

Summary
Controlling the abundance of a protein of interest in vivo is crucial to study its function. Here, we provide a step-by-step protocol for generating genetically engineered mouse (GEM) models harboring a degradation tag (dTAG) fused to endogenous proteins to enable their degradation. We discuss considerations for the overall design and details for vectors generation. Then, we include steps for generation and validations of edited mouse embryonic stem cells followed by mouse colony establishment via chimeric mouse generation.
For complete details on the use and execution of this protocol, please refer to Abuhashem et al. (2022c).
Subject areas: Cell culture, Developmental biology, Genetics, Microscopy, Model Organisms, Molecular Biology, CRISPR, Stem Cells
Graphical abstract
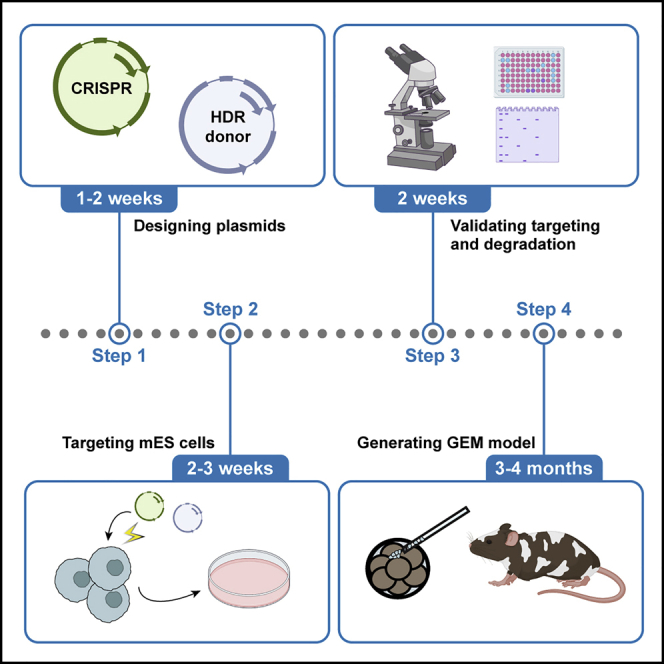
Highlights
-
•
Generation of endogenous tagged degradable proteins in mouse models using dTAG system
-
•
Generation of genetically edited homozygous mouse embryonic stem cells
-
•
Details of successive steps from vector design to mouse model generation and validation
-
•
Detailed recommendations and considerations for tagging any protein of interest
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Controlling the abundance of a protein of interest in vivo is crucial to study its function. Here, we provide a step-by-step protocol for generating genetically engineered mouse (GEM) models harboring a degradation tag (dTAG) fused to endogenous proteins to enable their degradation. We discuss considerations for the overall design and details for vectors generation. Then, we include steps for generation and validations of edited mouse embryonic stem cells followed by mouse colony establishment via chimeric mouse generation.
Before you begin
The protocol described here provides details on generating a genetically engineered mouse (GEM) model system harboring a FKBP12F36V knock-in at the C terminus of Negative Elongation Factor B (Nelfb) to use the dTAG degron system (Figure 1A). The dTAG system relies on mammalian ubiquitination systems, namely Cereblon (CRBN) or Von Hippel-Lindau (VHL), to achieve ubiquitination and degradation of a target protein (Figure 1B) (Nabet et al., 2018, 2020). Degradation of the fusion protein is induced by small molecules, such as dTAG-13 or dTAGv-1, which engage CRBN or VHL ubiquitination respectively. Specifically, the degradation small molecule serves as a molecular glue that brings the tag, and by extension the target protein, to the ubiquitination proteins which in turn ubiquitinate the target protein, resulting in its degradation. Most tissues express either of these systems, but it is worthwhile checking existing datasets for expression patterns since at least one is required. The protocol describes the process beginning with the generation of CRISPR knock-in mouse embryonic stem cells (mESCs), using these to generating germline transmitting chimeric mice, to performing in vivo degradation in the resulting GEM model. NELFB is a nuclear transcription protein, but this protocol is generalizable to any protein of interest (Abuhashem et al., 2022b). However, one will need to consider the properties of protein being targeted; its location within the cell and whether the C terminus is the most suitable position to introduce a tag. In cases where the N terminus is more suitable, the protocol is largely the same except for a modified donor plasmid design that is described below. The protocol is directly applicable to proteins that are expressed in mESCs. If your protein of interest is not expressed in mouse pluripotent embryonic stem (ES) cells, modifications to the donor plasmid or selection strategy may be required. To start the protocol, you need:
-
1.
Low passage, stable, germline-competent mESC line to perform genetic editing.
-
2.
Irradiated or Mitomycin-C inactivated drug-resistant DR4 mouse embryonic fibroblast feeders.
-
3.
Expertise or access to a transgenic mouse core facility to assist in generating chimeric mice via blastocyst or 8-cell embryo ESC injections or aggregations.
-
4.
An approved animal protocol that allows generating GEM models and intraperitoneal injections.
Figure 1.

Illustration of the dTAG system for protein tagging and degradation
(A) Required protein tagging for the dTAG system.
(B) Ubiquitination of the tagged protein in the presence of dTAG small molecules.
Cell lines and feeders can be acquired from ATCC or other commercial sources.
Plasmids and primers design
Timing: 1–2 days
-
5.Choose target locus and suitable terminus to tag your protein of interest.
-
a.Once a target gene is identified, check if previous studies successfully generated tagged versions of derivative proteins to get insights into the most suitable terminus to tag.
-
b.Alternatively, check previous studies for functional domains and structural studies to avoid tagging functionally relevant domains. Avoid tagging termini with alternative initiation/splicing sites, unless isoform degradation is desired.
-
c.Once a target gene and suitable terminus are identified, download the sequence of ∼2,000 base pairs (bp) around the desired insertion site from a genome browser. For this study, the sequence will be centered around the stop codon (Figure 2A).
-
i.This DNA piece will serve as right and left homology arms, each about 1,000 bp for Homology Directed Repair (HDR).
-
ii.We strongly encourage using plasmid/DNA viewing and annotating software to build a plasmid of interest in silico. An open-source software that we routinely use is Benchling.com.
-
i.
-
a.
-
6.Build necessary plasmids in silico. To carry out targeted knock-in, two plasmids are needed: (1) an HDR donor plasmid that will possess the desired final design of the gene, (2) a CRISPR-Cas9 vector with the appropriate guide RNA to target the region of insertion, ideally with a cut within 10 bp of insertion point (Figure 2).
-
a.For the HDR plasmid, the following pieces will be necessary:
-
i.A backbone containing an origin of replication for the plasmid. This can be amplified from available plasmids. We use a pBluescript plasmid as a backbone from Addgene #113097.
-
ii.The left arm for homology directed repair (∼1,000 bp). The last codon in this arm should be the last codon of the gene.
-
iii.The insert: FKBP12F36V-2×HA-P2A-Puro. This sequence can be imported directly from the Addgene plasmid #91796.Note: This sequence will produce an in-frame FKBP12F36V tag followed by an in-frame 2×HA epitope tag, followed by a cleaving peptide sequence (P2A) and a puromycin resistance cassette to enable efficient identification of targeted cells. The endogenous poly A site will serve a terminator of transcription.
-
iv.The right arm for homology directed repair (∼1,000 bp). The first codon in this arm should be the endogenous stop codon of the gene.
-
i.
-
b.For the CRISPR-Cas9 plasmid:
-
i.We use the PX459 pSpCas9(BB)-2A-Puro plasmid, Addgene #62988, which enables puromycin selection of successfully transfected cells.
