

Abstract
New 1H-pyrazolo[3,4-d]pyrimidine derivatives were designed and synthesised to act as epidermal growth factor receptor inhibitors (EGFRIs). The synthesised derivatives were assessed for their in vitro anti-proliferative activities against A549 and HCT-116 cancer cells. Compounds 8, 10, 12a, and 12b showed potent anti-proliferative activities. Compound 12b was the most promising member with IC50 values of 8.21 and 19.56 µM against A549 and HCT-116, respectively. Compounds 8, 10, 12a, and 12b were evaluated for their kinase inhibitory activities against wild EGFR (EGFRWT). Compound 12b was the most potent member showing an IC50 value of 0.016 µM. In addition, compound 12b showed noticeable activity against mutant EGFR (EGFRT790M) (IC50 = 0.236 µM). Flow cytometric analyses revealed that compound 12b is a good apoptotic inducer and can arrest the cell cycle at S and G2/M phases. Furthermore, it produced an 8.8-fold increase in BAX/Bcl-2 ratio. Molecular docking studies were carried out against EGFRWT and EGFRT790M.
Keywords: Anti-proliferative; apoptosis; EGFR inhibitors; molecular docking; 1H-pyrazolo[3,4-d]pyrimidine
Graphical Abstract

1. Introduction
Based on World Health Organisation International Agency for Research on Cancer (IARC), GLOBOCAN digital estimation confirmed the dramatically increased cancer incidence and mortality. The estimated value is about 19.3 million new cancer cases in 20201. In 2022, 1,918,030 new cancer cases and 609,360 cancer deaths are projected to occur in the United States, including approximately 350 deaths per day from lung cancer, the leading cause of cancer death2. Also, cancer is a serious health issue in Africa as almost half of the cancer incidences occur in developing countries3. Consequently, various drug innovations against cancer were recorded, despite that, the real cause of cancer is inevitably unclear till now. Yet, cancer is mainly referred to as uncontrolled cell proliferation and finally metastasis4,5. The regimen of cancer treatment is greatly modified by increasing the knowledge of molecular and tumour biology6. Noticeably, the selectivity of anticancer approaches has a low margin7. So, it is a serious concern to develop a new strategy of treating cancer that provides a high selectivity margin.
Regarding molecular targeted therapy against cancer, receptor tyrosine kinases (RTKs) play a vital role in cellular programs e.g. proliferation, migration, apoptosis, survival, and differentiation8. The role of RTKs is the phosphorylation of tyrosine residue via transferring a gamma phosphate group from ATP to it, so normal physiological cellular functions are maintained9–11. The general architecture of RTKs includes extracellular ligand-binding region, transmembrane helix, and cytoplasmic region that contains protein tyrosine kinase domain decides carboxy C-terminal and juxtamembrane regulatory regions. Abnormalities can alter the regulation of RTKs that become mutated or aberrantly activated, leading to different pathological conditions such as cancer8.
Epidermal growth factor receptor (EGFR) is one of the most important RTKs possessing a key role in cell growth12,13. There are many types of tumours with a high level of EGFR overexpression as breast cancer14, lung cancer (NSCLC)15, and hepatocellular carcinoma (HCC)16. EGFR was found to act as a strong prognostic indicator in head and neck, ovarian, cervical, bladder, and oesophageal cancers. In these cancers, increased EGFR expression was associated with reduced recurrence-free or overall survival rates in 70% of studies16. EGFRs are thought to be interesting targets for developing novel anticancer drugs17–21.
There are many FDA-approved EGFR-tyrosine kinase inhibitors (EGFR-TKIs). The first-generation as erlotinib I22 has a good effect against wild EGFR (EGFRWT). This class has many side effects23,24 in addition to the acquired drug resistance caused by EGFR-TK mutation25. The second-generation was discovered to overcome the resistance induced by EGFRT790M. Neratinib II26 is a one of the most famous drug in this generation. Unfortunately, latter class of drugs has a low maximal-tolerated dose producing inadequate clinical efficacy27,28. The third-generation EGFR-TKIs as olmutinib III and osimertinib IV29 showed enhanced actions against mutant EGFR (EGFRT790M). However, toxic epidermal necrolysis was associated with these drugs30. Hence, there is an urgent need to optimise the approved drugs to reach efficient and less harmful candidates.
EGFR-TKIs must possess some pharmacophoric features to bind efficiently the ATP binding site and hence exert their inhibitory activities. The first pharmacophore is the flat heteroaromatic system which can occupy the adenine binding pocket of the ATP binding site31. The second feature is the terminal hydrophobic head which can occupy the hydrophobic region I of the ATP binding site32. The third feature is the spacer moiety which is mainly an amino derivative to form a hydrogen bond in the linker region of the ATP-binding site33. The fourth feature is the hydrophobic tail which can occupy the hydrophobic region II of the ATP-binding site34,35. The fifth feature is the ribose binding moiety which can occupy the ribose binding pocket. Till now, there are limitations in research that target the ribose binding pocket36 (Figure 1).
Figure 1.

EGFR inhibitors and their pharmacophoric features.
1H-Pyrazolo[3,4-d]pyrimidine moiety is an important scaffold in the field of medicinal chemistry as it is a building block in many anticancer agents36 including EGFR-TKIs30. Compound V was approved as an ATP-competitive inhibitor showing EGFR inhibitory effect at a nanomolar concentration37. Compound VI is another example of 1H-pyrazolo [3,4-d]pyrimidine derivative with anti-EGFR activity38. Furthermore, our team synthesised 1H-pyrazolo[3,4-d]pyrimidine derivative (compound VII) as EGFR inhibitor. This compound showed good anti-proliferative activity with high inhibitory effect against wild and mutant EGFR39 (Figure 1). Due to the high similarity of this scaffold with the adenine moiety of ATP, it was used as a backbone for the design and synthesis of ATP competitive inhibitors, especially the compounds that target RTKs38,40.
Based on the previous reports including the high importance of EGFR as an anticancer target, the generated resistance against the FDA approved anticancer drugs, and the attractiveness of 1H-pyrazolo[3,4-d]pyrimidine moiety, it was decided to design and synthesise a new 1H-pyrazolo[3,4-d]pyrimidine derivatives that may have good inhibitory activities against EGFR. The synthesised compounds were designed to have the pharmacophoric features of EGFR inhibitors.
1.1. Rationale of molecular design
In this work, new 1H-pyrazolo[3,4-d]pyrimidine derivatives were designed and synthesised to have the main pharmacophoric features of EGFR-TKIs. In these compounds, many structural modifications for the reported EGFR-TKIs were carried out. The modification processes were achieved at five positions (Figure 2).
Figure 2.

ATP binding site of EGFR-TK cavity composed of five main features41 and summary of the possible modifications of EGFR-TK inhibitors.
Firstly,1H-pyrazolo[3,4-d]pyrimidine moiety was used as a heteroaromatic system to occupy the adenine binding region42,43. Second, different substituted phenyl or aliphatic structures were utilised as a hydrophobic head to occupy the hydrophobic region I of the ATP-binding site. The third modification was performed on the linker moiety. We used different linkers as imino group (compounds 7a,b, 8, and 9), hydrazone derivative (compounds 11a,b and 12a,b), and thiosemicarbazide moiety (compounds 13a,b). For the hydrophobic tail, we used a phenyl ring to occupy the hydrophobic region II of the ATP-binding site. To occupy the ribose-binding pocket, we used an aniline structure. The diversity of modifications gave us good results about the structure-activity relationship of the synthesised compounds as antiproliferative agents targeting EGFR. All modifications were clarified in Figure 3.
Figure 3.

Rationale of molecular design of the new proposed EGFR-TK inhibitors.
2. Results and discussion
2.1. Chemistry
The designed compounds were synthesised as outlined in Schemes 1–3. Ethoxymethylene malononitrile 141 was allowed to react with phenylhydrazine to produce 5-amino-1-phenyl-1H-pyrazole-4-carbonitrile 244. Compound 2 underwent partial hydrolysis using alcoholic NaOH to produce carboxamide derivative 345. Fusion of compound 3 with urea afforded 1-phenyl-1,7-dihydro-4H-pyrazolo[3,4-d]pyrimidine-4,6(5H)-dion 4. Chlorination of compound 4 using phosphorus oxychloride and phosphorus pentachloride produced 4,6-dichloro-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine 546. Stirring of compound 5 with aniline at room temperature afforded 4-chloro-N,1-diphenyl-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-6-amine 647. The obtained compound 6 was heated with commercially available different amines, namely ethylamine, propylamine, aniline, and cyclohexylamine in the presence of triethylamine afforded the target compounds 7a,b, 8, and 9, respectively. The IR spectra of 7a,b, and 9 demonstrated stretching bands at a range of 2950 − 2980 cm−1 corresponding to CH aliphatic groups. The 1H NMR spectra were characterised with abroad singlet at approximately 7–8 ppm due to the additional NH group.
Scheme 1.

Synthetic protocol of starting compounds. Reagents and conditions: (a) phenyl hydrazine, absolute ethanol, reflux, 2 h; (b) sodium hydroxide, absolute ethanol, reflux, 5 h; (c) urea, fusion; (d) POCl3, PCl5, reflux, 28 h; (e) aniline, absolute ethanol, room temperature.
Scheme 2.

Synthetic protocol of compounds 7a,b, 8, and 9. Reagents and conditions: (a) aliphatic amines, ethanol, reflux 4 h; (b) aniline, ethanol, reflux 6 h; (c) cyclohexylamine, ethanol, reflux 5 h.
Scheme 3.

