

ABSTRACT
Impairments in macroautophagy/autophagy, which degrades dysfunctional organelles as well as long-lived and aggregate proteins, are associated with several cardiomyopathies; however, the regulation of cardiac autophagy remains insufficiently understood. In this regard, ULK1 and ULK2 are thought to play primarily redundant roles in autophagy initiation, but whether their function is developmentally determined, potentially having an impact on cardiac integrity and function remains unknown. Here, we demonstrate that perinatal loss of ULK1 or ULK2 in cardiomyocytes (cU1-KO and cU2-KO mice, respectively) enhances basal autophagy without altering autophagy machinery content while preserving cardiac function. This increased basal autophagy is dependent on the remaining ULK protein given that perinatal loss of both ULK1 and ULK2 in cU1/2-DKO mice impaired autophagy causing age-related cardiomyopathy and reduced survival. Conversely, adult loss of cardiac ULK1, but not of ULK2 (i.e., icU1-KO and icU2-KO mice, respectively), led to a rapidly developing cardiomyopathy, heart failure and early death. icU1-KO mice had impaired autophagy with robust deficits in mitochondrial respiration and ATP synthesis. Trehalose ameliorated autophagy impairments in icU1-KO hearts but did not delay cardiac dysfunction suggesting that ULK1 plays other critical, autophagy-independent, functions in the adult heart. Collectively, these results indicate that cardiac ULK1 and ULK2 are functionally redundant in the developing heart, while ULK1 assumes a more unique, prominent role in the adult heart.
Abbreviations: ATG4: autophagy related 4, cysteine peptidase; ATG5: autophagy related 5; ATG7: autophagy related 7; ATG9: autophagy related 9; ATG13: autophagy related 13; CYCS: Cytochrome C; DNM1L, dynamin 1-like; MAP1LC3A: microtubule-associated protein 1 light chain 3 alpha; MAP1LC3B: microtubule-associated protein 1 light chain 3 beta; MFN1: mitofusin 1; MFN2: mitofusin 2; MT-CO1: mitochondrially encoded cytochrome c oxidase I; MYH: myosin, heavy polypeptide; NBR1: NBR1 autophagy cargo receptor; NDUFA9: NADH:ubiquinone oxidoreductase subunit A9; OPA1: OPA1, mitochondrial dynamin like GTPase; PPARGC1A, peroxisome proliferator activated receptor, gamma, coactivator 1 alpha; SDHA: succinate dehydrogenase complex, subunit A, flavoprotein (Fp); SQSTM1: sequestosome 1; ULK1: unc-51 like kinase 1; ULK2: unc-51 like kinase 2; UQCRC1: ubiquinol-cytochrome c reductase core protein 1
KEYWORDS: Age-related cardiomyopathy, dilated cardiomyopathy, heart failure, MAP1LC3, mitochondria, mitophagy, NBR1, SQSTM1
Introduction
Macroautophagy, from this point on referred to as autophagy, is a catabolic process conserved from yeast to mammals. Autophagy targets long-lived proteins and cellular organelles for degradation and is thereby required for protein quality control and normal cellular function [1]. Impaired or insufficient autophagy has been implicated in aging [2], and in the progression of chronic disease states including those associated with neurodegeneration [3] and obesity [4]. In the heart, deficient autophagy is a feature of several cardiomyopathies [5,6], while preserved or improved autophagy can be beneficial [7–9]. Accordingly, cardiomyocyte-specific deletion of the critical autophagy gene Atg5 leads to either adult or age-related heart failure [10,11]. Therefore, a better understanding of the molecular regulation of cardiac autophagy may reveal new therapeutic targets for preservation of cardiac function in various pathophysiological conditions.
ULK1 and ULK2, mammalian homologs of yeast Atg1 (autophagy related 1), are thought to function as metabolic rheostats stimulating autophagosome formation [12,13]. ULK1, by far the best characterized ULK, is sensitive to metabolic status as it is directly stimulated under low energy or low nutrient conditions via decreased inhibitory inputs from MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1) and increased stimulation by AMP-activated protein kinase (AMPK) [14,15]. Although ULK1 and ULK2 are considered to be functionally redundant, recent studies have uncovered distinct tissue-specific roles for these proteins in modulation of autophagy and other cellular processes [16,17]. Given the comprehensive nature of cellular structures targeted by autophagy, these findings indicate that the regulation of autophagy must be tailored to the specific proteostatic needs of each tissue. To this matter, the very high metabolic demands of uninterrupted, life-long contraction-relaxation cycles of the heart combined with the post-mitotic nature of cardiomyocytes represent a unique challenge to protein quality control processes such as autophagy. Moreover, autophagy might have distinct patterns of regulation in the adult versus developing heart [18]. Thus, it is critical to determine whether ULK1 and ULK2 play redundant roles in modulating cardiac autophagy, and if developmental differences exist in the adaptation of the heart to ULK1 or ULK2 deficiency.
The present study was designed to rigorously examine functional redundancies of ULK1 and ULK2 in the developing and the adult heart. Here, we addressed these questions by generating mouse models with perinatal or adult deletion of cardiac Ulk1 and/or Ulk2. Our findings demonstrate that cardiomyocyte expression of either ULK1 or ULK2 heightens autophagy and preserves cardiac function when the other ULK is lost perinatally. However, the adult loss of cardiac ULK1, but not of ULK2, compromises autophagy and mitochondrial function leading to heart failure.
Results
Perinatal loss of cardiomyocyte ULK1 or ULK2 enhances autophagy with maintenance of cardiac function
Ulk2 transcripts are more abundant than Ulk1 transcripts from birth to adulthood in mouse hearts (Figure S1A, Supplemental Materials and Methods). However, potential functional differences remain unknown. To establish their respective roles in regulating cardiomyocyte autophagy and function, we developed cardiac-specific knockouts of Ulk1 and Ulk2. Mice bearing floxed Ulk1 or Ulk2 alleles were crossed with transgenic mice expressing Cre recombinase under the control of the Myh6/α-myosin heavy chain promoter (Myh6-Cre) [19] generating ulk1flox/flox; Myh6-Cre (cU1-KO) and ulk2flox/flox; Myh6-Cre (cU2-KO) mice, respectively. MYH6-Cre becomes active at around embryonic day 8 [20], thereby causing the recombination of Ulk genes perinatally. cU1-KO mice were born with the same Mendelian frequency as wild-type littermates (Table S1) suggesting normal embryonic and perinatal development. cU2-KO mice, however, were born at slightly lower rates than wild-type littermates indicating a mildly negative impact of ulk2 deletion on early cardiac development (Table S2). After birth, no spontaneous deaths were observed in either cU1-KO or cU2-KO for up to 90 weeks of age when mice were euthanized. Successful deletion of Ulk1 and Ulk2 genes was confirmed in isolated cardiomyocytes from adult cU1-KO and cU2-KO hearts (Figures 1A and 2A). Phosphorylation of ATG13 S318 [21] was significantly decreased in both cU1-KO and cU2-KO hearts (Figures S1B and S2A). However, phosphorylation of BECN1 S30, another putative target of both ULK1 and ULK2 [22], was decreased in cU1-KO hearts but increased in cU2-KO hearts (Figures S1C and S2B) indicating that BECN1 S30 is preferentially targeted by ULK1 in the heart.
Figure 1.
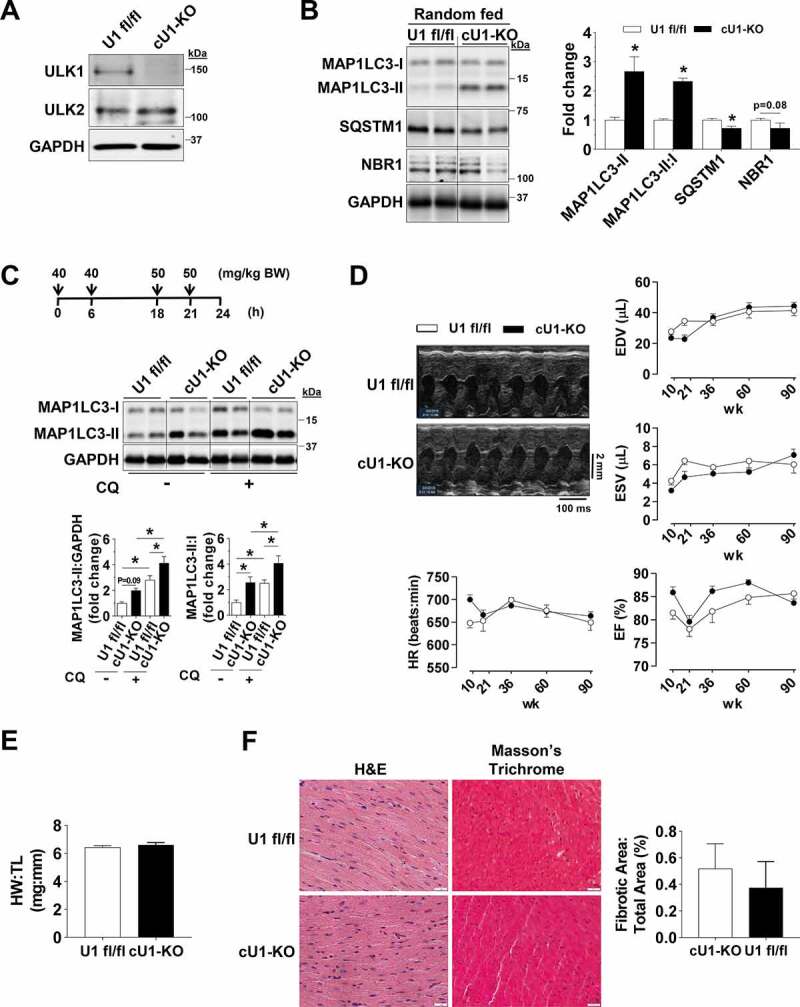
Perinatal deletion of ULK1 leads to a compensatory increase in basal autophagy. Data were obtained in 10 to 14-wk old mice unless otherwise specified. (A) Constitutive ulk1 deletion in isolated cardiomyocytes of cU1-KO mice. (B) Protein levels of MAP1LC3A-MAP1LC3B (MAP1LC3), SQSTM1, and NBR1 in randomly fed U1 fl/fl and cU1-KO mice. Representative immunoblots (Left) and summary of relative expression (Right) (n = 4–7). (C) Protein levels of MAP1LC3 analyzed after chloroquine administration as shown. Representative immunoblots (Top) and summary of relative expression (Bottom) (n = 5–8). (D) Representative B-mode images and serial cardiac function of U1 fl/fl and cU1-KO mice measured at 10, 21, 36, 60, and 90 weeks of age (n = 9–10). (E) Heart weight:tibia length reveals no overt cardiac hypertrophy (n = 7–8). (F) Representative H&E and Masson’s Trichrome images (Left) (scale: 20 μm) and quantification of fibrotic tissue (Right) (n = 4–6). Data are means ± SEM; *P < 0.05.
