

Abstract
In searching for new molecular drug targets, Carbonic Anhydrases (CAs) have emerged as valuable targets in diverse diseases. CAs play critical functions in maintaining pH and CO2 homeostasis, metabolic pathways, and much more. So, it is becoming attractive for medicinal chemists to design novel inhibitors for this class of enzymes with improved potency and selectivity towards the different isoforms. In the present study, three sets of carboxylic acid derivatives 5a–q, 7a–b and 12a–c were designed, developed and evaluated for the hCA inhibitory effects against hCA I, II, IX and XII. Compounds 5l, 5m, and 5q elicited the highest inhibitory activities against hCA II, IX and XII. In summary, structural rigidification, regioisomerism and structural extension, all played obvious roles in the degree of hCA inhibition. This present work could be a good starting point for the design of more non-classical selective hCA inhibitors as potential targets for several diseases.
Keywords: Carbonic anhydrase, H-NMR, enaminone, stopped-flow assay
1. Introduction
The Zn(II) metalloenzymes carbonic anhydrases (CA, EC 4.2.1.1) is a very significant family in humans and in most living organisms that catalyses the reversible CO2 hydration reaction to bicarbonate ion1. This fundamental reaction orchestrates several physiological processes requiring pH control and ion transport2. Fifteen humans (h) CA isoforms have been discovered so far, and they showed diverse distribution among tissues and cells.
Dysfunction of hCA activities results in several pathological consequences, highlighting these isozymes as promising drug targets for diverse therapeutic interventions with small molecule CA inhibitors (CAIs) 3. Accordingly, the pharmacological applications of several CAIs were reported for the management of different diseases including ophthalmologic problems4,5, human malignancies6, high-altitude sickness7, epilepsy8, peptic ulcers9, obesity10, and congestive heart failure11.
Carbonic anhydrases are classically inhibited by small molecules tethered with the primary sulphonamide-based zinc binding group (ZBG), as well as its bioisosteres such as sulfamides and sulfamates12–14. Although several chemotypes of CA inhibitors have been identified in the last few decades (such as coumarins, phenols, thiocarbamates, and carboxylates)15–17 only CA inhibitors based on sulphonamides could be used in the clinical setting for glaucoma treatment (such as acetazolamide, methazolamide and dorzolamide, Figure 1), and in clinical trials for the treatment of human malignancies (such as SLC-0111 and indisulam, Figure 1)18,19. Accordingly, the development of nonclassical CA inhibitors has emerged as a promising approach to discovering new efficient CA inhibitors for the management of various disorders.
Figure 1.
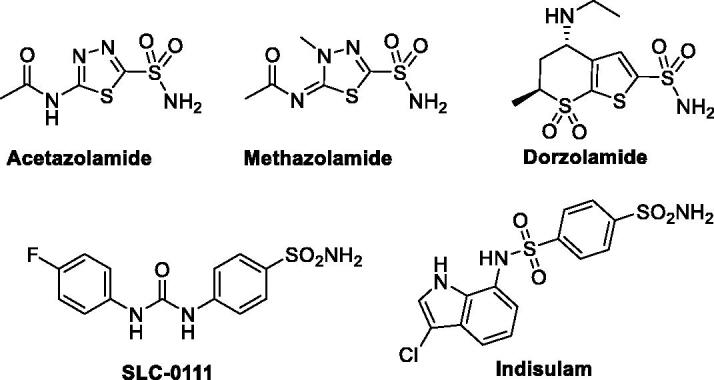
Structures of certain sulphonamide-based CAIs in clinical use and in clinical trials.
The carboxylic acid-bearing small molecules represent an important category of the non-classical carbonic anhydrase inhibitors20. Interestingly, they can inhibit the metalloenzymes via various modes of action. Firstly, they are able to bind directly to the catalytic zinc displacing bound water-hydroxide anion, similarly to sulphonamides. Alternatively, certain carboxylic acid derivatives can anchor to the zinc-bound water-hydroxide ion via a hydrogen bonding, similar to the binding mode noticed for phenol-based molecules. Lastly, carboxylates may bind within an adjacent pocket to the entrance which is located outside the active site of the carbonic anhydrase. This interaction results in a block of the proton shuttle His64 residue in its “out” conformation, leading to the inhibition of CA catalytic activity21–23.
In 2020, our research team has reported novel series of benzofuran-based carboxylic acid small molecules that tethered with benzoic or hippuric acid motifs as potential CA inhibitors24. These acid moieties are linked to 5-bromobenzofuran or 2-methylbenzofuran tail through an ureido linker. Among these acid derivatives, m-benzoic acid-bearing derivative (Compound 1, Figure 2) emerged as submicromolar hCA IX inhibitor (KI = 0.79 µM), as well as potent hCAXII inhibitor (KI = 2.3 µM). Moreover, we developed another ureides series of piperine-based carboxylic acid derivatives that incorporate benzoic acid moieties linked to the natural product piperine through an ureido spacer25. Acid derivative 2 (Figure 2) exerted moderate inhibition activities against both CA IX and XII isoforms (KI = 16.1 µM and 14.4 µM, respectively).
