

Abstract
Isocitrate dehydrogenase (IDH) is an essential metabolic enzyme in the tricarboxylic acid cycle (TAC). The high mutation frequency of the IDH gene plays a complicated role in gliomas. In addition to affecting gliomas directly, mutations in IDH can also alter their immune microenvironment and can change immune-cell function in direct and indirect ways. IDH mutations mediate immune-cell infiltration and function by modulating immune-checkpoint gene expression and chemokine secretion. In addition, IDH mutation-derived D2-hydroxyglutarate can be absorbed by surrounding immune cells, also affecting their functioning. In this review, we summarize current knowledge about the effects of IDH mutations as well as other gene mutations on the immune microenvironment of gliomas. We also describe recent preclinical and clinical data related to IDH-mutant inhibitors for the treatment of gliomas. Finally, we discuss different types of immunotherapy and the immunotherapeutic potential of IDH mutations in gliomas.
Keywords: IDH mutation, Tumor immune microenvironment, Immunotherapy, Glioma
Introduction
Gliomas are the most common and most lethal primary tumors of the central nervous system (CNS) [1]. The 2007 World Health Organization (WHO) classification system for CNS tumors categorizes gliomas from grade I to grade IV (glioblastomas, GBMs). Typically, patients with grade II or III gliomas (also known as lower-grade gliomas, or LGGs) survive from 2 to 3 years to more than 5 years, while those with GBMs generally survive no more than 1 year [2]. According to the data from the cancer genome atlas (TCGA)-LGG and TCGA-GBM cohorts, there are high frequencies of genetic alteration in gliomas [3]. Among these genes, mutation frequencies of gene isocitrate dehydrogenase (IDH), tumor protein P53 (TP53), phosphatase and tensin homolog (PTEN), ATRX chromatin remodeler (ATRX), titin (TTN), epidermal growth factor receptor (EGFR), and capicua transcriptional repressor (CIC) are each > 20% in LGGs or GBMs (Table 1). These mutations generate glioma heterogeneity and regulate the development, evolution, immune evasion, and therapeutic response of gliomas [4–9]. A better understanding of gene mutations in gliomas is crucial for tumor classification and therapy.
Table 1.
Top-30 Mutated Genes in LGGs and GBMs
| LGGs | GBMs | ||
|---|---|---|---|
| Gene names | Mutation frequency (%) | Gene names | Mutation frequency (%) |
| IDH1 | 76.80 | PTEN | 33.50 |
| TP53 | 48.40 | TP53 | 31.50 |
| ATRX | 37.70 | TTN | 25.70 |
| CIC | 21.00 | EGFR | 23.70 |
| TTN | 12.30 | MUC16 | 15.40 |
| FUBP1 | 9.30 | FLG | 13.60 |
| PIK3CA | 8.20 | NF1 | 11.60 |
| NOTCH1 | 7.40 | RYR2 | 10.80 |
| MUC16 | 7.00 | PIK3R1 | 9.80 |
| EGFR | 6.80 | PIK3CA | 9.60 |
| NF1 | 6.00 | SPTA1 | 9.60 |
| SMARCA4 | 4.90 | RB1 | 9.60 |
| PTEN | 4.70 | ATRX | 9.30 |
| FLG | 4.50 | SYNE1 | 8.60 |
| PIK3R1 | 4.30 | OBSCN | 7.60 |
| RYR2 | 4.10 | MUC17 | 7.30 |
| IDH2 | 4.10 | LRP2 | 7.30 |
| OBSCN | 3.90 | PCLO | 7.10 |
| ZBTB20 | 3.90 | HMCN1 | 6.80 |
| ARID1A | 3.70 | PKHD1 | 6.80 |
| NIPBL | 3.50 | COL6A3 | 6.50 |
| PCLO | 3.50 | AHNAK2 | 6.30 |
| HMCN1 | 3.30 | IDH1 | 6.30 |
| MUC17 | 3.10 | DNAH5 | 6.00 |
| APOB | 2.90 | DNAH2 | 6.00 |
| BCOR | 2.90 | USH2A | 5.50 |
| LRP2 | 2.90 | FLG2 | 5.50 |
| ADGRV1 | 2.70 | FAT2 | 5.50 |
| TCF12 | 2.70 | LAMA1 | 5.30 |
| ZNF292 | 2.50 | CFAP47 | 5.30 |
In 2016, the WHO classification system for CNS tumors incorporated molecular parameters into the definitions of tumor entities, with gliomas classified as IDH-mutant or wild-type [10]. Most LGGs and almost all secondary GBMs exhibit IDH mutations [11]. Due to the high frequency of IDH mutations and because they exhibit different biological characteristics, the new 2021 WHO classification of CNS tumors divides adult diffuse gliomas into IDH-mutant; IDH-mutant and 1p/19q-codeleted; and IDH-wild type [12]. In this classification, all IDH-mutant diffuse astrocytic tumors are considered a single type (astrocytoma) and are then graded as CNS WHO grade 2, 3, or 4. IDH-mutant and 1p/19q-co-deleted gliomas are regarded as oligodendroglioma (grade 2, 3). GBMs or IDH-wildtype LGGs in the presence of microvascular proliferation or necrosis or TERT promoter mutation or EGFR gene amplification or +7/−10 chromosome copy number changes are also diagnosed as IDH-wild type (GBMs, grade 4) [12]. Although glioma patients harboring IDH mutations have longer survival times than those without IDH mutations, these mutations are also believed to be an early event during the progression of LGGs to higher-grade gliomas [13, 14]. Hence, it is vital to explore the role of IDH mutations in the development and progression of glioma.
IDHs, including IDH1, IDH2, and IDH3, are essential metabolic enzymes in the tricarboxylic acid cycle (TAC), which converts isocitrate into α-ketoglutarate (α-KG), NAPDH, and CO2 [15]. IDH1 is localized to the cytoplasm and peroxisomes, while IDH2 and IDH3 are localized to the mitochondria [16]. Among these three IDHs, mutations in IDH1 and IDH2 have been found in gliomas, IDH1 being the most frequently mutated metabolic gene [17]. IDH mutations involve the replacement of a single amino-acid in IDH1 (arginine 132 residue, R132) and IDH2 (analogous residue arginine 172, R172; or arginine 140, R140) [18]. These are gain-of-function mutations, mediating the transformation of α-KG into D2-hydroxyglutarate (D2-HG) [19]. D2-HG is a homolog of α-KG, and functions as a competitive inhibitor of α-KG-dependent dioxygenases [5, 20].
It is known that D2-HG regulates gene expression via three mechanisms: transcriptional, post-transcriptional (including translation), and post-translational modifications (Fig. 1). First, D2-HG can inhibit the activity of the ten-eleven translocation (TET) family of methylcytosine hydroxylases, which mediates DNA demethylation by transforming 5-methylcytosine to 5-hydroxymethylcytosine [5]. Inhibition of the TET family of methylcytosine hydroxylases remodels the methylome to establish a CpG-island hypermethylated phenotype, which results in a reorganization of the methylome and transcriptome [21, 22]. Similarly, D2-HG can impair the activity of histone demethylases, thereby inhibiting histone demethylation, which, in turn, modulates gene transcription [5, 23]. Abnormalities in DNA and histone methylation regulate gene expression in a transcriptional manner. Messenger RNA N6-methyladenosine (m6A) methylation is the most common RNA modification [24]. N6-methyladenosine methylation is generally mediated by “writers” (methyltransferases, such as METTL3), “readers” (binding proteins, such as IGF2BP3), and “erasers” [demethylases, such as fat mass and obesity-associated protein (FTO)] [25]. D2-HG has been shown to increase the methylation levels of m6A by inhibiting the activity of demethylases like FTO, which eventually regulates gene expression in a post-transcriptional manner [26–28]. Our previous study found that mutations in IDH1 significantly upregulates the protein level of the transcription factor, hypoxia-inducible factor 1-alpha (HIF-1α) [29]. A subsequent study showed that D2-HG maintains HIF-1α protein stability by inhibiting its ubiquitination, which is mediated by the α-KG-dependent dioxygenase, EGLN, indicating that D2-HG can also regulate gene expression in a post-translational manner [30].
Fig. 1.

A model for the influence of IDH mutations on glioma cells and their surrounding immune microenvironment. In glioma cells, IDH mutations regulate gene expression via three mechanisms: transcriptional (via inhibition of DNA and histone demethylases), post-transcriptional (via inhibition of m6A demethylases), and post-translational (via inhibition of EGLN) modifications. In addition, IDH mutations affect the immune microenvironment of gliomas, modifying immune cell infiltration and functioning in direct (via regulating immune-related gene expression) and indirect ways (via IDH mutation-derived D2-HG).
In the immune microenvironment of gliomas, myeloid cells, including tumor-associated macrophages (TAMs), myeloid-derived suppressor cells, neutrophils, and dendritic cells, represent the largest immune subset [31]. Infiltration by these myeloid cells favors glioma progression and induces resistance to glioma treatment [32]. Although immunotherapies targeting inhibitory checkpoint molecules have been revolutionary for the treatment of solid tumors, the high infiltration of monocytes and low infiltration of lymphocytes induced by glioblastoma (GBM) cells establishes an immunosuppressive microenvironment that is responsible for resistance to immunotherapy [32–35]. Recent glioma studies have indicated that mutations in IDH also affect immune-cell infiltration, which, in turn, mediates the distinctive immune responses of IDH-mutant versus wild-type gliomas. Here, we summarize advances in our knowledge of the immune microenvironment and immunotherapy for IDH-mutant gliomas.