Figure 2.
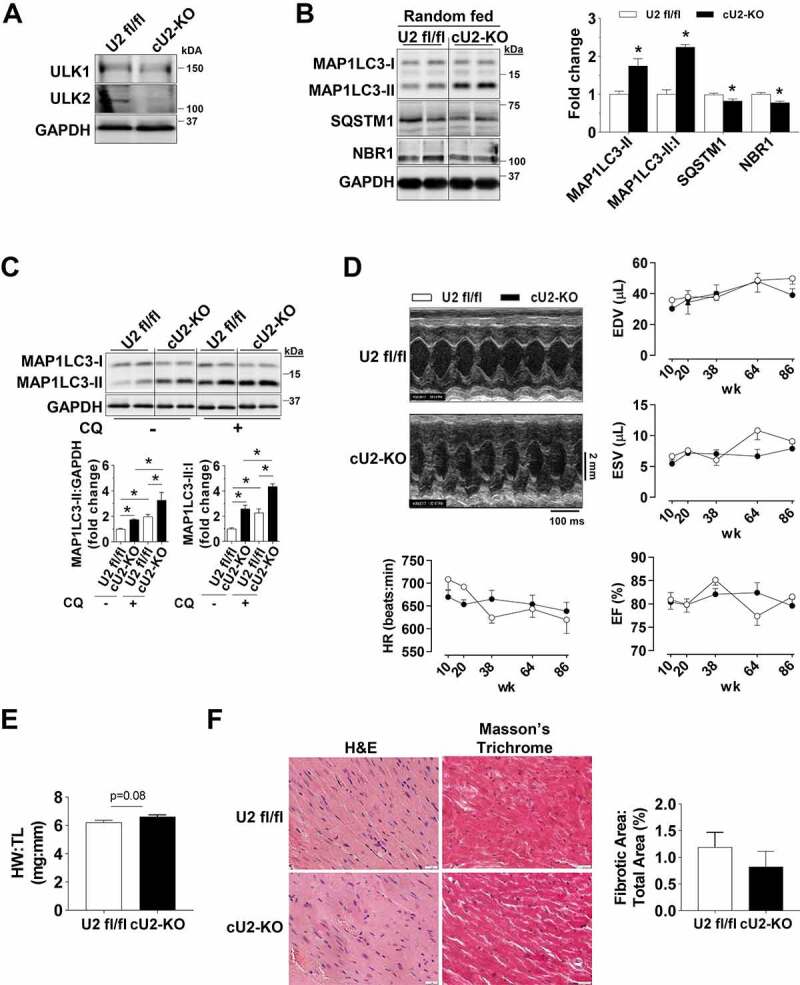
Perinatal deletion of ULK2 leads to a compensatory increase in basal autophagy. All data was obtained in 10- to 14-wk-old mice unless otherwise specified. (A) Constitutive ULK2 deletion in isolated cardiomyocytes of cU2-KO mice. (B) Protein levels of MAP1LC3A-MAP1LC3B (MAP1LC3), SQSTM1, and NBR1 in randomly fed U2 fl/fl and cU2-KO mice. Representative immunoblots (Left) and quantification of relative expression (Right) (n = 4–7). (C) Protein levels of MAP1LC3 analyzed after chloroquine administration. Representative immunoblots (Top) and summary of relative expression (Bottom) (n = 6–10). (D) Representative B-mode images and serial cardiac function of U2 fl/fl and cULK2-KO mice measured at 10, 38, 64, and 86 weeks of age (n = 5–13). (E) Heart weight:tibia length reveals mild cardiac hypertrophy in cU2-KO mice (n = 5–6). (F) Representative H&E and Masson’s Trichrome images (Left) (scale: 20 μm) and quantification of fibrotic tissue (Right) (n = 4–6). Data are means ± SEM; *P < 0.05.
Based on the previously described role of ULK1, and potentially ULK2, in initiating autophagy [23], we first examined protein levels of MAP1LC3A-MAP1LC3B (MAP1LC3) in randomly fed mouse hearts. Unexpectedly, in both cU1-KO and cU2-KO mice, MAP1LC3-II protein levels and the MAP1LC3-II:I ratio were significantly increased. These were paralleled by decreased levels of the autophagic receptors SQSTM1 and NBR1, which are degraded by autophagy (Figures 1B and 2B). Ubiquitinated proteins (i.e., covalently linked to UBA52, UBB, UBC, RPS27A), which commonly accumulate upon autophagy deficiency, were unaltered (Figures S1D and S2C), and transmission electron microscopy (TEM) images revealed increased numbers of autophagosomes and autolysosomes in both cU1-KO and cU2-KO hearts (Figures S1E and S2D). These findings suggested that perinatal loss of ULK1 or ULK2 could be stimulating cardiac autophagy. We then tested this possibility by assessing MAP1LC3 levels in mice after chloroquine (CQ) administration to inhibit autophagosome to lysosome fusion [24]. Indeed, CQ administration led to increased accumulation of MAP1LC3-II and significantly elevated MAP1LC3-II:I ratio in cU1-KO and cU2-KO mouse hearts when compared to WT littermates, indicating increased autophagic flux in hearts deficient of either ULK (Figures 1C and 2C). Taken together, these data indicate that perinatal loss of either ULK1 or ULK2 in cardiomyocytes increases autophagy in the postnatal heart. Although the hearts of cU2-KO trended to be slightly larger (~7%) than that of wild-type littermates, cardiac function and survival were unaltered and there was no increase in fibrosis in either adult cU1-KO or cU2-KO mice (Figures 1D–F and Figure2D–F, respectively). However, transcriptional markers of pathological remodeling were evident in cU2-KO but not in CU1-KO hearts (Figures S1F and S2E).
Given that induction of core autophagy proteins such as ATG7 could be sufficient to increase cardiac autophagy [25], we next tested if increased expression of ATG proteins contributed to the increased basal cardiac autophagy in cU1-KO and cU2-KO mice. However, expression levels of the essential core autophagy proteins ATG7, ATG12–ATG5, ATG9, ATG4 and ATG3 were unchanged in the hearts of these mice (Figures S1G and S2F).
Combined perinatal loss of ULK1 and ULK2 in cardiomyocytes impairs autophagy and leads to age-related cardiomyopathy
Because the increased basal autophagy seen in both cU1-KO and cU2-KO mice could not be explained by increases in the cardiac autophagy machinery, we hypothesized that the perinatal expression of a single ULK in cardiomyocytes (i.e., ULK1 or ULK2) was required for the enhanced autophagy observed. To this end, we generated cardiac-specific ulk1 and ulk2 double-KO (cU1/2-DKO) mice by crossing mice bearing both floxed Ulk1 and Ulk2 alleles with mice expressing Myh6-Cre [19]. Both ULK1 and ULK2 proteins were deleted in cU1/2-DKO adult mouse hearts (Figure 3A). cU1/2-DKO mice were born at a significantly lower rate than the expected Mendelian ratio (Table S3), indicating that the combined deletion of cardiomyocyte Ulk1 and Ulk2 compromised the development of some embryos. No spontaneous deaths occurred from birth to adulthood in cU1/2-DKO. Examination of adult cU1/2-DKO hearts revealed decreased MAP1LC3-II levels and decreased MAP1LC3-II:I ratio, which were accompanied by accumulation of SQSTM1 and NBR1 (Figure 3A), but without changes in ubiquitinated proteins and in the number of autophagosomes and autolysosomes in the myocardium (Figures S3A and S4A). We then assessed cardiac MAP1LC3 levels following CQ administration and observed decreased MAP1LC3-II accumulation and decreased MAP1LC3-II:I ratio in cU1/2-DKO compared to U1/2 fl/fl mice indicating impaired autophagic flux in the ULK deficient hearts (Figure 3B). The stimulation of autophagy by prolonged fasting was also substantially impaired in cU1/2-DKO hearts as evidenced by only a modest increase in MAP1LC3-II and MAP1LC3-II:I ratio and unchanged SQSTM1 and NBR1 (Figure S3B). These data indicated that basal autophagic flux was impaired in cU1/2-DKO mouse hearts. Total ATG13 was robustly increased in cU1/2-DKO mouse hearts, but the proportion of ATG13 S318 phosphorylation was decreased (Figure S3C). Phosphorylation of BECN1 S30 was substantially decreased in cU1/2-DKO mice (Figure S3D). Inspection of gross morphology in the hearts of young adult cU1/2-DKO mice indicated a ~ 30% increase in heart weight with increases in cardiomyocyte area without changes in fibrosis (Figures 3C, 3E and S4B). To address whether this cardiac hypertrophy portended to cardiomyopathy later in life, we monitored cU1/2-DKO and U1/U2 fl/fl mice until 100 weeks of age. cU1/2-DKO started to spontaneously die at 57 weeks of age, with death rates increasing substantially at 70 weeks of age (Figure 3D). Although cardiac function was normal in most adult cU1/2-DKO mice until about 69 weeks of age, significant increases in end-diastolic volume (EDV) and reductions in ejection fraction (EF) became obvious thereafter (Figure 3F). Cardiac fibrosis also dramatically increased in old cU1/2-DKO mice (Figure S4C). Collectively, these data indicate that simultaneous perinatal loss of cardiomyocyte ULK1 and ULK2 impaired autophagy in the adult heart leading to age-related cardiomyopathy and a shortened lifespan.
Figure 3.
