

Abstract

Glutaminyl cyclases (QC, isoQC) convert N-terminal glutamine or glutamate into pyroglutamate (pGlu) on substrates. IsoQC has recently been demonstrated to promote pGlu formation on the N-terminus of CD47, the SIRPα binding site, contributing to the “don’t eat me” cancer immune signaling of CD47-SIRPα. We developed new QC inhibitors by applying a structure-based optimization approach starting from fragments identified through library screening. Screening of metal binding fragments identified 5-(1H-benzimidazol-5-yl)-1,3,4-thiadiazol-2-amine (9) as a potent fragment, and further modification provided 5-(1-(3-methoxy-4-(3-(piperidin-1-yl)propoxy)benzyl)-1H-benzo[d]imidazol-5-yl)-1,3,4-thiadiazol-2-amine (22b) as a potent QC inhibitor. Treatment with 22b in A549 and H1975 lung cancer cells decreased the CD47/αhCD47-CC2C6 interaction, indicative of the CD47/SIRPα interaction, and enhanced the increased phagocytic activity of both THP-1 and U937 macrophages.
Keywords: QC, isoQC, inhibitor, metalloprotein, fragment, CD47, SIRPα, cancer immunotherapy, fibrosis, Alzheimer’s disease
Glutaminyl cyclases (QCs) include glutaminyl-peptide cyclotransferase (QPCT, QC) and glutaminyl-peptide cyclotransferase-like protein (QPCTL, isoQC).1 Both convert N-terminal glutamine (Gln) or glutamate (Glu) into pyroglutamate (pGlu) of substrates.2 Formation of pGlu at the N-terminus of a peptide or protein leads increased lipophilicity and loss of charge at the N-terminus, which results in maintenance of proteolytic resistance.3 Although both QCs are ubiquitously expressed and are structurally and functionally comparable, their subcellular localization, tissue distribution, and substrate specificity are different.4 QC proceeds in the secretory pathway mainly in neuronal tissue, whereas isoQC is exclusively localized within the Golgi. Although their amount of transcript did not fluctuate significantly in different tissues,5 both QCs show distinct substrate specificity according to a study.6 QC has been reported to predominantly engage in the regulation of neuropeptides, such as TRH7 and GnRH,8 and isoQC mostly engages chemoattractant chemokine ligand 2 (CCL2), collagen, and fibronectin.9 QCs are necessary for substrate stabilization and protein maturation. However, abnormal overexpression of QCs can induce several chronic diseases such as Alzheimer’s disease (AD)10 and fibrosis.11
QC, which is abundantly located in neuronal tissue, catalyzes the cyclization of the N-termini of truncated Aβ, leading to formation of pGlu-Aβ plaques by enhancing conformational stability.12 pGlu-Aβ peptides have been reported to be more neurotoxic than Aβ1–40 and Aβ1–42 and act as seeds for amyloid and tau plaques, because these hydrophobic peptides are more rapidly aggregated and resistant to proteolytic degradation.13 isoQC has also been shown to mature CCL2 via cyclization of the N-termini Glu into the more resistant pGlu form. Mature CCL2 act as a mediator of neurodegenerative diseases by accelerating Aβ oligomer formation in microglia thus aggravating AD.14 Therefore, QCs inhibition may be an effective strategy treating for AD.15
IsoQC and CCL2 are both widely expressed in a variety of different types of cells in tissues and involved in several inflammatory disorders.16 In addition, an advanced effect of CCL2 on fibrosis in the liver and lungs mediated by chronic inflammation has been reported.17 Since macrophages, which are derived from monocytes dependent on CCL2, play an essential role in initiation, mediation, and sustenance of an inflammatory signal in fibrosis,18 inhibition of isoQC contributes to perpetuating CCL2 as an innovative strategy to treat fibrosis.19
IsoQC has been recently recognized as an important modifier of CD47, which is considered a myeloid immune checkpoint known as the “don’t eat me” signal of cancer cells.20 CD47 is a surface glycoprotein that is broadly expressed on all normal cells and overexpressed on tumor cells, especially on non-small-cell lung cancer (NSCLC) tissues and cell lines.21 The pGlu N-terminus of CD47 has been shown to significantly contribute to the interaction between CD47 and SIRPα,22 and isoQC has been shown to be critical for pGlu formation of CD47.23 Biological analysis of the effect of isoQC deletion and isoQC inhibitors on CD47 activity has additionally identified isoQC as a modulator of the CD47-SIRPα pathway.20 These isoQC blockages eventually lead to reduced “don’t eat me” signals of CD47 on tumor cells. Therefore, development of a novel isoQC inhibitor may be a promising strategy for cancer immunotherapy.
Controlling QCs activity are a potential therapy for neurodegenerative disease, inflammatory disorder, and cancer because of the effect of QCs in the progression of Aβ aggregation and consequent increased neurotoxicity in the development of AD, differentiation of macrophages in the inflammatory mechanisms involved in fibrosis, and CD47 involvement in the immune system in cancer. Therefore, several researchers have developed potent QC or isoQC inhibitors. Most representative QC inhibitors are summarized in Figure 1 with their IC50 values.24−27
Figure 1.