Alterations in Immune Cell Infiltration in IDH-mutant Gliomas
Tumor-associated Macrophages
TAMs are the most common immune cells in the CNS, accounting for 30%–50% of all immune cells [36]. Originally, TAMs were subdivided into microglia (resident macrophages of the CNS) and peripheral blood-derived macrophages [37]. However, it was difficult to distinguish between these two subpopulations because of a lack of unique markers. In 2016, TMEM119 and CD49D/ITGA4 were identified as specific markers for microglia and peripheral blood-derived macrophages, respectively [38, 39]. TAMs play a critical role in both innate and adaptive immunity and have both pro- and anti-glioma functions. Traditionally, TAMs are divided into pro-inflammatory M1 signature and anti-inflammatory M2 signature. M1 signature TAMs are activated by IFN-γ, TNF-α, and TLR and contribute to inflammation [40]. In contrast, M2 signature TAMs occur after exposure to IL-4, IL-10, and IL-13 and promote an anti-inflammatory response, tissue repair, and tumor progression [36, 41]. Recently, results of single-cell RNA-sequencing of the TAMs have shown that both M1 and M2 signature genes are frequently co-expressed in individual cells [42]. Despite the fact that M2 signature TAMs secret TGF-β and IL-10 to promote glioma tumorigenesis and establish an immunosuppressive microenvironment, IL-1β and IL-6 derived from M1 signature TAMs have also been found to facilitate glioma cell growth and invasion [43–48]. In addition, high levels of TAM infiltration are indeed associated with an aggressive tumor subtype and predict a poor prognosis in glioma patients [49]. Hence, targeting TAMs may be a promising approach to the treatment of gliomas.
Mutant IDH1 has been shown to suppress immune-response-related pathways in an unbiased RNA-sequencing study, indicating that IDH1-mutant and wild-type gliomas exhibit different patterns of immune-cell infiltration and responses to immunotherapy [50, 51]. In both IDH1-mutant human and mouse glioma tissues, it has been reported that the infiltration of TAMs is lower than in IDH1-wild-type gliomas [52]. Another study found that, although the total number of TAMs is lower in IDH1-mutant GBM samples, the remaining TAMs are more pro-inflammatory [53]. In contrast, longitudinal single-cell profiling and mass cytometry studies have reported that TAMs from patients with IDH-mutant gliomas exhibit a more immunosuppressive phenotype than IDH-wild-type samples [54]. Apart from that, the microenvironment is also significantly different between IDH-mutant-1p/19q-co-deleted and IDH-mutant gliomas, and in particular in the abundance of TAMs. Gliomas harboring IDH-mutant-1p/19q-co-deleted present lower TAM infiltration with respect to the IDH-mutant [55–57].
Mutation in IDH1 not only alters the ratio of M1 and M2 signatures in TAMs, but also promotes TAM migration in vitro and in vivo. When glioma cells are co-cultured with human primary TAMs for 24 h, in vitro assays have showed that the IDH1 mutation increases the expression of M1 signature markers (CD40, CD80, TNF-α, and IL-12) and downregulates the expression of M2 signature markers (CD206, CD163, and IL10) in TAMs. Moreover, a conditioned medium derived from IDH1-mutant glioma cells significantly promotes TAM migration compared with that from IDH1-wild-type glioma cells. In orthotopic xenografts, isogenic human U87 IDH1-wild-type and -mutant glioma cells have been transplanted into BALB/c immunodeficient (SCID) mice and retain innate immune functions. Similar to the in vitro results, the IDH1-mutant glioma cells have increased TAM recruitment, which promotes the anti-tumor functions of TAMs in vivo [50]. Further results have suggested that IDH1 mutations increase TAM recruitment and the expression of phagocytosis markers through the inhibition of ICAM-1 expression by mediating hypermethylation in its promoter [50].
The oncometabolite D2-HG also plays an important role in mutant IDH1-mediated TAM activation. In human primary TAMs, D2-HG selectively increases the expression of IL-12, but not other markers, and also activates M1-type TAMs [50]. In murine BV2 microglial cells, co-culturing with conditioned media from IDH-wild-type GL261 glioma cells increases the expression of pro-inflammatory genes (IL-6, IL-1β, TNF-α, CCL2, and CXCL10) as well as anti-inflammatory markers, indicating activation of BV2 cells. D2-HG treatment abolishes this conditioned media-mediated pro-inflammatory response in activated BV-2 microglial cells by suppressing the AMPK/mTOR/NF-κB signaling pathway [58]. However, this study did not assess the effects of D2-HG on the anti-inflammatory response. Further studies have shown that D2-HG is taken up by TAMs through solute carrier 13A3 (SLC13A3). When taken up by TAMs, D2-HG acts as an allosteric activator of tryptophan-2,3-dioxygenase 2, which promotes the conversion of L-tryptophan into L-kynurenine (L-Kyn), a ligand of the aryl hydrocarbon receptor (AHR). Increased AHR activity then induces secretion of the immunosuppressive factors IL-10 and TGF-β in TAMs. Interestingly, co-culture of D2-HG-pretreated macrophages with T cells has been shown to promote L-Kyn accumulation and AHR activity in TAMs, resulting in a dose-dependent suppression of T-cell proliferation [54, 59].
CD8+ Cytotoxic T Lymphocytes
CD8+ T cells are one of the most vital immune cells in the adaptive immune response. CD8+ T cells can be classified into three types according to their state of differentiation: naïve, effector, and memory T cells [60]. Naïve CD8+ T cells have not yet received an antigen presentation signal, whereas effector and memory CD8+ T cells have been activated by antigens. During antigen presentation, CD8+ T cells carrying an antigen-specific T-cell receptor (TCR) specifically recognize tumor antigenic peptides on the cell surface, a process that is mediated by MHC class I molecules [61, 62]. Once activated, CD8+ T cells mainly function by secreting pro-inflammatory cytokines, binding to the Fas receptor on target cells via the Fas ligand, and releasing granzymes, thus causing the lysis of target cells [63]. CD8+ T cells obtained from glioma tissue have been shown to be phenotypically CD8+ CD25−, indicating a lack of T-cell activation. In addition, GBM patients with high CD8+ T-cell infiltration at the time of diagnosis are more likely to have a better overall survival than patients with focal CD8+ T-cell infiltration [64]. Therefore, triggering T-cell activation is a promising strategy for glioma treatment.
Decreased CD8+ T-cell numbers have been identified using immunofluorescence assays in IDH-mutant, lower-grade gliomas than in IDH-wild-type gliomas. Sequence data from the TCGA database has demonstrated that LGG tissue harboring IDH mutations has fewer infiltrated CD8+ T cells and lower IFN-γ–induced chemokine gene expression than IDH-wild-type tissue [6, 65]. In a murine glioma model, IDH-mutant gliomas also show lower infiltration of CD8+ cytotoxic T cells than IDH-wild-type gliomas [66]. An in vitro study found that co-culture with conditioned medium from IDHR132H gliomas reduces the migration of CD8+ T cells by inhibiting chemokine secretion mediated by signal transducer and activator of transcription 1 (STAT1) signaling [65].
There are two enantiomers of the metabolite 2-HG, named D2-HG and L2-HG. Unlike D2-HG driven by IDH1/2 mutations, L2-HG accumulation occurs in the context of hypoxia and mitochondrial dysfunction [67, 68]. Several studies have found that the two enantiomers play crucial roles in mediating the infiltration and function of CD8+ T cells. The D2-HG produced by IDH-mutant glioma cells can also be taken up by CD8+ T cells. The imported D2-HG impairs only the activation of these T cells but has no effects on T-cell apoptosis or proliferation [6]. When taken up by T cells, D2-HG interferes with Ca2+-dependent transcriptional activity of nuclear factor of activated T cells and inhibits ATP-dependent TCR signaling and polyamine biosynthesis in T cells, which results in a suppression of T-cell antitumor immunity. Similar results have also been reported in tumor models. L2-HG can be produced by CD8+ T cells in response to TCR triggering and environmental hypoxia. Adoptively transferred CD8+ T cells treated with L2-HG exhibit an increased capacity to proliferate in vitro and persistence in vivo, indicating enhanced anti-tumor efficacy [69]. In gliomas, D2-HG maintains HIF-1α protein stability by inhibiting EGLN [30]. In CD8+ T cells, HIF-1α activation increases their production of L2-HG. Autocrine L2-HG alters CD8+ T-cell differentiation by mediating hypermethylation [69].
Tumor-infiltrating CD4+ T Cells
CD4+ T cells represent a diverse cell population expressing CD4 cell surface markers that are associated with both innate and adaptive immune responses to pathogens and tumors [70]. CD4+ T cells are classified into Th1, Th2, Treg, Th17, and natural killer (NK) T cells based on their functions and cytokine secretion patterns [71]. Similar to TAMs, CD4+ T cells play both anti-tumor and pro-tumor roles depending on the cell subtype [72]. For example, CD4+ Th cells directly recognize antigens on MHC-II-expressing tumor cells and produce lymphokines that impair tumor growth and induce cell death. However, CD4+ Tregs function without this antigenic stimulation and mediate immune suppression by directly producing inhibitory cytokines or by influencing the state and function of dendritic cells (DCs) and other immune-cell subtypes [71]. During glioma progression and growth, CD4+ Tregs have been found to accumulate in both murine and human tumor tissues and to act as potent suppressors of anti-glioma immunity [73]. Targeting CD4+ T cells in combination with DC vaccination can lead to long-term immunity against experimental gliomas [74].