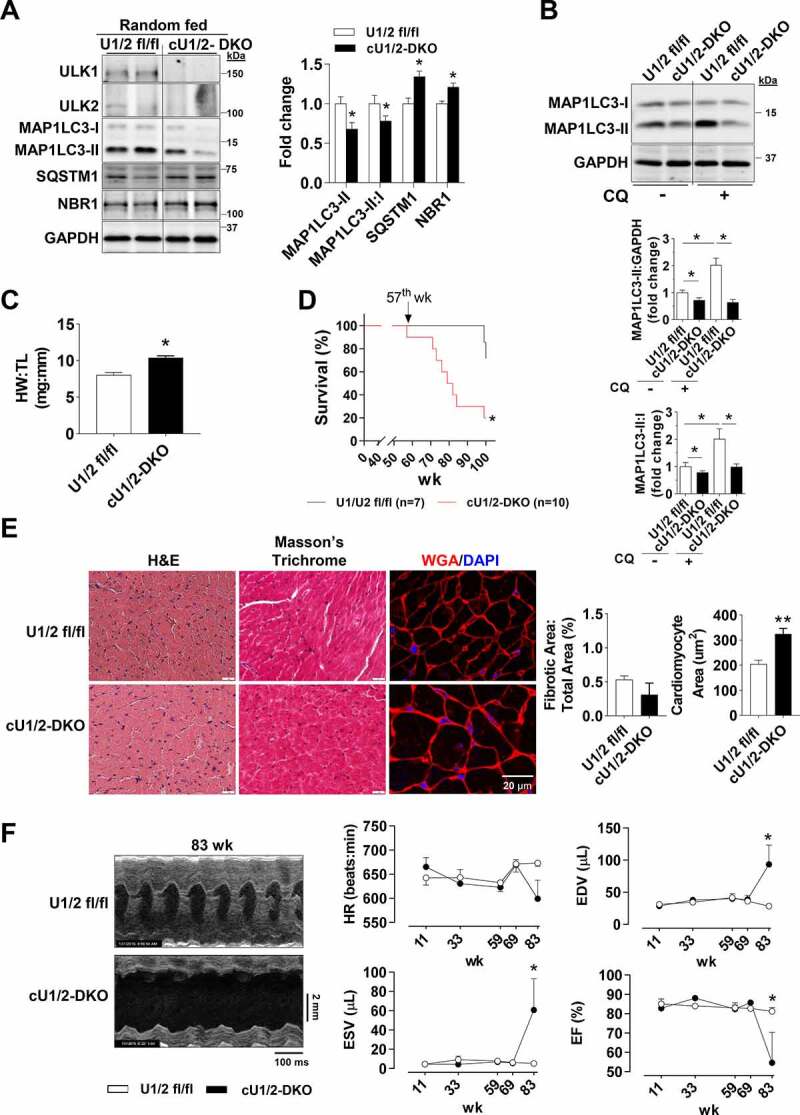
Perinatal deletion of ULK1 and ULK2 impairs basal autophagy and leads to age-associated cardiomyopathy. All data was obtained in 10 to14-wk old mice unless otherwise specified. (A) Constitutive ULK1 and ULK2 deletion in whole heart lysates and protein levels of MAP1LC3A-MAP1LC3B (MAP1LC3), SQSTM1, and NBR1 in cU1/2-DKO mice. Representative immunoblots (Left) and quantification (Right) (n = 4–12). (B) Protein levels of MAP1LC3 analyzed after chloroquine administration. Representative immunoblots (Top) and quantification of relative expression (Bottom) (n = 4–12). (C) Heart weight:tibia length reveals overt cardiac hypertrophy in cU1/2-DKO mice (n = 4–5). (D) Survival analysis of U1/2 fl/fl and cU1/2-DKO mice (n = 7–10). (E) Representative H&E, Masson’s Trichrome, and WGA/DAPI images (Left) and quantification (scale: 20 μm) of fibrotic tissue and cardiomyocyte area (Right) (n = 4–6). (F) Representative B-mode images and serial cardiac function of U1/2 fl/fl and cU1/2-DKO mice measured at 11, 33, 59, 69, and 83 weeks (n = 5–6 for U1/2 fl/fl throughout, and n = 3–6 for cU1/2-DKO). Data are means ± SEM; *P < 0.05.
Adult loss of ULK1, but not of ULK2, leads to heart failure and premature death
After observing that single perinatal deletions of ULK1 or ULK2 increased cardiac autophagy, we then examined whether this adaptation was developmentally determined. To test this hypothesis, we studied inducible cardiac-specific knockout mouse models of ULK1 and ULK2. Mice bearing either floxed Ulk1 or floxed Ulk2 alleles were crossed with transgenic mice expressing a tamoxifen-inducible Cre recombinase (Myh6-MerCreMer). Gene recombination in the Ulk1flox/flox; Myh6-MerCreMer+ mice (i.e., icU1-KO) and Ulk2flox/flox; Myh6-MerCreMer+ mice (i.e., icU2-KO) were induced at 8 weeks of age. Littermates not expressing Cre recombinase (i.e., Ulk1flox/flox and Ulk2flox/flox, respectively) were also injected with tamoxifen and used as controls. Effective loss of ULK proteins was confirmed in isolated cardiomyocytes of both icU1-KO and icU2-KO mice (Figures 4A and Figure4B). Robust early lethality was observed in icU1-KO mice with 100% of animals either dying or needing to be euthanized no later than 12 days after the initial tamoxifen administration (Figure 4C). In contrast, no spontaneous death was observed in icU2-KO mice up to 3 weeks post-tamoxifen administration, when animals were euthanized for further analyses (Figure 4D). Accordingly, only icU1-KO mice developed heart failure as evidenced by cardiac hypertrophy (~13%) with left ventricle enlargement, an >2-fold increase in end-diastolic volume (EDV) and markedly depressed ejection fraction (EF) (≤ 20%) (Figures 4E–4J). More detailed analyses of icU1-KO hearts revealed cardiomyocyte disorganization, increased expression of hypertrophy genes and prominent tissue fibrosis (Figures 4K, S5A and S5B), without obvious alterations in sarcomere length or structure (Figure S5C). Consistent with an overall impairment in autophagy, 5–8 days after the beginning of tamoxifen injections accumulation of SQSTM1, NBR1, and ubiquitinated proteins was obvious in icU1-KO, but not in icU2-KO hearts (Figures 4L, S6A and S6B). Despite accumulation of ATG13 and BECN1, ATG13 S318 and BECN1 S30 phosphorylation relative to total protein, were unchanged in icU2-KO hearts, and unexpectedly in icU1-KO hearts as well (Figures S6C-F) indicating that they might not reliably reflect ULK1 activity in failing hearts. Considering that cardiac function is heavily dependent on efficient energetics maintained primarily through oxidative metabolism, we next interrogated whether mitochondrial function and dynamics were defective in the hearts of icU1-KO. Assessment of mitochondrial respiration in permeabilized cardiac fibers revealed significant impairments in ADP-driven pyruvate/malate supported respiration and in both basal and ADP-driven palmitoyl-carnitine/malate supported respiration (Figure 5A), which were accompanied by proportional reductions in ATP synthesis rates (Figure 5B) in icU1-KO hearts. Additionally, we assessed expression of subunits present in each of the four electron transport chain complexes. NDUFA9, SDHA, and UQCRC1 were unchanged but expression of the mitochondrially encoded MTCO1, a subunit of the rate-limiting complex IV, was significantly decreased in icU1-KO (Figure 5C) suggesting that a defective complex IV could have contributed to the robust respiratory impairments observed. Because normal cardiac function is also contingent on a highly organized myocardium with electron-dense mitochondria compactly aligned between myofibrils [26,27], we then examined mitochondrial ultrastructure in the hearts of icU1-KO mice. Electron micrographs demonstrated that mitochondria were not tightly packed between myofibrils in icU1-KO hearts with clear signs of organelle degeneration, as indicated by mitochondria with disrupted and/or absent cristae structure as well as apparent remnants of cristae structure devoid of an outer mitochondrial membrane (Figure 5D). In addition, mitochondrial content and DNA were decreased along with Ppargc1a mRNA (Figures 5D–5F). Mitochondria from icU1-KO hearts were also significantly smaller both in terms of area and length (Figure 5D). Indeed, molecular inputs leading to hyper-fission were present in mitochondrial fractions of icU1-KO hearts as indicated by increased levels of phosphorylated DNM1L/DRP1 (p-DNM1L:DNM1L) without changes in MFN1 and MFN2 and OPA1 (Figure 5G). Collectively, these findings indicate that adult loss of ULK1, but not of ULK2, quickly leads to lethal dilated cardiomyopathy associated with major cellular and molecular abnormalities in the myocardium.
Figure 4.
