

Abstract

Sleeping sickness and leishmaniasis are neglected tropical diseases that threaten millions of people. The currently available therapies present several limitations, including high toxicity, lack of efficacy, and emerging drug resistance, prompting a search for novel therapeutic agents. In this work, we designed, synthesized, and in vitro evaluated the activity of new pyrimido[5,4-d]pyrimidines against Trypanosoma brucei and Leishmania infantum (promastigote and amastigote forms). The cytotoxicity of the compounds against the THP1 cell line was also assessed. Most tested compounds presented low micromolar activity against T. brucei with IC50 values in the range between 0.9 and 13.4 μM, and one compound also showed activity against L. infantum (IC50 = 3.13 μM). Several molecules presented a selectivity index higher than 10. The most active compound against booth parasites is derivative 4c, with IC50 = 0.94 μM (SI > 107) against T. brucei and IC50 = 3.13 μM (SI > 32) against L. infantum. This data enabled the identification of a new promising molecular scaffold for developing a novel class of antitrypanosomal and antileishmanial agents.
Keywords: novel class of antitrypanosomal agents; novel class of antileishmanial agents; pyrimido[5,4-d]pyrimidine derivatives; fused pyrimidine derivatives
Sleeping sickness or human African trypanosomiasis (HAT) and visceral leishmaniasis (VL) are infectious vector-borne diseases transmitted by flies that the WHO classifies as Neglected Tropical Diseases. HAT is confined to sub-Saharan Africa;1 however, VL is widespread in tropical and subtropical areas and is considered endemic in 79 countries in Europe, Africa, Asia, and America. HAT is caused by Trypanosoma brucei, and leishmaniasis is caused by several Leishmania spp. Together, these vector-borne parasites threaten millions of people.2
Presently, therapies available for HAT (Figure 1) have several limitations,1 such as being ineffective against CNS-stage disease (pentamidine and suramin), requiring parenteral administration (pentamidine, suramin, nifurtimox-eflornithine combination therapy (NECT), and melarsoprol), and having intrinsic toxicity (melarsoprol is an arsenic derivative).3,4 Fexinidazole is a new drug that has been recently approved;5 however, it showed cross-resistance to nifurtimox,6,7 which is an alert for the need to discover new agents against HAT.
Figure 1.

Current drugs for the treatment of human African trypanosomiasis. Pentamidine is also used in the treatment of leishmaniasis.
The available leishmaniasis treatment options (Figure 2) are pentavalent antimonials, amphotericin B and its formulations, pentamidine, miltefosine, and paromomycin.1 However, these drugs also present several limitations: high toxicity, lack of efficacy, parenteral route of administration affecting compliance, high costs, and emerging drug resistance.8 Indeed, pentavalent antimonials were used as the first choice to treat VL for decades until a significant number of therapeutic failures in the 1980s were reported in India.9,10
Figure 2.

Current drugs for leishmaniasis treatment.
Paromomycin was an effective VL treatment alternative in this area11 but failed when applied in East Africa.12−14 Miltefosine, the only available oral drug, was used as a first-line therapy in northeastern India;15 however, an increase in the number of relapses was noted after a few years.16 Furthermore, parasite resistance to miltefosine has been described in clinical isolates.17 Currently, WHO recommends the use of liposomal amphotericin B as the first choice in most areas or therapies that combine more than one of the previous drugs, a combination therapy, such as paromomycin plus antimonials in East Africa, miltefosine plus paromomycin, and miltefosine plus liposomal amphotericin B plus paromomycin in India.8,18 The combination therapies represent a last hope for the available therapeutic options; nevertheless, resistance may always emerge as these treatments become generalized.17 Thus, there is an urgent need to find new antileishmanial drugs.
The scientific community has been deeply involved in the search for new drugs for HAT and leishmaniasis.19−22 Different drug classes have been discovered, with aryl groups with hydroxy, methoxy, or alkyl substituents that decorate the fused aryl or heteroaryl nucleus, including quinolines.22,23 Some hydrazone derivatives were also described as being active against Leishmania. In our drug development program, the pyrimido[5,4-d]pyrimidine scaffold (Figure 3) has been used to generate new biologically active compounds.24
Figure 3.

