‐resolution flow cytometry (Figure S9b‐c) and western blot (Figure S9d). Furthermore, to ensure the capture antibodies do not produce biases in the detection levels, the expression levels of CD63 and CD9 were detected in the model EVs. There was an insignificant loss of CD9 with IFN‐γ stimulation, which was also observed with high‐resolution flow cytometry (P > 0.05, Figure S10) and western blot (Figure S9d). On the other hand, PD‐L1hi/PD‐1hi EVs had significantly higher expression of CD63 in comparison to PD‐L1lo/PD‐1lo, which was also observed with high‐resolution flow cytometry (P < 0.05, Figure S10) and western blot (Figure S9d). Therefore, utilizing both CD63 and CD9 for capture was necessary to avoid detection biases. Taken together, we effectively developed a highly sensitive AuSERP biochip to quantify four biomarkers (PD‐1/PD‐L1 proteins and mRNAs) on the surface and inside single EVs.
2.5. AuSERP enables single‐EV PD‐1/PD‐L1 protein and mRNA detection from NSCLC patient serum
Fifty‐four patients with NSCLC at stage IV were enrolled for this study regardless of their PD‐L1 IHC results (Table S1 and Table S2). Thirty‐seven patients underwent anti‐PD‐1 therapy with nivolumab, sixteen patients underwent anti‐PD‐1 therapy with pembrolizumab, and one patient underwent anti‐PD‐L1 therapy with atezolizumab. We classified the patients who showed a partial response (PR) or stable disease (SD) lasting more than 6 months as “responders” (n = 27), and the patients who showed progressive disease (PD) as “non‐responders” (n = 27). PR, SD, and PD were defined following the Response Evaluation Criteria in Solid Tumours (RECIST version 1.1) (Seymour et al., 2017). The patients’ tissue and serum samples were collected before starting immunotherapy. Tissue samples were characterized using the Dako PD‐L1 IHC 22C3 pharmDx platform. Tissue samples with tumour cells positive for PD‐L1 protein expression equal to or greater than 1% were positive for PD‐L1 IHC (Figure 4a). Of the patients within the cohort, 29 demonstrated a positive PD‐L1 IHC result, and 10 demonstrated a negative PD‐L1 IHC result. Meanwhile, 15 patients were marked as unknown since their PD‐L1 status was absent from their medical records, and their formalin‐fixed paraffin‐embedded samples were not available for testing (Figure 4b).
FIGURE 4.
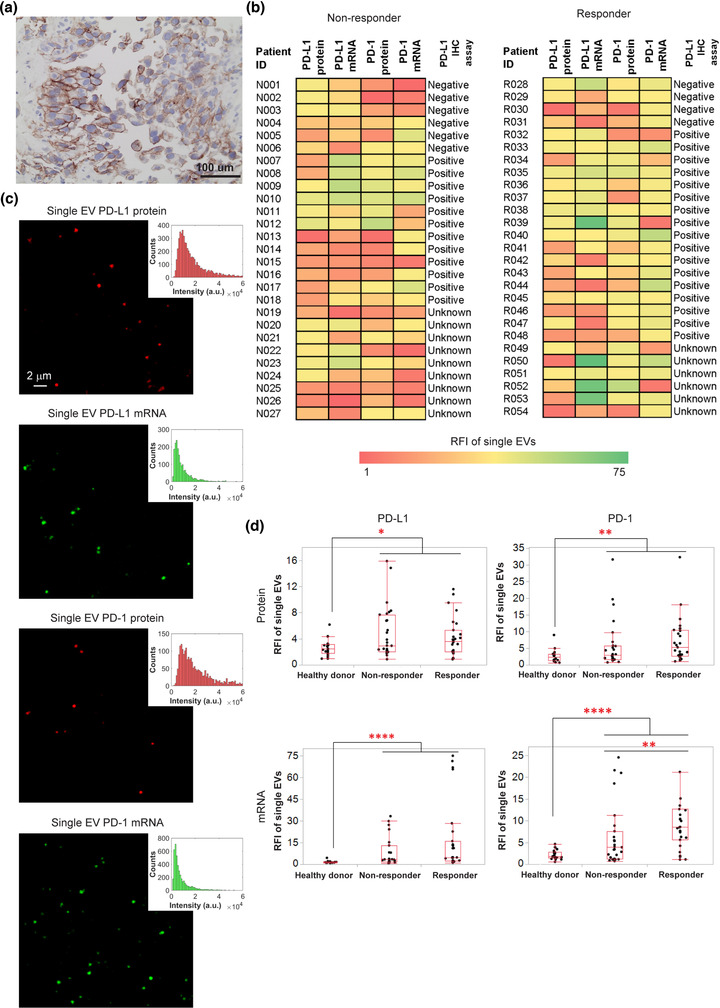
Measurements of PD‐1/PD‐L1 protein and mRNA biomarkers levels in single EVs from non‐small cell lung cancer (NSCLC) patient serum with AuSERP. (a) Representative IHC staining image of PD‐L1 protein in tissue biopsies. PD‐L1 protein was stained brown (by Dako PD‐L1 IHC 22C3 pharmDx), and cell nuclei were stained blue (by haematoxylin). (b) Heatmap of single‐EV PD‐1/PD‐L1 protein and mRNA expression levels measured with AuSERP compared to the corresponding IHC score for PD‐L1 expression in patient tissue (negative, positive, or unknown; detailed patient information is provided in Table S2). (c) Representative TIRF microscopy images and their corresponding histograms of single‐EV PD‐1/PD‐L1 protein and mRNA biomarkers characterized with AuSERP. The images were cropped and enlarged from their original images, which are provided in Figure S11. (d) Box plots of quantitative fluorescence intensities of PD‐1/PD‐L1 protein and mRNA expression levels (*P < 0.05, **P < 0.01, ****P < 0.0001, Mann‐Whitney U test). RFI, relative fluorescence intensity; a.u., arbitrary units. 54 patients were evaluated (27 responders and 27 non‐responders), along with 20 healthy donors
EVs were purified from patient serum samples for the characterization of single‐EV PD‐1/PD‐L1 protein and mRNA biomarkers with AuSERP. Using 20 μl of purified serum, all four biomarkers in the single EVs of the NSCLC patients were successfully detected with PD‐1/PD‐L1 antibodies and CLN‐MBs. Representative TIRF microscopy images and histograms of the PD‐1/PD‐L1 protein and mRNA fluorescence signals at the single‐EV level are shown in Figure 4(d) and Figure S11. Single‐EV PD‐1/PD‐L1 protein and mRNA expression levels of the NSCLC patients, in comparison with those of healthy donors, were quantified using the custom‐built MATLAB algorithm (Figure 4d). To indicate that the low abundance biomarkers detected by AuSERP are not solely derived from large vesicles that contain multiple copy numbers, we identified the presence of sEVs < 50 nm in purified serum via dynamic light scattering, which detects past the LOD of TRPS that indicated a mode of sEVs at 75.5 nm (Figure S12a‐b). To further prove AuSERP can detect these sEVs, Moloney murine leukemia virus, which is highly homogenous at ∼120 nm (Yeager et al., 1998), was detected with AuSERP for the V5 epitope (Figure S12c‐d) thereby indicating the capability of AuSERP to detect small vesicles with few copy numbers. Furthermore, the reproducibility of the assay at detecting PD‐L1+/PD‐1+ EVs in serum was demonstrated on a small cohort of three healthy donors, three non‐responders, and three responders for the four biomarkers on four separate AuSERP biochips (Figure S13). The capability of AuSERP to reproducibly detect single‐EV protein and mRNA signals at such a small volume of serum suggests its potential applications as a non‐invasive and sensitive approach to diagnose cancer patients and predict immunotherapy responses.
2.6. AuSERP enables single‐EV characterization in subpopulations
AuSERP offers excellent flexibility in capturing and sorting EVs into subpopulations based on their membrane protein compositions by incorporating the corresponding capture antibodies. Therefore, we wanted to know which EV subpopulation could provide the best predictions utilizing single‐EV PD‐1/PD‐L1 protein and mRNA detection on patient samples as EVs are highly heterogeneous and may not uniformly express CD63 and CD9. Given that PD‐L1 can be highly expressed in tumour cells, a subpopulation of tumour‐associated EVs was captured using anti‐EGFR/EpCAM antibodies, which were shown to be efficient in capturing lung circulating tumour cells (CTCs) (Reategui et al., 2015; Rima et al., 2020). Anti‐CD63/CD9 antibodies were also used to sort out the CD63+/CD9+ EV subpopulation. These two EV subpopulations derived from a small cohort of healthy donors, non‐responders, and responders were examined for single‐EV PD‐L1 protein and mRNA signals. The CD63+/CD9+ EV subpopulation provided stronger PD‐L1 signals for mRNA (P < 0.05 for mRNA, P > 0.05 for protein; Figure S14a). We also found high levels of PD‐L1 protein expression in the cytoplasm of H1568 cells colocalizing with CD63 proteins (Figure S14b). In addition to tumour cells, PD‐L1 is known to be robustly upregulated on tumour‐infiltrating immune cells (e.g., macrophages, dendritic cells, and T cells) (Diskin et al., 2020; Herbst et al., 2014). PD‐L1 expression on these immune cells has pleiotropic effects on innate and adaptive immune tolerance in cancer. The simultaneous detection of PD‐1 and PD‐L1 on single EVs from NSCLC patient serum indicates the presence of EV PD‐L1 derived from non‐tumour cells (Figure S14c). The colocalization further elucidates the greater efficacy of PD‐L1 protein and mRNA detection in single EVs captured by anti‐CD63/CD9 antibodies, since this cocktail captures EVs from all cellular sources, whereas anti‐EGFR/EpCAM antibodies only capture EVs from tumour cells.
Given that PD‐1 is highly expressed in activated T cells, T cell EV subpopulations were captured using either anti‐CD3, anti‐CD4, anti‐CD8, or anti‐CD4/CD8 antibodies. With a small cohort of healthy donors, non‐responders, and responders, we compared the single‐EV PD‐1 protein and mRNA signals. The CD63+/CD9+ EV subpopulation provided more consistent PD‐1 signals than the T cell‐specific subpopulation (P < 0.0005 for CD63+/CD9+, P > 0.05 for T‐cell specific; Figure S15). In addition to activated T cells, PD‐1 can be expressed on natural killer cells, T cells, B cells, dendritic cells, and activated monocytes (Keir et al., 2008). Our results suggest that anti‐CD63/CD9 antibodies that capture CD63+/CD9+ EVs from all cellular sources are preferable for single‐EV PD‐1/PD‐L1 protein and mRNA characterization. Therefore, we used anti‐CD63/CD9 antibodies to capture and sort EVs to detect all four biomarkers in single EVs for all NSCLC patient samples.