-
ii.Select a single guide RNA (sgRNA) producing a cut as close as possible to the stop codon. Several tools are available to select sgRNAs.Note: We routinely use the CHOPCHOP tool (chopchop.cbu.uib.no). Ideally, a cut will be made within 10 bp from the stop codon. Select sgRNA with the best predicted efficiency and lowest off-target cuts.
-
i.
-
a.
-
7.Design and order primers/oligos for both plasmids.
-
a.For the HDR plasmid, the in silico pieces are amplified by PCR and assembled using NEB’s HiFi/Gibson DNA assembly kit.
-
i.Design the primers for each piece using NEBuilder.neb.com.
-
ii.Simply, copy and paste each piece’s sequence from the in silico plasmid to the online tool in correct order to generate the required primers. The online tool will automatically generate overhangs to optimize the plasmid assembly reaction (Figure 2B).
-
i.
-
b.For the CRISPR-Cas9 plasmid, two oligos will be needed which correspond to the sgRNA.
-
i.Take the determined sgRNA from CHOPCHOP and remove the PAM sequence which is the 3′ NGG sequence in the CHOPCHOP result.
-
ii.The resulting 20 nucleotides will be used to generate two oligos.
-
iii.Oligo one: 5′-CACC-insert 20 nucleotides from step b-ii-3′.
-
iv.Oligo two: 5′-AAAC-insert 20 nucleotides reverse complement of step b-ii-3′.
-
i.
-
c.Order primers and oligos using any commercial DNA synthesis service. We use Integrated DNA Technologies (IDT).
-
a.
-
8.
Extract mouse genomic DNA to be used as a source to amplify the homology arms for the HDR donor plasmid.
Note: Ideally, this DNA should be extracted from the mESCs in which the editing will happen so that an isogenic targeting vector is generated. However, any other pure source of mouse DNA (cells or tissues), though preferably from the same genetic background as the mESCs you will be targeting, should suffice. DNA lysis buffer (available in materials and equipment) can be used. Simply, add 200 μL buffer/1 × 105 cells and incubate at 55°C overnight (∼16 h).
CRITICAL: Make sure that the desired insert design is not recognized and cut by the sgRNA. This can be achieved by choosing a guide that spans the left and right homology arms, or introducing silent mutations to the PAM sequence in the HDR plasmid using a kit such as the NEB site directed mutagenesis kit.
Optional: If you do not know which terminus of the protein would be suitable for tagging, we highly recommend construction of two transgenes tagging the N and C- termini followed by transient transfection and validation of fusions, prior to targeting the endogenous locus. We found this to be a quick and easy approach for determining degradation efficiency and protein integrity in the presence of FKBP12F36V at each terminus. In this approach, cDNA:tag fusions are inserted downstream of a constitutively expressed promoter/plasmid and transiently transfected into mESCs. Two days following transfection, protein integrity, localization, and degradation can be assayed for each terminus tagging using the validation approaches outlined later in this protocol.
Note: If the N terminus is to be tagged, the insert should be in-frame immediately after the ATG start codon. The order of pieces will be Puro-P2A-2×HA-FKBP12F36V which is available on Addgene #91793 (Figure 3). If the protein of interest is not expressed in mESCs, you may redesign the insert to either not include a selection marker, which will result in less efficient selection. Alternatively, the selection cassette can be replaced by a loxP-PGK-Puro-poly(A)-loxP sequence to get expression of puromycin resistance from a strong promoter which can be excised later using a Cre recombinase driver. This sequence is available on Addgene #171048 (Figure 3).
Note: A Cas9 vector other than PX459 may be used if selection using an alternative approach, for example a fluorescent marker, is desired.
Note: We provide the final maps and sequences of each of our vectors and are accessible in the key resources table.
Figure 2.

Constructs designed to tag the C terminus of a protein of interest
(A) The tagging strategy included causing a double-strand break using CRISPR, then HDR using a donor plasmid.
(B) PCR-amplified HDR donor plasmid fragments used to perform Gibson assembly.
(C) Structure of the complete HDR donor plasmid and PX459 Cas9 vector.
Figure 3.

Alternative HDR donor plasmid designs
(A) HDR donor plasmid design that can be used to tag the N terminus of a protein of interest.
(B) HDR donor plasmid design to tag the C terminus of proteins not expressed in mESCs.
mESCs tissue culture
-
9.One day before thawing mouse embryonic stem cells (ESCs), thaw a vial of mouse embryonic fibroblast (MEF) feeders.
-
a.Pregelatinize tissue culture plates with 0.1% gelatin in PBS−/− (no Magnesium, no Calcium) (wt/vol) for 10 min at room temperature (23°C–25°C). For one 6-well plate, 1 mL is sufficient.
-
b.Aspirate gelatin solution and replace with medium.
-
c.Thaw a vial of feeders in a 37°C water bath, dilute in 5 mL of maintenance medium, centrifuge at 350 × g for 5 min, and plate in the well at 2 × 104 cells/cm2.
-
d.Use mESCs maintenance medium throughout the entire procedure (medium composition in materials and equipment).
-
a.
-
10.
On the following day, change medium on the feeders and thaw a vial of mESCs as detailed in step 9c, and plate on top of the feeders at a concentration of 1 × 104 cells/cm2.
-
11.
Change medium every 1–2 days and passage cells every 2–3 days using 0.05% Trypsin when the cells reach 70%–80% confluency. Split with a ratio of 1:5. Media change and passaging times may vary across cell lines.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-COBRA1/NELFB Working dilution: 1:1000 immunoblot; 1:100 immunostaining | Invitrogen | Cat# PA5-54169, RRID: AB_2639998 |
| Rabbit anti-COBRA1 Working dilution: 1:1000 immunoblot | Cell Signaling Technology | Cat# 14894, RRID: AB_2798637 |
| Mouse anti-HA Working dilution: 1:1000 immunoblot; 1:500 immunostaining | Abcam | Cat# ab130275, RRID: AB_11156884 |
| Rabbit anti-HA Working dilution: 1:1000 immunoblot; 1:500 immunostaining | Cell Signaling Technology | Cat# 3724, RRID: AB_1549585 |
| Rabbit anti-NANOG Working dilution: 1:500 immunostaining | REPROCELL | Cat# RCAB001P, RRID: AB_1962694 |
| Rat anti-SOX2 Working dilution: 1:200 immunostaining | Thermo Fisher Scientific | Cat# 14-9811-82, RRID: AB_11219471 |
| Bacterial and virus strains | ||
| E. coli DH5a | NEB | Cat# C2987I |
| Chemicals, peptides, and recombinant proteins | ||
| Hoechst 33342 | Invitrogen | Cat# H3570 |
| T4 DNA ligase | NEB | Cat# M0202S |
| T4 PNK | NEB | Cat# M0201S |
| BbsI-HF | NEB | Cat# R3539S |
| Quick CIP | NEB | Cat# M0525S |
| Q5 High-Fidelity DNA Polymerase | NEB | Cat# M0491S |
| NEB 5-alpha Competent E. Coli | NEB | Cat# C2987H |
