

Abstract
Glycoconjugation strategies in anticancer drug discovery exploit the high expression of glucose transporters in malignant cells to achieve preferential uptake and hence attractive pharmacological characteristics of increased therapeutic windows and decreased unwanted toxicity. Here we present the design of glycoconjugated prochelators of aroylhydrazone AH1, an antiproliferative scavenger that targets the increased iron demand of rapidly proliferating malignant cells. The constructs feature a monosaccharide (d-glucose, d-glucosamine, or glycolytic inhibitor 2-deoxy-d-glucose) connected at the C2 or C6 position via a short linker, which masks the chelator through a disulfide bond susceptible to intracellular reduction. Cellular assays showed that the glycoconjugates rely on the GLUT1 transporter for uptake, lead to intracellular iron deprivation, and present antiproliferative activity. Ectopic overexpression of GLUT1 in malignant and normal cells increased the uptake and toxicity of the glycoconjugated prochelators, demonstrating that these compounds are well suited for targeting cells overexpressing glucose transporters and therefore for selective iron sequestration in malignant cells.
Keywords: iron, prochelator, glycoconjugation, GLUT1, cancer
Cancer cells primarily rely on aerobic glycolysis for energy production.1 This hallmark of cancer, known as the Warburg effect, is associated with high rates of glucose consumption and overexpression of glucose transporters (e.g., GLUT1) among cancer cells of different origins.2−4 In clinical practice, this characteristic of malignancy is exploited in positron emission tomography (PET), whereby accumulation of radiolabeled carbohydrate 2-deoxy-2-[18F]fluoro-d-glucose (18F-FDG, Chart 1) is used to detect and stage tumors.5
Chart 1. Structures of 18F-FDG and Examples of Glycoconjugates (top), and Chelators and Disulfide-Masked AH1 Prochelator (bottom).

The Warburg effect is also exploited in drug discovery through the conjugation of anticancer drugs to a carbohydrate moiety. Glycoconjugation is expected to impart tumor selectivity because malignant cells overexpress glucose transporters.6,7 For instance, glufosfamide (Chart 1), the 1-β-d-glucose conjugate of ifosfamide mustard, was the first glycoconjugate compound to be tested in clinical trials.8 This approach has been investigated for other anticancer agents including DNA alkylators (e.g., chlorambucil, Chart 1),9 taxoids,10 anthracyclines,11 and platinum-based compounds.12,13
Glycoconjugation has also been employed to mask the metal-binding units in prochelator systems that are designed for activation by enzymes (e.g., β-glucosidase)14 or intracellular reductants (e.g., glutathione).15 We have previously introduced glycoconjugation in the design of disulfide-masked thiosemicarbazone compounds that are reductively activated in cells to form high-affinity iron chelators.16 This strategy concurrently targets two metabolic characteristics of malignant cells–the elevated glucose consumption and the iron addiction that sustains rapid proliferation.
The high iron demand of cancer cells17 is currently viewed as a vulnerability and therapeutic opportunity pursued through the development and testing of iron-sequestering antiproliferative compounds.18,19 Several FDA-approved iron chelators, including siderophore deferoxamine (DFO, Chart 1), were tested in early clinical trials for cancer indications,20 and more recent clinical studies of iron-binding approaches have included tridentate thiosemicarbazones (e.g., DpC, COTI-2).21,22 We have shown that a disulfide linkage can be employed as a reductively activated switch for the intracellular release of tridentate thiosemicarbazone and aroylhydrazone chelators.23,24 For instance, the intracellular reduction of the disulfide bond in (AH1-S)2 (Chart 1) leads to the formation of thiolate AH1 featuring a tridentate (S, N, O) metal-binding unit. This approach is particularly attractive for anticancer applications because the intracellular environment of cancer cells is inherently more reducing due to elevated levels of reduced glutathione (GSH).25−27 These iron-sequestering constructs not only affect rapidly proliferating cancer cells but also tumor-associated macrophages, which promote cell proliferation and mobility in the tumor microenvironment in an iron-dependent fashion.28,29
Herein, we report the synthesis of glycoconjugate prochelators in which a disulfide linkage is employed to mask a metal-binding aroylhydrazone and connect it to a carbohydrate moiety (Chart 2). After transporter-mediated uptake of the glycoconjugate constructs, intracellular reduction of the disulfide bond in expected to release the tridentate aroylhydrazone chelator. Aroylhydrazones, such as pyridoxal isonicotinoyl hydrazone (PIH) and salicylaldehyde isonicotinoyl hydrazone (SIH, Chart 1), are chelators of high affinity for iron30 and well-documented antiproliferative activity.20 Aroylhydrazone binding units have also been incorporated in the design of prochelators that are activated under oxidative stress in cells.31
Chart 2. AH1 Glycoconjugates and Aglycone Control Compound Investigated in This Work.