2.7. AuSERP offers an excellent single‐EV characterization assay to diagnose NSCLC
We asked whether any single‐EV single biomarkers (PD‐1/PD‐L1 proteins and mRNAs) could be good predictors for NSCLC diagnosis with AuSERP. To address this question, EVs purified from serum samples of the 54 NSCLC patients (27 non‐responders and 27 responders) and 20 healthy donors were characterized for all four biomarkers with AuSERP. PD‐1/PD‐L1 protein and mRNA levels in patient samples (including responders and non‐responders) were significantly higher than those of healthy donors (P < 0.05 for PD‐L1 protein, P < 0.005 for PD‐1 protein, P < 0.0001 for PD‐1 mRNA and P < 0.00001 for PD‐L1 mRNA; Figure 4d). Despite the minute levels of PD‐1/PD‐L1 protein and mRNA present in healthy donor EVs, indicative of inflammatory events outside the NSCLC microenvironment consistent with previous investigations (Chen et al., 2018; Ricklefs et al., 2018; Wang et al., 2020), the differences in expression measured by AuSERP demonstrate its potential to diagnose NSCLC. Recent efforts in characterizing circulating EV PD‐L1 protein have also shown that PD‐L1 protein is a good biomarker to distinguish between cancer patients and healthy donors and can even determine the stage in cancer progression (Chen et al., 2018; Huang et al., 2020; Pang et al., 2020). A receiver operating characteristic (ROC) curve analysis (Figure 5a and Figure S16a) demonstrated that compared to single‐EV PD‐L1 protein (area under the curve (AUC) = 0.669), the single‐EV PD‐L1 mRNA biomarker exhibited considerably higher accuracy in differentiation between NSCLC patients and healthy donors (AUC = 0.838), suggesting the single‐EV PD‐L1 mRNA as a better biomarker for NSCLC diagnosis. An AUC of 0.7–0.8 is considered acceptable in diagnostic test assessment, while 0.8–0.9 is excellent, and more than 0.9 is outstanding (Mandrekar, 2010). The single‐EV PD‐L1 mRNA is, therefore, an excellent biomarker for NSCLC diagnosis.
FIGURE 5.
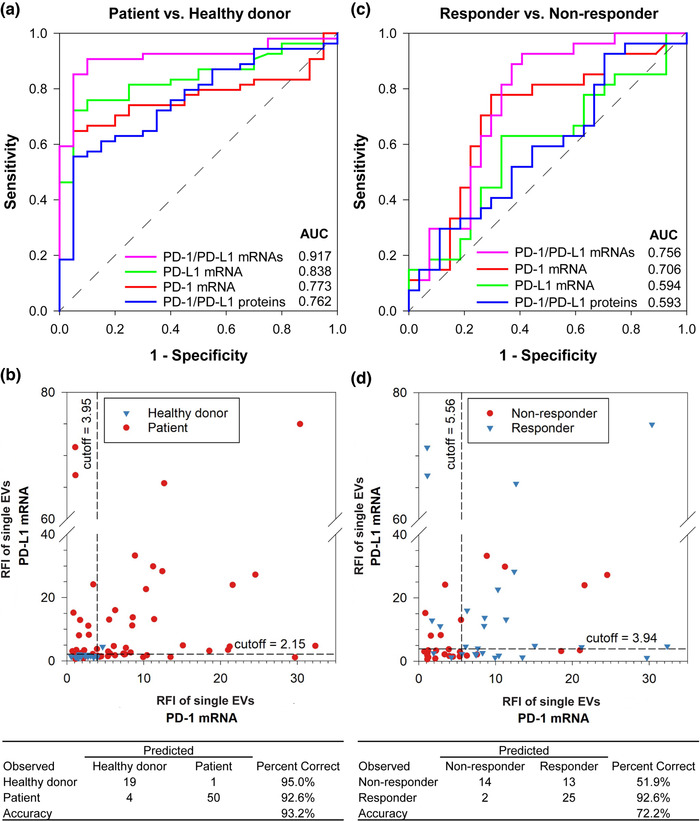
NSCLC diagnosis and prediction of immunotherapy response with AuSERP. (a) Receiver operating characteristic (ROC) curves for NSCLC diagnosis based on single‐EV analysis. (b) Scatter plot of fluorescence intensities of single‐EV dual PD‐1/PD‐L1 mRNA biomarkers. Optimal cut‐off values for NSCLC diagnosis based on single‐EV PD‐1/PD‐L1 mRNA signals were obtained from the ROC curves in Figure 5(a). An individual is diagnosed with NSCLC if either the single‐EV PD‐1 mRNA signal is larger than 3.95 or if the single‐EV PD‐L1 mRNA signal is larger than 2.15, lending a diagnostic accuracy of 93.2%. (c) ROC curves for predictions of NSCLC patient responses to anti‐PD‐1/PD‐L1 immunotherapy based on single‐EV analysis. (d) Scatter plot of fluorescence intensities of single‐EV dual PD‐1/PD‐L1 mRNA biomarkers. Optimal cut‐off values for immunotherapy prediction based on single‐EV PD‐1/PD‐L1 mRNA signals were obtained from the ROC curves in Figure 5(c). A patient is predicted as a responder if either the single‐EV PD‐1 mRNA signal is larger than 5.56 or if the single‐EV PD‐L1 mRNA signal is larger than 3.94, lending a prediction accuracy of 72.2%. 54 patients were evaluated (27 responders and 27 non‐responders), along with 20 healthy donors
Given the robustness of AuSERP and its potential to co‐quantify various single‐EV protein and mRNA biomarkers, we hypothesized that a combination of the single‐EV biomarkers (PD‐1/PD‐L1 proteins and mRNAs) would provide higher accuracy for NSCLC diagnosis. To test this hypothesis, a ROC curve analysis of different biomarker combinations was performed. Combining dual single‐EV PD‐1/PD‐L1 mRNA biomarkers further increased the AUC to 0.917 (Figure 5a), an outstanding level for cancer diagnosis. The data collected through ROC curve analyses were employed to determine cut‐offs for the single‐EV PD‐1 mRNA signal at 3.95 or the single‐EV PD‐L1 mRNA signal at 2.15 (Figure 5b). The accuracy of AuSERP for this diagnosis was as high as 93.2%. This suggests the advantage of single‐EV multi‐biomarker analysis with AuSERP. Therefore, we applied this multiplexed approach to predict patient responses to immunotherapy.
2.8. AuSERP single‐EV dual PD‐1/PD‐L1 mRNA biomarkers analysis outperforms PD‐L1 IHC at predicting responses to anti‐PD‐1/PD‐L1 immunotherapy
To evaluate the performance of AuSERP in predicting a patient's response to immunotherapy, we compared single‐EV PD‐1/PD‐L1 protein and mRNA signals from 54 NSCLC patients with their PD‐L1 IHC assay results (Figure 4b). Some responders demonstrated a positive PD‐L1 IHC result and high single‐EV PD‐1/PDL1 protein and mRNA signals (e.g., patient R033, patient R035). Interestingly, for a responder with a negative PD‐L1 IHC result (e.g., patient R028), the patient's single‐EV characterization with our platform indicated high PD‐1/PD‐L1 protein and mRNA signals. Furthermore, for a non‐responder with a positive PD‐L1 IHC score (e.g., patient N015), AuSERP showed extremely low single‐EV PD‐1/PD‐L1 protein and mRNA signals. These results suggest the potential of single‐EV PD‐1/PD‐L1 proteins and mRNAs as better predictive biomarkers in comparison to PD‐L1 IHC to anticipate patient responses to anti‐PD‐1/PD‐L1 treatment.
For single‐EV single biomarker analysis, there was no significant difference in PD‐L1 protein and mRNA and PD‐1 protein signals between responders and non‐responders (P > 0.05, Figure 4d). There were also no significant differences between the size distributions or concentrations of EVs in serum samples of responders and non‐responders (P > 0.05, Figure S16b‐c). Single‐EV PD‐1 mRNA was the only biomarker with significantly higher levels in responders than in non‐responders (P < 0.01, Figure 4d). A ROC curve analysis also demonstrated that single‐EV PD‐1 mRNA had the highest AUC value (AUC = 0.706) among other single‐EV biomarkers (Figure 5c and Figure S16a).
Interestingly, we found that mRNA biomarkers performed better for both PD‐1 and PD‐L1 than protein biomarkers as indicated by their higher AUC values (Figure S16a). The combination of single‐EV dual PD‐1/PD‐L1 mRNA biomarkers further increased the AUC to 0.756, which was significantly higher than that of single‐EV dual PD‐1/PD‐L1 protein biomarkers (AUC = 0.593, P < 0.05, Figure 5c). Following the previously introduced cut‐off approach, a patient could be predicted as a responder if either the single‐EV PD‐1 mRNA signal was larger than 5.56 or if the single‐EV PD‐L1 mRNA signal was larger than 3.94 (Figure 5d). The accuracy of AuSERP at predicting patient responses to immunotherapy was 72.2%, which is substantially higher than the commonly reported accuracy of the PD‐L1 IHC assay at 20%–40% (Camidge et al., 2019). As such, single‐EV dual PD‐1/PD‐L1 mRNA biomarkers characterized with AuSERP outperformed the FDA‐approved PD‐L1 IHC assay at predicting responses to anti‐PD‐1/PD‐L1 immunotherapy.
3. DISCUSSION
NSCLC represents 80%–85% of lung cancer, the second most common cancer and the leading cause of cancer‐related death in both men and women in the U.S. (Cancer Statistics Center, American Cancer Society, 2020). Until now, the FDA has approved three ICIs targeting PD‐L1 (atezolizumab, durvalumab, and avelumab) and two ICIs targeting PD‐1 (nivolumab and pembrolizumab) for NSCLC patients. However, the objective response rates (ORR) to PD‐L1 or PD‐1 blockade remain 20%–40%, even for patients with positive PD‐L1 expression (Camidge et al., 2019). This low ORR could be explained by the biology of PD‐L1 expression within tumours, which is known to be both spatially and temporally variable. PD‐L1 staining by IHC in one section at a given time does not reflect this variability and is referred to as a sampling error. Additionally, it is challenging to standardize PD‐L1 IHC testing due to multiple scoring criteria and the inevitable subjective interpretation of pathologist scoring (Laufer et al., 2017). Furthermore, using a single PD‐L1 protein biomarker might not be sufficient to predict a reliable response to immunotherapy (Camidge et al., 2019). EVs shed from primary and metastatic tumours that circulate in the bloodstream (Halvaei et al., 2018) can better represent the tumour burden. Present in a non‐invasive source, EVs can be readily collected at many time points before and after treatment to predict and surveil responses to immunotherapy. With our approach, single‐EV protein and mRNA signals, shown as bright spots in TIRF microscopy images, can be accurately quantified using a custom‐built algorithm that calculates the net fluorescence of every single EV, allowing for high‐throughput analyses of biomarker levels. Therefore, issues of standardization and subjectivity can be avoided with AuSERP. Moreover, AuSERP offers a unique approach to quantify mRNA and protein biomarkers for a better cancer prognosis on immunotherapy.
PD‐L1 and PD‐1 are cellular transmembrane proteins secreted as EV proteins or soluble proteins (Daassi et al., 2020; Zhu & Lang, 2017). In patients with cancer, extracellular PD‐L1 protein may serve as an agent of immune suppression by inhibiting T cell activation and/or counteracting biomolecules for immune activation (Chen et al., 2018; Daassi et al., 2020; Poggio et al., 2019). Meanwhile, extracellular PD‐1 protein plays an adjuvant role in enhancing antigen‐specific T cell immunity responses (Sorensen et al., 2016; Zhu & Lang, 2017). Previous studies have shown that PD‐L1 proteins are present on EV surfaces isolated from plasma/sera of patients with metastatic melanoma (Chen et al., 2018), head and neck squamous cell carcinoma (Theodoraki et al., 2018), glioma (Cumba Garcia et al., 2019), and NSCLC (Pang et al., 2020; Wu et al., 2019). In addition to PD‐L1 proteins, PD‐L1 mRNAs have been demonstrated to exist in EVs derived from saliva and plasma of patients with periodontitis and melanoma/NSCLC, respectively (Del Re et al., 2018; Yu et al., 2019). Compared to EV PD‐L1 protein and mRNA, studies on EV PD‐1 protein and mRNA are limited (Theodoraki et al., 2018). Furthermore, most of these studies used western blot, magnetic bead‐based flow cytometry, RT‐PCR, or ddPCR to measure EV protein and mRNA, which are bulk‐analysis methods with limited sensitivity in capturing the heterogeneity present in EVs. The technology we developed offers a highly sensitive method to quantify molecular contents in EVs at the single‐EV level by combining AuSERP with TIRF microscopy to simultaneously measure PD‐1/PD‐L1 protein and mRNA contents on the same device.