Figure 2.
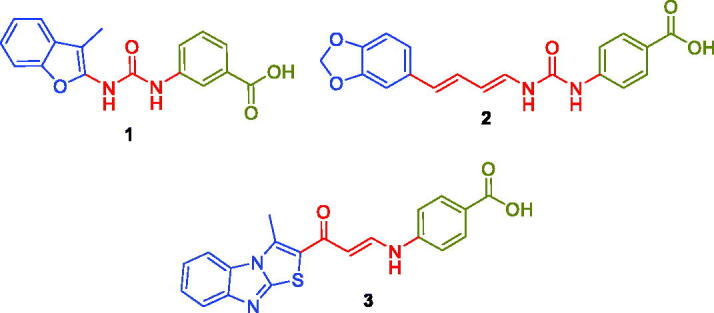
Structures for some reported carboxylic acid derivatives as non-classical CA inhibitors.
Also, in 2020 we designed and prepared a novel methylthiazolo[3,2-a]benzimidazole-tethered carboxylic acid derivatives as potential CA inhibitors26. In this work, the enaminone linker was utilised to link the ZBG benzoic acid motifs to the thiazolo[3,2-a]benzimidazole tail26. Enaminone-based carboxylic acid 3 (Figure 2) effectively inhibited hCA IX and XII isoforms (KI = 0.83 µM and 2.4 µM, respectively). In the current work, we devoted our effort to developing new non-classical CA inhibitors through the design of three sets of new carboxylic acid derivatives (5a–q, 7a–b and 12a–c), Figure 3.
Figure 3.

Design for herein reported carboxylic acid derivatives 5a–q, 7a–b and 12a–c.
The first (5a–q) and the second (7a–b) sets incorporated a benzoic acid moiety that is connected to different aryl tails through an enaminone linker, for the first set, or through the more flexible saturated 3-oxo-propylamine linker, for the second set, Figure 3. The incorporation of the enaminone linker is expected to provide more rigid molecules, than the corresponding molecules that tethered with the 3-oxo-propylamine linker. The expected conformational restriction in the enaminone-bearing carboxylic acids (5a–q) should be attributed to the unsaturation (the olefinic functional group), as well as due to the formation of an intramolecular hydrogen bonding that resulted in a pseudo-six-membered ring. Lastly, a structural extension approach was utilised to replace the benzoic acid motif with the hippuric acid to furnish the third series 12a–c (Figure 3). The potential ability of the target carboxylic acids (5a–q, 7a–b and 12a–c) to inhibit hCA I, II, IX and XII isoforms was assessed via the stopped-flow carbon dioxide hydrase assay.
2. Results and discussion
2.1. Chemistry
In this study, the synthesis of the new carboxylic acids-based carbonic anhydrase inhibitors 5a–q, 7a–b and 12a–c is outlined in Schemes 1 and 2. In Scheme 1, six aryl methyl ketones 1a–f were condensed with DMF-DMA to furnish the corresponding aryl enaminones intermediates 3a–f that subsequently reacted with ortho, meta and para aminobenzoic acids 4a–c in glacial acetic acid to produce the target carboxylic acids 5a–q. On the other hand, acetophenone 1a has been reacted with dimethyl amine and formaldehyde via Mannich reaction to get 3-(dimethylamino)-1-phenylpropan-1-one 6, which subsequently reacted with m- and p- aminobenzoic acids 4b–c in absolute ethanol to furnish targeted carboxylic acids 7a–b with saturated linker between the ZBG and the aryl tail.
Scheme 1.

Synthesis of benzoic acids-bearing enaminones 5a–q and 3/4-((3-oxo-3-phenylpropyl)amino)benzoic acids 7a–b.
Scheme 2.

Synthesis of hippuric acids-bearing enaminones 12a–c.
In Scheme 2, glycine 9 was acylated with 4-nitrobenzoyl chloride 8 in dry dioxan to get (4-nitrobenzoyl)glycine 10, which was then reduced to the corresponding (4-aminobenzoyl)glycine 11. The third set of the target carboxylic acids with the desired structural extension 12a–c was prepared through the nucleophilic substitution reaction of (4-aminobenzoyl)glycine 11 with the previously prepared enaminones 3a,d,f in a boiling glacial acetic acid (Scheme 2). The proposed structure for carboxylic acids enaminones 5a–q, 7a–b and 12a–c were supported by the elemental and spectral data.
