

Abstract
Our approach in predicting gene expression levels relates to codon usage differences among gene classes. In prokaryotic genomes, genes that deviate strongly in codon usage from the average gene but are sufficiently similar in codon usage to ribosomal protein genes, to translation and transcription processing factors, and to chaperone-degradation proteins are predicted highly expressed (PHX). By these criteria, PHX genes in most prokaryotic genomes include those encoding ribosomal proteins, translation and transcription processing factors, and chaperone proteins and genes of principal energy metabolism. In particular, for the fast-growing species Escherichia coli, Vibrio cholerae, Bacillus subtilis, and Haemophilus influenzae, major glycolysis and tricarboxylic acid cycle genes are PHX. In Synechocystis, prime genes of photosynthesis are PHX, and in methanogens, PHX genes include those essential for methanogenesis. Overall, the three protein families—ribosomal proteins, protein synthesis factors, and chaperone complexes—are needed at many stages of the life cycle, and apparently bacteria have evolved codon usage to maintain appropriate growth, stability, and plasticity. New interpretations of the capacity of Deinococcus radiodurans for resistance to high doses of ionizing radiation is based on an excess of PHX chaperone-degradation genes and detoxification genes. Expression levels of selected classes of genes, including those for flagella, electron transport, detoxification, histidine kinases, and others, are analyzed. Flagellar PHX genes are conspicuous among spirochete genomes. PHX genes are positively correlated with strong Shine-Dalgarno signal sequences. Specific regulatory proteins, e.g., two-component sensor proteins, are rarely PHX. Genes involved in pathways for the synthesis of vitamins record low predicted expression levels. Several distinctive PHX genes of the available complete prokaryotic genomes are highlighted. Relationships of PHX genes with stoichiometry, multifunctionality, and operon structures are discussed. Our methodology may be used complementary to experimental expression analysis.
Gene expression and protein abundances in prokaryotes are regulated at several levels: (i) initiation of transcription, promoter strength, promoter configuration, and transcription factors; (ii) transcription termination, mRNA stability, and turnover rates; (iii) codon usage; (iv) translation initiation and elongation; and (v) protein folding, degradation, and cellular localization. An accounting of high gene expression in prokaryotic genomes generally focuses on at least one of three criteria: (i) The gene possesses a potent promoter sequence sometimes associated with bent DNA and/or specific binding factors. However, the characterization of regulatory cis elements underlying gene transcription is largely an unresolved problem. (ii) The gene possesses a strong Shine-Dalgarno (SD) ribosome binding sequence, but recognition of SD sequences is not discriminating (10, 14, 34, 51, 53) (see also below). (c) The gene exhibits favorable codon usage; in rapidly dividing bacteria, this largely corresponds to the prevalent codon usage frequencies of ribosomal protein (RP) genes (20, 44, 54). Our approach to ascertaining gene expression levels relates to codon usage differences among gene classes. We show data suggesting that codon usage contributes importantly to setting the level of expression of the gene. Our data support the proposition that each genome has evolved a codon usage pattern accommodating “optimal” gene expression levels for most situations of its habitat, energy sources, and life style.
Gene codon preferences vary considerably within and between organisms (for reviews and perspectives, see references 25, 29, and 55). Variations in tRNA availabilities are interpreted by several authors as an important factor in generating codon biases of the “highly expressed genes” of yeast and Escherichia coli (24, 29, 32, 54, 55). Translational accuracy and efficiency and codon-anticodon interaction strength may contribute to codon choices (1, 38). Selective and nonselective substitutional biases operating during DNA replication, transcription, and repair also play key roles. Gene codon usages to some extent correlate with functional categories (29, 32), as exemplified by polypeptide synthesis and chaperone-degradation activities. Other factors that may influence codon choices include methylation effects of DNA, mRNA stability, tissue and cellular location, codon context, and species of origin (30).
It is generally recognized that in most prokaryotic genomes during exponential growth, RPs and translation and transcription processing factors (TF) are highly expressed. The major chaperone-degradation (CH) genes functioning in protein folding, trafficking, and secretion are also largely highly expressed (e.g., data in reference 65). The three classes RP, CH, and TF are consistent in that they record congruent high codon biases relative to the average gene, whereas the codon usage differences among these three gene classes are low. Specifically, for rapid division, many ribosomes are indispensable, augmented by abundant TF and CH proteins needed to assure properly translated, modified, and folded protein products. These proteins expedite and regulate cellular activities. From this perspective, we have used the three classes RP, CH, and TF as representative classes of highly expressed genes.
A gene is predicted highly expressed (PHX) if the gene has codon frequencies similar to the codon frequencies of the RP, TF, and CH genes but deviates significantly in codon usage from the average gene of the genome (see “definition I” below for precision). PHX genes in most prokaryotic genomes include, in addition to those for RP, TF, and CH proteins, the principal genes of energy metabolism and key genes involved in amino acid, nucleotide, and fatty acid biosyntheses. In the Synechocystis genome, many genes important in photosynthesis, respiration, and glycolysis are PHX, and among methanogens, those genes essential for methanogenesis are PHX.
MATERIALS AND METHODS
Data.
PHX genes are identified across the 22 complete prokaryotic genomes listed in Table 1. Information on genome sizes, doubling times, and life styles (e.g., parasite versus free living or extremeophilic versus mesophilic) are indicated where the information is available.
TABLE 1.
Statistics for highly expressed genes in diverse prokaryotic genomes
| Genome (doubling time) | Length (kb) | No. of genes ≥100 aa | No. (%) PHX | Max E(g) | Reference |
|---|---|---|---|---|---|
| Eubacteria | |||||
| Fast growing (<1 h) | |||||
| E. coli | 4,639 | 3,898 | 306 (8) | 2.66 | 6 |
| H. influenzae | 1,830 | 1,529 | 142 (9) | 2.01 | 15 |
| B. subtilis | 4,215 | 3,612 | 148 (4) | 2.34 | 37 |
| V. cholerae | 4,036 | 3,253 | 172 (5) | 2.25 | |
| V. cholorae long chromosome | 2,963 | 2,393 | 158 (7) | 2.25 | |
| Moderately fast (90 min) | |||||
| D. radiodurans | 3,284 | 2,923 | 337 (12) | 2.56 | 69 |
| D. radiodurans long chromosome | 2,649 | 2,421 | 307 (13) | 2.56 | |
| Obligate intracellular parasites | |||||
| R. prowazekii (10 h) | 1,112 | 770 | 42 (5) | 1.20 | 2 |
| C. trachomatis (3 h) | 1,043 | 829 | 52 (6) | 1.27 | 59 |
| C. pneumoniae (3 h) | 1,230 | 963 | 85 (9) | 1.38 | 26 |
| Surface parasites | |||||
| M. genitalium | 580 | 446 | 27 (6) | 1.25 | 17 |
| M. pneumoniae | 816 | 654 | 57 (9) | 1.36 | 23 |
| Spirochetes | |||||
| B. burgdorferi (11–12 h) | 1,231 | 1,007 | 76 (8) | 1.29 | 16 |
| T. pallidum (33 h) | 1,138 | 916 | 99 (11) | 1.35 | 18 |
| Pathogens | |||||
| H. pylori 26695 (4–12 h) | 1,668 | 1,388 | 73 (5) | 1.21 | 63 |
| M. tuberculosis (24 h) | 4,412 | 3,660 | 569 (16) | 1.68 | 9 |
| Cyanobacteria Synechocystis (6–18 h) | 3,573 | 2,896 | 380 (13) | 1.51 | 27 |
| Deep-branching gram-negative thermophiles | |||||
| A. aeolicus | 1,551 | 1,481 | 233 (16) | 1.62 | 11 |
| T. maritima | 1,861 | 1,681 | 175 (10) | 1.40 | 49 |
| Archaea | |||||
| Methanogens | |||||
| M. jannaschii (10 h) | 1,665 | 1,466 | 114 (8) | 1.59 | 7 |
| M. thermoautotrophicum (4 h) | 1,751 | 1,640 | 160 (10) | 1.38 | 57 |
| A. fulgidus (4 h) | 2,178 | 2,077 | 343 (17) | 1.49 | 34 |
| Hyperthermophiles | |||||
| P. horikoshii | 1,739 | 1,992 | 179 (9) | 1.47 | 33 |
| P. abyssi | 1,765 | 1,683 | 242 (14) | 1.60 |
Codon usage differences between gene classes.
We previously introduced a versatile way of assessing the codon biases of one group of genes (or a single gene) relative to a second group of genes (29, 32). Let G be a group of genes with average codon frequencies g(x, y, z) for the codon triplet (x, y, z) normalized for each amino acid codon family such that Σ(x,y,z) = ag(x,y,z) = 1, where the sum extends over all codons (x,y,z) translated to amino acid a. Let f(x,y,z) indicate the average codon frequencies for the gene group F, normalized to 1 in each amino acid codon family. The codon usage difference of the gene family F relative to the gene family G (codon bias relative to G) is calculated by the formula
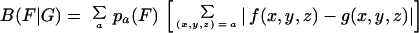 |
1 |
where {pa(F)} are the average amino acid frequencies of the genes of F. When no ambiguity is likely, we refer to B(F|G) as the codon bias of F with respect to G. The assessments implied by equation 1 can be made for any two gene groups from the same genome or from different genomes. Equation 1 can also be applied to a subset of amino acids (e.g., restricted to hydrophobic, charged, or aromatic types).
Measures of gene expression.
Let B(g|S), as above, denote the codon usage difference of the gene g relative to the gene class S as formalized in equation 1. The following gene classes are paramount: C, all protein genes; RP genes; CH genes; and TF genes. Qualitatively, a gene g is PHX if B(g|C) is high while B(g|RP), B(g|CH), and B(g|TF) are low. Predicted expression levels with respect to individual standards are based on the ratios
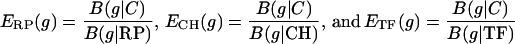 |
2 |
Combined, these produce the general expression measure
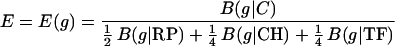 |
3 |
Other weighted combinations can also be used. For any class of genes S, a measure ES(g) for expression level of a gene g relative to S is calculated by B(g|C)/B(g|S).
Definition I.
A gene is PHX if the following two conditions are satisfied: at least two among the three expression values ERP(g), ECH(g), and ETF (g) exceed 1.05, and the general expression level E(g) is ≥1.00.
