

ABSTRACT
The intestinal epithelial tight junctions (TJs) provide barrier against paracellular permeation of lumenal antigens. Defects in TJ barrier such as increased levels of pore-forming TJ protein CLDN2 (claudin-2) is associated with inflammatory bowel disease. We have previously reported that starvation-induced macroautophagy/autophagy enhances the TJ barrier by degrading pore-forming CLDN2. In this study, we examined the molecular mechanism underlying autophagy-induced CLDN2 degradation. CLDN2 degradation was persistent in multiple modes of autophagy induction. Immunolocalization, membrane fractionation, and pharmacological inhibition studies showed increased clathrin-mediated CLDN2 endocytosis upon starvation. Inhibition of clathrin-mediated endocytosis negated autophagy-induced CLDN2 degradation and enhancement of the TJ barrier. The co-immunoprecipitation studies showed increased association of CLDN2 with clathrin and adaptor protein AP2 (AP2A1 and AP2M1 subunits) as well as LC3 and lysosomes upon starvation, signifying the role of clathrin-mediated endocytosis in autophagy-induced CLDN2 degradation. The expression and phosphorylation of AP2M1 was increased upon starvation. In-vitro, in-vivo (mouse colon), and ex-vivo (human colon) inhibition of AP2M1 activation prevented CLDN2 degradation. AP2M1 knockout prevented autophagy-induced CLDN2 degradation via reduced CLDN2-LC3 interaction. Site-directed mutagenesis revealed that AP2M1 binds to CLDN2 tyrosine motifs (YXXФ) (67–70 and 148–151). Increased baseline expression of CLDN2 and TJ permeability along with reduced CLDN2-AP2M1-LC3 interactions in ATG7 knockout cells validated the role of autophagy in modulation of CLDN2 levels. Acute deletion of Atg7 in mice increased CLDN2 levels and the susceptibility to experimental colitis. The autophagy-regulated molecular mechanisms linking CLDN2, AP2M1, and LC3 may provide therapeutic tools against intestinal inflammation.
Abbreviations: Amil: amiloride; AP2: adaptor protein complex 2; AP2A1: adaptor related protein complex 2 subunit alpha 1; AP2M1: adaptor related protein complex 2 subunit mu 1; ATG7: autophagy related 7; CAL: calcitriol; Cas9: CRISPR-associated protein 9; Con: control; CPZ: chlorpromazine; DSS: dextran sodium sulfate; EBSS: Earle’s balanced salt solution; IBD: inflammatory bowel disease; TER: trans-epithelial resistance; KD: knockdown; KO: knockout; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MβCD: Methyl-β-cyclodextrin; MET: metformin; MG132: carbobenzoxy-Leu-Leu-leucinal; MTOR: mechanistic target of rapamycin kinase; NT: non target; RAPA: rapamycin; RES: resveratrol; SMER: small-molecule enhancer 28; SQSTM1: sequestosome 1; ST: starvation; ULK1: unc-51 like autophagy activating kinase 1; WT: wild type.
KEYWORDS: Autophagy, CLDN2, inflammatory bowel disease, intestinal permeability, tight junction
Introduction
Inflammatory bowel disease (IBD) is a multifactorial intestinal disorder involving chronic inflammation of the gastrointestinal tract. IBD is considered to be caused by a complex interaction between genetics, environment, immune responses, and microbiome. The intestinal epithelial cells act as an epithelial barrier, which thereby limits the interaction between the host, and luminal contents along with intestinal microbiota and environmental antigens. The intestinal epithelial barrier against paracellular antigen permeation is mainly created by tight junction (TJ) complexes present in between two adjacent epithelial cells. The important transmembrane TJ proteins include claudins and OCLN (occludin), which are connected to the actin cytoskeleton via cytoplasmic plaque proteins such as TJP1/ZO-1, TJP2/ZO-2, TJP3/ZO-3 and CGN/cingulin [1]. As many as 26 distinct claudins are reported in mammals, which comprises both pore-forming claudins such as CLDN2/claudin 2, and barrier forming claudins such as CLDN1, CLDN3, and CLDN4 [2]. The disruption in this TJ barrier leads to increased intestinal permeability and increased host access to luminal antigens, resulting in persistent, unresolved inflammation. Though the pathogenesis of IBD is not clear, disruption of TJ barrier and particularly upregulation of pore-forming CLDN2 is consistently observed in various forms of IBD including ulcerative colitis (UC), Crohn disease (CD), microscopic colitis, as well as other intestinal disorders such as celiac disease, necrotizing enterocolitis, and ischemia-reperfusion injury [1,3]. CLDN2 is recognized as an important water and small cation-selective pore-forming TJ protein in the intestinal epithelium. Inactivation of CLDN2 has been shown to attenuate immune-mediated experimental colitis in mice, indicating CLDN2 as a potential target against intestinal inflammation [4].
Macroautophagy (autophagy) is an evolutionarily conserved, fundamental cellular process in which the cell eliminates defective organelles, toxic proteins, and aged and damaged cytoplasmic components via lysosome dependent degradation, in order to survive under hostile conditions such as nutrient starvation [5]. The major steps in autophagy are (i) initiation (formation of the phagophore), which is mediated by two key protein kinase complexes, ULK1/2-ATG13-RB1CC1/FIP200 complex, and the BECN1/beclin1-PIK3C3/Vps34-PIK3R4/Vps15-ATG14 complex; (ii) expansion of the phagophore, that involves ubiquitin-like proteins ATG12 and GABARAP/LC3; (iii) autophagosome formation (closure of phagophore) which requires LC3 modification (lipidation of cytosolic LC3-I to LC3-II) and binding to autophagosome membrane; and (iv) the fusion of autophagosome with the lysosome to form an autolysosome which degrades the cargo proteins [6]. Impairment in autophagy is associated extensively with Crohn disease. Genome wide association studies revealed association of genetic variants in autophagy related genes such as NOD2, IRGM, ULK1 and ATG16L1 with Crohn disease [7–9]. Autophagy is also associated with gut microbial composition, and impairment in autophagy has also been linked with intestinal dysbiosis [10]. Autophagy has been shown to play a role in dendritic-epithelial cell interactions, adaptive immune responses, NOD2-directed bacterial sensing, lysosomal destruction, and immune-mediated clearance, which are important processes in the pathogenesis of IBD [11–13]. Previously, we have demonstrated the role of autophagy in the regulation of paracellular TJ permeability. Nutrient starvation induced autophagy in intestinal epithelial cells enhanced the intestinal TJ barrier function by increasing the lysosomal degradation of the pore-forming TJ protein CLDN2 [14]. The aim of the present study was to delineate the molecular mechanisms of CLDN2 degradation upon autophagy induction. Our data showed that autophagy enhances intestinal epithelial TJ barrier by promoting CLDN2 degradation through clathrin-mediated endocytosis and demonstrates the crucial role of AP2M1 (adaptor related protein complex 2 subunit µ1) in this process.
Results
Autophagy-mediated reduction in CLDN2 levels
We have previously shown that starvation-induced autophagy reduces the levels of the pore-forming TJ protein CLDN2 in multiple epithelial cell lines [14]. In order to further examine if autophagy inducing agents, other than starvation, decrease CLDN2 levels, Caco-2 cells were plated on transwells and treated individually with rapamycin (an MTOR [mechanistic target of rapamycin kinase] inhibitor [15]), the small molecule compound SMER28 (autophagosome synthesis promoter [16]), resveratrol (MTOR inhibitor [15]), metformin (AMP-activated protein kinase activator [17]), and calcitriol (transcriptional activator of autophagy [18]), which induce autophagy via different mechanisms. Treatment with all the autophagy inducers used in the study caused a significant (p < 0.05) decrease in CLDN2 protein levels (Figure 1A). The intestinal epithelial TJ barrier permeability is measurable in terms of trans-epithelial electrical resistance (TER). Because CLDN2 is a cation-selective pore-forming TJ protein responsible for the flux of cations and small solutes, it is a primary determinant of TER [19,20]. Consistent with the decrease in CLDN2 levels, the Caco-2 TER was found to be increased upon treatment with autophagy inducers (Figure 1B). We also observed that, starvation induced autophagy induction was more effective in increasing the TER compared to other tested autophagy inducers.
Figure 1.
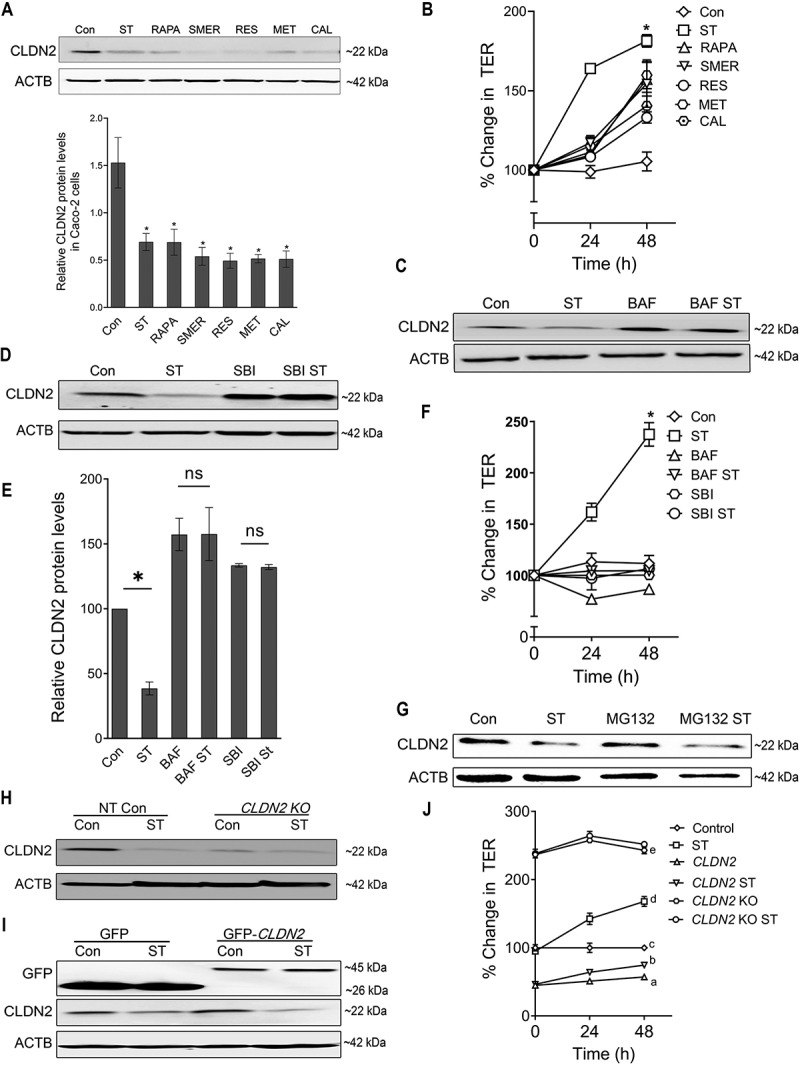
Autophagy-mediated reduction in CLDN2/Claudin-2 levels. (A) Confluent Caco-2 cells were incubated in EBSS for starvation (ST) or treated with known autophagy inducing compounds rapamycin (RAPA, 500 nM), small-molecule enhancer 28 (SMER, 50 µM), resveratrol (RES, 100 µM), metformin (MET, 100 µM), and calcitriol (CAL, 20 µM) for 24 h, and CLDN2 levels were measured by Western blotting. ACTB/β-actin is shown as a loading control. The protein levels of CLDN2 were reduced by all the autophagy inducers. ST: starvation. The densitometry analysis of CLDN2 expression was performed using ImageJ software to indicate the relative levels of CLDN2 after autophagy-inducing treatments. The graph is representative of ≥ 3 independent experiments (*, p < 0.005 versus control). (B) The autophagy-inducing treatments increased Caco-2 trans-epithelial resistance (TER) compared to untreated control group (*, p < 0.005 versus control). (C) Autophagy inhibitor bafilomycin A1 (BAF, 20 nM) treatment for 24 h alone increased CLDN2 levels and prevented starvation-induced decrease in CLDN2 levels. (D) Autophagy inhibitor SBI-0206965 (SBI, 30 µM) treatment for 24 h prevented starvation-induced decrease in CLDN2 levels. (E) Densitometry for CLDN2 expression in panel C and D. The graph is representative of ≥ 3 independent experiments (*, p < 0.05 versus control). BAF and SBI also prevented starvation-induced increase in TER (F) (*, p < 0.005 versus control). (G) Proteasomal inhibition with MG132 (10 µM) did not prevent starvation-induced reduction in CLDN2 levels. (H) Western blot showing CLDN2 knockout in CLDN2 KO Caco-2 cells. (I) Western blot showing GFP and CLDN2 GFP, confirming CLDN2 over expression in Caco-2 cells. Starvation induced no significant difference in the levels of exogenous CLDN2 but in contrast a significant decrease in endogenous CLDN2 upon starvation was seen in both Non-Target GFP and CLDN2 overexpressing Caco-2 cells. (J) CLDN2 overexpressed Caco-2 cells (CLDN2) showed reduced TER and CLDN2 KO showed increased TER at the baseline. CLDN2 KO cells did not show starvation-induced increase in TER, whereas CLDN2-overexpressing Caco-2 cells showed mild but significant increase in TER upon starvation. a, b, c, d, and e, P < 0.005 vs. each other in two-way ANOVA followed by Tukey’s multiple comparison test.