1H NMR spectra of 2-substituted carboxylic acids bearing enaminones 5a–e revealed they are existing in Z-form around the olefinic bond which was evidenced by up-field doublets of Z-Ha around δ 6.13–6.37 ppm with coupling constant (J) equals 8.0 Hz. The latter Z-form was assumed to be established through the formation of two intramolecular hydrogen bonds that resulted in two pseudo-six-membered rings. 1H NMR spectra of ortho-carboxylic acids bearing enaminones 5a–e exhibited a doublet D2O exchangeable signal that was integrated for one proton, attributable to the NH group of the Z-form, at more down-field region δ 13.2–13.7 ppm when compared with those of 3- and 4-substituted carboxylic acids bearing enaminones 5f–k and 5l–q, respectively, at δ 11.5–12.1 ppm (Figure 4). This negative shift could be explained by the presence of the two intramolecular hydrogen bonds. The coupling constant value of these doublets is 12.0 Hz which suggests the trans direction of NH around HN-CHb in enaminones 5a–e.
Figure 4.

The existence of ortho-substituted carboxylic acids enaminones 5a–e in Z-form and the existence of meta- and para-substituted carboxylic acids enaminones 5f–k and 5l–q in E/Z-forms (1:1) in DMSO.
1H NMR spectra of 3- and 4-substituted carboxylic enaminones 5f–k and 5l–q disclosed their presence in Z/E geometric conjugation around HaC=CHb in an equal ratio (Z:E = 1:1) according to the integrations of their signals. The presence of E-isomer was evidenced via the J constant for olefinic protons Ha and Hb (JHa-Hb = 12.00 Hz) which was determined by the down-field doublet signal of E-Ha around δ 6.4–6.7 ppm (Figures 4 and 5). In a similar way, the presence of the Z-form was revealed by determining the J constant for the olefinic protons Ha and Hb (J = 8.0 Hz) that came from the up-field doublet signals of Z-Ha around δ 6.13–6.41 ppm. Furthermore, the 1H NMR spectra displayed 2 doublet signal sets, integrated for one proton, each belonging to the (NH) proton for the E and Z isomers around δ 10.28–10.59 and 12.03–12.14 ppm (Figures 4 and 5).
Figure 5.

1H NMR of enaminone 5i showing the presence of Z-form [Z Ha (d), Z Hb (dd), E NH (d)] and E-form [E Ha (d), E Hb (overlapped dd), E NH (d)] in DMSO as a representative example for enaminones 5f–k and 5l–q.
The downfield signals of NH proton could be attributed to the Z isomer and the up-field signals for the same group to the E isomer according to their integration values as explained in our previous study27. The coupling constants for these doublets are 12.0 Hz which suggests the presence of NH of enaminones 5f–k and 5l–q in trans direction for Hb around HN-CHb bond in both forms E and Z (Figures 4 and 5). Moreover, Z (NH) appeared down-field, due to the formation of intra-molecular hydrogen bonding with the carbonyl oxygen. However, this hydrogen bond between C=O and NH groups was absolutely confirmed, in a previous study, by X-ray single crystal analysis of a sulphonamide analog for compound 5 which exhibited a Z configuration around HaC=CHb with the trans HN-CHb27.
Interestingly, the 1H NMR of meta-substituted carboxylic enaminones 5g and 5i, as well as 1H NMR for para-substituted carboxylic enaminones 5o, among 5f–k and 5l–q, revealed the appearance of down-field Z-Hb signal around δ 8.00 ppm as dd with J = 12.0 and 8.0 Hz due to JE-Hb-NH and JZ-Hb-Ha, respectively, whereas the up-field E-Hb signal appeared around δ 8.14 as overlapped dd with two J = 12.0 Hz (JE-Hb-NH = JE-Hb-Ha = 12.0 Hz). It is worthy to note that after the addition of D2O to the DMSO solution of these compounds (5g, 5i and 5o) in their NMR tube, the coupling of NH proton for Hb in both Z and E forms disappeared and the dd of Z-Hb was converted to doublet signal due to the coupling of Z-Ha only with J = 8.0 Hz parallel with the conversion of overlapped dd of E-Hb to doublet signal due to E-Ha coupling only with J = 12.0 Hz (Figure 6).
Figure 6.

The presence of meta- and para-substituted carboxylic enaminones 5f–k and 5l–q in Z/E-forms in DMSO and in D2O/DMSO (1H NMR).
Furthermore, the 1H NMR of 4-carboxamido enaminone 12b, as a representative example for enaminones 12a–c, showed the appearance of two sets of doublet signals, integrated for the two protons of –CH2- at δ 3.91 and δ 3.92 and two sets of triplet, for the proton of the amidic -NH group of at δ 8.74 and δ 8.79 beside two sets of doublet Z/E Ha and Z/E, Hb, and two dd of E NH, Figure 7.
Figure 7.