By combining PEG, BSA, and Tween‐20 to block non‐specific bindings, TIRF microscopy for high‐resolution imaging, a thin gold coating and gold NPs for SPR signal enhancement, and amplification methods for detection by antibodies (using TSA) and MBs (using CLNs), we engineered a highly sensitive platform. The presence of 30‐nm gold NPs significantly enhanced the sensitivity of single‐EV analyses with AuSERP by providing localized SPR. Spherical gold NPs have been widely used to amplify the signal and improve the sensitivity of SPR biosensors (Bedford et al., 2012; Mitchell et al., 2005; Springer et al., 2014). AuSERP exhibited a significantly high SNR of 482.02 ± 21.81, which enabled us to accurately quantify low expression biomarkers such as PD‐1/PD‐L1 proteins and mRNAs at a single‐EV level. While AuSERP cannot determine the specific copy number of proteins or RNAs in single EVs due to diffraction limitations, the assay instead measures the total fluorescence signals from proteins or RNAs present in single EVs. Whether these signals are derived from a few (∼ less than 10) copies or an abundance of molecules present in a single EV is a constraint of the assay. However, by quantifying the total fluorescence of single EVs, our platform was ∼ 1000 times more sensitive than the gold‐standard commercial methods to detect PD‐L1 protein and mRNA in bulk, ELISA and qRT‐PCR, respectively, indicating an incentive to conduct single‐vesicle analyses on non‐lysed EVs. Lysing EVs with heterogeneous expressions of protein and mRNA leads to the dilution of molecules‐of‐interest from high‐expressing EVs into the vast proteomic and transcriptomic landscape of low‐expressing EVs and EVs derived outside the NSCLC microenvironment. On the other hand, maintaining EV integrity via immobilization and detecting proteins and mRNA on individual EVs by immunofluorescence produces a focusing effect for the high‐expressing EVs whereby the molecules‐of‐interest are accentuated (Wu et al., 2013). Circumventing the artefact of dilution observed in the bulk characterization of EVs, AuSERP emphasized PD‐L1/PD‐1 events from single EVs derived from the NSCLC immunological landscape as bright spots, while minimizing signals from peripheral molecules or other inflammatory events. With such high sensitivity, our technology only required 20 μl of purified serum for protein and mRNA biomarker characterization. AuSERP is also capable of high‐throughput analyses with up to 256 individual samples per assay. In addition to being a sensitive and high‐throughput assay, AuSERP enabled multiplex detection of different protein and mRNA biomarkers simultaneously. Moreover, our platform offered great flexibility in capturing and sorting EVs into subpopulations to study the function of EV subpopulations. Both the subpopulation‐based sorting and multiplexed detection of protein and mRNA afforded by AuSERP exemplify the importance of detecting EVs at a single‐vesicle resolution to reveal EV heterogeneity.
EV PD‐L1 protein was previously reported as a potential predictive biomarker to distinguish patients with metastatic melanoma who respond to anti‐PD‐1 therapy (Chen et al., 2018). Elevated levels of pre‐treatment EV PD‐L1 following a ceased increase in on‐treatment PD‐L1 levels were observed in non‐responders, which may reflect the exhaustion of T cells. Meanwhile, responders displayed a larger increase in EV PD‐L1 levels as early as 3–6 weeks following the initial treatment, which correlates positively with T cell reinvigoration (Chen et al., 2018). However, for NSCLC, identifying responders from non‐responders using liquid biopsies is still very challenging (Daassi et al., 2020). With AuSERP, we successfully demonstrated a comprehensive profile of four immunotherapy biomarkers (PD‐1/PD‐L1 proteins and mRNAs) at a single‐EV resolution from EVs isolated from the serum of NSCLC patients. Using anti‐CD9 and anti‐CD63 antibodies to capture EVs after sized‐based separation with TFF, AuSERP characterized pure EVs without contaminating proteins or LPPs that are present in complex biofluids such as serum. A cohort of 27 non‐responders and 27 responders was examined in our study. Interestingly, a ROC curve analysis of four single‐EV biomarkers suggests that single‐EV mRNAs are better than single‐EV proteins for both cancer diagnosis and immunotherapy response predictions. We have a multifaceted hypothesis for the enhanced sensitivity mRNA provides. First, sEVs, which tend to be rich in CD63 and CD9 (Kowal et al., 2016), preferentially load fragmented mRNA in comparison to protein during EV biogenesis. Innately, sEVs contain more mRNA (Dong et al., 2016) and less protein (Kowal et al., 2016) than m/lEVs. Furthermore, CD63+/CD9+ sEVs load more mRNA than CD63–/CD9– m/lEVs via nanoporation (Yang et al., 2020); whereas transfected reporter proteins are loaded at higher efficiencies for CD63– m/lEVs (Kanada et al., 2015). Therefore, AuSERP, which captures CD63+/CD9+ EVs, may be sorting an mRNA‐rich/protein‐depleted sEV‐subpopulation. Second, the expression of cellular immune‐checkpoint proteins is spatiotemporally heterogeneous whereby the expression levels and efficacy of ICI vary depending on the pathway of T‐cell exhaustion, reinvigoration, and location (Blackburn et al., 2008; Budimir et al., 2022; Gueguen et al., 2021; Thommen et al., 2018); whereas, the indirect functional units simplify the complexities at the membrane surface as levels of cellular activation (Paré et al., 2018) that are, in turn, reflected in EVs (Figure S17). Third, EVs protect mRNAs within their phospholipid membrane from degradation in vivo, while EV surface proteins are exposed to protease degradation (Cheng et al., 2014; Kim et al., 2017; Skog et al., 2008). As such, we offer motivation to further investigate EV mRNA cargo as an alternative biomarker to enhance assay sensitivity and to determine its function in the immunosuppressive cascade. AuSERP offers a robust and highly sensitive approach to characterize single‐EV mRNA biomarkers in which single EVs are captured and directly detected using CLN‐MBs without the need for tedious RNA extraction, cDNA reverse transcription, and qRT‐PCR. Furthermore, while qRT‐PCR can detect very few copy numbers in high‐quality RNA, EV mRNA cargo tends to be fragmented (Wei et al., 2017). CLNs containing MB, which are short oligonucleotides that target sections of the mRNA strand, are less perturbed by fragmentation and thus lend higher sensitivity than qRT‐PCR. The dual single‐EV PD‐1/PD‐L1 mRNA biomarkers achieved AUC values of 0.917 and 0.756 to distinguish patients from healthy donors and responders from non‐responders. The accuracy of AuSERP to predict patient responses to immunotherapy was 72.2%, which exceeded the FDA‐approved PD‐L1 IHC assay. Our study, therefore, showed that pre‐treatment single‐EV dual PD‐1/PD‐L1 mRNA are good predictors to identify NSCLC patients that will benefit from anti‐PD‐1/PD‐L1 immunotherapy. Given the heterogeneity and dynamic changes of PD‐1/PD‐L1 expression in tumours and the invasive nature of tumour biopsies, developing a non‐invasive single‐EV assay with AuSERP is an attractive alternative to IHC scoring. Longitudinal assays before and shortly after the administration of ICIs would be an exciting avenue to explore to improve prediction accuracy.
A patient‐oriented approach to immunotherapy using predictive biomarkers is desired to maximize clinical benefit, improve cost‐effectiveness, and reduce the economic burden of the disease. A previous study showed that compared to treating all NSCLC patients with anti‐PD‐1/PD‐L1 immunotherapy, the selection of patients based on positive PD‐L1 IHC scores improved incremental quality‐adjusted life years by up to 183% and decreased the incremental cost‐effectiveness ratio by up to 65% (Aguiar et al., 2017). However, due to its limited precision, there are scenarios where patients with tumours scored as PD‐L1‐positive do not respond to anti‐PD‐1/PD‐L1 therapy (false‐positive testing) and vice versa (Camidge et al., 2019). In the case of false‐negative testing, it could diminish the number of potential life‐years saved. The success of this work in offering a more accurate prediction of NSCLC patient responses can, therefore, improve the survival of patients and minimize the cost of treatment. Furthermore, the success of AuSERP is a breakthrough in cancer therapy in which personalized cancer immunotherapy can be achieved by feasibly identifying patients most likely to benefit from immunotherapy and monitoring their responses throughout their treatment. In principle, our AuSERP technology applies to a broad spectrum of biomedical applications (e.g., early cancer diagnosis, neurodegenerative diseases such as Alzheimer's disease, traumatic brain injury, viral diseases, and cardiovascular diseases), in which any combination of antibodies and MBs could be used to detect disease‐specific proteins and RNAs of interest in specific membrane‐enveloped subpopulations of EVs.
4. MATERIALS AND METHODS
4.1. AuSERP fabrication
A high precision glass coverslip (#D 263 M Glass, 24 × 75 mm rectangle, 0.15 mm thickness Schott AG, Mainz, Germany) was cleaned with deionized (DI) water and ethanol two times alternately in an ultrasonic bath for 5 min each and then dried with nitrogen gas. Subsequently, the cleaned glass coverslip was activated using a UV‐ozone cleaner (UVO Cleaner Model 42, Jelight, Irvine, CA, USA) for 15 min. Thin layers of 2‐nm‐thick Ti and 10‐nm‐thick Au were then deposited, respectively, using a Denton e‐beam evaporator (DV‐502A, Moorestown, NJ, USA). After Au deposition, the freshly prepared Au‐coated glass was immersed into a linker solution containing β‐mercaptoethanol (βME, Sigma‐Aldrich, St. Louis, MO, USA), PEG‐SH (#MPEG‐SH‐2000, Laysan Bio Arab, AL, USA), and biotin‐PEG‐SH (#PG2‐BNTH‐2k, Nanocs, New York, NY, USA) (molar ratio = 95:3:2) in 200 proof ethanol (Fisher Scientific, Hampton, NH, USA) for overnight in the dark at 22°C. The glass coverslip was then rinsed with ethanol to remove any excess mixtures and subsequently air‐dried. The treated glass was attached to a 64‐well gasket (Grace Bio‐Labs ProPlate tray set, Sigma‐Aldrich), which has a working volume of 20 μl/well. Therefore, all incubation steps hereafter were performed with a volume of 20 μl/well. The wells were washed thoroughly with DI water. Next, 0.005% (w/v) streptavidin‐conjugated gold nanoparticles (NPs, Nanocs) in PBS were introduced into the wells for 2 h at room temperature (RT) on a rocker (24 rpm, Benchmark Scientific, Sayreville, NJ, USA). Different sized NPs (5, 30, and 50 nm) were used to test the EV capture efficacy and non‐specific binding of antibodies. After rinsing three times with PBS, the surface was incubated with a capture antibody cocktail for 1 h at RT on the rocker. The cocktail included 10 μg/ml each of a mouse anti‐CD63 monoclonal antibody (#MAB5048, R&D Systems, Minneapolis, MN, USA) and a mouse anti‐CD9 monoclonal antibody (#MAB1880, R&D Systems). These antibodies were biotinylated using an EZ‐Link micro Sulfo‐NHS‐biotinylation kit (ThermoFisher Scientific, Waltham, MA, USA) before the incubation. After the antibodies were tethered onto the gold surface, the free antibodies were washed away three times with PBS, and then blocked with 3% (w/v) BSA (Sigma‐Aldrich) and 0.05% (v/v) Tween 20 (Sigma‐Aldrich) in PBS for 1 h at RT before EV capture.