Currently developed QC inhibitors (A) and binding mode of 1 with QC (B).
Mammalian QC and isoQC, bacterial aminopeptidases, and a family of zinc hydrolases are closely related in their three-dimensional protein structures, but only mammalian QC and isoQC have a single zinc ion coordinated with Asp, Glu, and His proteins in their substrate binding sites.28 Mammalian QCs are approximately 42 kDa size metalloproteins, and the sequences are highly conserved (similarity: 80–90%). Although several sequences are different at the outer pocket of the metal binding sites in QC and isoQC, the amino acid sequence of QC at the drug binding pockets near the zinc site are identical to that of isoQC. Inspection of X-ray crystal structures of compound 1 with human QC (PDB: 3PBB) and isoQC (PDB: 3PB7)29 shows three distinguishing interactions between QCs and the inhibitors. As shown in Figure 1B, the zinc of QC binds with imidazole of 1, the NH in Gln304 provides a hydrogen bond to the thiourea of 1, and Phe325 is involved in π–π stacking with an aromatic ring of the inhibitor.
Since the zinc binding is regarded as the most potent interaction to decrease the catalytic reaction of QCs, current QC inhibitors use imidazole and benzimidazole as their key scaffolds. Although imidazole and benzimidazole are good zinc chelating groups, one concern is the potential of multiple interactions with other metalloproteins. Therefore, development of new scaffolds that efficiently bind to the zinc ion in the substrate binding site of QCs are necessary for the specific control of various metalloproteins.
In this study, we collected ∼40 potential metal binding fragments and screened them using a QC inhibition assay to identify new zinc binding scaffolds. After identifying a few new fragments, we attached a benzimidazole to the fragment for the additional π–π interaction with the nearby tryptophan residue on QC. An aromatic ring and alkylamine were also added to the molecules to explore additional pharmacophores. Using the developed inhibitor, we investigated whether the compound could block CD47 maturation and consequently the enhancement of macrophage phagocytosis by blocking the CD47/SIRPα axis in lung cancer cells.
To synthesize 10a–j and 11a–j, which have 1,3,4-thiadiazol-2-amine as a zinc binding motif, the intermediate 5-(1H-benzimidazol-5-yl)-1,3,4-thiadiazol-2-amine (9) was prepared via acyl chloride formation of 5-benzimidazolecarboxylic acid (7) with oxalyl chloride, followed by coupling with thiosemicarbazide and cyclization on a sulfuric acid catalyst condition as shown in Scheme 1. Benzylation of 9 with various benzyl bromide under K2CO3 in DMF provided an isomeric mixture of 10a–j and 11a–j. Each regioisomer was separated using prep-HPLC. For the synthesis of 13a–b, the common intermediate 8 was refluxed for 5 h with NaOH to obtain 5-(1H-benzo[d]imidazol-5-yl)-2,4-dihydro-3H-1,2,4-triazole-3-thione (12). In this case, the ring formation occurred via terminal amine condensation with the carbonyl in basic conditions. Further benzylation of 12 with benzyl bromide under K2CO3 in DMF provided the target product 13a–b (Scheme 2). The synthetic procedure for 17 was similar to the synthesis of 10a–j; intermediate 16 was obtained from 14 via a coupling reaction with thiosemicarbazide and cyclization in acidic conditions. The treatment of 3,4-dimethoxybenzyl chloride with 16 in DMF with K2CO3 provided the target compound 17 with a good yield (Scheme 3).
Scheme 1. Synthesis of 10a–j and 11a–j.

Reagents and conditions: (a) oxalyl chloride, cat. DMF, DCM, rt, 4 h; (b) thiosemicarbazide, DMF, rt, 24 h; (c) H2SO4, rt, 4 h; (d) benzyl bromide or chloride, K2CO3, DMF, 60 °C, 3 h.
Scheme 2. Synthesis of 13a,b.

Reagents and conditions: (a) NaOH, (b) benzyl bromide or chloride, K2CO3, DMF, 60 °C, 3 h.
Scheme 3. Synthesis of 17.

Reagents and conditions: (a) oxalyl chloride, cat. DMF, DCM, rt, 4 h; (b) thiosemicarbazide, DMF, rt, 24 h; (c) H2SO4, rt, 4 h; (d) benzyl chloride, K2CO3, DMF, 60 °C, 3 h.