| Trypsin-EDTA (0.05%), phenol red | Thermo Fisher Scientific | Cat# 25300054 |
| DMEM, high glucose | Thermo Fisher Scientific | Cat# 11995073 |
| Sodium Pyruvate (100 mM) | Thermo Fisher Scientific | Cat# 11360070 |
| MEM non-essential amino acids (100×) | Thermo Fisher Scientific | Cat# 11140050 |
| 2-Mercaptoethanol | Thermo Fisher Scientific | Cat# 21985023 |
| Fetal Bovine Serum | Thermo Fisher Scientific | Cat# 26140079 |
| Pen/Strep | Thermo Fisher Scientific | Cat# 15140122 |
| L-Glutamin (200 mM) | Thermo Fisher Scientific | Cat# 25030081 |
| LIF | Lab-made | N/A |
| dTAG-13 | Tocris | Cat# 6605 |
| dTAGv-1 | Tocris | Cat# 6914 |
| Cremophor EL | Sigma | Cat# 238470 |
| Critical commercial assays | ||
| NEBuilder HiFi assembly kit | NEB | Cat# E5520S |
| Pierce™ BCA protein assay kit | Thermo Fisher Scientific | Cat# 23225 |
| QIAprep Spin Miniprep Kit | QIAGEN | Cat# 27104 |
| QIAGEN Plasmid Maxi Kit | QIAGEN | Cat# 12162 |
| QIAquick Gel Extraction Kit | QIAGEN | Cat# 28704 |
| P3 Primary Cell 4D-Nucleofector X Kit L | Lonza | Cat# V4XP-3024 |
| Experimental models: Cell lines | ||
| Mouse: Embryonic stem cell line HK3i | (Kiyonari et al., 2010) | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: wild-type CD1 Purpose: expanding colony Females 7–15 weeks old | Charles River Laboratory | Cat# 022 |
| Oligonucleotides | ||
| Nelfb_sgRNA: CACCGTTCTCC ACAGCCTCACAGTG |
(Abuhashem et al., 2022c) | N/A |
| Nelfb_sgRNA_comp: AAACC ACTGTGAGGCTGTGGAGAAC |
(Abuhashem et al., 2022c) | N/A |
| LA-Nelfb_fwd: TCTGCTCACCCGGGTCTTG | (Abuhashem et al., 2022c) | N/A |
| LA-Nelfb_rev: ATGGTTTCCACCTG CACTCCGGCTGGAGCAGGCACGCT |
(Abuhashem et al., 2022c) | N/A |
| dTAG-Puro_fwd: GGAGTGC AGGTGGAAACCATCTC |
(Abuhashem et al., 2022c) | N/A |
| dTAG-Puro_rev: CTCACAGTGTTC AGGCACCGGGCTTGCG |
(Abuhashem et al., 2022c) | N/A |
| RA-Nelfb_fwd: CGGTGCCTG AACACTGTGAGGCTGTGGAG |
(Abuhashem et al., 2022c) | N/A |
| RA-Nelfb_rev: GCTCTAGAACTAGT GGATCCAGGCCCTACACCAGGTTAC |
(Abuhashem et al., 2022c) | N/A |
| BackBone-AmpR_fwd: GGAT CCACTAGTTCTAGAGC |
(Abuhashem et al., 2022c) | N/A |
| BackBone-AmpR_rev: GGT GTGGGGCAGATTACTGT |
(Abuhashem et al., 2022c) | N/A |
| Genotyping 1: GGTGTG GGGCAGATTACTGT |
(Abuhashem et al., 2022c) | N/A |
| Genotyping 2: CCTGCATAG TCCGGGACATCAT |
(Abuhashem et al., 2022c) | N/A |
| Genotyping 3: CCATGTCT GGTTTTCCTACAGA |
(Abuhashem et al., 2022c) | N/A |
| Genotyping 4: CTTGCCA AAGAACACCCCTC |
(Abuhashem et al., 2022c) | N/A |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-Puro (PX459) V2.0 | (Ran et al., 2013) | Addgene #62988 |
| pCRIS-PITChv2-dTAG-Puro | (Nabet et al., 2018) | Addgene #91796 |
| EasyFusion T2A-H2B-miRFP703 (Backbone source) | (Gu et al., 2018) | Addgene #113097 |
| NELFB_dTAG_Puro_HDR | (Abuhashem et al., 2022a, 2022b, 2022c) | https://benchling.com/s/seq-zeWBfm9fdRAzDVebANg6?m=slm-AwiRiQ5zbwSKYPZJdnek |
| PX459_Nelfb_sgRNA | (Abuhashem et al., 2022a, 2022b, 2022c) | https://benchling.com/s/seq-nEKilKKuDseDHqZFeD3D?m=slm-rqb2baHbGpRSPMfQDCbG |
| Software and algorithms | ||
| Fiji/ImageJ | (Schindelin et al., 2012) | https://imagej.nih.gov/ij/ |
| Benchling | Benchling | www.benchling.com |
| CHOPCHOP | (Labun et al., 2019) | chopchop.cbu.uib.no |
| NEB Tm calculator | NEB | tmcalculator.neb.com |
| NEBuilder | NEB | nebuilder.neb.com |
Materials and equipment
Maintenance medium: Serum/LIF medium
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM, high glucose | N/A | 500 mL |
| FBS | 15% | 75 mL |
| Sodium pyruvate | 1 mM | 6 mL |
| L-glutamine | 2 mM | 6 mL |
| MEM non-essential AA | 1× | 6 mL |
| Penicillin/streptomycin | 1× | 6 mL |
| 2-Mercaptoethanol | 0.05 mM | 600 μL |
| LIF | 10,000 U/mL | n/a |
| Total | N/A | 600 mL |
Once assembled, the medium is stable for up to three weeks at 4°C.
We generate LIF in house, but it’s widely available through commercial sources.
DNA lysis buffer: 10 mM Tris, pH 8.5, 50 mM KCl, 0.01% gelatin, 500 μg/mL proteinase K. This is stable at room temperature (23°C–25°C), but proteinase K has to be added fresh before use (proteinase K should be stored at −20°C).
Equipment: Amaxa 4D-nucleofector X Unit (Lonza, AAF-1002×), 4D-nucleofector Core Unit (Lonza, AAF-1002B). Use the ES cells program CG-104.
Alternatives: Other means of transfection can be used instead of the 4D nucleofector. These include other electroporation systems, as well as lipofectamine 2000 or 3000 which do not require any additional tools or equipment but may be less efficient. Follow the manufacturer’s guidelines for your choice of transfection method.
Step-by-step method details
Plasmids generation
This section describes how to generate both required plasmids for HDR genetic editing; the HDR donor plasmid using HiFi/Gibson assembly, and the CRISPR-Cas9 plasmid using restriction enzyme cloning. HiFi assembly is idea to assemble the HDR donor plasmid since several pieces from different sources can be ligated simultaneously (Gibson et al., 2009). For the CRISPR-Cas9 plasmid, PX459 includes an optimized restriction site to insert the sgRNA (Ran et al., 2013).
-
1.PCR amplify all required pieces for the HDR donor plasmid using Q5 DNA polymerase or an alternative high-fidelity DNA polymerase (day 1–3).
-
a.Use the online NEB Tm calculator (tmcalculator.neb.com) to estimate the annealing temperature for each primer pair acquired from the HiFi NEBuilder tool.
-
b.Follow the manufacturer’s guidelines to estimate extension times. Q5 polymerase generally amplifies 1,000 bp/20–40 s (difficult amplicons can take up to 40 s, while generally 20–30 s is sufficient. Manufacturer’s protocol available here).
-
c.The DNA template for each piece will be different. For homology arms, it will be extracted mouse genomic DNA. For other fragments, it will be the respective source plasmid.PCR reaction master mix
Reagent Amount DNA template Plasmid DNA: 1 pg-5 ng
Genomic DNA: 1 ng- 1 μgDNA Polymerase 0.5 μL Primer 1 (10 μM) 2.5 μL Primer 2 (10 μM) 2.5 μL dNTPs (10 mM) 1 μL 5× Q5 Reaction Buffer 5 μL 5× Q5 High GC Enhancer (optional) 5 μL ddH2O to 50 μL We use 1–3 ng for plasmid DNA, and 10–100 ng for genomic DNA templates. While the GC enhancer is optional, we usually find it helpful given that most assembly primers are long and have high annealing temperatures, which could be reduced with the GC enhancer.PCR cycling conditionsSteps Temperature Time Cycles Initial Denaturation 98°C 30 s 1 Denaturation 98°C 10 s 35 cycles Annealing 55°C–72°C (use Tm calc) 30 s Extension 72°C 30–40 s/kb Final extension 72°C 2 min 1 Hold 4°C N/A N/A -
d.Following amplification of all fragments, run PCR products on a 1.5% agarose gel to isolate the desired amplicon (band on the gel) from each reaction. Then, purify using QIAGEN gel purification kit or a similar kit (troubleshooting 1).
