

Abstract
Objective
To explore the composition of the intestinal microbiota in ulcerative colitis (UC) patients and to identify differences in the microbiota between patients with active disease and those in remission.
Methods
Between September 2020 and June 2021, we enrolled into our study, and collected stool samples from, patients with active UC or in remission and healthy control subjects. The diagnosis of UC was based on clinical, endoscopic, radiological, and histological findings. The composition of the intestinal microbiota was determined by sequencing of the 16S rRNA V3–V4 region and by bioinformatic methods. The functional composition of the intestinal microbiota was predicted using PICRUSt 2 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) software.
Results
We found that the intestinal flora was significantly less rich and diverse in UC patients than in healthy control subjects. Beta diversity analysis revealed notable differences in the intestinal flora compositions among the three groups, but there was no statistical difference in alpha diversity between UC patients with active disease and those in remission. At the phylum level, the relative abundances of Proteobacteria and Patescibacteria were significantly higher, and the relative abundances of Desulfobacterota and Verrucomicrobiota were lower, in UC patients with active disease than in the healthy control group. Higher levels of potential pathogens and lower levels of butyrate-producing bacteria were also detected in UC patients with active disease. Linear discriminant analysis Effect Size (LefSe) revealed that 71 bacterial taxa could serve as biomarkers, with 26 biomarkers at the genus level. In addition, network analysis showed that there was a positive correlation between Roseburia and Lachnospira. Functional predictions indicated that gene functions involving the metabolism of some substances, such as methane, lipopolysaccharide, geraniol, and ansamycins, were significantly different among the three groups.
Conclusion
The richness and diversity of the intestinal microbiota differed significantly among the three groups. Richness describes the state of being rich in number of intestinal bacteria, whereas diversity is the number of different species of intestinal bacteria. Different bacterial taxa could be used as biomarkers, expanding our understanding of the relationship between the intestinal microbiota microenvironment and UC in the future.
Keywords: ulcerative colitis, intestinal microbiota, bacterial, composition, diversity
Introduction
Ulcerative colitis (UC) and Crohn’s disease (CD) are clinical subtypes of inflammatory bowel disease (IBD) and are chronic, relapsing immune-related disorders of the gastrointestinal tract of unknown etiology. Individuals with UC and CD often present with symptoms such as persistent diarrhea, abdominal pain, and hematochezia (Malik et al., 2018). In 2017, it was estimated that over 6.8 million people worldwide suffered from IBD (GBD 2017 Inflammatory Bowel Disease Collaborators, 2020). The total number of cases of IBD is likely to continue to increase, and IBD is expected to become an enormous public health burden in the next decade (Ng et al., 2017). Using all means possible to identify the cause(s) of IBD and intervene appropriately will be the top priority of future IBD medical care.
Currently, it is widely believed that intestinal flora, which comprises over 100 trillion bacteria, as well as myriads of fungi, viruses (Saeedi et al., 2020), and protozoa, play a significant role in the pathogenesis of IBD (Sartor, 2008). Bacteria could influence intestinal inflammation through interaction with the immune system (Sabino et al., 2016), for example causing up-regulation of anti-inflammatory cytokines or down-regulation of pro-inflammatory cytokines (Kostic et al., 2014). It is known that the contribution of genetic factors to disease etiology is greater for CD than for UC, and so the impact of intestinal dysbiosis on UC may be worthy of further exploration (Tremelling et al., 2007). Previous studies have shown that, compared with healthy people, the gut of UC patients contains fewer Firmicutes organisms and more Bacteroidetes organisms and facultative anaerobes (Hansen et al., 2010). In addition, the relative abundance of Enterobacteriaceae, in particular Escherichia coli and Shigella species, is increased by the aggravation of intestinal inflammation, whereas the opposite is true of Lachnospiraceae and Ruminococcaceae (He et al., 2021). Increased understanding of the changes in the microbiota associated with UC has led to the development of microbiota-related therapeutic strategies, such as the widespread use of probiotic preparations and fecal microbiota transplantation. However, more research into the specific changes that occur in the enteric microbiota and the possible pathogenic mechanism is needed to clarify the relationship between the gut microbiota and the clinical manifestation of UC.
Using 16S rRNA gene amplicon sequencing, in this study we explored the diversity and compositional differences of gut bacterial communities in UC patients with active disease, UC patients in remission, and healthy individuals, which expanded our understanding of intestinal bacterial ecology in UC.
Materials and methods
Patients and specimens
For this study, 40 UC patients ranging in age from 18 to 75 years were enrolled at Beijing Friendship Hospital Affiliated to Capital Medical University from September 2020 to June 2021. To be eligible for the study, UC patients were required to have had a clinical disease for more than 3 months. The diagnosis of UC was based on clinical, endoscopic, radiological, and histological findings in accordance with consensus on the diagnosis and treatment of IBD (Nakase et al., 2021). In addition, the level of disease activity was determined by the Mayo score/Disease Activity Index, and UC patients were classified as having an active disease or being in remission (Schroeder et al., 1987). A total of 20 sex-matched healthy control subjects were also enrolled, that is, healthy volunteers unrelated to the patients in this study. None of the participants were taking antibiotics, antacids, non-steroidal anti-inflammatory drugs, antiplatelet drugs, anticoagulants, or glucocorticoids, and participants had a history of colectomy in the 3 months prior to stool sample collection. Fecal specimens collected from patients and healthy control subjects were immediately added to buffer solution and cryopreserved at –20°C until sequencing.