It is instructive to plot B(g|C) versus B(g|RP), B(g|TF), or B(g|CH) for all individual genes g (encoding proteins of ≥100-amino-acid [aa] length). This is done in Fig. 1 for the E. coli genome and for the Deinococcus radiodurans genome. The distribution of points reveals two horns. The upper left horn corresponds to the PHX genes, and the upper right horn is designated “alien” genes. Alien genes consist mostly of open reading frames (ORFs) of unknown function but also include genes encoding transposases, cryptic prophage sequences, and restriction or modification enzymes, which are often conjugatively transferred via plasmids. Other examples of alien genes in several genomes are genes associated with lipopolysaccharide biosynthesis and fimbrial-gene-like genes (29, 32, 47). The term “alien” was chosen because such genes with high codon bias might have been acquired through recent lateral gene transfer (39, 40, 50). The formal definition and an extensive analysis of alien genes in diverse prokaryotic genomes will be presented elsewhere. The focus of this paper concerns identification and interpretation of PHX genes across the 22 prokaryotic genomes at hand. Table 2 highlights the primary PHX genes of the gene classes TF and CH.
FIG. 1.

Genes of ≥100 codons in the E. coli (left) and D. radiodurans (right) genomes. Each gene is represented by a single point. Its position is determined by its bias relative to all genes [B(g|C)] and by its bias relative to the RP genes [B(g|RP)] (top). B(g|RP) is replaced by codon bias relative to the CH standard [B(g|CH)] (middle) and relative to TF standard [B[g|TF)] (bottom). RP genes are shown in red, CH genes are in blue, and TF are in green.
TABLE 2.
PHX genes in most bacterial genomes
| Gene class | Proteins |
|---|---|
| RP | All large-subunit and small-subunit RPs ≥100 aa |
| CH | GroEL (HSP60), DnaK (HSP70), GrpE, HtpG (HSP90), ClpP, ClpB, ClpC, ClpX, FtsH, HslU, trigger factor, PPIase, thioredoxin reductase, thermosomes in archaeal genomes |
| Protein synthesis processing factors | EF-Tu (Tuf), EF-G (Fus), EF-Ts, IF-2, IF-3, ATP-dependent RNA polymerase factors (β, β′, α), RpoB, RpoC, RpoA, transcription terminator-antiterminator protein (NusA, NusB, NusG), ribosome release factor (Rrf) |
RESULTS AND DISCUSSION
Statistics of PHX genes in prokaryotic genomes.
Implementation of definition I provides lists of PHX genes for each prokaryotic genome. The global statistics are displayed in Table 1. For genes encoding proteins of at least 100-aa length, the percentages of PHX genes across the different genomes range from 4 to 17%. In particular, the fast-growing E. coli and Haemophilus influenzae genomes (doubling time, ≤1 h) contain 8 to 9% PHX genes. Chromosome I of Vibrio cholerae contains about 7% PHX genes, and Bacillus subtilis contains only about 4% PHX genes. D. radiodurans, exhibiting the doubling time of 1 to 1.5 h, carries about 12 to 13% PHX genes. The slow-growing Mycobacterium tuberculosis (24- to 36-h doubling time) devotes 16% of its genome to PHX genes, with more than 80 of these genes acting in fatty acid biosynthesis or degradation. More than 40% of PHX genes in M. tuberculosis are ORFs of unknown function. The fraction of PHX genes of the archaeal genomes of Methanococcus jannaschii (∼10-h doubling time) is about 8%, and that for Methanobacterium thermoautotrophicum (4-h doubling time) is about 10%. Thus, the proportion of PHX genes does not correlate with growth rate (doubling time) or with genome size. There are no consistent PHX gene proportions among hyperthermophiles: Aquifex aeolicus, 16%; Thermotoga maritima, 10%; Pyrococcus abyssi, 14%; Pyrococcus horikoshii, 9%; and M. jannaschii, 8%. The pathogens Rickettsia prowazekii, Chlamydia trachomatis, and Helicobacter pylori contain only 5 to 6% PHX genes with expression levels (equation 3) only reaching 1.20. The archaea show many unidentified PHX genes ranging from 21% of all PHX genes in M. thermoautotrophicum to 47% in P. horikoshii. The highest expression levels are achieved for genomes of rapidly dividing organisms.
Special PHX genes of diverse prokaryotic genomes.
The complete lists of the PHX genes of the current complete genomes corresponding to Table 1 are available on our ftp site, gnomic.stanford.edu/pub/highlyexpressed. In this section, we highlight special PHX genes in several of the prokaryotic genomes.
(i) E. coli. (Table 3).
TABLE 3.
Prominent E. coli PHX genes
| Function class | Description | Specific PHX genes |
|---|---|---|
| 1 | RPs | All RPs ≥100 aa in length |
| 2 | Chaperonins and/or protein degradation | HSP70 (DnaK), HSP90 (HtpG), trigger factor, PPIase, HSP60 (GroEL), GroES, HslU, FtsH, thioredoxin, polynucleotide phosphorylase (mRNA degradation), protease DO (htrA) |
| 3 | Transcription and translation factors | EF-Tu (tufA; duplicated), EF-G (fusA), EF-Ts, ATP-dependent RNA helicase, DNA-dependent RNA polymerase β and β′, ς70 (rpoB, rpoC, rpoD), transcription terminator factor (rho), transcription antiterminator (NusG, NusB), ribosome release factor (rrf) |
| 4 | Detoxification | SodA, C, catalase, alkyl hydroperoxide reductase, thiol peroxidase, Dps (DNA protection during starvation) |
| 5 | Recombination and repair | RecA, single-stranded DNA binding protein (ssb) |
| 6 | Aminoacyl tRNA synthetases | ileS, proS, leuS, glnS, serS, asnS, pheS (α chain), pheT (β chain), thrS, aspS, argS, metG, gltX, alaS, lysS, valS, glyS (β chain), glyQ (α chain), glutamine amidotransferase (glmS) |
| 7 | Energy metabolism: mostly glycolysis and tricarboxylic acid (Krebs) cycle and several enzymes of anaerobic growth | Pyruvate dehydrogenase, g3pd (glyceraldehyde 3-phosphate dehydrogenase), triosephosphate isomerase, fructose 1,6-bisphosphate aldolase, dihydrolipoamide dehydrogenase, phosphoglycerate mutase, 6-phosphofructokinase aldolase, phosphate acetyltransferase, transketolase, deoxiribose-phosphate aldolase, succinyl-CoAa synthetase α and β, malate dehydrogenase, ATP synthetase F1α and F1β |
| 8 | Electron transport | Cytochrome o ubiquinol oxidase I, III, C; flavodoxin (fldA); cytochrome d ubiquinol oxidase I, II; fumarate and nitrate reductase I, II; NADH dehydrogenase subunits N, L, I, G, F, C, D; ferredoxin (fdx); NADH-nitrate oxidoreductase (nirB); fumarate reductase (frdA, B, D) |
| 9 | Outer membrane proteins | ompA, ompC, ompF, ompX, nmpC |
CoA, coenzyme A.
The polynucleotide phosphorylase (Pnp) gene attains the top expression level (E = 2.66) among all E. coli genes. Pnp is a multifunctional enzyme fundamental in RNA processing and mRNA degradation. The Pnp gene is also the gene with the highest expression level in Borrelia burgdorferi and is PHX in H. influenzae, V. cholerae, Synechocystis, M. tuberculosis, Treponema pallidum, Chlamydia pneumoniae, A. aeolicus, and T. maritima.
Peptidyl-prolyl cis-trans isomerases (PPIases) accelerate the folding of proteins. Their catalytic activity promotes cis-trans isomerization of proline bonds in oligopeptides. There are up to nine PPIases defined by sequence similarity in E. coli. The survival protein SurA version of PPIase concentrates on enhanced folding of periplasmic and outer membrane proteins. Some PPIases are PHX in H. influenzae, V. cholerae, B. subtilis, E. coli, D. radiodurans, H. pylori, R. prowazekii, M. tuberculosis, T. pallidum, A. aeolicus, P. horikoshii, and P. abyssi. Trigger factor is a ribosome-associated chaperone (exhibiting a PPIase activity) that can substitute for DnaK (12) and that also contributes to protein export. Trigger factor and DnaK cooperate in the folding of newly synthesized proteins. Simultaneous deletion of trigger factor and DnaK is lethal under the usual growth conditions (61). Trigger factor is broadly PHX, as seen in E. coli, H. influenzae, B. subtilis, V. cholerae, D. radiodurans, Synechocystis, T. pallidum, C. pneumoniae, and A. aeolicus.
The GAPDH (glyceraldehyde 3-phosphate dehydrogenase) protein is multifunctional, acting primarily in glycolysis; in eukaryotes it can also structurally bind actin filaments and microtubules. The GAPDH gene is PHX in almost all eubacterial genomes and also in the archaea M. jannaschii and P. abyssi. The data on expression levels of associated subunits of a protein complex are often variable. For example, in the UvrABC complex of E. coli, we find unit B to be PHX, whereas units A and C are not. Subunits may have activity separate from that of the total protein, which is the case with UvrB (56). From this perspective, the UvrB protein is multifunctional. By contrast, genes encoding ferredoxin oxidoreductase subunits in the genomes of A. aeolicus, M. jannaschii, P. horikoshii, and T. maritima tend to be incorporated in a single PHX operon at about equal expression levels.
The DNA binding protein Dps (DNA protection during starvation; labeled PexB in reference 43) is PHX (E = 1.13). Dps protects DNA from highly reactive oxygen radicals by forming a hollow spherical complex, where it sequesters DNA and iron to keep the reactive oxygen away (42). Not only is Dps highly expressed during rapid division, as shown by two-dimensional (2-D) gel assessments, it has been evaluated as being among the most abundant proteins in stationary phase (42).
(ii) B. subtilis.
PHX genes of the B. subtilis genome parallel the PHX genes of E. coli. These include mainstream glycolysis and respiration genes and the detoxification genes sodA and the catalase and alkyl hydroperoxide reductase genes. The chaperone thioredoxin catalyzes or removes disulfide bonds in implementing protein folding. The highest predicted expression level for thioredoxin occurs in B. subtilis (E = 1.35), followed by those in the other fast-growing bacteria in the order D. radiodurans, (1.23), V. cholerae (1.21), H. influenzae (1.11), and E. coli (1.06). Thioredoxin (trxA) and thioredoxin reductase (trxB) ordinarily carry multiple copies, with at least one of these PHX in most eubacterial genomes.
In contrast to E. coli, four flagellin genes (flagellin [hag], flagellar hook protein [flgE], flagellar hook-basal body [fliE], and flagellin homolog [yvzB]) of B. subtilis are PHX, whereas a lone flagellin gene of E. coli is PHX. Why this difference? Based on the assumption that soil is the major B. subtilis habitat and the human gut is primary for E. coli, the habitat localization may be relevant. The movements of B. subtilis mediated by PHX flagellar proteins may facilitate its acquisition of food from soil sources. By contrast, nutrition (many sugars) flows easily to E. coli in the human lower intestine. Moreover, flagellar genes in E. coli are strictly regulated and inducible, but they are constitutive in B. subtilis (58).
(iii) D. radiodurans.
