

Abstract
Autophagy plays essential roles in a wide variety of physiological processes, such as cellular homeostasis, metabolism, development, differentiation, and immunity. Selective pharmacological modulation of autophagy is considered a valuable potential therapeutic approach to treat diverse human diseases. However, development of such therapies has been greatly impeded by the lack of specific small molecule autophagy modulators. Here, we performed structure–activity relationship studies on a previously discovered weak Bcl-2 inhibitor SW076956, and developed a panel of small molecule compounds that selectively released Bcl-2-mediated inhibition of autophagy-related Beclin 1 compared to apoptosis-related Bax at nanomolar concentration. Our NMR analysis showed that compound 35 directly binds Bcl-2 and specifically inhibits the interaction between the Bcl-2 and Beclin 1 BH3 domains without disruption of the Bcl-2–Bax BH3 interaction. More broadly, this proof-of-concept study demonstrates that targeting protein–protein interactions of the intrinsic autophagy regulatory network can serve as a valuable strategy for the development of autophagy-based therapeutics.
Keywords: Autophagy, apoptosis, Bcl-2 inhibitors, Beclin 1, Bax, protein−protein interaction, NMR binding studies
Macroautophagy (hereafter referred to as autophagy) is an evolutionarily conserved catabolic pathway by which cells sequester unwanted cellular constituents (such as damaged organelles, protein aggregates ,and invading microbes) in double-membrane structures known as autophagosomes, and then target them for lysosomal degradation through formation of single-membrane structures called autolysosomes following the fusion between autophagosomes and lysosomes. Autophagy is conserved from plants to mammals and plays essential roles in a wide variety of physiological processes, such as cellular homeostasis, metabolism, development, differentiation, and immunity.1,2 A large amount of evidence demonstrates that autophagy and certain autophagy genes are closely associated with many human diseases including diabetes, cancer, neurodegenerative diseases, and infection.3,4 Whole-body or tissue-specific genetic disruption of autophagy in model animals leads to multiple pathologies, including tissue abnormalities, aberrant inflammation, impaired immunity, neurodegeneration, and susceptibility to tumorigenesis.3,4 In contrast, genetic disruption of autophagy suppressors (such as Bcl-2 and Rubicon) enhances basal autophagy levels in vivo and improved the lifespan and health of different model animals.5−8 Therefore, augmentation of autophagy through small molecule autophagy modulators is believed to be a promising novel candidate approach to treat diverse human diseases.4,9,10
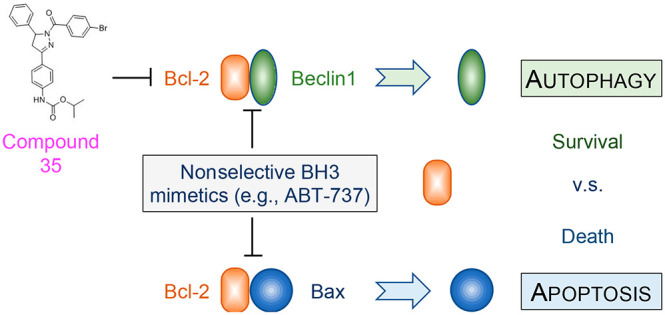
To date, there are no autophagy-specific modulating drugs available in clinical use or trials. Although several drugs (e.g., mTOR inhibitors and calcium channel inhibitors) also activate autophagy, their mode of action is indirect and primarily operates through other mechanisms unrelated to autophagy modulation. Thus, there is considerable interest in identifying specific agents as selective autophagy modulators. We predicted Bcl-2 to be a valuable cellular target as it is known to inhibit autophagy through its interaction with the essential autophagy modulator Beclin 1.11 Beclin 1 functions as a scaffold protein of the autophagy-specific class III phosphatidylinositol 3 kinase (PI3KC3 or PIK3C3) complexes that are required for autophagosomal biogenesis and maturation. Under basal conditions, Bcl-2 binds the BH3 domain of Beclin 1 and suppresses the assembly