In both human and mouse glioma tissues, fewer CD4+ T cells, including Tregs, are found in IDH-mutant samples than in IDH-wild-type samples [52, 66, 75]. A study reported that the migration, proliferation, differentiation, and cytokine secretion of Th1, Th17, and Treg cells are significantly inhibited by D2-HG [75]. In that study, CD4+ T cells from IDH-wild-type glioma mice were isolated, activated by monoclonal CD3 and CD28 antibodies, and then treated with or without D2-HG. It was found that the proliferation of polarized Th1s, Th17s, and Tregs was significantly inhibited in cells treated with D2-HG. D2-HG promoted the differentiation of these cells at a concentration of 30 mmol/L. In addition, D2-HG suppressed T-cell migration by downregulating CCL19 secretion [75]. Interestingly, the IDH1-mutant protein contains an immunogenic peptide, which has been shown to be suitable for mutation-specific vaccination. MHC-II molecules are responsible for presenting this specific immunogenic epitope to Th1 cells and inducing the Th1 cell response [76]. Since IDH mutations occur in the majority of LGGs and secondary GBMs, a mutation-specific anti-IDH vaccine may be a promising immunotherapeutic strategy for IDH-mutant gliomas.
Natural Killer Cells
NK cells are effective cytotoxic lymphocytes that fight various virus-infected and cancer cells [77]. NK cells are divided into five groups based on their migratory behavior and cytotoxic responses: i. NK cells that kill all target cells; ii. NK cells that do not kill target cells, iii. NK cells that interact with target cells; iv. NK cells that randomly kill target cells based on the net balance of stimuli detected by activating and inhibitory receptors, and v. exhausted NK cells [78]. The functioning of NK cells is modulated by an array of activating (NKp30, NKp44, NKp46, NKp80, NKG2D, CD2, and DNAM-1) and inhibitory (KIRs and NKG2A) receptors. Among these receptors, inhibitory receptors specifically bind to MHC-I molecules to inhibit NK cell cytotoxicity [79].
Unlike TAMs and T cells, a higher infiltration of NK cells has been reported in IDH-mutant gliomas, in which, D2-HG upregulates CX3CL1 expression, with a high expression level of this chemokine inducing NK-cell recruitment by targeting the CX3CL1 receptor. In addition, the higher infiltration of NK cells in IDH1-mutant gliomas is associated with a better prognosis [80]. Interestingly, IDH-mutant glioma cells and astrocytes are resistant to NK-cell-mediated cytolysis. Furthermore, IDH-mutant glioma cells escape NK-cell immune surveillance by downregulating NKG2D ligand expression. The inhibition of DNA methyltransferases increases NKG2D ligand expression in IDH-mutant glioma cells and restores the NK-mediated lysis of IDH-mutant glioma cells in an NKG2D-dependent manner [81]. Hence, targeting DNA methylation may represent a novel strategy to sensitize IDH-mutant gliomas to NK-cell-mediated immune lysis. Demethylation and epigenetic modifications via DNA methyltransferase has been shown to suppress IDH1-mutant glioma growth in combination with temozolomide in a mouse model [82].
Immune Checkpoint Alterations in IDH Mutant Gliomas
PD-1/PD-L1
Programmed cell death 1 (PD-1) is a receptor expressed in activated T cells. This immune inhibitory receptor is involved in regulating T-cell function and differentiation [83, 84]. Similarly, programmed cell death 1 ligand 1(PD-L1) is an immune receptor expressed in both hematopoietic and non-hematopoietic cells, including various tumors [85]. When PD-L1 interacts with its receptor, PD-1, T-cell activation is abolished [86, 87]. In tumors, this interaction provides a means for tumor cell immune escape via cytotoxic T-cell inactivation [88, 89]. Hence, targeting the PD-1/PD-L1 pathway is another potential treatment strategy for gliomas [90].
Anti-PD-1/PD-L1 immunotherapy has been a breakthrough that has prolonged survival times for patients with a variety of cancers [91]. However, patients with GBMs have had limited efficacy with anti-PD-1 therapy, except for in isolated case reports [92]. The low efficacy of immunotherapy for gliomas is likely due to multiple reasons, including the unique immune environment of the brain [91]. However, patients with recurrent GBMs have been shown to benefit from neoadjuvant anti-PD-1 immunotherapy, with intratumoral and systemic immune responses [92]. In IDH-mutant gliomas, the expression levels of PD-L1 and PD-1 are lower than in IDH-wild-type gliomas due to promoter methylation [93–96]. Whether this low PD-L1/PD-1 expression can alter the immune response to immunotherapy remains to be elucidated. Three clinical-phase studies (NCT03557359, NCT03718767, and NCT03925246) related to nivolumab (anti-PD-1) treatment for IDH-mutant, recurrent, or progressive gliomas and IDH-mutant gliomas with or without hypermutator phenotypes are ongoing.
CD47
Cluster of differentiation 47 (CD47), also known as integrin-associated protein, is a ubiquitously-expressed receptor belonging to the immunoglobulin (Ig) superfamily. CD47 possesses a single extracellular V-set IgSF domain, a five transmembrane domain, and a short cytoplasmic domain [97, 98]. Through interactions with its ligand, signal regulatory protein alpha (SIRPα), CD47 alters the phagocytosis of TAMs via the “don’t eat me” signal. In addition, CD47 regulates the activation of T cells, B cells, and dendritic cells by binding with other ligands, such as thrombospondin-1 and integrins [98–100]. In gliomas, CD47 is abnormally upregulated in tumor tissue and cell lines [101]. Disrupting the CD47/SIRPα axis has been shown to induce tumor phagocytosis and to elicit a potent anti-glioblastoma effect [102, 103]. Moreover, treatment with anti-CD47 antibody also has antitumor effects in gliomas and glioma stem cells, indicating that CD47 may be a target for tumor therapy [104].
Compared with IDH1-wild-type glioma cells, CD47 expression is decreased in IDH-mutant cell lines. In IDH1-wild-type glioma cells, the PKM2/β-catenin/BRG1/TCF4 co-factors bind to the CD47 promoter and modulate CD47 transcription. However, when IDH1 is mutated, recruitment of PKM2 and β-catenin to the TCF4 site is diminished, resulting in low CD47 expression. In addition, microglia co-cultured with IDH1-mutant glioma cells exhibit increased phagocytosis [105]. These findings reveal that mutations in IDH1 not only affect the microglial signature and infiltration but also alter microglial phagocytosis.
CTLA-4
Cytotoxic T-lymphocyte associated protein 4 (CTLA-4) is a member of the Ig superfamily and transmits an inhibitory signal to T cells [106]. In an orthotopic glioma model, CTLA-4 blockade has been shown to eradicate glioma cells by reshaping oligoclonal T-cell infiltration [107]. Furthermore, systemic CTLA-4 blockade ameliorates glioma-induced changes in the CD4+ T-cell compartment without affecting regulatory T-cell function [108]. In gliomas, lower CTLA-4 expression and higher methylation of the CTLA-4 promoter have been found in patients with IDH mutations [94, 109]. Whether lower expression of CTLA-4 affects the efficacy of anti-CTLA-4 immunotherapy in IDH-mutant gliomas requires further exploration.
Alterations in Immune Cell Infiltration and Immune Checkpoint in Other Gene-mutant Gliomas
TP53 Mutant Gliomas
The protein TP53 is known as a tumor suppressor and acts as a transcriptional regulator to regulate the tumor cell cycle, apoptosis, and autophagy [110, 111]. Somatic alterations in the TP53 gene in gliomas are of two types: loss-of-function (common) and gain-of-function (rare) [112, 113]. In TP53-mutant GBM pathologic specimens, immune checkpoint CTLA4 presents higher levels than in TP53-wild-type GBMs, indicating a correlation between TP53 mutation and immunologic markers [114]. Further research found that loss of TP53 by mutation might cooperate with the induction of SPARC to promote tumor cell escape from immune surveillance through modulating TAM recruitment and activation [115]. In contrast, gain-of-function mutation of TP53 is positively correlated with the tumor-associated myeloid signature via upregulating cytokines ccl2 and TNFα to promote TAM and other myeloid-derived immune cell infiltration [112].
PTEN Mutant Gliomas
PTEN protein acts as a tumor suppressor in gliomas, and suppressed expression of PTEN is prevalently found in PTEN-mutant gliomas [116]. Mutation in PTEN not only increases expression of the PD-L1 protein in gliomas, but also induces T-cell apoptosis [117, 118]. Subsequent results of a PD-1 immune checkpoint clinical trial have demonstrated that PTEN mutation promotes resistance to GBM immunotherapy via altering the immunosuppressive environment in GBM patients [119].
EGFR Mutant Gliomas
EGFR is a prominent driver in a variety of tumors as well as GBMs, and it has been reported that amplification and over-expression of EGFR is > 50% in GBMs [120]. Activated EGFR induced by EGF stimulates cell proliferation and migration, and inhibits apoptosis via various key signaling pathways such as the JAK/STAT3 and PI3K/AKT pathways [121]. Among EGFR amplifications, EGFRvIII (deletion of exons 2–7) mutation is the most prevalent and accounts for ~66% [122, 123]. Mutations in EGFR can remodel vessel walls and govern the recruitment of leukocytes, myeloid cells, and lymphocytes [124]. In addition, mutant EGFR protein also has immunogenic activity, and vaccines targeting EGFRvIII indeed induce potent T- and B-cell immunity in GBMs to eliminate tumor cells [121, 125].