-
e.Store all gel purified fragments at −20°C until assembly.
-
a.
-
2.Anneal and phosphorylate sgRNA oligos (day 1).
-
a.Prepare the following mix for the reaction:
Reagent Amount Oligo one (100 μM) 1 μL Oligo two (100 μM) 1 μL T4 PNK 1 μL T4 DNA Ligase reaction buffer 1 μL ddH2O 5 μL -
b.Incubate the mix in a thermocycler at 37°C for 30 min, then 95°C for 5 min, then ramp down to 25°C at an interval of 5°C/min.
-
c.The product can be stored at −20°C.
-
a.
-
3.Linearize and dephosphorylate CRISPR-Cas9 PX459 vector (day 2).
-
a.Start by purifying the plasmid from a bacterial exponential phase liquid culture using a kit such as the QIAGEN miniprep kit.
-
b.This vector is designed to be digested using BbsI, which will leave matching overhangs for the annealed sgRNA oligos to ligate (Ran et al., 2013). Set up the following digest:
Reagent Amount NEB CutSmart buffer 5 μL NEB BbsI-HF 1 μL PX459 plasmid 1 μg ddH2O To 50 μL -
c.Incubate in a thermocycler at 37°C for 40 min, followed by 65°C for 20 min to deactivate the enzyme.
-
d.Add 2 μL Quick CIP to the 50 μL product reaction and incubate at 37°C for 30 min to dephosphorylate the linearized plasmid.
-
e.Run the product on a 1.5% agarose gel. Cut and gel purify the linearized plasmid band.
-
f.This product can be stored at −20°C long term, and be used to future sgRNA ligations.Note: Dephosphorylation and gel purification are not required in this step. However, they will increase the efficiency of sgRNA ligation to this plasmid (∼100% efficient).
-
a.
-
4.Assemble/ligate both plasmids (day 4).
-
a.For the HDR donor plasmid:
-
i.Prepare the following mix using the NEB HiFi DNA Assembly Cloning Kit.
Reagent Amount NEBuilder HiFi DNA Assembly Master Mix 10 μL 1:1 molar ratios of all purified pieces from step 1 Total 0.2–0.5 pmoles, do not exceed 10 μL ddH2O To 20 μL Note: In our reaction, we have 4 pieces. Depending on available concentrations, we use 0.05–0.1 pmole/piece. For DNA fragments, pmoles = (weight in ng × 1000) / (length of piece in bp × 650). -
ii.Prepare a similar reaction, lacking only one insert piece to serve as a negative control for transformation.
-
iii.Incubate the mix in a thermocycler at 50°C for 60 min.
-
iv.The product is ready for transformation and can be stored at −20°C.
-
i.
-
b.For the PX459 plasmid:
-
i.Dilute the annealed sgRNA oligos from step 2: 1 μL in 200 μL ddH2O.
-
ii.Prepare the following ligation mix.
Reagent Amount T4 DNA Ligase 1 μL T4 DNA Ligase reaction buffer 2 μL Linear, dephosphorylated PX459 20 ng Diluted sgRNA oligos 2 μL ddH2O To 20 μL -
iii.Prepare a similar reaction, lacking the diluter sgRNA oligos to serve as a negative control for transformation.
-
iv.Incubate at room temperature (23°C–25°C) for 15 min, then place on ice.
-
i.
-
a.
-
5.Bacterial transformation of both plasmids (day 5).
-
a.Any transformation competent bacteria may be used. We routinely use NEB 5-alpha competent E. coli. Follow the manufacturer’s guidelines.
-
b.Use 2 μL of each assembled plasmid per 50 μL of bacteria.
-
c.Include a negative assembly mix plate, and a negative PX459 ligation control.
-
a.
-
6.Pick bacterial colonies and inoculate liquid cultures (day 6).
-
a.Note that the negative control plates will be very helpful at this stage. If performed correctly, they should contain no to very few colonies.
-
b.Pick using sterile pipette tips in a sterile field and inoculate 5 mL with the appropriate selection antibiotic of L-broth (LB) per colony (troubleshooting 2).
-
a.
Note: In our experience, both of these reactions are very (∼100%) efficient and should produce hundreds of colonies on a 10 cm bacterial L-agar plate. Picking 3–6 colonies per plate will be sufficient.
-
7.Purify plasmids using a kit such as a QIAGEN miniprep kit (day 7).
-
a.Save 1 mL of each culture and store at 4°C for inoculating a larger scale (maxi or midi) prep culture of your validated clones.
-
a.
-
8.Validate the HDR donor plasmid using restriction enzyme digestion (day 7).
-
a.Choose 2–3 enzymes that produce 2–3 cuts and result in a unique gel signature upon digestion.
-
b.We use Benchling.com to run a virtual digest of the in silico plasmid for easy design.
-
a.
-
9.Validate both plasmids by sequencing (day 7–9).
-
a.Any Sanger or long-read sequencing service would be suitable.
-
b.sgRNAs are too small to detect using restriction enzyme digests, thus sequencing is required.
-
c.For the HDR donor plasmid, the most important region to validate by sequencing is the insert and immediate left and right arm borders to ensure correct in-frame design.
-
a.
-
10.
Inoculate a large culture (100–200 mL) of one sequence-validated clone per plasmid, and purify plasmids using a kit such as the QIAGEN maxiprep kit (day 10–11).
Note: Aim for a concentration of 1–2 μg/μL for each plasmid to be ready for transfection. If concentration is less than 1 μg/μL, concentrate plasmid DNA using isopropanol precipitation or a kit.
Note: If a glycerol bacterial stock is desired, 0.5 mL of the large culture may be mixed with 0.5 mL of 50% glycerol in ddH2O (vol/vol) or LB (without antibiotics) and stored for long term at −80°C.
Pause point: Purified and concentrated plasmid DNAs can be stored at −20°C for short or long term until ready to proceed to the cell transfection step.
mESCs transfection and selection
This section details how to introduce the prepared plasmids into mESCs using an Amaxa 4D Nucleofector, to perform the desired genetic editing, and how to select the targeted cell population to enrich for cells having successfully integrated the HDR cassette. There are several alternative methods to achieve this, and we mention several of them throughout the protocol. In the following step, we use Lonza’s large cuvette/100 μL electroporation reaction. The manufacturer’s guidelines are available here.
-
11.
One day prior to transfection, prepare one 10-cm tissue culture plate by seeding MEFs feeders. Refer to the “before you begin” section for details on culture conditions and concentrations (day 1).
-
12.
Change medium on growing mESCs in culture. Aim to have cells 70%–80% confluent on the day of transfection. Refer to “before you begin” for details on culture conditions and concentrations (day 1).
-
13.Day of transfection (day 2):
-
a.Dissociate 70%–80% confluent mESCs using 0.05% Trypsin. One well of 6-well plate produces ∼1.5–3 × 106 cells, which is sufficient for one transfection.
-
b.Quench the Trypsin with medium and dissociate via pipetting to generate a single cell suspension.
-
c.Count cells using a hemocytometer or automated counter, then centrifuge 2 × 106 cells at 350 × g and aspirate medium.