Synthetic protocol of compounds 10, 11a,b, 12a-c, and 13a,b. Reagents and conditions: (a) hydrazine hydrate 99%, reflux, 6 h; (b) aromatic aldehydes, ethanol, glacial acetic acid, reflux 12 h; (c) acetophenones, ethanol, glacial acetic acid, reflux 14 h.
Compound 6 was heated with hydrazine hydrate to afford 4-hydrazinyl-N,1-diphenyl-1H-pyrazolo[3,4-d]pyrimidin-6-amine 10. The IR spectrum of 10 demonstrated stretching bands at 3444, 3352, and 3190 cm-1 corresponding to NH2 and NH, respectively. Moreover, 1H NMR of this compound showed two exchangeable signals at δ 4.73 and 9.89 ppm corresponding to NH2 and NH, respectively. Refluxing of 10 with commercially available aromatic aldehydes or acetophenones in the presence of glacial acetic acid afforded the target compounds 11a,b, and 12a-c. 1H NMR spectra of hydrazones revealed the presence of D2O exchangeable singlet signals of hydrazinyl NH at the range 11.76–12.24 and an increase in the integration of the aromatic protons indicating the presence of an additional aromatic ring.
2.2. Biological evaluation
2.2.1. In vitro antiproliferative activities
The cytotoxic activities of the synthesised compounds were assessed against two human cancer cell lines (lung, A549) and (colon HCT-116) using an MTT assay. These two cell lines were selected in this test due to the overexpression of EGFR in human lung and colon cancer cell lines48,49. The tested cells were reported to have a high expression level of EGFR.
As presented in Table 1, the tested compounds showed a wide range of anti-proliferative activities. This range varied from potent, moderate to weak cytotoxic effect. Comparing to erlotinib (IC50 = 6.77 and 19.22 µM against A549 and HCT-116, respectively), compounds 8 (IC50 = 16.75 and 24.16 µM), 10 (IC50 = 15.68 and 18.78 µM), 12a (IC50 = 13.72 and 23.33 µM), and 12b (IC50 = 8.21 and 19.56 µM) showed potent anti-proliferative activities. from these result, compounds 12b is considered the most promising member.
Table 1.
In vitro anti-proliferative activities of the tested compounds against human lung (A549) and colon (HCT-116) cancer cell lines.
| Comp. |
In vitro cytotoxicity IC50 (µM)a |
|
|---|---|---|
| A549 | HCT-116 | |
| 7a | 64.42 | 29.62 |
| 7b | 67.38 | 31.49 |
| 8 | 16.75 | 24.16 |
| 9 | 29.07 | 39.39 |
| 10 | 15.68 | 18.78 |
| 11a | 47.67 | 46.18 |
| 11b | 75.71 | 75.11 |
| 12a | 13.72 | 23.33 |
| 12b | 8.21 | 19.56 |
| 12c | 28.79 | 28.69 |
| 13a | 43.12 | 27.03 |
| 13b | 15.36 | 36.44 |
| Erlotinib | 6.77 | 19.22 |
aData are presented as the mean of the IC50 values from three different experiments.
Furthermore, compounds 7a, 7b, 12c, and 13a showed moderate activities against HCT-116 with IC50 values of 29.62, 31.49, and 27.03 µM, respectively. On the other hand, compounds 9, 11a, and 11b showed weak activities against the two tested cell lines while compounds 7a, 7b, 12c, and 12a exhibited weak activity against A549 cells.
2.2.2. Structure-activity relationship
Examining screening results for cytotoxicity assay (Table 1), it was found that the introduction of aliphatic amines as ethyl (compound 7a), propyl (compound 7b), and cyclohexyl (compound 9) in the 4-position of pyrazolo[3,4-d]pyrimidine scaffold was not beneficial for cytotoxic activity, particularly against A549 cells. On the contrary, the introduction of aniline moiety in the same position afforded compound 8 with enhanced anticancer activity. Additionally, a remarkable decline in cytotoxic activity against the two tested cancer cells was detected upon the condensation of hydrazine derivative 10 with aromatic aldehydes in compounds 11a,b. Conversely, the condensation of hydrazine 10 with acetophenone and p-chloroacetophenone furnished compounds 12a and 12b, respectively with better anticancer activity against A549 cells, compared to their parent hydrazine derivative 10. Compound 12b, bearing p-chlorophenyl moiety, stood out as the most potent derivative among the tested compounds, presenting excellent cytotoxic activity, comparable/equipotent to that of erlotinib against A549 and HCT-116 cells, respectively. On the other hand, the p-tolyl derivative 12c revealed a noticeable decrease in cytotoxic activity, relative to its parent hydrazine 10 as well as its p-chlorophenyl analog 12b. Finally, in agreement with the poor cytotoxic activity elicited by aliphatic amine derivatives 7a,b, the addition of aliphatic isothiocyanates as ethyl (compound 13a) and propyl (compound 13b) groups to hydrazine derivative 10, was not beneficial for anticancer activity (Figure 4).
Figure 4.
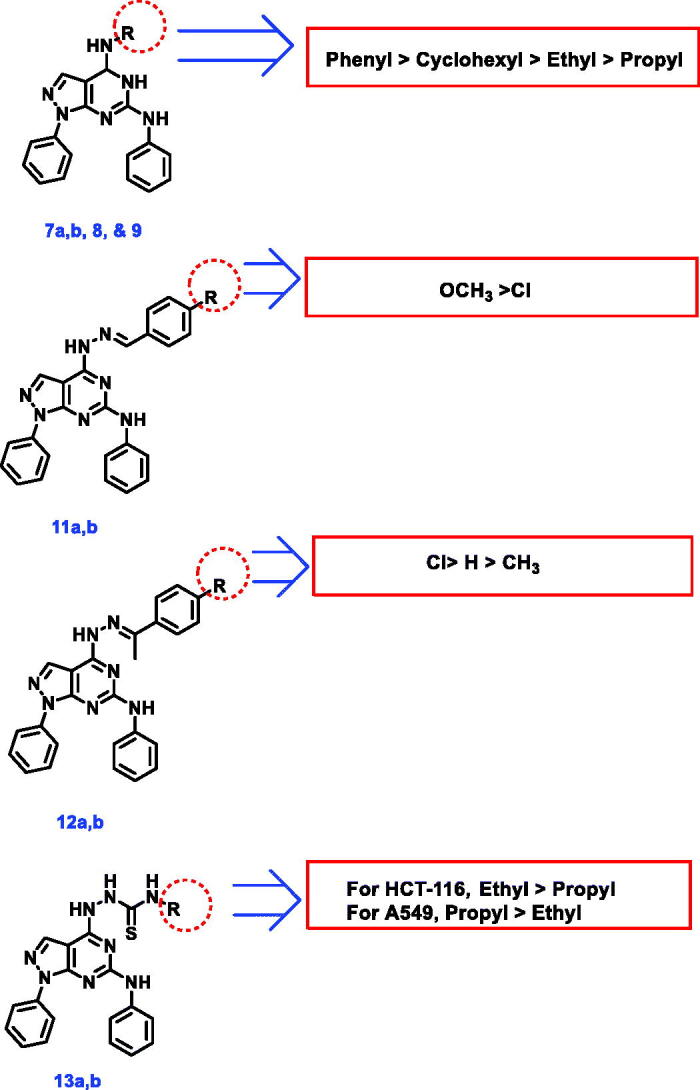
SAR study of the target compounds.
2.2.3. EGFRWT kinase inhibitory assay
The most promising compounds in the cytotoxicity assay (8, 10, 12a, and 12b) were further evaluated for their ability to inhibit the kinase activity of EGFRWT, employing erlotinib as a positive control. As shown in Table 2, the screened compounds significantly inhibited EGFRWT at low IC50 values ranging from 0.016 to 0.026 µM, relative to erlotinib (IC50 = 0.006 µM). Compound 12b was the most potent member showing an IC50 value of 0.016 µM.
Table 2.
the inhibitory activities of the tested compounds against EGFRWT and EGFRT790M kinases.
| Comp. | EGFRWT | EGFRT790M |
|---|---|---|
| IC50 (µM) a | IC50 (µM) a | |
| 8 | 0.026 | NTb |
| 10 | 0.023 | NTb |
| 12a | 0.021 | NTb |
| 12b | 0.016 | 0.236 |
| Erlotinib | 0.006 | 0.563 |
aData were expressed as the mean of three independent experiments.
bNT: Compounds not tested.
Consistent with the results obtained from the cytotoxicity assay, it was observed that the introduction of the chlorine atom in the 4-position of phenyl ring (compound 12b, (EGFRWT IC50 =0.016 µM) resulted in a noticeable enhancement of EGFRWT inhibitory activity, comparing to its unsubstituted analog 12a (IC50 =0.021 µM).
2.2.4. Egfrt790m kinase inhibitory assay
To assess the inhibitory activity of the synthesised compound against the mutant EGFR (EGFRT790M), the most promising member 12b was further screened against EGFRT790M utilising erlotinib as a positive control. Noticeably, it was found that compound 12b (IC50 =0.236 5 µM) was 2.4-fold more potent than erlotinib (IC50 = 0.563 µM) against EGFRT790M (Table 2).
2.2.5. Cell cycle analysis
To determine the biological phase at which the synthesised compounds can interfere with the cell growth, cell cycle analysis was carried out for the most active member 12b in A549 cells. The tested cells were treated with 12b at a concentration of 8.21 µM equal to its IC50 for 48 h. The results revealed a remarkable interference with the normal cell cycle distribution. The treated cells revealed about a 2-fold decrease in the percentage of cells in the G1 phase (from 53.87 to 28.04%), compared to untreated cells. Moreover, compound 12b induced a 1.5-fold increase in the percentage of cells in the S phase (from 28.70 to 42.39%) in addition to a 1.7-fold increase in % G2/M (from 16.56 to 28.55%). From these findings, it can be concluded that compound 12b can arrest the cell cycle at S and G2/M phases (Table 3 and Figure 5).
Table 3.
Effect of compound 12 b on cell cycle progression in A549 cells after 48 h treatment.
| Sample | Cell cycle distribution (%)a |
|||
|---|---|---|---|---|
| %Sub-G1 | %G1 | %S | % G2/M | |
| A549 | 0.87 ± 0.13 | 53.87 ± 0.58 | 28.70 ± 2.50 | 16.56 ± 2.30 |
| Compound 12 b/A549 | 0.89 ± 0.31 | 28.04 ± 2.78*** | 42.39 ± 3.50* | 28.55 ± 1.61* |
aValues are given as mean ± SEM of three independent experiments. *p < 0.05 ***p < 0.001 indicate statistically significant differences from the corresponding control (A549) group in unpaired t-tests.
Figure 5.