of PI3KC3 complexes. In response to diverse stress stimuli, Bcl-2-mediated inhibition is released through multiple mechanisms, including a multisite phosphorylation of the nonstructured loop of Bcl-2 and phosphorylation of the Beclin 1 BH3 domain to interrupt the Bcl-2–Beclin 1 protein–protein interaction, or the deployment of BH3-only proteins that competitively disrupt the Bcl-2–Beclin interaction.11 Recently, we and other investigators showed that genetically engineered mice carrying the point mutation F121A in Beclin 1 have decreased Bcl-2–Beclin 1 interaction and thereby increased autophagy in multiple tissues including brain, heart, muscle, liver, mammary gland, and kidney. These mice exhibited increased longevity and reduced neurodegeneration, and diminished aging-related renal and cardiac pathological changes and spontaneous tumorigenesis.5−8 These findings highlight the therapeutic potential of specific small molecule modulation of the Bcl-2–Beclin 1 protein–protein interaction as a new approach to activate autophagy to treat human disease (Figure 1). The potential difficulty with this approach is the design of specific Bcl-2–Beclin 1 protein interaction inhibitors. Notably, Bcl-2 is also a cellular suppressor of apoptosis through its interaction with the BH3 domain of the proapoptotic protein Bax. To date, known Bcl-2 modulators such as ABT-737 that act as BH3 mimetics activate both autophagy and apoptosis.12 Administration of such nonselective Bcl-2 modulators in vivo may thus raise substantial concern because of a potentially undesired induction of apoptosis. Therefore, an ideal Bcl-2-based autophagy modulator must have high selectivity for the suppression of the Bcl-2–Beclin 1 interaction while avoiding the disruption of the Bcl-2–Bax interaction (Figure 1). Here, we disclose a medicinal chemistry driven structure–activity relationship (SAR) study leading to a collection of small molecules that selectively suppress the Bcl-2–Beclin 1 protein–protein interaction at nanomolar concentration.
Figure 1.
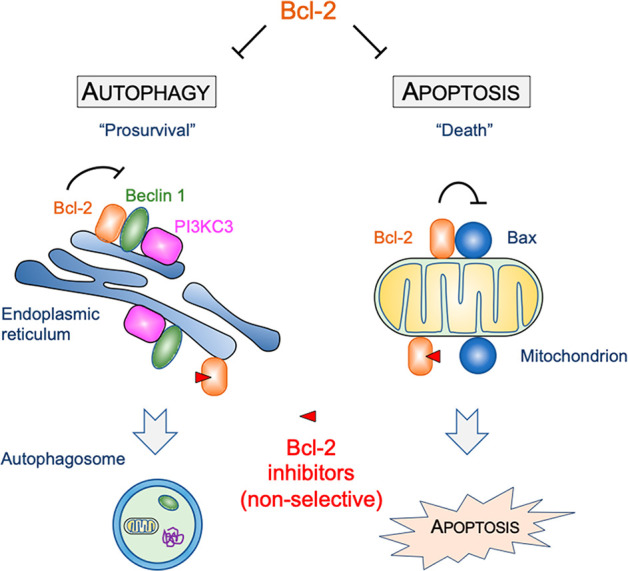
Bcl-2 is a key cellular regulator of both autophagy and apoptosis. Bcl-2 directly interacts with the essential autophagy modulator Beclin 1 to suppresses autophagy. Bcl-2 also suppresses apoptosis through binding to the proapoptotic protein Bax on the mitochondrion. Nonselective small molecules release Bcl-2-mediated inhibition of both autophagy and apoptosis. In contrast, small molecules that specifically target Bcl-2–Beclin 1 would be expected to activate autophagy without inducing apoptosis.
Previously, we reported a high-throughput screen (HTS) of the UTSW small molecule library and discovered compound SW076956 (1, Figure 2A) that suppressed the Bcl-2–Beclin 1 protein–protein interaction using an AlphaLISA protein–protein interaction assay (Figure 2B).13 Although SW076956 was not very potent at inhibiting the Bcl-2–Beclin 1 BH3 interaction (IC50 ∼ 16 μM), we were encouraged by the complete lack of inhibition of the corresponding Bcl-2–Bax BH3 interaction over a similar concentration range (Figure 2C, D). For comparison, the known Bcl-2 modulator ABT-737 was significantly more potent (IC50 = 3.6 nM, Figure 2C), but unfortunately also significantly inhibited the Bcl-2–Bax BH3 interaction (IC50 = 72 nM, Figure 2D). To obtain potent and selective autophagy inducers with sufficient drug-like properties for proof-of-concept studies, our initial studies around HTS hit 1 aimed at exploring SAR around the benzamide, furyl, and phenylmethanesulfonamide rings. All new analogues were evaluated in the AlphaLISA assay for both the Bcl-2–Beclin 1 and Bcl-2–Bax interaction.
Figure 2.