Pyrimido[5,4-d]pyrimidine scaffold used to generate new biologically active compounds
To generate new agents to combat HAT and leishmaniasis, we combined the pyrimido[5,4-d]pyrimidine nucleus, a fused heterocycle analogue of quinoline, with aryl units that have hydroxy and alkoxy substituents. A pyridine was also selected as the heteroaromatic substituent to evaluate hypothetic volume constrains in comparison with the alkoxy substituents. Furthermore, a hydrazone unit was used as one of the linkages between the heterocycle nucleus of pyrimidopyrimidine and one of the aryl subunits. As far as we know, pyrimido[5,4-d]pyrimidines were never reported as antitrypanosomal agents. Herein we report, for the first time, the synthesis of new derivatives of pyrimido[5,4-d]pyrimidines and their activity against T. brucei and L. infantum to assess their potential for drug development for HAT and leishmaniasis.
The target compounds were synthesized following a synthetic approach previously developed by the team,25,26 with some adaptations, as described in Scheme 1. The starting reagent, 6-cyanopurine 1, is not commercially available and was prepared following the procedures reported in refs (25) and (26). In those, an amidine derivative reacts with excess triethyl orthoformate at a temperature below 115 °C to avoid degradation. The alkylation of purines 1 was performed by reaction of 1 with the corresponding haloalkyl derivative in acetonitrile, and cesium carbonate was used as a base. Compound 2c was isolated from the reaction mixture; however, derivatives 2a,b were identified by TLC and used without isolation in the subsequent reaction. In the research group, compounds of structure 3 have been obtained from 6-cyanopurines 1 with a nucleophilic amine.24,26−28 Using similar reaction conditions, purine 2c reacted with excess hydrazine, in DMSO, at room temperature. The reaction was completed after 15 min, and the product was isolated by simple filtration from the reaction medium.
Scheme 1.

Reagents and conditions: (i) CH3CN, 4-(chloromethyl)pyridinium chloride (2 equiv), Cs2CO3 (4 equiv mol), 80 °C, 2 h. (ii) (a) CH3CN, Cs2CO3 (2 equiv mol), BrCH2CH2CH2OH (2 equiv), 80 °C, 1–2 h. (b) CH3CN/EtOH (3/1), H2NNH2·H2O (2.5 equiv), rt, 1 h. (iii) DMSO, H2NNH2·H2O (2.5 equiv), 27 °C, 15 min (iv) EtOH, piperidine (2 equiv), 80 °C, 8–25 h. (v) DMSO or EtOH, aldehyde (1.0–1.2 equiv), H2SO4 (cat.), 30 °C (10–60 min).
Derivatives 3a,b were obtained in a “one-pot” reaction from 1a,b after identifying the intermediates 2a,b in the reaction mixture by TLC, followed by the addition of hydrazine hydrate. Compounds of structure 4 are the Dimroth rearrangement products of 3. The conversion was achieved by reacting 3 with a convenient nucleophile at 80 °C, inspired by the reaction conditions reported previously.29 Finally, compounds 5a–u were obtained by reacting 4 with a slight excess of the adequate aldehyde, under acid catalysis, at room temperature. The products precipitated from the reaction medium or after water addition to the reaction medium. Compounds 5a–u were isolated by simple filtration and washing with water or a mixture of suitable solvents. The structures of all the synthesized compounds were supported by their physical, spectroscopic, and analytic properties.