4.2. Atomic force microscopy (AFM)
The topography of the surfaces coated with different‐sized streptavidin‐conjugated gold NPs was characterized using an AFM (Asylum Research MFP‐3D‐BIO AFM, Oxford Instruments, Abingdon, United Kingdom). Before imaging, the surfaces were rinsed thoroughly with DI water to avoid salt crystals and air‐dried.
4.3. Cell culture
H1568 cells (NCI‐H1568, ATCC CRL‐5876, Manassas, VA, USA) were cultured in a growth medium containing RPMI 1640 (ThermoFisher Scientific), 10% (v/v) fetal bovine serum (FBS, Sigma‐Aldrich), and 1% (v/v) penicillin‐streptomycin (PS, ThermoFisher Scientific). Mouse Embryonic Fibroblast (MEF; CF‐1, ATCC SCRC‐1040) were cultured in a growth medium containing Dulbecco's Modified Eagle Medium (DMEM, ThermoFisher Scientific), 10% (v/v) FBS and 1% (v/v) PS. U251 cells (U251‐MG, ECACC 09063001) were cultured in a growth medium containing RPMI, 10% (v/v) FBS and 1% (v/v) PS. The medium was replaced every 2 to 3 days, and cultures were maintained in a humidified incubator at 37°C with 5% CO2. When the cells reached 80%–90% confluence, they were detached using TrypLE express enzyme (ThermoFisher Scientific) and passaged at 1:3–1:6 ratios. H1568 cells at passages 6–10, MEF at passages 2 – 5, and U251 at passages 6 – 8 were used in this study.
To stimulate PD‐L1 expression, H1568 cells were first grown to 80% confluency in the growth medium in a cell culture flask (Corning Inc., Corning, NY, USA). The cells were then washed with PBS and changed to an RPMI medium supplemented with 10% (v/v) EV‐depleted FBS, 1% (v/v) PS, and 100 ng/ml recombinant human IFN‐γ (Peprotech, Rocky Hill, NJ, USA) for 48 hr. The cells cultured in the medium without IFN‐γ supplements were used as controls. EV‐depleted FBS was collected from the filtrate from FBS introduced through the TFF system with a 500 kDa MWCO hollow fiber filter (polysulfone, Repligen, Waltham, MA, USA). After 48 h, the culture supernatant was centrifuged at 2000 × g for 10 min (Centrifuge 5810R, Eppendorf, Hauppauge, NY, USA) to remove cell debris before EV purification. On the other hand, the adherent cells in the flasks were detached and then pelleted by centrifugation at 1000 rpm for 5 min. The cell pellets were used to characterize the PD‐L1 protein and mRNA in the cells using IHC staining, ELISA, and qRT‐PCR as described in the following sections.
4.4. EV purification from cell culture supernatants and clinical samples
10 ml of healthy donor and NSCLC patient blood samples were obtained with appropriate informed consent under approved Institutional Review Board (IRB) protocols #2018H0268 and #2015C0157, respectively, at The Ohio State University. The study was carried out following relevant guidelines and regulations. Blood samples from stage IV NSCLC patients were collected before they underwent immunotherapy. Serum was separated from blood using a BD Vacutainer Serum Separation Tube (SST, #367985, Becton Dickinson) according to the manufacturer's protocol.
Cell supernatants and sera were first filtered through 1‐μm filters (GE Healthcare Whatman Puradisc GMF, Fisher Scientific). 100 ml of cell supernatant or 150 μl of sera diluted into 7 ml of PBS were subsequently concentrated into 5 ml and diafiltrated using TFF with a 500 kDa filter for purification at a flow rate of 35 ml/min as previously described (Zhang et al., 2021). After TFF, the retentates with 99% of contaminants removed were concentrated using centrifugal units (10 kDa MWCO, MilliporeSigma Amicon Ultra Centrifugal Filter Unit, Fisher Scientific) at 3000 × g for 20 min to a final volume of 1 ml. The concentration and size distributions of EVs were quantified using a tunable resistive pulse sensing (TRPS) technology (qNano Gold instrument, Izon Science, Medford, MA, USA) with NP80 (target size range 40–255 nm), NP150 (target size range 70–420 nm), and NP600 (target size range 275–1570 nm) nanopore membranes.
4.5. CD63 detection of EVs
EVs harvested from H1568 cells without IFN‐γ stimulation were adjusted to a concentration of 1010 particles/ml. After that, the purified EVs were added onto AuSERP biochips coated with different NP sizes. PBS was used as a blank control. The following incubation and washing steps were performed at RT on a rocker. EVs were captured for 2 h, washed three times with PBS, and then blocked with 3% (w/v) BSA and 0.05% (v/v) Tween 20 in PBS for 1 h. The samples were subsequently incubated with a mouse anti‐CD63 monoclonal antibody (MX‐49.129.5) – Alexa Fluor 488 conjugate (#sc‐5275 AF488, Santa Cruz Biotechnology) at a dilution of 1:200 in 1% (w/v) BSA in PBS for 1 h. Next, the samples were rinsed three times with 0.05% (v/v) Tween 20 in PBS. Images were taken using a TIRF microscope (Nikon Eclipse Ti Inverted Microscope System). The images were recorded by an Andor iXon EMCCD camera with a 100x oil lens at the same laser power and exposure time. For each sample, 100 images (10 × 10 array) were collected.
4.6. PD‐L1 protein staining of cells and tissues
The cell pellets were fixed in a 4% formaldehyde solution in PBS (Fisher Scientific) for 2 h at RT and then washed with PBS by centrifugation. The pellets were subsequently blocked in agarose gel and embedded in paraffin. They were then sectioned into 5‐μm thick slices, stained for PD‐L1 protein, and then counterstained with haematoxylin using an automated Dako PD‐L1 IHC 22C3 pharmDx platform (Agilent Technologies, Santa Clara, CA, USA). The images were taken using a light microscope (CKX41 Inverted Microscope, Olympus, Tokyo, Japan).
Tissue biopsies were obtained with informed consent from NSCLC patients using the approved IRB protocol #2015C0157 at The Ohio State University. Tissue and blood samples were collected from each patient before starting immunotherapy. Classification of responders or non‐responders to immunotherapy and PD‐L1 IHC results were obtained from medical reports. PD‐L1 IHC of the tissue biopsies were performed using the automated Dako PD‐L1 IHC 22C3 pharmDx platform.
4.7. PD‐1/PD‐L1 protein detection of EVs
The purified EVs from H1568 cells (with and without IFN‐γ stimulation) and blood samples (healthy donors and NSCLC patients) were captured with AuSERP. PBS was used as a blank control, and a washing buffer. All incubation and washing steps were conducted at RT on the rocker. After capture, the samples were rinsed three times and stained for PD‐1/PD‐L1 proteins using an Alexa Fluor 647 Tyramide SuperBoost kit (#B40926, ThermoFisher Scientific). Firstly, the EVs were fixed with 4% formaldehyde solution in PBS for 10 min. After washing, a 3% hydrogen peroxide solution was added to quench the endogenous peroxidase activity of the samples for 15 min, followed by incubation with 3% (w/v) BSA and 0.05% (v/v) Tween 20 in PBS for 1 h. Rabbit anti‐PD‐L1 monoclonal antibody (#86744S, Cell Signalling Technology, Danvers, MA, USA) or rabbit anti‐PD‐1 monoclonal antibody (#86163S, Cell Signalling Technology) was then diluted 500‐fold in a blocking buffer and incubated for 1 h. Next, the samples were washed three times for 10 min each before incubation with a poly‐HRP‐conjugated secondary antibody for 1 h. After washing three times for 10 min each, a tyramide working solution was applied for 10 min. The reaction was stopped using a stop reagent. After that, the samples were rinsed three times and imaged using the TIRF microscope as mentioned above. The list of antibodies utilized in the study is provided in Table S5.
4.8. Design and fabrication of CLN‐MBs
MBs (listed 5′–3′) targeting PD‐1 and PD‐L1 mRNAs used in this study were +GGT +CCT /iCy3/ +CCT +TCA +GGG GCT GGC GCC CCT GAA GG /BHQ_2/ and +GGT +AGC /iCy3/ +CCT +CAG +CCT GAC ATG AGG CTG AGG /BHQ_2/, respectively. They were designed based on an NCBI reference sequence of PD‐1 (NM_005018.3) and PD‐L1 (NM_014143.4) using Primer3 and BLAST (Primer‐BLAST) provided by NCBI‐NIH. Locked nucleic acid nucleotides (positive sign (+) bases) were incorporated into oligonucleotide strands to improve the thermal stability and nuclease resistance of MBs for incubation at 37°C. The designed MBs were custom synthesized and purified by Integrated DNA Technologies (IDT, Coralville, IA, USA).
To fabricate CLN‐MBs, a lipid formulation of 1,2‐dioleoyl‐3‐trimethylammonium‐propane (DOTAP, Avanti Polar Lipids, AL, USA), Cholesterol (Sigma‐Aldrich), 1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine (DOPC, Avanti Polar Lipids), and Bis(1,2‐distearoyl‐sn‐glycero‐3‐phosphoethanolamine)‐N‐[(polyethylene glycol)‐2000] (Bis‐DSPE PEG2000, Avanti Polar Lipids) was prepared at a 50:30:18:2 molar ratio at a final concentration of 10 mg/ml in 200 proof ethanol. The aqueous solution consisted of 23.2 μl of Dulbecco's PBS (DPBS), 6.0 μl of 300 μM scramble RNA (miR‐54), and 0.8 μl of 100 μM PD‐L1/PD‐1 MB. Next, 20.0 μl of the lipid formulation was added to the aqueous solution, which was vortexed vigorously for 10 s and then sonicated for 5 min using an ultrasonic bath. The MB/lipid mixture was subsequently injected into 350 μl of DPBS, vortexed for 10 s, and sonicated for 5 min. Finally, the mixture was dialyzed with a 20 kDa MWCO to remove free MBs.
4.9. mRNA staining of cells
H1568 cells were seeded at a density of 105 cells/ml in 16‐well chambers (Grace Bio‐Labs ProPlate tray set) attached to a glass slide (Fisher Scientific). To stimulate PD‐L1 expression, the cells were incubated with 100 ng/ml recombinant human IFN‐γ in the growth medium for 48 h. Cells without IFN‐γ stimulation were employed as a control. The H1568 cells were then fixed in 4% formaldehyde solution in PBS for 30 min at RT and then permeabilized with 0.2% (v/v) Triton X‐100 (Sigma‐Aldrich) in PBS for 10 min at RT. The cells were subsequently rinsed with PBS and incubated with 0.5 μM free PD‐L1 MBs for 4 h at 37°C. After washing three times with PBS for 5 min each, the glass slide was detached and mounted onto a cover glass (Fisher Scientific) using ProLong Gold Antifade Mountant with DAPI (ThermoFisher Scientific). The images were taken using a confocal microscope (Olympus FV3000).