Ongoing Clinical Trials of IDH Mutant Inhibitors
Considering the crucial role of IDH mutations in both glioma cells and the tumor microenvironment, targeting mutant IDH protein is also a promising strategy for glioma therapy. In this section, we summarize the IDH-mutant inhibitors that are being tested in ongoing clinical trials of patients with gliomas. Other IDH-mutant inhibitors are listed in Table 2 [126–131].
Table 2.
Mutant IDH Inhibitors
| Drug names | Structure | Introductions |
|---|---|---|
| AGI-5198 |
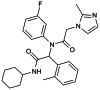
|
AGI-5198 (IDH-C35) is the first highly effective and selective mutant IDH1 inhibitor, with an IC50 = 0.07 and 0.16 μmol/L for IDH1R132H and IDH1R132C, respectively. |
| IDH1 Inhibitor 3 |
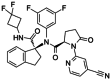
|
IDH1 Inhibitor 3 is a mutant IDH1 inhibitor, with an IC50 of 45 nmol/L for IDH1R132H. |
| IDH-889 |
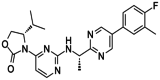
|
IDH-305 is an oral and brain-penetrating inhibitor of mutant IDH1, with an IC50 = 0.02, 0.072, and 1.38 mol/L for recombinant IDH1R132H, IDH1R132C, and wild-type IDH1, respectively. |
| Mutant IDH1-IN-1 |
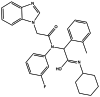
|
Mutant IDH1-IN-1 is a selective inhibitor of mutant IDH1R132H, with IC50 = 81.5 nmol/L. |
| α-Mangostin |
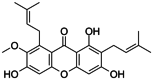
|
α-Mangostin is an inhibitor of mutant IDH1R132H with a Ki of 2.85 μmol/L; IC50 = 2.85 μmol/L. |
| FT-2102 |
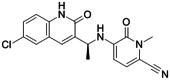
|
FT-2102 is a mutant IDH1 inhibitor, with an IC50 = 9 and 39 nmol/L for IDH1R132H and IDH1R132C, respectively, in U87 glioma cells. |
| AGI-6780 |
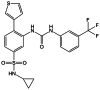
|
IDH2/R140Q mutation inhibitor |
| IDH-305 |
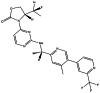
|
IDH-305 is an oral and brain-penetrating inhibitor of mutant IDH1, with an IC50 = 27, 28, and 6,140 nmol/L for recombinant IDH1R132H, IDH1R132C, and wild-type IDH1, respectively. |
| DS-1001b |
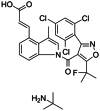
|
Seen in the main manuscript |
| BAY-1436032 |
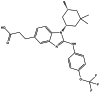
|
Seen in the main manuscript |
| AG-120 |
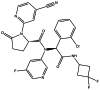
|
Seen in the main manuscript |
| (R,S)-AG-120 |
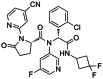
|
Seen in the main manuscript |
| AG-221 |
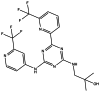
|
Seen in the main manuscript |
| AG-881 |
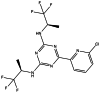
|
Seen in the main manuscript |
IDH-305
IDH-305 is an oral, selective, brain-penetrating IDH1 inhibitor. The IC50 values of IDH-305 for IDH1R132H, IDH1R132C, and IDH1-wild-type gliomas are 27, 28, and 6.14 μmol/L, respectively [132]. IDH-305 decreases D2-HG production and inhibits MCF-10A-IDH1R132H cell growth in a concentration-dependent manner. IDH305 has been shown to suppress D2-HG production in an HCT116-IDH1R132H mouse xenograft model at a dose of 200 mg/kg. Moreover, at 300 mg/kg, it reduces the concentration of D2-HG and inhibits tumor progression in an HMEX2838-IDH1R132C patient-derived melanoma mouse xenograft model [132]. These results indicate that IDH-305 may be a candidate for IDH1-mutant glioma treatment. A Phase I trial of IDH305 in patients with advanced malignancies harboring IDH1R132 mutations is currently ongoing (NCT02381886).
DS-1001b
DS-1001b is an oral selective inhibitor of mutant IDH1R132 that is designed to penetrate the blood-brain barrier and has potential anti-tumor activity. DS-1001b inhibits the proliferation of IDH1-mutant chondrosarcoma cell lines via demethylation of H3K9me3 and decreased D2-HG levels. Continuous administration of DS-1001b impairs tumor growth in xenograft mice [133, 134]. There have been two clinical trials of DS-1001b (NCT03030066 and NCT04458272) related to the treatment of IDH1-mutated gliomas. A first-in-human, multicenter, Phase I study (NCT03030066) included 45 eligible patients with recurrent/progressive IDH1-mutant gliomas who received DS-1001b twice daily (bid), continuously. Recurrent/progressive IDH1-mutant glioma patients responded to this treatment. During treatment, DS-1001b was well tolerated at dosages up to 1400 mg bid with a favorable brain distribution, and the maximum tolerated dose was not reached. Most adverse events (AEs) were grade 1 to 2 and no grade 4 or 5 AEs or serious drug-related AEs were reported [134]. A Phase II study of DS-1001b in patients with IDH1-mutated, WHO grade II gliomas is currently ongoing (NCT04458272). This study is being conducted to assess the efficacy and safety of DS-1001b in patients with chemotherapy- and radiotherapy-naïve, IDH1-mutated, WHO grade II gliomas.
AG881
AG-881 is an orally available, brain-penetrating, second-generation dual mutant IDH1/2 inhibitor. The IC50 of AG-881 ranges from 0.04 to 22 nmol/L against IDH1R132, 7 to 14 nmol/L against IDH2R140Q, and 130 nmol/L against IDH2R172K [135, 136]. AG-881 inhibits the transformation of α-KG into D2-HG, resulting in a reduction in D2-HG production. In vitro and in vivo studies have found that AG-881 readily crosses the blood-brain barrier, induces cell differentiation, and suppresses tumor growth via inhibition of D2-HG-mediated signals [136, 137]. At present, a clinical-phase study (NCT02481154) of orally administered AG-881 is being performed in patients with advanced solid tumors, including gliomas, with IDH1 and/or IDH2 mutations. The purpose of this Phase I, multicenter study is to evaluate the safety, pharmacokinetics, pharmacodynamics, and clinical activity of AG-881 in gliomas harboring an IDH1 and/or IDH2 mutation.
AG-120
AG-120 is an orally active IDH1 inhibitor with potential anti-tumor activity, while (R, S)-AG-120 is an enantiomer of AG-120 with less activity [138]. In IDH1-mutant TF-1 cells and ex vivo cultures of samples from primary human acute myeloid leukemia (AML) patients with mutant IDH1, AG-120 reduces intracellular D2-HG, inhibits growth factor-independent cell proliferation, and restores cell differentiation induced by erythropoietin [139]. In 2018, AG-120 was approved by the U.S. Food and Drug Administration for the treatment of patients with relapsed or refractory AML who have a susceptible IDH1 mutation. Meanwhile, clinical development for the treatment of AML, cholangiocarcinoma, glioma, myelodysplastic syndromes, and solid tumors is ongoing worldwide [140]. For gliomas, a multicenter, open-label, Phase I, dose-escalation and expansion study of AG-120 has been completed. In this study of 66 patients with advanced mutant-IDH1 gliomas, AG-120 was well tolerated at 500 mg once per day orally in 28-day cycles. The grade 3 and 4 AE rates were no more than 20%, and only two patients experienced AEs that were considered treatment-related. Among these AG-120-treated patients, stable disease was achieved in 85.7%, and the median progression-free survival was 13.6 months for non-enhancing gliomas. Furthermore, AG-120 reduced the volume and growth rates of non-enhancing tumors in an exploratory analysis. Hence, AG-120 has a favorable safety profile for IDH1-mutant advanced glioma patients [141]. Two Phase I clinical trials (NCT03343197 and NCT02073994) have been performed to further evaluate the safety and effectiveness of AG-120 in IDH-mutant glioma patients. Moreover, a Phase II study (NCT04056910) focused on the combination of the IDH1 inhibitor, AG-120, and the PD-1 inhibitor, nivolumab, in IDH1-mutant gliomas and advanced solid tumors is ongoing.
AG-221
AG-221 is a mutant IDH2 inhibitor with an IC50 of ~ 16 nmol/L. AG-221 reduces D2-HG by > 90%, reverses in vitro histone and DNA hypermethylation, and induces the differentiation of leukemia cells. In addition, human-specific CD45+ blast cells proliferate in a dose-dependent manner following treatment with AG-221 [142, 143]. The efficacy of AG-221 has been well studied in a primary human AML xenograft model with an IDH2 mutation. AG-221 potently reduces D2-HG levels in the plasma, bone marrow, and urine of engrafted mice. Moreover, treatment with AG-221 has a significant and dose-dependent survival benefit [143]. A Phase I/II, multicenter, open-label, dose-escalation study (NCT02273739) of AG-221 in patients with advanced solid tumors, including gliomas and angioimmunoblastic T-cell lymphomas who harbor an IDH2 mutation, was completed on February 23, 2021. The results of this multicenter study have not yet been published.
Conclusion and Perspectives
Mutations in IDH frequently occur in gliomas and can affect both glioma cells and the immune microenvironment. In glioma cells, IDH mutations alter cell proliferation, apoptosis, autophagy, and temozolomide sensitivity via transcriptional, post-transcriptional, and translational modifications. IDH mutations also affect the immune microenvironment of gliomas, modifying immune cell infiltration and functioning in direct and indirect ways. These IDH mutations affect immune cell migration and function via regulating the secretion of related chemokines and the expression of vital immune checkpoints in glioma cells, respectively. In contrast, IDH mutation-derived D2-HG can be taken up by immune cells, which, in turn, alters their functioning (Fig. 1).