Flow cytometric analysis of cell cycle phases after treatment with compound 12 b.
2.2.6. Annexin V-FITC apoptosis assay
As displayed in Table 4 and Figure 6, the treatment of A549 cells with compound 12b for 48 h resulted in a 3-fold decrease in the ratio of viable cells (Left bottom) from 93.43 to 31.57%. In addition, it exhibited an 11-fold increase in the early apoptosis ratio (Right Bottom) from 6.03 to 67.69% and a 1.5-fold increase in the late apoptosis ratio (Right Top) from 0.43 to 0.64% compared to untreated A549 cells. These results indicated that compound 12b is a good apoptotic inducer and that apoptosis is most probably the main mechanism by which compound causes cell death.
Table 4.
Effect of compound 12 b on stages of the cell death process in A549 cells after 48 h treatment.
| Sample | Viablea (Left Bottom) |
Apoptosisa |
Necrosisa (Left Top) |
|
|---|---|---|---|---|
| Early (Right Bottom) |
Late (Right Top) |
|||
| A549 | 93.43 ± 0.96 | 6.03 ± 0.76 | 0.43 ± 0.19 | 0.11 ± 0.03 |
| Comp. 12 b/ A549 | 31.57 ± 4.64 | 67.69 ± 4.86*** | 0.64 ± 0.30 | 0.10 ± 0.02 |
aValues are given as mean ± SEM of three independent experiments. ***p < 0.001 indicates a statistically significant difference from the corresponding control (A549) group in unpaired t-tests.
Figure 6.

Flow cytometric analysis of apoptosis in A549 cells exposed to compound 12 b.
2.2.7. Bax/bcl-2 ratio
The effect of the most active compound 12b on the expression levels of the apoptotic (BAX) and anti-apoptotic (Bcl-2) genes was evaluated. As shown in Table 5 and Figure 7, the treatment of A549 cells with compound 12b for 48 h resulted in a 3.3-fold increase in the level of BAX gene expression in addition to a 2.5-fold decrease in Bcl-2 gene expression. As a result, an 8.8-fold increase in BAX/Bcl-2 ratio was observed for 12b-treated A549 cells, compared to untreated A549 cells.
Table 5.
Effect of Compound 12 b on levels of BAX, and Bcl-2 genes expression in A549 cells treated for48 h.
| Sample | Gene expression (Fold Change)a |
||
|---|---|---|---|
| BAX | Bcl-2 | BAX/Bcl-2 ratio | |
| A549 | 1.00 ± 0.22 | 1.00 ± 0.13 | 1.00 ± 0.21 |
| 12b / A549 | 3.33 ± 0.37** | 0.40 ± 0.08* | 8.80 ± 1.41** |
aValues are given as changes from the corresponding control (A549) group, which is set to ‘1’. *p < 0.05 **p < 0.01 indicate statistically significant differences from the corresponding control in unpaired t-tests.
Figure 7.

Gene expression analysis for the expression levels of BAX, and Bcl-2 after treatment of A549 with compound 12 b for 48 h. Normalised data are expressed as the fold changes, with the control set to ‘1’. *p < 0.05 **p < 0.01 indicate statistically significant differences from the corresponding control in unpaired t-tests.
2.2.8. In vitro cytotoxicity against normal cell and selectivity index
The in vitro cytotoxic effect of the most active compound 12b against a normal cell line (WI-38) was assessed (Table 6). The results revealed that compound 12b has low toxicity against the tested cells with IC50 value of 39.15 μM. Erlotinib as a reference drug showed an IC50 value of 33.75 µM against WI-38 cell line.
Table 6.
In vitro cytotoxicity of 12 b and erlotinib against normal cells (WI-38).
| Comp. | Cytotoxicity WI-38 IC50 (µM)a |
Selectivity index (SI) |
|
|---|---|---|---|
| A549b | HCT-116c | ||
| 12b | 39.15 | 4.77 | 2.00 |
| Erlotinib | 33.75 | 4.99 | 1.76 |
aThe results were the mean of three replicates.
bSI = cytotoxicity against WI-38 cells/cytotoxicity against A549 cell line.
cSI = cytotoxicity against WI-38 cells/cytotoxicity against HCT-116 cell line.
The selectivity index (SI) of compound 12b against tumour cells were shown in Figure 8. This compound showed a SI of 4.77 and 2.00 against A549 and HCT-116, respectively. These indices are comparable to that of erlotinib (4.99 and 1.76) against A549 and HCT-116, respectively.
Figure 8.
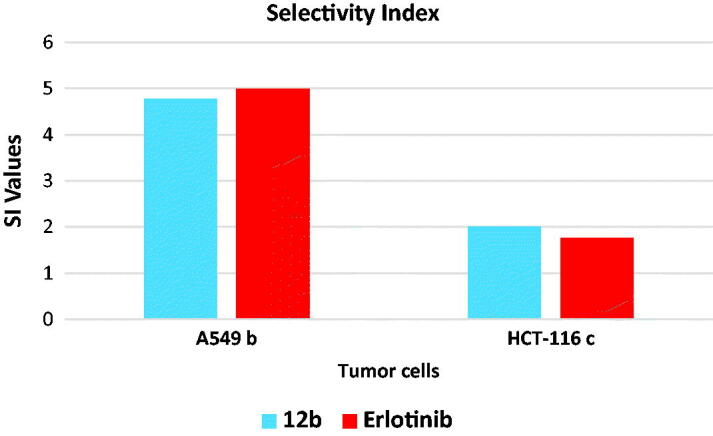
Selectivity indices of compound 12 b.
The results revealed that compound 12b has low toxicity against WI-38 cell line compared to erlotinib. In addition, it showed a high selectivity against the tumour cell lines.
2.3. In silico studies
2.3.1. Docking studies
To investigate the manner of binding with the hypothesised target, docking studies were performed for the synthesised compounds against the active site of the wild-type (EGFRWT, PDB: 4HJO)50 and the mutant type (EGFRT790M, PDB: 3W2O)51. The co-crystallised ligands erlotinib and TAK-285 of EGFRWT and EGFRT790M, respectively, were used as reference compounds. The docked compounds showed good binding affinities towards EGFRWT, with binding free energies ranging from −19.63 to −23.67 kcal/mol, according to the results of docking studies. For the docking against mutant type, the synthesised compounds showed binding energy ranging from −16.09 to −21.66 (Table 7).
Table 7.
The docking binding free energies of the synthesised compounds against EGFRWT and EGFRT790M
| Comp. | Binding free energy (kcal/mol) |
|
|---|---|---|
| EGFRWT | EGFRT790M | |
| 7a | −19.63 | −16.09 |
| 7b | −20.55 | −17.79 |
| 9 | −21.00 | −19.01 |
| 8 | −21.80 | −19.60 |
| 10 | −17.06 | −15.83 |
| 11a | −21.58 | −20.10 |
| 11b | −21.67 | −20.19 |
| 12a | −23.07 | −21.14 |
| 12b | −23.09 | −20.59 |
| 12c | −23.67 | −21.66 |
| 13a | −19.69 | −20.30 |
| 13b | −21.74 | −20.77 |
| Erlotinib | −22.59 | – |
| TAK-285 | – | −21.49 |
In these investigations, MOE 2019 software was used. The output figures were further visualised using Discovery Studio software 3.0. The docking mechanisms were initially validated by redocking each protein's co-crystallised ligands (Erlotinib and TAK-285) against the active sites of EGFRWT and EGFRT790M, respectively. The generated RMSD values between the re-docked conformers and the co-crystallised conformers were 1.18 and 1.66 Å for erlotinib and TAK-285, respectively. As reported, an RMSD value of less than 2 Å suggests that the docking operation is genuine. As a result, the obtained RMSD values confirmed the docking protocol's validity (Figure 9).
Figure 9.

(A) Superimposition of the re-docked conformer of erlotinib (pink) over the co-crystallised conformer (green) with an RMSD value of 1.18 Å. (B) Superimposition of the re-docked conformer of TAK-285 (pink) over the co-crystallised conformer (green) with an RMSD value of 1.66 Å.
The co-crystallised ligand (erlotinib) of EGFRWT produced a binding score of −22.59 kcal/mol. The binding mode of erlotinib against EGFRWT was shown in Figure 10. The heterocyclic quinazoline moiety was oriented into the adenine pocket forming a hydrogen bond with Met769. In addition, it formed four hydrophobic interactions with Lue694, Ala719, and Leu820. The ethynylphenyl moiety was oriented into the hydrophobic pocket I forming three hydrophobic interactions with Ala719, Lys721, and Val702. The two 2-methoxyethoxy groups occupied the hydrophobic region II forming a hydrogen bond with Cys773 in close contact with Gly772 and Leu694.
Figure 10.

(A) 3D interaction of erlotinib docked into the active site of EGFRWT. The hydrogen bonds were represented in green dashed lines. The pi interactions were represented in orange lines. (B) 2D interaction of erlotinib docked into the active site of EGFRWT.
Taking compound 12b as a representative example, it showed a similar binding pattern to erlotinib. Compound 12b exhibited a binding score of −23.07 kcal/mol. The 1H-pyrazolo[3,4-d]pyrimidin-6-amine moiety occupied the adenine pocket to form two hydrogen bonds with Met769 and Lys721. In addition, it formed four hydrophobic interactions with Val702, Ala719, and Leu820. The p-chlorophenyl moiety occupied the hydrophobic pocket I forming four hydrophobic interactions with Lys721, Leu764, and Leu834. In addition, it formed an electrostatic attraction with Asp831. Th hydrazinyl linker formed one hydrogen bond with2 Thr830. The phenyl ring at 1-position of 1H-pyrazolo[3,4-d]pyrimidine occupied the hydrophobic region II forming two hydrophobic interactions with Val702 and Cys773 (Figure 11).
Figure 11.