Fragment-based drug discovery (FBDD) is a basic strategy for the initial discovery of biologically active scaffolds. FBDD usually uses a mid-size library (100–1000 molecules) of small fragments (MW < 300 g/mol) to screen against the target protein. Recently, some studies have described the application of FBDD for metalloproteins as the drug target.30 In that study, several small metal chelators of the fragment libraries were shown to effectively inhibit metalloproteins. Here, we collected approximately 40 potential chelators with one to three donor atoms for binding the zinc of QC. The library includes imidazole, hydroxylpyridone, pyranone, pyranthione, quinolinols, quinolin, benzothiazolol, benzimidazole, thioxoimidazolidinone, triazolethione, pyrimidinetrione, β-ketoketones, and the well-known chelators, catechol, hydroxamic acid, and sulfonamide (Table 1). Evaluation of the inhibitory activity of each fragment toward QC was performed using a fluorogenic substrate, Gln-AMC (l-glutamine 7-amido-4-methylcoumarin), and an auxiliary enzyme, pyroglutamyl peptidase (pGAP). In this assay system, QC converts Gln-AMC to pGlu-AMC, and pGAP hydrolyzes pGlu-AMC to AMC. AMC is fluorescent; the quantity of produced AMC is accurately detected by measuring a 460 nm emission signal.
Table 1. QC IC50 Data for Metal Binding Fragments.

NE: no effect.
From the identified library members, eight molecules were found to weakly inhibit QC (IC50 < 500 μM), and two molecules were found to moderately inhibit QC (IC50 ≈ 5 μM). Interestingly, hydroxamic acid, boronic acid, and sulfonamide, which are the most famous metal chelators, did not show binding activity toward QC. This limited hit number and weak binding affinity were somewhat unexpected results since the fragments were collected from known metal chelator pools. However, the results also suggest that zinc ion coordination on the binding site of QC is a more specific chelation event than those of other metalloproteins and requires very well-arranged interactions between a chelator and the zinc metalloprotein.
These results prompted us to explore a new mid-size fragment library (Table 2) in which an aryl group was attached to the initial hit molecules aa, ab, or bh for an additional π–π stacking interaction with Trp207 of QC. Interestingly, the screening showed that a phenyl ring attached to ca and cb was less potent than their parent moieties aa and ab. Compounds cc and ce were also less potent than their parent, bh. However, 5-(1H-benzimidazol-5-yl)-1,3,4-thiadiazol-2-amine (cd, 9) was found to be the most potent inhibitor of QC (IC50 = 190 nM) among the tested fragments. This compound was accidentally synthesized from the intermediate 8 by adding an acid instead of a base during five-membered ring formation.
Table 2. QC IC50 Data of Mid-Size Fragments.

NE: no effect.
ND: not determined.
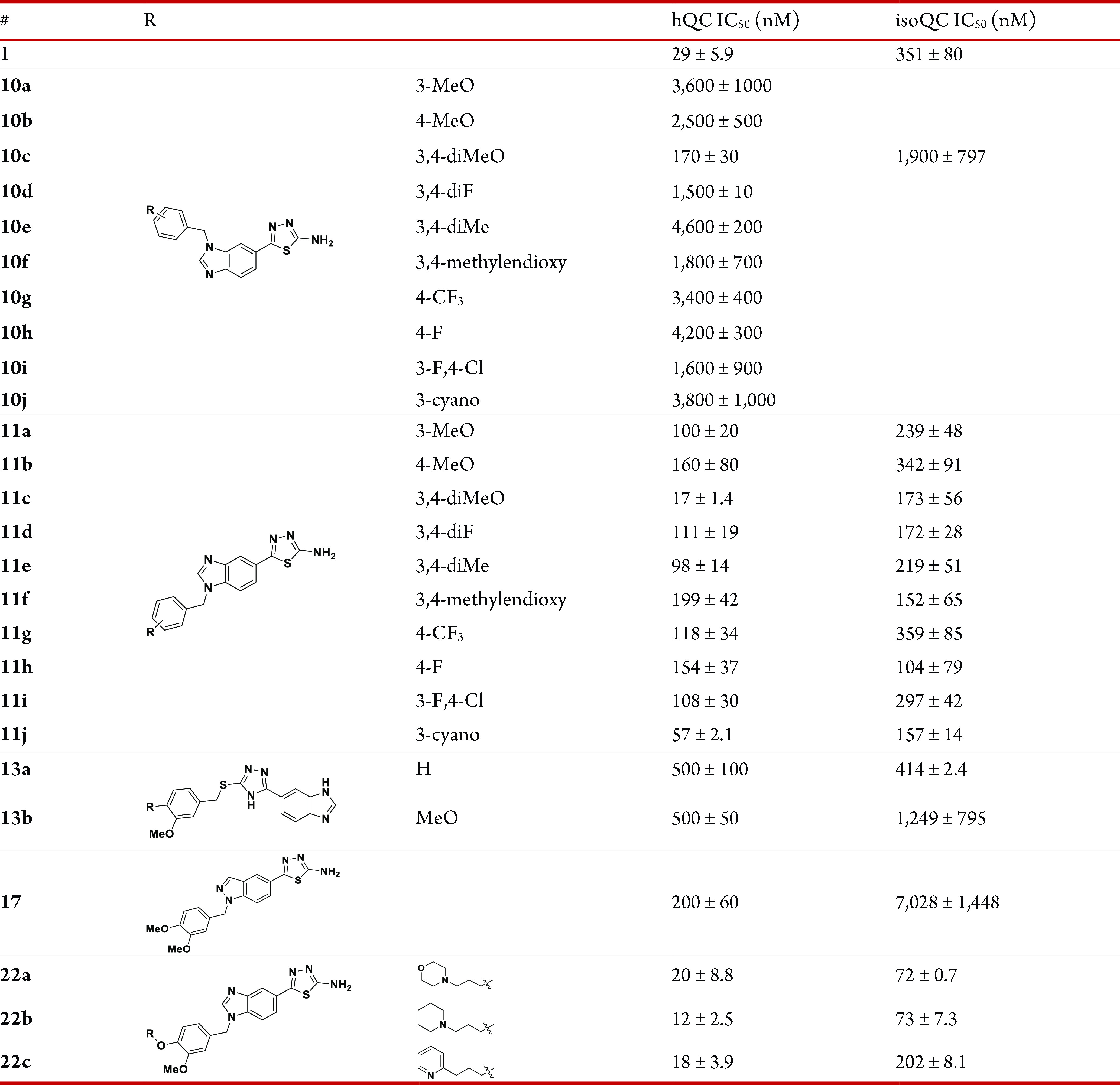