1H NMR of enaminone 12b which showed the existence of Z- and E-form in DMSO as a representative example for 12a–c.
2.2. Biological evaluation
2.2.1. CAS inhibition
In the current study, we have explored the inhibitory activities of the synthesised carboxylic acid derivatives 5a–q, 7a–b and 12a–c against different carbonic anhydrase isoforms (I, II, IX and XII) using acetazolamide (AAZ) as a reference CA inhibitor. The resulted inhibition constants were presented in Table 1.
Table 1.
hCA I, II, IX and XII inhibition results from the carboxylic acid derivatives 5a–q, 7a–b and 12a–c and acetazolamide (AAZ) as a reference CA inhibitor. 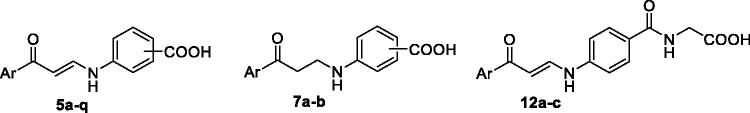
| Cmpd. | COOH | Ar | KI (μM)a |
|||
|---|---|---|---|---|---|---|
| hCA I | hCA II | hCA IX | hCA XII | |||
| 5a | ortho | -C6H5 | >100 | 39.4 | 10.6 | 6.4 |
| 5b | ortho | −4-F-C6H4 | >100 | 24.3 | 13.2 | 4.3 |
| 5c | ortho | −4-Cl-C6H4 | >100 | 32.7 | 17.7 | 12.5 |
| 5d | ortho | −4-NO2-C6H4 | >100 | 26.9 | 14.1 | 7.8 |
| 5e | ortho | −2-Naph | >100 | 49.0 | 19.3 | 15.7 |
| 5f | meta | -C6H5 | >100 | 72.8 | 15.9 | 9.5 |
| 5g | meta | −4-F-C6H4 | >100 | 68.2 | 9.6 | 10.3 |
| 5h | meta | −4-Cl-C6H4 | >100 | 77.1 | 19.2 | 8.6 |
| 5i | meta | −4-Br-C6H4 | >100 | 81.4 | 32.5 | 13.9 |
| 5j | meta | −4-NO2-C6H4 | >100 | 80.7 | 24.7 | 8.9 |
| 5k | meta | −2-Naph | >100 | 92.3 | 11.4 | 9.7 |
| 5l | para | -C6H5 | 75.3 | 9.8 | 1.2 | 0.97 |
| 5m | para | −4-F-C6H4 | 68.8 | 8.7 | 0.92 | 1.1 |
| 5n | para | −4-Cl-C6H4 | 81.5 | 13.5 | 3.4 | 4.6 |
| 5o | para | −4-Br-C6H4 | >100 | 15.3 | 5.6 | 6.2 |
| 5p | para | −4-NO2-C6H4 | 95.6 | 12.6 | 4.1 | 0.85 |
| 5q | para | −2-Naph | 79.4 | 7.4 | 0.76 | 1.5 |
| 7a | meta | -C6H5 | >100 | 64.3 | 23.7 | 7.4 |
| 7b | para | -C6H5 | 63.8 | 16.7 | 8.3 | 9.1 |
| 12a | – | -C6H5 | >100 | >100 | >100 | >100 |
| 12b | – | −4-Br-C6H4 | >100 | >100 | >100 | >100 |
| 12c | – | −2-Naph | >100 | >100 | >100 | >100 |
| AAZ | – | – | 0.250 | 0.012 | 0.025 | 0.006 |
aMean from 3 different assays (errors were in the range of ± 5–10% of the reported values).
The stopped-flow assay outputs disclosed that the off-target cytosolic hCA I exhibited the lowest inhibition in this study. Both the ortho and meta carboxylic acids bearing enaminones (5a–e and 5f–k) didn’t exert any noticeable inhibitory activity towards this isoform (KI > 100 µM), whereas the para carboxylic acids bearing enaminones 5l, 5m, 5n, 5p and 5q exhibited weak inhibitory effect (KI = 75.3, 68.8, 81.5, 95.6 and 79.4 µM, respectively), Table 1. In like manner, the target carboxylic acids 7a–b with a saturated 3-oxo-propylamine linker showed the same behaviour; the meta carboxylic acid derivative 7a emerged as inactive, whereas the para carboxylic acid derivative 7b emerged as a weak inhibitor for the hCA I isoform (KI = 63.8 µM), Table 1.