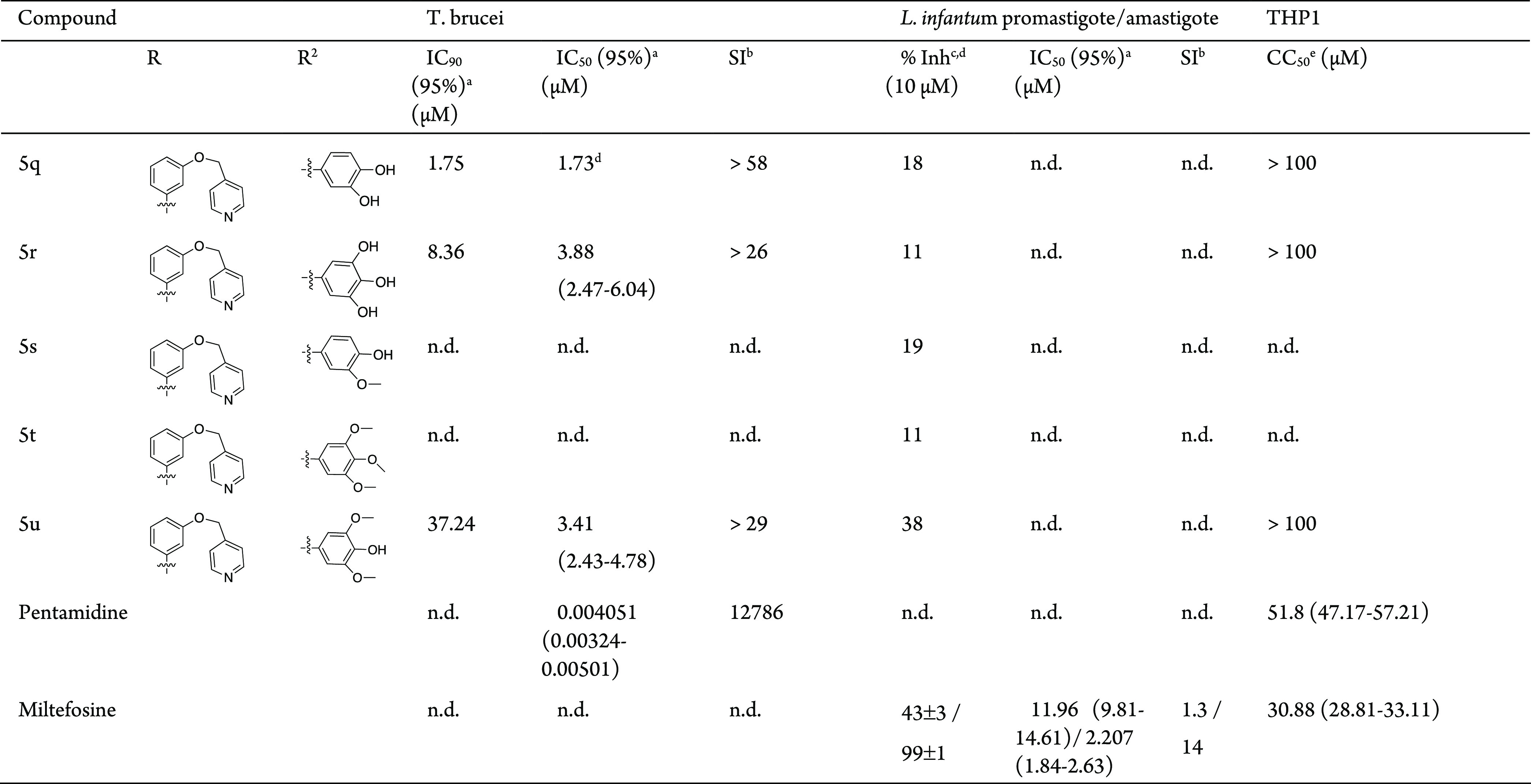
To evaluate the antitrypanosomal potential of the synthesized pyrimidopyrimidines derivatives, they were screened against T. brucei and the extracellular stage (promastigotes) and intracellular stage (amastigotes) of L. infantum parasites. Initially, all the synthesized derivatives were evaluated at 10 μM concentration against the parasites. Pentamidine and miltefosine were used as reference drugs and internal controls for T. brucei and L. infantum, respectively. When the percentage of parasite growth inhibition (% Inh) was greater than 50, the IC50 was determined. The IC90 for T. brucei was also determined for the most potent compounds (IC50 < 15 μM). When compounds showed antileishmanial activity against L. infantum promastigotes, the activity against the amastigote form was also evaluated at a 10 μM concentration. The cytotoxicity (CC50) of the active compounds was initially also assessed using the THP1 cell line. Compounds with no toxicity at 10 μM concentration (% Inhi < 20) were tested at 100 μM concentration. If at 100 μM concentration toxicity was observed, the CC50 was determined. The antitrypanosomal activity, cytotoxicity, and predicted selectivity index (SI) of compounds 5a–u and 4a–c are presented in Table 1.
Table 1. Biological Activity of Pyrimido[5,4-d]pyrimidines 4c and 5 against T. brucei, Wild Type L. infantum promastigote and Amastigote Forms, and the THP1 Cell Line.

Confidence interval 95% (μM) for the mean curve of all independent assays.
SI = CC50 (THP1)/IC50 (parasite).
Percent inhibition at 10 μM.
Data obtained from a single assay.
Cytotoxic concentration. n.a., inhibition lower than 20% at 10 μM. n.d., not determined.