4.10. Calibration of CLN‐MBs using artificial EVs
PD‐L1 ssDNA oligo (listed 5′–3′) used in this study was CTG ACA TGT CAG GCT GAG GGC TAC CCC AAG. The designed oligo was custom synthesized and purified by IDT. To fabricate artificial EVs, an aqueous solution of the PD‐L1 ssDNA oligo was mixed with a lipid formulation of 1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine (DOPE, Avanti Polar Lipids), linoleic acid (LA, Sigma‐Aldrich), and 1,2‐dimyristoyl‐rac‐glycero‐3‐methoxypolyethylene glycol‐2000 (DMG‐PEG 2000, Avanti Polar Lipids) (50:48:2 mole ratio in 200 proof ethanol). The mixture was subsequently sonicated for 5 min, injected into PBS, and then sonicated for another 5 min. After dialysis with a 20 kDa MWCO to remove free molecules, the suspension was spiked into healthy donor EVs at different concentrations of 0.625, 1.25, 2.5, 5, and 10%.
For the calibration experiment, the gold‐coated biochip was tethered with biotinylated PD‐L1 CLN‐MBs, which was made by replacing Bis‐DSPE PEG2000 with DSPE‐PEG(2000) Biotin (Avanti Polar Lipids) in the lipid formulation of CLN‐MB fabrication. The artificial EVs at different concentrations were then captured on the biochip by fusion with the PD‐L1 CLN‐MBs for 2 h at 37°C. After rinsing with PBS, the samples were imaged using the TIRF microscope.
4.11. PD‐1/PD‐L1 mRNA detection of EVs
The purified EVs from serum samples of healthy donors and NSCLC patients were captured with AuSERP for 2 h at RT. After washing with PBS, 1013 particles/ml of PD‐1/PD‐L1 CLN‐MBs were applied and incubated for 2 h at 37°C. The samples were rinsed with PBS and imaged using the TIRF microscope.
4.12. Multiplex detection of protein and mRNA
The purified EVs from serum samples of healthy donors and cancer patients were captured with AuSERP and detected for protein as previously described. After washing with PBS, CLN‐MBs were applied and incubated for 2 h at 37°C. The samples were rinsed with PBS and imaged using the TIRF microscope with sequential illumination of two lasers.
4.13. Cryogenic transmission electron microscopy (cryo‐TEM)
Cryo‐TEM was used to characterize purified EVs from IFN‐γ‐stimulated H1568 cells, their PD‐L1 proteins, PD‐L1 CLN‐MBs, and complexes of CLN‐MBs fused with EVs. A concentration of 1013 EVs/ml was necessary for this experiment. The presence of PD‐L1 protein on the surface membranes of EVs was verified using immunogold labelling. Goat PD‐L1 polyclonal antibody (#AF156, R&D Systems) was firstly conjugated with gold NPs using a gold conjugation kit (#ab201808, Abcam, Cambridge, United Kingdom) according to the manufacturer's instructions, and then incubated with the EVs for 1 h at RT. PD‐L1 CLN‐MBs were incubated with the EVs for 1 h at 37°C to observe their fusion.
Cryo‐TEM samples were prepared by applying a small aliquot (3 μl) of the samples to a specimen grid. After blotting away excess liquid, the grid was immediately plunged into liquid ethane to rapidly form a thin layer of amorphous ice using a Thermo Scientific Vitrobot Mark IV system. The grid was transferred under liquid nitrogen to a Thermo Scientific Glacios CryoTEM. Images were recorded by a Thermo Scientific Falcon direct electron detector.
4.14. Scanning electron microscopy (SEM)
EVs from IFN‐γ‐stimulated H1568 cells were fused with PD‐L1 CLN‐MBs for 2 h at 37°C. EVs from IFN‐γ‐stimulated H1568 cells after CLN‐MB fusion, sole EVs from IFN‐γ‐stimulated H1568 cells, and sole EVs from U251 cells were captured onto SEM coverslips. A 2% glutaraldehyde (MilliporeSigma, Burlington, MA, USA) and 0.1 M sodium cacodylate solution (Electron Microscopy Sciences, Hatfield, PA, USA) fixation buffer was added for 3 h to fix the EVs. Then, a mixture of 1% osmium tetraoxide (Electron Microscopy Sciences) and 0.1 M sodium cacodylate was added for 2 h to increase the contrast. EV samples were later dehydrated with increasing concentrations of ethanol (50, 70, 85, 95, and 100% (v/v)) for 30 min each. Lastly, a CO2 critical point dryer (Tousimis, Rockville, MD, USA) was applied to dry the sample. A 2‐nm layer of gold was sputtered onto the sample with a sputtering machine (Leica EM ACE 600, Buffalo Grove, IL, USA). The samples were then imaged using an Apreo 2 (FEI, ThermoFisher Scientific) SEM.
4.15. Image analysis
All the spots in the TIRF microscopy images were first located with distinguished edges, and background noise was removed by a Wavelet denoising method using a custom‐built Matlab algorithm. The net fluorescence intensity of the spot was then calculated by summing the intensities of all the pixels within the spot, which were subtracted by the mean intensity of pixels surrounding the spot. Subsequently, a histogram of net fluorescence intensities of all the spots was obtained from 100 TIRF images for each sample and their TFI was calculated as the area under the histogram. All spots falling outside a threshold of 3–8 pixels, set through a user interface, were not considered in the calculation. The relative fluorescence intensity (RFI) of an EV sample was defined as the ratio of the TFIEV to the TFIneg within the same AuSERP biochip.
4.16. ELISA
PD‐L1 protein expression levels in H1568 cells (with and without IFN‐γ stimulation) and on the surface of H1568 EVs (with IFN‐γ stimulation) were quantified using a PD‐L1 Human ELISA kit (#BMS2212, ThermoFisher Scientific). For the cells, their pellets were lysed in RIPA buffer (ThermoFisher Scientific) with the addition of Thermo Scientific Halt Protease and Phosphatase Inhibitor Cocktails on ice for 5 min, and then centrifuged at 14,000 × g for 15 min to remove cell debris. Similarly, for EVs, EVs from IFN‐γ‐stimulated H1568 cells were spiked in healthy donor serum at different concentrations ranging from 0 to 1011 particles/ml. The EVs were then purified via TFF and lysed with the RIPA and protease/phosphatase inhibitor cocktail on ice for 15 min as suggested by the manufacturer. All the samples were subsequently incubated in the ELISA plate, and their PD‐L1 expressions were quantitatively detected according to the manufacturer's instructions. The PD‐L1 concentration in the cell lysis was normalized to its total protein concentration, which was measured using a Pierce Rapid Gold BCA Protein Assay kit (ThermoFisher Scientific).
4.17. qRT‐PCR
PD‐L1 mRNA levels in H1568 cells (with and without IFN‐γ stimulation) and H1568 EVs (with IFN‐γ stimulation) were quantified using qRT‐PCR. Total RNA from the cells and EVs were first isolated and purified using an RNeasy Mini Kit and a miRNeasy Serum/Plasma kit (Qiagen, Hilden, Germany), respectively, according to the manufacturer's instructions. cDNA was then synthesized from the total RNA using a High‐Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA) on a thermal cycler (Veriti 96‐Well Thermal Cycler, Applied Biosystems). Subsequently, PD‐L1 mRNA expression was quantified using a TaqMan Gene Expression assay (ThermoFisher Scientific, Assay Id: Hs01125301_m1) on a Real‐Time PCR System (Applied Biosystems). The cellular PD‐L1 mRNA expression was normalized to GAPDH (Assay Id: Hs02758991_g1), a housekeeping gene.
4.18. Statistical analysis
All in vitro experiments and assays were repeated three times. All clinical samples were measured two times. JMP Pro 14, MATLAB R2019a, Sigma Plot 14.0, and IBM SPSS Statistics 25 were used for data analysis. For in vitro samples, a significance of mean differences was determined using a two‐tailed paired Student's t‐test. For clinical samples, a non‐parametric Mann–Whitney U test was used for analysis. Error bars shown in graphical data represent mean ± SD. A p‐value below 0.05 was considered a statistically significant difference. The performance of classification schemes was evaluated using ROC curve analyses. For single biomarker analyses, ROC curves were determined from the expression level of each biomarker. For multiple biomarker analyses, ROC curves were plotted following a binary logistic regression. Optimal cut‐off points were obtained from the ROC curves using the ROC curve analysis module in Sigma Plot. The accuracy of an assay was defined as
| (2) |
where TP, TN, FP, and FN represent the number of true positives, true negatives, false positives, and false negatives, respectively.
AUTHOR CONTRIBUTIONS
Luong T. H. Nguyen, Xilal Y. Rima, Jingjing Zhang, and Eduardo Reátegui prepared the figures and wrote the manuscript with input from all authors. Xilal Y. Rima and Jingjing Zhang revised the manuscript and prepared the rebuttal letter with the input of all the authors. Luong T. H. Nguyen, Kwang Joo Kwak, L. James Lee, and Eduardo Reátegui developed the technology. Luong T. H. Nguyen, Jingjing Zhang, Xilal Y. Rima, L. James Lee, and Eduardo Reátegui designed the study and analysed the data. Luong T. H. Nguyen and Xilal Y. Rima performed statistical analyses. Luong T. H. Nguyen and Kwang Joo Kwak optimized and fabricated the AuSERP biochip. Luong T. H. Nguyen, Jingjing Zhang, and Xilal Y. Rima performed cell culture experiments. Luong T. H. Nguyen performed ELISA, qRT‐PCR, and AFM. Luong T. H. Nguyen and Jingjing Zhang performed cryo‐TEM. Jingjing Zhang and Xilal Y. Rima performed SEM. Jingjing Zhang performed flow cytometry. Chi‐Ling Chiang performed western blot. Jingjing Zhang and Min Jin Yoon performed dynamic light scattering. Luong T. H. Nguyen and Xilal Y. Rima performed viral detection. Luong T. H. Nguyen and Jingjing Zhang performed biomarker characterization. Kwang Joo Kwak designed the MBs. Kwang Joo Kwak and Xinyu Wang fabricated the CLN‐MBs. Xinyu Wang, Xilal Y. Rima, and Donald Belcher developed the custom‐built algorithm for image analysis. Luong T. H. Nguyen, Jingjing Zhang, and Min Jin Yoon purified EVs using TFF. Jingjing Zhang, Min Jin Yoon, and Xilal Y. Rima performed TRPS measurements. Nicole Walters obtained healthy donor blood and isolated T cells. Tamio Okimoto, Joseph Amann, Takehito Shukuya, and David P. Carbone provided clinical samples, PD‐L1 IHC results, and patient clinical information. Yifan Ma performed confocal fluorescence imaging and assisted with qRT‐PCR. Xilal Y. Rima, Hong Li, and Andre F. Palmer assisted with AuSERP preparation, EV purification, and manuscript writing. Eduardo Reátegui supervised the project. All authors have read and approved the final manuscript.
CONFLICT OF INTERESTS
Luong T. H. Nguyen, Kwang Joo Kwak, L. James Lee., and Eduardo Reátegui have filed a patent application for the AuSERP technology.