Moreover, the stopped-flow assay outputs showed that the physiologically relevant hCA II isoform was affected by herein reported carboxylic acids bearing enaminones 5a–q and carboxylic acids 7a–b. Both the ortho and meta carboxylic acids bearing enaminones (5a–e and 5f–k) moderately inhibited hCA II isoform with inhibition constants spanning in the ranges 24.3–49.0 µM and 68.2–92.3 µM, respectively, whereas the para carboxylic acids bearing enaminones 5l–q effectively inhibited this isoform (KI range: 7.4–15.3 µM). In addition, carboxylic acids 7a–b with a saturated 3-oxo-propylamine linker displayed KI values equal to 64.3 µM and 16.7 µM, respectively. In particular, the best inhibitory activity in the CO2 hydrase assay was demonstrated by the para carboxylic acids bearing enaminones 5l, 5m and 5q that showed KI values in the single-digit micromolar range equal to 9.8 µM, 8.7 µM and 7.4 µM, respectively. Also, carboxylic acids 5n, 5o, 5p and 7b exhibited good activity against hCA II isoform with KI = 13.5 µM, 15.3 µM, 12.6 µM and 16.7 µM, respectively.
Regarding the effect of the regioisomerism with the enaminones series 5a–q, it was found that shifting the carboxylic acid group from the ortho position (compounds 5a–e) to the meta position (compounds 5f–k) resulted in decreasing activity. For example, the phenyl and naphthyl-bearing enaminones 5f and 5k displayed about 2-fold decreased potency (KI = 72.8 µM and 92.3 µM, respectively), in comparison to their ortho counterparts 5a and 5e (KI = 39.4 µM and 49.0 µM, respectively), Table 1. In turn, shifting the carboxylic acid functionality from the ortho position (enaminones 5a–e) to the para position (enaminones 5l–q) led to about 2–6.5 fold enhancement of the inhibitory activity against hCA II isoform. For example, the phenyl and naphthyl-bearing enaminones 5l and 5q emerged as more potent inhibitors (KI = 9.8 µM and 7.4 µM, respectively) than their ortho analogues 5a and 5e (KI = 39.4 µM and 49.0 µM, respectively). Similarly in the second set, the para carboxylic acid derivative 7b displayed better activity (KI = 16.7 µM) than its meta counterpart 7a (KI = 64.3 µM) against hCA II isoform, Table 1.
The trans-membrane hCA IX, the third examined isoform, was effectively inhibited by all carboxylic acids bearing enaminones 5a–q and carboxylic acids 7a–b evaluated here, with inhibition constants spanning in the range 0.76–32.5 µM. The best inhibitory action towards hCA IX isoform has been observed for enaminones 5m and 5q with sub-micromolar inhibition constants (0.92 µM and 0.76 µM, respectively), whereas compounds 5g, 5l, 5n, 5o, 5p and 7b exerted good inhibitory activity with KIs in the single-digit micromolar range equal to 9.6 µM, 1.2 µM, 3.4 µM, 5.6 µM, 4.1 µM and 8.3 µM, respectively.
As discussed above for the inhibitory activity against hCA II, grafting the carboxylic acid functionality at the para position (enaminones 5l–q) resulted in a much-enhanced activity (KI range: 0.76–5.6 µM) in comparison to the ortho (enaminones 5a–e; KI range: 10.6–19.3 µM) and meta (enaminones 5f–k; KI range: 11.4–32.5 µM) counterparts, Table 1. For example, the phenyl and naphthyl-bearing enaminones 5l and 5q showed about 9- and 25-fold enhanced potency (KI = 1.2 µM and 0.76 µM, respectively) in comparison to their ortho counterparts 5a and 5e (KI = 10.6 µM and 19.3 µM, respectively), and showed about 13- and 15-fold increased activity in comparison to their meta counterparts 5f and 5k (KI = 15.9 µM and 11.4 µM, respectively). In like manner, the para carboxylic acid derivative 7b exerted better hCA IX inhibitory activity (KI = 8.3 µM) than its meta counterpart 7a (KI = 23.7 µM), Table 1.
On the other hand, examining the impact of the substitution of the phenyl tail within the most potent enaminones series (5l–q) revealed that only grafting the small fluorine substituent (enaminone 5m; KI = 0.92 µM) can enhance the activity of the unsubstituted phenyl-bearing enaminone 5l (KI = 0.92 µM). In turn, substituting the phenyl tail with 4-Cl, 4-Br or 4-NO2 substituents reduced the activity. Moreover, the replacement of the phenyl tail with a naphthyl one resulted in the most potent hCA IX inhibitor in this work (enaminone 5m; KI = 0.76 µM), Table 1. Notably, the same SAR could be observed for the ortho carboxylic acids bearing enaminones 5f–k.
It is worthy to mention that the phenyl-bearing enaminones 5f and 5l exhibited better activity against hCA IX (KI = 15.9 µM and 1.2 µM, respectively) than their flexible counterparts 7a–b that bear the saturated 3-oxo-propylamine linker (KI = 23.7 µM and 8.3 µM, respectively) which hints out that the conformational restriction for herein reported compounds is more favourable for hCA IX inhibitory action.