We have previously shown that benzoylhydrazone AH1 forms low-spin ferric complexes of 2:1 ligand-to-metal stoichiometry,32 and that disulfide-masked prochelator (AH1-S)2 (Chart 1) exhibits micromolar toxicity and proapoptotic activity in breast cancer cells.24 With this study, we incorporate a tumor-targeting strategy through the conjugation of AH1 to three different carbohydrate moieties (i.e., d-glucose, d-glucosamine, 2-deoxy-d-glucose). Previous studies on glycoconjugate prochelators have shown that the constructs typically present enhanced hydrophilicity and that the glucose transporters (e.g., GLUT1) are at least in part responsible for their cellular uptake.16 Here, in addition to reporting on the synthesis, GLUT1-mediated uptake and antiproliferative activity of new AH1 glycoconjugate prochelators, we sought to probe more directly to what extent the GLUT1 expression has an impact on cellular uptake that is actually meaningful for toxicity. This type of investigation is key to reveal the potential of these glycoconjugates to target malignant cells expressing high GLUT1 levels in complex environments such as living organisms.
Our molecular design of AH1 prochelators (Chart 2) features the connection of glucopyranoses at the C2 or C6 positions because these substitutions are well tolerated by the glucose transporters.6 Examples of these targeting units include 2-d-glucose-conjugated paclitaxel,10 6-d-glucose-conjugated chlorambucil (Chart 1),9 and 2-d-glucosamine-conjugated adriamycin.11 Furthermore, we sought to examine the effect of 2-deoxy-d-glucose (2-DG), a competitive inhibitor of glycolysis,33,34 in our glycoconjugation strategy. 2-DG is an energy-restriction mimetic agent that interferes with glycolysis because its phosphorylation product (2-DG 6-phosphate) cannot be further metabolized to fructose 6-phosphate.33,34 The combination of 2-DG with anticancer agents (e.g., paclitaxel, etoposide) has been promising both in cultured cells and animal studies;35 however, 2-DG conjugation approaches remain rare.36 Indeed, 2-DG is an attractive glycoconjugation moiety as it is recognized by the glucose transporters and its uptake rate and affinity for GLUT1 are higher than those of d-glucose.37−39
For the preparation of the glucose conjugate at the C6 position (G6AH1) and the glucosamine conjugate at the C2 position (GA2AH1), we employed our previously reported precursors 1 and 3, respectively (Scheme 1), which are derived from the convenient 2-pyridyl disulfide cross-linker 3-(2-pyridyldithio)-propionic acid.16 In both cases, the carbohydrate is thus connected to the prochelator through an ester or amide bond, an ethylene spacer, and a disulfide switch for reductive activation of the aroylhydrazone chelator upon cellular uptake. As a control compound, we also prepared an aglycone analog featuring the same linker but lacking the carbohydrate moiety (AAH1, Chart 2, Scheme S1).
Scheme 1. Synthesis of Glycoconjugates G6AH1 and GA2AH1.

The synthesis of the pyridyl disulfide precursor (10, Scheme 2) for the preparation of the 2-DG conjugate at the C6 position (2DG6AH1) required careful optimization of our protection/deprotection sequence. Whereas 1,2,3,4,6-penta-trimethylsilyl-d-glucopyranose can be regioselectively deprotected at the C6 position, we observed persistent overdeprotection of trimethylsilyl (TMS) groups on the secondary alcohols of 1,3,4,6-tetra-trimethylsilyl-2-deoxy-d-glucopyranose in a variety of conditions. We therefore opted to protect the primary alcohol with triphenylmethane (trityl, Tr) chloride to obtain 5, and then protected the secondary alcohols with tert-butyldimethylsilyl (TBDMS) triflate. Compound 6 could be then deprotected selectively at the C6 position and the resulting primary alcohol 7 was connected to pyridyldithiol propionic acid 8 via Steglich esterification. Deprotection of the TBDMS groups in the presence of HF-pyridine (∼70% HF) in dry THF at 0 °C produced the key pyridyl disulfide precursor 10, which was converted into the desired prochelator 2DG6AH1 through disulfide exchange. To the best of our knowledge, the conjugation of 2-DG at the C6 position has not been previously reported. The precursors developed herein are amenable to the preparation of other conjugates, including a variety of chelators to be connected at the very last step.
Scheme 2. Synthesis of 2DG6AH1.

The newly synthesized compounds were fully characterized by NMR spectroscopy and electrospray ionization high-resolution mass spectrometry (ESI-HRMS), and the purity of the final glycoconjugates was confirmed by HPLC analysis (Figure S1). As expected for compounds featuring hydrophilic carbohydrate moieties, the calculated partition coefficients of the glycoconjugates (in the 0.8–1.8 range, Table 1) are significantly lower than that of the aglycone analog (3.4).