-
d.Resuspend cells in premixed 100 μL of electroporation buffer: 82 μL Nucleofector Solution and 18 μL Supplement. Pipette up and down gently several times until cells are completely resuspended. Avoid generating air bubbles.
-
e.Add the mix to a new tube containing 5 μg of PX459 and 5 μg of the HDR donor plasmids. Make sure the total volume of both plasmids does not exceed 10 μL (step 10).
-
f.Immediately add the mix to an electroporation cuvette and run ES cells program (GC-104) on a Lonza Amaxa 4D Nucleofector.
-
g.Gently add 500 μL of medium to the cuvette after electroporation. Do not pipette up and down.
-
h.Gently transfer the entire mix to the pre-warmed MEF feeders-covered 10-cm dish.
-
a.
-
14.
Change medium after 24 h. Avoid disturbing the cells as they recover from the electroporation (day 3).
-
15.
Alternatively, cells may be transfected using an alternative method established for use with mESCs, such as other electroporation techniques, or lipid-based approaches, such as Lipofectamine. Follow the manufacturer’s guidelines for each method.
-
16.Initiate puromycin selection 48 h after transfection, and change medium every 1–2 days (day 4- day 10).
-
a.Add puromycin to medium at 2 μg/mL, before medium change.
-
b.Change medium daily for the first 2–3 days of selection to wash off the significant dying cell debris resulting from the initiation of drug selection.
-
c.Notice the expansion of puromycin resistant colonies representing individual clones (troubleshooting 3).
-
a.
Note: Continuous puromycin selection will initially select for all cells that have acquired the PX459 vector. After one week, the puromycin resistance from this vector will be diminished, and only cells that have integrated the plasmid insert correctly in-frame will be puromycin resistant and selected for. Transfection with both plasmids in a circular state minimizes random integration events, and renders the selection extremely efficient. Given that HDR is inefficient, and we are selecting for it, relatively few surviving mESC colonies will be present in the dish after selection ends compared to starting cells. These cells will be sparse and will form ∼50–100 colonies per plate in our experience, that are virtually all clonal. This approach eliminates the need to perform single cell seeding following transfection, which ultimately decreases the time of the protocol and the in vitro culture of correctly targeted mESCs prior to chimera generation. Confirming that all cells within any one clone carry the insertion is also possible using immunostaining, as is described in the validation section below.
Picking, expanding, and freezing clones
This section discusses picking and expanding clones of targeted ESCs for carrying out downstream validation of targeted clones and cryopreservation. After 9–12 days, the colonies have well-demarcated bright edges and a dark center. The following steps will be carried in 96-well dishes to enable high-throughput analyses. Use of multi-channel pipettors is highly recommended, for speed and reproducibility.
-
17.
One day in advance, prepare one flat-bottom 96-well plate with MEF feeders (day 1).
-
18.Proceed to colony picking (day 2).
-
a.Prepare a U- or V-bottom 96-well plate by adding 25 μL 0.05% Trypsin into each well.
-
b.Aspirate medium from targeting/selection plates and wash twice with PBS−/−, then add 7 mL PBS−/− to the plate and proceed to colony picking.
-
c.Under a stereomicroscope in a sterile laminar flow hood, use a pipettor to pick large colonies with well-defined borders and place them in the prepared Trypsin-containing 96-well plate.CRITICAL: Do not pick colonies that are not singular (these are likely to be mixed clones) or too small (these may not proliferate). Do not pick colonies that have differentiation at borders. Additionally, pay attention not to add two colonies to the same well of the multi-well plate, and check under the microscope after picking to exclude any wells that may have more than one colony added. You should perform the colony picking as quickly as possible without stopping until you have picked colonies into all wells or a sufficient number (if you don’t have enough colonies to fill the plate) of the wells. Pick clones that are similar in size to have synchronized culture and splitting time across all wells.Note: In our experience, ∼50% of clones will have at least one correctly edited allele. Picking 36–48 clones consistently generated many correctly targeted clones, including both heterozygous and homozygous alleles.
-
d.Incubate the 96-well plate containing the picked colonies in a 37°C incubator for 10 min. Shake in the middle of the incubation to help dissociate the cells.
-
e.Add 75 μL of medium into each well and pipette up and down 10–15 times to gently dissociate the colonies into single cells.
-
f.Add the entire 100 μL of cells to the prepared 96-well plate with MEF feeders, which should contain 75 μL of medium. The final cells and medium volume in each well will be 175 μL. Return to the incubator.
-
a.
-
19.
Change medium every 1–2 days until the majority of wells have reached 80% confluency (usually 3–5 days).
-
20.Split/freeze the clones (day 6).
-
a.After dissociating the cells with 25 μL Trypsin, resuspend in a total of 200 μL medium.
-
b.Split into three 96-well plates, 50 μL each, for validation assays. Note that passaging these cells will be done without feeders.Note: Since these cells will be used for analysis, there is no need to culture them on feeders. A gelatinized dish will suffice. Refer to “before you begin”: step 9 for details of preparing gelatin-coated dishes.
-
c.Freeze the remaining 50 μL cell suspension by adding 50 μL of 2× freezing medium: 20% DMSO and 80% FBS, for a final freezing medium with 10% DMSO.
-
d.Cover the wells with 80 μL of mineral oil and place in a −80°C freezer for short term storage while clones are being screened and validated.
-
a.
-
21.
Change medium on analysis plates every 1–2 days (day 7–10).
Optional: If more downstream analyses are to be carried than detailed below, it is possible to keep expanding these clones as desired given that they are not being used for experiments requiring good developmental potential.
Validation of correctly targeted clones
This section describes methods used to screen and validate correct targeting of the clones and to determine hetero vs. homozygosity clones. The approach we use is aimed at maximizing speed and efficiency. As a result, we recommend taking advantage of the 2×HA tag as a readily detectable epitope to preliminarily assay correct targeting and degradation using immunofluorescence. Once several correct clones are identified, validation by western blotting for the fusion protein, and PCR for the edited allele can be performed.
-
22.
Once 70% confluent, take two of the three validation plates (step 20), and treat one of them with 500 nM dTAG-13 or dTAGv-1 in maintenance medium for 2 h. Maintain/passage the third plate as needed (day 1).
-
23.Fix the treated and control plates in 4% PFA for 10 min, and perform fluorescent immunostaining (day 2–3).
-
a.Stain both plates using an anti-HA antibody, and if available also an antibody to the target protein. Typical immunostaining procedure can be followed.
-
b.Check clones under an epifluorescence microscope. Take images of clones exhibiting fluorescence signal localized to the subcellular location expected for the target protein in the control plate and an absence of signal in the corresponding wells of the treated plate (in the case of NELFB, a nuclear signal is expected) (Figure 4A) (troubleshooting 4).
-
a.
Note: The strong affinity of anti-HA antibody makes this immunostaining more cost effective than any other analysis for 1st pass screening of clones. In situ immunostaining enables identification of positive clones for the HA-tagged knock-in, the degradation of this tag in the dTAG treated plate, and the correct subcellular localization of the anti-HA signal which should correspond to the localization of the tagged fusion protein (e.g., nucleus vs. cytoplasm vs. plasma membrane).
Note: Certain targets may be exclusively degraded using dTAG-13 or dTAGv-1. If HA staining is present in the correct compartment, but no degradation is observed with dTAG-13, this might be the case, and a repeat of this staining or trying dTAGv-1 in immunoblotting later is warranted. Alternatively, some targets may require several hours of incubation for significant degradation to occur.
-
24.Perform a validation PCR for clones that show HA staining in the expected subcellular compartment (day 4).
-
a.Design PCR primers that amplify both wild-type and edited alleles (Figure 4B).