On the other hand, we also examined the effect of autophagy inhibition on CLDN2 levels in starvation-induced autophagy model. For this, the Caco-2 cells were incubated with serum-free Earle’s balanced salt solution (EBSS) in the presence and absence of upstream autophagy inhibitor SBI-0206965 (ULK 1 inhibitor) [21] or downstream autophagy inhibitor bafilomycin A (autophagosome and lysosome fusion inhibitor) [14]. The treatment with these autophagy inhibitors prevented the starvation-induced reduction of CLDN2 levels (Figure 1C, D, and E). Moreover, the starvation-induced increase in TER was also found to be inhibited upon autophagy inhibitors treatment (Figure 1F). Since lysosomal and proteasomal degradation pathways are two important pathways in which proteins can be degraded, the role of proteasomal degradation pathway in starvation-induced reduction of CLDN2 levels was studied using MG132 (carbobenzoxy-Leu-Leu-leucinal). MG132 is a cell-permeable proteasome inhibitor [22], which effectively blocks the proteolytic activity of the 26S proteasome complex and disrupts the proteasome-regulated degradation of intracellular proteins. MG132 (10 µM) treatment on Caco-2 cells did not cause any significant difference in starvation-induced CLDN2 reduction, suggesting that CLDN2 is not degraded via the proteasomal degradation pathway (Figure 1G). These data showed that, CLDN2 reduction is a consistent feature of multiple modes of autophagy induction and is not mediated via proteasomes.
In order to demonstrate the role of CLDN2 in autophagy-mediated enhancement of TJ barrier, we studied the effect of CLDN2 knock overexpression and CRISPR-Cas9 mediated CLDN2 knockout (CLDN2 KO) on TER of Caco-2 cells. The CLDN2 KO cells showed increased baseline TER compared to WT Caco-2 cells but did not show starvation-induced increase in TER (Figure 1H, and J). On the other hand, as expected and consistent with previous report in MDCKI epithelial model [23], over expression of CLDN2 in Caco-2 cells caused a significant decrease in TER (Figure 1I, and J). When GFP-CLDN2 overexpressing Caco-2 cells were starved, we observed a small but significant increase in TER compared to corresponding untreated control. The Western blot images showed an insignificant difference in the expression levels of exogenous CLDN2 levels upon starvation (Figure 1I). In contrast, we observed a significant decrease in endogenous CLDN2 upon starvation in both NT GFP and CLDN2-overexpressing Caco-2 cells. These results point out the possibility that the abundance of CLDN2 in CLDN2-overexpressing Caco-2 cells prevails over the effect of autophagic degradation of CLDN2 upon starvation. This data reiterates the importance of CLDN2 in autophagy mediated increase in the TJ barrier.
Autophagy-induced CLDN2 reduction is mediated via clathrin
To study the localization of CLDN2 in cells upon starvation, we performed membrane fractionation studies, in which we found that the amount of CLDN2 in membrane fraction decreases gradually with an increase in starvation time period (Figure 2A). The localization of CLDN2 in the starved and control Caco-2 cells were also examined by immunogold transmission electron microscopy (TEM). Unlike control samples, CLDN2 was observed in vesicle like structures in the cytoplasm of starvation samples (Fig. S1). The reduction in CLDN2 protein levels, decreased membrane localization of CLDN2, and presence of CLDN2 in vesicle like structures in the cytoplasm upon autophagy induction, indicates that autophagy increases the endocytosis of CLDN2 from the membrane. In order to study the mode of endocytosis and degradation of CLDN2 upon autophagy induction, Caco-2 cell monolayers were incubated with EBSS in the presence and absence of pharmacological inhibitors of principal endocytic pathways. Amiloride inhibits sodium and hydrogen exchange and prevents macropinocytosis [24]. Methyl-β-cyclodextrin (MβCD), a selective inhibitor of clathrin-independent caveolae, acts by depleting cholesterol from plasma membrane [25]. Chlorpromazine (CPZ), a cationic amphipathic drug, inhibits clathrin-mediated endocytosis by engaging the adaptor proteins and clathrin on endosomal membranes thereby depleting them on cell surfaces [26]. Amiloride and MβCD treatment did not cause any significant difference in starvation-induced CLDN2 degradation, suggesting that CLDN2 is not degraded via micropinocytosis or caveolae mediated endocytosis pathway (Figure 2B). CPZ treatment inhibited CLDN2 degradation upon starvation, suggesting that the CLDN2 degradation is facilitated by clathrin-mediated endocytosis pathway (Figure 2C). Moreover, starvation-induced increase in TER was also found to be inhibited in presence of CPZ (Figure 2D). This data indicated the important role of clathrin-mediated endocytic pathway in autophagy mediated CLDN2 degradation and increase in TER.
Figure 2.
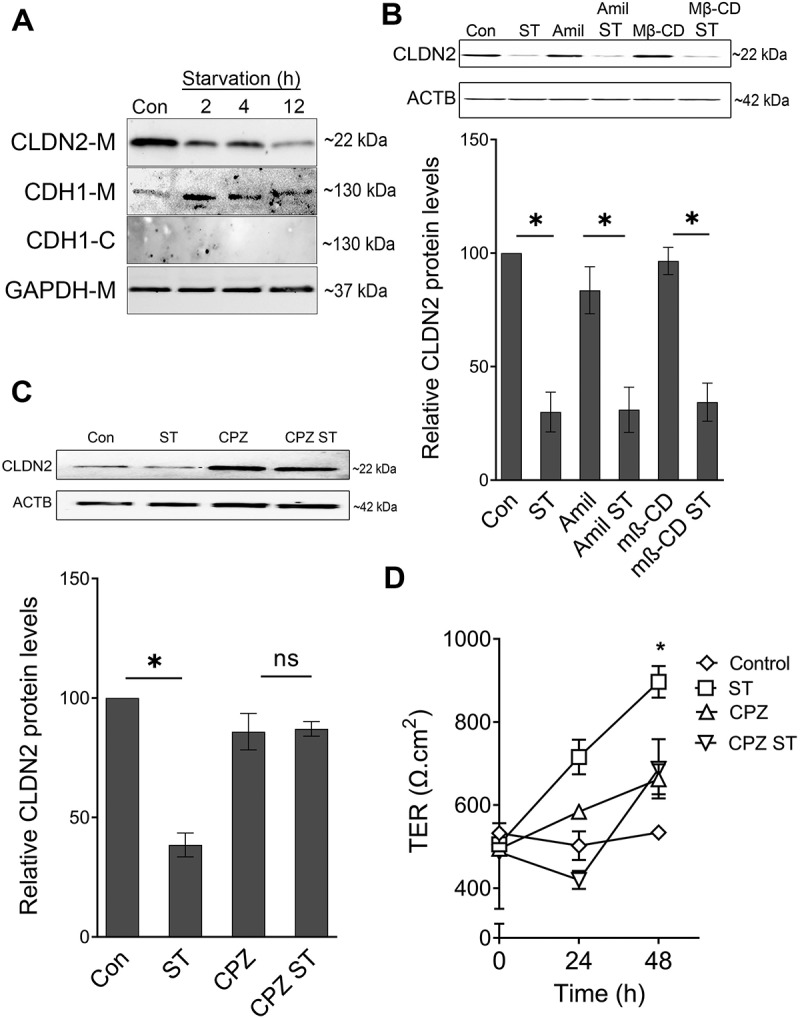
Autophagy-induced CLDN2 reduction is mediated via clathrin. (A) The amount of CLDN2 in membrane fraction is reduced gradually with increased starvation time period. E-cadherin-M and E-cadherin-C represents E-cadherin probes on cell membrane and cytoplasmic fractions, respectively. GAPDH is shown as a loading control for membrane fractions. The blots are representative of ≥ 3 independent experiments. (B) Amiloride (Amil, 10 µM) and Methyl-β-cyclodextrin (MβCD, 250 µM) treatment for 24 h did not alter starvation-induced reduction in CLDN2 levels. Densitometry for CLDN2 expression (*, p < 0.01 versus control). Clathrin inhibitor, chlorpromazine (CPZ, 10 µM) treatment for 24 h prevented starvation-induced reduction in CLDN2 levels (C). The densitometry representation (C) of CLDN2 expression after chlorpromazine treatment is also shown. (D) Chlorpromazine (CPZ, 10 µM) treatment for 24 h prevented starvation-induced increase in TER (*, p < 0.001 versus control) .
Interaction of CLDN2 with clathrin apparatus under starvation
Prompted by the involvement of clathrin-mediated endocytosis in autophagy induced CLDN2 degradation and increase in TER, we next investigated the interaction of CLDN2 with clathrin-mediated endocytosis apparatus proteins. Co-immunoprecipitation studies showed increased interaction of CLDN2 with clathrin, 6 to 12 h post-starvation (Figure 3A and B). AP2 (adaptor protein complex) is an essential component in clathrin-mediated endocytosis. The AP2 heterotetramer is composed of AP2A1/α, AP2B1/β, AP2M1/µ, and AP2S1/σ subunits among which, subunit AP2A1 and subunit AP2M1 participate directly in the clathrin coat assembly [27]. AP2 can bind to both clathrin as well as membrane cargo proteins and hence, it plays an important role in membrane associated protein endocytosis [27,28]. Because CLDN2 is also a membrane protein, we examined CLDN2 immunoprecipitates for AP2 subunits. The association of CLDN2 with AP2M1 and AP2A1 was also found to be markedly increased upon starvation. On the other hand, lipidation of ATG8/ LC3 is a hallmark of autophagosome biogenesis and hence, it acts as a specific marker for autophagy. Moreover, LC3 lipidation is triggered through direct interaction of the cargo receptors with multiple autophagy components [29]. In order to examine if CLDN2 is directly linked to autophagy, the interaction of CLDN2 with LC3 was also studied. The results showed increased interaction of CLDN2 with LC3 upon starvation. Consistent with our previous report of post-starvation localization of CLDN2 to lysosomes [14], we observed the interaction between CLDN2 and LAMP2 (lysosomal marker protein) to be increased at later time point (12 h) of starvation (Figure 3A and B). The interaction of CLDN2 with clathrin was further corroborated using confocal immunofluorescence microscopy. The results showed reduction in CLDN2 from the cell membrane and increased cytoplasmic colocalization of CLDN2 with clathrin upon starvation (Figure 3C). This data confirms the increased interaction of CLDN2 with clathrin apparatus proteins (clathrin, AP2A1, AP2M1) upon autophagy induction.
Figure 3.