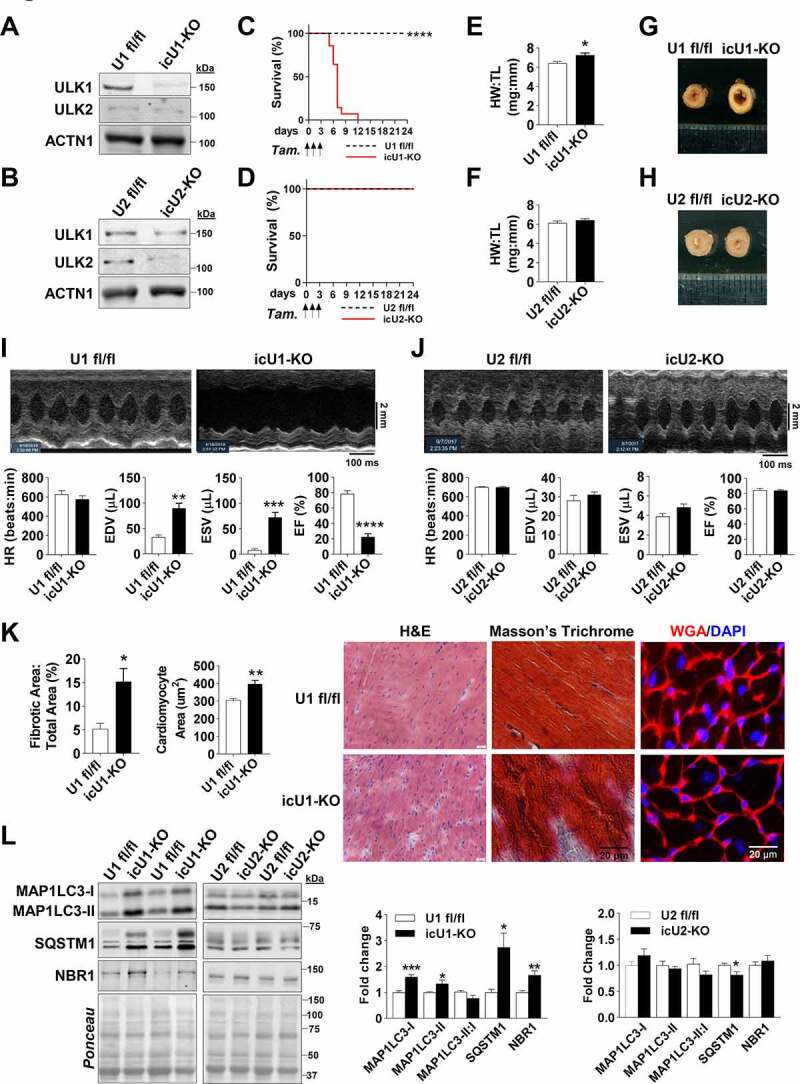
Adult deletion of ULK1, but not of ULK2, leads to a lethal, rapidly developing dilated cardiomyopathy and mitochondrial dysfunction. Data were obtained in 9-wk old mice 5–8 days after initial tamoxifen injection, unless otherwise specified. (A) Knockdown of ULK1 from isolated cardiomyocytes of icU1-KO mice. (B) Knockdown of ULK2 from isolated cardiomyocytes of icU2-KO mice. (C) Survival analysis of U1 fl/fl and icU1-KO mice (n = 13–14). (D) Survival analysis of U2 fl/fl and icU2-KO mice (n = 12–14). (E) Heart weight:tibia length reveals cardiac hypertrophy in icU1-KO mice (n = 7–15). (F) Heart weight:tibia length reveals no cardiac hypertrophy in icU2-KO mice (n = 5–8). (G) Representative left ventricle images from U1 fl/fl and icU1-KO mice. (H) Representative left ventricle images from U2 fl/fl and icU2-KO mice. (I) Echocardiographs of icU1-KO mice. Representative B-mode images and cardiac function of U1 fl/fl and icU1-KO mice (n = 7–10). (J) Echocardiographs of icU2-KO mice. Representative B-Mode Images (Top) and cardiac function (Bottom) (n = 6–10). (K) Representative H&E, Masson’s Trichrome, WGA/DAPI images (Right) and quantification of fibrotic tissue and cardiomyocyte area (Left). (L) Representative immunoblots of MAP1LC3A-MAP1LC3B (MAP1LC3), SQSTM1, and NBR1 (Left), and quantification of protein expression of MAP1LC3, SQSTM1, and NBR1 in icU1-KO hearts and icU2-KO hearts (Right) (n = 6–8). Data are means ± SEM; *P < 0.05, **P < 0.01 and ***P < 0.001.
Figure 5.
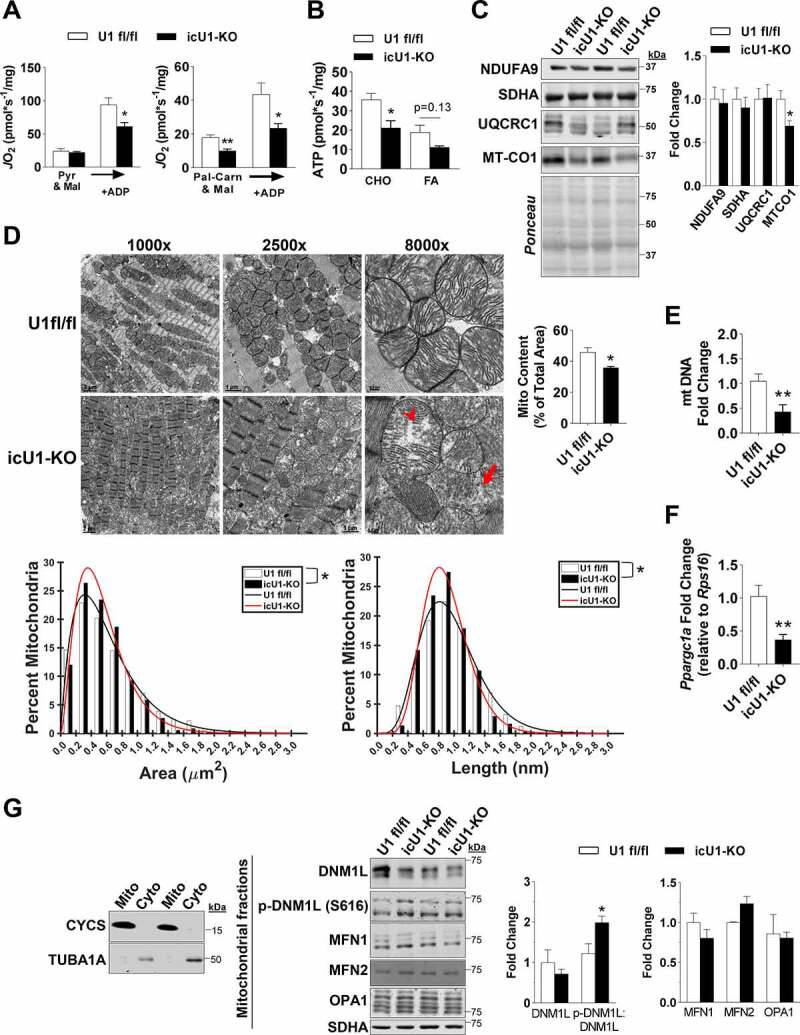
Mitochondrial function, morphology, and biogenesis in icU1-KO hearts. (A) Pyruvate-malate and palmitoyl-carnitine-malate driven oxygen flux (n = 4). (B) Pyruvate-malate and palmitoyl-carnitine-malate driven ATP production (n = 3–4). (C) Representative immunoblots of subunits of the electron transport chain (Left) and quantification (Right) (n = 6). (D) Representative TEM images of mitochondrial structure denoting a mitochondrion with aberrant and/or disrupted cristae structure (arrowhead) and remnants of a cristae structure devoid of outer mitochondrial membrane (arrow) (Top Left). Quantification of mitochondrial content in icU1-KO hearts expressed as a percentage of total area in TEM images (n = 4) (Top Right). Percentage distribution of mitochondria according to area and length assessed from TEM images (n = 3–4) (Bottom). (E) Mitochondrial DNA content quantification (n = 7). (F) Relative mRNA expression of Ppargc1a (n = 3–7). (G) Representative immunoblots of CYCS and TUBA1A in mitochondrial and cytosolic fractions from U1 fl/fl and icU1-KO hearts (Left). Representative immunoblots of DNM1L, MFN1, MFN2, and OPA1 (normalized to SDHA), and p-DNM1L (s616):DNM1L in enriched mitochondrial fractions from U1 fl/fl and icU1-KO hearts (Middle) and quantification (Right) (n = 3–4). Data are means ± SEM; *P < 0.05 and **P < 0.01.
Increasing autophagic flux following ULK1 loss in adult hearts does not delay cardiac dysfunction
To circumvent potential confounding effects of heart failure and to more precisely establish the impact of autophagy impairments on the development of cardiac dysfunction in icU1-KO mice, we performed additional experiments at day 4 after the initial tamoxifen injection. At this timepoint, icU1-KO mice exhibited normal behavior without any obvious signs of distress. ULK1 activity in lysates of isolated cardiomyocytes from icU1-KO hearts, estimated by in vitro HIS-BECN1 S15 phosphorylation [28], was also only modestly decreased (~35%) at this stage (Figures S7A and S7B, Supplemental Materials and Methods). We then administered CQ and observed that MAP1LC3-II and MAP1LC3-II:I ratio failed to increase in icU1-KO compared to U1 fl/fl mice consistent with impaired autophagy initiation (Figure 6A). Lysosomal content, as indicated by HEXB and CTSB protein levels, was unchanged, while CTSB activity was increased in icU1-KO hearts (Figures S7C and S7D, Supplemental Materials and Methods) suggesting that lysosomal function was at least preserved. Further, examination of TEMs demonstrated impairments in both autophagy and mitophagy, as the numbers of autophagosomes and mitophagosomes (i.e., autophagosomes containing mitochondrial remnants) failed to increase in icU1-KO hearts upon CQ administration (Figures 6B, 6C and S8). Next, we tested if ameliorating impaired autophagy would delay cardiac dysfunction in icU1-KO mice. Trehalose administration has been previously shown to increase autophagy in mice and to enhance mitochondrial function in cardiomyocytes and other cell types [29–31]. We confirmed that trehalose augmented cardiac autophagy flux in unstressed wild-type mice (Figure 7A), and then administered trehalose starting at day 1 of tamoxifen injections in icU1-KO mice. Our findings demonstrate that even though trehalose reduced the degree of autophagy impairments in icU1-KO mice, as indicated by decreasing accumulations of MAP1LC3, SQSTM1, and ubiquitinated proteins, cardiac dysfunction was not prevented or attenuated (Figures 7B–7D).
Figure 6.