A mesophilic bacterium, D. radiodurans can survive intense ionizing radiation at a dose of 5,000 Gy (4), which is lethal to virtually all other microorganisms. Such radiation causes DNA single- and double-strand breaks, generates DNA cross-links, and invokes a myriad of other types of DNA, RNA, and protein damage. It was hypothesized (69; but earlier in references 3 and 64) that although D. radiodurans possesses only the traditional prokaryotic repair repertoire, there probably are special mechanisms available that enhance repair. However, it seems paradoxical that recognized repair proteins show predominantly low expression levels, except for RecA (Table 4). RecA promotes and participates in many functions, including homologous recombination, DNA strand exchange, DNA repair and coprotease activity (reacting to DNA damage resulting in the SOS response), prophage induction, and/or mutagenesis subsequent to LexA cleavage (28, 36). Interestingly, RecA has the highest expression level in D. radiodurans of all the genomes shown in Table 1.
TABLE 4.
Repair genes of D. radiodurans
| E(g) | Length (aa) | Positiona | Gene |
|---|---|---|---|
| 0.76 | 1,015 | 1 1801990− | Excinuclease A (uvrA-1) |
| 0.52 | 921 | 2 197055− | Excinuclease A (uvrA-2) |
| 0.55 | 730 | 1 2272809+ | Excinuclease B (uvrB) |
| 0.57 | 616 | 1 1360069+ | Excinuclease C (uvrC) |
| 0.52 | 744 | 1 1804529+ | DNA helicase II (uvrD) |
| 0.58 | 1,053 | 1 1549685− | Transcription-repair coupling factor (mfd) |
| 0.52 | 325 | 1 1844356+ | UV damage endonuclease; putative (DR1819) |
| 0.56 | 246 | 1 705155+ | Uracil-DNA N-glycosylase (ung) |
| 0.64 | 198 | 1 733545− | G/U mismatch-specific DNA glycosylase (mug) |
| 0.57 | 290 | 1 493975+ | Formamidopyrimidine-DNA glycosylase (mutM) |
| 0.65 | 362 | 1 2283259− | A/G-specific adenine glycosylase (mutY) |
| 0.86 | 224 | 1 291364+ | Endonuclease III (nth-1) |
| 0.79 | 258 | 1 2439683− | Endonuclease III (nth-2) |
| 0.66 | 283 | 1 356790− | Exodeoxyribonuclease III (xthA) |
| 0.54 | 546 | 1 1718755+ | DNA mismatch repair protein MutL (mutL) |
| 0.45 | 765 | 1 1999856− | DNA mismatch repair protein MutS; putative (mutS) |
| 0.58 | 358 | 1 1098030− | recF protein (recF) |
| 0.51 | 563 | 1 1490126+ | DNA repair protein (recN) |
| 0.80 | 219 | 1 202795− | RecR protein (recR) |
| 0.40 | 823 | 1 1295908− | DNA helicase RecQ (recQ) |
| 0.49 | 714 | 1 1922060− | Exodeoxyribonuclease V; subunit RecD; putative |
| 0.71 | 908 | 1 1942746+ | Exonuclease SbcC (sbcC) |
| 0.55 | 415 | 1 1941499+ | Exonuclease SbcD; putative (DR1921) |
| 2.04b | 362 | 1 2337795+ | recA protein (recA) |
| 0.69 | 200 | 1 1281494− | Holliday junction binding protein (ruvA) |
| 1.06 | 332 | 1 609449− | Holliday junction DNA helicase (ruvB) |
| 0.88 | 178 | 1 436818+ | Holliday junction resolvase (ruvC) |
| 0.50 | 783 | 1 1936853+ | DNA helicase RecG (recG) |
| 0.57 | 955 | 1 1732431− | DNA-directed DNA polymerase (polA) |
| 0.61 | 699 | 1 2084844− | DNA ligase (dnlJ) |
| 0.76 | 170 | 1 80637− | MutT/nudix family protein (DR0079) |
| 0.76 | 177 | 1 276905+ | MutT/nudix family protein (DR0274) |
| 0.62 | 322 | 1 324367− | MutT/nudix family protein (DR0329) |
| 0.60 | 249 | 1 555381+ | MutT/nudix family protein (DR0550) |
| 0.73 | 166 | 1 991985− | MutT/nudix family protein (DR0975) |
| 0.94 | 165 | 1 1019945+ | MutT/nudix family protein (DR1007) |
| 0.83 | 158 | 1 1038101+ | MutT/nudix family protein (DR1025) |
| 0.78 | 175 | 1 1807313− | MutT/nudix family protein (DR1776) |
| 0.59 | 171 | 1 2270912+ | MutT/nudix family protein (DR2272) |
| 1.29b | 143 | 1 2354268+ | MutT/nudix family protein (DR2356) |
| 0.59 | 701 | 3 126894+ | Ribonucleoside-diphosphate reductase; alpha (nrdE) |
| 0.58 | 354 | 3 128990+ | Ribonucleoside-diphosphate reductase; beta (nrdF) |
| 0.89 | 140 | 3 126505+ | Ribonucleotide reductase; NrdI family (DRB0107) |
| 0.62 | 219 | 2 379822− | LexA repressor (lexA) |
| 0.87 | 502 | 1 1112934+ | DNA repair protein (radA) |
| 0.52 | 940 | 3 162296+ | ATP-dependent helicase HepA (hepA) |
| 0.61 | 1,066 | 3 78759+ | Extracellular nuclease; putative (DRB0067) |
| 1.02 | 143 | 1 100371+ | Single-stranded DNA binding protein (ssb) |
| 0.65 | 403 | 1 2335883+ | CinA protein (DR2338) |
| 0.83 | 343 | 3 124398+ | Integrase-recombinase XerD; putative (xerD) |
Position in the genome is indicated by a chromosome number (1, large chromosome; 2, small chromosome; 3, megaplasmid MP1) followed by position of the translation initiation site and orientation of the gene (+, direct strand; −, complementary strand).
Predicted highly expressed.
Our view of the viability of D. radiodurans suggested by the predictions of gene expression levels emphasizes three processes: (i) the many degradation and export vehicles for removing damaged DNA, RNA, and proteins; (ii) the surfeit of chaperonins, which putatively enhance the operations of the repair proteins; and (iii) the manifold detoxification facilities that neutralize and remove free oxygen radicals and other toxic substances.
Strikingly, Table 5 shows that, compared to the other prokaryotic genomes, D. radiodurans contains the greatest number of PHX detoxification genes. The major PHX CH genes in D. radiodurans include those encoding GroEL (E = 2.35), DnaK (2.24), the general stress protein Ctc (1.89), Lon (two copies: 1.69 and 1.41), FtsZ (1.67), trigger factor (1.48), FtsH (three copies, but only FtsH-3 is PHX) (1.47), DNA binding stress response protein (Dps family) (1.51), cyclophilin (1.46), PPIase (four copies: 1.23 to 1.51), grpE (1.24), fimbrial assembly (pili) (1.23), septum cell division protein MinD (1.14), HSP20 (1.39), ribonuclease PH (1.06), ribonuclease H (1.02), thioredoxin (1.23), and GroES (1.14).
TABLE 5.
Selected classes of PHX genes
| Genome | No. of genes
|
||||
|---|---|---|---|---|---|
| Flagellar | Histidine kinasea | Electron transferb | Detoxificationc | Aminoacyl tRNA synthetased | |
| E. coli | 1 | 0 | 20 | 8 | 20 |
| H. influenzae | 0 | 0 | 5 | 3 | 7 |
| B. subtilis | 4 | 0 | 5 | 4 | 4 |
| V. cholerae | 1 | 0 | 8 | 1 | 8 |
| D. radiodurans | 0 | 0 | 9 | 12 | 1 |
| R. prowazekii | 0 | 0 | 1 | 0 | 0 |
| C. trachomatis | 0 | 0 | 0 | 0 | 0 |
| C. pneumoniae | 0 | 0 | 0 | 0 | 1 |
| M. genitalium | 0 | 0 | 0 | 0 | 1 |
| M. pneumoniae | 0 | 0 | 0 | 0 | 1 |
| B. burgdorferi | 5 | 0 | 0 | 0 | 1 |
| T. pallidum | 7 | 1 | 0 | 1 | 1 |
| H. pylori | 2 | 1 | 5 | 2 | 0 |
| M. tuberculosis | 0 | 5 | 8 | 3 | 6 |
| Synechocystis | 0 | 7 | 16 | 4 | 1 |
| A. aeolicus | 4 | 0 | 37 | 5 | 9 |
| T. maritima | 3 | 0 | 11 | 2 | 5 |
| M. jannaschii | 1 | 0 | 5 | 0 | 0 |
| M. thermoautotrophicum | 0 | 2 | 6 | 1 | 2 |
| A. fulgidus | 1 | 0 | 24 | 4 | 7 |
| P. horikoshii | 3 | 0 | 10 | 1 | 6 |
| P. abyssi | 3 | 0 | 20 | 2 | 10 |
Also includes sensors and other two-component system proteins.
Includes cytochrome- and NADH-related proteins, ferredoxins, and flavodoxins.
Includes Sod, glutathione peroxidase, thiol-specific antioxidant-peroxidase, thiosulfate sulfurtransferase, cytochrome c peroxidase, glutathione-S-transferase, alkyl hydroperoxide reductase, catalase, LexA repressor, Dps, and rubrerythrin (peroxide oxidase).
Also includes amidotransferase.
D. radiodurans is distinctive among prokaryotic genomes in having genes for PHX proteases of many kinds, including three serine protease (E = 1.29 to 1.41), protease I (PfpI) (1.46), zinc metalloendopeptidase (1.28), carboxyl-terminal protease (1.48), ClpX (1.32), and signal peptidase (1.08). FtsH, an integral inner membrane protein, is a cell division metalloprotease that facilitates degradation and protein folding (52). FtsH is PHX in E. coli, D. radiodurans, Synechocystis, M. tuberculosis, and T. maritima. The archaeal genomes commonly feature their own PHX cell division control protein. FtsZ, generally PHX in eubacteria, also contributes to bacterial cell division; it is tubulin-like and very abundant.
D. radiodurans PHX detoxification genes include two catalase genes (1.92 and 1.55), sodA (1.75), two sodC (1.44 and 1.05), Dps (1.51), chloride peroxidase (1.14), singlet oxygen resistance protein (1.11), MutT-nudix (1.29), organic hydroperoxide resistance protein (1.12), and tellurium resistance protein TerD (two copies) (1.47 and 1.05). SodA and catalase protect cells against toxin components induced by oxygen radicals. Alkyl hydroperoxide reductase serves to protect the cell against DNA damage caused by alkyl hydroperoxides. It reduces organic hydroperoxide to its dithiol form.