Compound 9 has two potential zinc binding motifs: benzimidazole and amino-thiadiazole. These two motifs can individually undergo benzylation to form 5-(1-benzyl-1H-benzo[d]imidazol-5-yl)-1,3,4-thiadiazol-2-amine or 5-(1H-benzo[d]imidazol-5-yl)-N-benzyl-1,3,4-thiadiazol-2-amine. Since both benzylated products have good docking positions and scores with QC via additional interactions with neighboring residues, we tried to attach various substituted benzyl groups to 9. The docking study also predicted that the meta- or para-substituted benzyl would bind better than the ortho-substituted benzyl; thus, benzylation of 9 was mainly performed with para-, meta-, or both F, Me, OMe, CF3, Cl, or CN substituted benzyl bromide to yield 7-N-benzylated benzimidazole 10a–j and 9-N-benzylated benzimidazole 11a–j. Benzylation on the N of thiadiazole-2-amine was not observed. The products 11a–j, in which the thiadiazol-2-amine is the potential zinc binder, showed a generally improved binding affinity toward both QC and isoQC (Table 3). In particular, the dimethoxybenzyl product 11c was found to be 11-fold improved potency for QC and 2-fold improved potency for isoQC and displayed IC50 values of 17 and 173 nM, respectively. The 7-N-benzylated isomers 10a–j were generally >10-fold weaker than the 9-N-benzylated isomers 11a–j. Although meta-OMe substitution to the phenyl ring appears to advantageous for QC inhibition (11a,c), electronic or steric effects of meta- or para-substitution with OMe, Me, F, CF3, and CN were unclear for both QC and isoQC inhibition. Since direct benzylation to the N of 2-amino-thiadiazole of 9 is difficult, we synthesized the alternative compounds 13a,b in which the benzimidazole is the zinc binding motif, and the 1,3,4-thiadiazol-2-amine of 9 was replaced with 4H-1,2,4-triazole-3-thiol. However, 13a,b displayed reduced inhibitory activity, and IC50 values were approximately 500 nM for QC and 414–1249 nM for isoQC. Compound 17, which was derived from the fragment 16 and had an indazole instead of a benzimidazole of 11c, was 3-fold more potent than 16 for QC (IC50 = 200 nM), but its binding to isoQC was weak and much less potent than that of 11c. Compound 11c was further modified to 22a–c (Scheme 4) by adding 3-morpholinopropyl, 3-(piperidin-1-yl)propyl, and 3-(pyridin-2-nyl)propyl at the para-position of benzyl in 11c. The addition of an aminolic alkyl group on an aryl ring of the QC inhibitor 2 provided enhanced binding interactions to QCs in previous studies.23 The QC inhibitory activity of the synthesized compounds 22a–c was similar to that of their parent molecule 11c. Instead, 22a and 22b displayed 2-fold improved binding to isoQC with an IC50 value of ∼73 nM. It is presumed that the additional cationic aminolic alkyl binds better with isoQC since the binding site of the aminolic alkyl of isoQC is slightly larger than QC and has an anionic environment (Figure 2).
Table 3. QC and isoQC IC50 Data for Compounds 10a–j, 11a–j, 13a,b, 17, and 22a–c.
Scheme 4. Synthesis of 22a–c.

Reagents and conditions: (a) K2CO3, ACN, 50 °C, 12 h; (b) NaBH4, EtOH, rt, 2 h; (c) PBr3, DCM, 0 °C, 10 min; (d) K2CO3, DMF, 120 °C, 2 h.
Figure 2.

Docking results of 22b (orange) with (a) QC (green, PDB: 3PBB) and (b) isoQC (gray, PDB: 3PB7).