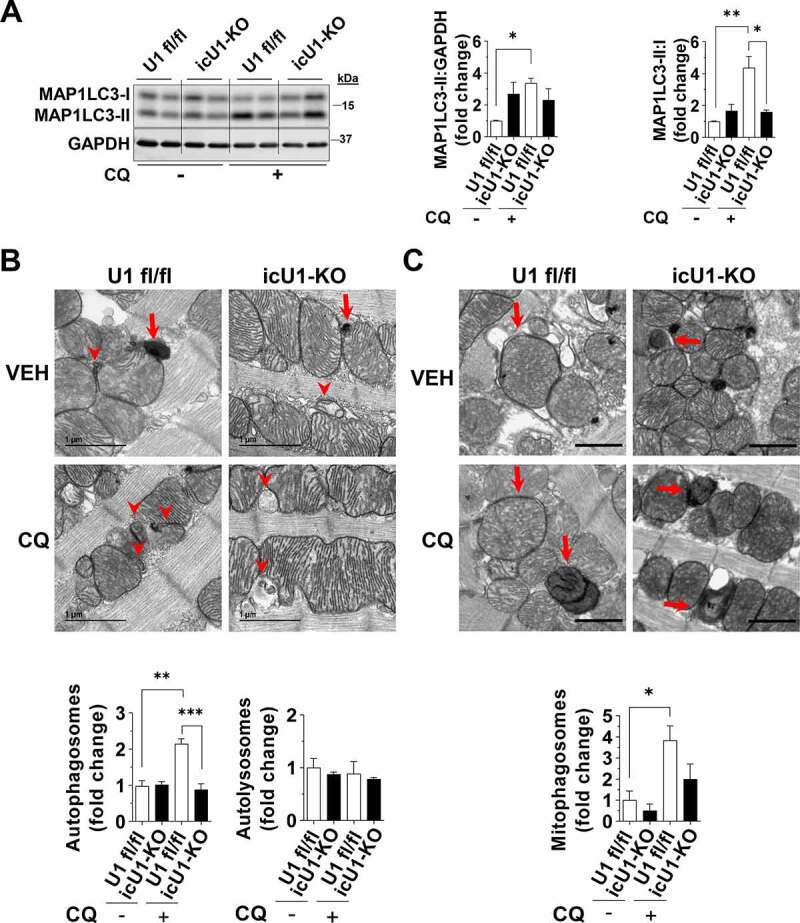
Adult deletion of ULK1 rapidly impairs cardiac autophagy. Data were obtained in 9-wk old mice 4 days after initial tamoxifen injection, unless otherwise specified. (A) Protein levels of MAP1LC3A-MAP1LC3B (MAP1LC3) analyzed after chloroquine administration. Representative immunoblots (Left) and quantification of relative expression (Right) (n = 5–6). (B) Representative TEM images (8000x magnification) of U1fl/fl and cU1-KO hearts treated with vehicle (VEH) or chloroquine (CQ) (Top). Red arrowheads indicate autophagosomes and red arrows indicate autolysosomes. Quantification of autophagosomes and autolysosomes relative to U1 fl/fl treated with vehicle (Bottom) (n = 4). (C) Representative TEM images (3x zoom of original 2000x magnification) of U1 fl/fl and icU1-KO hearts treated with vehicle (VEH) or chloroquine (CQ) (scale bar: 1 μm). Red arrows indicate mitophagosomes. Quantification of mitophagosomes relative to U1 fl/fl treated with vehicle (Bottom) (n = 4). Data are means ± SEM; *P < 0.05, **P < 0.01 and ***P < 0.001.
Figure 7.
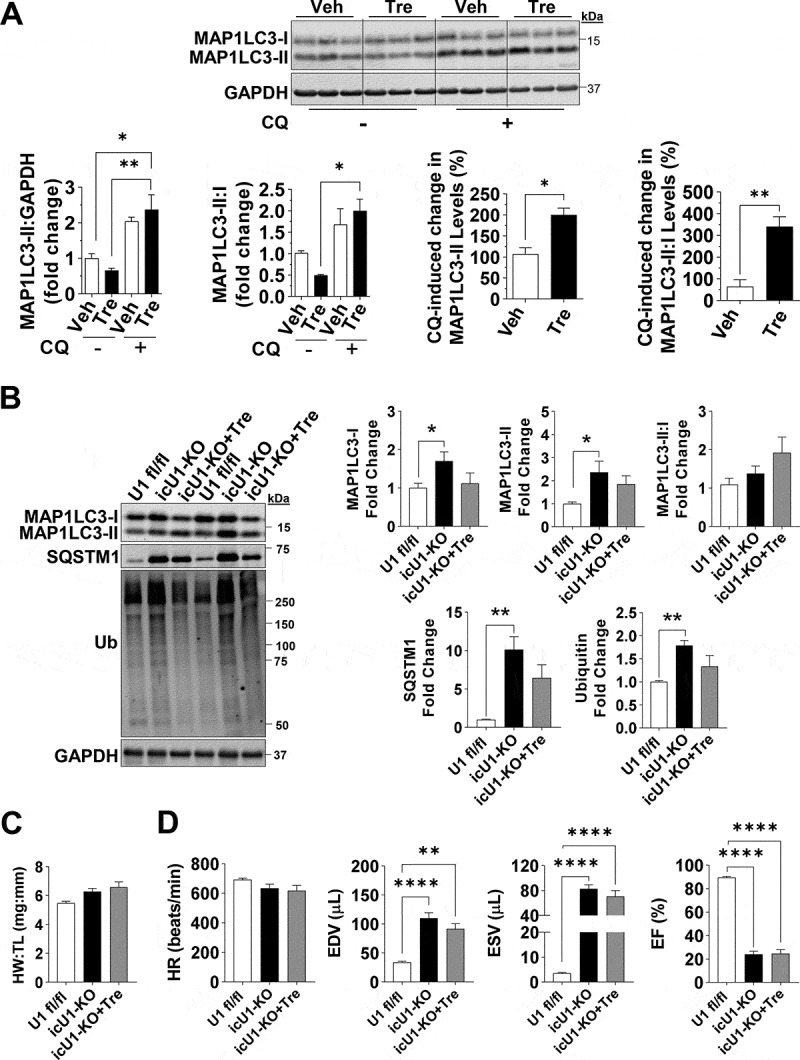
Trehalose administration restores impaired autophagic flux but does not prevent heart failure in icULK1 KO mice. (A) Autophagy flux measurements of vehicle (Veh) and trehalose (Tre) treated WT hearts. Representative immunoblots of MAP1LC3A-MAP1LC3B (MAP1LC3) and GAPDH (Top), quantification of changes in MAP1LC3-II:GAPDH and MAP1LC3-II:MAP1LC3-I and magnitude of change in MAP1LC3-II upon chloroquine (CQ) treatment (Bottom) (n = 3–4). (B) Representative immunoblots of MAP1LC3, SQSTM1, and ubiquitin (Left) and quantification (Right) in U1 fl/fl, icU1-KO, and icU1-KO+Tre mice 5–7 days after the first tamoxifen injection (n = 5–6). (C) Heart weight:tibia length of U1 fl/fl, icU1-KO, and icU1-KO+Tre mice (n = 4–6). (D) Cardiac function assessed from echocardiographs of U1 fl/fl, icU1-KO, and icU1-KO+Tre mice (n = 6–14). Data are means ± SEM; *P < 0.05, **P < 0.01 and ****P < 0.0001.
Discussion
Although mammalian ULK1 and ULK2 are believed to redundantly promote autophagy initiation, recent studies have begun to demonstrate tissue-specific roles for ULK1 and ULK2. For example, suppression of ULK1 in adipocytes decreases fatty acid oxidation and enhances fatty acid uptake, while suppression of ULK2 enhances fatty acid oxidation and decreases uptake [16]. In skeletal muscle, ULK2, but not ULK1, is required for the recognition and degradation of insoluble ubiquitinated protein aggregates [17]. However, the extent to which cardiac ULK1 and ULK2 have redundant functions on autophagy regulation to impact heart health was unknown. Furthermore, since autophagy plays an important role in cardiac development [32], it was plausible to hypothesize that different outcomes related to autophagy and cardiac function could ensue depending on the developmental stage at which loss of these genes occurred.
In this context, several key findings arise from the present study revealing that the roles of ULK1 and ULK2 in regulating cardiac autophagy are more complex than previously anticipated (Figure 8). First, perinatal loss of either ULK1 or ULK2 enhances autophagy in adult hearts, suggesting that the evolutionary advantage of expressing both ULKs is not to support high basal rates of cardiac autophagy. Second, we demonstrate that combined perinatal loss of ULK1 and ULK2 impairs autophagy leading to age-related cardiomyopathy and reduced lifespan. These findings are not due to a potential confounding effect of the Myh6-Cre transgene because we did not observe a similar phenotype in aging mice with perinatal cardiomyocyte loss of ULK2 or ULK1 respectively. In addition, age-related cardiomyopathy has been previously reported in mice with perinatal deletion of cardiac Atg5 [11] indicating that impaired autophagy seen in cU1/2-DKO mice is likely the primary reason for their gradual, late-onset cardiac dysfunction. Third, adult loss of ULK1, but not of ULK2, rapidly induces a fatal dilated cardiomyopathy, a hyper-fissed mitochondrial reticulum and low respiratory capacity in cardiomyocytes. Interestingly, even though autophagy is impaired very early after gene recombination in icU1-KO hearts, attenuating autophagy impairments with trehalose does not protect against or delay cardiac dysfunction. These findings implicate both redundant and distinct functions for ULK1 and ULK2 in the developing and adult heart and demonstrate that the ability of cardiomyocytes to adapt to losses of these genes is developmentally determined.
Figure 8.
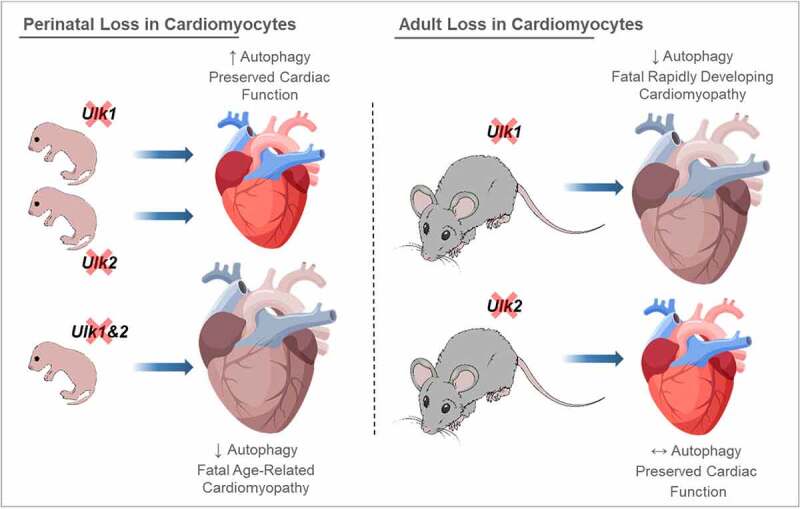
Differential consequences of perinatal vs. adult loss of ULK1 and/or ULK2 on cardiac autophagy and function. Perinatal deletion of ULK1 or ULK2 enhances basal cardiac autophagy. This adaptation requires the preserved expression of the remaining ULK (i.e., ULK1 in cU2-KO and ULK2 in cU1-KO) because perinatal loss of both ULK1 and ULK2 leads to impaired autophagy in the adult heart, age-related cardiomyopathy and early death. However, adult loss of cardiac ULK1 (i.e., in icU1-KO mice) rapidly impairs autophagy leading to dilated cardiomyopathy and heart failure, whereas adult loss of cardiac Ulk2 (i.e., in icU2-KO mice) does not impair autophagy or cardiac function. Attenuation of impaired cardiac autophagy following adult ULK1 loss (with trehalose; not depicted) does not delay cardiac dysfunction suggesting that ULK1 plays other important roles in the heart. Collectively, these findings indicate that ULK1 and ULK2 redundantly modulate autophagy and cardiac function early in development, but not in the adult unstressed heart.