Virtually all of the DNA repair genes identified in D. radiodurans have functional analogs in other bacterial species. It is surmised that D. radiodurans possesses high redundancy in repair, but except for the nudix family (69) of nucleoside triphosphate pyrophosphorylases, this has not been verified. Actually, the degree of similarity claimed for the nudix genes is tenuous. Growing D. radiodurans cells contain 4 to 10 genome equivalents, which putatively provide facile opportunities for recombination and repair. However, most rapidly growing bacteria, e.g., E. coli and V. cholerae, contain at least three genome equivalents but fail to be resistant to ionizing radiation. Does D. radiodurans possess novel repair proteins more effective than those in other species or use the common repair machinery in new ways? None of this is established. On the contrary, Harsojo et al. (22) reported that with varied genome numbers in D. radiodurans there is no difference in radiation resistance.
We propose that the proliferation of PHX chaperones and degradation and detoxification proteins helps intrinsically in maintaining the survival and stability of the D. radiodurans cell subject to severe conditions of ionizing and/or UV radiation. We speculate that chaperones affect D. radiodurans cells when needed to expedite repair. Along these lines, there are precedents for chaperone influences that enhance the UvrA function in removing DNA damage during the process of nucleotide excision repair (75). Following massive UV radiation damage, D. radiodurans rapidly degrades at least 40% of its DNA and expels it from the cell. Faulty chromosomes could also be expelled, probably harmlessly. The chromosomes are fractured into many subgenomic fragments, but in less than 3 h, they can be accurately reassembled without loss of viability (3). The elaborate CH protein ensemble allows D. radiodurans to maintain the integrity of its essential macromolecules. Along these lines, D. radiodurans contains a preponderance of cell division, degrading, and recycling proteins. The proliferation of antioxidative stress proteins can putatively also mitigate desiccation and negative thermal effects. Most PHX genes of the small chromosome of D. radiodurans are duplicated in the large chromosome.
There are substantial parallels between the principal PHX genes of E. coli and D. radiodurans for the RP, TF, and CH gene classes. However, D. radiodurans possesses a collection of PHX S-layer (surface structure) proteins which may provide environmental protection (e.g., against desiccation). D. radiodurans also has an abundance of PHX ATP-binding cassette transporters for various peptides, branched-chain amino acids (LivK), phosphates, and maltose (periplasmic maltose-binding protein). Intriguingly, the top PHX gene is the multifunctional tricarboxylic acid cycle gene aconitate hydratase (aconitase).
(iv) M. tuberculosis.
The PHX isocitrate lyase enzyme (AceA) is very abundant when M. tuberculosis inhabits macrophages (5). Concomitant to infection of activated macrophages, M. tuberculosis exhibits an increased expression of isocitrate lyase, preferred over isocitrate dehydrogenase, the first enzyme of the glyoxylate shunt pathway that yields a net carbon gain in metabolism of fatty acids (5). This is consistent with the elevated expression levels of this gene. When M. tuberculosis enters macrophages, induction of stress proteins also results. Many fatty acid biosynthesis genes (e.g., fas; fadA; fadB; fadE4, -E5, and -E7; fadD3 and others) are PHX. Also, mycolic acid synthases 2 and 3 (both PHX) are abundant on the bacterial outer cell wall. Apart from M. tuberculosis, of all the genomes in Table 1, the AceA gene appears only in D. radiodurans (PHX) and E. coli (not PHX). Thirty-six genes labeled PE-PGRS (9) and distinguished by a preponderance of glycine-glycine doublets and anomalous repetitive structures are PHX. These may obstruct the host immune system (cf. references 31 and 46).
The bacterial cell has developed complex mechanisms to deal with membrane translocation, secretion of polypeptides, and subsequent folding. SecA, essential and unique to eubacteria (i.e., not found in archaea), is fundamental for protein translocation to the periplasm. Secretion-specific chaperones include SecB and the signal recognition particle. In these activities, the major chaperones GroEL, DnaK, and the trigger factor are also involved (13). In addition to structural subunits, such as SecY, SecE, and SecG, the translocase has a mechanical motor device, the SecA ATPase, that binds to SecYEG to establish the functional translocase core. M. tuberculosis possesses two SecA paralogs with distinct substrate specificities. The SecA gene is also PHX in V. cholerae, E. coli, Synechocystis, Mycoplasma pneumoniae, T. pallidum, B. burgdorferi, and A. aeolicus. The secretion pathway is used by many protein substrates. The cellular destination of all secretory polypeptides is governed by a 20- to 30-residue amino-terminal sequence, the leader peptide, which also helps guide SecA binding to the substrate. SecA, SecB, and SecG are all involved in protein export and chaperonin activity. Gram-negative bacteria also secrete a variety of proteins into the extracellular milieu mediated by secretion apparatus types I to IV (13). These proteins can influence bacterium-host interactions.
(v) Synechocystis (Cyanobacterium).
In the Synechocystis genome, the chaperonin GroEL-2 attains the highest expression level (E = 1.51) and the duplicate GroEL-1 (E = 1.47) and DnaK (E = 1.40) also have high expression levels. Apart from GroEL and DnaK, several CH proteins are among the most highly expressed, including ClpC (E = 1.46) and three copies of FtsH (1.49, 1.30, and 1.17). A fourth FtsH has an expression level of 0.82. In many genomes, duplicated genes have only a single copy that is PHX (S. Karlin, A. D. Kaiser, A. M. Campbell, and J. Mrázek, unpublished data).
The majority of primary photosynthesis genes attain very high expression levels, e.g., the phycobilisome LCM core-membrane linker polypeptide (ApcE) gene records the highest predicted expression level (E = 1.51) in Synechocystis. The PHX genes include more than 30 genes contributing to photosynthesis. The large and small subunits of rubisco are both highly expressed. Interestingly, rubisco is also PHX in the archaeal genome of Archaeoglobus fulgidus. There are several glycolysis and gluconeogenesis genes in Synechocystis that satisfy our criteria as PHX, including those for phosphoglycerate kinase, fructose-1,6-bisphosphate aldolase, GAPDH, enolase, fructose-1,6-bisphosphatase, phosphofructokinase, and pyruvate kinase. These genes also act in photosynthesis. The PHX “giant” ribosomal protein S1, weakly homologous to S1 of E. coli, is only 327 aa, much reduced from the usual size exceeding 500 aa. There are many PHX genes for aerobic respiration and many contributing to electron transport. This is consistent with the proposition that respiration and photosynthesis are linked in Synechocystis.
(vi) H. pylori.
H. pylori lives in the thick mucus lining that protects the stomach from its own digestive juices. Among the most PHX genes are those for urease α (UreA), urease β (UreB), and the accessory ureI (all occurring as a cluster, or operon), which convert urea from gastric juices into bicarbonate and ammonia (NH3), which help to neutralize the highly acidic stomach environment and allow H. pylori to safely traverse the mucus layer to the epithelium surface. Ammonia could also serve as a nitrogen source for amino acids (19, 45). Other accessory proteins, UreE, UreF, UreG, and UreH, that are not part of the urease enzyme and are not PHX help to incorporate Ni2+ ions required for urease enzyme assembly and activity. UreI pumps urea from the outside to the inside of the cell.
The H. pylori genome is rife with genes encoding a family of outer membrane proteins encompassing more than 32 members (many duplicated). The genes encoding outer membrane proteins 2, 9, 11, 14, 21, and 28 of this family are PHX. Some porins may be involved importantly in the antibiotic susceptibility of H. pylori. The PHX chaperone genes of H. pylori include those for GroEL and the two DnaK cochaperones GrpE and DnaJ (the expression level of DnaK is 1.00, at the boundary of PHX genes); the general stress gene ctc and the thioredoxin (trxA) and thioredoxin reductase (trxB) genes are in a cluster. The two chemotaxis genes cheW and cheA as an operon are PHX, as are the two flagellar genes motB and flaE. The cheA gene controls flagellar motor on-off changes. Urease plays a key role in chemotactic motility (48).
The mycoplasma Ureaplasma urealyticum genome has just been sequenced. It is distinguished by an operon of three urease complex components, ureG, ureE, and ureC, among the most PHX genes in this genome (data not shown). Several fatty acid biosynthesis genes (those for FadA, biotin carboxylase, and cyclopropane fatty acid synthase), a spectrum of cytochrome genes, and the genes for two antioxidants (catalase and alkylhydroperoxide reductase) carry high expression values.
(vii) C. trachomatis and C. pneumoniae (mammalian obligate intracellular parasites).
Chlamydia live in vacuoles which burst to spread. Their PHX genes include one of two ATP-ADP exchange translocase genes. This antiporter takes ATP from host cytoplasmic sources and releases ADP from the bacterial cell; the standard mitochondrial exchange is reversed. The ATP-ADP translocase is very uncommon among bacteria and has been found only in Chlamydia and Rickettsia bacteria and in a spectrum of plant plastids. The two C. trachomatis ATP-ADP translocases function differently (71). One exchanges ATP and ADP as described above, and the other is a nucleotide transporter (62). NusA and NusG contribute to termination and antitermination as components of the transcription process (41). The highest expression level of NusA (E = 1.28) is applicable to C. pneumoniae. The Nus proteins are PHX in E. coli, V. cholerae, D. radiodurans, Synechocystis, M. pneumoniae, T. pallidum, B. burgdorferi, C. pneumoniae, M. jannaschii, and P. abyssi.
The PHX OmpA receptor of C. pneumoniae makes up more than 60% of all its membrane proteins (R. S. Stephens, personal communication). There are three GroEL chaperonins each in C. trachomatis and C. pneumoniae, of which only GroEL-1 is PHX. The chaperonin trigger factor and another PPIase are significantly PHX in C. pneumoniae.
(viii) R. prowazekii.
Unlike C. trachomatis, the human obligate intracellular parasite R. prowazekii is not able to metabolize glucose (70). A distinctive PHX gene encodes the cell division protein FtsZ. In contrast to C. trachomatis, R. prowazekii contains five ATP-ADP exchange translocase genes, but individually none is PHX. Perhaps the redundancy of five suffices. There are no PHX glycolysis genes in R. prowazekii and C. trachomatis. R. prowazekii does engage in some respiration, but these microbes apparently extract substantial energy straight from the host.
(ix) T. pallidum.
Spirochetes in general, but not T. pallidum in particular, are mostly free living and are found in soil and freshwater, but they are also commensal with clams and other animals. T. pallidum is restricted to human hosts. Its genome stands out, with the greatest number of PHX flagellar genes among the genomes shown in Table 5 (see also reference 8). These include the genes for the flagellar filament outer layer proteins FlaA-1 and FlaA-2, flagellar motor switch protein, flagellar filament 33-kDa core protein, and flagellar basal-body rod protein FlgG-2. Flagellar proteins in spirochetes operate in the periplasm. This is different from most other genomes, where flagella are extracellular at the surface. Why is T. pallidum so mobile? The bacterium (which causes syphilis) invades all parts of the human body, including the brain. The abundance of highly expressed flagellar genes in T. pallidum could facilitate its movement and enhance its survival by spreading. The genes encoding the response regulator CheY and the purine binding CheW are also PHX genes. The recombination-repair proteins RecA, RecX, and Ssb (single-stranded binding) are PHX. T. pallidum features the longest S1 RP (862 aa) among all complete genomes (Table 6).