The newly reported carboxylic acids (5a–q and 7a–b) efficiently inhibited the trans-membrane hCA XII isoform with KI range of 0.85–15.7 µM. Uniquely, the para carboxylic acids bearing enaminones 5l and 5p induced inhibition in the submicromolar range, with KIs of 0.97 µM and 0.85 µM, respectively. Similarly, to the inhibition profile for both hCA II and IX isoforms, the para carboxylic acids bearing enaminones (5l–q) exerted more potent activity (KI range: 0.85–6.2 µM) than their ortho (enaminones 5a–e; KI range: 4.3–15.7 µM) and meta (enaminones 5f–k; KI range: 8.6–13.9 µM) counterparts, Table 1. With an exception for the 4-NO2 substitution (enaminone 5p; KI = 0.85 µM), neither substitution of the phenyl tail nor its bioisosteric replacement with the naphthyl moiety enhanced the hCA XII inhibitory activity.
Finally, the third set of hippuric acid-bearing enaminones 12a–c couldn’t inhibit any of the examined CA isoforms up to 100 µM, highlighting that the structural extension approach is not appropriate for the CA inhibitory activity (Table 1).
3. Conclusion
Three sets of our designed non-classical CA inhibitors (5a–q, 7a–b and 12a–c) were synthesised and evaluated for their CA inhibitory activity. The first (5a–q) and the second (7a–b) sets incorporated a benzoic acid moiety that is connected to different aryl tails through an enaminone linker, for the first set, or through the more flexible saturated 3-oxo-propylamine linker, for the second set. For the third set, a structural extension approach was utilised to replace the benzoic acid motif with the hippuric acid to furnish the carboxylic acid derivatives 12a–c. The inhibitory activity for the prepared carboxylic acids was variable across the four tested CA isoforms, where for example, most compounds showed no or weak inhibition towards hCA I. Furthermore, compounds 5q, 5m, and 5l exhibited a very similar inhibition pattern towards hCA II and IX with KI values equal to 7.4, 8.7, 9.8 and 0.76, 0.92, 1.2 µM respectively. Regarding hCA XII, the para nitro-substituted derivative 5p showed the highest inhibitory activity. Structural extension as done in the third set 12a–c, showed no inhibitory actions towards all tested hCA isoforms, which suggested that this extension might lead to undesired interactions within the CA active site. Generally, ortho/meta carboxylic acid substituted derivatives exhibited weaker inhibitory activities against the tested hCAs. Future studies could reveal the desired pharmacophoric features which could enhance hCA inhibitory actions, as well as selectivity across different isoforms, that belong to this family of enzymes.
4. Experimental
4.1. Chemistry
4.1.1. General
Stuart melting point apparatus was used for melting point measurements and were uncorrected. Schimadzu FT-IR 8400S spectrophotometer was used to record Infra-red (IR) spectra as KBr discs. NMR Spectra were recorded on a Bruker NMR spectrometer, at 400 and 100 MHz for 1H spectrum and 13 C spectrum, respectively, and were run at 100 MHz in deuterated dimethylsulphoxide (DMSO-d6). HRMS spectra were recorded by a Bruker MicroTOF spectrometer.
4.1.2. Synthesis of intermediates 3, 6 and 10
3-(Dimethylamino)-1-phenylprop-2-en-1-ones (3a–f28) 3-(dimethylamino)-1-phenylpropan-1-one (629) and (4-aminobenzoyl)glycine (1130) were prepared according to the literature procedures.
4.1.3. Synthesis of the target carboxylic acids enaminones 5a–q
An amount of 0.6 mmol from the appropriate enaminone intermediate 3a–f was dissolved in 4 ml glacial acetic acid, then the required amount (0.8 g, 0.6 mmol) of o-, m-, p-aminobenzoic acid 4a–c was added to the reaction mixture. Reflux was continued for 3 h, and then the precipitated solid was filtered off while hot, washed with hot 70% ethanol (3 × 2 ml), dried and recrystallized from DMF/methanol mixture (1:4) to yield the target enaminones 5a–q (Supporting Information).
4.1.4. Synthesis of target 3/4-((3-oxo-3-phenylpropyl)amino)benzoic acids 7a–b
To a hot solution of 3-(dimethylamino)-1-phenylpropan-1-one 6 (0.25 g, 1.41 mmol) in absolute ethanol (12 ml), an equivalent amount of m- or p-aminobenzoic acid 4b–c (0.19 g, 1.41 mmol) was added, and the mixture was heated under reflux for 4 h. The formed solid, after cooling, was collected and washed with aq. potassium bicarbonate solution and cold methanol (2 × 3 ml), then recrystallized from isopropanol to get the 3/4-((3-oxo-3-phenylpropyl)amino)benzoic acids 7a–b, respectively (Supporting Information).