Table 1. Partition Coefficients and Antiproliferative Activities of Test Compounds.
| IC50values (μM) |
||||
|---|---|---|---|---|
| logPo/wa | A2780 (ovary) | MDA-MB-231 (breast) | MRC-5 (normal lung) | |
| G6AH1 | 1.07 | 5.2 ± 0.8 | 17 ± 2 | 26 ± 4 |
| GA2AH1 | 0.85 | 5.3 ± 0.8 | 17 ± 3 | 33 ± 3 |
| 2DG6AH1 | 1.85 | 2.5 ± 0.6 | 22 ± 2 | 29 ± 4 |
| AAH1 | 3.40 | 2.1 ± 0.2 | 12 ± 2 | 12 ± 2 |
| DFO | 3.5 ± 0.4 | 6.1 ± 0.4 | 9 ± 1 | |
Consensus lipophilicity calculated by SwissADME.40
Prior to biological testing, the stability of the AH1 glycoconjugates and the corresponding aglycone was evaluated in cell growth media supplemented with fetal bovine serum (10% v/v) by monitoring their optical absorbance at 305 nm. Only minor hydrolytic degradation of the glycoconjugates was observed within the first 8 h. The aglycone was generally less stable than the glycoconjugates; nevertheless, 58% of the aglycone and more than 70% of the glycoconjugates remained intact after 24 h (Figure S2). The compounds were therefore found suitable for testing in cultured cells and indeed the cellular uptake was found to be considerably faster than 8 h (vide infra).
Antiproliferative activities were assessed using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium) assay in MDA-MB-231 (breast) and A2780 (ovary) cancer cells because an increased iron demand has been widely documented in breast and ovarian cancer.41−43 In addition, MRC-5 lung fibroblasts were included as a comparison to normal cells. The antiproliferative activities for all compounds are summarized in Table 1. After 72 h, the IC50 values of AH1 glycoconjugates were in the range of 2.5 to 5 μM in A2780 cells and 17 to 22 μM in MDA-MB-231 cells, whereas the toxicity was lower for the normal MRC-5 cells (with IC50 values from 26 to 33 μM). Conversely, the aglycone analog AAH1 and siderophore DFO, both lacking the carbohydrate moiety, presented similar toxicity in the malignant and normal cell lines. The observed antiproliferative activity of DFO (in the low micromolar range) is attributed to iron sequestration and consistent with other reported cancer cell panels.23,44
Overall, the AH1 glycoconjugates displayed antiproliferative activities similar to those of other disulfide-masked prochelators,23,24 including thiosemicarbazone glycoconjugates,16 and a higher selectivity toward cancer cell lines when compared to the aglycone control. Interestingly, we found that the ovarian malignant cell line A2780 was the most susceptible to all the compounds. This cell line was chosen for further testing of cell death, iron sequestration, and transporter-mediated uptake.
The type of cell death induced by the glycoconjugates was assessed by flow cytometry in A2780 cells stained with propidium iodide (PI) and fluorescein-conjugated Annexin V (FITC-AnnV) (Figure S3). Our previously reported disulfide-based prochelators and several iron chelators such as SIH and DFO are known to initiate apoptotic pathways of cell death in cancer cells.24 For all the AH1 glycoconjugates, the percent of cells undergoing early apoptosis (5–6%) was comparable to that of cells exposed to SIH (7.8%) (Figure 1). The fractions of cells in late apoptosis were generally smaller for the glycoconjugates than for SIH (38%), with 2DG6AH1 presenting the largest percentage (20%). 2-DG alone (at the same concentration) caused some early apoptosis (3.6%) and late apoptosis (3.4%). Interestingly, synergistic effects with 2-DG have been employed to enhance the sensitivity of cancer cells to traditional antineoplastic agent.45 Pending further investigation, the slightly higher toxicity and apoptosis induction of 2DG6AH1 in A2780 cells could be attributable to metabolic effects of 2-DG or also to improved cellular uptake of this particular glycoconjugate. Overall, these experiments indicated that all the AH1-derived compounds (including the aglycone control AAH1) have proapoptotic effects consistent with their antiproliferative activity and with the effects of other iron chelators and prochelators.
Figure 1.

Apoptotic cell death in the presence of AH1 glycoconjugates and control compounds in A2780 cells. The compounds (20 μM in all cases) were incubated for 48 h. Following treatment with FITC-AnnV and PI, the cells were analyzed by flow cytometry. Experiments were conducted in triplicate and the values shown are averages ± standard deviation. ** p < 0.01, *** p < 0.001, **** p < 0.0001.
The ability of the glycoconjugate prochelators to elicit intracellular iron sequestration was confirmed using the calcein assay in A2780 cells. The fluorescence emission of intracellular calcein is partially quenched by the coordination of paramagnetic cations, primarily cytosolic high-spin Fe(II), and therefore the uptake of high-affinity chelators that compete for metal ions results in an increase in fluorescence intensity.46 Indeed, upon incubation with all the glycoconjugates and the aglycone control (20 μM), we observed a 20–25% increase in calcein fluorescence, comparable to the effect of cell-permeant chelator SIH (Figure S4). There was no significant change of fluorescence over an incubation period of 20 min or 1 h, indicating rapid cellular uptake and saturation of the calcein response in the chosen experimental conditions (0.1 μM calcein). These findings confirmed that all the disulfide-based prochelators reach the intracellular milieu, undergo reduction/activation, and effectively coordinate iron. In spite of their significantly lower lipophilicity relative to AAH1 (Table 1), the glycoconjugates are efficiently taken up by cells.