(A) 3 D interaction of compound 12 b docked into the active site of EGFRWT. The hydrogen bonds were represented in green dashed lines. The pi interactions were represented in orange lines. (B) 2 D interaction of compound 12 b docked into the active site of EGFRWT.
Docking of the synthesised compounds against the mutant EGFR(EGFRT790M) gave a good insight into its binding pattern. The synthesised compounds showed binding scores ranging from −16.09 to −21.66 kcal/mol (Table 7).
2The reference compound (TAK-285) produced a binding score of −21.49 kcal/mol. The heterocyclic 5H-pyrrolo[3,2-d]pyrimidine moiety(the main neuclus) was oriented into the adenine pocket to form one hydrogen bond with Met793. In addition, it formed many hydrophobic interactions with Leu844 and Ala743. The 1-chloro-2–(3-(trifluoromethyl)phenoxy)benzene moiety occupied the hydrophobic pocket I to form many hydrophobic interactions with Lys745, Ile759. Lys745, Glu762, Met766, Met790, Val726, Leu788, and Ala743. Additionally, it formed two hydrogen bonds with Lys745 and Glu762. The N-ethyl-3-hydroxy-3-methylbutanamide moiety was buried in the hydrophobic region II to form one hydrogen bond with Ser720 (Figure 12).
Figure 12.

(A) 3 D interaction of TAK-285 docked into the active site of EGFRT790M. The hydrogen bonds were represented in green dashed lines. The pi interactions were represented in orange lines. (B) 2 D interaction of TAK-285docked into the active site of EGFRT790M.
Compound 12b, as a representative example, showed a binding mode like that of TAK-285 against the mutant EGFR with a binding score of −20.59 kcal/mol. The 1H-pyrazolo[3,4-d]pyrimidin-6-amine moiety occupied the adenine pocket to form one hydrogen bond with Met793. Also, it formed five hydrophobic interactions with Leu718, Leu844, Val726, and Ala743. The p-chlorophenyl moiety occupied the hydrophobic pocket I forming seven hydrophobic interactions with Ala743, Met790, Leu788, Lys745, and Ile759. The phenyl ring at 1-position of 1H-pyrazolo[3,4-d]pyrimidine occupied the hydrophobic region II forming two hydrophobic interactions with Gly796 and Leu718 (Figure 13).
Figure 13.

(A) 3 D interaction of compound 12 b docked into the active site of EGFRT790M. The hydrogen bonds were represented in green dashed lines. The pi interactions were represented in orange lines. (B) 2 D interaction of compound 12 b docked into the active site of EGFRT790M.
2.3.2. In silico ADMET analysis
Discovery studio 4.0 was used to predict ADMET descriptors for all compounds. The predicted descriptors are listed in Table 8. Blood-Brain Barrier (BBB) penetration studies predicted that compounds 8, 11a, 11b, 12a, 12b, 12c, 13a, and 13b have very low BBB penetration levels. Accordingly, such compounds were expected to be safe for CNS. All the tested compounds showed low to very low range levels of ADMET aqueous solubility and have good to moderate intestinal absorption levels. Additionally, all compounds were predicted to be cytochrome P450 2D6 non-inhibitors. Consequently, the liver dysfunction side effect maybe not expected upon administration of these compounds. Due to the high planarity of the synthesised compounds, all of them are expected to bind plasma protein over 90% (Table 8 & Supplementary data).
Table 8.
Predicted ADMET for the designed compounds and reference drugs
| Comp. | BBB levela | Solubility levelb | Absorption levelc | CYP2D6 predictiond | PPB predictione |
|---|---|---|---|---|---|
| 7a | 2 | 2 | 0 | false | false |
| 7b | 2 | 2 | 0 | false | true |
| 9 | 1 | 2 | 0 | false | true |
| 8 | 4 | 1 | 1 | false | true |
| 10 | 3 | 2 | 0 | false | true |
| 11a | 4 | 1 | 1 | false | true |
| 11b | 4 | 1 | 2 | false | true |
| 12a | 4 | 1 | 1 | false | true |
| 12b | 4 | 1 | 2 | false | true |
| 12c | 4 | 1 | 2 | false | true |
| 13a | 4 | 2 | 0 | false | true |
| 13b | 4 | 1 | 1 | false | true |
| Erlotinib | 1 | 2 | 0 | false | true |
aBBB level, blood brain barrier level, 0 = very high, 1 = high, 2 = medium, 3 = low, 4 = very low.
bSolubility level, 1 = very low, 2 = low, 3 = good, 4 = optimal.
cAbsorption level, 0 = good, 1 = moderate, 2 = poor, 3 = very poor.
dCYP2D6, cytochrome P2D6, TRUE = inhibitor, FALSE = non inhibitor.
ePBB, plasma protein binding, FALSE means less than 90%, TRUE means more than 90%.
2.3.3. In silico toxicity studies
In this work, six toxicity parameters were estimated computationally depending on the constructed toxicity models in Discovery studio software. The results of in silico toxicity studies were depicted in Table 9,
Table 9.
In silico toxicity studies of the synthesised compounds and erlotinib
| Comp. | Ames mutagenicity | Developmental Toxicity Potential | Carcinogenic Potency TD50 (Rat)a |
Rat Maximum Tolerated Dose (Feed)b |
Ocular Irritancy | Skin Irritancy |
|---|---|---|---|---|---|---|
| 7a | Non-Mutagen | Non-Toxic | 34.965 | 0.287 | Mild | None |
| 7b | Non-Mutagen | Non-Toxic | 27.147 | 0.401 | Mild | None |
| 9 | Non-Mutagen | Non-Toxic | 4.054 | 0.266 | Mild | None |
| 8 | Non-Mutagen | Non-Toxic | 1.528 | 0.213 | Mild | None |
| 10 | Non-Mutagen | Non-Toxic | 18.673 | 0.291 | Mild | None |
| 11a | Non-Mutagen | Non-Toxic | 1.655 | 0.152 | Mild | None |
| 11b | Non-Mutagen | Non-Toxic | 2.675 | 0.391 | Mild | None |
| 12a | Non-Mutagen | Non-Toxic | 3.791 | 0.294 | Mild | None |
| 12b | Non-Mutagen | Non-Toxic | 2.321 | 0.358 | Mild | None |
| 12c | Non-Mutagen | Non-Toxic | 2.98 | 0.139 | Mild | None |
| 13a | Non-Mutagen | Non-Toxic | 32.038 | 0.530 | Mild | None |
| 13b | Non-Mutagen | Non-Toxic | 24.690 | 0.735 | Mild | None |
| Erlotinib | Non-Mutagen | Non-Toxic | 8.057 | 0.083 | Mild | None |
aUnit: mg/kg body weight/day.
bUnit: g/kg body weight.
In general, most of the synthesised compounds showed decreased toxicity potential. In detail, all compounds were predicted to be non-mutagenic and non-toxic against Ames mutagenicity and developmental toxicity potential models. In addition, all compounds were anticipated to be non-irritant and mild irritant against Skin Irritancy and Ocular Irritancy models, respectively. For, compounds 7a, 7b, 10, 13a, and 13b showed carcinogenic potency TD50 values ranging from 18.673 to 34.965 mg/kg body weight/day, which were higher than that of erlotinib (8.057 mg/kg body weight/day). the other compounds showed less carcinogenic potency TD50 values. In addition, the tested compounds showed rat maximum tolerated dose values ranging from 0.139 to 0.735 g/kg body weight. This range is higher than the rat's maximum tolerated dose value of erlotinib (0.083 g/kg body weight).
2.3.4. Molecular dynamic simulations
To study the stability and the binding strength of the protein-compound 12b complex, GROMACS 2021 was used to run a 100 ns classical molecular dynamics simulation, and the trajectory was analysed using VMD. RMSD for the protein alone, compound 12b alone, and the complex (Figure 14(A)), RMSF (Figure 14(B)), SASA (Figure 14(C)), RoG (Figure 14(D)), and the change in the hydrogen bonds for the protein in the protein-compound 12b complex (Figure 14(E)) were calculated. The distance between the centre of mass of protein and the centre of mass of compound 12b (Figure 14(F)) was measured throughout the trajectory.
Figure 14.
The analyses performed on the trajectory using VMD. (A) The RMSD values of the protein only, compound 12 b only, and protein-compound 12 b complex during the trajectory. (B) The RMSF of the amino acids along the whole trajectory. (C) The SASA values of the protein. (D) The radius of gyration of the protein. (E) The change in the numbers of the hydrogen bonds between amino acids of the protein. (F) The change in the distance from the centre of mass of compound 12 b and the protein.


RMSD values show that the system was stable throughout the trajectory with no drastic fluctuation and an average of 2.26 Å for the protein alone. For compound 12b alone, the RMSD showed a stable trend in almost all the trajectory with two exceptions. The duration from 44.5 ns to 54.6 ns and from 86 ns to 91.6 ns show RMSD of values larger than 2 Å. The RMSD of the complex showed a similar trend to the RMSD of the protein only with slightly larger values. In addition, the amino acids fluctuation depicted in the RMSF values showed that most of the amino acids have fluctuations of less than 2 Å except for the C-terminal (around 6 Å) and the loop from E842:P853 reaching a maximum of 3.5 Å. The SASA (average = 15301 Å2), RoG (average = 19.58 Å), and the change in the number of H-bonds (average = 58 bonds) showed that the protein is stable and did not undergo a change in its folded state. The change in the distance between the centre of mass of compound 12b and that of the protein showed a stable binding with an average distance of 9.95 Å.
The binding free energy between the protein and compound 12b was calculated using MM-GBSA implemented in the gmx_MMPBSA tool. Figure 15 showed the values of different energy components produced from MM-GBSA. The predominant type of interaction is the Van Der Waals interactions with an average of −59.14 Kcal/mol followed by electrostatic interactions with an average of −17.22 Kcal/mol. Decomposition analysis was performed to give information on the contribution of amino acids to the binding. Figure 16 showed that L694 (−1.36 Kcal/mol), V702 (−1.96 Kcal/mol), A719 (−1.12 Kcal/mol), K721 (−2.27 Kcal/mol), L764 (−1.19 Kcal/mol), C773 (−1.27 Kcal/mol), L820 (−1.32 Kcal/mol), and T830 (−2.63 Kcal/mol) have a contribution of values stronger (more negative) than −1 Kcal/mol. Only one amino acid (D831) shows a positive contribution to the binding with a value of +1.4 Kcal/mol.
Figure 15.

values of different energy components obtained from MM-GBSA analysis. Bars represent the standard deviation values.
Figure 16.