The observation that perinatal loss of either ULK1 or ULK2 enhances basal autophagy in the adult heart was unexpected. Although we have not established the precise mechanisms involved, our results provide important insights indicating that heightened autophagy in these ULK deficient hearts: a) is not driven by increased expression of core autophagy proteins; b) relies on the preserved expression of one of the other ULK (i.e., ULK1 in cU2-KO and ULK2 in cU1-KO); and c) occurs independently of ATG13 S318 phosphorylation, which has been shown to be required for ULK1-mediated mitophagy of depolarized mitochondria [33]. Additionally, our findings also suggest that signals culminating to enhance autophagy could be different depending on which ULK is preserved. For example, BECN1 S30 phosphorylation was increased when only ULK1 was expressed (i.e., in cU2-KO), but decreased when only ULK2 was expressed (i.e., in cU1-KO). Even though further studies are required to establish how cardiac autophagy is regulated when either ULK is lost, collectively our findings support a model where cardiac ULK1 and ULK2 may exert mutual feedback inhibition that keeps basal autophagy at relatively lower (and likely optimal) levels. Thus, when unopposed early in development, either ULK1 or ULK2 can accelerate basal autophagy flux. A potential shortcoming, however, could be that this enhanced autophagy might not be as adaptive (and likely not as selective) potentially reducing the ability of cardiomyocytes to endure stress (e.g., hemodynamic, metabolic, etc.). Indeed, adult hearts deficient in ULK1 early in development are more prone to cardiac dysfunction following ischemia-induced cardiac injury in association with impaired mitophagy [34], and in obesity in association with impaired autophagic degradation of cardiac LPL (lipoprotein lipase) [35]. Therefore, future studies are required to delineate this potential cross-regulatory role of cardiomyocyte ULK1 and ULK2 during development, as it appears to have significant implications for stress responses during adulthood.
Deletion of the Ulk1 and Ulk2 genes in the adult heart elicited very different outcomes, with loss of ULK1 causing a rapid onset dilated cardiomyopathy and heart failure, while loss of ULK2 did not alter cardiac function. These findings also have important implications. First, they demonstrate that the adaptive mechanisms required for the enhanced autophagy observed when either Ulk1 or Ulk2 was lost perinatally were no longer in place in the adult heart. This should be considered in future pre-clinical studies using perinatal versus adult deletion of these genes. Second, it indicates that endogenous ULK2 cannot functionally compensate for the loss of ULK1 in the adult heart. It is important to note that the dilated cardiomyopathy observed upon adult loss of cardiac ULK1 (i.e., in icU1-KO hearts) developed very quickly with 50% of the mice dying between 5–8 days after initial tamoxifen injection. Therefore, determining the precise cellular and molecular events directly dependent on the loss of ULK1 was challenging. Nevertheless, at this timepoint, loss of ULK1 led to major morphological and functional disruptions in cardiac mitochondria including decreased content and respiratory capacity, and a hyper-fissed reticulum. Because mitochondrial fission is important for segregation of damaged/depolarized mitochondria so that they can be degraded through mitophagy [36], these observations suggested that early defects in mitophagy could have led to this outcome. In fact, ULK1 has been shown to be important for mitophagy in both cardiac and skeletal muscles [34,37]. To assess this prospect more directly, we then assessed autophagic flux and quantified autophagosome, lysosome and mitophagosome numbers in TEM micrographs of U1 fl/fl and icU1-KO hearts only 4 days after the initial tamoxifen injections (i.e., when animals were not showing any sign of distress). We then confirmed that even though ~65% of ULK1 activity was preserved in icU1KO hearts at this timepoint, autophagy flux and mitophagy were already clearly impaired. Interestingly, augmenting autophagic flux via trehalose treatment did not attenuate cardiac dysfunction in these mice. Collectively, these observations indicate that even though impaired autophagy impacting mitochondrial function could occur very early upon adult loss of cardiac ULK1, other, yet to be determined mechanisms dependent on ULK1 play a major role in the rapid cardiac dysfunction that ensues. Future studies identifying additional processes and signals modulated by cardiac ULK1 may reveal therapeutic targets for various conditions where autophagy and mitochondrial function are impaired in the heart.
In conclusion, we report that ULK1 and ULK2 play redundant functions in modulating MAP1LC3-dependent basal autophagy in the developing heart, and that the loss of one of these ULKs leads to enhanced basal autophagy that is dependent on the remaining ULK. Perinatal loss of both ULK1 and ULK2, however, impairs basal autophagy leading to age-related cardiac dysfunction. In the adult heart, ULK2 is unable to compensate for the loss of ULK1 leading to a rapidly developing cardiomyopathy involving major disruptions of the autophagy-lysosomal pathway as well as of mitochondrial respiratory function. Moreover, ameliorating autophagy impairments caused by adult loss of ULK1 does not delay cardiac dysfunction, indicating that ULK1 plays other critical unknown roles in the heart. Given the central role that ULK1 plays in preserving autophagy, mitochondrial function, and cardiac homeostasis in the adult heart, increasing ULK1 activity might represent a potential therapeutic approach for several cardiomyopathies. Conversely, while ULK2 was not required to preserve homeostasis in the unstressed adult heart, its potential role in cardiac adaptations to physiologic and/or pathologic stimuli deserves future investigation.
Materials and methods
Animal models
All mouse lines used in this study were on the C57BL6/J background. The mouse line expressing Cre recombinase under the control of the α-myosin heavy chain promoter (Myh6-Cre) has been previously described [19]. ulk1 and ulk2 floxed mice, and mice expressing a tamoxifen-inducible Cre-recombinase under the control of the inducible cardiac-specific Myh6 promoter (Myh6-MerCreMer; B6.FVB(129)-A1cfTg(Myh6-cre/Esr1*)1Jmk/J) were obtained from The Jackson Laboratory (00565). Perinatal cardiac-specific ulk1 knockout (cU1-KO), ulk2 knockout (cU2-KO) and ulk1 ulk2 double-knockout (cU1/2-DKO) mice were generated by crossing mice bearing floxed ulk1 alleles (B6.129-Ulk1tm1Thsn/J)(017976), floxed ulk2 alleles (B6.129-Ulk2tm1Thsn/J)(017977) or floxed ulk1 and ulk2 alleles, respectively, with Myh6-Cre mice. Wild type littermates harboring the same floxed genes, but not expressing Myh6-Cre were used as controls. These mice were studied between 10 and ~90 weeks of age.
ulk1 and ulk2 inducible cardiac-specific knockout mice (icU1-KO and icU2-KO, respectively) were generated by crossing mice with either floxed Ulk1 or floxed Ulk2 alleles with mice expressing Myh6-MerCreMer. Adult gene recombination was induced by injecting mice intraperitoneally with 30 mg/kg of tamoxifen (Sigma-Aldrich, T5648) per day for 3 days starting at 8 weeks of age, as previously described [38]. For experimental consistency, all mice in icU1-KO and icU2-KO groups with incomplete gene recombination resulting in preserved expression of cardiac Ulk1 and Ulk2, respectively, were excluded from the study. Most data analyses were performed in these mice at 5–8 days after the initial tamoxifen injection. Autophagy flux, however, was performed 4 days after the initial tamoxifen injection when the general behavior of icU1-KO mice was indistinguishable from that of their wild-type littermates (i.e., U1 fl/fl).
Mice were housed in the Medical Laboratories vivarium (University of Iowa, Iowa City, IA) in temperature-controlled (21°C) quarters with a 12:12 h light-dark cycle and free access to water and chow. Euthanasia was performed by cervical dislocation after mice were anesthetized by 2% isoflurane gas with an inflow rate of 1 mL/min. Hearts were immediately removed and rinsed in ice-cold phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.2) before being snap-frozen in liquid nitrogen. For starvation experiments, mouse hearts were harvested following 48 h of food deprivation starting early in the light cycle (i.e., 9:00 AM). All animal protocols were approved by the University of Iowa Institutional Animal Care and Use Committee. All efforts were made to comply with the ARRIVE guidelines [39].
Echocardiography
Echocardiography was performed at the University of Iowa Mouse Cardiovascular Imaging Core by an experienced operator blinded to the mouse genotypes. Cardiac function was evaluated in conscious, sedated mice (Midazolam, 0.3 mg/kg body weight). Left-sided chest hair was removed. Parasternal long and short axis views were obtained using a high frequency echocardiography (30 MHz) linear array transducer (Vevo 2100; Visual Sonics, Toronto, ON, Canada).
Chloroquine (CQ) administration
For CQ treatment, CQ (Sigma-Aldrich, C6628) was dissolved in PBS, and mice were injected intraperitoneally at 24, 18, 6, and 3 h prior to harvesting hearts. The doses were 40, 40, 50, and 50 mg CQ/kg body weight, respectively. Mice were then euthanized, hearts were immediately removed, rinsed in ice-cold PBS, and then frozen in liquid nitrogen.
RNA extraction and quantitative real-time polymerase chain reaction (qPCR)
Total RNA was extracted from hearts with TRIzol reagent (Invitrogen, 15596018), and 1 μg was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, 4368814). qPCR was performed with a mixture containing the resulting cDNA, primers, and the Power SYBR Green PCR Master Mix (Applied Biosystems, 4367659) in a 7500 Fast Real-time PCR System (Applied Biosystems, Foster City, CA). The primer sequences used are listed in Table S4. The purity of each amplified product was confirmed by melting curve inspections after amplification, and Rps16s/16S was used as an internal control for cDNA quantification and normalization of amplified products.
