

Abstract
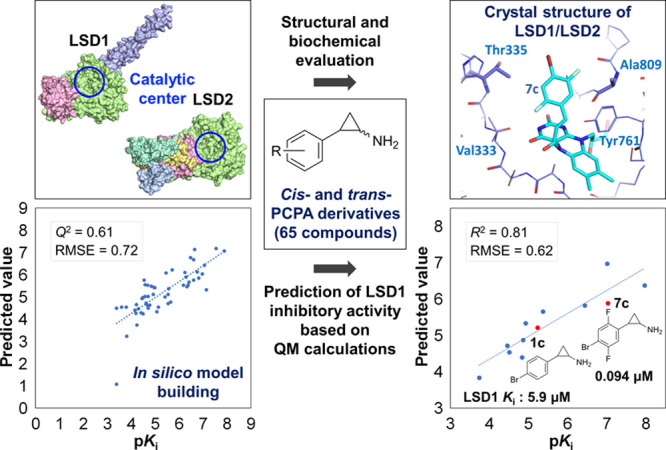
trans-2-Phenylcycloproylamine (trans-PCPA) has been used as the scaffold to develop covalent-binding inhibitors against lysine-specific demethylase 1 (LSD1/KDM1A), a therapeutic target for several cancers. However, the effects of different structural moieties on the inhibitory activity, selectivity, and reactivity of these derivatives, including the cis isomers, against LSD1 and its paralogue LSD2/KDM1B are not fully understood. Here we synthesized 65 cis- and trans-PCPA derivatives and evaluated their inhibitory activity against LSD1 and LSD2. One of the derivatives, 7c (cis-4-Br-2,5-F2-PCPA; S1024), inhibited LSD1 and LSD2 with Ki values of 0.094 μM and 8.4 μM, respectively, and increased the level of dimethylated histone H3 at K4 in CCRF-CEM cells. A machine learning-based regression model (Q2 = 0.61) to predict LSD1-inhibitory activity was also constructed and showed a good prediction accuracy (R2 = 0.81) for 12 test-set compounds, including 7c. The present methodology would be useful when designing covalent-binding inhibitors for other enzymes.
Keywords: epigenetics, fragment molecular orbital, histone, inhibitor, X-ray crystallography
Lysine-specific demethylase 1 (LSD1) is a flavin adenine dinucleotide (FAD)-dependent amine oxidase that primarily demethylates mono- and dimethylated lysine at the fourth (H3K4) and ninth (H3K9) position in histone H3. The catalytic domain of LSD1 is homologous to that of the monoamine oxidases (MAOs), which also use FAD to oxidize substrate amines.1 Aberrant expression of LSD1 is implicated in the maintenance of cancer stem cells, making LSD1 a potential therapeutic target for the treatment of several cancers,2 including T-cell acute lymphoblastic leukemia (T-ALL).3 LSD2, a paralogue of LSD1, also demethylates H3K4 and possesses a catalytic domain homologous to that of LSD1,4 although LSD2 interacts with the nucleosome differently from LSD1,1,5 and plays distinct biological roles with its protein partners. LSD2 is implicated in a diverse range of cellular processes including maternal germline imprinting and carcinogenesis.1
Many inhibitors of LSD1 have been developed, such as trans-2-phenylcyclopropylamine (trans-PCPA), which covalently binds to FAD at the bottom of the catalytic pocket thereby arresting FAD-dependent oxidative demethylation by LSD1.6 Although trans-PCPA may have the advantage of being a drug as an MAO inhibitor,7,8 it is a weak nonselective inhibitor of LSD1. To enhance its potency and selectivity toward LSD1, researchers have introduced substituents to the phenyl ring, cyclopropyl ring, and amine group of trans-PCPA.1,9−12 However, there are currently several knowledge gaps regarding these derivatives. For example, although PCPA has two chiral centers, yielding four stereoisomers (i.e., two cis and two trans enantiomers), almost all of the PCPA-based LSD1 inhibitors reported so far have been in the trans form; exceptions being the compounds used in studies of the four stereoisomers each of PCPA, 4-Br-PCPA,13 and benzoylamino-PCPA derivatives,14 and studies of derivatives with substituents on the cyclopropyl ring.15,16 Similarly, studies examining the inhibition of LSD2 by such PCPA derivatives are also limited.13,17−19 Here, to elucidate the structural determinants of the inhibitory activity of PCPA derivatives against LSD1 and LSD2, we synthesized 65 PCPA derivatives in both their cis and trans forms and examined the relationships between their inhibitory activities and quantum chemical properties. 4-Br-PCPA was selected as the scaffold from which to construct the derivatives because it has several times stronger inhibitory activity against both LSD1 and LSD2 compared with PCPA.13 The structures of the compounds are shown in Table 1, Tables S1 and S2, and Figure S7 (compounds 1c/1t to 35c/35t, where c = cis and t = trans). The synthesized derivatives were all racemic (i.e., (±)-cis isomers and (±)-trans isomers).