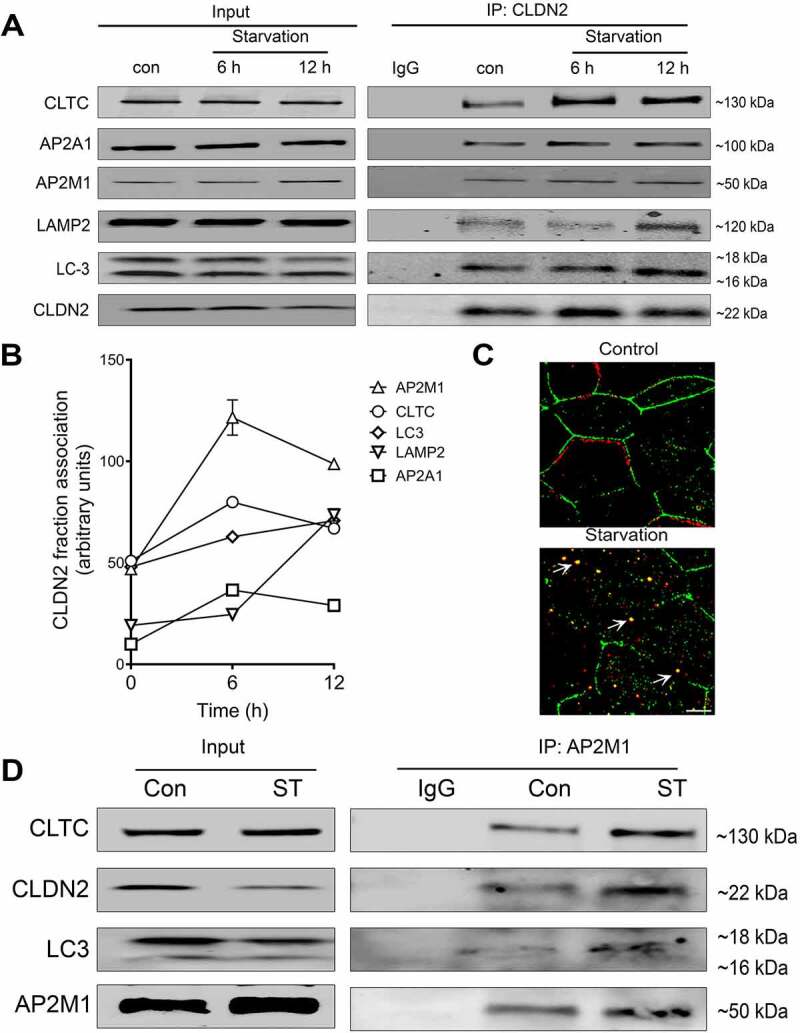
Interaction of CLDN2 with clathrin and autophagy apparatus. (A) Co-immunoprecipitation studies showed an increased association of CLDN2 with AP2M1, AP2A1, clathrin, LC3 during early starvation and lysosomal marker protein LAMP2 at the later 12-h time point. The negative control includes immunoprecipitation with control IgG. (B) Quantification of CLDN2 fraction associated with various clathrin and autophagy proteins, as shown in panel A. (C) Confocal immunofluorescence examination showed that, CLDN2 (green) migrated away from the cell membrane and increased cytoplasmic colocalization with clathrin (red) after starvation (yellow). White bar: 5 µm. (D) AP2M1 immunoprecipitates showed increased presence of CLDN2, LC3 and clathrin after starvation. Representation of ≥ 3 independent experiments.
Since AP2M1 is the adaptor protein involved in membrane associated protein endocytosis and given that CLDN2 is observed predominantly on the membrane, we further pursued the role of AP2M1 in autophagy-medicated CLDN2 degradation. In co-immunoprecipitation studies, CLDN2 and clathrin were found to be increasingly present in AP2M1 immunoprecipitates after starvation, suggesting increased interaction between AP2M1, clathrin, and CLDN2 upon starvation (Figure 3D). To examine if the observed association between CLDN2 and AP2M1 is also present in in-vivo conditions, colonic tissue from wild type mice were examined by confocal immunofluorescence microscopy. In mouse colon, too, the baseline colocalization of CLDN2 and AP2M1 was observed on the apical membrane of colonocytes (Fig. S2). Over all, these data clearly suggested an important role of AP2M1 in autophagy-mediated clathrin endocytosis of CLDN2. In agreement with the property of AP2 to bind with both clathrin, as well as membrane proteins, we hypothesized that AP2M1 may act as an anchor between CLDN2 and clathrin.
AAK1 mediated activation of AP2M1 upon autophagy induction
AP2M1 gets activated upon phosphorylation at the T156 residue by AAK1/Adaptor-associated kinase. AAK1 has been shown to bind to AP2 complex and mediate phosphorylation of the AP2M1 [30]. The activated AP2M1 (phospho-AP2M1) has high affinity for binding to tyrosine signals within cargo membrane proteins, leading to cargo recruitment, and internalization [31,32]. To further understand the importance of AP2M1 and its activation in the study, the protein expression of AP2M1 and Phospho-AP2M1 was studied along with AP2A1 upon starvation. The results showed no significant difference in AP2A1 levels whereas a significant increase in the protein expression of AP2M1 and phospho-AP2M1 was observed upon starvation (Figure 4A and B). We asked if inhibition of starvation-induced AP2M1 activation could prevent CLDN2 degradation upon starvation. Caco-2 cells were treated with sunitinib, an inhibitor of AAK1-mediated AP2M1 activation [33,34]. Indeed, sunitinib treatment during starvation prevented starvation-induced reduction in CLDN2 levels (Figure 4C). Sunitinib also prevented starvation-induced increase in TER and reduction in urea flux in Caco-2 cells (Fig. S3). In order to specifically study the role of AAK1 in AP2M1 activation and reduction in CLDN2 levels, we generated CRISPR-Cas9 mediated AAK1 KO in Caco-2 cells (AAK1 KO). In comparison with non-target control cells, AAK1 KO cells showed a significant decrease in activation of AP2M1 (Phospho-AP2M1) and a mild but significant increase in CLDN2 levels at the base line (Figure 4D and E). Additionally, AAK1 KO cells did not exhibit starvation-induced reduction in CLDN2 levels (Figure 4D and E), increase in TER (Figure 4F) and reduction in urea flux (Figure 4G).
Figure 4.
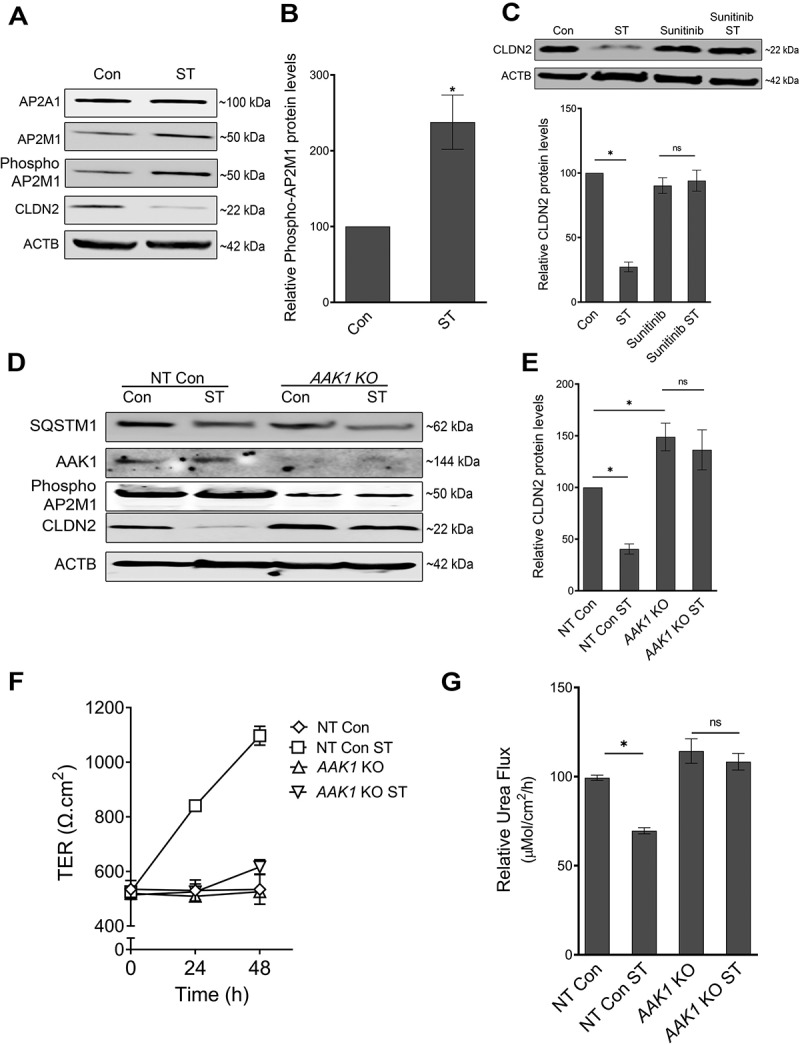
Role of AP2M1 in regulation of CLDN2 levels. (A) AP2M1 is increasingly phosphorylated after starvation. ACTB is shown as a loading control. The blots are representative of 3 independent experiments. (B) Densitometry for phospho-AP2M1 expression in panel A (*, p < 0.01 versus control). (C) Inhibition of AP2M1 activation with Sunitinib (25 µM) treatment prevented starvation-induced reduction in CLDN2 levels. Densitometry for CLDN2 levels in Sunitinib treatment (*, p < 0.001 versus control). (D) Western blot showing efficiency of CRISPR-Cas9-mediated knockout of AAK1 in Caco-2 cells. AAK1 KO led to a significant increase in baseline CLDN2 levels and abolished starvation-induced CLDN2 reduction. (E) Densitometry for CLDN2 levels in untreated and starved non target control (NT) and AAK1 KO cells, as shown in panel D (*, p < 0.05 versus control). AAK1 knockout also significantly inhibited starvation-induced increase in TER (F) and reduction in urea flux (G) (*, p < 0.05 versus control) .
Considering the unexplored role of AAK1 and AP2M1 in intestinal TJ barrier, we also examined the effect of sunitinib on mouse colonic CLDN2 levels and colonic TJ barrier. Sunitinib treatment for 48 h led to a significant increase in CLDN2 levels in mouse colonic mucosa (Figure 5A). To study the impact of sunitinib on colonic TJ barrier function, we conducted ex-vivo studies where excised mouse colon were mounted in Ussing chambers. In these studies, the colon from sunitinib treated mice showed reduced TER (Figure 5B) and increased colonic small molecule urea flux, compared to vehicle treated mice (Figure 5C). Also, sunitinib treated mice colon showed a reduction in phospho-AP2M1 and an increase in CLDN2 immunolocalization in the colonic epithelium (Figure 5D and E). Overall, these data further emphasized the important role of the AP2M1 activation in CLDN2 degradation, and thereby strengthened our hypothesis that AP2M1 anchors CLDN2 to clathrin-coated vesicles upon autophagy induction.
Figure 5.
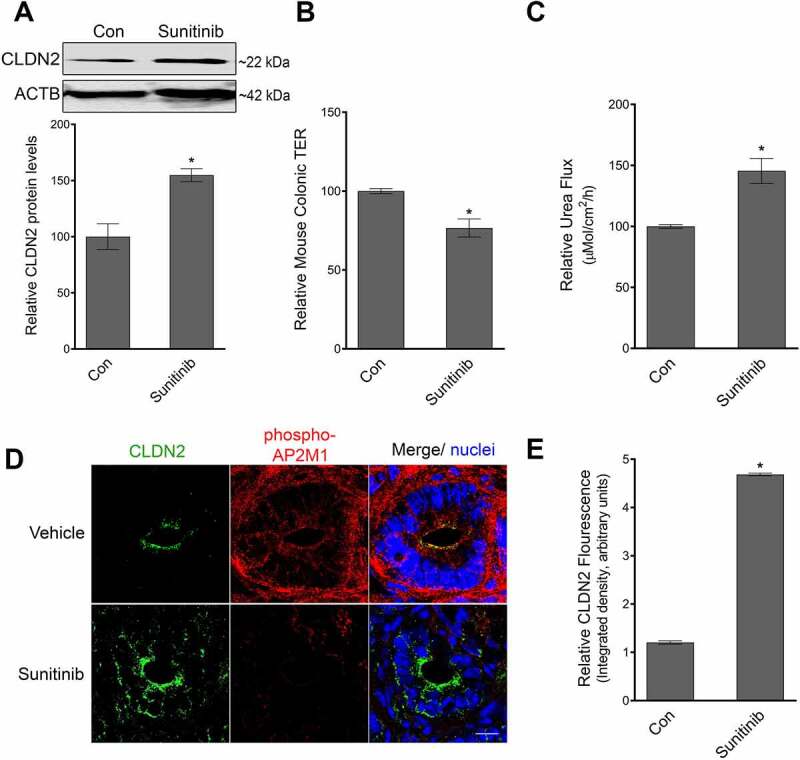
Role of AP2M1 in mice colonic TJ barrier. (A) Sunitinib administration (40 mg/kg/day for 2 days, oral gavage), causes an increase in CLDN2 levels in mouse colonocytes. Densitometry for CLDN2 levels upon sunitinib treatment (*, p < 0.005 versus control). In ex-vivo experiments in Ussing chambers, the colon of Sunitinib administered mice showed reduction in TER (*, p < 0.005 versus control) (B) and increase in urea (small molecule) flux (C) p < 0.005, compared to control mice. (D) In confocal immunofluorescence examination, sunitinib administered mice showed reduced staining for phospho-AP2M1 (red) and increased amount of CLDN2 (green) on the apical membrane of colonocytes in comparison with control mice. White bar: 20 µm. (E) Quantification of CLDN2 fluorescence from panel D (*, p < 0.01 versus control) .