An induced-fit docking study using the Schrodinger software package was performed to understand the binding positions of the most potent compound 22b with QC (PDB: 3PBB) and isoQC (PDB: 3PB7).27 The binding modes of 22b with both QCs were highly conserved. The 2-N of 1,3,4-thiadiazol-2-amine was identified as a substituent of water molecules and was coordinated with a single zinc ion for both QC and isoQC (Figure 2). The NH2 of 1,3,4-thiadiazol-2-amine contributes to high potency for QC by interacting with COOH of two Asp (D159 and D248 of QC, D186 and D269 of isoQC). Benzimidazole participates in the π–π stacking interaction, leading to improving binding affinity of 22b for both QCs. In particular, the effect of benzimidazole on the enhanced potency for isoQC may be caused by maintaining an inward orientation of W231 of isoQC, which has an outward orientation only in the isoQC crystal structure. Furthermore, the positively charged piperidine group of 22b bound to the phenyl ring of F325 and F346 of both QCs with cation−π interaction. Although the conformations of 22b for both QCs were highly identical, 22b was approximately 6-fold more effective for QC than isoQC. QC has a bulkier side chain, I303, near the binding site compared with V324 of isoQC. The relatively narrow binding pocket of QC compared with that of isoQC may lead to a better binding with 22b. However, introduction of a posit ively charged piperidine at the para-position of the benzyl ring may also improve the binding affinity with a negatively charged surface around the active site of isoQC compared with 10c without any substituent at the benzyl ring.
Accumulating evidence that CD47 expression is not restricted for myeloid cells and that its overexpression in tumor cells inhibits phagocytosis and evades immune surveillance suggests that CD47 is a promising therapeutic target.31 We investigated the effect of 22b on the viability of human lung cancer cells together with HY-112269 (5) and SEN177 (6) as positive controls for QC inhibitor.32 As shown in Figure 3a, 22b and the two positive controls at the indicated concentrations had no effect on cell growth in A549 and H1975 cells. Considering the 10–80 nM IC50 values of the QC inhibitors, direct cytotoxicity of 22b on lung cancer cells could be excluded.
Figure 3.

Effect of 22b on the expression of mature CD47 on human lung cancer cells. (a) Effect of 22b on human lung cancer cell viability was determined by MTT assay. (b) Expression of CD47 on human lung cancer cells were measured by two human fluorescence-labeled antibodies CD47-CC2C6 and CD47-B6H12. The inhibitory effect of 22b, HY-112269, and SEN177 on αhCD47-CC2C6 (c) and αhCD47-B6H12 (d) expression was examined by flow cytometry. The graph represents means ± SEM of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 vs control.
To examine whether CD47 is expressed in human lung cancer cells, two commercially available anti-human CD47 antibody clones (αhCD47-CC2C6 and αhCD47-B6H12) were used for FACS analysis. αhCD47-CC2C6 is known to bind to the SIRPα recognition site of mature CD47,13 whereas αhCD47-B6H12 is an antibody for the general cell surface of CD47. Both antibodies were detected on lung cancer cells, and high expression of CD47 was observed in A549 and H1975 cells (Figure 3b).
IsoQC inhibition is also known to affect binding of αhCD47-CC2C6 or SIRPα to CD47 by interfering with the CD47 maturation process and pGlu N-terminus formation.13 As shown in Figure 3c, the surface expression level of αhCD47-CC2C6 on A549 and H1975 cells was decreased by 22b, HY-112269, and SEN177 at 24 h and persisted up to 48 h although it was slightly attenuated. Treatment with the isoQC inhibitor 22b did not interfere with αhCD47-B6H12 binding with CD47 (Figure 3d). These results demonstrated that 22b could selectively interfere with the interaction of CD47/SIRPα through isoQC inhibition.
To investigate whether 22b enhances macrophage phagocytosis by blocking the CD47/SIRPα axis, a “don’t eat me” signal in lung cancer cells, we determined phagocytosis of human macrophage THP-1 or U937 cells against agent-treated cancer cells. As shown in Figure 4, the double-positive population in the upper right quadrant region (Q2) indicates phagocytosed CTFR-labeled cancer cells by CFSE-labeled macrophages. Treatment of A549 and H1975 cells with 22b increased the phagocytic activity of both THP-1 and U937 macrophages in a dose-dependent manner. Thus, 22b effectively inhibited pGlu formation of the N-terminus of CD47 in lung cancer cells, consequently blocking the CD47-SIRPα interaction, a “don’t eat me” signal, thereby increasing phagocytosis. Treatment of cancer cells with SEN177 or HY-112269 also increased the phagocytic activity of THP-1 and U937 cells to a similar extent to that of 22b treatment.
Figure 4.