TABLE 6.
RPs
| Genome | No. of RP genesa | S1 length (aa) | S2 isolatedj | L7-L12g | P0f |
|---|---|---|---|---|---|
| E. coli | 55 | 556 | + | + | − |
| H. influenzae | 55 | 548 | + | + | − |
| B. subtilis | 57 | 381b | + | + | − |
| V. cholerae | 49c | 555 | + | + | − |
| D. radiodurans | 52 | 628 | + | + | − |
| R. prowazekii | 54 | 567 | + | + | − |
| C. trachomatis | 52 | 568 | + | + | − |
| C. pneumoniae | 52 | 579 | + | + | − |
| M. genitalium | 50 | Missing | + | + | − |
| M. pneumoniae | 50 | Missing | + | + | − |
| B. burgdorferi | 53 | 552 | + | + | − |
| T. pallidum | 52 | 862 | + | + | − |
| H. pylori | 52 | 555 | + | + | − |
| M. tuberculosis | 55 | 480 | + | + | − |
| Synechocystis | 53 | 327, 304h | + | + | − |
| A. aeolicus | 53 | 534 | + | + | − |
| T. maritima | 55 | 542i | + | + | − |
| M. jannaschii | 60 | Missing | − | −d | + |
| M. thermoautotrophicum | 61 | Missing | −e | −d | + |
| A. fulgidus | 61 | Missing | −e | −d | + |
| P. horikoshii | 54 | Missing | −e | −d | + |
| P. abyssi | 61 | Missing | −e | −d | − |
Including duplicated genes.
S1 homolog.
More RP genes may be present in V. cholerae that failed to be identified by similarity searches.
The archaeal genomes have distinct RP L7 and L12, but none is similar to E. coli L7-L12.
M. thermoautotrophicum RP Sa (homolog of E. coli S2) gene is 2 kb downstream from S16 (E. coli S9) separated by genes for DNA-dependent RNA polymerase subunit N, DNA-dependent RNA polymerase subunit K, and enolase. Exactly the same sequence of genes occurs in A. fulgidus, P. horikoshii, and P. abyssi (with the only exception that in P. horikoshii, two ORFs replace the subunit K and enolase genes).
P0 is functionally equivalent to E. coli L10 (SwissProt) and is sometimes called L10E. P0 has a hyperacidic charge run at the C terminus. Eukaryotes also have acidic RPs P1 and P2 (not present in the archaeal genomes at hand). +, present; −, absent.
Unlike most bacterial RPs, which are basic (mostly >20% basic residues), L7-L12 is acidic. For example, E. coli L7-L12 contains 17% acidic residues. +, present; −, absent.
Synechocystis has two copies of S1, and both are significantly smaller than the typical S1 from most other eubacterial genomes.
T. maritima S1 contains a frameshift.
+, isolated; −, proximal to other RP genes.
(x) A. aeolicus.
On the basis of the 16S RNA sequence, the A. aeolicus genome is classified as that of a deeply branching gram-negative hyperthermophile. However, with respect to PHX genes, there is much resemblance to E. coli. A. aeolicus, like a classical gram-negative bacterium, contains an S1 RP of 534 aa which is very highly expressed in this genome. Many electron transport proteins stand out as PHX, including cytochrome c oxidase, cytochrome b, cytochrome c552, several NADH dehydrogenase subunits, and most subunits (α, β, and γ) of the iron sulfur ferredoxin oxidoreductase. The detoxification genes sodA, the alkyl hydroperoxide reductase gene, ahpC1, and ahpC2 and four flagellar genes are PHX. Biotin carboxylase is a PHX protein important in fatty acid biosynthesis. Biotin acts as a coenzyme covalently linked to carboxylase (67). Its highest expression level occurs in A. aeolicus. Biotin carboxylase is also PHX in E. coli, H. influenzae, V. cholerae, H. pylori, Synechocystis, C. trachomatis, and A. fulgidus.
(xi) M. jannaschii.
M. jannaschii (strictly anaerobic) carries out no fermentation. Energy conservation proceeds exclusively by the conversion of H2 plus CO2 to CH4. Special PHX genes are those for thermosome (ths) (this applies to all archaea) and flagellin (flaB1). As expected, the PHX genes include more than 20 participating in methanogenesis. Actually, the three genes with the highest expression levels participate in methanogenesis. The thermosome “homolog” of GroEL is very highly expressed (E = 1.56). The absence of the giant S1 RP applies to all archaeal genomes. The top PHX genes correlate with the greatest protein abundances verified by 2-D-gel analysis (C. Giometti, Argonne National Laboratory, personal communication).
(xii) M. thermoautotrophicum.
The predominant PHX genes in M. thermoautotrophicum are the thermosome subunits thsA (E = 1.33) and thsB (1.38). Again, most PHX genes of M. thermoautotrophicum are involved in methanogenesis. DnaK, missing from M. jannaschii, is PHX in M. thermoautotrophicum.
(xiii) A. fulgidus.
In A. fulgidus, both thermosome units α and β are PHX. The elaborate proteasome complex is PHX in A. fulgidus and P. abyssi. Intriguingly, A. fulgidus contains two PHX copies of rubisco (rbcL-1 and rbcL-2). A. fulgidus has more than 300 PHX genes, compared to about 150 in M. jannaschii and M. thermoautotrophicum. Many NADH dehydrogenases and general anaerobic respiration proteins (electron acceptors) are based on nitrate and sulfate. A. fulgidus grows using sulfate or thiosulfate as an electron acceptor and H2 as an electron donor. Although A. fulgidus is not a methanogen, there are several methanogenesis homologs among the PHX genes. Cells are regular to irregular spheres and have flagella at one end for motility; flaB1 is PHX. A. fulgidus seems to have much more metabolic flexibility with organic and inorganic sources than the methanogens. The polyamine spermidine-putrescine transporter in the periplasm is PHX in H. influenzae, V. cholerae, M. pneumoniae, T. maritima, and A. fulgidus. These apparently help to maintain charge homeostasis. The polyamines are small organic molecules generally present in all living organisms. They are synthesized by a highly regulated pathway from arginine or ornithine and can also be transported in and out of cells. Polyamines influence the transcriptional and translational stages of protein synthesis, stabilize membranes, and, in mammalian systems, modulate neurophysiological functions and may act as intracellular messengers. The five archaeal genomes are rife with PHX genes which conduct electron transfer as needed with anaerobic respiration.
Selected classes of highly expressed genes.
Three gene groups are prominently PHX: RP, CH, and TF. This finding is consistent with protein abundance assessments deduced from 2-D-gel assays for E. coli (65) (see below). These results support the choice of the RP, CH, and TF gene classes as representative standards for PHX genes in prokaryotes. Five specialized classes of genes were examined in Table 5 for PHX genes.
(i) Flagellar genes.
Assembly of a flagellum, the motive organelle produced by many bacteria, requires export of protein subunits from the cytoplasm to the outer surface of the cell by a mechanism resembling type III secretion (74). Flagella generally consist of three main components: the basal body, the hook, and the filament. Flagellum biogenesis and chemotaxis occur in coordination with flagellum assembly and in response to environmental signals. In this context, class I flagellar genes, consisting of flhD and flhC, are first produced. Class II genes encode structural and accessory proteins needed for assembly of the basal body and hook components. Class III proteins are required for maturation of the flagellum and the chemosensory system. This and recent evidence indicate that the flagellum regulon can influence bacterium-host interactions independent of motility (74). There is also an established selective connection of flagellar motion and chemotaxis responses. The flagellum secretion apparatus may be viewed as part of the chaperone family essential for bacterial viability. Flagella are generally absent in nonmotile prokaryotes.
Why do flagellar PHX genes proliferate among the spirochete genomes of B. burgdorferi and T. pallidum? It is known that the flagella of spirochetes are enclosed in a compartment inside the periplasm, whereas in most other bacteria they are attached to a cell surface receptor outside the cell. Moreover, the flagellar genes of the spirochetes respond to a specialized sigma factor, ς28, whereas the flagellar genes of E. coli are commonly activated by the standard ς70. Several flagellar PHX genes of H. pylori have mixed controls, e.g., ς28 and ς54. The flagellar export apparatus in E. coli also functions as a protein secretion system (74).
(ii) Detoxification genes.
PHX genes acting in detoxification are preponderant in D. radiodurans and significant in E. coli, A. aeolicus, and Synechocystis. We suggested that the high levels of CH and detoxification proteins in D. radiodurans contribute to its capacities for prevention of damage to DNA and RNA and for repair of DNA, RNA, and protein damage caused by severe ionizing radiation.
(iii) Electron transfer genes.
Many electron transfer PHX genes are prominent in fast-growing bacteria, in the archaeal genomes, in the deeply branching thermophilic eubacteria A. aeolicus and T. maritima, and in Synechocystis, M. tuberculosis, and H. pylori. By contrast, parasitic eubacteria apparently do not possess electron transfer PHX genes. This is to be expected, since they derive much of their energy from the host.
(iv) Histidine kinase genes.
None of the rapidly dividing bacteria possess PHX genes for sensors, histidine kinases, regulatory protein kinases, or chemotaxis proteins (see also Table 7). Of course, there are many metabolic kinases which are highly expressed. By contrast, several PHX histidine kinase genes are contained in Synechocystis and M. tuberculosis.
TABLE 7.
Expression levels of two-component system genes of E. coli and B. subtilis
| E(g) | Length (kb) | Positiona | Gene and comments |
|---|---|---|---|
| E. coli | |||
| 0.39 | 430 | 417113+ | Phosphate regulon sensor protein (phoR) |
| 0.37 | 893 | 723637− | Sensor protein (kdpD) |
| 0.40 | 485 | 1188999− | Sensor protein (phoQ) |
| 0.40 | 597 | 1276841− | Nitrate-nitrite sensor protein (narX) |
| 0.39 | 653 | 1973348− | Chemotaxis protein (cheA) |
| 0.36 | 565 | 2583751+ | Nitrate-nitrite sensor protein (narQ) |
| 0.47 | 449 | 3533506− | Osmolarity sensor protein (envZ) |
| 0.39 | 500 | 3847766− | Sensor protein (uhpB) |
| 0.43 | 348 | 4053919− | Negative regulation of glnA (glnL or ntrB) |
| 0.70 | 456 | 4102556− | Chemosensory transducer (cpxA) |
| 0.38 | 473 | 4634265+ | Sensor protein (creC) |
| B. subtilis | |||
| 0.42 | 605 | 1469428+ | Histidine kinase (kinA) |
| 0.55 | 428 | 3229144+ | Histidine kinase (kinB) |
| 0.65 | 145 | 2443877− | Anti-sigma factor and serine kinase (spoIIAB) |
Position in the genome is shown as position of the translation initiation site and orientation of the gene (+, direct strand; −, complementary strand).