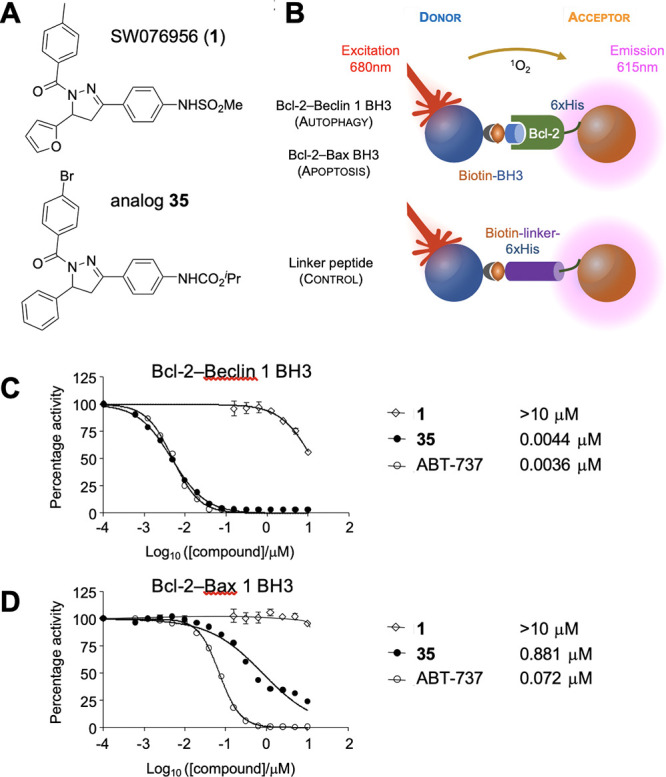
Compound structures, AlphaLISA assay, and representative dose–response quantification of Bcl-2–Beclin 1 BH3 and Bcl-2–Bax BH3 protein–protein interactions. (A) Structures of SW076956 (1) and analogue 35. (B) Design of the AlphaLISA assay. Details can be found in ref (13). (C and D) Quantification of the Bcl-2–Beclin 1 (C) and the Bcl-2–Bax interaction (D). Dose–response curves for compounds 1, 35, and the nonselective Bcl-2 inhibitor ABT-737 for the indicated interactions. Dose–response curves were analyzed using the nonlinear regression function in Prism 7.0 (GraphPad).
As shown in Table 1, removing the 4-Me substituent (Ar1) killed activity (9) as did replacement with a 4-MeSO2, 4-MeC(O), 4-CF3C(O), or 4-tBu substituent (analogues 26-29). Other para-substitutions either had little effect (4-F, 4-CN, and 4-Me; analogues 2, 7, and 8) or modestly improved activity 5.5–8-fold (4-Cl, 4-Br, 4-CF3, 4-NO2, analogues 3–6). Meta-substituted benzamides 10 and 11 were inactive, as were furyl (12) and pyridinyl analogues 30–32. Linking Ar1 via a sulfonamide or methylene instead of an amide, or replacement of Ar1 with a cyclohexyl resulted in inactive compounds (Table S1), as did substituting the methylsulfonamide in the right-hand ring with a trifluoromethylsulfonamide (13), acetamide (14), or primary amine (15). The corresponding tert-butylcarbamates improved activity but not dramatically (2–5-fold, e.g., 16, 17 vs 1 and 4). Comparing compounds 18–23 to analogue 17 indicates that exchanging the 2-furanyl ring (Ar2) with other heterocyclic rings did not improve the activity significantly, whereas eliminating this ring altogether resulted in loss of activity (Table S1). However, a phenyl replacement enhanced activity 5-fold (24 vs 17). Selected other 2-, 3-, and 4-substituted phenyls (Br, Me, NO2) significantly impaired activity (Table S1). Thus far, a combination of 4-BrPh (Ar1), Ph (Ar2) and a tert-butylcarbamate (R) provided the most potent Bcl-2–Beclin 1 antagonist (analogue 24) with an in vitro AlphaLISA IC50 of 262 nM and no activity against the corresponding Bcl-2–Bax BH3 interaction.
Table 1. SAR of Trisubstituted 4,5-Dihydropyrazoles 1–44.