AP2M1 is required for autophagy-mediated reduction in CLDN2.
In order to confirm the requirement of AP2M1 in autophagy mediated CLDN2 down-regulation, CRISPR-Cas9 mediated AP2M1 knockout (AP2M1 KO) were generated. The AP2M1 KO showed near complete depletion of AP2M1 compared to scrambled sgRNA transfected cells (Figure 6A). Importantly, AP2M1 KO led to a significant increase in baseline CLDN2 levels (Figure 6A and B). Moreover, unlike control cells, starvation did not reduce CLDN2 levels in AP2M1 KO cells. In consistent with a number of previous studies [35,36], we observed an increase in accumulation of SQSTM1/p62 in untreated AP2M1 KO cells. SQSTM1/p62 is an autophagosome cargo protein, which gets accumulated upon autophagy inhibition. The reduction of SQSTM1/p62 levels upon starvation showed that the AP2M1 KO cells were autophagy competent (Figure 6A). Consistent with the role of AP2M1 in CLDN2 removal from the TJ membrane, AP2M1 deletion significantly inhibited starvation-induced increase in TER and reduction in urea flux (Figure 6C and D). Finally, AP2M1 being a key anchor protein in this study, we asked if AP2M1 deletion would disrupt starvation-induced association of CLDN2 with LC3. Indeed, we found that starvation induced marked increase in CLDN2-LC3 colocalization in non-target control cells while AP2M1 KO significantly inhibited starvation-associated colocalization of CLDN2 and LC3. (Figure 6E and F). These data showed the role of AP2M1 in CLDN2 endocytosis under constitutive and autophagic condition and further confirms that AP2M1 plays a principal role in bridging CLDN2 cargo with the autophagy pathway.
Figure 6.
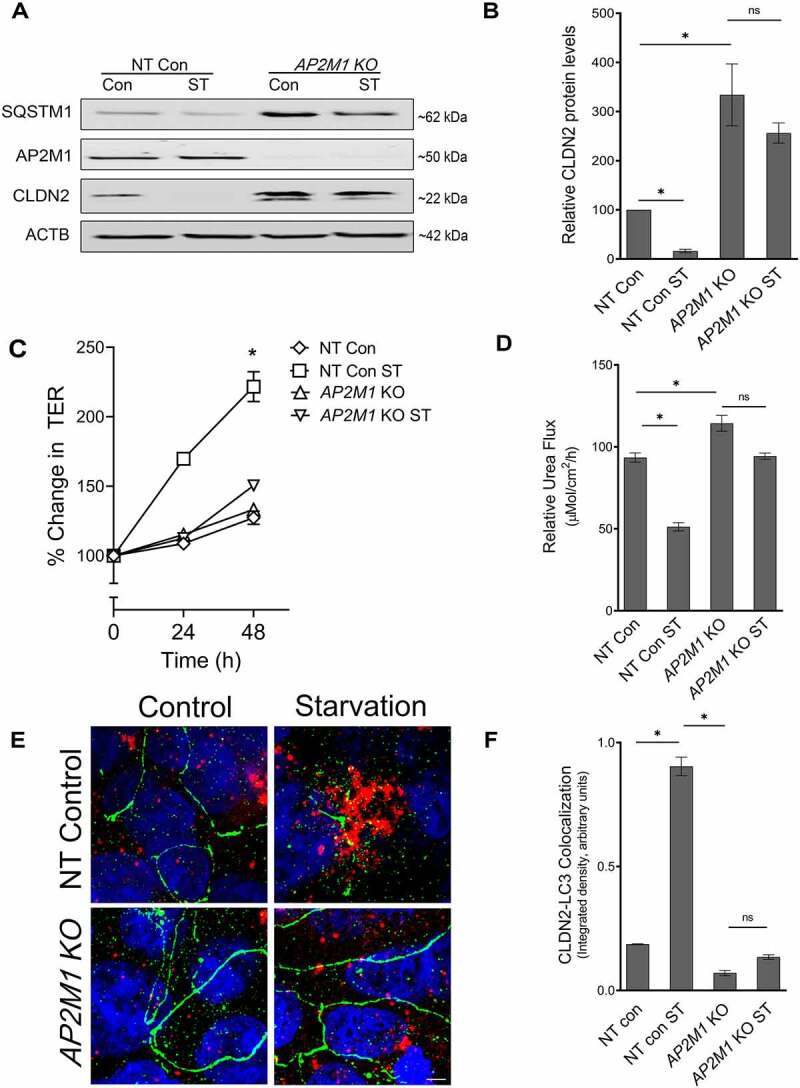
AP2M1 is required for autophagy-mediated reduction in CLDN2. (A) Western blot showing efficiency of CRISPR-Cas9 mediated deletion of AP2M1 in Caco-2 cells. AP2M1 KO led to a marked increase in baseline CLDN2 levels and abolished starvation-induced CLDN2 reduction. (B) Densitometry for CLDN2 levels in untreated and starved non target control (NT) and AP2M1 KO cells, as shown in panel A (*, p < 0.05). AP2M1 deletion also significantly inhibited starvation-induced increase in TER (C) (*, p < 0.001) and reduction in urea flux (D) (*, p < 0.001). (E) In confocal immunofluorescence examination, starvation induced increased colocalization of CLDN2 (green) and LC3 (red) when compared to control Caco-2 cells. AP2M1 KO cells showed prominent presence of CLDN2 (green) on the membrane with least colocalization with LC3 (red), even upon starvation. Nuclei: blue. White bar: 5 µm. (F) Quantification of CLDN2 and LC3 colocalization from panel E (*, p < 0.01) .
Identification of AP2M1 anchoring region in CLDN2.
AP2M1 recognizes the tyrosine signals YXXΦ motifs (Φ is a bulky hydrophobic residue – L/I/M/V/F and X is any amino acid) on the transmembrane cargo proteins for trafficking into clathrin-coated vesicles [37]. Inspection of the CLDN2 protein sequence revealed two conserved YXXΦ motifs at the amino acid regions 67–70 and 148–151 (Figure 7A). We speculated that the presence of two YXXΦ motifs in CLDN2 protein may play a critical role in clathrin-mediated CLDN2 endocytosis. To examine this possibility, the two YXXΦ on CLDN2 were mutated together as well as individually to generate a double motif mutant or single motif mutants CLDN2 respectively, by site directed mutagenesis. The Y and Φ amino acids were substituted with alanine as depicted in Figure 7A. To determine whether AP2M1 binds CLDN2 via YXXΦ region, wild type GFP-CLDN2 and the mutated GFP-CLDN2 carrying plasmids were transfected individually into HEK293 cells which have minimal endogenous CLDN2 expression. The cell extracts were immunoprecipitated using an anti-GFP antibody. AP2M1 was co-immunoprecipitated only with wild type CLDN2 and not with either of the single mutated motifs or the double motif mutant CLDN2 (Figure 7B). Thus, AP2M1 binds with the conserved YXXΦ region at 67–70 and 148–151 region of CLDN2 and substituting the Y and Φ amino acids with alanine (A) inhibits the AP2M1 binding with CLDN2. We also observed similar phenomenon upon confocal immunofluorescence examination where mutations in AP2M1 anchoring motifs in CLDN2 abrogated CLDN2-AP2M1 colocalization (Figure 7C). In addition, to study the importance of AP2M1 binding site on autophagy induced CLDN2 degradation, the wild type GFP-CLDN2 and the mutated GFP-CLDN2 transfected HEK293 cells were assessed for the levels of exogenous CLDN2 protein expression in control and starvation conditions. The results showed starvation induced degradation of wild type GFP-CLDN2 alone but not in any of the mutated GFP-CLDN2 (Figure 7D). Together these data suggests the importance of AP2M1 binding site in CLDN2 on autophagy mediated CLDN2 reduction, and further reiterates the significant role of AP2M1 in autophagy-mediated CLDN2 reduction.
Figure 7.
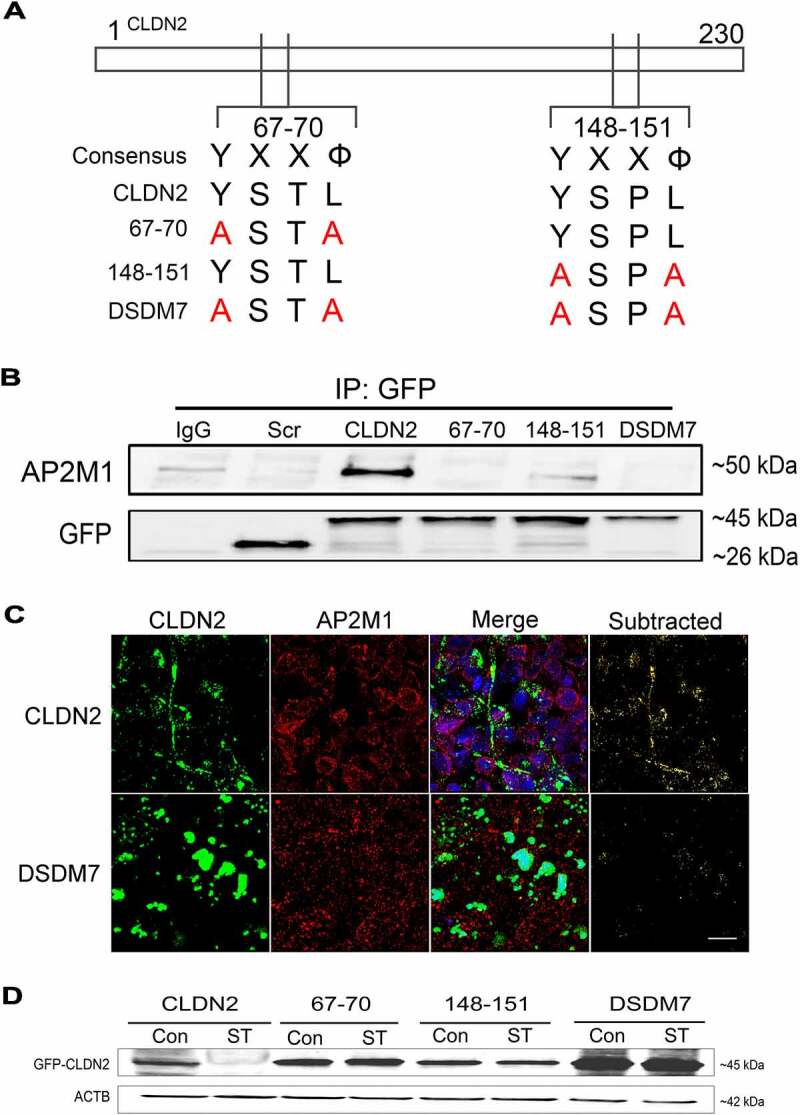
Identification of AP2M1 anchoring region in CLDN2. (A) CLDN2 has two YXXΦ (Φ, a bulky hydrophobic residue – L/I/M/V/F) AP2M1 anchoring tyrosine motifs at amino acid 67–70 and 148–151 region. These motifs were mutated individually (67–70: CLDN2Y67A,L70A; 148–151: CLDN2Y148A,L151A) or together (Double mutant, DSDM7: CLDN2Y67A,L70A,Y148A,L151A) by site-directed mutagenesis, substituting the Y and Φ amino acids with alanine, as shown in panel A. (B) Wild type GFP-CLDN2 and the mutated GFP-CLDN2 carrying plasmids were transfected individually into HEK293 cells and co-immunoprecipitated using anti-GFP antibody. The immunoprecipitates when probed for AP2M1, showed AP2M1 being co-immunoprecipitated only with wild type CLDN2 and not with CLDN2 mutants. GFP bands are shown as loading control. IgG: Normal IgG control. Scr: scrambled control plasmid.CLDN2: wild type GFP-CLDN2. (C) In confocal immunofluorescence examination, HEK293 cells transfected with wild-type GFP-CLDN2 (CLDN2) showed strands of CLDN2 (green) and AP2 (red) colocalization. In subtracted panel, only yellow color of CLDN2-AP2 colocalization was retained. Mutations in AP2M1 anchoring motif in CLDN2 (Double mutant) altered CLDN2 localization and reduced CLDN2-AP2 colocalization, compared to wild-type CLDN2 transfected cells. Representation of 3 fields from 3 separate HEK293 monolayer samples. White bar: 10 µm. (D) Western blot showing that starvation reduces wild type GFP-CLDN2 compared to untreated control but all other mutated GFP-CLDN2 showed no significant difference compared to their corresponding untreated controls.