4.1.5. Synthesis of target carboxylic acids enaminones 12a–c
Enaminones 12a–c were prepared via the same method described previously for enaminones 5a–q, utilising (4-aminobenzoyl)glycine 11 instead of aminobenzoic acids 4a–c (Supporting Information).
4.2. Biological evaluation
4.2.1. Ca inhibitory assay
The target carbonic anhydrase inhibitors reported in this work were evaluated for their potential CA-catalysed CO2 hydration activities utilising an Applied Photophysics stopped-flow instrument as described previously31–35 (Supporting Materials).
Supplementary Material
Funding Statement
The authors extend their appreciation to the Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2022R25), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors acknowledge financial support from the Researchers Supporting Project number (RSP-2021/103), King Saud University, Riyadh, Saudi Arabia.
Disclosure statement
CT Supuran is Editor-in-Chief of the Journal of Enzyme Inhibition and Medicinal Chemistry. He was not involved in the assessment, peer review, or decision-making process of this paper. The authors have no relevant affiliations of financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
- 1.Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68. [DOI] [PubMed] [Google Scholar]
- 2.Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88. [DOI] [PubMed] [Google Scholar]
- 3.Mishra CB, Tiwari M, Supuran CT.. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: where are we today? Med Res Rev 2020;40:2485–565. [DOI] [PubMed] [Google Scholar]
- 4.Mincione F, Nocentini A, Supuran CT.. Advances in the discovery of novel agents for the treatment of glaucoma. Expert Opin Drug Discov 2021;16:1209–25. [DOI] [PubMed] [Google Scholar]
- 5.Supuran CT. Acetazolamide for the treatment of idiopathic intracranial hypertension. Epxert Rev Neurother 2015;15:851–6. [DOI] [PubMed] [Google Scholar]
- 6.Nerella SG, Singh P, Arifuddin M, Supuran CT.. Anticancer carbonic anhydrase inhibitors: a patent and literature update 2018-2022. Expert Opin Ther Pat 2022;32:833–47. [DOI] [PubMed] [Google Scholar]
- 7.Swenson ER. Carbonic anhydrase inhibitros and high altitude ilnesses. Subcell Biochem 2014;75:361–86. [DOI] [PubMed] [Google Scholar]
- 8.Ciccone L, Cerri C, Nencetti S, Orlandini E.. Carbonic anhydrase and epilepsy: state of the art and future perspectives. Molecules 2021;26:6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buzás GM, Supuran CT.. The history and rationale of using carbonic anhydrase inhibitors in the treatment of peptic ulcers. In memoriam Ioan Puşcaş (1932–2015). J Enzyme Inhib Med Chem 2016;31:527–33. [DOI] [PubMed] [Google Scholar]
- 10.Costa G, Carta F, Ambrosio FA, et al. A computer-assisted discovery of novel potential anti-obesity compounds as selective carbonic anhydrase VA inhibitors. Eur J Med Chem 2019;181:111565. [DOI] [PubMed] [Google Scholar]
- 11.Wongboonsin J, Thongprayoon C, Bathini T, et al. Acetazolamide therapy in patients with heart failure: a meta-analysis. J Clin Med 2019;8:349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moi D, Nocentini A, Deplano A, et al. Structure-activity relationship with pyrazoline-based aromatic sulfamates as carbonic anhydrase isoforms I, II, IX and XII inhibitors: synthesis and biological evaluation. Eur J Med Chem 2019;182:111638. [DOI] [PubMed] [Google Scholar]
- 13.Bozdag M, Poli G, Angeli A, et al. N-aryl-N’-ureido-O-sulfamates: potent and selective inhibitors of the human carbonic anhydrase VII isoform with neuropathic pain relieving properties. Bioorg Chem 2019;89:103033. [DOI] [PubMed] [Google Scholar]
- 14.Zaraei S-O, Abduelkarem AR, Anbar HS, et al. Sulfamates in drug design and discovery: pre-clinical and clinical investigations. Eur J Med Chem 2019;179:257–71. [DOI] [PubMed] [Google Scholar]
- 15.Al-Warhi T, Sabt A, Elkaeed EB, Eldehna WM.. Recent advancements of coumarin-based anticancer agents: an up-to-date review. Bioorg Chem 2020;103:104163. [DOI] [PubMed] [Google Scholar]
- 16.Eldeeb AH, Abo-Ashour MF, Angeli A, et al. Novel benzenesulfonamides aryl and arylsulfone conjugates adopting tail/dual tail approaches: synthesis, carbonic anhydrase inhibitory activity and molecular modeling studies. Eur J Med Chem 2021;221:113486. [DOI] [PubMed] [Google Scholar]
- 17.Lomelino CL, Supuran CT, McKenna R.. Non-classical inhibition of carbonic anhydrase. Int J Mol Sci 2016;17:1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masini E, Carta F, Scozzafava A, Supuran CT.. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:705–16. [DOI] [PubMed] [Google Scholar]
- 19.A study of SLC-0111 and gemcitabine for metastatic pancreatic ductal cancer in subjects positive for CAIX (SLC-0111-17-01). [cited 2022 May 30]. Available from: https://clinicaltrials.gov/ct2/show/NCT03450018.