Western blot analysis
Heart samples were snap-frozen in liquid nitrogen. Hearts were pulverized, and an aliquot of ~15 mg was used for protein analysis. Tissues were processed using glass homogenizers in ~17 μL/mg of ice-cold protein loading buffer containing 50 mM Tris·HCl, pH 6.8, 1% sodium dodecyl sulfate (SDS), 10% glycerol, 20 mM dithiothreitol, 127 mM 2-mercaptoethanol, and 0.01% bromophenol blue, supplemented with Complete Mini protease inhibitor mixture (Roche Applied Biosciences, 11836153001) and Phosphatase Inhibitor Cocktail 2 (Sigma-Aldrich, P5726). Tissue lysates were subsequently heated for 5 min at 95°C, centrifuged for 5 min at 15,000 x g at room temperature to precipitate debris, and then stored in a new tube at −80°C. Protein concentration of each sample was determined using the RC DC assay (Bio-Rad, 5000111). An equal amount of protein from each sample was subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose or polyvinylidene difluoride (PVDF) membranes. Anti-mouse or rabbit secondary antibodies conjugated to 4X PEG fluorescent dye were used for Western blotting at 1:10,000 (Cell Signaling Technologies, 5151S, 5257S, 5366S, and 5470S). Bands of interest were then analyzed using the Image Studio Software (Licor, Lincoln, NE). GAPDH (1:2000; 2118), ATG13 (1:1000; 13468), ATG7 (1:1000; 8558), ATG5 (1:1000; 12994), ATG9A (1:1000; 13509), ATG4B (1:1000; 13507), ULK1 (1:1000; 8054), SQSTM1 (1:1000;23214), MAP1LC3A-MAP1LC3B (1:1000;4108), ACTN1 (1:1000; 3134), CTSB (1:1000; 31718), DNM1L (1:1000; 8570), DNM1L S616 (1:1000; 3455), ubiquitin (i.e., UBA52, UBB, UBC and RPS27A; 1:1000; 3936), BECN1 (1:1000; 3738), BECN1 S30 (1:1000; 54101), HIS-TAG (1:1000; 2366), and BECN1 S15 (1:1000; 84966) from Cell Signaling Technologies, GAPDH (1:2000; sc-32233), NBR1 (1:1000; sc-130380) from Santa Cruz Biotechnology, ATG13 S318 (1:1000; 600–401-C49S) from Rockland Immunochemicals, ATG3 (1:1000; A3231), ULK1 (1:1000; A7481), SQSTM1 (1:1000; P0067), MAP1LC3A-MAP1LC3B (1:1000; L7543), and MFN1 (1:1000; SAB2106161) from Sigma Aldrich, ULK2 (1:500; NBP1-33136) from Novus Biologicals, NDUFA9 (1:10000; ab14713), SDHA (1:10000; ab14715), UQCRC1 (1:10000; ab110252), MTCO1 (1:10000; ab14705), MFN2 (1:1000; ab56889), CYCS (1:1000; ab13576), and TUBA1A (1:5000; ab7291) from Abcam, HEXB (1:1000; orb161301) from Biorbyt, ubiquitin (i.e., mono- and polyubiquitinated conjugates; 1:1000; BML-PW8810) from Enzo Life Sciences, and OPA1 (1:1000; 612606) from Biosciences were the primary antibodies used. Results for protein expression were normalized to GAPDH, Coomassie Brilliant Blue stain, or Ponceau stain unless otherwise specified.
Cardiomyocyte isolation
Adult cardiomyocytes were isolated as described previously [40]. Briefly, upon removal of the heart, the aorta was clamped to a cannula and ligated with surgical silk. Cannulated hearts were initially perfused for 4–5 min with perfusion buffer containing 113 mM NaCl, 4.7 mM KCl, 0.6 mM KH2PO4, 0.6 mM Na2HPO4, 1.2 mM MgSO4, 10 mM HEPES, 12 mM NaHCO3, 10 mM KHCO3, 30 mM taurine, 10 mM 2,3-butanedione monoxime (BDM; Sigma-Aldrich, B0753), 5.5 mM glucose, pH 7.0. The perfusion buffer was then replaced with digestion buffer (perfusion buffer supplemented with 300 U/mL of type II collagenase [Gibco, 17101015] and 50 µM of CaCl2) for 10–12 min. Following digestion, the ventricles were dissected into small pieces in perfusion buffer containing BSA (100 mg/mL; Research Products International, A30075). The resulting cell suspension was then centrifuged for 3 min at 2,000 x g. The supernatant was removed, and the resulting cardiomyocyte pellet was saved. Cardiomyocytes were then processed for Western blots, or for in vitro kinase assay of ULK1 activity (refer to the kinase assay description for details). For Western blots, cardiomyocytes were homogenized in lysate buffer containing 50 mM HEPES, pH 7.2, 10 mM sodium pyrophosphate, 10 mM sodium fluoride, 2 mM EDTA, 2 mM sodium orthovanadate, 1% Triton X-100 (Fisher Scientific, BP151-100), 10% glycerol, supplemented with HaltTM protease and phosphatase inhibitor cocktail (ThermoFisher Scientific, 78446).
Mitochondria respiration and ATP synthesis
Mitochondria respiratory function was assessed using an Oroboros Instruments Oxygraph 2 K (Innsbruck, Austria). Cardiac fibers from a small piece of the endocardium (~15 mg) were dissected and separated into bundles in ice-cold BIOPS buffer (7.23 mM K2EGTA, 2.77 mM CaK2EGTA, 20 mM imidazole [Sigma-Aldrich, I2399], 0.5 mM DTT, 20 mM taurine, 5.7 mM ATP [Sigma-Aldrich, A7699], 14.3 mM phosphocreatine [Sigma-Aldrich, P7936], 6.56 mM MgCl2 · 6H2O, and 50 mM MES hydrate, pH 7.1). For permeabilization, fibers were then transferred to 1 mL of BIOPS buffer containing 40 µg/mL of saponin (Sigma-Aldrich, S7900) and were gently mixed for 30 min at 4°C. After this point, fibers were then transferred to 1 mL of respiration buffer (105 mM K-MES, 30 mM KCl, 10 mM K2HPO4, 5 mM MgCl2 · 6H2O, 1 mM EGTA, 20 µm blebbistatin [Sigma-Aldrich, B0560] 2.5 mg/mL fatty acid free BSA [Sigma-Aldrich, A8806], pH 7.4) and gently mixed for additional 10 min at 4°C. Following incubation, 1–1.5 mg of tissue was added into the O2K chamber filled with 2 mL of respiration buffer. Both pyruvate-malate and palmitoyl-carnitine malate supported respiration were assessed. Oxygen flux was normalized to wet tissue weight. Aerobic ATP re-synthesis was assessed from 10 µL buffer aliquots of pyruvate-malate+ADP-dependent and palmitoyl-carnitine-malate+ADP-dependent respiration collected every 10 s for 1 min. These respiration buffer aliquots were quenched in 190 µL of dimethyl sulfoxide (DMSO) stored at −20°C prior to collection. ATP production was measured using the Enliten™ ATP Assay system with bioluminescence detection kit (Promega, FF2000) according to the manufacturer’s instructions.
Histological analysis and immunofluorescence
After dissection, heart sections were directly frozen in optimum cutting temperature compound (OCT) or fixed in 4% paraformaldehyde, dehydrated in 70% ethanol, and embedded in paraffin. Following freezing, 10-µm serial sections were cut from samples using a Microm cryostat HM505E (Microm International, Waldorf, Germany) and placed onto microscope slides. Following paraffin embedding, 10-µm serial sections were cut from samples using an HM 355S Automatic Microtome (Microm International, Waldorf, Germany) and placed onto microscope slides. Histological analysis was performed in The University of Iowa Central Microscopy Core. Heart sections were post-fixed in pre-cooled zinc formalin. Sections were stained with H&E to assess cardiomyocyte organization and morphological changes. Heart sections stained with Masson’s trichrome were used to assess fibrosis and collagen deposition. To determine cardiomyocyte size, sections were labeled with fluorescein isothiocyanate (FITC)-labeled wheat germ agglutinin (WGA; Sigma Aldrich, L4895). Slides were imaged using an Olympus BX61 Microscope (Olympus, Center Valley, PA) or a Zeiss 710 Confocal Microscope (Oberkochen, Germany). Quantification of fibrosis was done using ImageJ software as described previously using the color deconvolution plugin [41]. The Masson Trichrome vector was selected, and the green section was used for analysis. Each image’s threshold was set between 0, for the lower slider, and 200, for the upper slider. A minimum of 3 randomly selected images from the left ventricle of each heart were analyzed for fibrosis area (i.e., % of total area), and their average was considered representative of that animal. Quantification of cardiomyocyte size in WGA-stained sections was performed using the ImageJ software.
Transmission electron microscopy (TEM)
TEM was performed on heart sections at the University of Iowa Central Microscopy Core. Briefly, hearts were cut into small pieces (~1 mm3) and fixed in 2.5% glutaraldehyde solution in 0.1 M cacodylate buffer at 4°C until further processing. Tissue was post fixed in 1% osmium tetroxide (OsO4) and 1.5% potassium ferrocyanide in 0.2 M cacodylate buffer for 1–2 h at room temperature. Heart pieces were then stained with 2.5% uranyl acetate for 20 min, dehydrated with a series of acetone dilutions (50–100%), embedded in EPON resin (Ted Pella Inc., 18010), and baked (70°C oven) overnight. Embedded tissue was sectioned using Leica EM UC6 Ultramicrotome (Leica Microsystems, Buffalo Grove, IL, USA) and 90‐nm sections were mounted on grids, stained with uranyl acetate and lead citrate. Images were observed and captured using the JEOL JEM-1230 Transmission Electron Microscope (Jeol, Peabody, MA, USA). Analysis of mitochondrial length and area was performed using Matlab software (The MathWorks, Inc. Natick, MA, USA) as previously described [42]. Mitochondrial length and area were binned, normalized to compute percentages, and then plotted via histogram. Unbinned data was fit to a gamma distribution curve. Statistical analysis was performed using Kruskal Wallis and Student’s t test to detect significant changes in distribution or the mean, respectively.
In mouse models where there was biochemical evidence for changes in autophagy, autophagosome and autolysosome numbers were quantified as previously described [43]. Because of the densely packed sarcomeres and high mitochondrial content present in cardiomyocytes, autophagosomes and autolysosomes were always found in close proximity to mitochondria. Therefore, we conservatively named these structures as “autophagosomes and autolysosomes” and presented their quantification combined. Due to the pronounced phenotype seen in icU1-KO hearts, TEM micrographs were also analyzed after chloroquine treatment. Under these conditions, the number of autophagosomes, autolysosomes (for the most part indistinguishable from lysosomes) and mitophagosomes (when a mitochondrion and/or cristae remnants were observed inside double-membrane structures) were determined in a blinded fashion. These analyses were performed by determining the average number of structures observed in three random 2000x myocardium images from each animal.