Supporting information
Supplementary information
ACKNOWLEDGEMENTS
We acknowledge all the patients and healthy volunteers who participated in this study. We thank Dr. Xiaokui Mo for helpful advice on the statistical tests. Electron microscopy was performed at the Center for Electron Microscopy and Analysis (CEMAS) at The Ohio State University. This work was supported by the U.S. National Institutes of Health (NIH) grants UG3/UH3 TR002884 and U18TR003807 (Eduardo Reátegui & L. James Lee), R01HL126945, R01HL138116, R01EB021926 (Andre F. Palmer), and UG1CA233259 (David P. Carbone). Additional support for Eduardo Reátegui was provided by the William G. Lowrie Department of Chemical and Biomolecular Engineering and the James Comprehensive Cancer Center at The Ohio State University. Confocal fluorescence microscopy images were generated using the instruments and services at the Campus Microscopy and Imaging Facility (CMIF), The Ohio State University. This facility is supported in part by grant P30 CA016058, National Cancer Institute, Bethesda, MD, USA.
Nguyen, L. T. H. , Zhang, J. , Rima, X. Y. , Wang, X. , Kwak, K. J. , Okimoto, T. , Amann, J. , Yoon, M. J. , Shukuya, T. , Chiang, C.‐L. , Walters, N. , Ma, Y. , Belcher, D. , Li, H. , Palmer, A. F. , Carbone, D. P. , Lee, L. J. , & Reátegui, E. (2022). An immunogold single extracellular vesicular RNA and protein (AuSERP) biochip to predict responses to immunotherapy in non‐small cell lung cancer patients. Journal of Extracellular Vesicles, 11, e12258. 10.1002/jev2.12258
Luong T. H. Nguyen, Jingjing Zhang and Xilal Y. Rima contributed equally to this work.
DATA AVAILABILITY STATEMENT
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
REFERENCES
- Abiko, K. , Matsumura, N. , Hamanishi, J. , Horikawa, N. , Murakami, R. , Yamaguchi, K. , Yoshioka, Y. , Baba, T. , Konishi, I. , & Mandai, M. (2015). IFN‐gamma from lymphocytes induces PD‐L1 expression and promotes progression of ovarian cancer. British Journal of Cancer, 112, 1501–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguiar, P. N. Jr. , Perry, L. A. , Penny‐Dimri, J. , Babiker, H. , Tadokoro, H. , de Mello, R. A. , & Lopes, G. L. Jr. (2017). The effect of PD‐L1 testing on the cost‐effectiveness and economic impact of immune checkpoint inhibitors for the second‐line treatment of NSCLC. Annals of Oncology, 28, 2256–2263. [DOI] [PubMed] [Google Scholar]
- Ancevski Hunter, K. , Socinski, M. A. , & Villaruz, L. C. (2018). PD‐L1 testing in guiding patient selection for PD‐1/PD‐L1 inhibitor therapy in lung cancer. Molecular diagnosis & therapy, 22, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford, E. E. , Spadavecchia, J. , Pradier, C. M. , & Gu, F. X. (2012). Surface plasmon resonance biosensors incorporating gold nanoparticles. Macromolecular Bioscience, 12, 724–739. [DOI] [PubMed] [Google Scholar]
- Blackburn, S. D. , Shin, H. , Freeman, G. J. , & Wherry, E. J. (2008). Selective expansion of a subset of exhausted CD8 T cells by αPD‐L1 blockade. PNAS, 105, 15016–15021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budimir, N. , Thomas, G. D. , Dolina, J. S. , & Salek‐Ardakani, S. (2022). Reversing T‐cell exhaustion in cancer: Lessons learned from PD‐1/PD‐L1 immune checkpoint blockade. Cancer Immunology Research, 10, 146–153. [DOI] [PubMed] [Google Scholar]
- Camidge, D. R. , Doebele, R. C. , & Kerr, K. M. (2019). Comparing and contrasting predictive biomarkers for immunotherapy and targeted therapy of NSCLC. Nature Reviews Clinical Oncology, 16, 341–355. [DOI] [PubMed] [Google Scholar]
- Chen, G. , Huang, A. C. , Zhang, W. , Zhang, G. , Wu, M. , Xu, W. , Yu, Z. , Yang, J. , Wang, B. , Sun, H. , Xia, H. , Man, Q. , Zhong, W. , Antelo, L. F. , Wu, B. , Xiong, X. , Liu, X. , Guan, L. , Li, T. , … Guo, W. (2018). Exosomal PD‐L1 contributes to immunosuppression and is associated with anti‐PD‐1 response. Nature, 560, 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, L. , Sharples, R. A. , Scicluna, B. J. , & Hill, A. F. (2014). Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell‐free blood. Journal of Extracellular Vesicles, 3, 23743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumba Garcia, L. M. , Peterson, T. E. , Cepeda, M. A. , Johnson, A. J. , & Parney, I. F. (2019). Isolation and analysis of plasma‐derived exosomes in patients with glioma. Frontiers in Oncology, 9, 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daassi, D. , Mahoney, K. M. , & Freeman, G. J. (2020). The importance of exosomal PDL1 in tumour immune evasion. Nature Reviews Immunology, 20, 209–215. [DOI] [PubMed] [Google Scholar]
- Del Re, M. , Marconcini, R. , Pasquini, G. , Rofi, E. , Vivaldi, C. , Bloise, F. , Restante, G. , Arrigoni, E. , Caparello, C. , & Bianco, M. G. (2018). PD‐L1 mRNA expression in plasma‐derived exosomes is associated with response to anti‐PD‐1 antibodies in melanoma and NSCLC. British Journal of Cancer, 118, 820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diskin, B. , Adam, S. , Cassini, M. F. , Sanchez, G. , Liria, M. , Aykut, B. , Buttar, C. , Li, E. , Sundberg, B. , Salas, R. D. , Chen, R. , Wang, J. , Kim, M. , Farooq, M. S. , Nguy, S. , Fedele, C. , Tang, K. H. , Chen, T. , Wang, W. , … Miller, G. (2020). PD‐L1 engagement on T cells promotes self‐tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nature Immunology, 21, 442–454. [DOI] [PubMed] [Google Scholar]
- Dong, L. , Lin, W. , Qi, P. , Xu, M.‐d. , Wu, X. , Ni, S. , Huang, D. , Weng, W.‐w. , Tan, C. , Sheng, W. , Zhou, X. , & Du, X. (2016). Circulating long RNAs in serum extracellular vesicles: Their characterization and potential application as biomarkers for diagnosis of colorectal cancer. Cancer Epidemiology, Biomarkers & Prevention, 25, 1158–1166. [DOI] [PubMed] [Google Scholar]
- EL Andaloussi, S. , Mager, I. , Breakefield, X. O. , & Wood, M. J. (2013). Extracellular vesicles: Biology and emerging therapeutic opportunities. Nature Reviews. Drug discovery, 12, 347–357. [DOI] [PubMed] [Google Scholar]
- Emelyanov, A. , Shtam, T. , Kamyshinsky, R. , Garaeva, L. , Verlov, N. , Miliukhina, I. , Kudrevatykh, A. , Gavrilov, G. , Zabrodskaya, Y. , Pchelina, S. , & Konevega, A. (2020). Cryo‐electron microscopy of extracellular vesicles from cerebrospinal fluid. Plos One, 15, e0227949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faget, L. , & Hnasko, T. S. (2015). Tyramide signal amplification for immunofluorescent enhancement. In ELISA: Methods and protocols, Hnasko, R. (ed.), Springer New York, pp. 161–172. [DOI] [PubMed] [Google Scholar]
- Fan, J. , Yin, Z. , Xu, J. , Wu, F. , Huang, Q. , Yang, L. , Jin, Y. , & Yang, G. (2020). Circulating microRNAs predict the response to anti‐PD‐1 therapy in non‐small cell lung cancer. Genomics, 112, 2063–2071. [DOI] [PubMed] [Google Scholar]
- Fang, Y. (2015). Total internal reflection fluorescence quantification of receptor pharmacology. Biosensors (Basel), 5, 223–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser, K. , Jo, A. , Giedt, J. , Vinegoni, C. , Yang, K. S. , Peruzzi, P. , Chiocca, E. A. , Breakefield, X. O. , Lee, H. , & Weissleder, R. (2019). Characterization of single microvesicles in plasma from glioblastoma patients. Neuro‐oncol, 21, 606–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garon, E. B. , Rizvi, N. A. , Hui, R. , Leighl, N. , Balmanoukian, A. S. , Eder, J. P. , Patnaik, A. , Aggarwal, C. , Gubens, M. , & Horn, L. (2015). Pembrolizumab for the treatment of non–small‐cell lung cancer. New England Journal of Medicine, 372, 2018–2028. [DOI] [PubMed] [Google Scholar]
- Goodman, A. , Patel, S. P. , & Kurzrock, R. (2017). PD‐1–PD‐L1 immune‐checkpoint blockade in B‐cell lymphomas. Nature Reviews Clinical Oncology, 14, 203. [DOI] [PubMed] [Google Scholar]
- Gueguen, P. , Metoikidou, C. , Dupic, T. , Lawand, M. , Goudot, C. , Baulande, S. , Lameiras, S. , Lantz, O. , Girard, N. , Seguin‐Givelet, A. , Lefevre, M. , Mora, T. , Walczak, A. M. , Waterfall, J. J. , & Amigorena, S. (2021). Contribution of resident and circulating precursors to tumor‐infiltrating CD8+T cell populations in lung cancer. Science Immunology, 6, eabd5778. [DOI] [PubMed] [Google Scholar]
- Gulley, J. L. , Berzofsky, J. A. , Butler, M. O. , Cesano, A. , Fox, B. A. , Gnjatic, S. , Janetzki, S. , Kalavar, S. , Karanikas, V. , Khleif, S. N. , Kirsch, I. , Lee, P. P. , Maccalli, C. , Maecker, H. , Schlom, J. , Seliger, B. , Siebert, J. , Stroncek, D. F. , Thurin, M. , … Butterfield, L. H. (2017). Immunotherapy biomarkers 2016: Overcoming the barriers. Journal for ImmunoTherapy of Cancer, 5, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haes, A. J. , & Van Duyne, R. P. (2004). A unified view of propagating and localized surface plasmon resonance biosensors. Analytical and Bioanalytical Chemistry, 379, 920–930. [DOI] [PubMed] [Google Scholar]
- Halvaei, S. , Daryani, S. , Eslami, S. Z. , Samadi, T. , Jafarbeik‐Iravani, N. , Bakhshayesh, T. O. , Majidzadeh, A. K. , & Esmaeili, R. (2018). Exosomes in cancer liquid biopsy: A Focus on Breast Cancer. Molecular Therapy. Nucleic Acids, 10, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst, R. S. , Soria, J. C. , Kowanetz, M. , Fine, G. D. , Hamid, O. , Gordon, M. S. , Sosman, J. A. , McDermott, D. F. , Powderly, J. D. , Gettinger, S. N. , Kohrt, H. E. , Horn, L. , Lawrence, D. P. , Rost, S. , Leabman, M. , Xiao, Y. , Mokatrin, A. , Koeppen, H. , Hegde, P. S. , … Hodi, F. S. (2014). Predictive correlates of response to the anti‐PD‐L1 antibody MPDL3280A in cancer patients. Nature, 515, 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, X. , Sullivan, R. J. , Kalinich, M. , Kwan, T. T. , Giobbie‐Hurder, A. , Pan, S. , LiCausi, J. A. , Milner, J. D. , Nieman, L. T. , Wittner, B. S. , Ho, U. , Chen, T. , Kapur, R. , Lawrence, D. P. , Flaherty, K. T. , Sequist, L. V. , Ramaswamy, S. , Miyamoto, D. T. , Lawrence, M. , … Haber, D. A. (2018). Molecular signatures of circulating melanoma cells for monitoring early response to immune checkpoint therapy. PNAS, 115, 2467–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, J. , Sheng, Y. , Kwak, K. J. , Shi, J. , Yu, B. , & Lee, L. J. (2017). A signal‐amplifiable biochip quantifies extracellular vesicle‐associated RNAs for early cancer detection. Nature Communication, 8, 1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, M. , Yang, J. , Wang, T. , Song, J. , Xia, J. , Wu, L. , Wang, W. , Wu, Q. , Zhu, Z. , Song, Y. , & Yang, C. J. (2020). Homogeneous, low‐volume, efficient and sensitive quantitation of circulating exosomal PD‐L1 for Cancer diagnosis and immunotherapy response prediction. Angewandte Chemie (International ed in English), 59, 4800–4805. [DOI] [PubMed] [Google Scholar]