All the tested compounds showed activity against T. brucei and low cytotoxicity against the THP1 cell line (Table 1). The global analysis of the three series, 4a, 5a–h; 4b, 5i–p; and 4c, 5q–u, suggests that the activity against T. brucei and cytotoxicity against the THP1 cell line depends on the substituent groups present at position C8 and C4 of the heterocyclic nucleus (Scheme 1, Table 1). Although these substituents affect the activity, a clear relationship between the activity and substituents could not be established.
In the series 4a, 5a–h that has R = 4-(HO(CH2)3O)C6H4 and different substituents at C4 (Scheme 1, Table 1), the most potent compound, 5b, has R2 = 3,4-(HO)2C6H3. A Low micromolar IC50 was also registered for compounds 5d, 5e and 5g. The lesser cytotoxic compounds were 5c and 5g with CC50 > 100 μM. In this series, the most selective compound was 5g (SI > 30).
In the series 4b, 5i–p that has R = 3-(HO(CH2)3O)C6H4, the most active compound 5j also has R2 = 3,4-(HO)2C6H3; however, the activity and selectivity are slightly lower than those of the corresponding compound 5b of the previous series. Low micromolar activities were also determined for compounds 5k and 5n with IC50 values of, respectively, 3.26 and 2.72 μM. The lower cytotoxicity of compound 5n led to a higher selectivity index for this compound (SI > 37).
In the series 4c, 5q–u where R = 3-(4′-pyridinylCH2O)C6H4, the highest active compound was 4c with IC50 = 0.94 μM followed by compound 5q with IC50 = 1.73 μM. The other tested compounds of this series, 5r and 5u, also presented low micromolar activities. All the compounds of this series showed reduced cytotoxicity (CC50 > 100 μM). The high potency of the compounds combined with reduced cytotoxicity led to compounds with the highest selectivity indexes, compound 5q (SI > 58) and compound 4c (SI > 107).
From all tested compounds against L. infantum promastigotes at 10 μM, only one compound showed percentage growth inhibition greater than 50%: compound 4c that presents IC50 = 3.13 μM. Compounds 4a, 5i, 5j, 5n, and 5u also showed some ability to inhibit the growth of L. infantum promastigotes, with values of percentage inhibition ranging from 21 to 38%. These results suggest that substituent groups at C4 and C8 affect the activity. Comparing the results from series 5a–h with series 5i–p, it is possible to observe the importance of the position of the alkyl unit of substituent R. The position meta, for the alkyl subunit, seems to be more favorable for activity against L. infantum promastigotes than position para. The most active compound was 4c, having the smaller substituent group present at C4 of the heterocycle and the less bulky group at C8. This result seems to indicate that the aryl unit R2 present in derivatives 5 does not favor the activity; however, this substituent is involved in the modulation of the activity, as can be observed by the results of derivatives 5i, 5j, 5n, and 5u. The most potent compound 4c was also tested against L. infantum amastigote form; however, it did not present activity in the tested concentration ranges.
An initial SAR model can be proposed from the collected information (Figure 4). Considering the pyrimidopyrimidine unit as a scaffold, toxicity is minimized when the substituent of the 8-arylamine is at the meta position and the alkoxy group (green) is pyridine. The planar heteroaryl group (green) increased activity in comparison with a linear hydroxyalkyl group, indicating possible volume constraints or preferential HBA/ionic interactions. While activity against L. infantum is higher when the 8-arylamine is meta-substituted, the activity against T. brucei is similar for either meta- or para-substituents. The group X = pyridine, together with R2 (red), is crucial for activity against L. infantum. The combination of a planar HBA/ionizable unit (X) and a small hydrophilic/ionizable group led to an active compound; however, bulky hydrophilic and hydrophobic groups reduce activity, suggesting volume constraints. The group R2 (red) also is important for activity against T. brucei; however, bulky and small groups are allowed. Good activity is observed when R2 (red) is a substituted benzylidene amino group, being better when hydroxy groups are at 3,4-positions, but the higher activity was registered for the hydrophilic/ionizable small substituent NH2.
Figure 4.

Structure–activity relationship for pyrimido[5,4-d]pyrimidines against L. infantum and T. brucei.