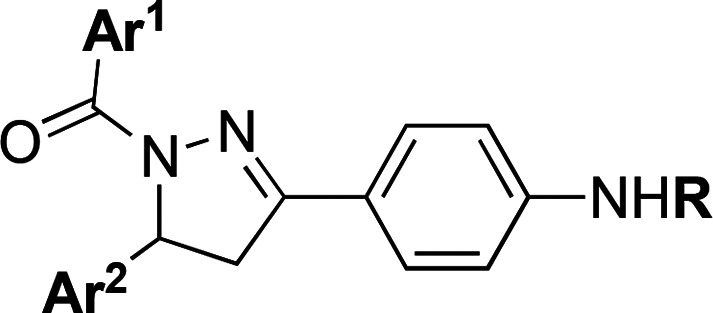
| compda | Ar1 | Ar2 | R | IC50 (μM)b |
|---|---|---|---|---|
| 1 | 4-MePh | 2-furyl | SO2Me | 15.9 ± 0.8 |
| 2 | 4-FPh | 2-furyl | SO2Me | 12.66 ± 0.22 |
| 3 | 4-ClPh | 2-furyl | SO2Me | 2.72 ± 0.11 |
| 4 | 4-BrPh | 2-furyl | SO2Me | 2.47 ± 0.36 |
| 5 | 4-CF3Ph | 2-furyl | SO2Me | 2.88 ± 0.06 |
| 6 | 4-NO2Ph | 2-furyl | SO2Me | 1.97 ± 0.12 |
| 7 | 4-CNPh | 2-furyl | SO2Me | 7.44 ± 0.21 |
| 8 | 4-MeOPh | 2-furyl | SO2Me | 10.11 ± 0.30 |
| 9 | Ph | 2-furyl | SO2Me | inactive |
| 10 | 3-MePh | 2-furyl | SO2Me | inactive |
| 11 | 3-FPh | 2-furyl | SO2Me | inactive |
| 12 | 2-furyl | 2-furyl | SO2Me | inactive |
| 13 | 4-MePh | 2-furyl | SO2CF3 | inactive |
| 14 | 4-MePh | 2-furyl | C(O)Me | inactive |
| 15 | 4-MePh | 2-furyl | H | inactive |
| 16 | 4-MePh | 2-furyl | CO2tBu | 3.29 ± 0.16 |
| 17 | 4-BrPh | 2-furyl | CO2tBu | 1.26 ± 0.03 |
| 18 | 4-BrPh | 3-furyl | CO2tBu | 3.82 ± 0.09 |
| 19 | 4-BrPh | thiazol-2-yl | CO2tBu | 1.61 ± 0.01 |
| 20 | 4-BrPh | thiazol-5-yl | CO2tBu | 4.36 ± 0.08 |
| 21 | 4-BrPh | 2-pyridinyl | CO2tBu | 0.65 ± 0.04 |
| 22 | 4-BrPh | 3-pyridinyl | CO2tBu | 1.63 ± 0.11 |
| 23 | 4-BrPh | 4-pyridinyl | CO2tBu | 2.78 ± 0.13 |
| 24 | 4-BrPh | Ph | CO2tBu | 0.262 ± 0.039 |
| 25 | 4-NO2Ph | Ph | CO2tBu | 0.439 ± 0.016 |
| 26 | 4-MeSO2Ph | Ph | CO2tBu | inactive |
| 27 | 4-MeC(O)Ph | Ph | CO2tBu | inactive |
| 28 | 4-tBuPh | Ph | CO2tBu | inactive |
| 29 | 4-CF3C(O)Ph | Ph | CO2tBu | inactive |
| 30 | 2-pyridinyl | Ph | CO2tBu | inactive |
| 31 | 3-pyridinyl | Ph | CO2tBu | 14.5 ± 3.7 |
| 32 | 4-pyridinyl | Ph | CO2tBu | inactive |
| 33 | 4-BrPh | Ph | CO2Me | 2.10 ± 0.07 |
| 34 | 4-BrPh | Ph | CO2Et | 0.0328 ± 0.0006 |
| 35 | 4-BrPh | Ph | CO2iPr | 0.0044 ± 0.0002 |
| 36 | 4-BrPh | Ph | CO2cPr | 0.0441 ± 0.0028 |
| 37 | 4-BrPh | Ph | CO2iBu | 0.164 ± 0.033 |
| 38 | 4-BrPh | Ph | CO2allyl | 0.0318 ± 0.0028 |
| 39 | 4-BrPh | Ph | CO2(CH2)2C2H | 0.065 ± 0.022 |
| 40 | 4-BrPh | Ph | C(O)NHMe | 5.53 ± 0.12 |
| 41 | 4-BrPh | Ph | C(O)NMe2 | 2.41 ± 0.03 |
| 42 | 4-BrPh | Ph | C(O)pyrrolidine | inactive |
| 43 | 4-BrPh | Ph | C(O)NHiPr | 0.350 ± 0.009 |
| 44 | 4-BrPh | Ph | C(O)NHtBu | 9.22 ± 0.51 |
All compounds are racemates.