Although mutant IDH affects the immune microenvironment of gliomas, its role in immune cells is complicated. The interaction of PD-L1 with its receptor, PD-1, inhibits CD8+ T-cell activation and cytokine production, with PD-L1 expression decreased in IDH-mutant gliomas. Though IDH-mutant glioma samples with lower PD-L1 expression should have higher levels of activated CD8+ T cells, other studies have found that exogenous D2-HG taken up by CD8+ T cells impairs T-cell activation and antitumor immunity. Interestingly, apart from IDH-mutant gliomas, activated CD8+ T cells also auto-secrete D2-HG to regulate CD8+ T cells differentiation.
Similar to CD8+ T cells, the role of IDH mutations in TAMs is also controversial. Several studies have found that mutations in IDH decrease TAM infiltration, while the remaining cells are more commonly of the M1 type and express higher levels of phagocytosis markers. In addition, mutations in IDH also decrease TAM-inhibitory molecular CD47 expression, and microglia co-cultured with IDH1-mutant glioma cells exhibit increased phagocytosis. In contrast, D2-HG can also be taken up by TAMs via SLC13A3. In TAMs, D2-HG induces the secretion of the immunosuppressive factors IL-10 and TGF-β to establish an immunosuppressive environment, which results in resistance to immunotherapy. Moreover, co-culture of D2-HG-pretreated TAMs with T cells suppresses T-cell proliferation in a dose-dependent manner. It seems that blockade of D2-HG secretion by IDH-mutant glioma cells is a promising immunotherapy target. Despite the fact that IDH-mutant inhibitors, which decrease D2-HG secretion, have been designed to suppress glioma growth in vitro and in vivo, the role of these inhibitors in influencing the immune environment of gliomas requires further exploration.
Current immunotherapeutic strategies for glioma can be divided into four types: vaccination, immune checkpoint blockade, chimeric antigen receptor (CAR)-T cell therapy, and oncolytic viral therapy [33]. Vaccine therapy depends on dendritic cell-mediated presentation of the released lysate such as peptides and antigens derived from tumors to induce the activation of CD8+ cytotoxic T cells, which eventually kills glioma cells [144]. It has been reported that the IDH1-mutant protein contains an immunogenic peptide, which has been shown to be suitable for mutation-specific vaccination [76]. Results of a multicenter, single-arm, open-label, first-in-humans phase I trial demonstrated that IDH1R132H vaccine indeed induces immune responses and improves the two-year progression-free rate in most IDH1-mutant glioma patients [145]. A mutation-specific anti-IDH vaccine is a feasible immunotherapeutic approach for IDH-mutant gliomas. Furthermore, mutations in IDH also alters immune checkpoint molecular expression and immune-cell infiltration, and combination of IDH inhibitors with immune checkpoint inhibitors is also promising for IDH-mutant glioma immunotherapy. Apart from vaccines and immune checkpoint inhibitors, genetically-modified T cells is another interesting immunotherapeutic strategy for gliomas. Briefly, T cells are engineered to express CARs, which recognize domains of antibodies linked to the T cell receptor CD3 ζ-chain and co-stimulatory receptors (such as CD28 and/or TNFRSF9). The engineered CAR T cells also possess antigen recognition domains that are specific for tumor-associated antigens [33]. As previously described, IDH-mutant protein harbors an immunogenic peptide, and CAR T cells targeting IDH-mutant also have potential for glioma immunotherapy. Hence, further exploration of the roles of IDH mutations in glioma cells and in the immune environment of gliomas are vital for future IDH-mutant glioma therapy.
Acknowledgements
We thank Taylor & Francis (www.tandfeditingservices.cn) for its linguistic assistance during the preparation of this manuscript. This review was supported by the Translational Medicine Research Fund of Zhongnan Hospital of Wuhan University (ZLYNXM202011 and ZNLH201901) and the National Health Commission of China (2018ZX-07S-011).
Conflict of interest
The authors declare that they have no conflicts of interest.
Footnotes
Feng Tang and Zhiyong Pan have contributed equally to this work.
Contributor Information
Zefen Wang, Email: wangzf@whu.edu.cn.
Zhiqiang Li, Email: Lilizhiqiang@whu.edu.cn.
References
- 1.Gravendeel LAM, Kouwenhoven MCM, Gevaert O, de Rooi JJ, Stubbs AP, Duijm JE, et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009;69:9065–9072. doi: 10.1158/0008-5472.CAN-09-2307. [DOI] [PubMed] [Google Scholar]
- 2.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cerami E, Gao JJ, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eskilsson E, Røsland GV, Solecki G, Wang QH, Harter PN, Graziani G, et al. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro-oncology. 2018;20:743–752. doi: 10.1093/neuonc/nox191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bunse L, Pusch S, Bunse T, Sahm F, Sanghvi K, Friedrich M, et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med. 2018;24:1192–1203. doi: 10.1038/s41591-018-0095-6. [DOI] [PubMed] [Google Scholar]
- 7.Chen PW, Zhao D, Li J, Liang X, Li JX, Chang A, et al. Symbiotic macrophage-glioma cell interactions reveal synthetic lethality in PTEN-null glioma. Cancer Cell. 2019;35:868–884.e6. doi: 10.1016/j.ccell.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma JH, Benitez JA, Li J, Miki S, de Albuquerque CP, Galatro T, et al. Inhibition of nuclear PTEN tyrosine phosphorylation enhances glioma radiation sensitivity through attenuated DNA repair. Cancer Cell. 2019;36:690–691. doi: 10.1016/j.ccell.2019.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, Yang J, Zheng HR, Tomasek GJ, Zhang P, McKeever PE, et al. Expression of mutant p53 proteins implicates a lineage relationship between neural stem cells and malignant astrocytic glioma in a murine model. Cancer Cell. 2009;15:514–526. doi: 10.1016/j.ccr.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016;131:803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 11.Ichimura K, Pearson DM, Kocialkowski S, Bäcklund LM, Chan R, Jones DTW, et al. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro-oncology. 2009;11:341–347. doi: 10.1215/15228517-2009-025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro-oncology. 2021;23:1231–1251. doi: 10.1093/neuonc/noab106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller JJ, Loebel F, Juratli TA, Tummala SS, Williams EA, Batchelor TT, et al. Accelerated progression of IDH mutant glioma after first recurrence. Neuro-oncology. 2019;21:669–677. doi: 10.1093/neuonc/noz016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan H, Parsons DW, Jin GL, McLendon R, Rasheed BA, Yuan WS, et al. IDH1andIDH2Mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu S, Cadoux-Hudson T, Schofield CJ. Isocitrate dehydrogenase variants in cancer—cellular consequences and therapeutic opportunities. Curr Opin Chem Biol. 2020;57:122–134. doi: 10.1016/j.cbpa.2020.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reitman ZJ, Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J Natl Cancer Inst. 2010;102:932–941. doi: 10.1093/jnci/djq187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waitkus MS, Diplas BH, Yan H. Isocitrate dehydrogenase mutations in gliomas. Neuro-oncology. 2016;18:16–26. doi: 10.1093/neuonc/nov136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pirozzi CJ, Yan H. The implications of IDH mutations for cancer development and therapy. Nat Rev Clin Oncol. 2021;18:645–661. doi: 10.1038/s41571-021-00521-0. [DOI] [PubMed] [Google Scholar]
- 19.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhavya B, Anand CR, Madhusoodanan UK, Rajalakshmi P, Krishnakumar K, Easwer HV, et al. To be wild or mutant: Role of isocitrate dehydrogenase 1 (IDH1) and 2-hydroxy glutarate (2-HG) in gliomagenesis and treatment outcome in glioma. Cell Mol Neurobiol. 2020;40:53–63. doi: 10.1007/s10571-019-00730-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huisman B, Manske G, Carney S, Kalantry S. Functional dissection of the m6A RNA modification. Trends Biochem Sci. 2017;42:85–86. doi: 10.1016/j.tibs.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y, Hsu PJ, Chen YS, Yang YG. Dynamic transcriptomic m 6 A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018;28:616–624. doi: 10.1038/s41422-018-0040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao Y, Ouyang X, Zuo L, Xiao Y, Sun Y, Chang C, et al. R-2HG downregulates ERα to inhibit cholangiocarcinoma via the FTO/m6A-methylated ERα/miR16-5p/YAP1 signal pathway. Mol Ther Oncolytics. 2021;23:65–81. doi: 10.1016/j.omto.2021.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su R, Dong L, Li CY, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m 6 A/MYC/CEBPA signaling. Cell. 2018;172:90–105.e23. doi: 10.1016/j.cell.2017.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qing Y, Dong L, Gao L, Li CY, Li YC, Han L, et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m 6 A/PFKP/LDHB axis. Mol Cell. 2021;81:922–939.e9. doi: 10.1016/j.molcel.2020.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao SM, Lin Y, Xu W, Jiang WQ, Zha ZY, Wang P, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324:261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Semukunzi H, Roy D, Li HY, Khan GJ, Lyu XD, Yuan ST, et al. IDH mutations associated impact on related cancer epidemiology and subsequent effect toward HIF-1α. Biomed Pharmacother. 2017;89:805–811. doi: 10.1016/j.biopha.2017.02.083. [DOI] [PubMed] [Google Scholar]