The cellular uptake of the AH1 glycoconjugates was investigated by a competition experiment utilizing the fluorescent d-glucose derivative 2-NBDG (i.e., 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxy-glucose). The cells were incubated in glucose-free media for 12 h and then the competition experiments were conducted in growth media with a d-glucose content (1 g/L) similar to that of human blood.47 The competition of the glycoconjugates (50 μM) for glucose transporters resulted in decreased uptake of 2-NBDG (100 μM), which was recorded as a decreased fluorescence intensity by flow cytometry (Figure 2). The effect was greater than 20% for all three glycoconjugates, comparable to the uptake suppression caused by phloretin (50 μM), a GLUT1 inhibitor48 employed as a positive control. Conversely, the presence of aglycone AAH1 did not affect the uptake of 2-NBDG as expected for a negative control lacking the carbohydrate moiety.
Figure 2.

Cellular uptake of fluorescent glucose analog 2-NBDG in the presence of glycoconjugate competitors and aglycone control in A2780 cells. Tested compounds (50 μM) were coincubated with 2-NBDG probe (100 μM) for 20 min. GLUT1 inhibitor phloretin (50 μM) was used as a positive control. Experiments were conducted in triplicate and values shown are averages ± standard deviation. * p < 0.05, ** p < 0.01.
To assess the importance of transporter-mediated uptake for cytotoxicity, we evaluated the ability of GLUT1 inhibitor phloretin to rescue A2780 cells treated with the AH1 glycoconjugates. Indeed, coincubation with phloretin (12.5 μM) led to increased IC50 values and therefore decreased toxicity of the glycoconjugates in MTT assays. Notably, phloretin alone did not significantly affect the growth of A2780 cells at the chosen concentration (Figure S5), but these data indicate that it partially inhibited the uptake of AH1 glycoconjugates. For instance, the IC50 of 2DG6AH1 is more than 5 times higher in the presence of phloretin, whereas the aglycone AAH1 maintained the same toxicity (Figure 3).
Figure 3.

Effects of GLUT1 inhibitor phloretin (12.5 μM) on the IC50 values (72 h, MTT assays) of AH1 glycoconjugates in A2780 cells. Experiments were conducted in triplicate and the values shown are averages ± standard deviation. **** p < 0.0001.
Collectively, these experiments indicated that the AH1 glycoconjugates are antiproliferative prochelators that undergo, at least in part, transporter-mediated cellular uptake and interfere with intracellular iron availability. With this information at hand, we sought to examine whether the expression levels of the GLUT1 transporter have an impact on antiproliferative activities.
The relative GLUT1 expression levels in our cultured cells were estimated through Western blotting. Although the GLUT1 protein band is expected at 55 kDa, the range from 50 kDa to 70 kDa was monitored to take into account potential glycosylation (Figure 4). After normalizing the GLUT1 protein to the loading control β-actin, we observed that the GLUT1 expression level in A2780 cells was higher than in MDA-MB-231 cells, which in turn was higher than that in the normal MRC-5 cells. As such, the GLUT1 expression reflects the measured antiproliferative activities (Table 1), with lowest IC50 values for the cell line with highest GLUT1 levels and potentially highest uptake capacity for glycoconjugate prochelators. The susceptibility to iron sequestration, however, could also be ascribed to other factors in this comparison across cell lines. To determine whether the AH1 glycoconjugates are indeed more toxic to cells expressing more GLUT1 transporters, we generated isogenic cell pairs differing only in GLUT1 expression levels.
Figure 4.

Expression of GLUT1 in different cell lines. (a) Western blot of GLUT1 in MRC-5, A2780, MDA-MB-231 with β-actin as a loading control. (b) GLUT1 expression levels normalized to the control as averages of four replicates ± standard deviation.
Isogenic cell line pairs, ectopically overexpressing GLUT1 and control ectopically expressing Turbo-635 fluorescent protein, were generated from the MDA-MB-231 and MRC-5 parental cell lines, which intrinsically have lower basal GLUT1 expression (Figure 4). Western blot analyses confirmed that the overexpression derivative lines were indeed expressing GLUT1 at a much higher level than their corresponding control lines (i.e., transduced with the same vector but no GLUT1 plasmid) (Figure 5a). Glucose uptake assays employing the fluorescent probe 2-NBDG confirmed that the GLUT1 overexpressing derivative lines take up more probe compared to their corresponding control lines (Figure 5b).
Figure 5.