IC50 values represent the half maximal (50%) inhibitory concentration for the Bcl-2–Beclin 1 BH3 interaction as determined in the AlphaLISA assay. Error represents SD (n = 3). All compounds were inactive (at 10 μM) against the Bcl-2–Bax BH3 interaction except compounds 34 (IC50 = 6.62 ± 0.27 μM), 35 (IC50 = 0.88 μM ± 0.04 μM), 36 (IC50 = 15.1 ± 0.04 μM), and 38 (IC50 = 8.83 ± 0.39 μM).
As we advanced analogues for improved potency, we also evaluated a select subset for metabolic stability (Table S2). All sulfonamides tested (1, 4–6, 61) displayed poor stability with T1/2 of 2.9–18.1 min upon incubation with mouse microsomes, whereas the corresponding tert-butylcarbamates improved stability 5–7-fold. The two most potent analogues (24 and 25) coincidently were also metabolically the most stable with an excellent T1/2 of 120 min (Table S2). Because a nitrophenyl represent a known toxicophore (25), we focused our next round of SAR optimization on exploring various carbamates. Using compound 24 as the reference (IC50 = 262 nM), we found that sterically less encumbered alkylcarbamates 34-39 improved activity significantly, up to 60-fold for isopropylcarbamate 35 (IC50 = 4.4 nM). The smallest carbamate (R = NHCO2Me, 33) was an exception and was 8-fold less active than 24. Replacing carbamate functionality with ureas (40-44) was unfruitful and resulted in significant loss of activity. Although we started to observe some inhibition of the undesired Bcl-2–Bax protein–protein interaction with some of the more potent carbamates 34-36, and 38 (Table 1 footnotes), a significant selectivity for disruption of the Bcl-2–Beclin 1 interaction was maintained (>200-fold). Dose–response curves and selectivity for the most potent analogue 35 are included in Figure 1.
In Table 2, we explore some additional miscellaneous SAR related to the right-hand aromatic ring, or additional substitution in the central dihydropyrazole ring. First, transposing the carbamate from the para to the meta-position was detrimental for activity (45 vs 24). In an attempt to improve polarity (compound 24 has a Clog P of 6.2), we prepared more polar pyridine analogues 46 and 47 with an acceptable ClogP of 4.7 below the Lipinski Rule-of-Five recommended threshold. Fortunately, this polarity improvement did not come at a cost of potency. Introduction of an additional methyl substituent on the central dihydropyrazole ring creates a new stereogenic center and resulted in a 10-fold improvement of activity for compound 48 versus comparator 24, and was also beneficial for the more polar pyridine analogue 50 (IC50 = 2 nM; ClogP 4.8). However, the selectivity of compound 50 for the Bcl-2–Beclin 1 interaction dropped to 71-fold (Bcl-2–Bax, IC50 157 nM; vs 200-fold selectivity for 35). Finally, other functionality including acids, esters, carbonates, and amides (51-57) worsened activity and did not lead to new avenues for improved analogue design.
Table 2. Additional Miscellaneous SAR.
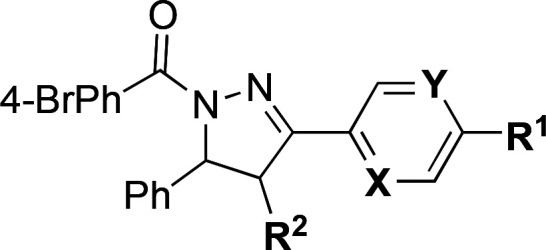
| compda | R1 | R2 | X | Y | IC50 (μM)b |
|---|---|---|---|---|---|
| 24 | NHCO2tBu | H | CH | CH | 0.262 ± 0.039 |
| 45 | H | H | CH | CNHCO2tBu | 11.06 ± 0.33 |
| 46 | NHCO2tBu | H | CH | N | 0.309 ± 0.010 |
| 47 | NHCO2tBu | H | N | CH | 0.372 ± 0.038 |
| 48 | NHCO2tBu | Mec | CH | CH | 0.0274 ± 0.0012 |
| 49 | NHCO2tBu | Phec | CH | CH | 0.172 ± 0.075 |
| 50 | NHCO2iPr | Mec | CH | N | 0.002 ± 0.001 |
| 51 | oxazolidinone | H | CH | CH | 5.16 ± 0.07 |
| 52 | CO2H | H | CH | CH | inactive |
| 53 | CO2Me | H | CH | CH | 1.64 ± 0.27 |
| 54 | CO2iPr | H | CH | CH | 0.110 ± 0.049 |
| 55 | OCOiPr | H | CH | CH | 5.45 ± 0.23 |
| 56 | CONMe2 | H | CH | CH | 5.91 ± 0.27 |
| 57 | CONHtBu | H | CH | CH | inactive |
All compounds are racemates.