Table 1. In Vitro Histone Demethylation Inhibition Assay of PCPA Derivativesa.
 |
Ki (μM) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| ID | R2 | R3 | R4 | R5 | R6 | cis/trans | LSD1 | LSD2 | selectivity |
| 1c | H | H | Br | H | H | cis | 5.9 ± 0.65 | 80 ± 5.6 | 14 |
| 1t | trans | 2.9 ± 0.40 | 740 ± 88 | 260 | |||||
| 2c | H | F | Br | H | H | cis | 0.66 ± 0.048 | 55 ± 4.3 | 83 |
| 2t | trans | 0.50 ± 0.079 | 280 ± 40 | 550 | |||||
| 3c | H | OH | Br | H | H | cis | 34 ± 5.4 | 88 ± 14 | 2.6 |
| 3t | trans | 28 ± 5.1 | 290 ± 30 | 10 | |||||
| 4c | F | H | Br | H | H | cis | 0.47 ± 0.040 | 29 ± 2.3 | 62 |
| 4t | trans | 0.42 ± 0.040 | 270 ± 32 | 640 | |||||
| 5c | CF3 | H | Br | H | H | cis | 0.38 ± 0.033 | 250 ± 41 | 660 |
| 5t | trans | 0.24 ± 0.023 | 390 ± 37 | 1600 | |||||
| 6c | F | F | Br | H | H | cis | 0.069 ± 0.011 | 17 ± 1.4 | 240 |
| 6t | trans | 0.22 ± 0.017 | 99 ± 13 | 450 | |||||
| 7c | F | H | Br | F | H | cis | 0.094 ± 0.0057 | 8.4 ± 0.43 | 89 |
| 7t | trans | 0.098 ± 0.0071 | 180 ± 17 | 1800 | |||||
| 8c | H | F | Br | F | H | cis | 0.011 ± 0.0047 | 7.0 ± 0.72 | 640 |
| 8t | trans | 0.11 ± 0.013 | 130 ± 13 | 1300 | |||||
| 9c | Cl | H | Br | F | H | cis | 0.013 ± 0.010 | 20 ± 1.2 | 1600 |
| 9t | trans | 0.027 ± 0.0092 | 100 ± 10 | 3700 | |||||
| 10c | CH3 | H | Br | F | H | cis | 0.56 ± 0.050 | 48 ± 3.5 | 86 |
| 10t | trans | 0.32 ± 0.055 | 280 ± 30 | 860 | |||||
| 11c | OCH2C6H5 | H | Br | H | F | cis | 8.3 ± 0.85 | >500 | >60 |
| 11t | trans | 12 ± 1.6 | 490 ± 46 | 41 | |||||
| 12c | F | H | H | H | H | cis | 11 ± 0.93 | 70 ± 4.8 | 6.1 |
| 12t | trans | 20 ± 1.9 | >500 | >24 | |||||
| 13c | F | H | H | F | H | cis | 3.4 ± 0.37 | 53 ± 4.0 | 16 |
| 13t | trans | 1.9 ± 0.14 | >500 | >260 | |||||
| 14c | OCH2C6H5 | H | H | H | F | cis | 61 ± 8.3 | 370 ± 26 | 6.0 |
| 14t | trans | 58 ± 8.6 | 240 ± 13 | 4.1 | |||||
| 15c | OCH2C6H5 | H | H | F | F | cis | 10 ± 1.1 | >500 | >49 |
| 15t | trans | 14 ± 1.4 | >500 | >36 | |||||
| 16c | OCH2C6H5 | F | H | H | F | cis | 7.3 ± 0.67 | >500 | >68 |
| 16t | trans | 26 ± 2.4 | 200 ± 6.5 | 7.5 | |||||
| 17c | OC6H5 | H | H | H | H | cis | 12 ± 1.5 | >200 | >17 |
| 17t | trans | 19 ± 2.8 | >1000 | >53 | |||||
| 18c | H | F | OCH3 | H | H | cis | 35 ± 3.8 | >200 | >5.7 |
| 18t | trans | 32 ± 3.8 | >200 | >6.3 | |||||
| 19c | H | F | OCH2CH3 | H | H | cis | 31 ± 3.2 | 290 ± 24 | 9.4 |
| 19t | trans | 19 ± 2.0 | >500 | >26 | |||||
| 20c | H | F | CF3 | H | H | cis | NA | NA | NA |
| 20t | trans | 0.15 ± 0.017 | 180 ± 17 | 1200 | |||||
| 21c | F | H | Cl | F | H | cis | 0.37 ± 0.028 | 35 ± 2.3 | 95 |
| 21t | trans | 0.25 ± 0.036 | >500 | >2000 | |||||
| 22c | F | H | (3-OCH3)C6H4 | F | H | cis | 0.18 ± 0.027 | 7.0 ± 1.0 | 40 |
| 22t | trans | 0.087 ± 0.016 | 86 ± 15 | 990 | |||||
NA, not available.