Activation of macrophage phagocytosis by interfering 22b. (a) A549 cells or (b) H1975 cells were labeled with CTFR. Lung cancer cells were co-cultured with CFSE-labeled human macrophages (THP-1 or U937 cells) for 30 min and then treated with 22b, HY-112269 and SEN177. Phagocytosis was determined by flow cytometry. Double-positive Q2 proportion indicates engulfed human cancer cells by macrophages. The graph represents means ± SEM of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 vs control.
In summary, recent research has demonstrated that isoQC is a crucial modifier of CD47 and consequently a regulator of the CD47-SIRPα “don’t eat me” pathway for cancer immune therapy. Thus, isoQC inhibition can promote tumor control by activation of cancer cell phagocytosis.24 In the work described herein, we screened various metal binding fragments and identified 5-(1H-benzimidazol-5-yl)-1,3,4-thiadiazol-2-amine (9) as a new QC binding fragment. Further modification of 9 provided 22b as a potent QC inhibitor with IC50 values of 73 and 17 nM for isoQC and QC, respectively. Treatment of A549 and H1975 cells with 22b decreased the surface expression level of αhCD47-CC2C6 without interfering in αhCD47-B6H12 binding with CD47, representing effective interference of the CD47-SIRPα interaction in lung cancer cells. Treatment of A549 and H1975 cells with compound 22b enhanced the increased phagocytic activity of both THP-1 and U937 macrophages in a dose-dependent manner.
Acknowledgments
This work was supported by National Research Foundation of Korea (NRF) grants funded by MSIT (NRF-2018R1A5A2025286 and NRF-2019R1A2C2004142) and Korea Basic Science Institute (National Research Facilities and Equipment Center) grant funded by the Ministry of Education (2021R1A6C101A442).
Glossary
Abbreviations
- CD47
cluster of differentiation 47
- SIRPα
signal regulatory protein α
- Aβ
amyloid beta
- CCL2
CC chemokine ligand 2
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00256.
Protocol for testing and data analysis of compounds in the QC/isoQC inhibition assay, cell culture, phagocytosis assay, computational chemistry, synthesis methods, experimental data for compounds, NMR spectra of final compounds 22a–c, and HPLC traces of final compounds (PDF)
Author Contributions
E.P., K.-H.S., and D.K. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Becker A.; Kohlmann S.; Alexandru A.; Jagla W.; Canneva F.; Bäuscher C.; Cynis H.; Sedlmeier R.; Graubner S.; Schilling S.; Demuth H. U.; von Hörsten S. Glutaminyl cyclase-mediated toxicity of pyroglutamate-beta amyloid induces striatal neurodegeneration. BMC Neurosci. 2013, 14, 108. 10.1186/1471-2202-14-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling S.; Manhart S.; Hoffmann T.; Ludwig H. H.; Wasternack C. W.; Demuth H. U. Substrate specificity of glutaminyl cyclases from plants and animals. Biol. Chem. 2003, 384, 1583–1592. 10.1515/BC.2003.175. [DOI] [PubMed] [Google Scholar]
- Cynis H.; Hoffmann T.; Friedrich D.; Kehlen A.; Gans K.; Kleinschmidt M.; Rahfeld J. U.; Wolf R.; Wermann M.; Stephan A.; Haegele M.; Sedlmeier R.; Graubner S.; Jagla W.; Müller A.; Eichentopf R.; Heiser U.; Seifert F.; Quax P. H. A.; de Vries M. R.; Hesse I.; Trautwein D.; Wollert U.; Berg S.; Freyse E. J.; Schilling S.; Demuth H. U. The isoenzyme of glutaminyl cyclase is an important regulator of monocyte infiltration under inflammatory conditions. EMBO Mol. Med. 2011, 3, 545–558. 10.1002/emmm.201100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayan D. K.; Zhang K. Y. J. Human glutaminyl cyclase: Structure, function, inhibitors and involvement in Alzheimer’s disease. Pharmacol. Res. 2019, 147, 104342. 10.1016/j.phrs.2019.104342. [DOI] [PubMed] [Google Scholar]
- Cynis H.; Rahfeld J. U.; Stephan A.; Kehlen A.; Koch B.; Wermann M.; Demuth H. U.; Schilling S. Isolation of an isoenzyme of human glutaminyl cyclase: retention in the Golgi complex suggests involvement in the protein maturation machinery. J. Mol. Biol. 2008, 379 (5), 966–980. 10.1016/j.jmb.2008.03.078. [DOI] [PubMed] [Google Scholar]