The decomposition of the free binding energy of amino acids around 10 Å of compound.12b.
The trajectory was clustered using TTClust to obtain the different clusters and a representative frame for each one. To know the different types and numbers of interactions, PLIP was utilised to detect the interactions between compound 12b and the protein in the representative frames for each cluster. Table 10 showed the types and numbers of interactions produced from PLIP. Most of the interactions are hydrophobic with only one amino acid forming a hydrogen bond with the compound 12b. Figure 17 showed the 3D conformations for the complex in representative frames of each cluster.
Table 10.
The number and types of interactions between compound 12 b and EGFR as obtained from PLIP webserver for the representative frame of each cluster. Amino acids in bold are common in all of the clusters representative.
| Cluster number | Number of hydrophobic interactions | Amino acids in receptor | Number of hydrogen bonds | Amino acids in receptor |
|---|---|---|---|---|
| C1 | 9 | L694 - V702 - K721 (2) - L753 - L764 - R817 - L820 - T830 | 1 | T830 |
| C2 | 9 | L694 - V702 - K721 (2) - L764 - T766 - L768 - R817 - T830 | 1 | T830 |
| C3 | 11 | L694 - V702 - K721 - L764 (2) - T766 - L768 - R817 - L820 - T830 - L834 | 1 | T830 |
Figure 17.