To begin, we conducted a pair interaction energy decomposition analysis (PIEDA) with fragment molecular orbital (FMO) calculation20 to obtain the interaction energies between the amino acid residues of LSD1 and (+)-cis-4-Br-PCPA (an enantiomer of 1c, Table 1). For the interaction between LSD1 and (+)-cis-4-Br-PCPA (PDB ID: 2XAF),13 the largest attraction was found for Tyr761, with the CH/π interaction between the p-orbital of Tyr761 and the hydrogen atom of the 4-bromophenyl ring being −5.4 kcal/mol (Figure 1A, B and Figures S1 and S2). Thr810 had a weak attractive CH/π interaction with the 4-bromophenyl ring that was offset by an exchange repulsion. Asn540 and Asp555 both had weak electrostatic attractions with the 4-bromophenylethyl fragment. No other notable interactions involving residues near to the 4-bromophenylethyl fragment, such as Val334 and Ala809, were observed, indicating that molecular modification could potentially yield derivatives with more residue-level interactions.
Figure 1.

Residue-level interactions between LSD1 or LSD2 and cis-4-Br-PCPA (PDB ID: 2XAF(13) and 7XE1, respectively). (A) Interactions between LSD1 and (+)-cis-4-Br-PCPA. The (+)-cis-4-Br-PCPA-derived portion of the adduct is colored yellow, FAD is colored gray, and the LSD1 residues are colored according to the interaction energy color key. ES, electrostatic; EX, exchange repulsion; CT+mix, charge transfer with higher-order mixed terms; DI, dispersion interaction. (B) Interaction energies stratified by interaction term, as determined by the FMO method. (C) Crystal structure of LSD2 and (±)-cis-4-Br-PCPA (1c) (PDB ID: 7XE1). The adduct and the LSD2 residues are colored in magenta and salmon, respectively. The omit mFo-DFc map contoured at +3.0 σ is shown as a green mesh. The anomalous difference map contoured at 5.0 σ is shown as an orange mesh. (D) Superimposition of the structures of LSD1–(+)-cis-Br-PCPA and LSD2–(±)-cis-4-Br-PCPA (1c). The adduct and the residues in LSD1 are colored cyan and blue, respectively. The LSD2 structure is depicted as in panel C.
We then determined the crystal structure of LSD2 complexed with (±)-cis-4-Br-PCPA (1c) and compared it with that of LSD1 complexed with (+)-cis-4-Br-PCPA. In the structure of LSD2 in complex with racemic 1c, the phenyl ring was found to stick out into the catalytic pocket (Figure 1C), which was basically the same inhibitor–FAD adduct structure as that of LSD1 with (+)-cis-4-Br-PCPA (Figure 1D). This phenyl ring orientation was different from that in the complex of LSD2 with unsubstituted trans-PCPA (PDB ID: 4GUU; Figure S3). We also determined the crystal structure of LSD2 complexed with 1t (PDB ID: 7XE2; Figure S4). The phenyl ring was oriented mainly in a lateral direction, and the derivative appeared to be connected via the benzyl carbon atom to the C4a atom of the flavin, which is a configuration observed in the structure of MAO-B in complex with trans-PCPA.21
Next, we examined the effects of a variety of substituents on the inhibitory activity of the parent compound, 1c/1t. We introduced different substituents at the meta and ortho positions of the phenyl ring of 4-Br-PCPA (Table 1), which is where the interaction analysis indicated that there was room for modification to improve the binding with LSD1. Fluorine was used as the main substituent because fluorine substitutions have been shown to enhance the potency of PCPA.22,23 Inhibition of LSD1 and LSD2 was measured by means of the peroxidase-coupled method, using K4-dimethylated H3 (1–20) peptide as the substrate.24 The Ki of the parent compounds, 1c and 1t, was 5.9 and 2.9 μM, respectively.
Single substitution with a fluorine atom at the meta- or ortho-position of 4-Br-PCPA (2c/2t and 4c/4t) provided markedly increased inhibitory activity against LSD1 (Ki = 0.66/0.50 and 0.47/0.42 μM, respectively). In both cases, the cis and trans forms exhibited nearly the same activity levels, which was not the case for the parent compounds for which the trans form exhibited stronger inhibitory activity than the cis form. The same substitution did not much enhance the inhibitory activity against LSD2. Substituting with a hydroxy group (3c/3t) at the meta position decreased the inhibitory activity against LSD1. Substituting with a trifluoromethyl group at the ortho position increased the inhibitory activity against LSD1 of both the cis (5c) and trans (5t) forms.