-
b.DNA can be acquired for any clone of interest from one of the immunostained plates.
-
i.After finishing immunostaining, wash the plate 3× with PBS+/+ (with Magnesium and Calcium).
-
ii.Add 40 μL of DNA lysis buffer to each well (see materials and equipment).
-
iii.Incubate overnight (∼16 h) at 55°C in a humidified chamber.
-
i.
-
a.
Note: Due to the presence of MEF feeders, homozygous clones may show a faint wild-type band in the PCR assay, but it will be of lower intensity than the wild-type band observed in heterozygous clones. If a cleaner PCR is required for validation, cells may be passaged 2–3 times from the remaining validation plate prior to DNA lysis which will be sufficient to dilute out the feeders.
-
25.Identify 3–4 heterozygous and homozygous clones from the PCR and perform a western blot (day 4–7).
-
a.Expand these clones from the remining analysis plate, each into one well in a 24-well plate, then into 2 wells in a 12-well plate. Ultimately, a minimum of two, 12-well plate wells are required for the following immunoblotting analysis. Expand to more wells if other analyses are desired.
-
b.Treat one of the 12-well plates with 500 nM dTAG-13 or dTAG-v1 for 2 h (see step 24 note).
-
c.Isolate protein from these plates and perform immunoblotting analysis using anti-HA and antibody against the target protein. Typical immunoblotting procedure can be followed.
-
d.Tagged alleles will express a fusion protein and show a size shift corresponding to the molecular weight of FKBP12F36V (∼12 kDa) in the target protein blot. The HA blot will show higher (two-fold) intensity for homozygous clones. dTAG treated colonies should show near complete degradation (Figure 4C) (troubleshooting 5, troubleshooting 6).
-
a.
Figure 4.

Validation of correct knock-in of the tag at the endogenous locus encoding a protein of interest, and degradation efficiency of the tagged fusion protein
(A) Immunofluorescence of stained mESC colonies showing examples of negative, heterozygous, and homozygous clones. Scale bars 50 μm.
(B) (top) Structure of edited endogenous allele with validation primers highlighted. (bottom) PCR validation of targeted knock-in of the tag.
(C) Immunoblotting assay showing negative, heterozygous, and homozygous clones +/- dTAG-13.
Chimeric mouse generation and colony establishment
This section discusses the steps to generate a transgenic mouse from targeted mESCs. Generating mice is most commonly achieved by injection or aggregation of correctly targeted mESCs into host 8-cell or blastocyst stage mouse embryos, followed by transfer of embryos into pseudo-pregnant surrogate females (Poueymirou et al., 2007). Alternatively, direct injection of the validated plasmids to mouse zygotes or 2-cell embryos could be pursued (Chen et al., 2016; Gu et al., 2018). Performing microinjections and transferring embryos require technical skills and specialized equipment, and are typically performed as a service by an institutional transgenic core facility or a company. Once a date for injections is arranged with a facility, information on the parental mESCs regards the strain from which they are derived, in particular their coat color, is needed to determine the host wild-type embryo genotype and its derivative coat color (which should contrast with that of the mESCs). 7–10 days in advance, thaw the clones.
-
26.
One day in advance, seed MEFs onto a 12-well plate (day 1).
-
27.Thaw 2–3 of the clones identified as correctly targeted (day 2).
-
a.Take out the freezing plate and thaw in a 37°C incubator for 10–15 min.
-
b.Once thawed, transfer each clone of interest to one medium-containing well. This should be a well in a 24-well plate with 1 mL of medium to dilute the freezing medium as much as possible. The cells will be sparse initially, which is not a problem.
-
a.
-
28.
Change medium daily and passage as needed on feeders (day 3–7) (troubleshooting 7).
-
29.
On the day of injection, dissociate cells into a single cell suspension using Trypsin and submit 1–2 clones for injection/aggregation into wild-type embryos (Figure 5A).
Note: The facility performing your mESC injections should provide you with instruction on how to prepare your cells. Typically, maintenance medium with a cell suspension prepared on ice is suitable. However, adding HEPES buffer may be beneficial for cell survival during injection experiments. We typically submit 2 clones for injection into embryos; one heterozygous and one homozygous. Note that homozygotes should be developmentally neutral if the fusion is behaving as your wild-type protein of interest, which is what you expect. Either heterozygous or homozygous would be sufficient to establish a colony. We prefer 8-cell diploid host embryo injections or 8-cell tetraploid host embryo aggregations over blastocyst injections as they produce completely mESC-derived embryos, expediting breeding and establishing a GEM colony (Eakin and Hadjantonakis, 2006; Nagy et al., 1993; Poueymirou et al., 2007).
-
30.
Once litters are delivered, observe coat color to determine extent of mESC contribution (5 weeks) (troubleshooting 8).
-
31.
Once sexually mature, initiate mouse breeding with the highest percentage male chimeric mice to the desired genetic background towards generating a homozygous colony of animals (4 months) (troubleshooting 9).
Note: Given that the used mESC are typically XY, high percentage chimeras that are female are typically karyotypically abnormal, XO, and often sterile. Unless no viable high contribution males were retrieved, these mice should be avoided.
-
32.
Validate homozygosity and heterozygosity of the targeted protein in the colony using immunoblotting (Figure 5B).
Note: In our experience, outbred female mice (e.g., CD1, ICR, or Swiss Webster) are ideal to cross with chimeric males due to their diversified genetics and large litter size, which enable quick and reliable establishment of a GEM colony. Additionally, F1 mice should be screened for any morphological/behavioral abnormalities that may reflect unintended phenotypes of the tagged protein.
Optional: We highly recommend functional validation of the newly generated colony as soon as possible. This can be done by western immunoblotting for the tagged protein from tissues obtained from heterozygous adult mice. Dissection of embryos or isolation of target cell populations in culture can enable dTAG treatment of these tissues in vitro to ensure proper functionality.
Figure 5.

Generation of a GEM model with NELFB dTAG knock-in from targeted mESCs
(A) Illustration of 8-cell embryo injection and resulting chimeric mouse.
(B) Immunoblotting validation of the GEM colony once established. The samples represent liver tissue lysates.
(C) Intraperitoneal injections in dTAG in mice, and degradation across tissues (other than the brain) shown via immunoblotting.
(D) Left: illustration of the crossing and injections paradigm to recapitulate Nelfb null allele phenotype. Middle: Brightfield image of littermate embryos retrieved at E7.5. Scale bar 250 μm. Right: Optical sections of immunostained embryos from this experiment. Nuclei are visualized using Hoechst. Maximum intensity projection (MIP) of the same embryos provided as well. Scale bar 250 μm. (B) and (C) panels are from (Abuhashem et al., 2022c) and permission to reuse were granted.
In vivo degradation of the targeted protein
Once a dTAG GEM model is generated, it is possible to expose mice or tissues to dTAG small molecules to induce degradation rapidly and reversibly. In the case of the pre-implantation developmental period or any ex vivo culture of cells or tissues derived from post-implantation embryos or adult mice, dTAG-13 or dTAGv-1 can be added to the culture medium (Abuhashem et al., 2022c). For in vivo studies, direct delivery of dTAG-13 or dTAGv-1 can be achieved via simple intraperitoneal injections. In the case of the central nervous system, direct injection (e.g., stereotactic injection into the brain) is required given that neither dTAG-13 nor dTAGv-1 traverse the blood brain barrier.
-
33.
Dissolve dTAG small molecules in DMSO at a concentration of 1 mg/25 μL. Aliquot 25 μL per 1.5 mL tube.
-
34.
Add 475 μL of 10% Cremophor EL in PBS+/+ (vol/vol) to each 25 μL of dTAG small molecules.