Intersection of CLDN2 endocytosis and autophagy
Once we have established the role of AP2M1 in clathrin-mediated CLDN2 endocytosis and degradation during autophagy, we next examined the link between AP2M1-CLDN2 cargo and autophagy. Because ATG7, an autophagy-related E1-like enzyme, promotes ATG12 conjugation and LC3 lipidation, which is essential for the autophagic process [38], we first studied the effect of genetic deletion of ATG7 on CLDN2 levels. CRISPR-Cas9 mediated ATG7 knockout (ATG7 KO) in Caco-2 cells led to near complete depletion of ATG7 compared to scrambled sgRNA-transfected cells (Figure 8A). Accumulation of SQSTM1/p62 in ATG7 KO cells indicated abrogation of autophagy in these cells. ATG7 KO led to an increase in baseline CLDN2 protein levels and significantly prevented starvation-induced reduction in CLDN2 levels (Figure 8A and B). Since CLDN2 was increasingly found to be associated with autophagosome marker LC3 during starvation (Figure 3A and B), we examined CLDN2-LC3 colocalization in ATG7 KO Caco-2 cells. In confocal immunofluorescence examination following starvation, more CLDN2 was present on the membrane and CLDN2-LC3 colocalization was diminished in ATG7 KO cells compared to control scrambled sgRNA transfected cells (Fig. S4). These data demonstrate proximity between CLDN2 and LC3 during autophagic flux.
Figure 8.
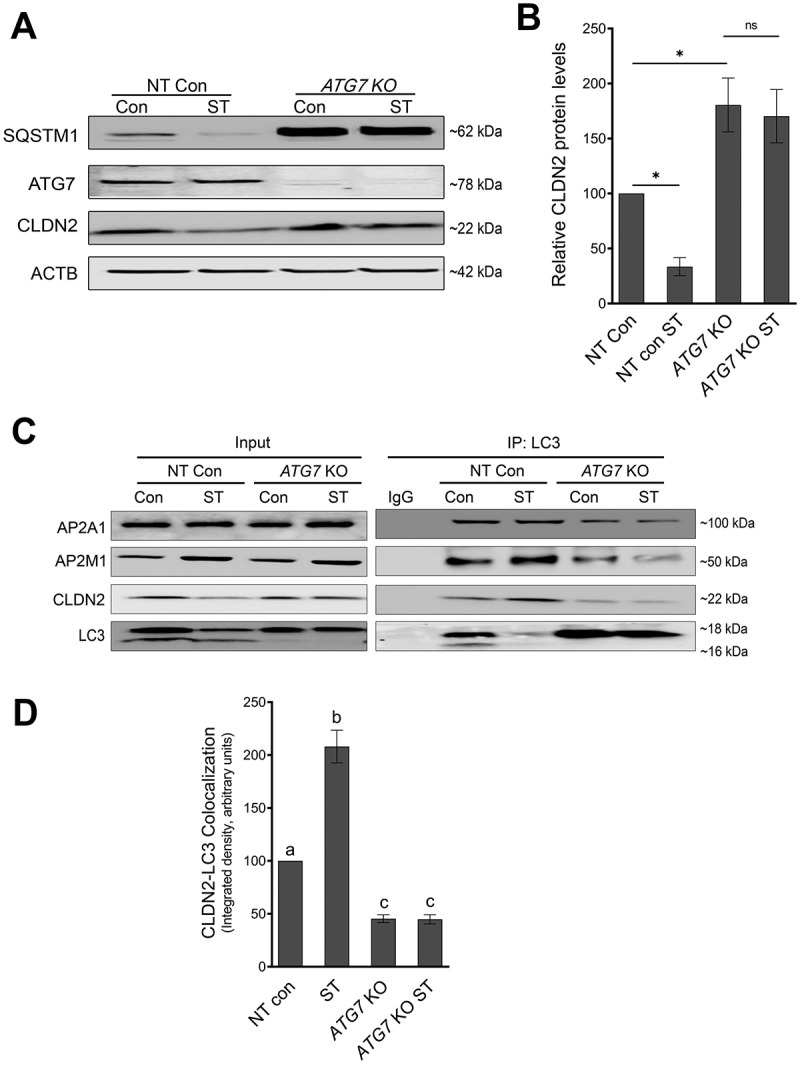
AP2M1 at the intersection of endocytosis and autophagy. (A) Western blot shows efficacy of ATG7 knockout (ATG7 KO) in Caco-2 cells using CRISPR-Cas9. ATG7 deletion led to baseline increase in SQSTM1/p62 and CLDN2. (B) Densitometry for CLDN2 levels in panel (A) showed reduction of CLDN2 levels after starvation in non-target (NT) control cells but not in ATG7 KO cells. *, p < 0.05 versus control, NS: non-significant. (C) Co-immunoprecipitation studies using anti-LC3 antibody showed increased interaction of AP2M1 and CLDN2, with LC3 during starvation in non-target control cells. (D) Densitometry ratio of CLDN2: LC3 in panel C showed increased CLDN2-LC3 interaction after starvation in non-target (NT) control cells in comparison with ATG7 KO cells. The negative control includes immunoprecipitation with control IgG. The blots are representative of 3 independent experiments. a, b, c, and d, P < 0.01 vs. each other in two-way ANOVA followed by Tukey’s multiple comparison test.
AP2, is known to interact with LC3 through the LC3 interacting region (LIR) motif in the AP2A1 subunit and the presence of AP2M1 in LC3 coimmunoprecipitates from mice brain tissues [28] shows the interaction of both AP2A1 and AP2M1 with LC3. Moreover, AP2 is also reported to function as an LC3 receptor, which shuttles proteins from the endocytic pathway to autophagosomes for degradation [28]. In order to clarify the role of AP2 in linking CLDN2 to the autophagic pathway, we studied the interaction of LC3 with CLDN2, AP2A1, and AP2M1 in non-target control and ATG7 KO cells under normal and starvation conditions. The results showed an increase in the interaction of CLDN2 and AP2M1 with LC3 upon starvation in control cells (Figure 8C and D), which is consistent with increased interaction of AP2M1 with eGFP-LC3 upon starvation previously reported in HeLa cells [28]. This interaction of AP2M1 with LC3 was found to be reduced in ATG7 KO cells in the baseline as well as starvation conditions (Figure 8C). The confocal immunofluorescence studies further confirmed the increased colocalization between AP2M1 and LC3 after starvation. A multi-fold increase in AP2M1 and LC3 colocalization was observed after starvation compared to control (Fig. S5). This result further reinforced the important role of AP2M1 in linking CLDN2 to LC3 and thereby leading to autophagic degradation of CLDN2.
Autophagy deficiency increases CLDN2 levels in-vivo
Next, we examined the relation between autophagy and CLDN2 levels in-vivo in autophagy deficient atg7 conditional knockout (atg7 cKO) mice. Acute deletion of Atg7 in adult mice as well as inhibition of autophagy in atg7 cKO mice, in terms of increased accumulation of SQSTM1/p62, is shown in Figure 9A. atg7 cKO showed an increase in constitutive CLDN2 levels in colonic epithelial cells (Figure 9A and B). In agreement with increased baseline CLDN2 levels, the atg7 cKO mice also showed reduced baseline colonic TER and increased colonic urea flux compared to Atg7fl/fl mice (Figure 9C and D). Moreover, when subjected to experimental dextran sodium sulfate (DSS) colitis, atg7 cKO mice showed increased reduction in colonic TER and significantly increased colonic urea flux compared to control Atg7fl/fl DSS mice (Figure 9C and D). Consistent with a previous report [38], atg7 cKO mice were found to have increased susceptibility to experimental DSS colitis which was reflected in higher disease activity index and severe colonic inflammation compared to control Atg7fl/fl DSS mice (Fig. S6). These data provided evidence for in-vivo autophagy-mediated reduction in CLDN2 levels as well as the role of CLDN2 mediated colonic TJ barrier in health and disease.
Figure 9.
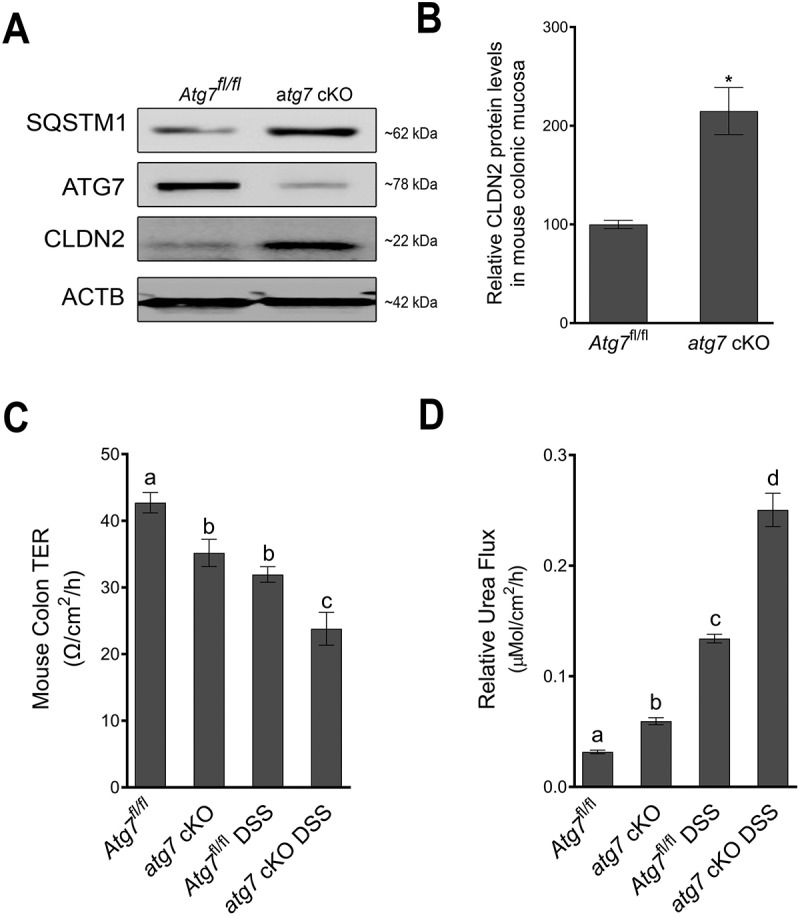
Autophagy deficiency increases CLDN2 levels in-vivo. (A) Tamoxifen treatment of adult mice with Atg7 floxed and Ubc-CreERT2 alleles resulted in the loss of ATG7 protein in colonic mucosa (atg7 cKO mice) compared to ATG7 floxed (Atg7fl/fl) control mice. The Western blot also showed disruption of autophagy in atg7 cKO mice in terms of accumulation of SQSTM1/p62. Acute deletion of Atg7 also caused an increase in constitutive CLDN2 levels in the colonic epithelial cells. (B) Densitometry for CLDN2 levels in atg7 cKO mice, as shown in panel A (*, p < 0.005 versus control). The atg7 cKO mice showed reduced baseline colonic TER (C) and increased colonic urea flux (D) compared to control Atg7fl/fl mice. In acute dextran sodium sulfate (DSS) colitis model, atg7 cKO mice showed increased reduction in colonic TER and markedly increased colonic urea flux compared to Atg7fl/fl DSS mice (C and D). a, b, c, and d, P < 0.01 vs. each other in two-way ANOVA followed by Tukey’s multiple comparison test.
Role of AP2M1 in human colonic TJ barrier
Considering the central role of AP2M1 activation in autophagy-mediated CLDN2 endocytosis and degradation, we examined the function of AP2M1 in intestinal TJ barrier in human colonic mucosa. Apparently healthy human colonic tissue from surgical resection were stripped of serosal muscle layers and pair-cultured on gelatin sponge with or without sunitinib, an inhibitor of AAK1-mediated AP2M1 activation. Following overnight culture, the colonic mucosa were mounted on Ussing chambers to study the TJ barrier. We found that sunitinib treatment of human colonic mucosa significantly decreased the TER and increased urea flux in the paired samples (Figure 10A and B). Moreover, sunitinib treatment reduced phospho-AP2M1 levels and significantly increased CLDN2 levels in human colonic mucosal samples (Figure 10C). These experiments demonstrated specific and vital involvement of AP2M1 in regulation of intestinal TJ barrier function via modulation of CLDN2 levels. Considering the role of AP2M1 in autophagic degradation of CLDN2, this study also provides a conceptual basis for modulation of the human intestinal TJ barrier via autophagy.