IC50 values represent the half maximal (50%) inhibitory concentration for the Bcl-2–Beclin 1 BH3 interaction as determined in the AlphaLISA assay. Error represents SD (n = 3). All compounds were inactive (at 10 μM) against the Bcl-2–Bax BH3 interaction except compounds 50 (IC50 = 0.157 ± 0.048 μM) and 54 (IC50 = 4.36 ± 0.03 μM).
Single diastereomer; configuration not assigned.
Compound 35 was selected for further follow-up. It provided the best balance of potency, selectivity, and in vitro metabolic stability. Compound 35 had a 3600-fold improved activity in suppressing autophagy-related Bcl-2–Beclin 1 BH3 interaction by the AlphaLISA assay (IC50 = 4.4 nM) compared to HTS hit 1 (IC50 ∼ 16 μM), which is comparable to the potency of the commercial Bcl-2 inhibitor ABT-737 (IC50 = 3.6 nM; Figure 2C). Notably, ABT-737 also disrupted the apoptosis-related Bcl-2–Bax BH3 interaction with an IC50 of 72.2 nM (Figure 2D), thus providing only ∼20-fold selectivity. In contrast, compound 35 was 200-fold more selective at suppressing the Bcl-2–Beclin 1 BH3 interaction. Gratifyingly, compound 35 was also one of the most stable analogues with a T1/2 of 120 min in murine microsomal fractions (Table S2). Compound 35 also showed favorable in vivo pharmacokinetics after intraperitoneal delivery, with marginally better exposure than the corresponding tert-butylcarbamate 24 and excellent oral bioavailability (Table S3).
To determine whether compound 35 directly binds to Bcl-2, we acquired 1H–15N transverse relaxation optimized (TROSY) heteronuclear single quantum coherence (HSQC) spectra of a 15N-labeled fragment corresponding to the cytoplasmic domain of Bcl-2 (Bcl-2 aa1–218) in the absence and presence of compound 35. We found that compound 35 induced dramatic changes in the spectrum (Figure 3A), demonstrating that compound 35 directly interacts with Bcl-2. As expected, a peptide corresponding to the BH3 domain of Beclin 1 also resulted in cross-peak changes in the 1H–15N TROSY-HSQC spectrum of Bcl-2 (Figure 3B) which is consistent with the prior structure biology studies indicating a direct interaction between the Beclin 1 BH3 domain and Bcl-2 family members.14−16 Notably, the shifts of the Bcl-2 spectrum caused by the Beclin 1 BH3 peptide were clearly distinct from those caused by compound 35 (when comparing red contours in Figure 3A with light blue contours in Figure 3B). This indicates that Beclin 1 BH3 peptide and compound 35 likely bind to somewhat different confirmations of Bcl-2. Furthermore, incubation with Beclin 1 BH3 peptide failed to alter the 1H–15N TROSY-HSQC spectrum of compound 35-bound Bcl-2 (Figure 3C), showing that compound 35 prevents binding of the Beclin 1 BH3 peptide to Bcl-2.
Figure 3.

Compound 35 displaces the Beclin 1 BH3 domain from Bcl-2, but not the Bax BH3 domain. (A–F) Superpositions of 1H–15N TROSY-HSQC spectra of (A) Bcl-2 alone (black contours) or in the presence of compound 35 (red contours); (B) Bcl-2 alone (black contours) or in the presence of the Beclin 1 BH3 peptide (light blue contours); (C) Bcl-2 in the presence of compound 35 and the absence (red contours) or presence (blue contours) of the Beclin 1 BH3 peptide; (D) Bcl-2 alone (black contours) or in the presence of the Bax BH3 peptide (orange contours); (E) Bcl-2 in the presence of the Beclin 1 BH3 peptide (light blue contours) or the Bax BH3 peptide (orange contours); (F) Bcl-2 in the presence of the Bax BH3 peptide and the absence (orange contours) or presence (gray contours) of compound 35.