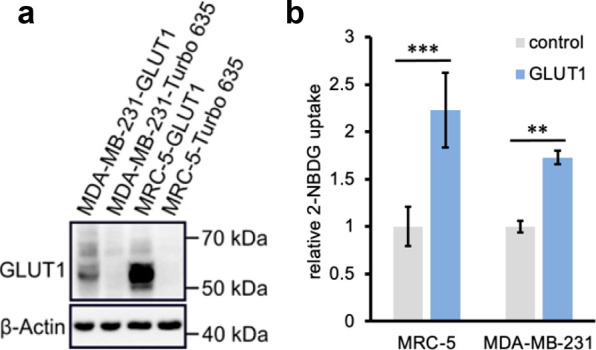
Efficiency of GLUT1 overexpression in isogenic MDA-MB-231 and MRC-5 derivative lines. (a) Western blot of GLUT1-transduced vs Turbo-635-transduced control cell lines. β-Actin is used as a loading control. (b) Glucose uptake capacity of the transduced cell lines as indicated by the relative uptake of fluorescent glucose analog 2-NBDG (100 μM, 20 min). Experiments were conducted in triplicate and the values shown are averages ± standard deviation. ** p < 0.01, *** p < 0.001.
In comparative MTT assays (Figure 6), we found that the glycoconjugate prochelators had consistently higher antiproliferative activity in the cell lines with GLUT1 overexpression, and the effect was particularly pronounced for the normal cell line that was transduced to bring GLUT1 expression to levels comparable to those of malignant cells. This increase in toxicity was not observed for the aglycone AAH1, which does not rely on the glucose transporters for cellular uptake. These experiments therefore indicate that the described glycoconjugation approach for the design of iron prochelators is suitable to target cancer phenotypes presenting elevated expression of glucose transporters (Figure 6).
Figure 6.
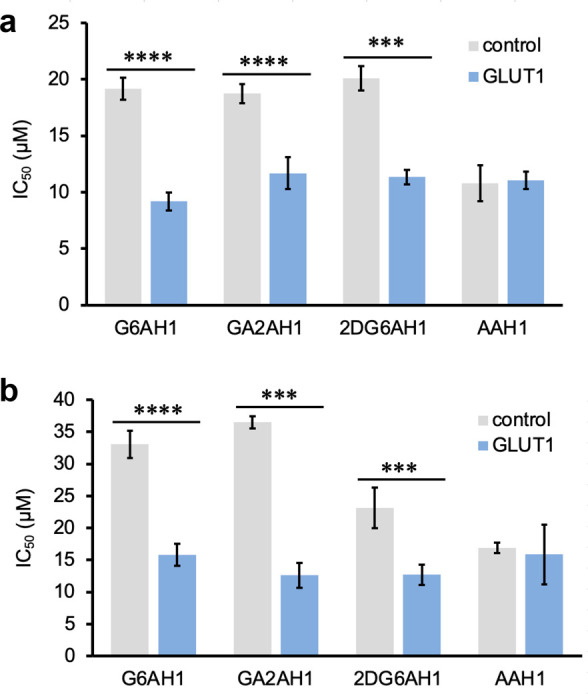
Effects of GLUT1 amplification on the antiproliferative activity of AH1 glycoconjugates in transduced (a) MDA-MB-231 and (b) MRC-5 cell lines. IC50 values were obtained from MTT assays (72 h). Experiments were conducted in triplicate and the values shown are averages ± standard deviation. *** p < 0.001, **** p < 0.0001.
In summary, we report the syntheses of three monosaccharide-conjugated prochelators of aroylhydrazone AH1, including a new route to the conjugation of 2-deoxy-d-glucose (2-DG), a competitive inhibitor of glycolysis that is recognized with high affinity by the glucose transporters. The carbohydrate moiety significantly decreases the lipophilicity of these disulfide-based prochelators, which therefore have to rely on glucose transporters, particularly the most abundant GLUT1, for cellular uptake. The reduction/activation of the prochelators to release iron-binding chelator AH1 was confirmed in cells, along with the expected pro-apoptotic response. Critically, the toxicity of the glyco-AH1 prochelators is reduced when GLUT1 is blocked by inhibitor phloretin and in turn it is enhanced when the GLUT1 is overexpressed. Our experiments in isogenic cell line pairs differing only in GLUT1 expression indicate that these compounds are more toxic to cells with higher GLUT1 expression levels. Overall, by relying significantly on transporter-mediated uptake, glycoconjugated prochelators have the potential to target iron in cancer cells overexpressing GLUT1 and hence to display favorable therapeutic indexes in vivo.
Acknowledgments
We thank John C. Fitch for assistance with data acquisition and analysis at the UArizona Cancer Center Flow Cytometry Shared Resource.
Glossary
Abbreviations
- GLUT1
glucose transporter 1
- DFO
deferoxamine
- GSH
reduced glutathione
- 2-DG
2-deoxy-d-glucose
- TMS
trimethylsilyl
- TBDMS
tert-butyldimethylsilyl
- DMSO
dimethyl sulfoxide
- THF
tetrahydrofuran
- DCC
N,N′-dicyclohexylcarbodiimide
- DMAP
4-dimethylaminopyridine
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
- PI
propidium iodide
- FITC-AnnV
fluorescein-conjugated Annexin V
- 2-NBDG
2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxy-glucose
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00250.