- 31.Kwok D, Okada H. T-Cell based therapies for overcoming neuroanatomical and immunosuppressive challenges within the glioma microenvironment. J Neurooncol. 2020;147:281–295. doi: 10.1007/s11060-020-03450-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng ZN, Zhang JX, Jiang JZ, He Y, Zhang WY, Mo XP, et al. Remodeling tumor immune microenvironment (TIME) for glioma therapy using multi-targeting liposomal codelivery. J Immunother Cancer. 2020;8:e000207. doi: 10.1136/jitc-2019-000207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamran N, Alghamri MS, Nunez FJ, Shah D, Asad AS, Candolfi M, et al. Current state and future prospects of immunotherapy for glioma. Immunotherapy. 2018;10:317–339. doi: 10.2217/imt-2017-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma QQ, Long WY, Xing CS, Chu JJ, Luo M, Wang HY, et al. Cancer stem cells and immunosuppressive microenvironment in glioma. Front Immunol. 2018;9:2924. doi: 10.3389/fimmu.2018.02924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson DB, Sullivan RJ, Menzies AM. Immune checkpoint inhibitors in challenging populations. Cancer. 2017;123:1904–1911. doi: 10.1002/cncr.30642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19:20–27. doi: 10.1038/nn.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haage V, Semtner M, Vidal RO, Hernandez DP, Pong WW, Chen ZH, et al. Comprehensive gene expression meta-analysis identifies signature genes that distinguish microglia from peripheral monocytes/macrophages in health and glioma. Acta Neuropathol Commun. 2019;7:20. doi: 10.1186/s40478-019-0665-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. 2016;113:E1738–E1746. doi: 10.1073/pnas.1525528113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep. 2016;17:2445–2459. doi: 10.1016/j.celrep.2016.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Franco R, Fernández-Suárez D. Alternatively activated microglia and macrophages in the central nervous system. Prog Neurobiol. 2015;131:65–86. doi: 10.1016/j.pneurobio.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 41.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–555. doi: 10.1016/S1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 42.Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017;18:234. doi: 10.1186/s13059-017-1362-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qi L, Yu HQ, Zhang Y, Zhao DH, Lv P, Zhong Y, et al. IL-10 secreted by M2 macrophage promoted tumorigenesis through interaction with JAK2 in glioma. Oncotarget. 2016;7:71673–71685. doi: 10.18632/oncotarget.12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wesolowska A, Kwiatkowska A, Slomnicki L, Dembinski M, Master A, Sliwa M, et al. Microglia-derived TGF-β as an important regulator of glioblastoma invasion—an inhibition of TGF-β-dependent effects by shRNA against human TGF-β type II receptor. Oncogene. 2008;27:918–930. doi: 10.1038/sj.onc.1210683. [DOI] [PubMed] [Google Scholar]
- 45.Ye XZ, Xu SL, Xin YH, Yu SC, Ping YF, Chen L, et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J Immunol. 2012;189:444–453. doi: 10.4049/jimmunol.1103248. [DOI] [PubMed] [Google Scholar]
- 46.Sørensen MD, Kristensen BW. Tumour-associated CD204 + microglia/macrophages accumulate in perivascular and perinecrotic niches and correlate with an interleukin-6-enriched inflammatory profile in glioblastoma. Neuropathol Appl Neurobiol. 2022;48:e12772. doi: 10.1111/nan.12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang YJ, Yu GZ, Chu HY, Wang XJ, Xiong LL, Cai GQ, et al. Macrophage-associated PGK1 phosphorylation promotes aerobic glycolysis and tumorigenesis. Mol Cell. 2018;71:201–215.e7. doi: 10.1016/j.molcel.2018.06.023. [DOI] [PubMed] [Google Scholar]
- 48.Kai K, Komohara Y, Esumi S, Fujiwara Y, Yamamoto T, Uekawa K, et al. Macrophage/microglia-derived IL-1β induces glioblastoma growth via the STAT3/NF-κB pathway. Hum Cell. 2022;35:226–237. doi: 10.1007/s13577-021-00619-8. [DOI] [PubMed] [Google Scholar]
- 49.Sørensen MD, Dahlrot RH, Boldt HB, Hansen S, Kristensen BW. Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol Appl Neurobiol. 2018;44:185–206. doi: 10.1111/nan.12428. [DOI] [PubMed] [Google Scholar]
- 50.Ma D, Zhan DQ, Fu Y, Wei S, Lal B, Wang J, et al. Mutant IDH1 promotes phagocytic function of microglia/macrophages in gliomas by downregulating ICAM1. Cancer Lett. 2021;517:35–45. doi: 10.1016/j.canlet.2021.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wei S, Wang J, Oyinlade O, Ma D, Wang SY, Kratz L, et al. Heterozygous IDH1R132H/WT created by “single base editing” inhibits human astroglial cell growth by downregulating YAP. Oncogene. 2018;37:5160–5174. doi: 10.1038/s41388-018-0334-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amankulor NM, Kim Y, Arora S, Kargl J, Szulzewsky F, Hanke M, et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017;31:774–786. doi: 10.1101/gad.294991.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Poon CC, Gordon PMK, Liu K, Yang RZ, Sarkar S, Mirzaei R, et al. Differential microglia and macrophage profiles in human IDH-mutant and-wild type glioblastoma. Oncotarget. 2019;10:3129–3143. doi: 10.18632/oncotarget.26863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Friedrich M, Sankowski R, Bunse L, Kilian M, Green E, Ramallo Guevara C, et al. Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat Cancer. 2021;2:723–740. doi: 10.1038/s43018-021-00201-z. [DOI] [PubMed] [Google Scholar]
- 55.Lin WZ, Qiu XX, Sun P, Ye YL, Huang QT, Kong L, et al. Association of IDH mutation and 1p19q co-deletion with tumor immune microenvironment in lower-grade glioma. Mol Ther Oncolytics. 2021;21:288–302. doi: 10.1016/j.omto.2021.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. 2017;355:eaai8478. doi: 10.1126/science.aai8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barthel FP, Johnson KC, Varn FS, Moskalik AD, Tanner G, Kocakavuk E, et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature. 2019;576:112–120. doi: 10.1038/s41586-019-1775-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han CJ, Zheng JY, Sun L, Yang HC, Cao ZQ, Zhang XH, et al. The oncometabolite 2-hydroxyglutarate inhibits microglial activation via the AMPK/mTOR/NF-κB pathway. Acta Pharmacol Sin. 2019;40:1292–1302. doi: 10.1038/s41401-019-0225-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Dierendonck XAMH, de Goede KE, van den Bossche J. IDH-mutant brain tumors hit the Achilles' heel of macrophages with R-2-hydroxyglutarate. Trends Cancer. 2021;7:666–667. doi: 10.1016/j.trecan.2021.06.003. [DOI] [PubMed] [Google Scholar]
- 60.Gupta SS, Wang J, Chen M. Metabolic reprogramming in CD8 + T cells during acute viral infections. Front Immunol. 2020;11:1013. doi: 10.3389/fimmu.2020.01013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kelly A, Trowsdale J. Genetics of antigen processing and presentation. Immunogenetics. 2019;71:161–170. doi: 10.1007/s00251-018-1082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shastri N, Cardinaud S, Schwab SR, Serwold T, Kunisawa J. All the peptides that fit: The beginning, the middle, and the end of the MHC class I antigen-processing pathway. Immunol Rev. 2005;207:31–41. doi: 10.1111/j.0105-2896.2005.00321.x. [DOI] [PubMed] [Google Scholar]
- 63.van Duijn J, Kuiper J, Slütter B. The many faces of CD8+ T cells in atherosclerosis. Curr Opin Lipidol. 2018;29:411–416. doi: 10.1097/MOL.0000000000000541. [DOI] [PubMed] [Google Scholar]
- 64.Gieryng A, Pszczolkowska D, Walentynowicz KA, Rajan WD, Kaminska B. Immune microenvironment of gliomas. Lab Invest. 2017;97:498–518. doi: 10.1038/labinvest.2017.19. [DOI] [PubMed] [Google Scholar]
- 65.Kohanbash G, Carrera DA, Shrivastav S, Ahn BJ, Jahan N, Mazor T, et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J Clin Invest. 2017;127:1425–1437. doi: 10.1172/JCI90644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Richardson LG, Nieman LT, Stemmer-Rachamimov AO, Zheng XS, Stafford K, Nagashima H, et al. IDH-mutant gliomas harbor fewer regulatory T cells in humans and mice. Oncoimmunology. 2020;9:1806662. doi: 10.1080/2162402X.2020.1806662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Intlekofer AM, Dematteo RG, Venneti S, Finley LWS, Lu C, Judkins AR, et al. Hypoxia induces production of L-2-hydroxyglutarate. Cell Metab. 2015;22:304–311. doi: 10.1016/j.cmet.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mullen AR, Hu ZP, Shi XL, Jiang L, Boroughs LK, Kovacs Z, et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 2014;7:1679–1690. doi: 10.1016/j.celrep.2014.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Veliça P, et al. S-2-hydroxyglutarate regulates CD8+ T-lymphocyte fate. Nature. 2016;540:236–241. doi: 10.1038/nature20165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Accogli T, Bruchard M, Végran F. Modulation of CD4 T cell response according to tumor cytokine microenvironment. Cancers. 2021;13:373. doi: 10.3390/cancers13030373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kennedy R, Celis E. Multiple roles for CD4+ T cells in anti-tumor immune responses. Immunol Rev. 2008;222:129–144. doi: 10.1111/j.1600-065X.2008.00616.x. [DOI] [PubMed] [Google Scholar]