The 3 D interaction between compound 12 b and EGFR in each of the representative frame for each cluster. Amino acids are shown as blue sticks. compound 12 b is shown as brown sticks. Grey dashed lines: hydrophobic interaction. Blue solid lines: hydrogen bonds.
3. Conclusion
Twelve 1H-pyrazolo[3,4-d]pyrimidine derivatives having the essential pharmacophoric features of EGFR inhibitors have been designed and synthesised. Four compounds 8, 10, 12a, and 12b showed potent anti-proliferative activities against A549 and HCT-116 cell lines. Compound 12b (IC50 = 8.21 and 19.56 µM) exhibited the highest activity against A549 and HCT-116, respectively. The inhibitory activities of compound 12b against EGFRWT and EGFRT790M were 0.016 and 0.236 µM, respectively compared to erlotinib (IC50 = 0.006 and 0.563 µM, respectively). SAR study revealed that the introduction of aliphatic amines in the 4-position of pyrazolo[3,4-d]pyrimidine scaffold was not beneficial for cytotoxic activity. On the contrary, the introduction of aniline moiety in the same position enhanced the anticancer activity. Additionally, the condensation of hydrazine derivative 10 with p-chloroacetophenone gave better anticancer activity against A549 cells. The effect of compound 12b on cell cycle distribution and apoptosis induction was analysed. Such a compound provoked apoptosis and arrested the cell cycle at S and G2/M phases. Moreover, compound 12b induced a high expression level of BAX and a low expression level of Bcl-2 in A549 cells indicating its apoptotic behaviour. Compound 12b showed low toxicity against WI-38 cell line compared to erlotinib. In addition, it showed a high selectivity against the tumour cell lines. Docking studies suggested that the synthesised compounds have good binding modes against EGFRWT and EGFRT790M crystal structures.
4. Experimental
4.1. Chemistry
4.1.1. General
1H NMR spectra were run at 400 MHz and 13 C NMR spectra were determined at 101 MHz in deuterated dimethyl sulfoxide (DMSO-d6) on a Varian Mercury VX-400 NMR spectrometer. Chemical shifts are given in parts per million (ppm) on the delta (d) scale. Chemical shifts were calibrated relative to those of the solvents. The progress of reactions was monitored with Merck silica gel IB2-F plates (0.25 mm thickness). The infra-red spectra were recorded in potassium bromide discs on Pye Unicam SP 3300 and Shimadzu FT IR 8101 PC infra-red spectrophotometer. Elemental analyses (C, H, N) were performed on a CHN analyser, and all compounds were within ± 0.4 of the theoretical values. Compounds 2, 3, 4, and 5 were prepared according to reported procedures 44–46.
4.1.2. 4-Chloro-N,1-diphenyl-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-6-amine (6)
A solution of 4,6-dichloro-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine 5 (2.65 g, 0.01 mol) and aniline (0.01 mol) in absolute ethanol (20 ml) was heated under reflux for 6 h. Then, the solvent was evaporated under reduced pressure. The resulting precipitate was filtered, dried, and crystallised from absolute ethanol to afford the corresponding target compound 6.
4.1.3. General procedure for the synthesis of compounds 7a,b, 8, and 9
A mixture of 4-chloro-N,1-diphenyl-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-6-amine 6 (0.5 g, 1.54 mmol) and appropriate amines namely, ethylamine, propylamine, aniline, and cyclohexylamine in the presence of TEA (15 ml) was refluxed for 4 h. Then, the solvent was evaporated under reduced pressure. The resulting precipitate was filtered, dried, and crystallised from absolute ethanol to afford the corresponding target compounds 7a,b, 8, and 9, respectively.
4.1.3.1. N4-Ethyl-N6,1-diphenyl-1H-pyrazolo[3,4-d]pyrimidine-4,6-diamine (7a)
White solid, yield: 85%; m.p. 213–215 °C. IR (KBr) cm−1: 3282 (NH), 3028 (CH aromatic), 2924 (CH aliphatic); 1H NMR (400 MHz, DMSO-d6) δ 8.64 (s, 1H), 8.35 (s, 1H), 8.26 − 8.19 (m, 2H), 7.76 (dd, J = 20.5, 6.8 Hz, 3H), 7.56 − 7.48 (m, 2H), 7.35 (t, J = 7.7 Hz, 2H), 7.25 (t, J = 7.4 Hz, 1H), 6.95 (t, J = 7.3 Hz, 1H), 3.68 − 3.57 (m, 2H), 1.26 (t, J = 7.1 Hz, 3H); 13 C NMR (101 MHz, DMSO-d6) δ 157.13, 156.56, 153.52, 144.58, 142.39, 139.64, 129.47(2 C), 129.29 (2 C), 125.27, 121.10, 120.25(2 C), 117.91(2 C), 93.51, 35.73, 15.09; DEPT (DMSO-d6) (ppm)δ 157.13, 129.47, 129.29, 125.27, 121.10, 120.25, 117.91, 35.73, 15.09; (C19H18N6) (M.W. = 331).
4.1.3.2. N4-Propyl-N6,1-diphenyl-1H-pyrazolo[3,4-d]pyrimidine-4,6-diamine (7b)
White solid, Yield: 82%; m.p. 222–224 °C; IR (KBr) cm-1: 3275 (NH), 3099 (CH aromatic), 2927 (CH aliphatic); 1H NMR (400 MHz, DMSO-d6) δ 8.65 (s, 1H), 8.35 (s, 1H), 8.22 (d, J = 8.1 Hz, 2H), 7.74 (dd, J = 17.4, 6.9 Hz, 3H), 7.56 − 7.48 (m, 2H), 7.39 − 7.31 (m, 2H), 7.25 (t, J = 7.4 Hz, 1H), 6.95 (t, J = 7.3 Hz, 1H), 3.54 (dd, J = 7.9, 5.9 Hz, 2H), 1.67 (h, J = 7.4 Hz, 2H), 0.95 (t, J = 7.4 Hz, 3H); 13 C NMR (101 MHz, DMSO-d6) δ 157.12, 156.73, 153.55, 144.58, 142.41, 139.63, 129.47(2 C), 129.30(2 C), 125.28, 121.12, 120.27 (2 C), 117.92 (2 C), 93.51, 42.58, 22.66, 11.88; DEPT (DMSO-d6) (ppm)δ 157.12, 129.47, 129.30, 125.28, 121.12, 120.27, 117.92, 42.58, 22.66, 11.88; Anal. Calc. for: (C20H20N6) (M.W. = 344).
4.1.3.3. N4,N6,1-Triphenyl-1H-pyrazolo[3,4-d]pyrimidine-4,6-diamine (8)
White solid, Yield: 87%; m.p. 255–257 °C. IR (KBr) cm−1: 3387 (NH), 3086 (CH aromatic). 1H NMR (400 MHz, DMSO-d6) δ 9.28 (s, 1H), 8.94 (s, 1H), 8.43 (s, 1H), 8.24 (d, J = 8.1 Hz, 2H), 7.74 (d, J = 8.1 Hz, 2H), 7.68 (d, J = 7.9 Hz, 2H), 7.55 (t, J = 7.8 Hz, 2H), 7.40 (dt, J = 24.4, 7.7 Hz, 4H), 7.29 (t, J = 7.3 Hz, 1H), 7.20 (t, J = 7.4 Hz, 1H), 6.97 (t, J = 7.3 Hz, 1H); 13 C NMR (101 MHz, DMSO-d6) δ 156.78, 155.16, 153.80, 144.40, 142.37, 139.43, 138.87, 129.57 (2 C), 129.36, 129.09 (2 C), 125.61, 124.87, 123.79 (2 C), 121.23, 120.46(2 C), 117.93 (2 C), 94.58; DEPT (DMSO-d6) (ppm) δ 156.78, 129.57, 129.36, 129.09, 125.60, 124.87, 123.79, 121.23, 120.46, 117.93; (C23H18N6) (M.W. = 378).
4.1.3.4. N4-Cyclohexyl-N6,1-diphenyl-1H-pyrazolo[3,4-d]pyrimidine-4,6-diamine (9)
White solid, Yield: 80%; m.p. 235–237 °C. IR (KBr) cm−1: 3294 (NH), 3066 (CH aromatic), 2947 (CH aliphatic); 1H NMR (400 MHz, DMSO-d6) δ 8.84 (s, 1H), 8.35 (s, 1H), 8.22 (d, J = 8.1 Hz, 2H), 7.66 (d, J = 8.0 Hz, 2H), 7.52 (t, J = 7.8 Hz, 2H), 7.35 (t, J = 7.8 Hz, 2H), 7.26 (t, J = 7.4 Hz, 1H), 7.19 (d, J = 7.7 Hz, 1H), 6.96 (t, J = 7.5 Hz, 1H), 4.22 (s, 1H), 2.52 (s, 2H), 1.99 (s, 2H), 1.76 (s, 2H), 1.66 (d, J = 12.9 Hz, 1H), 1.35 (d, J = 9.8 Hz, 2H), 1.19 (s, 1H); 13 C NMR (101 MHz, DMSO-d6) δ 157.12, 155.93, 153.73, 144.52, 142.54, 139.58, 129.48(2 C), 129.40(2 C), 125.36, 121.16, 120.33(2 C), 117.89(2 C), 93.75, 49.89, 32.64(2 C), 25.78, 25.35(2 C); (C23H24N6) (M.W. = 384).
4.1.3.5. 4-Hydrazinyl-N,1-diphenyl-1H-pyrazolo[3,4-d]pyrimidin-6-amine (10)
A mixture of 4-chloro-1,6-diphenyl-1H-pyrazolo[3,4-d]pyrimidine (3.21 g, 0.01 mol) and hydrazine hydrate (99%, 5 ml, 0. 1 mol) was heated under reflux for 4 h. After cooling, the formed solid was collected by filtration, washed with hot ethanol (95%, 10 ml), and crystallised form isopropanol to yield the desired product 10.
4.1.4. General procedure for synthesis of compounds 11a,b, 12a,b, and 13a,b
A mixture of hydrazide derivative 10 (0.31 g, 0.001 mol), appropriate aromatic aldehydes or acetophenones (0.001 mol), and a catalytic amount of glacial acetic acid (0.5 ml) was heated under reflux in absolute ethanol (20 ml) for a specific time. The formed precipitate was filtered and crystallised from ethanol to yield the title compounds 11a,b, 12a,b, and 13a,b.
4.1.4.1. 4–(2-(4-Methoxybenzylidene)hydrazinyl)-N,1-diphenyl-1H-pyrazolo[3,4-d] pyrimidin-6-amine (11a)
White solid, Yield: 80%; m.p. 265–267 °C; IR (KBr) cm−1: 3290 (NH), 3055 (CH aromatic), 2935 (CH aliphatic). 1H NMR (400 MHz, DMSO-d6) δ 12.11 (s, 1H), 9.91 (s, 1H), 8.44 (d, J = 11.5 Hz, 1H), 8.28 (d, J = 8.1 Hz, 1H), 8.14 (d, J = 8.5 Hz, 1H), 7.77 − 7.66 (m, 2H), 7.59 − 7.46 (m, 4H), 7.38 (t, J = 8.0 Hz, 1H), 7.34 − 7.23 (m, 3H), 7.09 − 7.02 (m, 2H), 6.99 − 6.90 (m, 1H), 3.84 (s, 3H); 13 C NMR (101 MHz, DMSO-d6) δ 161.76, 161.27, 149.42, 147.77, 147. 49, 144.81, 139.50, 132, 28, 129.84, 129.73, 129.58, 129.50, 129.24, 128.38, 126.10, 125.45, 121.19, 120.63, 120.44, 118.06, 116.81, 115.17, 114.62, 92.87, 55.95; DEPT (DMSO-d6) (ppm) δ 153.77, 149.42, 129.84, 129.73, 129.50, 129.24, 125.45, 121.19, 120.63, 120.44, 118.06, 116.81, 115.17, 114.62, 55.95; (C25H21N7O) (M.W. = 435).
4.1.4.2. 4–(2-(4-Chlorobenzylidene)hydrazinyl)-N,1-diphenyl-1H-pyrazolo[3,4-d]pyrimidin-6-amine (11b)
White solid, Yield: 78%; m.p. 274–276 °C; IR (KBr) cm−1: 3390 (NH), 3074 (CH aromatic), 2981 (CH aliphatic). 1H NMR (400 MHz, DMSO-d6) δ 12.23 (s, 1H), 9.69 (s, 1H), 8.59 (s, 1H), 8.49 (d, J = 15.0 Hz, 1H), 8.28 (d, J = 8.1 Hz, 1H), 8.20 (s, 1H), 8.14 (d, J = 8.0 Hz, 1H), 8.03 − 7.98 (m, 1H), 7.76 (t, J = 7.1 Hz, 2H), 7.56 (d, J = 7.9 Hz, 3H), 7.40 (dd, J = 14.8, 7.4 Hz, 2H), 7.33 − 7.24 (m, 2H), 6.95 (dt, J = 13.9, 7.3 Hz, 1H); 13 C NMR (101 MHz, DMSO-d6) δ 152.65, 149.38, 148.81, 147.53, 141.08, 140.94, 139.14, 135.58, 134.73, 129.79, 129.75, 129.67, 129.61, 129.54, 129.25, 129.21, 126.10, 121.27, 120.69, 120.51, 118.01, 116.85, 93.37, 93.13; DEPT (DMSO-d6) (ppm) δ 152.65, 149.39, 129.75, 129.67, 129.61, 129.54, 129.25, 126.10, 125.57, 121.27, 121.07, 120.69, 120.51, 118.00, 116.85. (C24H18ClN7) (M.W. = 439).
4.1.4.3. N,1-Diphenyl-4–(2-(1-phenylethylidene)hydrazinyl)-1H-pyrazolo[3,4-d]pyrimidin-6-amine (12a)
White solid, Yield: 70%; m.p. 244–246 °C; IR (KBr) cm−1: 3402 (NH), 3032 (CH aromatic), 2982 (CH aliphatic). 1H NMR (400 MHz, DMSO-d6) δ 11.72 (s, 1H), 10.46 (s, 1H), 9.98 (s, 1H), 8.37 (d, J = 28.7 Hz, 1H), 8.29 − 8.20 (m, 2H), 8.11 (dd, J = 21.8, 7.7 Hz, 2H), 7.97 (s, 1H), 7.73 (s, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.52 (t, J = 8.0 Hz, 3H), 7.39 − 7.35 (m, 1H), 7.29 − 7.24 (m, 1H), 7.00 − 6.94 (m, 1H), 6.77 (d, J = 8.3 Hz, 1H), 1.93 (s, 3H); 13 C NMR (101 MHz, DMSO-d6) δ 172.49, 156.24, 149.42, 148.00, 144.57, 141.42, 141.01, 139.13, 130.18, 129.57, 128.81, 128.60, 127.77, 127.14, 125.89, 125.25, 120.93, 120.55, 120.31, 117.22, 117.19, 117.13, 116.57, 93.39, 21.52; (C25H21N7) (M.W. = 419).