Substitution of two fluorines (6c/6t, 7c/7t, and 8c/8t) considerably increased the inhibitory activity against LSD 1 (Ki = 0.011–0.22 μM). 7c/7t both exhibited strong inhibitory activity (Ki < 0.1 μM). 6c/6t and 8c/8t showed stronger inhibitory activity in the cis form than in the trans form. Of these six derivatives, 8c showed the strongest inhibitory activity (Ki = 0.011 μM), which was a sixty-fold enhancement over the single meta-fluorine derivative 2c and a 10-fold enhancement over the trans counterpart 8t. Introducing a chlorine atom at the ortho position also considerably increased the inhibitory activity of both the cis (9c) and trans (9t) forms. Together, these results show the potential for cis-PCPA derivatives to be potent LSD1 inhibitors. Against LSD2, double fluorine substitutions markedly increased the inhibitory activity of the cis forms (7c and 8c; Ki = 8.4 and 7.0 μM, respectively), but not the trans forms (7t and 8t; Ki = 180 and 130 μM, respectively), into the single micromolar range, which is a one-order-of-magnitude enhancement compared with the parent compound 1c. Other than halogens, addition of a methyl group (10c/10t) to 4-Br-3-F-PCPA (2c/2t) did not change the inhibitory activity against LSD1 or LSD2, and addition of benzyloxy group (11c/11t), a part of the LSD1 inhibitor S2101,22 to the other ortho position in 4-Br-2-F-PCPA (4c/4t) did not improve the inhibitory activity against LSD1 or LSD2.
Derivatives debrominated at the para position (12c/12t to 17c/17t) showed weak inhibition against LSD1 and residual to no inhibition against LSD2, indicating the significance of the bromine atom at the para position. Trifluoromethyl substitution at the para position of 3-F-PCPA (20t), and chlorine or 3-methoxylphenyl substitution at the para position of 2,5-F2–PCPA (21c/21t and 22c/22t), showed submicromolar inhibitory activity against LSD1 (Table 1). For 22, the cis form, but not the trans form, showed micromolar inhibitory activity against LSD2. Extended substitutions at the para position of PCPA (23t, 24c, 25t, and 26c/26t to 30c/30t, Table S1) did not improve inhibitory activity for LSD1. The Ki values of another 10 derivatives, which were used in the in silico calculation described below, are shown in Table S2 (31c/31t to 35c/35t).
Generally, the cis derivatives showed stronger inhibitory activity against LSD2 than their trans counterparts, which can be attributed to steric hindrance in LSD2. Several residues at the catalytic sites in LSD2 are bigger than those in LSD1 (Figure 1D). Indeed, the adduct formed by trans-Br-PCPA with FAD had a different configuration from the adduct formed by cis-Br-PCPA (Figure S4), reflecting a different positioning of the molecule with respect to FAD before the reaction. However, this does not preclude the possibility of developing potent trans-PCPA-based LSD2 inhibitors, because trans-PCPA derivatives with a bulky benzamide substituent at the para position of the phenyl ring still inhibit LSD2 with a single-digit micromolar IC50.18
Compounds 7c/7t and 8c/8t, which showed strong inhibitory activity against both LSD1 and LSD2, did not inhibit peroxidase (IC50 > 500 μM; Figure S5), indicating that the Ki values measured in the LSD1 and LSD2 assays reflect the derivative-mediated inhibition of demethylation activity.
Next, we evaluated the effects of the derivatives exhibiting relatively potent LSD1-inhibitory activity on cell proliferation in two human T-ALL cell lines (CCRF-CEM and Jurkat) in which overexpression of LSD1 is associated with cancer malignancy25 (Table 2). Compound 7c inhibited proliferation of the T-ALL cell lines with an IC50 of 12 μM and 16 μM, respectively, without inhibiting the human normal fibroblast cell line WI-38. Among the tested derivatives, the cis isomers more favorably and weakly inhibited hERG in vitro than the trans ones, except for 7 (Table 2). Treatment of CCRF-CEM cells with 20 μM 7c (S1024) for 24 h significantly increased the level of dimethylated H3K4 (H3K4me2) 2.9-fold (**p < 0.01) compared with untreated cells (Figure 2). In contrast, 7t (S1025) had no effect on the H3K4me2 level, suggesting that substantial chemical inhibition of both LSD1 and LSD2 is important for increasing the H3K4me2 level in these cells.
Table 2. Cell Proliferation Inhibition Assay or Br-PCPA and Its Fluorinated Derivativesa.
| cell
assay IC50 (μM) |
||||
|---|---|---|---|---|
| ID | CCRF-CEM | Jurkat | WI-38 | hERG |
| 1c | ND | ND | ND | 18 ± 2.7 |
| 1t | ND | ND | ND | 13 ± 1.2 |
| 2c | 110 ± 1.6 | 57 ± 1.9 | 89 ± 0.9 | 44 ± 7.8 |
| 2t | 49 ± 0.6 | 44 ± 0.7 | 67 ± 0.8 | 16 ± 2.3 |
| 6c | 14 ± 0.3 | 18 ± 2.9 | >50 | 35 ± 4.7 |
| 6t | 25 ± 0.4 | 28 ± 1.2 | >50 | 15 ± 2.4 |
| 7c | 12 ± 0.1 | 16 ± 1.3 | >50 | 7.3 ± 1.8 |
| 7t | 23 ± 0.8 | 26 ± 1.7 | >50 | 7.7 ± 1.3 |
| 8c | 25 ± 0.5 | 27 ± 0.3 | >50 | ND |
| 8t | >50 | >50 | >50 | ND |
ND, not determined.