Synthetic procedures and chemical characterization data, protocols for cell-based assays, Western blotting, plasmid preparation, and transduction experiments (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the US National Institutes of Health (awards R01 GM127646 to E.T. and R01 CA226920 to A.O.). The Bruker NEO-500 spectrometer in the UArizona Department of Chemistry and Biochemistry NMR Facility was purchased thanks to support from the National Science Foundation (MRI award CHE-1920234). The UArizona Cancer Center Flow Cytometry Shared Resource is supported by the National Cancer Institute (award P30 CA023074).
The authors declare no competing financial interest.
Supplementary Material
References
- Vander Heiden M. G.; Cantley L. C.; Thompson C. B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szablewski L. Expression of glucose transporters in cancers. Biochim. Biophys. Acta, Rev. Cancer 2013, 1835, 164–169. 10.1016/j.bbcan.2012.12.004. [DOI] [PubMed] [Google Scholar]
- Macheda M. L.; Rogers S.; Best J. D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 2005, 202, 654–662. 10.1002/jcp.20166. [DOI] [PubMed] [Google Scholar]
- Ancey P.-B.; Contat C.; Meylan E. Glucose transporters in cancer - from tumor cells to the tumor microenvironment. FEBS J. 2018, 285, 2926–2943. 10.1111/febs.14577. [DOI] [PubMed] [Google Scholar]
- Paydary K.; Seraj S. M.; Zadeh M. Z.; Emamzadehfard S.; Shamchi S. P.; Gholami S.; Werner T. J.; Alavi A. The Evolving Role of FDG-PET/CT in the Diagnosis, Staging, and Treatment of Breast Cancer. Mol. Imaging Biol. 2019, 21, 1–10. 10.1007/s11307-018-1181-3. [DOI] [PubMed] [Google Scholar]
- Calvaresi E. C.; Hergenrother P. J. Glucose conjugation for the specific targeting and treatment of cancer. Chem. Sci. 2013, 4, 2319–2333. 10.1039/c3sc22205e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J.; Yang J.; Seeberger P. H.; Yin J. Glycoconjugates for glucose transporter-mediated cancer-specific targeting and treatment. Carbohydr. Res. 2020, 498, 108195. 10.1016/j.carres.2020.108195. [DOI] [PubMed] [Google Scholar]
- Briasoulis E.; Pavlidis N.; Terret C.; Bauer J.; Fiedler W.; Schöffski P.; Raoul J. L.; Hess D.; Selvais R.; Lacombe D.; Bachmann P.; Fumoleau P. Glufosfamide administered using a 1-h infusion given as first-line treatment for advanced pancreatic cancer. A phase II trial of the EORTC-new drug development group. Eur. J. Cancer 2003, 39, 2334–2340. 10.1016/S0959-8049(03)00629-4. [DOI] [PubMed] [Google Scholar]
- Halmos T.; Santarromana M.; Antonakis K.; Scherman D. Synthesis of glucose-chlorambucil derivatives and their recognition by the human GLUT1 glucose transporter. Eur. J. Pharmacol. 1996, 318, 477–484. 10.1016/S0014-2999(96)00796-0. [DOI] [PubMed] [Google Scholar]
- Lin Y.-S.; Tungpradit R.; Sinchaikul S.; An F.-M.; Liu D.-Z.; Phutrakul S.; Chen S.-T. Targeting the Delivery of Glycan-Based Paclitaxel Prodrugs to Cancer Cells via Glucose Transporters. J. Med. Chem. 2008, 51, 7428–7441. 10.1021/jm8006257. [DOI] [PubMed] [Google Scholar]
- Cao J.; Cui S.; Li S.; Du C.; Tian J.; Wan S.; Qian Z.; Gu Y.; Chen W. R.; Wang G. Targeted cancer therapy with a 2-deoxyglucose-based adriamycin complex. Cancer Res. 2013, 73, 1362–1373. 10.1158/0008-5472.CAN-12-2072. [DOI] [PubMed] [Google Scholar]
- Patra M.; Johnstone T. C.; Suntharalingam K.; Lippard S. J. A Potent Glucose-Platinum Conjugate Exploits Glucose Transporters and Preferentially Accumulates in Cancer Cells. Angew. Chem., Int. Ed. 2016, 55, 2550–2554. 10.1002/anie.201510551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Wang Q.; Huang Z.; Yang X.; Nie Q.; Hao W.; Wang P. G.; Wang X. Glycosylated Platinum(IV) Complexes as Substrates for Glucose Transporters (GLUTs) and Organic Cation Transporters (OCTs) Exhibited Cancer Targeting and Human Serum Albumin Binding Properties for Drug Delivery. J. Med. Chem. 2017, 60, 5736–5748. 10.1021/acs.jmedchem.7b00433. [DOI] [PubMed] [Google Scholar]
- Oliveri V.; Giuffrida M. L.; Vecchio G.; Aiello C.; Viale M. Gluconjugates of 8-hydroxyquinolines as potential anti-cancer prodrugs. Dalton Trans. 2012, 41, 4530–4535. 10.1039/c2dt12371a. [DOI] [PubMed] [Google Scholar]