- 20.D'Ambrosio K, Carradori S, Monti SM, et al. Out of the active site binding pocket for carbonic anhydrase inhibitors. Chem Comm 2015;51:302–5. [DOI] [PubMed] [Google Scholar]
- 21.Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60. [DOI] [PubMed] [Google Scholar]
- 22.Supuran CT. Novel carbonic anhydrase inhibitors. Future Med Chem 2021;13:1935–7. [DOI] [PubMed] [Google Scholar]
- 23.Cau Y, Vullo D, Mori M, et al. Potent and selective carboxylic acid inhibitors of tumor-associated carbonic anhydrases IX and XII. Molecules 2017;23:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eldehna WM, Nocentini A, Elsayed ZM, et al. Benzofuran-based carboxylic acids as carbonic anhydrase inhibitors and antiproliferative agents against breast cancer. ACS Med Chem Letters 2020;11:1022–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elimam DM, Elgazar AA, Bonardi A, et al. Natural inspired piperine-based sulfonamides and carboxylic acids as carbonic anhydrase inhibitors: design, synthesis and biological evaluation. Eur J Med Chem 2021;225:113800. [DOI] [PubMed] [Google Scholar]
- 26.Alkhaldi AA, Al-Sanea MM, Nocentini A, et al. 3-Methylthiazolo [3, 2-a] benzimidazole-benzenesulfonamide conjugates as novel carbonic anhydrase inhibitors endowed with anticancer activity: design, synthesis, biological and molecular modeling studies. Eur J Med Chem 2020;207:112745. [DOI] [PubMed] [Google Scholar]
- 27.Eldehna WM, Abo-Ashour MF, Berrino E, et al. SLC-0111 enaminone analogs, 3/4-(3-aryl-3-oxopropenyl) aminobenzenesulfonamides, as novel selective subnanomolar inhibitors of the tumor-associated carbonic anhydrase isoform IX. Bioorg Chem 2019;83:549–58. [DOI] [PubMed] [Google Scholar]
- 28.Xu R-S, Yue L, Pan Y-J.. Regioselective copper(I)-catalyzed C–H hydroxylation/C–S coupling: expedient construction of 2-(styrylthio)phenols. Tetrahedron 2012;68:5046–52. [Google Scholar]
- 29.Xu Z, Tice CM, Zhao W, et al. Structure-based design and synthesis of 1,3-oxazinan-2-one inhibitors of 11β-hydroxysteroid dehydrogenase type 1. J Med Chem 2011;54:6050–62. [DOI] [PubMed] [Google Scholar]
- 30.Glas C, Dietschreit JCB, Wössner N, et al. Identification of the subtype-selective Sirt5 inhibitor balsalazide through systematic SAR analysis and rationalization via theoretical investigations. Eur J Med Chem 2020;206:112676. [DOI] [PubMed] [Google Scholar]
- 31.Abo-Ashour MF, Eldehna WM, Nocentini A, et al. 3-Hydrazinoisatin-based benzenesulfonamides as novel carbonic anhydrase inhibitors endowed with anticancer activity: synthesis, in vitro biological evaluation and in silico insights. Eur J Med Chem 2019;184:111768. [DOI] [PubMed] [Google Scholar]
- 32.Abdelrahman MA, Ibrahim HS, Nocentini A, et al. Novel 3-substituted coumarins as selective human carbonic anhydrase IX and XII inhibitors: synthesis, biological and molecular dynamics analysis. Eur J Med Chem 2021;209:112897. [DOI] [PubMed] [Google Scholar]
- 33.Elimam DM, Eldehna WM, Salem R, et al. Natural inspired ligustrazine-based SLC-0111 analogues as novel carbonic anhydrase inhibitors. Eur J Med Chem 2022;228:114008. [DOI] [PubMed] [Google Scholar]
- 34.Abdelrahman MA, Eldehna WM, Nocentini A, et al. Novel benzofuran-based sulfonamides as selective carbonic anhydrases IX and XII inhibitors: synthesis and in vitro biological evaluation. J Enzyme Inhib Med Chem 2020;35:298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ibrahim HS, Abdelrahman MA, Nocentini A, et al. Insights into the effect of elaborating coumarin-based aryl enaminones with sulfonamide or carboxylic acid functionality on carbonic anhydrase inhibitory potency and selectivity. Bioorg Chem 2022;126:105888. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.