4.1.4.4. 4–(2-(1–(4-Chlorophenyl)ethylidene)hydrazinyl)-N,1-diphen yl-1H-pyrazolo[3,4-d]pyrimidin-6-amine (12b)
White solid; Yield: 80%; m.p. 248–250 °C; IR (KBr) cm−1: 3387 (NH), 3098 (CH aromatic), 2961 (CH aliphatic); 1H NMR (400 MHz, DMSO-d6) δ 12.06 (s, 1H), 9.76 (s, 1H), 8.47 (d, J = 13.8 Hz, 1H), 8.23 (s, 1H), 8.14 (dd, J = 8.2, 5.6 Hz, 2H), 8.02 (s, 1H), 7.78 (d, J = 8.1 Hz, 1H), 7.66 (d, J = 8.0 Hz, 1H), 7.53 (dd, J = 21.4, 8.2 Hz, 3H), 7.39 (t, J = 7.7 Hz, 1H), 7.32 (t, J = 7.6 Hz, 1H), 7.05 (t, J = 7.8 Hz, 1H), 6.97 (t, J = 7.3 Hz, 1H), 6.80 (d, J = 8.2 Hz, 1H), 2.52 (s, 3H); 13 C NMR (101 MHz, DMSO-d6) δ 156.80, 149.51, 148.70, 147.63, 147.55, 140.99, 139.19, 137.82, 134.32, 130.57, 129.71, 129.63, 129.60, 129.52, 129.26, 128.89, 128.79, 128.63, 126.04, 121.06, 120.60, 117.19, 116.62, 94.07, 14.25; DEPT (DMSO-d6) (ppm) δ 149.51, 130.57, 129.71, 129.60, 129.52, 129.26, 128.89, 128.79, 128.63, 126.04, 121.05, 120.73, 120.60, 117.19, 116.62, 14.24; (C25H20ClN7) (M.W. = 453).
4.1.4.5. N,1-Diphenyl-4–(2-(1-(p-tolyl)ethylidene)hydrazinyl)-1H-pyrazolo [3,4-d] pyrimidin-6-amine (12c)
White solid, Yield: 85%; m.p. 251–253 °C; IR (KBr) cm−1: 3367 (NH), 3047 (CH aromatic), 2968 (CH aliphatic). 1H NMR (400 MHz, DMSO-d6) δ 11.87 (s, 1H), 10.50 (s, 1H), 8.38 (s, 1H), 8.28 (d, J = 8.0 Hz, 2H), 7.76 (d, J = 6.8 Hz, 3H), 7.55 (t, J = 7.4 Hz, 3H), 7.39 (t, J = 7.1 Hz, 3H), 7.28 (t, J = 7.3 Hz, 2H), 6.96 (d, J = 7.4 Hz, 1H), 2.31 − 2.12 (m, 6H); 13 C NMR (101 MHz, DMSO-d6) δ 156.36, 154.03, 144.62, 144.58, 141.40, 139.53, 136.31, 129.67, 129.60, 129.50, 129.27, 128.80, 128.71, 125.38, 121.04, 212.55, 121.04, 120.45, 120.55, 120.37, 117.58, 117.46, 117.24, 92.34, 18.87, 18.49; DEPT (DMSO-d6) (ppm) δ 129.67, 129.60, 129.50, 129.20, 125.39, 121.55, 121.04, 120.37, 117.58, 117.51, 117.24, 18.87, 18.49; (C26H23N7) (M.W. = 433).
4.1.4.6. N-Ethyl-2–(1-phenyl-6-(phenylamino)-1H-pyrazolo[3,4-d]pyrimidin-4-yl) hydrazine-1-carbothioamide (13a)
Yellowish solid, Yield: 72%; m.p. 250–252 °C. IR (KBr) cm−1: 3250 (NH), 3060 (CH aromatic), 2950 (CH aliphatic). 1H NMR (300 MHz, DMSO-d6) δ 9.89 (s, 1H), 9.65 (s, 1H), 8.50 (s, 1H), 8.28 − 8.17 (m, 2H), 8.10 (dd, J = 12.6, 6.2 Hz, 1H), 7.85 (s, 1H), 7.58 − 7.51 (m, 3H), 7.36 − 7.27 (m, 3H), 7.03 − 6.85 (m, 2H), 3.50 (m, 2H), 1.24 − 1.12 (t, 3H); (C20H20N8S) (M.W = 404).
4.1.4.7. 2–(1-Phenyl-6-(phenylamino)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)-N-propyl hydrazine-1-carbothioamide (13b)
Yellowish solid, yield: 70%; m.p. 244–246 °C. IR (KBr) cm−1: 3256 (NH), 3035 (CH aromatic), 2934 (CH aliphatic). 1H NMR (300 MHz, DMSO-d6) δ 9.65 (s, 1H), 8.51 (s, 1H), 8.41 (s, 1H), 8.25 (d, J = 8.4 Hz, 1H), 8.14 − 8.04 (m, 1H), 7.89 − 7.81 (m, 1H), 7.76 (d, J = 6.0 Hz, 1H), 7.55 (dt, J = 13.8, 6.8 Hz, 3H), 7.42 − 7.25 (m, 4H), 7.01 − 6.88 (m, 1H), 3.62 − 3.38 (t, 2H), 1.69 − 1.43 (m, 2H), 0.97 − 0.71 (t, 3H); (C21H22N8S) (M.W = 418).
4.2. Biological evaluation
4.2.1. In vitro cytotoxic activity
In vitro cytotoxicity was carried out for the synthesised compounds against A549, HCT-116, and WI-38 cell lines using the MTT assay protocol52–55 as described in Supplementary data.
4.2.2. In vitro EGFR kinase assay
In vitro EGFR inhibitory activity was assessed using a Homogeneous time-resolved fluorescence (HTRF) assay56 as described in Supplementary data.
4.2.3. Cell cycle analysis
The effect of compound 12b on cell cycle distribution was performed using the propidium iodide (PI) staining technique as described in Supplementary data56–58.
4.2.4. Apoptosis analysis
The effect of compound 12b on cell apoptosis was investigated as described in Supplementary data59.
4.2.5. Quantitative Real-Time Reverse-Transcriptase PCR (qRT-PCR) technique
The effect of compound 12b on the expression of BAX and Bcl‐2 was determined using qRT-PCR as described in Supplementary data60–62.
4.3. In silico studies
4.3.1. Docking studies
Molecular docking studies of the synthesised compounds were carried out against EGFRWT (PDB ID: 4HJO, resolution 2.75 Å and EGFRT790M (PDB ID: 3W2O, resolution 2.35 Å) as described in Supplementary data38.
4.3.2. ADMET studies
ADMET descriptors were determined using Discovery studio 4.0 according to the reported method63,64 (Supplementary data).
4.3.3. Toxicity studies
The toxicity parameters of the synthesised compounds were calculated using Discovery studio 4.065,66 as described in Supplementary data.
4.3.4. M D Simulations and MM-GBSA
CHARMM-GUI web server was employed and GROMACS 2021 was used as an MD engine as outlined thoroughly in Supplementary data. The Gmx_MMPBSA package was used as outlined thoroughly in Supplementary data67–70.
Supplementary Material
Funding Statement
This paper is based upon work supported by Science, Technology & Innovation Funding Authority (STIFA) under grant number 43327.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA 2021;71:209–49. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Fuchs HE, Jemal A.. Ahmedin Jemal, Cancer statistics, 2022. CA 2022;72:7–33. [DOI] [PubMed] [Google Scholar]
- 3.Ogunbiyi JO, Stefan DC, Rebbeck TR.. African Organization for Research and Training in Cancer: position and vision for cancer research on the African Continent. Infect Agents Cancer 2016;11:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Michor F, Iwasa Y, Nowak MA.. Dynamics of cancer progression. Nat Rev Cancer 2004;4:197–205. [DOI] [PubMed] [Google Scholar]
- 5.Quail DF, Joyce JA.. Microenvironmental regulation of tumor progression and metastasis. Nature Medicine 2013;19:1423–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eastern Cooperative Oncology Group, Schiller D, Harrington CP, Belani C, et al. Comparison of four chemotherapy regimens for advanced non–small-cell lung cancer. N Engl J Med 2002;346:92–8. [DOI] [PubMed] [Google Scholar]
- 7.Mongre RK, Mishra CB, Shukla AK, et al. Emerging importance of tyrosine kinase inhibitors against cancer: quo vadis to cure? Int J Molecul Sc 2021;22:11659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2000;103:211–25. [DOI] [PubMed] [Google Scholar]
- 9.Metibemu DS, Akinloye OA, Akamo AJ, et al. Exploring receptor tyrosine kinases-inhibitors in Cancer treatments. Egyptian J Med Human Genet 2019;20:1–16. [Google Scholar]
- 10.Yamaoka T, Kusumoto S, Ando K, et al. Receptor tyrosine kinase-targeted cancer therapy. Int J Molecul Sci 2018;19:3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butti R, Das S, Gunasekaran VP, et al. Receptor tyrosine kinases (RTKs) in breast cancer: signaling, therapeutic implications and challenges. Molecular Cancer 2018;17:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olayioye MA, Neve RM, Lane HA, Hynes NE.. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J 2000;19:3159–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Castro Barbosa ML, Lima LM, Tesch R, et al. Novel 2-chloro-4-anilino-quinazoline derivatives as EGFR and VEGFR-2 dual inhibitors. Eur J Med Chem 2014;71:1–14. [DOI] [PubMed] [Google Scholar]
- 14.Chang J, Ren H, Zhao M, et al. Development of a series of novel 4-anlinoquinazoline derivatives possessing quinazoline skeleton: design, synthesis, EGFR kinase inhibitory efficacy, and evaluation of anticancer activities in vitro. Eur J Med Chem 2017;138:669–88. [DOI] [PubMed] [Google Scholar]
- 15.Chen Y, Wu J, Wang A, et al. Discovery of N-(5-((5-chloro-4-((2-(isopropylsulfonyl) phenyl) amino) pyrimidin-2-yl) amino)-4-methoxy-2-(4-methyl-1, 4-diazepan-1-yl) phenyl) acrylamide (CHMFL-ALK/EGFR-050) as a potent ALK/EGFR dual kinase inhibitor capable of overcoming a variety of ALK/EGFR associated drug resistant mutants in NSCLC. Eur J Med Chem 2017;139:674–97. [DOI] [PubMed] [Google Scholar]
- 16.Nicholson RI, Gee JM, Harper ME.. EGFR and cancer prognosis. Eur J Cancer 2001;37:9–15. [DOI] [PubMed] [Google Scholar]
- 17.Zhang H-Q, Gong F-H, Ye J-Q, et al. Design and discovery of 4-anilinoquinazoline-urea derivatives as dual TK inhibitors of EGFR and VEGFR-2. Eur J Med Chem 2017;125:245–54. [DOI] [PubMed] [Google Scholar]
- 18.Song Z, Huang S, Yu H, et al. Synthesis and biological evaluation of morpholine-substituted diphenylpyrimidine derivatives (Mor-DPPYs) as potent EGFR T790M inhibitors with improved activity toward the gefitinib-resistant non-small cell lung cancers (NSCLC). Eur J Med Chem 2017;133:329–39. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Chen L, Xu H, et al. 6, 7-Dimorpholinoalkoxy quinazoline derivatives as potent EGFR inhibitors with enhanced antiproliferative activities against tumor cells. Eur J Med Chem 2018;147:77–89. [DOI] [PubMed] [Google Scholar]
- 20.Abdelsalam EA, Zaghary WA, Amin KM, et al. Synthesis and in vitro anticancer evaluation of some fused indazoles, quinazolines and quinolines as potential EGFR inhibitors. Bioorganic Chemistry 2019;89:102985. [DOI] [PubMed] [Google Scholar]
- 21.Ismail RS, Abou-Seri SM, Eldehna WM, et al. Novel series of 6-(2-substitutedacetamido)-4-anilinoquinazolines as EGFR-ERK signal transduction inhibitors in MCF-7 breast cancer cells. Eur J Med Chem 2018;155:782–96. [DOI] [PubMed] [Google Scholar]
- 22.Bonomi P. Erlotinib: a new therapeutic approach for non-small cell lung cancer. Expert Opin Investigat Drugs 2003;12:1395–401. [DOI] [PubMed] [Google Scholar]
- 23.Celik T, Kosker M.. Ocular side effects and trichomegaly of eyelashes induced by erlotinib: a case report and review of the literature. Contact Lens Anterior Eye 2015;38:59–60. [DOI] [PubMed] [Google Scholar]
- 24.Muhsin M, Graham J, Kirkpatrick P.. Gefitinib. Berlin, Germany: Nature Publishing Group; 2003. [Google Scholar]
- 25.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005;2:e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.U.S. Food & Drug Administration. FDA approves neratinib for extended adjuvant treatment of early stage HER2-positive breast cancer. 2017. [DOI] [PubMed] [Google Scholar]