Figure 2.

Dimethylation at H3K4 in human CCRF-CEM cells treated with 7c or 7t. (A) Western blotting. (B) Relative intensity. Cells were cultured for 24 h with a 20 μM concentration of the indicated compound containing 0.5% DMSO, and nuclear extracts were analyzed by Western blotting. -, 0.5% DMSO alone. Data are shown as mean ± standard error (n = 3) and were statistically analyzed by using the independent-samples one-sided t-test (**p < 0.01).
To confirm the binding mode of the derivative that showed high potency, we determined the crystal structures of LSD1 and LSD2 in complex with 7c (PDB ID: 7W3L and 7XE3, respectively). The adduct formed from 7c and FAD in LSD1 had basically the same structure as that of (+)-cis-4-Br-PCPA and FAD (PDB ID: 2XAF)13 with respect to the position and orientation of the 4-bromophenyl ring (Figure 3A). The meta-fluorine was orientated to Ala809 and the ortho-fluorine on the other side of the phenyl ring was oriented to Val333. The adduct formed from 7c and FAD in LSD2 had the same overall structure as that found for LSD1; however, the meta-fluorine in the LSD2 complex was oriented to Asn440 (Figure 3B). The electron density of the side chain of Asn440 was not clearly visible, suggesting that its conformation was not fixed. We calculated the interaction energies by PIEDA between LSD1 and the 7c–FAD (Figure 3C and 3D). Compared with the interactions between LSD1 and (+)-cis-4-Br-PCPA (Figure 1A, B), 7c retained the interaction with Tyr761 and stabilized the interaction with neighboring residues, such as Ala809, Val333, and Thr810 (Figure 3C, D and Figure S6A, B), of which the contribution of Thr810 increased the most. Based on interfragment interaction energy (IFIE) analysis, the sum of the interaction energies between LSD1 and the 1c–FAD and 7c–FAD adducts was −24.9 kcal/mol and −29.2 kcal/mol, respectively, demonstrating that 7c interacted more favorably with LSD1 than did 1c.
Figure 3.

Crystal structures of LSD1 or LSD2 with 7c–FAD. (A) Structure of 7c–FAD in complex with LSD1 (PDB ID: 7W3L). The adduct molecule and the residues are colored in cyan and blue, respectively. The omit mFo-DFc map contoured at +4.0 σ is shown as a green mesh. The anomalous difference map contoured at 6.0 σ is shown as an orange mesh. (B) Structure of 7c–FAD in complex with LSD2 (PDB ID: 7XE3). The molecules and the maps are depicted as in Figure 1C. The side chain of Asn440 was not modeled because of a lack of electron density. (C) Interactions between individual LSD1 residues and 7c. The 7c-derived portion of the adduct is colored yellow, and the LSD1 residues are colored according to the interaction energy color key. (D) Interaction energies stratified by interaction term, as determined by the FMO method.
The concomitant enhancement of the LSD1-inhibitory activity of the cis and trans forms suggested that the reactivity of the derivatives was a more dominant determinant of inhibitory activity than stereoisomerism. This reactivity is related to the quantum chemical properties of the molecule and depends on the substituents in the derivative. Therefore, to further investigate the structure–activity relationship, we constructed a regression model by machine learning using the 65 compounds listed in Table 1 and Tables S1 and S2. The compounds were divided into 53 compounds as the training set and 12 as the test set. Using 12 physical property values as the descriptors (i.e., the atomic charges of 10 heavy atoms on the PCPA scaffold, HOMO, and LUMO; Table S3), the regression model was constructed by a support vector machine. In the 5-fold cross-validation, the model showed good performance (Q2 = 0.61 and RMSE = 0.72; Figure 4A). The prediction accuracy of the constructed regression model was evaluated using the 12 compounds in the test set, which included the starting compound 1c and potent compounds 7c and 8c. The correlation of pKi between experimental and predicted values (R2 = 0.81 and RMSE = 0.62; Figure 4B) indicated that the constructed regression model had good prediction performance. The contributions of the descriptors in the constructed model (Table S3) confirmed that changes in the atomic charges of the ipso and para carbons (C5 and C8) on the phenyl ring affected the activity the most and that these changes were the result of a change in the overall electronic state by addition of the substituent to the phenyl ring. The atomic charges of the cyclopropylamine moiety (N1, C2, C3, and C4), which are involved in the reaction with FAD, were also major contributors to the activity.
Figure 4.