Figure 10.
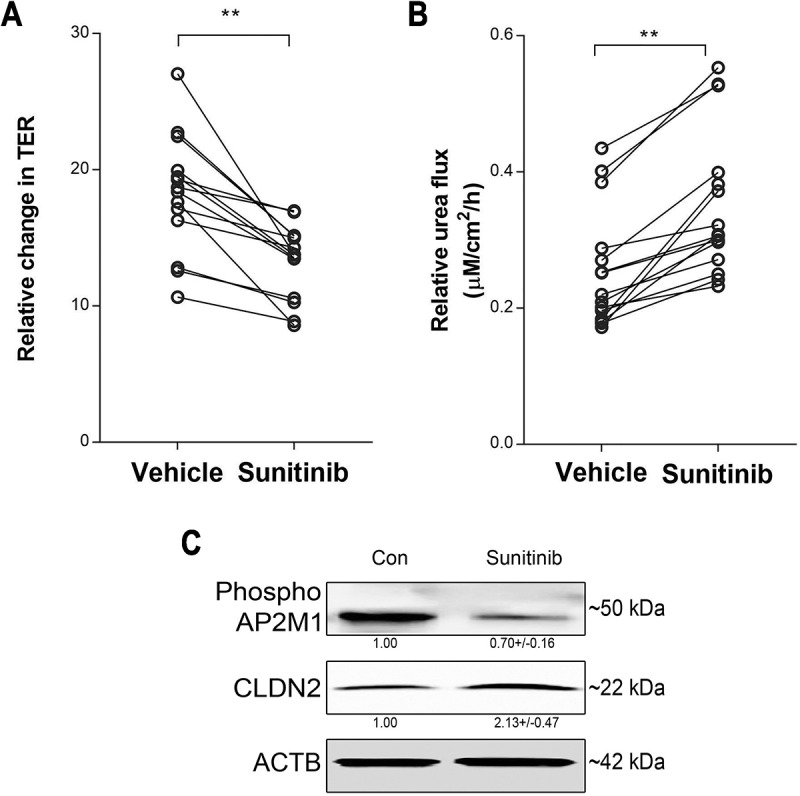
Role of AP2M1 in human colonic TJ barrier. Sunitinib (25 µM, 18 h) significantly decreased TER (A) and increased urea flux (B) in human colonic mucosal samples. N = 15, (*, p < 0.001 versus vehicle). (C) Sunitinib treatment reduced phospho-AP2M1 levels and increased CLDN2 levels in human colonic mucosal samples. The numbers below the individual bands indicate densitometry in terms of phospho-AP2M1:ACTB and CLDN2:ACTB ratio. (*, p < 0.01 versus vehicle) .
Discussion
We have previously reported that nutrient starvation-induced autophagy reduces intestinal epithelial TJ permeability and enhances TJ barrier function via degradation of cation-selective, pore-forming TJ protein CLDN2 [14]. Building on our previous findings, the present study was focused on deciphering the molecular mechanism underlying autophagy-mediated CLDN2 degradation. Collectively, our data showed that autophagy facilitates clathrin-mediated endocytosis of CLDN2 from the membrane. The AAK1 mediated activation of adaptor protein (AP2) µ subunit AP2M1, plays a crucial role in anchoring membrane CLDN2 to clathrin and LC3, for ultimate autophagic degradation (Figure 11). This study on CLDN2, an important regulator of epithelial TJ permeability, demonstrates a central role for autophagy in regulating epithelial TJ barrier function. The impact of CLDN2 knockout and over expression on Caco-2 TJ barrier further substantiated the important role of CLDN2 in regulating intestinal epithelial TJ barrier function under constitutive and autophagic conditions. The intestinal epithelial TJ barrier plays an important role in maintaining intestinal homeostasis as it allows movement of select solutes while restricting passage of luminal antigens and toxins, which can activate host immune system and cause intestinal inflammation. CLDN2 forms a pore pathway within the TJs and is responsible for the flux of small solutes [19,20,39]. While exogenous expression of CLDN2 is known to increase TJ permeability [23,40], we have shown previously that CLDN2 depletion alone is sufficient to enhance TJ barrier function in Caco-2 and MDCK II cells [14]. Beside genetic regulation, various modes of intracellular vesicular transport have been shown to regulate the composition and function of TJs [41,42]. We, and others, have previously demonstrated constitutive internalization, recycling, and sorting to the lysosomal pathway of TJ proteins including CLDN2 [14,43–45]. Using several autophagy inducers which are reported to induce autophagy via different mechanisms, we clearly showed that autophagic induction leads to degradation of CLDN2 and this phenomenon is not limited to starvation. Similarly, by examination of major endocytic pathways including clathrin, caveolae, and micropinocytosis, we determined that autophagy-mediated CLDN2 degradation occurs via clathrin-mediated endocytic pathway. Consistent with our observations, a previous study has shown EGF-induced CLDN2 endocytosis via clathrin pits that were positive for AP2A1 [46].
Figure 11.
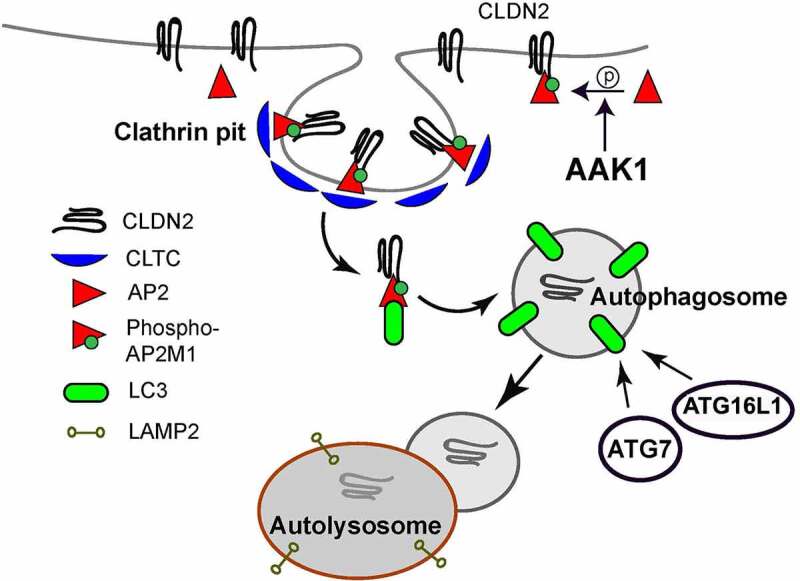
Schematic model of autophagy-induced CLDN2/Claudin-2 degradation. Autophagy and AAK1 (AP2 associated kinase 1) activates AP2/Adaptor protein-2 via phosphorylation of AP2M1 (adaptor related protein complex 2 subunit mu 1). Activated AP2M1 binds to YXXΦ (AP2M1 binding site) region of CLDN2 and facilitate its CLTC/clathrin-mediated endocytosis. AP2 also contains LC3 interacting region and acts as a bridge in connecting endocytosed CLDN2 to LC3. ATG7 dependent LC3 lipidation is required for internalization of CLDN2 into autophagosomes. The CLDN2-containing autophagosome fuses with lysosome, resulting in degradation of CLDN2.
Our study has advanced the understanding of CLDN2 trafficking in many ways. Foremost, by demonstrating the inhibition of CLDN2 degradation by mutation of AP2M1 binding motifs (YXXФ) on CLDN2, we confirmed clathrin-AP2 as a major route for CLDN2 endocytosis. We also showed a key role of AAK1 mediated activation of AP2M1 in clathrin-mediated CLDN2 endocytosis. Furthermore, the reduction in CLDN2 levels caused by various autophagy inducers and the increase in CLDN2 levels resulting from knockout of AAK1, AP2M1 and ATG7 demonstrate an important role of autophagy in regulating CLDN2 levels. The increase in colonocytes CLDN2 level in autophagy deficient atg7 cKO mice, resulting into increased TJ permeability and increased susceptibility to experimental colitis, support the in-vitro findings of autophagy regulation of the TJ barrier via CLDN2.
In this study, AP2M1 was established to be a central player in autophagy-mediated CLDN2 degradation. AP2M1 was found to be activated and required during autophagy-mediated CLDN2 degradation. Our studies support a model where on one hand AP2 secures CLDN2 cargo within the clathrin pits via AP2M1 binding sites in CLDN2 and on the other hand it connects the cargo to the autophagy pathway by its interaction with LC3. This was particularly supported by the observation that CLDN2-LC3 association was disrupted when AP2M1 was deleted in Caco-2 cells. Autophagy and endocytosis are inter-dependent processes and share molecular machinery at the several intermediate steps before culminating in the lysosomes [47]. Various tethering factors regulated by Rab-family GTPases and SNARE proteins provide specificity in membrane trafficking and fusion events [48]. The process of autophagophore formation and expansion involves multiple membrane fusions via endocytosis pathway. For instance, the octameric tethering complex TRAPPIII is involved in autophagosome biogenesis [49] while HOPS tethering complex is reported to play a role in autophagosome maturation [50]. The endocytic vesicles which contribute to phagophore formation are also known to undergo homotypic fusion mediated by a VAMP7 containing SNARE complex [51]. RAB11-positive recycling endosomes constituting autophagy proteins ULK1 and ATG9 also contribute to the LC3 positive early autophagosome [52]. RAB7, a late endosomal RAB protein is reported to be found in nascent autophagosomes and proposed to play a key role in fusion of autophagosomes with lysosomes [53]. Thus the temporal intersection of autophagy and endocytosis must be tightly controlled for proper execution of both the processes. If clathrin-bound CLDN2 cargo directly fuses with autophagosome or the involvement of other intermediate endocytic compartments, remains a subject for future investigations. Identification of the role of upstream AAK1-mediated activation of AP2M1 in autophagy mediated CLDN2 degradation is another important highlight of this study. Using in-vitro cell culture, in-vivo mouse, and ex-vivo human colonic mucosa, we showed that AAK1 KO or inhibition of AAK1-mediated activation of AP2M1 with sunitinib increases epithelial CLDN2 levels. While AAK1-mediated activation of AP2 results into increased affinity between AP2 and membrane protein sorting signals [31], AAK1 also interacts with ATG8 protein and has LC3 interacting region (LIR) motifs [47,54]. The exact role of AAK1 in autophagy-mediated increase in CLDN2 endocytosis needs further investigation.
Among instances of autophagy-mediated degradation of membrane proteins, one prominent example is of Alzheimer disease. Autophagy has been shown to target membrane bound amyloid precursor protein (APP, a precursor for β-amyloid) for degradation [28], indicating therapeutic potential of the autophagy pathway. Notch1 [55], focal adhesions [56], and E‐cadherin [57] are some other examples of plasma membrane protein targets of autophagy. Since the plasma membrane has been shown to contribute to the formation of pre-autophagosomal structures [35], autophagic degradation of CLDN2 may be thought of as a passive degradation along with the plasma membrane components merging into autophagosomal structures. However, CLDN2 and no other claudins were found to be degraded during autophagy induction [14], indicating selective nature of autophagic CLDN2 degradation. In recent studies, transgenic CLDN2 expression was shown to exacerbate while genetic CLDN2 deficiency dampens immune-mediated colitis in T cell transfer model [4]. Several studies have shown that the inflamed intestinal mucosa in patients with active IBD has increased CLDN2 expression [58–60]. Moreover, pro-inflammatory cytokines including TNF-α, IL-13, IL-17, and IL-6, whose levels are increased markedly in IBD patients, are known to increase CLDN2 expression and cause a CLDN2-dependent increase in TJ permeability [59,61–63]. In addition, CLDN2 has also been shown to promote cell proliferation and tumorigenicity in IBD-associated dysplasia and colitis-associated carcinogenesis [64,65]. Thus, our findings of autophagy regulation of CLDN2, are highly significant in view of the role of CLDN2 mediated TJ permeability in intestinal inflammation and the defects in autophagy reported in IBD. In conclusion, our studies, for the first time, directly link an important TJ protein CLDN2 to autophagy. Our data indicates that clathrin adaptor protein AP2M1 subunit plays a crucial role in autophagy-mediated enhancement of the intestinal TJ barrier function via degradation of the pore-forming TJ protein CLDN2, and autophagy may provide a therapeutic tool against intestinal inflammation.