All specimen studies were approved by the Medical Ethical Committee of the Beijing Friendship Hospital Affiliated to the Capital University of Medical Sciences (number 2019-P2-233-01) and conducted in accordance with the principles expressed in the Declaration of Helsinki. Written informed consent was obtained from all patients and healthy individuals participating in this study.
DNA extraction and 16S ribosomal RNA gene sequencing
DNA was extracted from fecal specimens (approximately 150 mg per stool sample) using a QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) in accordance with the manufacturer’s protocols. The V3–V4 region of the bacterial 16S rRNA gene was amplified by PCR (94 °C for 5 min, followed by 30 cycles at 94 °C for 30 s, 50 °C for 30 s, 72°C for 60 s, then 72 °C for 7 min) with the forward primer 338F (5′-ACTCCTACGGGAGGCAGCA-3′) and the reverse primer 806R (5′-GGACTACHVGGGTWTCTAAT-3′). PCR products were electrophoresed on a 1% agarose gel to identify the size of the amplified target band and purified using the Agencourt AMPure XP Nucleic Acid Purification Kit (Beijing, China). Sequencing was performed using the MiSeqPE300 platform from Illuminate.
Bioinformatics analyses
The pyrosequencing data were processed using Quantitative Insights Into Microbial Ecology (QIIME) v. 1.8.0 (Nakase et al., 2016) and VSEARCH (v. 2.7.1) software (Rognes et al., 2016). Operational taxonomic units (OTUs) with a threshold of 97% sequence similarity were clustered using UPARSE (version 7.1) (Edgar, 2013). The taxonomy of each OTU representative sequence was analyzed using the Ribosomal Database Project Classifier (Cole et al., 2009; Sabino et al., 2016) and compared against the SILVA database (release 138) (Quast et al., 2013) using a confidence threshold of 0.7. FastTree (Price et al., 2009) software was used to construct a phylogenetic tree, and Python was applied to visualize their abundance and evolutionary relationship. The microbial alpha and beta diversities were calculated in QIIME 1.8.0 and visualized in the R environment. Meanwhile, hierarchical clustering analysis was carried out according to the unweighted UniFrac distance, and a dendrogram was constructed using the unweighted pair group method with arithmetic mean (UPGMA).
Statistical analysis
MetaStats, the Kruskal–Wallis test, analysis of molecular variance, the Wilcoxon rank-sum test, and analysis of similarities (ANOSIM) were all appropriately used to test the significance of differences in species composition and community structure between the groups. Linear discriminant analysis Effect Size (LEfSe) analysis (Segata et al., 2011) was employed to identify species whose abundance differed significantly between the three groups (i.e. biomarkers). Spearman’s test method and the top 20 genus-level results of absolute abundance in all samples were selected for correlation analysis. The calculated results were plotted after filtering out p-values above 0.05 or correlation values | R | < 0.6. Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt 2.0) (Langille et al., 2013) was applied to predict metagenome function. All significance thresholds were set at p-values < 0.05.
Results
Characteristics and microbiota basic analysis of the subjects
Forty patients with UC and 20 healthy control subjects were enrolled in this study. The UC patients comprised 20 with active disease and 20 in remission. The following basic clinical characteristics were recorded: age, sex, duration of disease, and disease type distribution (E1/E2/E3) ( Table 1 ). No significant differences in the baseline characteristics were observed between UC patients and healthy control subjects.
Table 1.
Clinical characteristics of the subjects.
| Characteristics | UC patients (n=40) | Healthy controls (n=20) | |
|---|---|---|---|
| Active stage (n=20) | Remission stage (n=20) | ||
| Age (years) | 46.55±15.04 | 47.25 ± 16.18 | 50.65 ± 9.43 |
| Gender (male/female) | 12/8 | 12/8 | 10/10 |
| Duration (months) | 69.85 ± 53.18 | 73.40 ± 67.34 | – |
| Extent | |||
| Ulcerative proctitis (E1) | 6 | 10 | – |
| Left sided colitis (E2) | 3 | 6 | – |
| Pancolitis (E3) | 11 | 4 | – |
Data are presented as mean ± standard error of mean.
*p< 0.05 compared with healthy controls group.
From the stool samples from the 40 UC patients and healthy control subjects, we obtained a total of 4,825,757 high-quality sequences from the original MiSeq sequencing data ( Supplementary Figure 1A ). A total of 1325 OTUs were identified using 97% sequence similarity as the taxonomic standard at the species level. The number of unique and common OTUs between groups is depicted in a Venn diagram ( Supplementary Figure 1B ).
The rarefaction curve tended to remain flat, and the Shannon–Wiener curve was saturated, which indicated that the number of sequencing data was reasonable and the data can be considered to comprehensively reflect species richness ( Supplementary Figures 2A, B ). The flattening species accumulation curves indicated that the sample size was sufficient. The rank abundance curve demonstrated that the samples from the healthy control group were more species-rich than samples from UC patients, and the species composition of microbial communities was more uniform in samples from healthy control subjects ( Supplementary Figures 2C, D ).
Decrease in diversity of intestinal flora in ulcerative colitis patients
Community richness was evaluated by Chao1. Community diversity was evaluated by the Simpson Diversity Index, the Shannon Diversity Index, and PD Whole Tree ( Figure 1 ) using alpha diversity analysis. In addition, Good’s coverage was used to assess sequencing depth, and observed species was used to represent the number of OTUs actually observed with the increase in sequencing depth. Compared with the healthy control subjects [Shannon Diversity Index 4.795 (95% CI 4.33 to 5.42)], the diversity of the intestinal flora among UC patients in remission [Shannon Diversity Index 4.19 (95% CI 3.695 to 4.57)] was significantly lower (p < 0.05) ( Figure 1D ). Chao1 and observed species showed that the diversity of the intestinal flora was considerably reduced in UC patients in remission ( Figures 1A, B ).
Figure 1.