- 72.Ahrends T, Borst J. The opposing roles of CD4 + T cells in anti-tumour immunity. Immunology. 2018;154:582–592. doi: 10.1111/imm.12941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Grauer OM, Nierkens S, Bennink E, Toonen LWJ, Boon L, Wesseling P, et al. CD4+FoxP3+ regulatory T cells gradually accumulate in gliomas during tumor growth and efficiently suppress antiglioma immune responses in vivo. Int J Cancer. 2007;121:95–105. doi: 10.1002/ijc.22607. [DOI] [PubMed] [Google Scholar]
- 74.Maes W, Rosas GG, Verbinnen B, Boon L, de Vleeschouwer S, Ceuppens JL, et al. DC vaccination with anti-CD25 treatment leads to long-term immunity against experimental glioma. Neuro Oncol. 2009;11:529–542. doi: 10.1215/15228517-2009-004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang LJ, Sorensen MD, Kristensen BW, Reifenberger G, McIntyre TM, Lin F. D-2-hydroxyglutarate is an intercellular mediator in IDH-mutant gliomas inhibiting complement and T cells. Clin Cancer Res. 2018;24:5381–5391. doi: 10.1158/1078-0432.CCR-17-3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. 2014;512:324–327. doi: 10.1038/nature13387. [DOI] [PubMed] [Google Scholar]
- 77.Maher DP, Walia D, Heller NM. Suppression of human natural killer cells by different classes of opioids. Anesth Analg. 2019;128:1013–1021. doi: 10.1213/ANE.0000000000004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu YL, Li J, Jabbarzadeh Kaboli P, Shen J, Wu X, Zhao YS, et al. Natural killer cells as a double-edged sword in cancer immunotherapy: a comprehensive review from cytokine therapy to adoptive cell immunotherapy. Pharmacol Res. 2020;155:104691. doi: 10.1016/j.phrs.2020.104691. [DOI] [PubMed] [Google Scholar]
- 79.Ogbomo H, Cinatl J, Jr, Mody CH, Forsyth PA. Immunotherapy in gliomas: limitations and potential of natural killer (NK) cell therapy. Trends Mol Med. 2011;17:433–441. doi: 10.1016/j.molmed.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 80.Ren FF, Zhao QT, Huang L, Zheng YJ, Li LF, He QY, et al. The R132H mutation in IDH1 promotes the recruitment of NK cells through CX3CL1/CX3CR1 chemotaxis and is correlated with a better prognosis in gliomas. Immunol Cell Biol. 2019;97:457–469. doi: 10.1111/imcb.12225. [DOI] [PubMed] [Google Scholar]
- 81.Zhang XR, Rao A, Sette P, Deibert C, Pomerantz A, Kim WJ, et al. IDH mutant gliomas escape natural killer cell immune surveillance by downregulation of NKG2D ligand expression. Neuro-oncology. 2016;18:1402–1412. doi: 10.1093/neuonc/now061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yamashita AS, da Costa Rosa M, Borodovsky A, Festuccia WT, Chan T, Riggins GJ. Demethylation and epigenetic modification with 5-azacytidine reduces IDH1 mutant glioma growth in combination with temozolomide. Neuro-oncology. 2019;21:189–200. doi: 10.1093/neuonc/noy146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dong H, Zhu G, Tamada K, Chen L. B7–H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 84.Xi X, Liu JM, Guo JY. Correlation of PD-1/PD-L1 signaling pathway with treg/Th17 imbalance from asthmatic children. Int Arch Allergy Immunol. 2018;176:255–267. doi: 10.1159/000489338. [DOI] [PubMed] [Google Scholar]
- 85.Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48:434–452. doi: 10.1016/j.immuni.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jiang XJ, Wang J, Deng XY, Xiong F, Ge JS, Xiang B, et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. 2019;18:10. doi: 10.1186/s12943-018-0928-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: Seven steps to success (or failure) Ann Oncol. 2016;27:1492–1504. doi: 10.1093/annonc/mdw217. [DOI] [PubMed] [Google Scholar]
- 90.Xue S, Hu M, Iyer V, Yu JM. Blocking the PD-1/PD-L1 pathway in glioma: a potential new treatment strategy. J Hematol Oncol. 2017;10:81. doi: 10.1186/s13045-017-0455-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Khasraw M, Reardon DA, Weller M, Sampson JH. PD-1 inhibitors: Do they have a future in the treatment of glioblastoma? Clin Cancer Res. 2020;26:5287–5296. doi: 10.1158/1078-0432.CCR-20-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25:477–486. doi: 10.1038/s41591-018-0337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Berghoff AS, Kiesel B, Widhalm G, Wilhelm D, Rajky O, Kurscheid S, et al. Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro-oncology. 2017;19:1460–1468. doi: 10.1093/neuonc/nox054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Röver LK, Gevensleben H, Dietrich J, Bootz F, Landsberg J, Goltz D, et al. PD-1 (PDCD1) promoter methylation is a prognostic factor in patients with diffuse lower-grade gliomas harboring isocitrate dehydrogenase (IDH) mutations. EBioMedicine. 2018;28:97–104. doi: 10.1016/j.ebiom.2018.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mu LY, Long Y, Yang CL, Jin LC, Tao HP, Ge HT, et al. The IDH1 mutation-induced oncometabolite, 2-hydroxyglutarate, may affect DNA methylation and expression of PD-L1 in gliomas. Front Mol Neurosci. 2018;11:82. doi: 10.3389/fnmol.2018.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang Z, Zhang CB, Liu X, Wang ZL, Sun LH, Li GZ, et al. Molecular and clinical characterization of PD-L1 expression at transcriptional level via 976 samples of brain glioma. Oncoimmunology. 2016;5:e1196310. doi: 10.1080/2162402X.2016.1196310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001;11:130–135. doi: 10.1016/S0962-8924(00)01906-1. [DOI] [PubMed] [Google Scholar]
- 98.Logtenberg MEW, Scheeren FA, Schumacher TN. The CD47-SIRPα immune checkpoint. Immunity. 2020;52:742–752. doi: 10.1016/j.immuni.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu XJ, Kwon H, Li ZH, Fu YX. Is CD47 an innate immune checkpoint for tumor evasion? J Hematol Oncol. 2017;10:12. doi: 10.1186/s13045-016-0381-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hayat SMG, Bianconi V, Pirro M, Jaafari MR, Hatamipour M, Sahebkar A. CD47: role in the immune system and application to cancer therapy. Cell Oncol (Dordr) 2020;43:19–30. doi: 10.1007/s13402-019-00469-5. [DOI] [PubMed] [Google Scholar]
- 101.Liu XJ, Wu X, Wang YM, Li YH, Chen XL, Yang WC, et al. CD47 promotes human glioblastoma invasion through activation of the PI3K/Akt pathway. Oncol Res. 2019;27:415–422. doi: 10.3727/096504018X15155538502359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hutter G, Theruvath J, Graef CM, Zhang M, Schoen MK, Manz EM, et al. Microglia are effector cells of CD47-SIRPα antiphagocytic axis disruption against glioblastoma. Proc Natl Acad Sci U S A. 2019;116:997–1006. doi: 10.1073/pnas.1721434116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang XY, Chen W, Fan JJ, Wang SF, Xian ZS, Luan JY, et al. Disrupting CD47-SIRPα axis alone or combined with autophagy depletion for the therapy of glioblastoma. Carcinogenesis. 2018;39:689–699. doi: 10.1093/carcin/bgy041. [DOI] [PubMed] [Google Scholar]
- 104.Li F, Lv BK, Liu Y, Hua T, Han JB, Sun CM, et al. Blocking the CD47-SIRPα axis by delivery of anti-CD47 antibody induces antitumor effects in glioma and glioma stem cells. Oncoimmunology. 2017;7:e1391973. doi: 10.1080/2162402X.2017.1391973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gowda P, Patrick S, Singh A, Sheikh T, Sen E. Mutant isocitrate dehydrogenase 1 disrupts PKM2-β-catenin-BRG1 transcriptional network-driven CD47 expression. Mol Cell Biol. 2018;38:e00001–e00018. doi: 10.1128/MCB.00001-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang CF, Chen JN, Song Q, Sun XY, Xue MJ, Yang ZY, et al. Comprehensive analysis of CTLA-4 in the tumor immune microenvironment of 33 cancer types. Int Immunopharmacol. 2020;85:106633. doi: 10.1016/j.intimp.2020.106633. [DOI] [PubMed] [Google Scholar]
- 107.Field CS, Hunn MK, Ferguson PM, Ruedl C, Ancelet LR, Hermans IF. Blocking CTLA-4 while priming with a whole cell vaccine reshapes the oligoclonal T cell infiltrate and eradicates tumors in an orthotopic glioma model. OncoImmunology. 2018;7:e1376154. doi: 10.1080/2162402X.2017.1376154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fecci PE, Ochiai H, Mitchell DA, Grossi PM, Sweeney AE, Archer GE, et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res. 2007;13:2158–2167. doi: 10.1158/1078-0432.CCR-06-2070. [DOI] [PubMed] [Google Scholar]