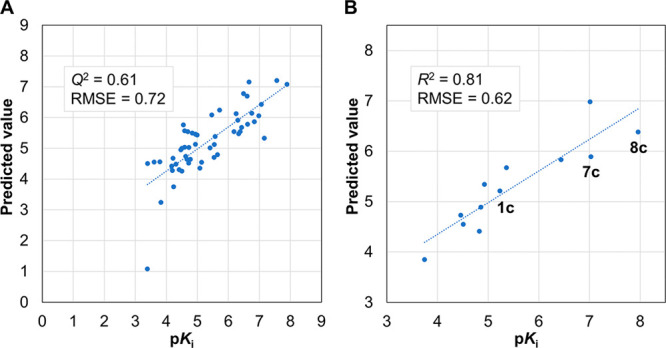
Correlation of experimental and predicted pKi values for PCPA derivatives. (A) Five-fold cross-validation for the training set. (B) Correlation using the test set based on the regression model.
The results of the inhibitory activity prediction support that the high inhibitory activity of the present derivatives against LSD1 was mainly a result of their reactivity, which was dependent on the substituents in each derivative. In addition, as the interaction energy analysis for LSD1 and 7c–FAD demonstrates, the stability of the adduct also contributes to increasing the inhibitory activity. When a covalent-binding compound inactivates its target, it must first access the reaction site and position itself in a suitable orientation with respect to the reaction partner. For better prediction of inhibitory activity, the current in silico analysis could be integrated with molecular docking analysis because the interactions in the prereaction stage are presumably important determinants for the activity of the inhibitor. Studies that integrate all three stages—molecular docking analysis for the prereaction stage, quantum chemical analysis for the reaction stage, and tertiary structural analysis for the postreaction stage—will be pivotal for the rational design and evaluation of potent covalent-binding enzyme inhibitors.
In conclusion, we synthesized PCPA derivatives in cis and trans forms and evaluated their inhibitory activity against LSD1 and LSD2. Both the cis and trans forms of Br-PCPA derivatives with a fluorine and either another fluorine or a chlorine atom showed high inhibitory activity against LSD1. A significant increase in H3K4me2 level induced by 7c (S1024) in CCRF-CEM cells suggested the importance of dual inhibition of LSD1 and LSD2. A machine learning-based model constructed by using quantum chemical descriptors for predicting the inhibitory activity of a derivative agreed well with the experimentally determined inhibitory activity for LSD1. We expect the present methodology to be applicable for predicting the inhibitory activity of other covalent-binding inhibitors.
Acknowledgments
We thank Mai Yamazaki (Kogakuin University), Shun Hashimoto (Kogakuin University), and Hideki Watanabe (Tokyo Chemical Industry Co., Ltd.) for compound synthesis; Shiori Toyama (Kogakuin University), Kanta Nakayama (Kogakuin University), and Hideki Mizutani (Kogakuin University) for compound analysis; Mariko Ikeda (RIKEN), Sayako Kohno (RIKEN), and Noboru Ohsawa (RIKEN) for preparation of LSD2; Tomohiro Sato (RIKEN) for machine learning; and the staff at BL26B2 (No. 20140020) of SPring-8 (Hyogo, Japan) and X06DA (20172015) of the Swiss Light Source (Paul Scherrer Institut, Villigen, Switzerland) for X-ray data collection. This study was supported by the Japan Society for the Promotion of Science (JSPS) (JP21K06461 to K.M.; JP20H03388 and JP20K21406 to T.U.); the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research: BINDS) of the Japan Agency for Medical Research and Development (AMED) (JP21am0101113 to T.H.); and the Drug Discovery Informatics project of AMED to T.H.
Glossary
Abbreviations
- PCPA
2-phenylcycloproylamine
- LSD1
lysine-specific demethylase 1
- LSD2
lysine-specific demethylase 2
- MAO
monoamine oxidase
- T-ALL
T-cell acute lymphoblastic leukemia
- PIEDA
pair interaction energy decomposition analysis
- FMO
fragment molecular orbital
- PDB
protein data bank
- ES
electrostatic
- EX
exchange-repulsion
- CT+mix
charge transfer with higher-order mixed terms
- DI
dispersion interaction
- IFIE
interfragment interaction energy
- RMSE
root-mean-square error
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00294.
Figures S1–S7, Tables S1 and S2 for compounds 23t, 24c, 25t, and 26c/26t to 35c/35t, and Tables S3 and S4; experimental details for biochemical procedures, crystallographic methods, in silico calculations, and chemistry (PDF)
Author Contributions
† H.N. and C.W. contributed equally to this work.
The authors declare no competing financial interest.
Special Issue
Published as part of the ACS Medicinal Chemistry Letters special issue “Epigenetics 2022”.