In conclusion, a set of new pyrimido[5,4-d] pyrimidine-based compounds was synthesized, characterized, and in vitro evaluated against T. brucei and L. infantum, two parasites responsible for two neglected tropical diseases. The cytotoxicity of the compounds was also assessed against the THP1 cell line. All the tested compounds showed low micromolar activity against T. brucei (0.9 μM < IC50 < 13.4 μM), and one compound presented activity against L. infantum (IC50 = 3.13 μM). The activity against both parasites seems to depend on the substituent’s groups present around the heterocycle nucleus at positions C4 and C8; however, it was not possible to establish a clear relationship between those groups and the activity. The most active compound against both parasites was compound 4c, which has R = 3-(4′-pyridinylCH2O)C6H4 at C8 and the group NHNH2 at C4. Most compounds showed low or very low cytotoxicity against the THP1 cell line, 100 μM > CC50 > 25 μM or CC50 > 100 μM, respectively, and it should be highlighted that all the compounds of the series 4c, 5q–u present CC50 > 100 μM. The high potency combined with very low cytotoxicity presented by compound 4c allowed us to present this compound as a hit compound for further development of a novel class of antitrypanosomal and antileishmanial agents.
Acknowledgments
This work and N.S. were supported by Fundação para a Ciência e Tecnologia (FCT)/Ministério da Educação e Ciência (MEC) cofunded by the FEDER through the COMPETE 2020 - Operacional Programme for Competitiveness and Internationalisation (POCI) ref POCI-01-0145-FEDER-031013 and funds in the framework of the Strategic Funding of CQUM (UID/QUI/00686/2020), (UID/QUI/00686/2018) and Rede Nacional de RMN (PINFRA/22161/2016).
Glossary
Abbreviations
- TLC
thin layer chromatography
- SI
selectivity index
- SAR
structure–activity relationship
- HBA
hydrogen bond acceptor
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00170.
Synthetic procedures, biological assay protocols, physical, analytical, IR, and 1H and 13C NMR characterization of all the compounds, and 1H and 13C NMR spectra (PDF)
Author Contributions
M.A.C. designed the compounds, interpreted the results, and wrote the manuscript. A.L. synthesized the compounds, performed their characterization, performed the biological assays, and analyzed the results. A.C.S. and N.S. conceived the Leishmania, T. brucei, and THP1 studies and interpreted the results. All authors gave significant contributions in discussion and revision. All authors agreed to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Field M. C.; Horn D.; Fairlamb A. H.; Ferguson M. A.; Gray D. W.; Read K. D.; De Rycker M.; Torrie L. S.; Wyatt P. G.; Wyllie S.; Gilbert I. H. Anti-trypanosomatid drug discovery: an ongoing challenge and a continuing need. Nat. Rev. Microbiol. 2017, 15 (4), 217–231. 10.1038/nrmicro.2016.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. https://www.who.int/news-room/fact-sheets (accessed January 2022).
- Pyana Pati P.; Van Reet N.; Mumba Ngoyi D.; Ngay Lukusa I.; Karhemere Bin Shamamba S.; Buscher P. Melarsoprol sensitivity profile of Trypanosoma brucei gambiense isolates from cured and relapsed sleeping sickness patients from the Democratic Republic of the Congo. PLoS Neglected Trop. Dis. 2014, 8 (10), e3212. 10.1371/journal.pntd.0003212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steverding D. The development of drugs for treatment of sleeping sickness: a historical review. Parasites Vectors 2010, 3 (1), 15. 10.1186/1756-3305-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A. FDA approves first all-oral sleeping sickness drug. Nat. Rev. Drug Discovery 2021, 20, 658. 10.1038/d41573-021-00140-5. [DOI] [PubMed] [Google Scholar]
- Sokolova A. Y.; Wyllie S.; Patterson S.; Oza S. L.; Read K. D.; Fairlamb A. H. Cross- resistance to nitro drugs and implications for treatment of human African trypanosomiasis. Antimicrob. Agents Chemother. 2010, 54 (7), 2893–2900. 10.1128/AAC.00332-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie S.; Foth B. J.; Kelner A.; Sokolova A. Y.; Berriman M.; Fairlamb A. H. Nitroheterocyclic drug resistance mechanisms in Trypanosoma brucei. J. Antimicrob. Chemother. 2016, 71 (3), 625–634. 10.1093/jac/dkv376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . Control of the leishmaniasis: report of a meeting of the World Health Organization Expert Committee on the Control of Leishmaniases, Geneva, 22–26 March 2010; World Health Organization Technical Report Series 949; 2010; pp 1–186. https://apps.who.int/iris/bitstream/handle/10665/44412/WHO_TRS_949_eng.pdf.