- Pujol A. M.; Cuillel M.; Jullien A.-S.; Lebrun C.; Cassio D.; Mintz E.; Gateau C.; Delangle P. A Sulfur Tripod Glycoconjugate that Releases a High-Affinity Copper Chelator in Hepatocytes. Angew. Chem., Int. Ed. 2012, 51, 7445–7448. 10.1002/anie.201203255. [DOI] [PubMed] [Google Scholar]
- Akam E. A.; Tomat E. Targeting Iron in Colon Cancer via Glycoconjugation of Thiosemicarbazone Prochelators. Bioconjugate Chem. 2016, 27, 1807–1812. 10.1021/acs.bioconjchem.6b00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torti S. V.; Torti F. M. Iron and cancer: more ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. 10.1038/nrc3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbasi U.; Abbina S.; Gill A.; Takuechi L. E.; Kizhakkedathu J. N. Role of Iron in the Molecular Pathogenesis of Diseases and Therapeutic Opportunities. ACS Chem. Biol. 2021, 16, 945–972. 10.1021/acschembio.1c00122. [DOI] [PubMed] [Google Scholar]
- Jung M.; Mertens C.; Tomat E.; Brüne B. Iron as a Central Player and Promising Target in Cancer Progression. Int. J. Mol. Sci. 2019, 20, 273. 10.3390/ijms20020273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinowski D. S.; Richardson D. R. The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol. Rev. 2005, 57, 547–583. 10.1124/pr.57.4.2. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Kalinowski D. S.; Kovacevic Z.; Siafakas A. R.; Jansson P. J.; Stefani C.; Lovejoy D. B.; Sharpe P. C.; Bernhardt P. V.; Richardson D. R. Thiosemicarbazones from the Old to New: Iron Chelators That Are More Than Just Ribonucleotide Reductase Inhibitors. J. Med. Chem. 2009, 52, 5271–5294. 10.1021/jm900552r. [DOI] [PubMed] [Google Scholar]
- Heffeter P.; Pape V. F. S.; Enyedy É. A.; Keppler B. K.; Szakacs G.; Kowol C. R. Anticancer Thiosemicarbazones: Chemical Properties, Interaction with Iron Metabolism, and Resistance Development. Antioxid. Redox Signal. 2019, 30, 1062–1082. 10.1089/ars.2017.7487. [DOI] [PubMed] [Google Scholar]
- Chang T. M.; Tomat E. Disulfide/thiol switches in thiosemicarbazone ligands for redox-directed iron chelation. Dalton Trans. 2013, 42, 7846–7849. 10.1039/c3dt50824b. [DOI] [PubMed] [Google Scholar]
- Akam E. A.; Utterback R. D.; Marcero J. R.; Dailey H. A.; Tomat E. Disulfide-masked iron prochelators: Effects on cell death, proliferation, and hemoglobin production. J. Inorg. Biochem. 2018, 180, 186–193. 10.1016/j.jinorgbio.2017.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamcsik M. P.; Kasibhatla M. S.; Teeter S. D.; Colvin O. M. Glutathione levels in human tumors. Biomarkers 2012, 17, 671–691. 10.3109/1354750X.2012.715672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akam E. A.; Chang T. M.; Astashkin A. V.; Tomat E. Intracellular reduction/activation of a disulfide switch in thiosemicarbazone iron chelators. Metallomics 2014, 6, 1905–1912. 10.1039/C4MT00153B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. H.; Sessler J. L.; Kim J. S. Disulfide-Based Multifunctional Conjugates for Targeted Theranostic Drug Delivery. Acc. Chem. Res. 2015, 48, 2935–2946. 10.1021/acs.accounts.5b00406. [DOI] [PubMed] [Google Scholar]
- Mertens C.; Akam E. A.; Rehwald C.; Brüne B.; Tomat E.; Jung M. Intracellular Iron Chelation Modulates the Macrophage Iron Phenotype with Consequences on Tumor Progression. PLoS One 2016, 11, e0166164 10.1371/journal.pone.0166164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnetz M.; Meier K. J.; Rehwald C.; Mertens C.; Urbschat A.; Tomat E.; Akam A. E.; Baer P.; Roos C. F.; Brüne B.; Jung M. The Disturbed Iron Phenotype of Tumor Cells and Macrophages in Renal Cell Carcinoma Influences Tumor Growth. Cancers 2020, 12, 530. 10.3390/cancers12030530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.-L.; Kong X.; Xie Y.; Hider R. C. The interaction of pyridoxal isonicotinoyl hydrazone (PIH) and salicylaldehyde isonicotinoyl hydrazone (SIH) with iron. J. Inorg. Biochem. 2018, 180, 194–203. 10.1016/j.jinorgbio.2017.12.007. [DOI] [PubMed] [Google Scholar]