Highly expressed RP genes in prokaryotic genomes.
In our original predictions of PHX genes for each genome, the RP genes served as a representative group. Following the analysis based on definition I and equations 2 and 3, we observed that practically all RP genes of all sizes qualify as highly expressed. Those with the highest expression levels (arranged by decreasing predicted levels in E. coli) are the genes encoding L2, S2, L4, S3, S1, L1, L3, S9, L20, L5, L13, S4, L14, and S13. Among the prokaryotic genomes at hand, distinct RPs number from 50 to 60 (Table 6), and in eukaryotes they number 79 (in yeast, 78) (68, 72, 73). Special cases and distributional properties of RPs stand out, as described below.
The eubacterial RP genes generally feature a large cluster (operon) encompassing 20 to 40% of all RP genes. Some of the main TF including Tuf, Fus, RpoA, RpoB, and RpoC are often encoded within or proximal to the large RP gene operon. Other operons usually consist of two to five RP genes. For example, the cluster of L7-L12, L10, L1, L11, rpoB, and rpoC stands out. B. subtilis unites in its genome the equivalents of the two largest E. coli clusters. In many genomes (e.g., Synechocystis and M. tuberculosis), several major CH proteins are proximal to the major RP operons. It is tempting to speculate that these chaperones may contribute to ribosomal assembly. In the presence of a unique oriC, the bulk of eubacterial RP clusters are positioned near the origin of replication.
A giant RP (labeled S1, RpsA, or Rps1 and generally exceeding 500 aa) is recognized in most eubacteria. The S1 gene is essential in E. coli and putatively contributes to the initiation of protein synthesis. In Synechocystis, S1 (327 aa) occurs as a drastically reduced version of the typical S1. The major RP cluster in Synechocystis has the genes for RpoB, RpoC, and GroEL-1 nearby. In B. subtilis, there is a putative S1 homolog of 380 aa, and S1 is definitely missing from the mycoplasma genomes Mycobacterium genitalium, M. pneumoniae, and U. urealyticum. S1, when extant, is isolated (not part of an RP operon) and tends to score among the highest expression levels. The deeply branching gram-negative A. aeolicus encodes a giant S1. T. maritima, allowing for a frameshift, also encodes an S1 homolog. None of the archaeal genomes possesses an S1 homolog, and eukaryote genomes also lack an S1 homolog.
Unlike the giant eubacterial S1, Saccharomyces cerevisiae RP genes are all less than 350 aa in length (mostly between 50 and 250 aa) and are randomly distributed over the 16 yeast chromosomes. This is consistent with the general absence of operons from yeast. Most yeast RPs are duplicated and achieve impressively high expression levels (68).
The S2 RP gene in eubacterial genomes is separated from other RPs. However, S2 in the archaeal genomes (those of M. thermoautotrophicum, A. fulgidus, P. abyssi, and P. horikoshii) is proximal to RP clusters.
RPs are generally very cationic and tend to bind nucleic acids, particularly RNA. The acidic RPs (containing a carboxyl hyperacidic residue run) P0, P1, and P2 are found in eukaryotes; P0 is generally part of the RP repertoire in the archaeal genomes. P. horikoshii contains a ribosomal P0-like acidic protein of 341 aa. Acidic RPs have not been detected in eubacterial genomes, except for L7-L12.
Comparison of predicted expression levels with 2-D gel patterns.
For some E. coli proteins, 2-D gel electrophoresis data on protein abundances under different growth conditions are available (65, 66). We compared the molar abundances (protein abundance divided by protein molecular weight) of 96 proteins of ≥100 aa with the set of PHX genes. The genes for the 20 most molar abundant of the 96 proteins include (in decreasing abundances) tufA, metE, rplL, ompA, fabB, rpsA, rpsF, groEL, eno, fusA, hns, purC, glyA, ilvE, tsf, folA, dnaK, tig, atpA, and glnA. Seventeen of these genes were identified as PHX by our method. The three that were not identified as PHX are metE (methionine synthase), ilvE (branched-chain amino acid aminotransferase), and folA (dihydrofolate reductase). Interestingly, all three are involved in amino acid or nucleotide biosynthesis. At the other extreme, among the 20 least abundant proteins of the 96, only five qualify as highly expressed. These include the aminoacyl tRNA synthetases LeuS and ValS, the RP RplI, N utilization substance protein B (NusB), and phosphoenolpyruvate carboxykinase (PckA). The results for the remaining 56 proteins of intermediate molar abundance include 28 identified as highly expressed.
PHX genes and SD signals.
Initiation of gene translation in E. coli and in many eubacteria involves interactions between a conserved SD sequence immediately upstream of the initiation codon in the mRNA leader and an equally conserved anti-SD sequence at the 3′ end of the 16S rRNA. Not all mRNAs possess a recognizable SD sequence. The consensus SD sequence features at its core the purine run GGAGG, generally traversing positions −9 to −5 relative to the initiation codon and the 16S rRNA gene which persistently carries the anti-SD sequence CACCTCCTTTC at its 3′ end. The bulk of genomes, including those of all five archaea, have at least one copy of 16S rRNA that has the CCTCCT terminal motif. Two bacterial genomes, those of B. burgdorferi and R. prowazekii, do not have rRNA genes with this motif, and two bacterial genomes (those of Synechocystis and D. radiodurans) have an additional copy of a 16S rRNA gene with a different 3′ end. In several genomes, we investigated the proportion of genes in possession of a strong SD sequence among three groups of genes: PHX genes, genes with predicted moderate expression levels (PMX), and genes with predicted low expression levels (PLX). The statistics are displayed in Table 8. The collection of PHX genes examined is complete. A random sample of the PMX and PLX genes was investigated. The data show that more PHX genes than genes with an average or low expression level tend to possess a strong SD sequence, indicating a significant positive correlation between predicted expression levels of genes and the existence of a strong SD sequence.
TABLE 8.
Shine-Dalgarno (SD) sequence in prokaryotic genomesa
| Genome | Type | Expression [E(g)] value | No. of genes examined | No. (%) with SD | No. (%) of strong SD sequences with GAGG or GGAG |
|---|---|---|---|---|---|
| E. coli | PHX | 1.00–2.66 | 306 | 248 (81) | 161 (53) |
| PMX | 0.50–0.99 | 258b | 156 (66) | 82 (34) | |
| PLX | 0.27–0.37 | 113 | 64 (57) | 35 (31) | |
| V. cholerae | PHX | 1.00–2.25 | 238 | 156 (66) | 52 (45) |
| PLX | 0.27–0.37 | 113b | 65 (57) | 48 (25) | |
| H. pylori | PHX | 1.00–1.21 | 73 | 67 (92) | 30 (41) |
| PMX | 0.74–0.99 | 154b | 128 (83) | 32 (21) | |
| T. maritima | PHX | 1.00–1.40 | 181 | 177 (97) | 146 (81) |
| PMX | 0.74–0.99 | 209b | 192 (92) | 148 (71) | |
| A. fulgidus | PHX | 1.00–1.39 | 176 | 106 (66) | 79 (45) |
| PMX | 0.58–0.99 | 188b | 105 (56) | 53 (28) |
Putative SD sequences are defined as purine runs ≥5 bp long within 20 bp upstream of the translation start. A strong SD sequence includes GAGG or GGAG.
A random sample of genes in this category was examined.
Gene classes not highly expressed.
Proteins required in few copies per cell cycle are not expected to be highly expressed. In fact, the following gene groups are seldom highly expressed: (i) specific regulatory proteins, (ii) specific transcription factors, and (iii) strict replication proteins. We display in Table 7 the expression levels for several two-component sensor genes (histidine kinases) in E. coli and B. subtilis. In all the examples, the expression levels are emphatically low, ranging from 0.30 to 0.70, with most values about 0.40. A second gene group with prevalent low expression levels are those for the repair proteins of D. radiodurans (Table 4). Only the paramount recombination protein, RecA, is significantly highly expressed (E = 2.04). However, the bulk of repair proteins of D. radiodurans score in the interval E = 0.4 to 0.8 (Table 4). The repair protein repertoire of E. coli (again with the exception of RecA and RuvB) and those in almost all prokaryotic genomes are not PHX.
Pathways for the synthesis of vitamins, of which only small amounts are generally needed to achieve adequate function, also record low E values, about 0.4 to 0.8 (Karlin et al., unpublished). Exceptionally, RibH (riboflavin synthase β subunit) of E. coli, in a pathway of vitamin synthesis, is PHX. RibE (riboflavin synthase α subunit; not PHX) forms a complex with RibH composed of three units of RibE joined with 60 units of RibH (52a). This stoichiometric anomaly on RibH makes it likely that RibH furnishes structural support and in this purview may be used in multiple capacities.
Perspectives.
Why are CH proteins outstandingly PHX? Their functional attributes are far ranging. Chaperones are vitally needed during both rapid growth and stationary phase. In normal cell physiology, these proteins contribute decisively to ensuring proper protein folding, to correcting misfolded structures, to coordinating protein transport, and to directing protein secretion. Chaperones also contribute to conformational changes and to minimizing protein damage during stress. Accordingly, during starvation, molecular chaperones reduce protein denaturation. Starvation is accompanied by toxic metabolites and oxidative stress. Dps also controls proteins concerned with oxidative protection. A large number of starvation proteins are involved in protein and DNA repair (43). Overall, the three protein families—RPs, TFs, and CHs—are needed in large quantities at many stages of the life cycle, and putatively the organism has evolved codon usages to promote, as needed, growth, stability, and plasticity. From this perspective, codon usage has evolved to accommodate most situations of the cell's existence.
Protein synthesis can be divided into four essential steps: initiation, elongation, termination, and ribosome disassembly. The major highly expressed proteins involved in these processes rely on initiation factors (InfB and InfC), elongation factors (EF-Tu, EF-G, and EF-Ts), and the ribosome release factor (Rrf). The principal genes of the transcriptome also feature the prime components of RNA polymerase, RpoA, RpoB, and RpoC, sometimes supplemented by the sigma factors RpoD, RpoE, and RpoH and terminators-antiterminators (NusA, NusB, and NusG) (Table 2). However, not all transcription factors are PHX. For example, RpoA is selectively highly expressed, and most ς factors are not PHX. EF-Tu and EF-G genes often localize in eubacterial genomes among RP clusters, but these are not found near to archaeal RP operons. For eubacteria with a unique bidirectional origin of replication, PHX genes are predominantly encoded from the leading strand.
More than 80% of PHX genes possess an unambiguous SD sequence compared to genes of average or lower expression levels, with percentages indicating a positive association of E(g) values and an extant strong SD sequence. Generally, PHX genes exploit favorable codon usages, tend to possess a strong SD sequence, and are probably endowed with a strong promoter sequence.