Note: Other solubilizers such as Kolliphor can be used in place of Cremophor.
-
35.
Immediately, pipette up and down until the solution becomes clear.
Note: dTAG small molecules are poorly water-soluble and will precipitate once 10% Cremophor EL in PBS is added. Continued pipetting ensures quick and complete resuspension of the precipitated dTAG small molecules.
-
36.
Load 500 μL in 1 mL syringe for injection. Inject per animal facility guidelines.
-
37.
Validate degradation in tissue of interest using assay of choice, such as immunoblotting or immunostaining.
In our experience, a concentration of dTAG-13 or dTAGv-1 of ∼35 mg/kg, is sufficient to induce degradation within ∼4 h in all assayed tissues. However, it is critical to validate and titrate this concentration for one’s protein fusion and tissue of interest (Figure 5C). Intravenous injections can also be performed if higher concentration is needed in tissues and a more acute onset of degradation is desired (troubleshooting 10).
Expected outcomes
The result of this complete protocol will be two models each harboring an endogenously targeted degradable protein of interest: an engineered mESC line, and a derivative GEM model. These two models facilitate experiments in vitro and in vivo respectively, as needed to study the functions of the tagged degradable protein of interest. With respect to the degradation dynamics, many studies have established the power of the dTAG system to deplete proteins to undetectable levels within a few hours in vitro, and our experience with NELFB suggests that these dynamics are also reflected in the in vivo efficiency (Bensimon et al., 2020; Nabet et al., 2018, 2020).
More importantly, the protocol that we present here is modular. It can be adapted to tag and validate a wide variety of proteins. In our experience, the generation of homozygous tagged mESCs is relatively efficient, with each targeting round resulting in at least several homozygous clones. Thus, the streamlined protocol we present here makes it possible to produce these powerful pluripotent stem cell-based models in two to three months. Of note, the efficiency of degradation and targeting may be protein dependent, thus it might be helpful to consider generating constructs that tag the N- and C- termini initially to validate proper folding and degradation of a protein of interest. This can be achieved quickly by assembling plasmids for transgenic expression of the tagged protein of interest at each terminus and performing transient transfections. This is particularly helpful for proteins without known structures/functional domains, and those where no previous studies having tagged them.
With respect to the GEM colony, once the targeting and efficiency are validated in vitro, it is expected that the resulting animals will be homozygous viable and fertile, and not exhibit any overt morphological or behavioral phenotypes, that result from tagging the protein. GEM models are expected to be normal in the absence of degradation.
In the presence of continuous degradation, the resulting tissue is expected to phenocopy a mouse model with a null allele. We recently reported that null Nelfb mouse embryos exhibit reduced growth and failure to specify the primed posterior epiblast at embryonic day (E) 6.5 (Abuhashem et al., 2022a). To recapitulate this defect, we crossed NelfbdTAG/dTAG male with NelfbdTAG/+ female. This crossing strategy ensures producing heterozygous littermates for comparison purposes and avoids potential deleterious effects on a homozygous mother with NELFB depletion. Subsequently, the female received four 1 mg dTAGv-1 injections every 12 h between 5.5 and 7.0 days post conception (dpc). Embryos were then dissected at E7.5. The homozygous embryos exhibited smaller size compared to heterozygous littermates (Figure 5D). Additionally, immunostaining revealed that, while the homozygous embryos exhibit SOX2 staining, a marker of anterior epiblast at E7.5, they lack NANOG staining on the posterior side, a marker of the posterior primed epiblast (Figure 5D). These defects closely resemble the null allele defects we previously reported (Abuhashem et al., 2022a). Note that degradation and recovery kinetics for each protein can be different, and they should be defined prior to attempting recapitulating a null defect. Additionally, this system is suboptimal for long-term studies as repeated injections can become tedious and expensive.
Limitations
There are some limitations associated with applying the dTAG system to a protein of interest for in vivo degradation. The limitations that are specific to our protocol detailed here. With respect to the system, any tag that is introduced into an endogenous protein has the potential to affect its folding and function. This especially applies to proteins that have not been tagged previously, or ones that are known to have critical interactions at N- and C- termini. Introducing the FKBP12F36V to these proteins may affect their folding or the tag, resulting in an unusable system. This issue can be addressed by attempting to introduce the tag at either end initially using a transgene approach before performing HDR targeting. Additionally, introducing linkers can be helpful in creating some disordered separation between the tag and a protein of interest, allowing each to fold properly.
Another limitation of this system is the relatively rapid clearance of dTAG small molecules from mice in vivo. In our experience with mouse embryos developing in utero and with a robustly and widely expressed protein of interest (NELFB), protein expression can recover to normal levels within 24 h of dTAG injection. This recovery was observed to be longer for the liver, suggesting that rates of recovery also depend on the organ/tissue. Thus, continuous or repeated injections might be necessary for long-term experiments of robustly and widely expressed proteins, which may not be ideal for this system, and should be tested empirically. Repeated injections with large volumes could also be difficult or not possible depending on the animal protocol and institution/country. Additionally, dTAGv-1 has been shown to have improved in vivo pharmacokinetics over dTAG-13. Thus, some dTAG molecules may be more effective than others depending on the target protein, tissue, and experimental paradigm. Testing the speed of reversibility in cells in vitro can provide valuable information regarding the feasibility of continuous degradation in vivo (rapid reversibility within a few hours upon dTAG-13 or dTAGv-1 removal suggests that it will be difficult to sustain degradation of a target protein in vivo).
With respect to our protocol, our tagging strategy is primarily for proteins expressed in mESCs, which readily enable antibiotic or fluorescence selection for efficient targeting. For proteins not expressed in mESCs, we recommend a strategy with a selection cassette that is driven by a strong promoter such as PGK, as illustrated (Figure 3).
Troubleshooting
Problem 1
PCR of fragments for the generation of plasmids is not working (step 1).
Potential solution
Optimize the annealing temperature. NEB Tm calculator generally suggests high temperatures for Q5 DNA Polymerase. Attempt temperature gradients with lower temperatures. Additionally, ensure the purity and integrity of the DNA template.
Problem 2
HiFi assembly of the HDR donor plasmid is not working (step 6).
Potential solution
Gel-purify all fragments to ensure clean input into the reaction. Ensure adding the recommended mass of each fragment. Maximize the reaction time to 60 min. Use commercial NEB bacteria, as we noticed better efficiency when we use it.
Problem 3
No colonies on plate after continuous puromycin selection, consequently no knock-in is achieved (step 16).
Potential solution
Ensure that your method of transfection is working using a control (e.g., GFP-expressing) plasmid. Determine the sensitivity of your mESC line to puromycin (e.g., perform a killing curve) to avoid killing cells that have acquired the resistance gene. Given that in our design long-term resistance can only be acquired by colonies which have a correct integration at the endogenous locus, check that the Cas9 vector produces double strand breaks efficiently. This can be achieved via transient transfection of cells in the absence of HDR plasmid, selection with puromycin, collection of DNA after 4 days and PCR a 500 bp region surrounding the cut area, followed by sequencing.
If a cut is achieved, ensure proper design of the HDR donor plasmid and the expression of puromycin in-frame with the targeted gene.
Problem 4
Degradation could not be achieved or is inefficient with dTAG-13 (step 23).
Potential solution
Attempt using dTAGv-1 which recruits the VHL ubiquitination system and may be more effective for certain targets. Alternatively, try other dTAG small molecules, such as dTAG-47, which may be more effective in certain scenarios (Nabet et al., 2018, 2020). It is possible that tagging the other terminus of the protein will result in a better outcome. To avoid repeating all steps, test this possibility initially using a transient transgene approach. Lastly, it is possible that the dTAG system may not be the ideally suited for targeting all proteins of interest, in which case an alternative degradation tagging system such as the AID system should be considered (Yesbolatova et al., 2020).