- Schilling S.; Kohlmann S.; Bäuscher C.; Sedlmeier R.; Koch B.; Eichentopf R.; Becker A.; Cynis H.; Hoffmann T.; Berg S.; Freyse E. J.; von Hörsten S.; Rossner S.; Graubner S.; Demuth H.-U. Glutaminyl cyclase knock-out mice exhibit slight hypothyroidism but no hypogonadism implication for enzyme function and drug development. J. Biol. Chem. 2011, 286, 14199–14208. 10.1074/jbc.M111.229385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waniek A.; Hartlage-Rübsamen M.; Höfling C.; Kehlen A.; Schilling S.; Demuth H. U.; Roßner S. Identification of thyrotropin-releasing hormone as hippocampal glutaminyl cyclase substrate in neurons and reactive astrocytes. Biochim. Biophys. Acta. Mol. Basis. Dis. 2015, 1852, 146–155. 10.1016/j.bbadis.2014.11.011. [DOI] [PubMed] [Google Scholar]
- Pohl T.; Zimmer M.; Mugele K.; Spiess J. Primary structure and functional expression of a glutaminyl cyclase. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 10059–10063. 10.1073/pnas.88.22.10059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker A.; Eichentopf R.; Sedlmeier R.; Waniek A.; Cynis H.; Koch B.; Stephan A.; Bauscher C.; Kohlmann S.; Hoffmann T.; Kehlen A.; Berg S.; Freyse E.-J.; Osmand A.; Plank A.-C.; Roßner S.; von Horsten S.; Graubner S.; Demuth H.-U.; Schilling S. IsoQC (QPCTL) knock-out mice suggest differential substrate conversion by glutaminyl cyclase isoenzymes. Biol. Chem. 2016, 397, 45–55. 10.1515/hsz-2015-0192. [DOI] [PubMed] [Google Scholar]
- Schilling S.; Zeitschel U.; Hoffmann T.; Heiser U.; Francke M.; Kehlen A.; Holzer M.; Hutter-Paier B.; Prokesch M.; Windisch M.; Jagla W.; Schlenzig D.; Lindner C.; Rudolph T.; Reuter G.; Cynis H.; Montag D.; Demuth H. U.; Rossner S. Glutaminyl cyclase inhibition attenuates pyroglutamate Aβ and Alzheimer’s disease–like pathology. Nat. Med. 2008, 14, 1106–1111. 10.1038/nm.1872. [DOI] [PubMed] [Google Scholar]
- Morawski M.; Schilling S.; Kreuzberger M.; Waniek A.; Jäger C.; Koch B.; Cynis H.; Kehlen A.; Arendt T.; Hartlage-Rübsamen M.; Demuth H. U.; Roßner S. Glutaminyl cyclase in human cortex: correlation with (pGlu)-amyloid-β load and cognitive decline in Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 39, 385–400. 10.3233/JAD-131535. [DOI] [PubMed] [Google Scholar]
- Bayer T. A.; Wirths O. Focusing the amyloid cascade hypothesis on N-truncated Abeta peptides as drug targets against Alzheimer’s disease. Acta neuropathol. 2014, 127, 787–801. 10.1007/s00401-014-1287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbaum J. M.; Schilling S.; Cynis H.; Silva A.; Swanson E.; Wangsanut T.; Tayler K.; Wiltgen B.; Hatami A.; Ronicke R.; Reymann K.; Hutter-Paier B.; Alexandru A.; Jagla W.; Graubner S.; Glabe C. G.; Demuth H. U.; Bloom G. S. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-beta. Nature 2012, 485, 651–655. 10.1038/nature11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling M. F.; Luster A. D. Novel approach to inhibiting chemokine function. EMBO Mol. Med. 2011, 3, 510–512. 10.1002/emmm.201100161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu A.; He F.; Yu C.; Qu Y.; Zhang Q.; Lv J.; Zhang X.; Ran Y.; Wei C.; Wu J. The Development of Small Molecule Inhibitors of Glutaminyl Cyclase and Isoglutaminyl Cyclase for Alzheimer’s Disease. ChemistrySelect. 2019, 4, 10591–10600. 10.1002/slct.201902852. [DOI] [Google Scholar]
- Rose C. E. Jr; Sung S. -S. J.; Fu S. M. Significant involvement of CCL2 (MCP-1) in inflammatory disorders of the lung. Microcirculation 2003, 10, 273–288. 10.1080/mic.10.3-4.273.288. [DOI] [PubMed] [Google Scholar]
- Li L.; Wei W.; Li Z.; Chen H.; Li Y.; Jiang W.; Chen W.; Kong G.; Yang J.; Li Z. The spleen promotes the secretion of CCL2 and supports an M1 dominant phenotype in hepatic macrophages during liver fibrosis. Cell. Physiol. Biochem. 2018, 5, 557–574. 10.1159/000495276. [DOI] [PubMed] [Google Scholar]
- Krenkel O.; Tacke F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. 10.1038/nri.2017.11. [DOI] [PubMed] [Google Scholar]