Questions for future study include the following. Are the prime prokaryotic PHX genes “ancient,” meaning significantly conserved across many genomes? How does proteome content (in terms of protein abundances) correlate with transcriptome data? This also concerns correlations of 2-D-gel assessments with mRNA levels. Several reports depict these correlations as weak (21, 60). What are the core (essential) numbers and types of genes that genomes require for fast growth? The rapidly growing bacteria E. coli, H. influenzae, V. cholerae, B. subtilis, and D. radiodurans attain the highest expression levels [E(g) values] of genes among bacterial genomes (Table 1). What is the relation of induced versus constitutive protein expression to PHX genes? What is the influence of stoichiometry of subunits or the half-life of a protein on expression levels? Do operons and complexes entail components concordant or discordant with respect to their PHX status? In these contexts, PHX ORFs are attractive targets for knockout studies.
ACKNOWLEDGMENTS
We thank B. E. Blaisdell, L. Brocchieri, A. M. Campbell, A. Danchin, D. Kaiser, J. Ma, G. Miklos, and A. Spormann for valuable discussions and comments on the manuscript.
S.K. was supported in part by NIH grants 5R01GM10452-35 and 5R01HG00335-11 and NSF grant DMS9704552.
REFERENCES
- 1.Andersson S G E, Kurland C G. Codon preferences in free-living microorganisms. Microbiol Rev. 1990;54:198–210. doi: 10.1128/mr.54.2.198-210.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersson S G E, Zomorodipour A, Andersson J O, Sicheritz-Ponten T, Alsmark U C M, Podowski R M, Naeslund A K, Eriksson A S, Winkler H H, Kurland C G. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396:133–140. doi: 10.1038/24094. [DOI] [PubMed] [Google Scholar]
- 3.Battista J R. Against all odds: the survival strategies of Deinococcus radiodurans. Annu Rev Microbiol. 1997;51:203–224. doi: 10.1146/annurev.micro.51.1.203. [DOI] [PubMed] [Google Scholar]
- 4.Battista J R, Earl A M, Park M J. Why is Deinococcus radiodurans so resistant to ionizing radiation? Trends Microbiol. 1999;7:362–365. doi: 10.1016/s0966-842x(99)01566-8. [DOI] [PubMed] [Google Scholar]
- 5.Bentrup K H Z, Miczak A, Swenson D L, Russell D G. Characterization of activity and expression of isocitrate lyase in Mycobacterium avium and Mycobacterium tuberculosis. J Bacteriol. 1999;181:7161–7167. doi: 10.1128/jb.181.23.7161-7167.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blattner F R, Plunkett III G, Bloch C A, Perna N T, Burland V, Riley M, Collado-Vides J, Glasner J D, Rode C K, Mayhew G F, Gregor J, Davis N W, Kirkpatrick H A, Goeden M A, Rose D J, Mau B, Shao Y. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 7.Bult C J, White O, Olsen G J, Zhou L, Fleischmann R D, Sutton G G, Blake J A, FitzGerald L M, Clayton R A, Gocayne J D, et al. Complete genome sequence of the methanogenic archeon, Methanococcus jannaschii. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 8.Charon N W, Goldstein S F, Block S M, Curci K, Ruby J D, Kreiling J A, Limberger R J. Morphology and dynamics of protruding spirochete periplasmic flagella. J Bacteriol. 1992;174:832–840. doi: 10.1128/jb.174.3.832-840.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cole S T, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon S V, Eiglmeier K, Gas S, Barry III C E, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Daulin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Barnell B G, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 10.Condo I, Ciammaruconi A, Benelli D, Ruggero D, Londei P. Cis-acting signals controlling translational initiation in the thermophilic archaeon Sulfolobus solfataricus. Mol Microbiol. 1999;34:377–384. doi: 10.1046/j.1365-2958.1999.01615.x. [DOI] [PubMed] [Google Scholar]
- 11.Deckert G, Warren P V, Gaasterland T, Young W G, Lenox A L, Graham D E, Overbeek R, Snead M A, Keller M, Aujay M, Huber R, Feldman R A, Short J M, Olson G J, Swanson R V. The complete genome of the hyperthermophilic bacterium Aquifex aeolicus. Nature. 1998;392:353–358. doi: 10.1038/32831. [DOI] [PubMed] [Google Scholar]
- 12.Deuerling E, Schulze-Specking A, Tomoyasu T, Mogk A, Bukau B. Trigger factor and DnaK cooperate in folding of newly synthesized proteins. Nature. 1999;400:693–696. doi: 10.1038/23301. [DOI] [PubMed] [Google Scholar]
- 13.Economou A. Following the leader: bacterial protein export through the Sec pathway. Trends Microbiol. 1999;7:315–320. doi: 10.1016/s0966-842x(99)01555-3. [DOI] [PubMed] [Google Scholar]
- 14.Etchegaray J P, Inouye M. Translational enhancement by an element downstream of the initiation codon in Escherichia coli. J Biol Chem. 1999;274:10079–10085. doi: 10.1074/jbc.274.15.10079. [DOI] [PubMed] [Google Scholar]
- 15.Fleischmann R D, Adams M D, White O, Clayton R A, Kirkness E F, Kerlavage A R, Bult C J, Tomb J-F, Dougherty B A, Merrick J M, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 16.Fraser C M, Casjens S, Huang W M, Sutton G G, Clayton R A, Lathigra R, White O, Ketchum K A, Dodson R, Hickey E K, Gwinn M, Dougherty B, Tomb J F, Fleischmann R D, Richardson D, Peterson J, Kerlavage A R, Quackenbush J, Salzberg S, Hanson M, van Vugt R, Palmer N, Adams M D, Gocayne J, Venter J C, et al. Genomic sequence of a Lyme disease spirochete, Borrelia burgdorferi. Nature. 1997;390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- 17.Fraser C M, Gocayne J D, White O, Adams M D, Clayton R A, Fleischmann R D, Bult C J, Kerlavage A R, Sutton G, Kelley J M, et al. The minimal gene complement of Mycoplasma genitalium. Science. 1995;270:397–403. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- 18.Fraser C M, Norris S J, Weinstock G M, White O, Sutton G G, Dodson R, Gwinn M, Hickey E K, Clayton R, Ketchum K A, et al. Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science. 1998;281:375–388. doi: 10.1126/science.281.5375.375. [DOI] [PubMed] [Google Scholar]
- 19.Ge Z, Taylor D E. Contributions of genome sequencing to understanding the biology of Helicobacter pylori. Annu Rev Microbiol. 1999;53:353–387. doi: 10.1146/annurev.micro.53.1.353. [DOI] [PubMed] [Google Scholar]
- 20.Guerdoux-Jamet P, Henaut A, Nitschke P, Risler J L, Danchin A. Using codon usage to predict gene origin: is the Escherichia coli outer membrane a patchwork of products from different genomes? DNA Res. 1997;4:257–265. doi: 10.1093/dnares/4.4.257. [DOI] [PubMed] [Google Scholar]
- 21.Gygi S P, Rochon Y, Franza B R, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–1730. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harsojo, Kitayama S, Matsuyama A. Genome multiplicity and radiation resistance in Micrococcus radiodurans. J Biochem. 1981;90:877–880. doi: 10.1093/oxfordjournals.jbchem.a133544. [DOI] [PubMed] [Google Scholar]
- 23.Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li B-C, Herrmann R. Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 1996;24:4420–4449. doi: 10.1093/nar/24.22.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J Mol Biol. 1981;151:389–409. doi: 10.1016/0022-2836(81)90003-6. [DOI] [PubMed] [Google Scholar]
- 25.Irwin B, Heck J D, Hatfield G W. Codon pair utilization biases influence translational elongation step times. J Biol Chem. 1995;270:22801–22806. doi: 10.1074/jbc.270.39.22801. [DOI] [PubMed] [Google Scholar]
- 26.Kalman S, Mitchell W, Marathe R, Lammel C, Fan L, Hyman R W, Olinger L, Grimwood L, Davis R W, Stephens R S. Comparative genomes of Chlamydia pneumoniae and C. trachomatis. Nat Genet. 1999;21:385–389. doi: 10.1038/7716. [DOI] [PubMed] [Google Scholar]
- 27.Kaneko T, Sato S, Kotani H, Tanaka A, Asamizu E, Nakamura Y, Miyajima N, Hirosawa M, Sugiura M, Sasamoto S, Kimura T, Hosouchi T, Matsuno A, Muraki A, Nakazaki N, Naruo K, Okumura S, Shimpo S, Takeuchi C, Wada T, Watanabe A, Yamada M, Yasuda M, Tabata S. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 1996;3:109–136. doi: 10.1093/dnares/3.3.109. [DOI] [PubMed] [Google Scholar]
- 28.Karlin S, Brocchieri L. Evolutionary conservation of RecA genes in relation to protein structure and function. J Bacteriol. 1996;178:1881–1894. doi: 10.1128/jb.178.7.1881-1894.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karlin S, Campbell A M, Mrázek J. Comparative DNA analysis across diverse genomes. Annu Rev Genet. 1998;32:185–225. doi: 10.1146/annurev.genet.32.1.185. [DOI] [PubMed] [Google Scholar]
- 30.Karlin S, Mrázek J. What drives codon choices in human genes? J Mol Biol. 1996;262:459–472. doi: 10.1006/jmbi.1996.0528. [DOI] [PubMed] [Google Scholar]
- 31.Karlin S, Mrázek J, Campbell A M. Frequent oligonucleotides and peptides of the Haemophilus influenzae genome. Nucleic Acids Res. 1996;24:4263–4272. doi: 10.1093/nar/24.21.4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karlin S, Mrázek J, Campbell A M. Codon usages in different gene classes of the Escherichia coli genome. Mol Microbiol. 1998;29:1341–1355. doi: 10.1046/j.1365-2958.1998.01008.x. [DOI] [PubMed] [Google Scholar]
- 33.Kawarabayasi Y, Sawada M, Horikawa H, Haikawa Y, Hino Y, Yamamoto S, Sekine M, Baba S, Kosugi H, Hosoyama A, Nagai Y, Sakai M, Ogura K, Otsuka R, Nakazawa H, Takamiya M, Ohfuku Y, Funahashi T, Tanaka T, Kudoh Y, Yamazaki J, Kushida N, Oguchi A, Aoki K, Kikuchi H. Complete sequence and gene organization of the genome of a hyper-thermophilic archaebacterium, Pyrococcus horikoshii OT3. DNA Res. 1998;5:55–76. doi: 10.1093/dnares/5.2.55. [DOI] [PubMed] [Google Scholar]
- 34.Klenk H P, Clayton R A, Tomb J, White O, Nelson K E, Ketchum K A, Dodson R J, Gwinn M, Hickey E K, Peterson J D, et al. The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature. 1997;390:364–370. doi: 10.1038/37052. [DOI] [PubMed] [Google Scholar]
- 35.Kochetov A V, Ponomarenko M P, Frolov A S, Kisselev L L, Kolchanov N A. Prediction of eukaryotic mRNA translational properties. Bioinformatics. 1999;15:704–712. doi: 10.1093/bioinformatics/15.7.704. [DOI] [PubMed] [Google Scholar]