- Jotatsu, T. , Oda, K. , & Yatera, K. (2018). PD‐L1 immunohistochemistry in patients with non‐small cell lung cancer. Journal of Thoracic Disease, 10, S2127–S2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanada, M. , Bachmann, M. H. , Hardy, J. W. , Frimannson, D. O. , Bronsart, L. , Wang, A. , Sylvester, M. D. , Schmidt, T. L. , Kaspar, R. L. , Butte, M. J. , Matin, A. C. , & Contag, C. H. (2015). Differential fates of biomolecules delivered to target cells via extracellular vesicles. PNAS, 112, E1433–E1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keir, M. E. , Butte, M. J. , Freeman, G. J. , & Sharpe, A. H. (2008). PD‐1 and its ligands in tolerance and immunity. Annual Review of Immunology, 26, 677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kil, S. H. , Estephan, R. , Sanchez, J. , Zain, J. M. , Kadin, M. E. , Young, J. W. , Rosen, S. T. , & Querfeld, C. (2017). PD‐L1 Is Regulated By Interferon Gamma and Interleukin 6 through STAT1 and STAT3 Signaling in Cutaneous T‐Cell Lymphoma. Blood, 130, 1458–1458. [Google Scholar]
- Kim, K. M. , Abdelmohsen, K. , Mustapic, M. , Kapogiannis, D. , & Gorospe, M. (2017). RNA in extracellular vesicles. WIREs RNA, 8, e1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. Y. , Khanal, D. , Tharkar, P. , Kalionis, B. , & Chrzanowski, W. (2018). None of us is the same as all of us: Resolving the heterogeneity of extracellular vesicles using single‐vesicle, nanoscale characterization with resonance enhanced atomic force microscope infrared spectroscopy (AFM‐IR). Nanoscale Horizons, 3, 430–438. [DOI] [PubMed] [Google Scholar]
- Ko, J. , Wang, Y. , Carlson, J. C. T. , Marquard, A. , Gungabeesoon, J. , Charest, A. , Weitz, D. , Pittet, M. J. , & Weissleder, R. (2020). Single extracellular vesicle protein analysis using immuno‐droplet digital polymerase chain reaction amplification. Advanced Biosystems, 4, 1900307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal, J. , Arras, G. , Colombo, M. , Jouve, M. , Morath, J. P. , Primdal‐Bengtson, B. , Dingli, F. , Loew, D. , Tkach, M. , & Théry, C. (2016). Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proceedings of the National Academy of Sciences, 113, E968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudalkar, E. M. , Deng, Y. , Davis, T. N. , & Asbury, C. L. (2016). Coverslip cleaning and functionalization for total internal reflection fluorescence microscopy. Cold Spring Harbor Protocols, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufer, S. , D'Angelo, A. D. , Kwan, C. , Ray, R. D. , Yudkowsky, R. , Boulet, J. R. , McGaghie, W. C. , & Pugh, C. M. (2017). Rescuing the clinical breast examination: Advances in classifying technique and assessing physician competency. Annals of Surgery, 266, 1069–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, K. , Fraser, K. , Ghaddar, B. , Yang, K. , Kim, E. , Balaj, L. , Chiocca, E. A. , Breakefield, X. O. , Lee, H. , & Weissleder, R. (2018). Multiplexed profiling of single extracellular vesicles. ACS Nano, 12, 494–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon, K. M. , Wakefield, D. L. , Maddox, A. L. , Brehove, M. S. , Willner, A. N. , Garcia‐Mansfield, K. , Meechoovet, B. , Reiman, R. , Hutchins, E. , Miller, M. M. , Goel, A. , Pirrotte, P. , Van Keuren‐Jensen, K. , & Jovanovic‐Talisman, T. (2019). Single molecule characterization of individual extracellular vesicles from pancreatic cancer. Journal of Extracellular Vesicles, 8, 1685634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liangsupree, T. , Multia, E. , & Riekkola, M. ‐ L. (2020). Modern isolation and separation techniques for extracellular vesicles. Journal of Chromatography A, 11636, 461773. [DOI] [PubMed] [Google Scholar]
- Liu, C. , Xu, X. , Li, B. , Situ, B. , Pan, W. , Hu, Y. , An, T. , Yao, S. , & Zheng, L. (2018). Single‐exosome‐counting immunoassays for cancer diagnostics. Nano Letters, 18, 4226–4232. [DOI] [PubMed] [Google Scholar]
- Liu, C. , Zhao, J. , Tian, F. , Chang, J. , Zhang, W. , & Sun, J. (2019). lambda‐DNA‐ and aptamer‐mediated sorting and analysis of extracellular vesicles. Journal of the American Chemical Society, 141, 3817–3821. [DOI] [PubMed] [Google Scholar]
- Lyon, L. A. , Peña, D. J. , & Natan, M. J. (1999). Surface plasmon resonance of au colloid‐modified Au films: Particle size dependence. The Journal of Physical Chemistry B, 103, 5826–5831. [Google Scholar]
- Mandrekar, J. N. (2010). Receiver operating characteristic curve in diagnostic test assessment. Journal of Thoracic Oncology, 5, 1315–1316. [DOI] [PubMed] [Google Scholar]
- Meng, X. , Liu, X. , Guo, X. , Jiang, S. , Chen, T. , Hu, Z. , Liu, H. , Bai, Y. , Xue, M. , Hu, R. , Sun, S. C. , Liu, X. , Zhou, P. , Huang, X. , Wei, L. , Yang, W. , & Xu, C. (2018). FBXO38 mediates PD‐1 ubiquitination and regulates anti‐tumour immunity of T cells. Nature, 564, 130–135. [DOI] [PubMed] [Google Scholar]
- Mitchell, J. S. , Wu, Y. , Cook, C. J. , & Main, L. (2005). Sensitivity enhancement of surface plasmon resonance biosensing of small molecules. Analytical Biochemistry, 343, 125–135. [DOI] [PubMed] [Google Scholar]
- Motzer, R. J. , Escudier, B. , McDermott, D. F. , George, S. , Hammers, H. J. , Srinivas, S. , Tykodi, S. S. , Sosman, J. A. , Procopio, G. , & Plimack, E. R. (2015). Nivolumab versus everolimus in advanced renal‐cell carcinoma. New England Journal of Medicine, 373, 1803–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien, K. , Breyne, K. , Ughetto, S. , Laurent, L. C. , & Breakefield, X. O. (2020). RNA delivery by extracellular vesicles in mammalian cells and its applications. Nature Reviews Molecular Cell Biology, 21, 585–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang, Y. , Shi, J. , Yang, X. , Wang, C. , Sun, Z. , & Xiao, R. (2020). Personalized detection of circling exosomal PD‐L1 based on Fe3O4@TiO2 isolation and SERS immunoassay. Biosensors & Bioelectronics, 148, 111800. [DOI] [PubMed] [Google Scholar]
- Paré, L. , Pascual, T. , Seguí, E. , Teixidó, C. , Gonzalez‐Cao, M. , Galván, P. , Rodríguez, A. , González, B. , Cuatrecasas, M. , Pineda, E. , Torné, A. , Crespo, G. , Martin‐Algarra, S. , Pérez‐Ruiz, E. , Reig, Ò. , Viladot, M. , Font, C. , Adamo, B. , Vidal, M. , … Prat, A. (2018). Association between <em>PD1</em>mRNA and response to anti‐PD1 monotherapy across multiple cancer types. Annals of Oncology, 29, 2121–2128. [DOI] [PubMed] [Google Scholar]
- Poggio, M. , Hu, T. , Pai, C. C. , Chu, B. , Belair, C. D. , Chang, A. , Montabana, E. , Lang, U. E. , Fu, Q. , Fong, L. , & Blelloch, R. (2019). Suppression of exosomal PD‐L1 induces systemic anti‐tumor immunity and memory. Cell, 177, 414–427.e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reategui, E. , Aceto, N. , Lim, E. J. , Sullivan, J. P. , Jensen, A. E. , Zeinali, M. , Martel, J. M. , Aranyosi, A. J. , Li, W. , Castleberry, S. , Bardia, A. , Sequist, L. V. , Haber, D. A. , Maheswaran, S. , Hammond, P. T. , Toner, M. , & Stott, S. L. (2015). Tunable nanostructured coating for the capture and selective release of viable circulating tumor cells. Advanced Materials, 27, 1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas, A. (2015). Adaptive immune resistance: How cancer protects from immune attack. Cancer Discovery, 5(9), 915–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklefs, F. L. , Alayo, Q. , Krenzlin, H. , Mahmoud, A. B. , Speranza, M. C. , Nakashima, H. , Hayes, J. L. , Lee, K. , Balaj, L. , & Passaro, C. (2018). Immune evasion mediated by PD‐L1 on glioblastoma‐derived extracellular vesicles. Science Advances, 4, eaar2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rima, X. Y. , Walters, N. , Nguyen, L. T. H. , & Reategui, E. (2020). Surface engineering within a microchannel for hydrodynamic and self‐assembled cell patterning. Biomicrofluidics, 14, 014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliminejad, K. , Khorshid, H. R. K. , & Ghaffari, S. H. (2019). Why have microRNA biomarkers not been translated from bench to clinic? Future Oncology, 15, 801–803. [DOI] [PubMed] [Google Scholar]
- Seymour, L. , Bogaerts, J. , Perrone, A. , Ford, R. , Schwartz, L. H. , Mandrekar, S. , Lin, N. U. , Litière, S. , Dancey, J. , Chen, A. , Hodi, F. S. , Therasse, P. , Hoekstra, O. S. , Shankar, L. K. , Wolchok, J. D. , Ballinger, M. , Caramella, C. , & de Vries, E. G. E. (2017). iRECIST: Guidelines for response criteria for use in trials testing immunotherapeutics. The Lancet Oncology, 18, e143–e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, W. , Guo, K. , Adkins, G. B. , Jiang, Q. , Liu, Y. , Sedano, S. , Duan, Y. , Yan, W. , Wang, S. E. , Bergersen, K. , Worth, D. , Wilson, E. H. , & Zhong, W. (2018). A Single Extracellular Vesicle (EV) flow cytometry approach to reveal EV heterogeneity. Angewandte Chemie (International ed in English), 57, 15675–15680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava, A. , & Gupta, V. (2011). Methods for the determination of limit of detection and limit of quantitation of the analytical methods. Chronicles of Young Scientists, 2, 21. [Google Scholar]