Supplementary Material
References
- Niwa H.; Umehara T. Structural insight into inhibitors of flavin adenine dinucleotide-dependent lysine demethylases. Epigenetics 2017, 12, 340–352. 10.1080/15592294.2017.1290032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakaidos P.; Verigos J.; Magklara A. LSD1/KDM1A, a gate-keeper of cancer stemness and a promising therapeutic target. Cancers 2019, 11, 1821. 10.3390/cancers11121821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T.; Koyama D.; Kikuchi J.; Honda H.; Furukawa Y. Overexpression of the shortest isoform of histone demethylase LSD1 primes hematopoietic stem cells for malignant transformation. Blood 2015, 125, 3731–3746. 10.1182/blood-2014-11-610907. [DOI] [PubMed] [Google Scholar]
- Karytinos A.; Forneris F.; Profumo A.; Ciossani G.; Battaglioli E.; Binda C.; Mattevi A. A novel mammalian flavin-dependent histone demethylase.. J. Biol. Chem. 2009, 284, 17775–17782. 10.1074/jbc.M109.003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marabelli C.; Marrocco B.; Pilotto S.; Chittori S.; Picaud S.; Marchese S.; Ciossani G.; Forneris F.; Filippakopoulos P.; Schoehn G.; Rhodes D.; Subramaniam S.; Mattevi A. A tail-based mechanism drives nucleosome demethylation by the LSD2/NPAC multimeric complex. Cell Rep. 2019, 27, 387–399. 10.1016/j.celrep.2019.03.061. [DOI] [PubMed] [Google Scholar]
- Schmidt D. M.; McCafferty D. G. trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 2007, 46, 4408–4416. 10.1021/bi0618621. [DOI] [PubMed] [Google Scholar]
- Khan M. N.; Suzuki T.; Miyata N. An overview of phenylcyclopropylamine derivatives: biochemical and biological significance and recent developments. Med. Res. Rev. 2013, 33, 873–910. 10.1002/med.21269. [DOI] [PubMed] [Google Scholar]
- Ulrich S.; Ricken R.; Adli M. Tranylcypromine in mind (Part I): Review of pharmacology. Eur. Neuropsychopharmacol. 2017, 27, 697–713. 10.1016/j.euroneuro.2017.05.007. [DOI] [PubMed] [Google Scholar]
- Dai X. J.; Liu Y.; Xiong X. P.; Xue L. P.; Zheng Y. C.; Liu H. M. Tranylcypromine based lysine-specific demethylase 1 inhibitor: summary and perspective. J. Med. Chem. 2020, 63, 14197–14215. 10.1021/acs.jmedchem.0c00919. [DOI] [PubMed] [Google Scholar]
- Schulz-Fincke J.; Hau M.; Barth J.; Robaa D.; Willmann D.; Kürner A.; Haas J.; Greve G.; Haydn T.; Fulda S.; Lübbert M.; Lüdeke S.; Berg T.; Sippl W.; Schüle R.; Jung M. Structure-activity studies on N-substituted tranylcypromine derivatives lead to selective inhibitors of lysine specific demethylase 1 (LSD1) and potent inducers of leukemic cell differentiation. Eur. J. Med. Chem. 2018, 144, 52–67. 10.1016/j.ejmech.2017.12.001. [DOI] [PubMed] [Google Scholar]
- Niwa H.; Sato S.; Handa N.; Sengoku T.; Umehara T.; Yokoyama S. Development and structural evaluation of N-alkylated trans-2-phenylcyclopropylamine-based LSD1 inhibitors. ChemMedChem. 2020, 15, 787–793. 10.1002/cmdc.202000014. [DOI] [PubMed] [Google Scholar]
- Koda Y.; Sato S.; Yamamoto H.; Niwa H.; Watanabe H.; Watanabe C.; Sato T.; Nakamura K.; Tanaka A.; Shirouzu M.; Honma T.; Fukami T.; Koyama H.; Umehara T. Design and synthesis of tranylcypromine-derived LSD1 inhibitors with improved hERG and microsomal stability profiles. ACS Med. Chem. Lett. 2022, 13, 848–854. 10.1021/acsmedchemlett.2c00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda C.; Valente S.; Romanenghi M.; Pilotto S.; Cirilli R.; Karytinos A.; Ciossani G.; Botrugno O. A.; Forneris F.; Tardugno M.; Edmondson D. E.; Minucci S.; Mattevi A.; Mai A. Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J. Am. Chem. Soc. 2010, 132, 6827–6833. 10.1021/ja101557k. [DOI] [PubMed] [Google Scholar]
- Valente S.; Rodriguez V.; Mercurio C.; Vianello P.; Saponara B.; Cirilli R.; Ciossani G.; Labella D.; Marrocco B.; Monaldi D.; Ruoppolo G.; Tilset M.; Botrugno O. A.; Dessanti P.; Minucci S.; Mattevi A.; Varasi M.; Mai A. Pure enantiomers of benzoylamino-tranylcypromine: LSD1 inhibition, gene modulation in human leukemia cells and effects on clonogenic potential of murine promyelocytic blasts. Eur. J. Med. Chem. 2015, 94, 163–174. 10.1016/j.ejmech.2015.02.060. [DOI] [PubMed] [Google Scholar]