Alpha and beta diversity analysis of the intestinal flora in stool samples from ulcerative colitis patients. (A–D). Results of alpha diversity analysis of intestinal flora as evaluated by Chao 1, observed species, PD Whole Tree, and the Shannon Diversity Index. The diversity of intestinal flora in UC patients in remission was found to be significantly reduced. (E, F). Results of beta diversity analysis. PCA based on OTU leve, and PCoA based on Bray–Curtis distance. Points of different colors or shapes represent different sample groupings, and the scales of the horizontal and vertical axes are relative distances. AUC, UC patients with active disease; RUC, UC patients in remission; and HC, healthy control subjects.
Euclidean distance and Bray–Curtis distance were determined by principal component analysis (PCA) and principal coordinates analysis (PCoA) respectively, which are types of beta diversity analysis. Both PCA ( Figure 1E ) and PCoA ( Figure 1F ) revealed clear differences in the structure of flora structure between the two UC groups and the control group. Both Adonis (PERMANOVA) analysis (p = 0.001) and the ANOSIM test (r = 0.0982, p = 0.001) indicated significant intergroup differences.
Taxonomic differences in the gut microbiota of ulcerative colitis patients
The microbiota composition of the stool samples at the phylum and genus levels were determined by OTU abundance ( Figure 2 ). As shown in Figure 2 , the main phyla in all three groups were Firmicutes, Bacteroidota, Actinobacteriota, and Proteobacteria, but their relative abundances were different in each group. In particular, Firmicutes was more abundant than Bacteroidota and Actinobacteriota. The abundance of Proteobacteria and Patescibacteria was considerably higher in UC patients with active disease than in healthy control subjects (p < 0.05), but the opposite was true for Desulfobacterota and Verrucomicrobiota ( Figure 3A ). The relative abundance of Patescibacteria was higher, and the relative abundance of Desulfobacterota, Synergistota, and Verrucomicrobiota was lower, in UC patients in remission than in healthy control subjects ( Figure 3B ). And the abundance of Proteobacteria in those with active disease than in those in remission ( Figure 3C ).
Figure 2.