- Ehling J.; Bartneck M.; Wei X.; Gremse F.; Fech V.; Möckel D.; Baeck C.; Hittatiya K.; Eulberg D.; Luedde T.; Kiessling F.; Trautwein C.; Lammers T.; Tacke F. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut 2014, 63, 1960–1971. 10.1136/gutjnl-2013-306294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logtenberg E. M.; Jansen M. J.; Raaben M.; Toebes M.; Franke K.; Brandsma M. A.; Matlung H. L.; Fauster A.; Gomez-Eerland R.; Bakker M. N. A.; van der Schot S.; Marijt K. A.; Verdoes M.; Haanen A. G. J. B.; van den Berg J. H.; Neefjes J.; van den Berg T. K.; Brummelkamp T. R.; Leusen W. J. H.; Scheeren F. A.; Schumacher T. N. Glutaminyl cyclase is an enzymatic modifier of the CD47-SIRPα axis and a target for cancer immunotherapy. Nat. Med. 2019, 25, 612–619. 10.1038/s41591-019-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S.; Jamieson C. H.; Pang W. W.; Park C. Y.; Chao M. P.; Majeti R.; Traver D.; van Rooijen N.; Weissman I. L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009, 138, 271–285. 10.1016/j.cell.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatherley D.; Graham S. C.; Turner J.; Harlos K.; Stuart D. I.; Barclay A. N. Paired receptor specificity explained by structures of signal regulatory proteins alone and complexed with CD47. Mol. Cell. 2008, 31, 266–277. 10.1016/j.molcel.2008.05.026. [DOI] [PubMed] [Google Scholar]
- Matlung H. L.; Szilagyi K.; Barclay N. A.; van den Berg T. K. The CD47-SIRPα signaling axis as an innate immune checkpoint in cancer. Immunol. Rev. 2017, 276, 145–164. 10.1111/imr.12527. [DOI] [PubMed] [Google Scholar]
- Buchholz M.; Hamann A.; Aust S.; Brandt W.; Böhme L.; Hoffmann T.; Schilling S.; Demuth H. U.; Heiser U. Inhibitors for human glutaminyl cyclase by structure based design and bioisosteric replacement. J. Med. Chem. 2009, 52, 7069–7080. 10.1021/jm900969p. [DOI] [PubMed] [Google Scholar]
- Hoang V. H.; Tran P. T.; Cui M.; Ngo V. T. H.; Ann J.; Park J.; Lee J.; Choi K.; Cho H.; Kim H.; Ha H. J.; Hong H. S.; Choi S.; Kim Y. H.; Lee J. Discovery of potent human glutaminyl cyclase inhibitors as anti-Alzheimer’s agents based on rational design. J. Med. Chem. 2017, 60, 2573–2590. 10.1021/acs.jmedchem.7b00098. [DOI] [PubMed] [Google Scholar]
- Ramsbeck D.; Buchholz M.; Koch B.; Böhme L.; Hoffmann T.; Demuth H. U.; Heiser U. Structure–activity relationships of benzimidazole-based glutaminyl cyclase inhibitors featuring a heteroaryl scaffold. J. Med. Chem. 2013, 56, 6613–6625. 10.1021/jm4001709. [DOI] [PubMed] [Google Scholar]
- Hoffmann T.; Meyer A.; Heiser U.; Kurat S.; Böhme L.; Kleinschmidt M.; Bühring K. U.; Hutter-Paier B.; Farcher M.; Demuth H. U.; Lues L.; Schilling S. Glutaminyl Cyclase Inhibitor PQ912 Improves Cognition in Mouse Models of Alzheimer’s Disease—Studies on Relation to Effective Target Occupancy. J. Pharmacol. Exp. Ther. 2017, 362, 119–130. 10.1124/jpet.117.240614. [DOI] [PubMed] [Google Scholar]
- Stephan A.; Wermann M.; von Bohlen A.; Koch B.; Cynis H.; Demuth H. U.; Schilling S. Mammalian glutaminyl cyclases and their isoenzymes have identical enzymatic characteristics. FEBS J. 2009, 276, 6522–6536. 10.1111/j.1742-4658.2009.07337.x. [DOI] [PubMed] [Google Scholar]
- Huang K. F.; Liaw S. S.; Huang W. L.; Chia C. Y.; Lo Y. C.; Chen Y. L.; Wang A. H. J. Structures of human Golgi-resident glutaminyl cyclase and its complexes with inhibitors reveal a large loop movement upon inhibitor binding. J. Biol. Chem. 2011, 286, 12439–12449. 10.1074/jbc.M110.208595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S. M. A Bioinorganic Approach to Fragment-Based Drug Discovery Targeting Metalloenzymes. Acc. Chem. Res. 2017, 50, 2007–2016. 10.1021/acs.accounts.7b00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong B.; Wang M. CD47 is a novel potent immunotherapy target in human malignancies: current studies and future promises. Future Oncol. 2018, 14, 2179–2188. 10.2217/fon-2018-0035. [DOI] [PubMed] [Google Scholar]
- Li M.; Dong Y.; Yu X.; Li Y.; Zou Y.; Zheng Y.; He Z.; Liu Z.; Quan J.; Bu X.; Wu H. Synthesis and Evaluation of Diphenyl Conjugated Imidazole Derivatives as Potential Glutaminyl Cyclase Inhibitors for Treatment of Alzheimer’s Disease. J. Med. Chem. 2017, 60, 6664–6677. 10.1021/acs.jmedchem.7b00648. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.