Next, we studied the effect of compound 35 on the Bcl-2–Bax BH3 interaction, which is essential for the apoptotic regulation. Incubation of a peptide corresponding to the BH3 domain of Bax with Bcl-2 led to dramatic shifts in the 1H–15N TROSY-HSQC cross-peaks of Bcl-2 (Figure 3D), which is consistent with prior findings regarding the direct interaction between Bcl-2 and the Bax BH3 domain.17 Notably, the resulting spectrum of Bcl-2 bound to the Bax BH3 peptide was almost identical to that observed for Bcl-2 bound to the Beclin 1 BH3 peptide (Figure 3E). This finding is consistent with the fact that Beclin 1 BH3 domain and Bax BH3 domain both bind to the same evolutionarily conserved hydrophobic groove on Bcl-2 family members.18 Since the spectrum of Bcl-2 bound to the Beclin 1 BH3 peptide had fewer cross-peaks, we speculated that the moderate affinity of the Beclin 1 BH3 peptide with Bcl-2 likely causes exchange broadening, while the Bax BH3 peptide binds to Bcl-2 with higher affinity that does not lead to exchange broadening. Importantly, compound 35 did not cause marked changes in the 1H–15N TROSY-HSQC spectrum of Bcl-2 bound to the Bax BH3 peptide (Figure 3F). These findings show that compound 35 did not displace the Bax BH3 peptide from Bcl-2 under the conditions of these experiments, even though compound 35 completely displaced the Becling 1 BH3 peptide at the same concentrations. These differences are consistent with the AlphaLisa results (Figure 2C,D) and can be attributed to the much higher affinity of the Bax BH3 peptide for Bcl-2 than the Beclin 1 BH3 peptide.
In summary, we identified a set of new Bcl-2 inhibitors that can potently suppress the desired Bcl-2–Beclin 1 BH3 interaction while avoiding to disrupt the Bcl-2–Bax BH3 interaction that could lead to off-target induction of apoptosis. Compound 35 provided for a reasonable balance of high potency (IC50 = 4.4 nM), selectivity (200-fold), metabolic stability and in vivo pharmacokinetic properties. Mechanistically, the selectivity of the compound in inhibiting the Bcl-2-Beclin 1 interaction can be explained by the differences in affinity of the Bax and Beclin 1 BH3 domains for Bcl-2. Hence, this proof-of-concept study demonstrates Bcl-2, an essential cellular regulator of both autophagy and apoptosis, as an excellent cellular target for the development of agents to specifically augment autophagy without induction of apoptosis. More broadly, our study highlighted the importance in thorough characterization of protein–protein interaction networks and the development of selective small molecule compounds that can specifically modulate one protein–protein interaction among distinct protein complexes that share a common partner protein.19
Acknowledgments
Financial support was provided by the NIH (AI142784). J.K.D.B. acknowledges support of the Welch Foundation (Grant I-1422) and holds the Julie and Louis Beecherl, Jr., Chair in Medical Science. We thank Liwei Wang for assistance with the expression and purification of Bcl-2 proteins for AlphaLISA and NMR experiments, and Zhongju Zou, Yuting Yang, and Sangita Singha for kindly providing critical reagents for this study, and Shuguang Wei for assistance with the luminescence plate reader.
Glossary
Abbreviations
- Bax
Bcl-2 associated X
- Bcl-2
B-cell lymphoma 2
- Beclin 1
Coiled-coil moesin-like Bcl-2-interacting protein 1
- BH3
Bcl-2 homology domain 3
- HSQC
heteronuclear single quantum coherence
- NMR
nuclear magnetic resonance
- PI3KC3
class III phosphatidylinositol 3 kinase
- TROSY
transverse relaxation optimized spectroscopy
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00309.