- Thakur C. P. Epidemiological, clinical and therapeutic features of Bihar kala-azar (including post kala-azar dermal leishmaniasis). Transactions of the Royal Society of Tropical Medicine and Hygiene 1984, 78, 391–398. 10.1016/0035-9203(84)90131-7. [DOI] [PubMed] [Google Scholar]
- Thakur C. P.; Kumar M.; Singh S. K.; Sharma D.; Prasad U. S.; Singh R. S.; Dhawan P. S.; Achari V. Comparison of regimens of treatment with sodium stibogluconate in kala-azar. British Medical Journal (Clinical Research Ed.) 1984, 288, 895–897. 10.1136/bmj.288.6421.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundar S.; Jha T. K.; Thakur C. P.; Sinha P. K.; Bhattacharya S. K. Injectable paromomycin for Visceral leishmaniasis in India. N. Engl. J. Med. 2007, 356, 2571–2581. 10.1056/NEJMoa066536. [DOI] [PubMed] [Google Scholar]
- Hailu A.; Musa A.; Wasunna M.; Balasegaram M.; Yifru S.; Mengistu G.; Hurissa Z.; Hailu W.; Weldegebreal T.; Tesfaye S.; Makonnen E.; Khalil E.; Ahmed O.; Fadlalla A.; El-Hassan A.; Raheem M.; Mueller M.; Koummuki Y.; Rashid J.; Mbui J.; Mucee G.; Njoroge S.; Manduku V.; Musibi A.; Mutuma G.; Kirui F.; Lodenyo H.; Mutea D.; Kirigi G.; Edwards T.; Smith P.; Muthami L.; Royce C.; Ellis S.; Alobo M.; Omollo R.; Kesusu J.; Owiti R.; Kinuthia J. Geographical variation in the response of visceral leishmaniasis to paromomycin in East Africa: a multicentre, open- label, randomized trial. PLoS Negl. Trop.Dis. 2010, 4, e709. 10.1371/journal.pntd.0000709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musa A. M.; Younis B.; Fadlalla A.; Royce C.; Balasegaram M.; Wasunna M.; Hailu A.; Edwards T.; Omollo R.; Mudawi M.; Kokwaro G.; El-Hassan A.; Khalil E. Paromomycin for the treatment of visceral leishmaniasis in Sudan: a randomized, open- label, dose-finding study. PLoS Negl. Trop. Dis. 2010, 4, e855. 10.1371/journal.pntd.0000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailu A.; Musa A.; Wasunna M.; Balasegaram M.; Yifru S.; Mengistu G.; Hurissa Z.; Hailu W.; Weldegebreal T.; Tesfaye S.; Makonnen E.; Khalil E.; Ahmed O.; Fadlalla A.; El-Hassan A.; Raheem M.; Mueller M.; Koummuki Y.; Rashid J.; Mbui J.; Mucee G.; Njoroge S.; Manduku V.; Musibi A.; Mutuma G.; Kirui F.; Lodenyo H.; Mutea D.; Kirigi G.; Edwards T.; Smith P.; Muthami L.; Royce C.; Ellis S.; Alobo M.; Omollo R.; Kesusu J.; Owiti R.; Kinuthia J. Sodium stibogluconate (SSG) & paromomycin combination compared to SSG for visceral leishmaniasis in East Africa: a randomised controlled trial. PLoS Negl. Trop. Dis. 2012, 6, e1674. 10.1371/journal.pntd.0001674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundar S.; Jha T. K.; Thakur C. P.; Engel J.; Sindermann H.; Fischer C.; Junge K.; Bryceson A.; Berman J. Oral miltefosine for Indian visceral leishmaniasis. N. Engl. J. Med. 2002, 347, 1739–1746. 10.1056/NEJMoa021556. [DOI] [PubMed] [Google Scholar]
- Dorlo T. P.; Rijal S.; Ostyn B.; de Vries P. J.; Singh R.; Bhattarai N.; Uranw S.; Dujardin J. C.; Boelaert M.; Beijnen J. H.; Huitema A. D. Failure of miltefosine in visceral Leishmaniasis is associated with low drug exposure. J. Inf. Dis. 2014, 210, 146–153. 10.1093/infdis/jiu039. [DOI] [PubMed] [Google Scholar]