- 36.Kowalczykowski S C, Dixon D A, Eggleston A K, Lauder S D, Rehrauer W M. Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev. 1994;58:401–465. doi: 10.1128/mr.58.3.401-465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kunst F, Ogasawara N, Moszer I, Albertini A M, Alloni G, Azevedo V, Bertero M G, Bessieres P, Bolotin A, Borchert S, et al. The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature. 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- 38.Kurland C G. Major codon preference: theme and variations. Biochem Soc Trans. 1993;21:841–846. doi: 10.1042/bst0210841. [DOI] [PubMed] [Google Scholar]
- 39.Lawrence J G, Ochman H. Amelioration of bacterial genomes: rates of change and exchange. J Mol Evol. 1997;44:383–397. doi: 10.1007/pl00006158. [DOI] [PubMed] [Google Scholar]
- 40.Lawrence J G, Ochman H. Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci USA. 1998;95:9413–9417. doi: 10.1073/pnas.95.16.9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J, Mason S W, Greenblatt J. Elongation factor NusG interacts with termination factor rho to regulate termination and antitermination of transcription. Genes Dev. 1993;7:161–172. doi: 10.1101/gad.7.1.161. [DOI] [PubMed] [Google Scholar]
- 42.Martinez A, Kolter R. Protection of DNA during oxidative stress by the nonspecific DNA-binding protein Dps. J Bacteriol. 1997;179:5188–5194. doi: 10.1128/jb.179.16.5188-5194.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matin A, Baetens M, Pandza S, Park C H, Waggoner S. Survival strategies in stationary phase. In: Rosenberg E, editor. Microbial ecology and infectious diseases. Washington, D.C.: American Society for Microbiology; 1999. pp. 32–48. [Google Scholar]
- 44.Médigue C, Rouxel T, Vigier P, Henaut A, Danchin A. Evidence for horizontal gene transfer in Escherichia coli speciation. J Mol Biol. 1991;222:851–856. doi: 10.1016/0022-2836(91)90575-q. [DOI] [PubMed] [Google Scholar]
- 45.Merrick M J, Edwards R A. Nitrogen control in bacteria. Microbiol Rev. 1995;59:604–622. doi: 10.1128/mr.59.4.604-622.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moxon E R, Rainey P B, Nowak M A, Lenski R E. Adaptive evolution of highly mutable loci in pathogenic bacteria. Curr Biol. 1994;4:24–33. doi: 10.1016/s0960-9822(00)00005-1. [DOI] [PubMed] [Google Scholar]
- 47.Mrázek J, Karlin S. Detecting alien genes in bacterial genomes. Ann N Y Acad Sci. 1999;870:314–329. doi: 10.1111/j.1749-6632.1999.tb08893.x. [DOI] [PubMed] [Google Scholar]
- 48.Nakamura H, Yoshiyama H, Takeuchi H, Mizote T, Okita K, Nakazawa T. Urease plays an important role in the chemotactic motility of Helicobacter pylori in a viscous environment. Infect Immun. 1998;66:4832–4837. doi: 10.1128/iai.66.10.4832-4837.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nelson K E, Clayton R A, Gill S R, Gwinn M L, Dodson R J, Haft D H, Hickey E K, Peterson J D, Nelson W C, Ketchum K A, et al. Evidence for lateral gene transfer between Archaea and Bacteria from genome sequence of Thermotoga maritima. Nature. 1999;399:323–329. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- 50.Ochman H, Lawrence J G, Groisman E A. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- 51.Osada Y, Saito R, Tomita M. Analysis of base-pairing potentials between 16S rRNA and 5′ UTR for translation initiation in various prokaryotes. Bioinformatics. 1999;15:578–581. doi: 10.1093/bioinformatics/15.7.578. [DOI] [PubMed] [Google Scholar]
- 52.Pfanner N. Who chaperones nascent chains in bacteria? Curr Biol. 1999;9:R720–R724. doi: 10.1016/s0960-9822(99)80467-9. [DOI] [PubMed] [Google Scholar]
- 52a.Ritsert K, Huber R, Turk D, Landenstein R, Schmidt-Base K, Bacher A. Studies on the lumazine synthase/riboflavin synthase complex of Bacillus subtilis: crystal structure analysis of reconstituted, icosahedral beta-subunit capsids with bound substrate analogue inhibitor at 2.4 A resolution. J Mol Biol. 1995;253:151–167. doi: 10.1006/jmbi.1995.0542. [DOI] [PubMed] [Google Scholar]
- 53.Saito R, Tomita M. Computer analyses of complete genomes suggest that some archaebacteria employ both eukaryotic and eubacterial mechanisms in translation initiation. Gene. 1999;238:79–83. doi: 10.1016/s0378-1119(99)00254-1. [DOI] [PubMed] [Google Scholar]
- 54.Sharp P M, Li W-H. The codon adaptation index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sharp P M, Matassi G. Codon usage and genome evolution. Curr Opin Genet Dev. 1994;4:851–860. doi: 10.1016/0959-437x(94)90070-1. [DOI] [PubMed] [Google Scholar]
- 56.Shizuya H, Dykhuizen D. Conditional lethality of deletions which include uvrB in strains of Escherichia coli lacking deoxyribonucleic acid polymerase I. J Bacteriol. 1972;112:676–681. doi: 10.1128/jb.112.2.676-681.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith D R, Doucette-Stamm L A, Deloughery C, Lee H-M, Dubois J, Aldredge T, Bashirzadeh R, Blakely D, Cook R, Gilbert K, et al. Complete genome sequence of Methanobacterium thermoautotrophicum delta H: functional analysis and comparative genomics. J Bacteriol. 1997;179:7135–7155. doi: 10.1128/jb.179.22.7135-7155.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Soutourina O, Kolb A, Krin E, Laurent-Winter C, Rimsky S, Danchin A, Bertin P. Multiple control of flagellum biosynthesis in Escherichia coli: role of H-NS protein and the cyclic AMP-catabolite activator protein complex in transcription of the flhDC master operon. J Bacteriol. 1999;181:7500–7508. doi: 10.1128/jb.181.24.7500-7508.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stephens R S, Kalman S, Lammel C J, Fan J, Marathe R, Aravind L, Mitchell W P, Olinger L, Tatusov R L, Zhao Q, Koonin E V, Davis R W. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science. 1998;282:754–759. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- 60.Tao H, Bausch C, Richmond C, Blattner F R, Conway T. Functional genomics: expression analysis of Escherichia coli growing on minimal and rich media. J Bacteriol. 1999;181:6425–6440. doi: 10.1128/jb.181.20.6425-6440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Teter S A, Houry W A, Ang D, Tradler T, Rockabrand D, Fischer G, Blum P, Georgopoulos C, Hartl F U. Polypeptide flux through bacterial Hsp70: DnaK cooperates with trigger factor in chaperoning nascent chains. Cell. 1999;97:755–765. doi: 10.1016/s0092-8674(00)80787-4. [DOI] [PubMed] [Google Scholar]
- 62.Tjaden J, Winkler H H, Schwoppe C, Van der Laan M, Mohlmann T, Neuhaus H E. Two nucleotide transport proteins in Chlamydia trachomatis, one for net nucleoside triphosphate uptake and the other for transport of energy. J Bacteriol. 1999;181:1196–1202. doi: 10.1128/jb.181.4.1196-1202.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tomb J-F, White O, Kerlavage A R, Clayton R A, Sutton G G, Fleischmann R D, Ketchum K A, Klenk H P, Gill S, Dougherty B A, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 64.Udupa K S, O'Cain P A, Mattimore V, Battista J R. Novel ionizing radiation-sensitive mutants of Deinococcus radiodurans. J Bacteriol. 1994;176:7439–7446. doi: 10.1128/jb.176.24.7439-7446.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.VanBogelen R A, Abshire K Z, Pertsemlidis A, Clark R L, Neidhardt F C. Gene-protein database of Escherichia coli K-12, edition 6. In: Neidhardt F C, Curtiss III R, Ingraham J L, Lin E C C, Low K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Escherichia coli and Salmonella: cellular and molecular biology. 2nd ed. Washington, D.C.: ASM Press; 1996. pp. 2067–2117. [Google Scholar]
- 66.VanBogelen R A, Schiller E E, Thomas J D, Neidhardt F C. Diagnosis of cellular states of microbial organisms using proteomics. Electrophoresis. 1999;20:2149–2159. doi: 10.1002/(SICI)1522-2683(19990801)20:11<2149::AID-ELPS2149>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 67.Waldrop G L, Rayment I, Holden H M. Three-dimensional structure of the biotin carboxylase subunit of acetyl-CoA carboxylase. Biochemistry. 1994;33:10249–10256. doi: 10.1021/bi00200a004. [DOI] [PubMed] [Google Scholar]
- 68.Warner J R. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24:437–440. doi: 10.1016/s0968-0004(99)01460-7. [DOI] [PubMed] [Google Scholar]
- 69.White O, Eisen J A, Heidelberg J F, Hickey E K, Peterson J D, Dodson R J, Haft D H, Gwinn M L, Nelson W C, Richardson D L, et al. Genome sequence of the radioresistant bacterium Deinococcus radiodurans R1. Science. 1999;286:1571–1577. doi: 10.1126/science.286.5444.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Winkler H H, Daugherty R M. Acquisition of glucose by Rickettsia prowazekii through the nucleotide intermediate uridine 5′-diphosphoglucose. J Bacteriol. 1986;167:805–808. doi: 10.1128/jb.167.3.805-808.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Winkler H H, Neuhaus H E. Non-mitochondrial ATP transport. Trends Biochem Sci. 1999;24:64–68. doi: 10.1016/s0968-0004(98)01334-6. [DOI] [PubMed] [Google Scholar]
- 72.Wool I G. Extraribosomal functions of ribosomal proteins. Trends Biochem Sci. 1996;21:164–165. [PubMed] [Google Scholar]
- 73.Wool I G, Chan Y L, Gluck A. Structure and evolution of mammalian ribosomal proteins. Biochem Cell Biol. 1995;73:933–947. doi: 10.1139/o95-101. [DOI] [PubMed] [Google Scholar]
- 74.Young G M, Schmiel D H, Miller V L. A new pathway for the secretion of virulence factors by bacteria: the flagellar export apparatus functions as a protein-secretion system. Proc Natl Acad Sci USA. 1999;96:6456–6461. doi: 10.1073/pnas.96.11.6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zou Y, Crowley D J, Van Houten B. Involvement of molecular chaperonins in nucleotide excision repair. DnaK leads to increased thermal stability of UvrA, catalytic UvrB loading, enhanced repair, and increased UV resistance. J Biol Chem. 1998;273:12887–12892. doi: 10.1074/jbc.273.21.12887. [DOI] [PubMed] [Google Scholar]