- Vianello P.; Botrugno O. A.; Cappa A.; Ciossani G.; Dessanti P.; Mai A.; Mattevi A.; Meroni G.; Minucci S.; Thaler F.; Tortorici M.; Trifiro P.; Valente S.; Villa M.; Varasi M.; Mercurio C. Synthesis, biological activity and mechanistic insights of 1-substituted cyclopropylamine derivatives: a novel class of irreversible inhibitors of histone demethylase KDM1A. Eur. J. Med. Chem. 2014, 86, 352–363. 10.1016/j.ejmech.2014.08.068. [DOI] [PubMed] [Google Scholar]
- Borrello M. T.; Schinor B.; Bartels K.; Benelkebir H.; Pereira S.; Al-Jamal W. T.; Douglas L.; Duriez P. J.; Packham G.; Haufe G.; Ganesan A. Fluorinated tranylcypromine analogues as inhibitors of lysine-specific demethylase 1 (LSD1, KDM1A). Bioorg. Med. Chem. Lett. 2017, 27, 2099–2101. 10.1016/j.bmcl.2017.03.081. [DOI] [PubMed] [Google Scholar]
- Kakizawa T.; Mizukami T.; Itoh Y.; Hasegawa M.; Sasaki R.; Suzuki T. Evaluation of phenylcyclopropylamine compounds by enzymatic assay of lysine-specific demethylase 2 in the presence of NPAC peptide. Bioorg. Med. Chem. Lett. 2016, 26, 1193–1195. 10.1016/j.bmcl.2016.01.036. [DOI] [PubMed] [Google Scholar]
- Vianello P.; Botrugno O. A.; Cappa A.; Dal Zuffo R.; Dessanti P.; Mai A.; Marrocco B.; Mattevi A.; Meroni G.; Minucci S.; Stazi G.; Thaler F.; Trifiro P.; Valente S.; Villa M.; Varasi M.; Mercurio C. Discovery of a novel inhibitor of histone lysine-specific demethylase 1A (KDM1A/LSD1) as orally active antitumor agent. J. Med. Chem. 2016, 59, 1501–1517. 10.1021/acs.jmedchem.5b01209. [DOI] [PubMed] [Google Scholar]
- Li C.; Su M.; Zhu W.; Kan W.; Ge T.; Xu G.; Wang S.; Sheng L.; Gao F.; Ye Y.; Wang J.; Zhou Y.; Li J.; Liu H. Structure-activity relationship study of indolin-5-yl-cyclopropanamine derivatives as selective lysine specific demethylase 1 (LSD1) inhibitors. J. Med. Chem. 2022, 65, 4335–4349. 10.1021/acs.jmedchem.1c02156. [DOI] [PubMed] [Google Scholar]
- Watanabe C.; Watanabe H.; Okiyama Y.; Takaya D.; Fukuzawa K.; Tanaka S.; Honma T. Development of an automated fragment molecular orbital (FMO) calculation protocol toward construction of quantum mechanical calculation database for large biomolecules. Chem-Bio Inf. J. 2019, 19, 5–18. 10.1273/cbij.19.5. [DOI] [Google Scholar]
- Bonivento D.; Milczek E. M.; McDonald G. R.; Binda C.; Holt A.; Edmondson D. E.; Mattevi A. Potentiation of ligand binding through cooperative effects in monoamine oxidase B. J. Biol. Chem. 2010, 285, 36849–36856. 10.1074/jbc.M110.169482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimasu S.; Umezawa N.; Sato S.; Higuchi T.; Umehara T.; Yokoyama S. Structurally designed trans-2-phenylcyclopropylamine derivatives potently inhibit histone demethylase LSD1/KDM1. Biochemistry 2010, 49, 6494–6503. 10.1021/bi100299r. [DOI] [PubMed] [Google Scholar]
- Miyamura S.; Araki M.; Ota Y.; Itoh Y.; Yasuda S.; Masuda M.; Taniguchi T.; Sowa Y.; Sakai T.; Suzuki T.; Itami K.; Yamaguchi J. C-H activation enables a rapid structure-activity relationship study of arylcyclopropyl amines for potent and selective LSD1 inhibitors. Org. Biomol. Chem. 2016, 14, 8576–8585. 10.1039/C6OB01483F. [DOI] [PubMed] [Google Scholar]
- Kitagawa H.; Kikuchi M.; Sato S.; Watanabe H.; Umezawa N.; Kato M.; Hisamatsu Y.; Umehara T.; Higuchi T. Structure-based identification of potent lysine-specific demethylase 1 inhibitor peptides and temporary cyclization to enhance proteolytic stability and cell growth-inhibitory activity. J. Med. Chem. 2021, 64, 3707–3719. 10.1021/acs.jmedchem.0c01371. [DOI] [PubMed] [Google Scholar]
- Saito S.; Kikuchi J.; Koyama D.; Sato S.; Koyama H.; Osada N.; Kuroda Y.; Akahane K.; Inukai T.; Umehara T.; Furukawa Y. Eradication of central nervous system leukemia of T-cell origin with a brain-permeable LSD1 inhibitor. Clin. Cancer Res. 2019, 25, 1601–1611. 10.1158/1078-0432.CCR-18-0919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.