- Kielar F.; Helsel M. E.; Wang Q.; Franz K. J. Prochelator BHAPI protects cells against paraquat-induced damage by ROS-triggered iron chelation. Metallomics 2012, 4, 899–909. 10.1039/c2mt20069d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astashkin A. V.; Utterback R. D.; Sung Y.-S.; Tomat E. Iron Complexes of an Antiproliferative Aroyl Hydrazone: Characterization of Three Protonation States by Electron Paramagnetic Resonance Methods. Inorg. Chem. 2020, 59, 11377–11384. 10.1021/acs.inorgchem.0c01120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz S.; Mazerbourg S.; Boisbrun M.; Cerella C.; Diederich M.; Grillier-Vuissoz I.; Flament S. Energy restriction mimetic agents to target cancer cells: Comparison between 2-deoxyglucose and thiazolidinediones. Biochem. Pharmacol. 2014, 92, 102–111. 10.1016/j.bcp.2014.07.021. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Li J.; Wang F.; Hu J.; Wang S.; Sun Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. 10.1016/j.canlet.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Pajak B.; Siwiak E.; Sołtyka M.; Priebe A.; Zieliński R.; Fokt I.; Ziemniak M.; Jaśkiewicz A.; Borowski R.; Domoradzki T.; Priebe W. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int. J. Mol. Sci. 2020, 21, 234. 10.3390/ijms21010234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi Q.; Ma Y.; Gao X.; Liu R.; Liu P.; Mi Y.; Fu X.; Gao Q. 2-Deoxyglucose conjugated platinum (II) complexes for targeted therapy: design, synthesis, and antitumor activity. J. Biomol. Struct. Dyn. 2016, 34, 2339–2350. 10.1080/07391102.2015.1114972. [DOI] [PubMed] [Google Scholar]
- Mueckler M.; Makepeace C. Identification of an Amino Acid Residue That Lies between the Exofacial Vestibule and Exofacial Substrate-binding Site of the Glut1 Sugar Permeation Pathway. J. Biol. Chem. 1997, 272, 30141–30146. 10.1074/jbc.272.48.30141. [DOI] [PubMed] [Google Scholar]
- Jung C. Y. Inactivation of Glucose Carriers in Human Erythrocyte Membranes by 1-Fluoro-2,4-dinitrobenzene. J. Biol. Chem. 1974, 249, 3568–3573. 10.1016/S0021-9258(19)42610-0. [DOI] [PubMed] [Google Scholar]
- Krupka R. M.; Devés R. Evidence for allosteric inhibition sites in the glucose carrier of erythrocytes. Biochim. Biophys. Acta 1980, 598, 127–133. 10.1016/0005-2736(80)90270-9. [DOI] [PubMed] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinnix Z. K.; Miller L. D.; Wang W.; D’Agostino R.; Kute T.; Willingham M. C.; Hatcher H.; Tesfay L.; Sui G.; Di X.; Torti S. V.; Torti F. M. Ferroportin and Iron Regulation in Breast Cancer Progression and Prognosis. Sci. Transl. Med. 2010, 2, 43ra56. 10.1126/scitranslmed.3001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basuli D.; Tesfay L.; Deng Z.; Paul B.; Yamamoto Y.; Ning G.; Xian W.; McKeon F.; Lynch M.; Crum C. P.; Hegde P.; Brewer M.; Wang X.; Miller L. D.; Dyment N.; Torti F. M.; Torti S. V. Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene 2017, 36, 4089–4099. 10.1038/onc.2017.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockfield S.; Raffel J.; Mehta R.; Rehman N.; Nanjundan M. Iron overload and altered iron metabolism in ovarian cancer. Biol. Chem. 2017, 398, 995–1007. 10.1515/hsz-2016-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung Y.-S.; Wu W.; Ewbank M. A.; Utterback R. D.; Marty M. T.; Tomat E. Albumin Conjugates of Thiosemicarbazone and Imidazole-2-thione Prochelators: Iron Coordination and Antiproliferative Activity. ChemMedChem. 2021, 16, 2764–2768. 10.1002/cmdc.202100278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laussel C.; Léon S. Cellular toxicity of the metabolic inhibitor 2-deoxyglucose and associated resistance mechanisms. Biochem. Pharmacol. 2020, 182, 114213. 10.1016/j.bcp.2020.114213. [DOI] [PubMed] [Google Scholar]
- Ma Y.; Abbate V.; Hider R. C. Iron-sensitive fluorescent probes: monitoring intracellular iron pools. Metallomics 2015, 7, 212–222. 10.1039/C4MT00214H. [DOI] [PubMed] [Google Scholar]
- O’Neil R. G.; Wu L.; Mullani N. Uptake of a fluorescent deoxyglucose analog (2-NBDG) in tumor cells. Mol. Imaging Biol. 2005, 7, 388–392. 10.1007/s11307-005-0011-6. [DOI] [PubMed] [Google Scholar]
- Xintaropoulou C.; Ward C.; Wise A.; Marston H.; Turnbull A.; Langdon S. P. A comparative analysis of inhibitors of the glycolysis pathway in breast and ovarian cancer cell line models. Oncotarget 2015, 6, 25677–25695. 10.18632/oncotarget.4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.