Materials and methods
Chemicals and antibodies
Rapamycin (Life Technologies, PH21235), MG132 (carbobenzoxy-Leu-Leu-leucinal; EMD Millipore, 474,790), Bafilomycin A1 (Santa Cruz Biotechnology, sc-201,550), Amiloride hydrochloride hydrate (A7410), methyl-β-cyclodextrin (C4555), chlorpromazine (CPZ, C8138), sunitinib (PZ0012) and SBI-0206965 (SML1540) from Sigma-Aldrich were purchased from the indicated companies. [14C] Urea (specific activity 56.5 mCi/ mmol) was purchased from Moravek Inc. (MC141). The primary antibodies used included anti-CLDN2 (Abcam, ab53032), anti-LC3 (Sigma, L7543), anti-AP2A1 (Gene Tex, GTX22807), anti-phospho-AP2M1 (Cell signaling Technologies, 73,995), anti-clathrin, anti-AP2M1, anti-ATG7, anti-ATG16L1, anti-SQSTM1/p62, anti-ACTB/β
-actin (ProteinTech, 26,523-1-AP, 27,355-1-AP, 10,088-2-AP, 19,812-1-AP, 18,420-1-AP, HRP-60008, respectively), and anti-tGFP (OriGene, TA150041). The secondary antibodies included anti-rabbit HRP (Invitrogen, 31,460) and anti-mouse HRP (Invitrogen, 31,430). Primers used in the study were synthesized using Sigma Genosys (Woodlands, TX, USA).
Cell culture
Human intestinal epithelial Caco-2 cells (ATCC, HTB-37) were maintained in DMEM (Dulbecco’s Modified Eagle’s Medium) – High Glucose (Gibco, 11,965,118) supplemented with 10% heat inactivated fetal bovine serum (R&D systems, S11150H) and Penicillin-Streptomycin antibiotics (Gibco, 15,140,122) at 37°C in a 5% CO2 incubator. Caco-2 cells were grown on 0.4 μm pore size, 12 mm diameter filter inserts. The trans-epithelial electrical resistance (TER) of the filter-grown cells was measured by an epithelial voltohmeter (World Precision Instruments, Sarasota, FL, USA), and monolayers with a TER of 450–500 Ω/cm2 were used for experiments. Nutrient Starvation was induced in filter-grown Caco-2 monolayers by replacing DMEM with serum-free EBSS (Earle’s Balanced Salt Solution; Sigma, E3024).
Determination of Caco-2 paracellular flux
Caco-2 paracellular permeability was determined using the smaller size paracellular marker urea (14C, Mr = 60). The apical-to-basal flux rates of the paracellular markers were determined by adding them to the apical solution and radioactivity was measured in the basal solution at 30 min and 60 min using a scintillation counter, as described by us previously [66].
Western blot analysis for assessment of protein expression
Caco-2 monolayers were rinsed twice with ice-cold PBS (Corning, 21–040-CV) and lysed using RIPA buffer (Sigma, R0278) containing cOmplete mini, protease inhibitor (Sigma, 11,836,170,001). The cell lysates were centrifuged at 9600 G, for 10 min in order to remove cell debris and the clear supernatant was used for protein quantification. Protein quantification of the extracted aliquots was performed (BCA protein assay kit; Pierce, 23,225), and Laemmli gel loading buffer (Invitrogen, NP007) was added to the lysate and boiled at 70°C for 10 min. An equal amount of protein was loaded in SDS-PAGE gel, separated, and transferred to a nitrocellulose membrane. The membrane was incubated for 1 h in blocking solution (5% nonfat dry milk (Bio-Rad, 1,706,404) in TBS (Bio-Rad, 1,706,435)-0.1% Tween 20 (Bio-Rad, 1,706,531) buffer, followed by incubation with the appropriate primary antibody in blocking solution. After incubation with primary antibody, the membrane was washed in TBS-0.1% Tween 20 buffer, incubated in the appropriate secondary antibody and developed using SuperSignal West Pico PLUS kit (Thermo Scientific, 34,580). For membrane and cell fractionation, Mem-PER™ Plus Membrane Protein Extraction Kit (Thermo Scientific, 89,842) was used as per manufacturer’s instructions. The densitometry analysis was performed using ImageJ software [67].
Transmission electron microscopy
Caco-2 cell monolayers grown on 0.4 µm membrane were subjected to starvation as mentioned above. The control and starvation samples were then fixed in 4% paraformaldehyde (Electron Microscopy Sciences, 15,710) and 0.1% glutaraldehyde Electron Microscopy Sciences, 16,000) in 0.1 M 1,4-piperazinediethanesulfonic acid (PIPES) buffer pH 7.4, and rinsed four times in 0.1 M PIPES buffer (Electron Microscopy Sciences, 19,230). The samples were dehydrated with ethanol in increasing concentrations (30, 50, and 70%), incubated in mixture of 100% LR White resin (Electron Microscopy Sciences, 14,381) and 70% ethanol (2:1) for 30 min at room temperature, followed by overnight incubation in 100% LR White. The samples were then placed in the gelatin capsules (Ted Pella Inc., 130–1), incubated at 50°C for 24 h to polymerize the resin and cut into thin sections of 60–70 nm to be mounted onto nickel grids by Diatome diamond knife using Leica UC7 Ultramicrotome (Leica Microsystems Inc., Buffalo Grove, IL, USA). The mounted sections were incubated overnight in anti-CLDN2 antibody at 4°C, washed with TBS followed by incubation with 10-nm gold particle tagged secondary antibody (Sigma, G3779). The finished grids were stained by uranyl acetate and lead citrate and viewed at 60 kV under JEOL JEM 1400 transmission electron microscope (Penn State College of Medicine).
Confocal immunofluorescence
Confocal Immunofluorescence for CLDN2, AP2M1, clathrin, and LC3 on Caco-2 cell monolayers or mice colonic tissue was performed by standard methods. Caco-2 monolayers were washed twice with cold PBS, fixed with 2% paraformaldehyde for 20 min. The colon cryosections on the other hand were fixed in acetone. The cell monolayers or cryosections were permeabilized with 0.1% Triton X-100 (Sigma, X100) in PBS at room temperature for 5 min. The cell monolayers or cryosections were then blocked in normal serum (Invitrogen, 50197Z) and labeled with primary antibodies in blocking solution overnight at 4°C. After PBS washes, the sections were incubated in Alexa Fluor-488, Cy-3, or Alexa Fluor-647-conjugated secondary antibodies (Invitrogen, A11078, A10521, and A21244). ProLong Gold antifade reagent (Invitrogen, P36931) containing DAPI as a nuclear stain was used to mount the sections on glass slides. The slides were examined using a confocal fluorescence microscope Leica SP8. Images were processed with LAS X software (Leica Microsystems, Penn State College of Medicine).
Co-immunoprecipitation
The co-immunoprecipitation experiments were performed using protein G dynabeads from Invitrogen (10004D) on cell lysates according to the manufacturer’s protocol. Briefly, antibodies conjugated to protein G dynabeads were incubated overnight at 4°C with cell lysates solubilized in RIPA buffer. The dynabeads were washed, eluted, and separated using SDS-PAGE and analyzed by Western blot.
Lentiviral mediated CRISPR-Cas9 knockout of AP2M1, ATG7.
The plasmid Cas9 nuclease CP-LVC9NU (Genecopoeia) was used individually with a single guide RNA (sgRNA) targeting the region CAACTTGTGGGCTACATCCT of CLDN2, TTTCGACTCCCGGCGAGAGC of AAK1, GATGTCATCTCGGTAGACTC of AP2M1, a sgRNA targeting AAATAATGGCGGCAGCTACG of ATG7 and scrambled sgRNA for control in pCRISPR-LVSG03 (Genecopoeia) to generate respective lentiviral particles packaged in Lenti-X 293 T cells using PMD2.G and pPAX2 (Addgene, 12,259, 12,260; deposited by Didier Trono). The lentiviral particles obtained were used to transduce Caco-2 cells in presence of polybrene (EMD Millipore, TR-1003-G), and were selected in their respective antibiotic selection media to generate stable knockout Caco-2 cells. The gene knockout was further confirmed using Western blot analysis. CLDN2 ORF in pCMV6-AC-GFP (Origene, RG204199) and corresponding control plasmid were used to transfect Caco-2 cells using Lipofectamine 2000 (Invitrogen, 11,668,027) as per manufacturer’s instructions and the transfected cells were selected using respective antibiotic selection media to generate stable CLDN2-overexpressing Caco-2 cells.
Site directed mutagenesis
Single and multiple point mutations were introduced into the CLDN2 gene of above mentioned CLDN2 ORF plasmid using the QuikChange lightning site-directed mutagenesis kit (Agilent, 200,518) as instructed by the manufacturer. Primers were designed with the desired mutation (Table S1) according to manufacturer’s guidelines, and a thermocycling reaction permitted in-vitro synthesis of the plasmid DNA was performed with the high-fidelity polymerase PfuTurbo. Parental template DNA was digested away by the methylase specific DNase, DpnI, and the resultant mutated plasmid mixture was used to transform XL10-gold ultra-competent cells. The mutated plasmids were sequenced to confirm site specific mutations. The sequenced mutated plasmids were transfected individually for further analysis.
Experimental animals
Experimental methodologies used in the study were approved by the Institutional Animal Care and Use Committee of Pennsylvania State University College of Medicine. Adult mice were engineered with floxed alleles of Atg7 and a transgene expressing the TAM-regulated Cre recombinase fusion protein under the control of the ubiquitously expressed ubiquitin C (Ubc) promoter [68]. ATG7 deficiency was created by providing tamoxifen to 10-week old mice leading to Cre activation only in mice with Atg7 floxed and Ubc-Cre-ESRT2/Ubc-CreERT2 alleles, producing loss of ATG7 protein (atg7 cKO mice). Tamoxifen-treated mice with only floxed alleles of Atg7 (Atg7fl/fl) were used as controls. Tamoxifen (Sigma, T5648; 20 mg/ml suspended in 98% sunflower seed oil [Spectrum Chemical, S1929] and 2% ethanol mixture) was injected i. p. (200 µl per 25 g of mice body weight) into 8 to 10 week old UBC-CreERT2+/+; Atg7fl/fl mice once per day for 5 days and the mice were used for experiment after 2 weeks. Control animals Atg7fl/fl mice also received the same amount of tamoxifen. The wild-type C57BL/6 J mice (Jackson Laboratory, 000664) were treated with sunitinib (40 mg/kg/day for 2 days, oral gavage). In the dextran sulfate sodium (DSS) model, mice were treated with 2.5% DSS (MP Biomedicals, 160,110) in drinking water for 7-days, as detailed previously [69].
Human tissue samples and treatment
The surgically resected human colon samples were obtained freshly from the Department of Surgery, Division of Colon and Rectal Surgery as per the protocols approved by Institutional Review Board (STUDY00010256). Human colonic tissue were washed, the muscular layer was stripped, and the isolated mucosal epithelial tissue were incubated in supplemented DMEM, overnight, on gelatin sponge (Ethicon, 1975), in the presence and absence of sunitinib (25 µg/ml).
Measurement of paracellular permeability and trans-epithelial electrical resistance (TER) of murine and human colon
The trans-epithelial resistance of the mice and human colonic tissue were measured by mounting the colonic tissue on 0.03 cm2-aperture Ussing chambers (Physiologic Instruments, CA, USA). Trans-epithelial electrical resistance (TER, Ω·cm2) was calculated from the spontaneous potential difference and short-circuit current. The paracellular permeability was assessed by mucosal-to-serosal flux of [14C]-urea, as described by us previously [70].
Statistical analysis
Data are reported as means ± SE. Whenever needed, data were analyzed by using an ANOVA for repeated measures (SigmaStat, Systat Software, San Jose, CA). Tukey’s test was used for post-hoc analysis between treatments following ANOVA (P < 0.05).
Acknowledgments
The authors thank the IBD and Colorectal Diseases Biobank, Transmission Electron Microscopy, Confocal Microscopy, and Animal Facility cores at the Penn State College of Medicine for their excellent technical assistance.