- Shukuya, T. , Ghai, V. , Amann, J. M. , Okimoto, T. , Shilo, K. , Kim, T. K. , Wang, K. , & Carbone, D. P. (2020). Circulating microRNAs and extracellular vesicle‐containing microRNAs as response biomarkers of anti‐programmed cell death protein 1 or programmed death‐ligand 1 therapy in NSCLC. Journal of Thoracic Oncology, 15, 1773–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva, A. M. , Lázaro‐Ibáñez, E. , Gunnarsson, A. , Dhande, A. , Daaboul, G. , Peacock, B. , Osteikoetxea, X. , Salmond, N. , Friis, K. P. , Shatnyeva, O. , & Dekker, N. (2021). Quantification of protein cargo loading into engineered extracellular vesicles at single‐vesicle and single‐molecule resolution. Journal of Extracellular Vesicles, 10, e12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skog, J. , Würdinger, T. , van Rijn, S. , Meijer, D. H. , Gainche, L. , Curry, W. T. , Carter, B. S. , Krichevsky, A. M. , & Breakefield, X. O. (2008). Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nature Cell Biology, 10, 1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen, S. F. , Demuth, C. , Weber, B. , Sorensen, B. S. , & Meldgaard, P. (2016). Increase in soluble PD‐1 is associated with prolonged survival in patients with advanced EGFR‐mutated non‐small cell lung cancer treated with erlotinib. Lung Cancer, 100, 77–84. [DOI] [PubMed] [Google Scholar]
- Springer, T. , Ermini, M. L. , Spackova, B. , Jablonku, J. , & Homola, J. (2014). Enhancing sensitivity of surface plasmon resonance biosensors by functionalized gold nanoparticles: Size matters. Analytical Chemistry, 86, 10350–10356. [DOI] [PubMed] [Google Scholar]
- Sun, W. , Marchuk, K. , Wang, G. , & Fang, N. (2010). Autocalibrated scanning‐angle prism‐type total internal reflection fluorescence microscopy for nanometer‐precision axial position determination. Analytical Chemistry, 82, 2441–2447. [DOI] [PubMed] [Google Scholar]
- Ter‐Ovanesyan, D. , Kowal, E. J. K. , Regev, A. , Church, G. M. , & Cocucci, E. (2017). In Extracellular Vesicles: Methods and Protocols, Kuo, W. P. & Jia, S. (eds.) Springer New York, pp. 233–241. [DOI] [PubMed] [Google Scholar]
- Theodoraki, M. N. , Yerneni, S. S. , Hoffmann, T. K. , Gooding, W. E. , & Whiteside, T. L. (2018). Clinical significance of PD‐L1(+) exosomes in plasma of head and neck cancer patients. Clinical Cancer Research, 24, 896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thery, C. , Witwer, K. W. , Aikawa, E. , Alcaraz, M. J. , Anderson, J. D. , Andriantsitohaina, R. , Antoniou, A. , Arab, T. , Archer, F. , Atkin‐Smith, G. K. , Ayre, D. C. , Bach, J. M. , Bachurski, D. , Baharvand, H. , Balaj, L. , Baldacchino, S. , Bauer, N. N. , Baxter, A. A., Bebawy, M. , … Zuba‐Surma, E. K. (2018). Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles, 7, 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thommen, D. S. , Koelzer, V. H. , Herzig, P. , Roller, A. , Trefny, M. , Dimeloe, S. , Kiialainen, A. , Hanhart, J. , Schill, C. , Hess, C. , Savic Prince, S. , Wiese, M. , Lardinois, D. , Ho, P. ‐ C. , Klein, C. , Karanikas, V. , Mertz, K. D. , Schumacher, T. N. , & Zippelius, A. (2018). A transcriptionally and functionally distinct PD‐1+ CD8+ T cell pool with predictive potential in non‐small‐cell lung cancer treated with PD‐1 blockade. Nature Medicine, 24, 994–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, Q. , He, C. , Liu, G. , Zhao, Y. , Hui, L. , Mu, Y. , Tang, R. , Luo, Y. , Zheng, S. , & Wang, B. (2018). Nanoparticle counting by microscopic digital detection: Selective quantitative analysis of exosomes via surface‐anchored nucleic acid amplification. Analytical Chemistry, 90, 6556–6562. [DOI] [PubMed] [Google Scholar]
- Tian, Y. , Gong, M. , Hu, Y. , Liu, H. , Zhang, W. , Zhang, M. , Hu, X. , Aubert, D. , Zhu, S. , Wu, L. , & Yan, X. (2020). Quality and efficiency assessment of six extracellular vesicle isolation methods by nano‐flow cytometry. Journal of Extracellular Vesicles, 9, 1697028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uludag, Y. , & Tothill, I. E. (2012). Cancer biomarker detection in serum samples using surface plasmon resonance and quartz crystal microbalance sensors with nanoparticle signal amplification. Analytical Chemistry, 84, 5898–5904. [DOI] [PubMed] [Google Scholar]
- Verdegaal, E. M. , De Miranda, N. F. , Visser, M. , Harryvan, T. , Van Buuren, M. M. , Andersen, R. S. , Hadrup, S. R. , Van Der Minne, C. E. , Schotte, R. , & Spits, H. (2016). Neoantigen landscape dynamics during human melanoma–T cell interactions. Nature, 536, 91. [DOI] [PubMed] [Google Scholar]
- Wang, J. , Zhang, H. , Sun, X. , Wang, X. , Ren, T. , Huang, Y. , Zhang, R. , Zheng, B. , & Guo, W. (2020). Exosomal PD‐L1 and N‐cadherin predict pulmonary metastasis progression for osteosarcoma patients. Journal of Nanobiotechnology, 18, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Khan, A. , Huang, R. , Ye, S. , Di, K. , Xiong, T. , & Li, Z. (2020). Recent advances in single extracellular vesicle detection methods. Biosensors & Bioelectronics, 154, 112056. [DOI] [PubMed] [Google Scholar]
- Wei, Z. , Batagov, A. O. , Schinelli, S. , Wang, J. , Wang, Y. , El Fatimy, R. , Rabinovsky, R. , Balaj, L. , Chen, C. C. , Hochberg, F. , Carter, B. , Breakefield, X. O. , & Krichevsky, A. M. (2017). Coding and noncoding landscape of extracellular RNA released by human glioma stem cells. Nature Communications, 8, 1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisman, S. , Hirsch‐Lerner, D. , Barenholz, Y. , & Talmon, Y. (2004). Nanostructure of cationic lipid‐oligonucleotide complexes. Biophysical Journal, 87, 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal, M. , & Lamszus, K. (2015). Circulating biomarkers for gliomas. Nature Reviews Neurology, 11, 556. [DOI] [PubMed] [Google Scholar]
- Wolchok, J. D. , Kluger, H. , Callahan, M. K. , Postow, M. A. , Rizvi, N. A. , Lesokhin, A. M. , Segal, N. H. , Ariyan, C. E. , Gordon, R. ‐ A. , & Reed, K. (2013). Nivolumab plus ipilimumab in advanced melanoma. New England Journal of Medicine, 369, 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, D. , Yan, J. , Shen, X. , Sun, Y. , Thulin, M. , Cai, Y. , Wik, L. , Shen, Q. , Oelrich, J. , Qian, X. , Dubois, K. L. , Ronquist, K. G. , Nilsson, M. , Landegren, U. , & Kamali‐Moghaddam, M. (2019). Profiling surface proteins on individual exosomes using a proximity barcoding assay. Nature Communication, 10, 3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, F. , Yin, Z. , Yang, L. , Fan, J. , Xu, J. , Jin, Y. , Yu, J. , Zhang, D. , & Yang, G. (2019). Smoking induced extracellular vesicles release and their distinct properties in non‐small cell lung cancer. Journal of Cancer, 10, 3435–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Y. , Kwak, K. J. , Agarwal, K. , Marras, A. , Wang, C. , Mao, Y. , Huang, X. , Ma, J. , Yu, B. , Lee, R. , Vachani, A. , Marcucci, G. , Byrd, J. C. , Muthusamy, N. , Otterson, G. , Huang, K. , Castro, C. E. , Paulaitis, M. , Nana‐Sinkam, S. P. , & Lee, L. J. (2013). Detection of extracellular RNAs in cancer and viral infection via tethered cationic lipoplex nanoparticles containing molecular beacons. Analytical Chemistry, 85, 11265–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, R. , Rai, A. , Chen, M. , Suwakulsiri, W. , Greening, D. W. , & Simpson, R. J. (2018). Extracellular vesicles in cancer ‐ implications for future improvements in cancer care. Nature Reviews Clinical Oncology, 15(10), 617–638. [DOI] [PubMed] [Google Scholar]
- Yang, Z. , Shi, J. , Xie, J. , Wang, Y. , Sun, J. , Liu, T. , Zhao, Y. , Zhao, X. , Wang, X. , Ma, Y. , Malkoc, V. , Chiang, C. , Deng, W. , Chen, Y. , Fu, Y. , Kwak, K. J. , Fan, Y. , Kang, C. , Yin, C. , … Lee, L. J. (2020). Large‐scale generation of functional mRNA‐encapsulating exosomes via cellular nanoporation. Nature Biomedical Engineering, 4, 69–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager, M. , Wilson‐Kubalek Elizabeth, M. , Weiner Scott, G. , Brown Patrick, O. , & Rein, A. (1998). Supramolecular organization of immature and mature murine leukemia virus revealed by electron cryo‐microscopy: Implications for retroviral assembly mechanisms. Proceedings of the National Academy of Sciences, 95, 7299–7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekula, A. , Muralidharan, K. , Kang, K. M. , Wang, L. , Balaj, L. , & Carter, B. S. (2020). From laboratory to clinic: Translation of extracellular vesicle based cancer biomarkers. Methods (San Diego, Calif.), 177, 58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, J. , Lin, Y. , Xiong, X. , Li, K. , Yao, Z. , Dong, H. , Jiang, Z. , Yu, D. , Yeung, S. J. , & Zhang, H. (2019). Detection of exosomal PD‐L1 RNA in saliva of patients with periodontitis. Frontiers in Genetics, 10, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuana, Y. , Koning, R. I. , Kuil, M. E. , Rensen, P. C. , Koster, A. J. , Bertina, R. M. , & Osanto, S. (2013). Cryo‐electron microscopy of extracellular vesicles in fresh plasma. Journal of Extracellular Vesicles, 2, 10.3402/jev.v2i0.21494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , & Lyden, D. (2019). Asymmetric‐flow field‐flow fractionation technology for exomere and small extracellular vesicle separation and characterization. Nature Protocols, 14, 1027–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Nguyen, L. T. H. , Hickey, R. , Walters, N. , Wang, X. , Kwak, K. J. , Lee, L. J. , Palmer, A. F. , & Reátegui, E. (2021). Immunomagnetic sequential ultrafiltration (iSUF) platform for enrichment and purification of extracellular vesicles from biofluids. Scientific Reports, 11, 8034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, X. , & Lang, J. (2017). Soluble PD‐1 and PD‐L1: Predictive and prognostic significance in cancer. Oncotarget, 8, 97671. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.