Mitochondrial fractionation
Frozen powdered heart tissue was homogenized in 1 mL of ice-cold buffer containing 225 mM D-mannitol (Research Products International, M22080), 75 mM sucrose (Research Products International, S24065), 5 mM HEPES, pH 7.4 supplemented with 10 μL of HaltTM protease and phosphatase inhibitor cocktail (ThermoFisher Scientific, 78446). Centrifugation was performed for 4 min at 4,000 x g at 4°C, after which the pellet was discarded, and the supernatant was kept for further centrifugation for 10 min at 10,000 x g at 4°C. The remaining pellet was saved as the mitochondrial fraction and the supernatant saved as the cytosolic fraction.
Mitochondrial DNA (mtDNA)
Mitochondrial DNA content was quantified by real‐time polymerase chain reaction (RT–PCR). Briefly, total DNA was extracted and purified from heart tissue with the DNeasy Kit (Qiagen Inc., 69504). Five ng of DNA was used to quantify mitochondrial and nuclear DNA markers. RT–PCR was performed using an ABI Prism 7900HT instrument (Applied Biosystems, Foster City, CA, USA) in 384‐well plate format with SYBR Green I chemistry and ROX internal reference (Invitrogen). Analysis of results was automated using scripting with SDS 2.1 (Applied Biosystems), Microsoft Access, and Microsoft Excel. Mitochondrial DNA content was determined by quantification of D-loop region mitochondrial DNA (forward: GGTTCTTACTTCAGGGCCATCA, reverse: GATTAGACCCGATACCATCGAGAT) relative to genomic Rpl13a gene (forward: GAGGCCCCTACCATTTCCGA, reverse: GAGGCCCCTACCATTTCCGA).
Trehalose treatment protocol
A trehalose treatment protocol was first established in wild-type mice, which were treated with trehalose (Cayman Chemical, 6138–23-4; 2 mg/g intraperitoneally twice daily for 48 h and 2% in the drinking water) or vehicle (saline IP injections for the initial 48 h and access to regular drinking water) for 1 week. Autophagic flux was assessed at day 7 of treatment. In the studies reducing autophagy impairments in mice with adult loss of cardiac ULK1 (i.e., in icU1-KO), trehalose treatments started at the beginning of tamoxifen administration. These included 3 groups: one group of icU1-KO mice was treated with trehalose, while one group of icU1-KO mice and one group of U1 fl/fl mice were treated with vehicle. Both echocardiographic and biochemical analyses were performed 5 days after the beginning of tamoxifen and trehalose administrations.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 9 (Graphpad Software Inc., San Diego, CA). Results are presented as means ± SEM, and the number of samples/group are provided in figure legends. Chloroquine, fasting, and trehalose experiments were analyzed by a two-way ANOVA followed by Tukey-Kramer test. Survival was assessed using the Kaplan-Meier method and log-rank test. The Chi-square test was used to determine Mendelian ratios of live births of the perinatal cardiac-specific knockout mouse models. Unpaired t test was used for all remaining data analysis. Values of P< 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We would like to acknowledge use of the University of Iowa Central Microscopy Research Facility, a core resource supported by the University of Iowa Vice President for Research, and the Carver College of Medicine. The JEOL JEM-1230 Transmission Electron Microscope was purchased with funding from the NIH SIG grant 1S10 RR018998-01. We would like to acknowledge the University of Iowa Mouse Cardiovascular Imaging Core and the associated NIH grants S10 OD019941 and S10 RR026293. We would also like to acknowledge Dr. Coleen Mitchell, a professor in the Department of Mathematics at the University of Iowa, for her assistance in the analysis of mitochondrial area and length.
Funding Statement
This work was supported by the American Heart Association Grant [16SDG30360001 to V.A.L.], by the National Institutes of Health [R56AG063820 to V.A.L., and R01HL108379 and U01 HL087947 to E.D.A], and grant support from the Fraternal Order of Eagles Diabetes Research Center. E.D.A. is an established investigator of the American Heart Association.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. PMID: 18305538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rubinsztein DC, Mariño G, Kroemer G.. Autophagy and aging. Cell. 2011;146:682–695. PMID: 21884931. [DOI] [PubMed] [Google Scholar]
- [3].Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. PMID: 23921753. [DOI] [PubMed] [Google Scholar]
- [4].Choi AMK, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–662. PMID: 23406030. [DOI] [PubMed] [Google Scholar]
- [5].Pattison JS, Osinska H, Robbins J. Atg7 induces basal autophagy and rescues autophagic deficiency in CryAB R120G cardiomyocytes. Circ Res. 2011;109:151–160. PMID: 21617129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sciarretta S, Zhai P, Shao D, et al. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation. 2012;125:1134–1146. PMID: 22294621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–29787. PMID: 16882669. [DOI] [PubMed] [Google Scholar]
- [8].Xie Z, Lau K, Eby B, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770–1778. PMID: 21562078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Eisenberg T, Abdellatif M, Schroeder S, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. 2016;22:1428–1438. PMID: 27841876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nakai A, Yamaguchi O, Takeda T, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. PMID: 17450150. [DOI] [PubMed] [Google Scholar]
- [11].Taneike M, Yamaguchi O, Nakai A, et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy. 2010;6:600–606. PMID: 20431347. [DOI] [PubMed] [Google Scholar]
- [12].Chan EYW, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem. 2007;282:25464–25474. PMID: 17595159. [DOI] [PubMed] [Google Scholar]
- [13].Chan EYW, Longatti A, McKnight NC, et al. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol. 2009;29:157–171. PMID: 18936157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. PMID: 21258367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. PMID: 21205641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ro S-H, Jung CH, Hahn WS, et al. Distinct functions of Ulk1 and Ulk2 in the regulation of lipid metabolism in adipocytes. Autophagy. 2013;9:2103–2114. PMID: 24135897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fuqua JD, Mere CP, Kronemberger A, et al. ULK2 is essential for degradation of ubiquitinated protein aggregates and homeostasis in skeletal muscle. FASEB J. 2019;33:11735–12745. PMID: 31361156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823–830. PMID: 20811354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Abel ED, Kaulbach HC, Tian R, et al. Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J Clin Invest. 1999;104:1703–1714. PMID: 10606624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lyons GE, Schiaffino S, Sassoon D, et al. Developmental regulation of myosin gene expression in mouse cardiac muscle. J Cell Biol. 1990;111:2427–2436. PMID: 2277065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ganley IG, Lam DH, Wang J, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–12305. PMID: 19258318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Park JM, Seo M, Jung CH, et al. ULK1 phosphorylates Ser30 of BECN1 in association with ATG14 to stimulate autophagy induction. Autophagy. 2018;14:584–597. PMID: 29313410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lee E-J, Tournier C. The requirement of uncoordinated 51-like kinase 1 (ULK1) and ULK2 in the regulation of autophagy. Autophagy. 2011;7:689–695. PMID: 21460635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Iwai-Kanai E, Yuan H, Huang C, et al. A method to measure cardiac autophagic flux in vivo. Autophagy. 2008;4:322–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bhuiyan S, Pattison JS, Osinska H, et al. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123:5284–5297. PMID: 18216495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Milner DJ, Mavroidis M, Weisleder N, et al. Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J Cell Biol. 2000;150:1283–1298. PMID: 10995435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kuznetsov A, Margreiter R, Kuznetsov AV, et al. Heterogeneity of mitochondria and mitochondrial function within cells as another level of mitochondrial complexity. Int J Mol Sci. 2009;10:1911–1929. PMID: 19468346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15:741–750. PMID: 23685627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tang Q, Zheng G, Feng Z, et al. Trehalose ameliorates oxidative stress-mediated mitochondrial dysfunction and ER stress via selective autophagy stimulation and autophagic flux restoration in osteoarthritis development. Cell Death Dis. 2017;8(10):e3081–e3081. PMID: 28981117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sciarretta S, Yee D, Nagarajan N, et al. Trehalose-induced activation of autophagy improves cardiac remodeling after myocardial infarction. J Am Coll Cardiol. 2018;71:1999–2010. PMID: 29724354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhu L, Yuan Y, Yuan L, et al. Activation of TFEB-mediated autophagy by trehalose attenuates mitochondrial dysfunction in cisplatin-induced acute kidney injury. Theranostics. 2020;10:5829–5844. PMID: 32483422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lee E, Koo Y, Ng A, et al. Autophagy is essential for cardiac morphogenesis during vertebrate development. Autophagy. 2014;10:572–587. PMID: 24441423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Joo JH, Dorsey FC, Joshi A, et al. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol Cell. 2011;43:572–585. PMID: 21855797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Saito T, Nah J, Oka S, et al. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J Clin Invest. 2019;129:802–819. PMID: 30511961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].An M, Ryu D-R, Won Park J, et al. ULK1 prevents cardiac dysfunction in obesity through autophagy-meditated regulation of lipid metabolism. Cardiovasc Res. 2017;113:1137–1147. PMID: 28430962. [DOI] [PubMed] [Google Scholar]
- [36].Twig G, Elorza A, Molina AJA, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. PMID: 18200046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Laker RC, Drake JC, Wilson RJ, et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun. 2017;8(1):548. PMID: 28916822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bersell K, Choudhury S, Mollova M, et al. Moderate and high amounts of tamoxifen in α-MHC-MerCreMer mice induce a DNA damage response, leading to heart failure and death. Disease Models & Mechanisms. 2013;6:1459–1469. PMID: 23929941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kilkenny C, Browne WJ, Cuthill IC, et al. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:e1000412. PMID: 20613859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Li D, Wu J, Bai Y, et al. Isolation and culture of adult mouse cardiomyocytes for cell signaling and in vitro cardiac hypertrophy. J Vis Exp. 2014;87:51357. PMID: 24894542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chen Y, Yu Q, Xu CB. A convenient method for quantifying collagen fibers in atherosclerotic lesions by imagej software. Int J Clin Exp Med. 2017;10:14904–14910. [Google Scholar]
- [42].Ponce JM, Coen G, Spitler KM, et al. Stress‐induced cyclin C translocation regulates cardiac mitochondrial dynamics. J Am Heart Assoc. 2020;9:e014366. PMID: 32248761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sun Y, Yao X, Zhang Q-J, et al. Beclin-1-dependent autophagy protects the heart during sepsis. Circulation. 2018;138:2247–2262. PMID: 29853517. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.