- Mondelaers A.; Hendrickx S.; Van Bockstal L.; Maes L.; Caljon G. Miltefosine-resistant Leishmania infantum strains with an impaired MT/ROS3 transporter complex retain amphotericin B susceptibility. J. Antimicrob. Chemother. 2018, 73, 392–394. 10.1093/jac/dkx407. [DOI] [PubMed] [Google Scholar]
- Atia A. M.; Mumina A.; Tayler-Smith K.; Boulle P.; Alcoba G.; Elhag M. S.; Alnour M.; Shah S.; Chappuis F.; Van Griensven J.; Zachariah R. Sodium stibogluconate and paromomycin for treating visceral leishmaniasis under routine conditions in eastern Sudan. Trop. Med. Int. Health 2015, 20, 1674–1684. 10.1111/tmi.12603. [DOI] [PubMed] [Google Scholar]
- Kapil S.; Singh P. K.; Silakari O. An update on small molecule strategies targeting leishmaniasis. Eur. J. Med. Chem. 2018, 157, 339–367. 10.1016/j.ejmech.2018.08.012. [DOI] [PubMed] [Google Scholar]
- Ferreira L. L. G.; de Moraes J.; Andricopulo A. D. Approaches to advance drug discovery for neglected tropical diseases. Drug Discov Today 2022, 27, 2278–2287. 10.1016/j.drudis.2022.04.004. [DOI] [PubMed] [Google Scholar]
- Drugs for Neglected Diseases initiative. https://dndi.org/research-development/portfolio/ (accessed 5 July 2022).
- Mahender T.; Pankaj W.; Kumar S. P.; Ankur V.; Kumar S. S. Some Scaffolds as Anti-leishmanial Agents: A Review. Mini-reviews in Med. Chem. 2022, 22, 743–757. 10.2174/1389557521666210913115116. [DOI] [PubMed] [Google Scholar]
- Barbolla I.; Hernández-Suárez L.; Quevedo-Tumailli V.; Nocedo-Mena D.; Arrasate S.; Dea-Ayuela M. A.; González-Díaz H.; Sotomayor N.; Lete E. Palladium-mediated synthesis and biological evaluation of C-10b substituted Dihydropyrrolo[1,2-b]isoquinolines as antileishmanial agents. Eur. J. Med. Chem. 2021, 220, 113458. 10.1016/j.ejmech.2021.113458. [DOI] [PubMed] [Google Scholar]
- Bacelar A. H.; Carvalho M. A.; Proença M. F. Synthesis and in vitro evaluation of substituted pyrimido[5,4-d]pyrimidines as a novel class of Antimycobacterium tuberculosis agents. Eur. J. Med. Chem. 2010, 45 (7), 3234–3239. 10.1016/j.ejmech.2010.03.047. [DOI] [PubMed] [Google Scholar]
- Alves M. J.; Booth B. L.; Al-Duaj O. Kh.; Eastwood P.; Nezhat L.; Proença M. F.; Ramos A. S. J. Chem. Res. (S) 1993, 402. [Google Scholar]; J. Chem. Res. (M) 1993, 2701.
- Al-Azmi A.; Booth B. L.; Carpenter R. A.; Carvalho A.; Marrelec E.; Pritchard R. G.; Proença M.F.J.R.P. Facile Synthesis of 6-Cyano-9-Substituted-9H-Purines and Their Ring Expansion to 8-(Arylamino)-4-Imino-3-Methylpyrimidino[5,4-d]Pyrimidines. J. Chem. Soc. Perkin Trans. 1 2001, 2532–2537. 10.1039/b106539b. [DOI] [Google Scholar]
- Ribeiro A.; Carvalho M. A.; Proença M. F. A mild approach to the synthesis of 4-amino-8-(arylamino)pyrimido[5,4-d] pyrimidine 3-oxides. Eur. J. Org. Chem. 2009, 2009, 4867–4872. 10.1002/ejoc.200900216. [DOI] [Google Scholar]
- Carvalho M. A.; Esperança S.; Esteves T.; Proença M. F. 2007. An Efficient Synthesis of 7,8-Dihydropyrimido[5,4-d]pyrimidines. Eur. J. Org. Chem. 2007, 2007, 1324–1331. 10.1002/ejoc.200600883. [DOI] [Google Scholar]
- Rocha A.; Bacelar A. H.; Fernandes J.; Proença M. F.; Carvalho M. A. 6-Carbohydrazonamidepurines as convenient precursors to 4,8-disubstituted pyrimido[5,4-d]pyrimidines. Synlett 2014, 343–348. 10.1055/s-0033-1340344. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.