Problem 5
The tagged protein is less stable than wild-type protein/basal degradation present (step 25).
Potential solution
The folding of a protein of interest may be affected. Attempt tagging the other terminus or separate the FKBP12F36V tag from the protein using a peptide linker.
Problem 6
No homozygous knock-in colonies recovered (step 25).
Potential solution
The tagged protein may not be functional and homozygous tagging results in cell lethality. Consider modifying the tagging strategy, as discussed in the previous troubleshooting points. Alternatively, if yield is low, attempt tagging with two HDR plasmids each having different antibiotic selection (e.g., puromycin and blasticidin), to enable selection and enrichment for homozygous clones.
Problem 7
mESCs are unstable and differentiate before introduction into host embryos for chimera production (step 28).
Potential solution
Ensure culturing of mESCs on a layer of good quality MEF feeders and maintaining proper confluency in the dish (20%–80%). mESCs should not become over confluent. Avoid using medium older that 3-weeks. Use the most stable colonies to move forward.
Problem 8
Low chimeric contribution (step 30).
Potential solution
Ensure proper handling when preparing mESCs for introduction into host mouse embryos. Test selected mESC clones for chromosomal abnormalities (e.g., perform karyotyping). We always recommend performing the editing in a parental mESC line that has good developmental potential, having been previously established to contribute extensively in chimeras with germline transmission, to rule out the possibility that the cell line itself is not fit.
Problem 9
Litters of reduced size result from crossing homozygous mice (step 31).
Potential solution
Outbreed the colony at least 3× prior to homozygosing. Outbred mouse strains generally produce larger and healthier litter sizes than inbred strains.
Problem 10
No in vivo degradation (step 37).
Potential solution
Ensure proper suspension of dTAG small molecules prior to injection. Check genotyping and validate presence of tagged protein using immunoblotting. Alternatively, recovery may be achieved very quickly after a single injection. Confirm speed of recovery in vitro after removal of dTAG small molecules from tissue culture. A complete recovery within few hours suggests that it may be difficult to achieve strong and continuous depletion in vivo due to quick recovery dynamics of target protein and short half-life of dTAG small molecules in vivo. In this case, repeated injections may be attempted if allowed by animal regulations or pursuing alternative methods such as ex vivo cultures.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Anna-Katerina Hadjantonakis (hadj@mskcc.org).
Materials availability
Plasmids, cell lines, and mice generated in this study are available and will be fulfilled upon request.
Acknowledgments
We thank members of the Hadjantonakis lab for constructive feedback on the manuscript and stimulating discussions on the design and application of degron alleles in genetically engineered mouse and pluripotent stem cell-based models. We also thank Elizabeth Robertson and members of her lab for sharing unpublished observations, comments on the manuscript, and discussions. We thank MSK’s Mouse Genetics Core Facility for assistance with injecting targeted mESCs and generating the NelfbdTAG GEM model. A.A. is supported by a MSTP training grant from the NIH (T32GM007739) awarded to the Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD-PhD Program and NIH F30HD103398. Work in A.K.H.’s lab is supported by the NIH (R01HD094868, R01DK127821, R01HD086478, and P30CA008748). Illustrations were created with BioRender.com.
Author contributions
A.A. performed experiments and wrote the original manuscript. A.K.H. supervised the work.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Abderhman Abuhashem, Email: aba2036@med.cornell.edu.
Anna-Katerina Hadjantonakis, Email: hadj@mskcc.org.
Data and code availability
This protocol did not generate or analyze any datasets.
References
- Abuhashem A., Chivu A.G., Zhao Y., Rice E.J., Siepel A., Danko C.G., Hadjantonakis A.-K. RNA Pol II pausing facilitates phased pluripotency transitions by buffering transcription. Preprint at bioRxiv. 2022 doi: 10.1101/2022.04.21.489065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abuhashem A., Garg V., Hadjantonakis A.-K. RNA polymerase II pausing in development: orchestrating transcription. Open Biol. 2022;12:210220. doi: 10.1098/rsob.210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abuhashem A., Lee A.S., Joyner A.L., Hadjantonakis A.-K. Rapid and efficient degradation of endogenous proteins in vivo identifies stage-specific roles of RNA Pol II pausing in mammalian development. Dev. Cell. 2022;57:P1068–P1080.e6. doi: 10.1016/j.devcel.2022.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensimon A., Pizzagalli M.D., Kartnig F., Dvorak V., Essletzbichler P., Winter G.E., Superti-Furga G. Targeted degradation of SLC transporters reveals amenability of multi-pass transmembrane proteins to ligand-induced proteolysis. Cell Chem. Biol. 2020;27:728–739.e9. doi: 10.1016/j.chembiol.2020.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Lee B., Lee A.Y.-F., Modzelewski A.J., He L. Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. J. Biol. Chem. 2016;291:14457–14467. doi: 10.1074/jbc.M116.733154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eakin G.S., Hadjantonakis A.-K. Production of chimeras by aggregation of embryonic stem cells with diploid or tetraploid mouse embryos. Nat. Protoc. 2006;1:1145–1153. doi: 10.1038/nprot.2006.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson D.G., Young L., Chuang R.-Y., Venter J.C., Hutchison C.A., 3rd, Smith H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Gu B., Posfai E., Rossant J. Efficient generation of targeted large insertions by microinjection into two-cell-stage mouse embryos. Nat. Biotechnol. 2018;36:632–637. doi: 10.1038/nbt.4166. [DOI] [PubMed] [Google Scholar]
- Kiyonari H., Kaneko M., Abe S., Aizawa S. Three inhibitors of FGF receptor, ERK, and GSK3 establishes germline-competent embryonic stem cells of C57BL/6N mouse strain with high efficiency and stability. Genesis. 2010;48:317–327. doi: 10.1002/dvg.20614. [DOI] [PubMed] [Google Scholar]
- Labun K., Montague T.G., Krause M., Torres Cleuren Y.N., Tjeldnes H., Valen E. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic. Acids Res. 2019;47:W171–W174. doi: 10.1093/nar/gkz365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B., Ferguson F.M., Seong B.K.A., Kuljanin M., Leggett A.L., Mohardt M.L., Robichaud A., Conway A.S., Buckley D.L., Mancias J.D., et al. Rapid and direct control of target protein levels with VHL-recruiting dTAG molecules. Nat. Commun. 2020;11:4687. doi: 10.1038/s41467-020-18377-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B., Roberts J.M., Buckley D.L., Paulk J., Dastjerdi S., Yang A., Leggett A.L., Erb M.A., Lawlor M.A., Souza A., et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018;14:431–441. doi: 10.1038/s41589-018-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A., Rossant J., Nagy R., Abramow-Newerly W., Roder J.C. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc. Natl. Acad. Sci. USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poueymirou W.T., Auerbach W., Frendewey D., Hickey J.F., Escaravage J.M., Esau L., Doré A.T., Stevens S., Adams N.C., Dominguez M.G., et al. F0 generation mice fully derived from gene-targeted embryonic stem cells allowing immediate phenotypic analyses. Nat. Biotechnol. 2007;25:91–99. doi: 10.1038/nbt1263. [DOI] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yesbolatova A., Saito Y., Kitamoto N., Makino-Itou H., Ajima R., Nakano R., Nakaoka H., Fukui K., Gamo K., Tominari Y., et al. The auxin-inducible degron 2 technology provides sharp degradation control in yeast, mammalian cells, and mice. Nat. Commun. 2020;11:5701. doi: 10.1038/s41467-020-19532-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This protocol did not generate or analyze any datasets.