Data for additional compounds; in vitro metabolic stability; pharmacokinetic data for compounds 24 and 35; materials and methods; procedures and characterization of synthetic compounds; copies of 1H and 13C NMR spectra (PDF)
Author Contributions
‡ X.D. and Q.L. contributed equally. X.D., B.L., and J.K.D.B. conceived and designed the study; Q.L. synthesized and characterized all the synthetic compounds in the lab of J.K.D.B.; N.S.W. and X.W. performed all the in vitro and in vivo pharmacology; X.D. and W.-C.C. performed all the biological assays in the lab of B.L.; Y.-Z.P., Y.-C.K., X.Z., and J.R. performed the NMR studies and analyzed the corresponding data; X.D., J.R., B.L., and J.K.D.B. analyzed the data; X.D., N.S.W., J.R., and J.K.D.B. wrote and edited the manuscript. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Levine B.; Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X.; Levine B. Autophagy and viruses: adversaries or allies?. J. Innate. Immun. 2013, 5, 480–493. 10.1159/000346388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B.; Kroemer G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. 10.1016/j.cell.2018.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N.; Levine B. Autophagy in Human Diseases. N. Engl. J. Med. 2020, 383, 1564–1576. 10.1056/NEJMra2022774. [DOI] [PubMed] [Google Scholar]
- Fernández Á.F.; Sebti S.; Wei Y.; Zou Z.; Shi M.; McMillan K. L.; He C.; Ting T.; Liu Y.; Chiang W. C.; Marciano D. K.; Schiattarella G. G.; Bhagat G.; Moe O. W.; Hu M. C.; Levine B. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 2018, 558, 136–140. 10.1038/s41586-018-0162-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocchi A.; Yamamoto S.; Ting T.; Fan Y.; Sadleir K.; Wang Y.; Zhang W.; Huang S.; Levine B.; Vassar R.; He C. A Becn1 mutation mediates hyperactive autophagic sequestration of amyloid oligomers and improved cognition in Alzheimer’s disease. PLoS Genet. 2017, 13, e1006962 10.1371/journal.pgen.1006962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega-Rubín-de-Celis S.; Zou Z.; Fernández Á.F.; Ci B.; Kim M.; Xiao G.; Xie Y.; Levine B. Increased autophagy blocks HER2-mediated breast tumorigenesis. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 4176–4181. 10.1073/pnas.1717800115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S.; Oba M.; Suzuki M.; Takahashi A.; Yamamuro T.; Fujiwara M.; Ikenaka K.; Minami S.; Tabata N.; Yamamoto K.; Kubo S.; Tokumura A.; Akamatsu K.; Miyazaki Y.; Kawabata T.; Hamasaki M.; Fukui K.; Sango K.; Watanabe Y.; Takabatake Y.; Kitajima T. S.; Okada Y.; Mochizuki H.; Isaka Y.; Antebi A.; Yoshimori T. Suppression of autophagic activity by Rubicon is a signature of aging. Nat. Commun. 2019, 10, 847. 10.1038/s41467-019-08729-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein D. C.; Codogno P.; Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discovery 2012, 11, 709–730. 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L.; Bravo-San Pedro J. M.; Levine B.; Green D. R.; Kroemer G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat. Rev. Drug Discovery 2017, 16, 487–511. 10.1038/nrd.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B.; Liu R.; Dong X.; Zhong Q. Beclin orthologs: integrative hubs of cell signaling, membrane trafficking, and physiology. Trends Cell Biol. 2015, 25, 533–544. 10.1016/j.tcb.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-San Pedro J. M.; Wei Y.; Sica V.; Chiara Maiuri M.; Zou Z.; Kroemer G.; Levine B. BAX and BAK1 are dispensable for ABT-737-induced dissociation of the BCL2-BECN1 complex and autophagy. Autophagy 2015, 11, 452–459. 10.1080/15548627.2015.1017191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang W. C.; Wei Y.; Kuo Y. C.; Wei S.; Zhou A.; Zou Z.; Yehl J.; Ranaghan M. J.; Skepner A.; Bittker J. A.; Perez J. R.; Posner B. A.; Levine B. High-Throughput Screens To Identify Autophagy Inducers That Function by Disrupting Beclin 1/Bcl-2 Binding. ACS Chem. Biol. 2018, 13, 2247–2260. 10.1021/acschembio.8b00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W.; Huang S.; Wu H.; Zhang M. Molecular basis of Bcl-xL’s target recognition versatility revealed by the structure of Bcl-xL in complex with the BH3 domain of Beclin-1. J. Mol. Biol. 2007, 372, 223–235. 10.1016/j.jmb.2007.06.069. [DOI] [PubMed] [Google Scholar]
- Oberstein A.; Jeffrey P. D.; Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J. Biol. Chem. 2007, 282, 13123–13132. 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- Lee E. F.; Smith N. A.; Soares da Costa T. P.; Meftahi N.; Yao S.; Harris T. J.; Tran S.; Pettikiriarachchi A.; Perugini M. A.; Keizer D. W.; Evangelista M.; Smith B. J.; Fairlie W. D. Structural insights into BCL2 pro-survival protein interactions with the key autophagy regulator BECN1 following phosphorylation by STK4/MST1. Autophagy 2019, 15, 785–795. 10.1080/15548627.2018.1564557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku B.; Liang C.; Jung J. U.; Oh B. H. Evidence that inhibition of BAX activation by BCL-2 involves its tight and preferential interaction with the BH3 domain of BAX. Cell Res. 2011, 21, 627–641. 10.1038/cr.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B.; Sinha S.; Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600–606. 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parts of this work have been patented, see: De Brabander J. K.; Liang Q.; Levine B.; Chiang W.-C.. Small molecule inducers of autophagy. US patent WO 2019236433, December 12, 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.