Funding Statement
This research work was supported in part by National institute of diabetes and digestive and kidney diseases grant DK100562(PN), DK114024 (PN), National institute of diabetes and digestive and kidney diseases DK-106072(TM), National Institute of General Medical Sciences grant P20-GM-121176, and Crohn’s & Colitis Foundation Award Crohn’s and Colitis Foundation of America Crohn’s and Colitis Foundation of America 694583(MN). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies. The authors also acknowledge support by the Peter and Marshia Carlino Fund for IBD Research.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
References
- [1].Ma TY, Nighot P, Al-Sadi R.. Chapter 25 - tight junctions and the intestinal barrier. In: Said HM, editor. Physiology of the gastrointestinal tract. Sixth ed. Academic Press; 2018. p. 587–639. [Google Scholar]
- [2].Garcia-Hernandez V, Quiros M, Nusrat A. Intestinal epithelial claudins: expression and regulation in homeostasis and inflammation. Ann N Y Acad Sci. 2017;1397(1):66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9(11):799–809. [DOI] [PubMed] [Google Scholar]
- [4].Raju P, Shashikanth N, Tsai PY, et al. Inactivation of paracellular cation-selective claudin-2 channels attenuates immune-mediated experimental colitis in mice. J Clin Invest. 2020. Oct 1;130(10):5197–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013. Oct;13(10):722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39(2):207–211. [DOI] [PubMed] [Google Scholar]
- [8].Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39(5):596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Parkes M, Barrett JC, Prescott NJ, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39(7):830–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lavoie S, Conway KL, Lassen KG, et al. The Crohn’s disease polymorphism, ATG16L1 T300A, alters the gut microbiota and enhances the local Th1/Th17 response. Elife. 2019. Jan 22;8. DOI: 10.7554/eLife.39982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456(7219):264–268. [DOI] [PubMed] [Google Scholar]
- [12].Strisciuglio C, Duijvestein M, Verhaar AP, et al. Impaired autophagy leads to abnormal dendritic cell-epithelial cell interactions. J Crohns Colitis. 2013. Aug;7(7):534–541. [DOI] [PubMed] [Google Scholar]
- [13].Cooney R, Baker J, Brain O, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16(1):90–97. [DOI] [PubMed] [Google Scholar]
- [14].Nighot PK, Hu CA, Ma TY. Autophagy enhances intestinal epithelial tight junction barrier function by targeting claudin-2 protein degradation. J Biol Chem. 2015. Mar 13;290(11):7234–7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Park D, Jeong H, Lee MN, et al. Resveratrol induces autophagy by directly inhibiting mTOR through ATP competition. Sci Rep. 2016. Feb 23;6(1):21772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Renna M, Jimenez-Sanchez M, Sarkar S, et al. Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases. J Biol Chem. 2010. Apr 9;285(15):11061–11067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shaw RJ, Lamia KA, Vasquez D, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005. Dec 9;310(5754):1642–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yuk J-M, Shin D-M, Lee H-M, et al. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe. 2009. 2009/09/17/;6(3):231–243. [DOI] [PubMed] [Google Scholar]
- [19].Van Itallie CM, Holmes J, Bridges A, et al. The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J Cell Sci. 2008;121(Pt 3):298–305. [DOI] [PubMed] [Google Scholar]
- [20].Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol. 2009;1(2):a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Egan DF, Chun MG, Vamos M, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell. 2015. Jul 16;59(2):285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ikari A, Taga S, Watanabe R, et al. Clathrin-dependent endocytosis of claudin-2 by DFYSP peptide causes lysosomal damage in lung adenocarcinoma A549 cells. Biochim Biophys Acta. 2015. Oct;1848(10 Pt A):2326–2336. [DOI] [PubMed] [Google Scholar]
- [23].Furuse M, Furuse K, Sasaki H, et al. Conversion of zonulae occludentes from tight to leaky strand type by introducing claudin-2 into Madin-Darby canine kidney I cells. J Cell Biol. 2001;153(2):263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Koivusalo M, Welch C, Hayashi H, et al. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J Cell Biol. 2010;188(4):547–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hailstones D, Sleer LS, Parton RG, et al. Regulation of caveolin and caveolae by cholesterol in MDCK cells. J Lipid Res. 1998. 1998/02/01/;39(2):369–379. [PubMed] [Google Scholar]
- [26].Wang LH, Rothberg KG, Anderson RG. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J Cell Biol. 1993. Dec;123(5):1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Traub LM. Sorting it out: AP-2 and alternate clathrin adaptors in endocytic cargo selection. J Cell Biol. 2003. Oct 27;163(2):203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tian Y, Chang JC, Fan EY, et al. Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer’s APP-CTF for terminal degradation via autophagy. Proc Natl Acad Sci U S A. 2013;110(42):17071–17076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chang C, Shi X, Jensen LE, et al. Reconstitution of cargo-induced LC3 lipidation in mammalian selective autophagy. Sci Adv. 2021. Apr;7(17). doi: 10.1126/sciadv.abg4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Conner SD, Schmid SL. Identification of an adaptor-associated kinase, AAK1, as a regulator of clathrin-mediated endocytosis. J Cell Biol. 2002. Mar 4;156(5):921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ricotta D, Conner SD, Schmid SL, et al. Phosphorylation of the AP2 mu subunit by AAK1 mediates high affinity binding to membrane protein sorting signals. J Cell Biol. 2002. Mar 4;156(5):791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Neveu G, Ziv-Av A, Barouch-Bentov R, et al. AP-2-associated protein kinase 1 and cyclin G-associated kinase regulate hepatitis C virus entry and are potential drug targets. J Virol. 2015. Apr;89(8):4387–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Karaman MW, Herrgard S, Treiber DK, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008. 2008/01/01;26(1):127–132. [DOI] [PubMed] [Google Scholar]
- [34].Bekerman E, Neveu G, Shulla A, et al. Anticancer kinase inhibitors impair intracellular viral trafficking and exert broad-spectrum antiviral effects. J Clin Invest. 2017. Apr 3;127(4):1338–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ravikumar B, Moreau K, Jahreiss L, et al. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010. Aug;12(8):747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Popovic D, Dikic I. TBC1D5 and the AP2 complex regulate ATG9 trafficking and initiation of autophagy. EMBO Rep. 2014. Apr;15(4):392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72(1):395–447. [DOI] [PubMed] [Google Scholar]
- [38].Tsuboi K, Nishitani M, Takakura A, et al. Autophagy protects against colitis by the maintenance of normal gut microflora and secretion of mucus. J Biol Chem. 2015;290(33):20511–20526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Van Itallie CM, Mitic LL, Anderson JM. Claudin-2 forms homodimers and is a component of a high molecular weight protein complex. J Biol Chem. 2010;286(5):3442–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Amasheh S, Meiri N, Gitter AH, et al. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J Cell Sci. 2002;115(Pt 24):4969–4976. [DOI] [PubMed] [Google Scholar]
- [41].Terry SJ, Zihni C, Elbediwy A, et al. Spatially restricted activation of RhoA signalling at epithelial junctions by p114RhoGEF drives junction formation and morphogenesis. Nat Cell Biol. 2011;13(2):159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shin K, Fogg VC, Margolis B. Tight junctions and cell polarity. Annu Rev Cell Dev Biol. 2006;22(1):207–235. [DOI] [PubMed] [Google Scholar]
- [43].Wong M, Ganapathy AS, Suchanec E, et al. Intestinal epithelial tight junction barrier regulation by autophagy-related protein ATG6/beclin 1. Am J Physiol Cell Physiol. 2019. May 1;316(5):C753–C765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dukes JD, Whitley P, Chalmers AD. The PIKfyve inhibitor YM201636 blocks the continuous recycling of the tight junction proteins claudin-1 and claudin-2 in MDCK cells. PLoS One. 2012;7(3):e28659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lu R, Johnson DL, Stewart L, et al. Rab14 regulation of claudin-2 trafficking modulates epithelial permeability and lumen morphogenesis. Mol Biol Cell. 2014;25(11):1744–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ikari A, Takiguchi A, Atomi K, et al. Epidermal growth factor increases clathrin-dependent endocytosis and degradation of claudin-2 protein in MDCK II cells. J Cell Physiol. 2011;226(9):2448–2456. [DOI] [PubMed] [Google Scholar]
- [47].Birgisdottir ÅB, Johansen T. Autophagy and endocytosis - interconnections and interdependencies. J Cell Sci. 2020. May 22;133(10): 1–16. [DOI] [PubMed] [Google Scholar]
- [48].Tooze SA, Abada A, Elazar Z. Endocytosis and Autophagy: exploitation or Cooperation? Cold Spring Harb Perspect Biol. 2014. May 1;6(5):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lynch-Day MA, Bhandari D, Menon S, et al. Trs85 directs a Ypt1 GEF, TRAPPIII, to the phagophore to promote autophagy. Proc Natl Acad Sci U S A. 2010. Apr 27;107(17):7811–7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Liang C, Lee JS, Inn KS, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. 2008. Jul;10(7):776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Moreau K, Ravikumar B, Renna M, et al. Autophagosome precursor maturation requires homotypic fusion. Cell. 2011. Jul 22;146(2):303–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Longatti A, Lamb CA, Razi M, et al. TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J Cell Biol. 2012. May 28;197(5):659–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Jäger S, Bucci C, Tanida I, et al. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004. Sep 15;117(Pt 20):4837–4848. [DOI] [PubMed] [Google Scholar]
- [54].Loi M, Muller A, Steinbach K, et al. Macroautophagy proteins control MHC class i levels on dendritic cells and shape anti-viral CD8(+) T cell responses. Cell Rep. 2016. May 3;15(5):1076–1087. [DOI] [PubMed] [Google Scholar]
- [55].Wu X, Fleming A, Ricketts T, et al. Autophagy regulates Notch degradation and modulates stem cell development and neurogenesis. Nat Commun. 2016. Feb 3;7(1):10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kenific CM, Stehbens SJ, Goldsmith J, et al. NBR1 enables autophagy-dependent focal adhesion turnover. J Cell Biol. 2016. Feb 29;212(5):577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014. Mar;15(3):178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zeissig S, Burgel N, Gunzel D, et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut. 2007;56(1):61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Heller F, Florian P, Bojarski C, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129(2):550–564. [DOI] [PubMed] [Google Scholar]
- [60].Schmitz H, Barmeyer C, Fromm M, et al. Altered tight junction structure contributes to the impaired epithelial barrier function in ulcerative colitis. Gastroenterology. 1999;116(2):301–309. [DOI] [PubMed] [Google Scholar]
- [61].Suzuki T, Yoshinaga N, Tanabe S. Interleukin-6 (IL-6) regulates claudin-2 expression and tight junction permeability in intestinal epithelium. J Biol Chem. 2011;286(36):31263–31271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kinugasa T, Sakaguchi T, Gu X, et al. Claudins regulate the intestinal barrier in response to immune mediators. Gastroenterology. 2000;118(6):1001–1011. [DOI] [PubMed] [Google Scholar]
- [63].Mankertz J, Amasheh M, Krug SM, et al. TNFalpha up-regulates claudin-2 expression in epithelial HT-29/B6 cells via phosphatidylinositol-3-kinase signaling. Cell Tissue Res. 2009;336(1):67–77. [DOI] [PubMed] [Google Scholar]
- [64].Weber CR, Nalle SC, Tretiakova M, et al. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab Invest. 2008;88(10):1110–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Dhawan P, Ahmad R, Chaturvedi R, et al. Claudin-2 expression increases tumorigenicity of colon cancer cells: role of epidermal growth factor receptor activation. Oncogene. 2011;30(29):3234–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Nighot PK, Blikslager AT. Chloride channel ClC-2 modulates tight junction barrier function via intracellular trafficking of occludin. Am J Physiol Cell Physiol. 2012;302(1):C178–87. [DOI] [PubMed] [Google Scholar]
- [67].Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012. Jul;9(7):671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Karsli-Uzunbas G, Guo JY, Price S, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4(8):914–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Nighot M, Ganapathy AS, Saha K, et al. Matrix metalloproteinase MMP-12 promotes macrophage transmigration across intestinal epithelial tight junctions and increases severity of experimental colitis. J Crohns Colitis. 2021Apr;9:jjab064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Nighot P, Al-Sadi R, Rawat M, et al. Matrix metalloproteinase 9-induced increase in intestinal epithelial tight junction permeability contributes to the severity of experimental DSS colitis. Am J Physiol Gastrointest Liver Physiol. 2015. Dec 15;309(12):G988–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.