- 27.Sequist LV, Besse B, Lynch TJ, et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of a phase II trial in patients with advanced non–small-cell lung cancer. J Clin Oncol 2010;28:3076–83. [DOI] [PubMed] [Google Scholar]
- 28.Kim Y, Ko J, Cui Z, et al. The EGFR T790M mutation in acquired resistance to an irreversible second-generation EGFR inhibitor. Mol Cancer Ther 2012;11:784–91. [DOI] [PubMed] [Google Scholar]
- 29.Jänne PA, Yang JC-H, Kim D-W, et al. AZD9291 in EGFR inhibitor–resistant non–small-cell lung cancer. N Engl J Med 2015;372:1689–99. [DOI] [PubMed] [Google Scholar]
- 30.Carroll J. Following lethal tox report, Boehringer scraps plans for high-speed development, kills $730M Hanmi deal. 2019. https://endpts.com/following-lethal-tox-report-boehringer-scraps-plans-for-high-speed-development-kills-730m-hanmi-deal/
- 31.Zhao Z, Wu H, Wang L, et al. Exploration of type II binding mode: a privileged approach for kinase inhibitor focused drug discovery? ACS Chem Biol 2014;9:1230–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mowafy S, Galanis A, Doctor ZM, et al. Toward discovery of mutant EGFR inhibitors; Design, synthesis and in vitro biological evaluation of potent 4-arylamino-6-ureido and thioureido-quinazoline derivatives. Biorg. Med. Chem 2016;24:3501–12. [DOI] [PubMed] [Google Scholar]
- 33.Furet P, Caravatti G, Lydon N, et al. Modelling study of protein kinase inhibitors: binding mode of staurosporine and origin of the selectivity of CGP 52411. J. Comput. Aided Mol. Des 1995;9:465–72. [DOI] [PubMed] [Google Scholar]
- 34.Gandin V, Ferrarese A, Dalla Via M, et al. Targeting kinases with anilinopyrimidines: discovery of N-phenyl-N’-[4-(pyrimidin-4-ylamino) phenyl] urea derivatives as selective inhibitors of class III receptor tyrosine kinase subfamily. Scientific Reports 2015;5:16750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y, Gray NS.. Rational design of inhibitors that bind to inactive kinase conformations. Nature Chem Biol 2006;2:358–64. [DOI] [PubMed] [Google Scholar]
- 36.Ismail NS, Ali EM, Ibrahim DA, et al. Pyrazolo [3, 4-d] pyrimidine based scaffold derivatives targeting kinases as anticancer agents. Future J Pharmaceut Sci 2016;2:20–30. [Google Scholar]
- 37.Traxler P, Bold G, Frei J, et al. Use of a pharmacophore model for the design of EGF-R tyrosine kinase inhibitors: 4-(phenylamino) pyrazolo [3, 4-d] pyrimidines. J. Med. Chem 1997;40:3601–16. [DOI] [PubMed] [Google Scholar]
- 38.Gaber AA, Bayoumi AH, El-Morsy AM, et al. Design, synthesis and anticancer evaluation of 1H-pyrazolo [3, 4-d] pyrimidine derivatives as potent EGFRWT and EGFRT790M inhibitors and apoptosis inducers. Bioorg Chem 2018;80:375–95. [DOI] [PubMed] [Google Scholar]
- 39.Abbas SES, Aly EI, Awadallah FM, Mahmoud WR.. 4‐Substituted‐1‐phenyl‐1H‐pyrazolo [3, 4‐d] pyrimidine derivatives: design, synthesis, antitumor and EGFR tyrosine kinase inhibitory activity. Chem Biol Drug Design 2015;85:608–22. [DOI] [PubMed] [Google Scholar]
- 40.Abbas SES, Aly EI, Awadallah FM, Mahmoud WR.. Design, 4‐substituted‐1‐phenyl‐1H‐pyrazolo [3, 4‐d] pyrimidine derivatives: design. Synthesis, Antitumor EGFR Tyrosine Kinase Inhibitory Activity. Chem Biol Drug Des 2015;85:608–22. [DOI] [PubMed] [Google Scholar]
- 41.Li R, Tang H, Fu H, et al. Arynes double bond insertion/nucleophilic addition with vinylogous amides and carbodiimides. J Org Chem 2014;79:1344–55. [DOI] [PubMed] [Google Scholar]
- 42.Sharma VK, Nandekar PP, Sangamwar A, et al. Structure guided design and binding analysis of EGFR inhibiting analogues of erlotinib and AEE788 using ensemble docking, molecular dynamics and MM-GBSA. RSC Adv 2016;6:65725–35. [Google Scholar]
- 43.Traxler P, Furet P.. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol Ther 1999;82:195–206. [DOI] [PubMed] [Google Scholar]
- 44.Schenone S, Radi M, Musumeci F, et al. Biologically driven synthesis of pyrazolo [3, 4-d] pyrimidines as protein kinase inhibitors: an old scaffold as a new tool for medicinal chemistry and chemical biology studies. Chem. Rev 2014;114:7189–238. [DOI] [PubMed] [Google Scholar]
- 45.He H-Y, Zhao J-N, Jia R, et al. Novel pyrazolo [3, 4-d] pyrimidine derivatives as potential antitumor agents: exploratory synthesis, preliminary structure-activity relationships, and in vitro biological evaluation. Molecules 2011;16:10685–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng C, ROBINS RK.. Potential Purine Antagonists. XII. Synthesis of 1-Alkyl (aryl)-4, 6-disubstituted Pyrazolo [3, 4-d] pyrimidines1. J Org Chem 1958;23:852–61. [Google Scholar]
- 47.Ibrahim M, Taghour M, Metwaly A, et al. Design, synthesis, molecular modeling and anti-proliferative evaluation of novel quinoxaline derivatives as potential DNA intercalators and topoisomerase II inhibitors. Eur J Med Chem 2018;155:117–34. [DOI] [PubMed] [Google Scholar]
- 48.Yang J, Qu X, Russell P, Goldstein D.. Regulation of epidermal growth factor receptor in human colon cancer cell lines by interferon α. Gut 2004;53:123–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qian Y, Qiu M, Wu Q, et al. Enhanced cytotoxic activity of cetuximab in EGFR-positive lung cancer by conjugating with gold nanoparticles. Scientific Reports 2014;4:7490–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park JH, Liu Y, Lemmon MA, Radhakrishnan R.. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem J 2012;448:417–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sogabe S, Kawakita Y, Igaki S, et al. Structure-based approach for the discovery of pyrrolo [3, 2-d] pyrimidine-based EGFR T790M/L858R mutant inhibitors. ACS Med Chem Lett 2013;4:201–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63. [DOI] [PubMed] [Google Scholar]
- 53.Denizot F, Lang R.. Rapid colorimetric assay for cell growth and survival: modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 1986;89:271–7. [DOI] [PubMed] [Google Scholar]
- 54.Thabrew MI, Hughes RD, McFarlane IG.. Screening of hepatoprotective plant components using a HepG2 cell cytotoxicity assay. J Pharm Pharmacol 2011;49:1132–5. [DOI] [PubMed] [Google Scholar]
- 55.Al-Rashood ST, Hamed AR, Hassan GS, et al. Antitumor properties of certain spirooxindoles towards hepatocellular carcinoma endowed with antioxidant activity. J Enzyme Inhibit Med Chem 2020;35:831–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eldehna WM, Hassan GS, Al-Rashood ST, et al. Synthesis and in vitro anticancer activity of certain novel 1-(2-methyl-6-arylpyridin-3-yl)-3-phenylureas as apoptosis-inducing agents. J Enzyme Inhib Med Chem 2019;34:322–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sabt A, Abdelhafez OM, El-Haggar RS, et al. Novel coumarin-6-sulfonamides as apoptotic anti-proliferative agents: synthesis, in vitro biological evaluation, and QSAR studies. J Enzyme Inhib Med Chem 2018;33:1095–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang J, Lenardo MJ.. Roles of caspases in apoptosis, development, and cytokine maturation revealed by homozygous gene deficiencies. J Cell Sci 2000;113:753–7. [DOI] [PubMed] [Google Scholar]
- 59.Al-Sanea MM, Al-Ansary GH, Elsayed ZM, et al. Development of 3-methyl/3-(morpholinomethyl) benzofuran derivatives as novel antitumor agents towards non-small cell lung cancer cells. J Enzyme Inhib Med Chem 2021;36:987–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Balah A, Ezzat O, Akool E-S.. Vitamin E inhibits cyclosporin A-induced CTGF and TIMP-1 expression by repressing ROS-mediated activation of TGF-β/Smad signaling pathway in rat liver. Int Immunopharmacol 2018;65:493–502. [DOI] [PubMed] [Google Scholar]
- 61.Aborehab NM, Elnagar MR, Waly NE.. Gallic acid potentiates the apoptotic effect of paclitaxel and carboplatin via overexpression of Bax and P53 on the MCF‐7 human breast cancer cell line. J Biochem Mol Toxicol 2021;35:e22638. [DOI] [PubMed] [Google Scholar]
- 62.Elnagar MR, Walls AB, Helal GK, et al. Functional characterization of α7 nicotinic acetylcholine and NMDA receptor signaling in SH-SY5Y neuroblastoma cells in an ERK phosphorylation assay. Eur J Pharmacol 2018;826:106–13. [DOI] [PubMed] [Google Scholar]
- 63.El-Adl K, Ibrahim M-K, Alesawy MS, Eissa IH.. [1, 2, 4] Triazolo [4, 3-c] quinazoline and bis ([1, 2, 4] triazolo)[4, 3-a: 4′, 3′-c] quinazoline derived DNA intercalators: design, synthesis, in silico ADMET profile, molecular docking and anti-proliferative evaluation studies. Bioorg Med Chem 2021;30:115958. [DOI] [PubMed] [Google Scholar]
- 64.Alanazi MM, Mahdy HA, Alsaif NA, et al. New bis ([1, 2, 4] triazolo)[4, 3-a: 3′, 4′-c] quinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers: design, synthesis, in silico studies, and anticancer evaluation. Bioorg Chem 2021;112:104949. [DOI] [PubMed] [Google Scholar]
- 65.El-Metwally SA, Abou-El-Regal MM, Eissa IH, et al. Discovery of thieno [2, 3-d] pyrimidine-based derivatives as potent VEGFR-2 kinase inhibitors and anti-cancer agents. Bioorg Chem 2021;112:104947. [DOI] [PubMed] [Google Scholar]
- 66.Parmar DR, Soni JY, Guduru R, et al. Discovery of new anticancer thiourea-azetidine hybrids: design, synthesis, in vitro antiproliferative, SAR, in silico molecular docking against VEGFR-2, ADMET, toxicity, and DFT studies. Bioorg Chem 2021;115:105206. [DOI] [PubMed] [Google Scholar]
- 67.Suleimen YM, Jose RA, Suleimen RN, et al. Isolation and in silico anti-SARS-CoV-2 papain-like protease potentialities of two rare 2-phenoxychromone derivatives from Artemisia spp. Molecules 2022;27:1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jo S, Kim T, Iyer VG, Im W.. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Computat Chem 2008;29:1859–65. [DOI] [PubMed] [Google Scholar]
- 69.Brooks BR, Brooks CL, III, Mackerell AD, Jr, et al. CHARMM: the biomolecular simulation program. J Computat Chem 2009;30:1545–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee J, Cheng X, Swails JM, et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J Chem Theory Comput 2016;12:405–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.