- 109.Liu FK, Huang J, Liu XM, Cheng Q, Luo CK, Liu ZX. CTLA-4 correlates with immune and clinical characteristics of glioma. Cancer Cell Int. 2020;20:7. doi: 10.1186/s12935-019-1085-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yin YL, Stephen CW, Luciani MG, Fåhraeus R. p53 Stability and activity is regulated by Mdm2-mediated induction of alternative p53 translation products. Nat Cell Biol. 2002;4:462–467. doi: 10.1038/ncb801. [DOI] [PubMed] [Google Scholar]
- 111.Sabapathy K, Lane DP. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat Rev Clin Oncol. 2018;15:13–30. doi: 10.1038/nrclinonc.2017.151. [DOI] [PubMed] [Google Scholar]
- 112.Ham SW, Jeon HY, Jin X, Kim EJ, Kim JK, Shin YJ, et al. TP53 gain-of-function mutation promotes inflammation in glioblastoma. Cell Death Differ. 2019;26:409–425. doi: 10.1038/s41418-018-0126-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pfaff E, Remke M, Sturm D, Benner A, Witt H, Milde T, et al. TP53 mutation is frequently associated with CTNNB1 mutation or MYCN amplification and is compatible with long-term survival in medulloblastoma. J Clin Oncol. 2010;28:5188–5196. doi: 10.1200/JCO.2010.31.1670. [DOI] [PubMed] [Google Scholar]
- 114.Rahman M, Kresak J, Yang CL, Huang JP, Hiser W, Kubilis P, et al. Analysis of immunobiologic markers in primary and recurrent glioblastoma. J Neurooncol. 2018;137:249–257. doi: 10.1007/s11060-017-2732-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Thomas SL, Schultz CR, Mouzon E, Golembieski WA, El Naili R, Radakrishnan A, et al. Loss of sparc in p53-null astrocytes promotes macrophage activation and phagocytosis resulting in decreased tumor size and tumor cell survival. Brain Pathol. 2015;25:391–400. doi: 10.1111/bpa.12161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fults D, Pedone C. Immunocytochemical mapping of the phosphatase and tensin homolog (PTEN/MMAC1) tumor suppressor protein in human gliomas. Neuro-oncology. 2000;2:71–79. doi: 10.1093/neuonc/2.2.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7–H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84–88. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 118.Waldron JS, Yang I, Han S, Tihan T, Sughrue ME, Mills SA, et al. Implications for immunotherapy of tumor-mediated T-cell apoptosis associated with loss of the tumor suppressor PTEN in glioblastoma. J Clin Neurosci. 2010;17:1543–1547. doi: 10.1016/j.jocn.2010.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhao JF, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. 2019;25:462–469. doi: 10.1038/s41591-019-0349-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fan QW, Cheng CK, Gustafson WC, Charron E, Zipper P, Wong RA, et al. EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer Cell. 2013;24:438–449. doi: 10.1016/j.ccr.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chistiakov DA, Chekhonin IV, Chekhonin VP. The EGFR variant III mutant as a target for immunotherapy of glioblastoma multiforme. Eur J Pharmacol. 2017;810:70–82. doi: 10.1016/j.ejphar.2017.05.064. [DOI] [PubMed] [Google Scholar]
- 122.Binder ZA, Thorne AH, Bakas S, Wileyto EP, Bilello M, Akbari H, et al. Epidermal growth factor receptor extracellular domain mutations in glioblastoma present opportunities for clinical imaging and therapeutic development. Cancer Cell. 2018;34:163–177.e7. doi: 10.1016/j.ccell.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 124.Segura-Collar B, Garranzo-Asensio M, Herranz B, Hernández-Sanmiguel E, Cejalvo T, Casas BS, et al. Tumor-derived pericytes driven by EGFR mutations govern the vascular and immune microenvironment of gliomas. Cancer Res. 2021;81:2142–2156. doi: 10.1158/0008-5472.CAN-20-3558. [DOI] [PubMed] [Google Scholar]
- 125.Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Bigner DD. Tumor-specific immunotherapy targeting the EGFRvIII mutation in patients with malignant glioma. Semin Immunol. 2008;20:267–275. doi: 10.1016/j.smim.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zheng QG, Tang S, Fu XL, Chen ZQ, Ye Y, Lan XJ, et al. Discovery and structure-activity-relationship study of novel conformationally restricted indane analogues for mutant isocitric dehydrogenase 1 (IDH1) inhibitors. Bioorg Med Chem Lett. 2017;27:5262–5266. doi: 10.1016/j.bmcl.2017.10.029. [DOI] [PubMed] [Google Scholar]
- 128.Levell JR, Caferro T, Chenail G, Dix I, Dooley J, Firestone B, et al. Optimization of 3-pyrimidin-4-yl-oxazolidin-2-ones as allosteric and mutant specific inhibitors of IDH1. ACS Med Chem Lett. 2016;8:151–156. doi: 10.1021/acsmedchemlett.6b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Deng GJ, Shen JQ, Yin M, McManus J, Mathieu M, Gee P, et al. Selective inhibition of mutant isocitrate dehydrogenase 1 (IDH1) via disruption of a metal binding network by an allosteric small molecule. J Biol Chem. 2015;290:762–774. doi: 10.1074/jbc.M114.608497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kim HJ, Fei X, Cho SC, Choi BY, Ahn HC, Lee K, et al. Discovery of α-mangostin as a novel competitive inhibitor against mutant isocitrate dehydrogenase-1. Bioorg Med Chem Lett. 2015;25:5625–5631. doi: 10.1016/j.bmcl.2015.10.034. [DOI] [PubMed] [Google Scholar]
- 131.Caravella JA, Lin J, Diebold RB, Campbell AM, Ericsson A, Gustafson G, et al. Structure-based design and identification of FT-2102 (olutasidenib), a potent mutant-selective IDH1 inhibitor. J Med Chem. 2020;63:1612–1623. doi: 10.1021/acs.jmedchem.9b01423. [DOI] [PubMed] [Google Scholar]
- 132.Cho YS, Levell JR, Liu G, Caferro T, Sutton J, Shafer CM, et al. Discovery and evaluation of clinical candidate IDH305, a brain penetrant mutant IDH1 inhibitor. ACS Med Chem Lett. 2017;8:1116–1121. doi: 10.1021/acsmedchemlett.7b00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nakagawa M, Nakatani F, Matsunaga H, Seki T, Endo M, Ogawara Y, et al. Selective inhibition of mutant IDH1 by DS-1001b ameliorates aberrant histone modifications and impairs tumor activity in chondrosarcoma. Oncogene. 2019;38:6835–6849. doi: 10.1038/s41388-019-0929-9. [DOI] [PubMed] [Google Scholar]
- 134.Natsume A, Wakabayashi T, Miyakita Y, Narita Y, Mineharu Y, Arakawa Y, et al. Phase I study of a brain penetrant mutant IDH1 inhibitor DS-1001b in patients with recurrent or progressive IDH1 mutant gliomas. J Clin Oncol. 2004;2019:37. doi: 10.1093/neuonc/noac155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ma TF, Zou FX, Pusch S, Xu YG, von Deimling A, Zha XM. Inhibitors of mutant isocitrate dehydrogenases 1 and 2 (mIDH1/2): an update and perspective. J Med Chem. 2018;61:8981–9003. doi: 10.1021/acs.jmedchem.8b00159. [DOI] [PubMed] [Google Scholar]
- 136.Fujii T, Khawaja MR, DiNardo CD, Atkins JT, Janku F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med. 2016;21:373–380. [PubMed] [Google Scholar]
- 137.Upadhyay VA, Brunner AM, Fathi AT. Isocitrate dehydrogenase (IDH) inhibition as treatment of myeloid malignancies: progress and future directions. Pharmacol Ther. 2017;177:123–128. doi: 10.1016/j.pharmthera.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 138.Popovici-Muller J, Lemieux RM, Artin E, Saunders JO, Salituro FG, Travins J, et al. Discovery of AG-120 (ivosidenib): a first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med Chem Lett. 2018;9:300–305. doi: 10.1021/acsmedchemlett.7b00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hansen E, Quivoron C, Straley K, Lemieux RM, Popovici-Muller J, Sadrzadeh H, et al. AG-120, an oral, selective, first-in-class, potent inhibitor of mutant IDH1, reduces intracellular 2HG and induces cellular differentiation in TF-1 R132H cells and primary human IDH1 mutant AML patient samples treated ex vivo. Blood. 2014;124:3734. doi: 10.1182/blood.V124.21.3734.3734. [DOI] [Google Scholar]
- 140.Dhillon S. Ivosidenib: first global approval. Drugs. 2018;78:1509–1516. doi: 10.1007/s40265-018-0978-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mellinghoff IK, Ellingson BM, Touat M, Maher E, de la Fuente MI, Holdhoff M, et al. Ivosidenib in isocitrate dehydrogenase 1–mutated advanced glioma. J Clin Oncol. 2020;38:3398–3406. doi: 10.1200/JCO.19.03327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shih AH, Shank KR, Meydan C, Intlekofer AM, Ward P, Thompson CB, et al. AG-221, a small molecule mutant IDH2 inhibitor, remodels the epigenetic state of IDH2-mutant cells and induces alterations in self-renewal/differentiation in IDH2-mutant AML model in vivo. Blood. 2014;124:437. doi: 10.1182/blood.V124.21.437.437. [DOI] [Google Scholar]
- 143.Yen K, Travins J, Wang F, David MD, Artin E, Straley K, et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 2017;7:478–493. doi: 10.1158/2159-8290.CD-16-1034. [DOI] [PubMed] [Google Scholar]
- 144.Weller M, Roth P, Preusser M, Wick W, Reardon DA, Platten M, et al. Vaccine-based immunotherapeutic approaches to gliomas and beyond. Nat Rev Neurol. 2017;13:363–374. doi: 10.1038/nrneurol.2017.64. [DOI] [PubMed] [Google Scholar]
- 145.Platten M, Bunse L, Wick A, Bunse T, le Cornet L, Harting I, et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature. 2021;592:463–468. doi: 10.1038/s41586-021-03363-z. [DOI] [PMC free article] [PubMed] [Google Scholar]