The microbiological compositions at the phylum and genus levels. The stacked bars show the average relative abundances of all phyla and the most common genera and species identified in the three groups. The histograms show species distribution at the phylum (A) and genus (B) levels in the three groups. AUC, UC patients with active disease; RUC, UC patients in remission; and HC, healthy control subjects.
Figure 3.

(A) Comparison of AUC and HC at the phylum level. (B) Comparison of HC and RUC at the phylum level. (C) Comparison of AUC and RUC at the phylum level. (D) Comparison of AUC and HC at the genus level. (E) Comparison of HC and RUC at the genus level. (F) Comparison of AUC and RUC at the genus level.
At the genus level, the relative abundances of Rothia, Streptococcus, and Veillonella were significantly higher (p < 0.05) in UC patients with active disease than in healthy control subjects ( Figure 3D ). In contrast, the relative abundances of Butyricicoccus, Dorea, Eubacterium hallii group, Fusicatenibacter, Incertae sedis, Lachnoclostridium, Lachnospira, Lactobacillus, Monoglobus, Parabacteroides, Phascolarctobacterium, Roseburia, Ruminococcus, Ruminococcus torques group, Subdoligranulum, and Tyzzerella were significantly lower in the UC patients with active disease than in the healthy control group ( Figure 3D ). Compared with healthy control subjects, UC patients in remission demonstrated higher relative abundances of Blautia, Faecalitalea, and Flavonifractor and lower relative abundances of Agathobacter, Alistipes, Christensenellaceae R-7 group, Coprococcus, Enterococcus, Eubacterium hallii group, Fusicatenibacter, Incertae sedis, Lachnospiraceae ND3007 group, Lachnospiraceae UCG-001, Monoglobus, Phascolarctobacterium, Ruminococcus, Ruminococcus torques group, Subdoligranulum, UCG-002, and UCG-005 ( Figure 3E ). In addition, there was an increase in the relative abundances of Finegoldia magna, Gordonibacter, Haemophilus, human gut metagenome, Klebsiella, Kroppenstedtia, Lachnoanaerobaculum, Lautropia, Pantoea, Pediococcus, Peptostreptococcus, Rothia, and Sutterella. A decrease in the relative abundances of Anaerostignum, Butyricicoccus, Lachnoclostridium, Monoglobus, Ruminococcus gauvreauii group, and Tyzzerella was observed in both groups of UC patients, that is, both those with active disease and those in remission ( Figure 3F ).
LEfSe analysis (a logarithmic LDA score cut-off point of 3.0 and p-value < 0.05) revealed remarkable differences among the three groups in 71 bacterial taxa (26 at the genus level) ( Figure 4 ). The phylogenetic tree of OTUs at the genus taxonomic level is shown in Figure 5 . Based on the unweighted UniFrac distance matrix, the UPGMA method was used for cluster analysis and dendrogram, and the clustering results were integrated with the relative abundance of species at the phylum ( Figure 6A ) and genus ( Figure 6B ) levels in each sample.
Figure 4.

LEfSe analysis of the bacterial communities. LEfSe bar plot of the bacterial communities. AUC, UC patients with active disease; RUC, UC patients in remission; and HC, healthy control subjects.
Figure 5.

Genus-level evolutionary tree. The first column is the grouping information, and the second column is the phylum level corresponding to the genus level. The representative sequences corresponding to the most abundant OTUs were selected by genus as a unit to build the dendrogram, and the outer circle of the evolutionary tree shows the relative abundance of each genus in each of the different groups. The length of the color blocks represents the relative abundance. AUC, UC patients with active disease; RUC, UC patients in remission; and HC, healthy control subjects.
Figure 6.

Cluster analysis and dendrogram of microbiota. Left: hierarchical clustering analysis among samples; right: histogram of the community structure of the samples. The abscissa shows the relative abundance of species in the sample, and the ordinate sample name. (A) At the phylum level. (B) At the genus level. AUC, UC patients with active disease; RUC, UC patients in remission; and HC, healthy control subjects.
The gut microbiota network
Network analysis clearly revealed the interactions among the top 20 gut microbiota in absolute abundance at the genus level. As shown in Supplementary Figure 3 , there was a positive correlation between Roseburia and Lachnospira (p < 0.05).
Predicted functional composition of fecal microbial communities
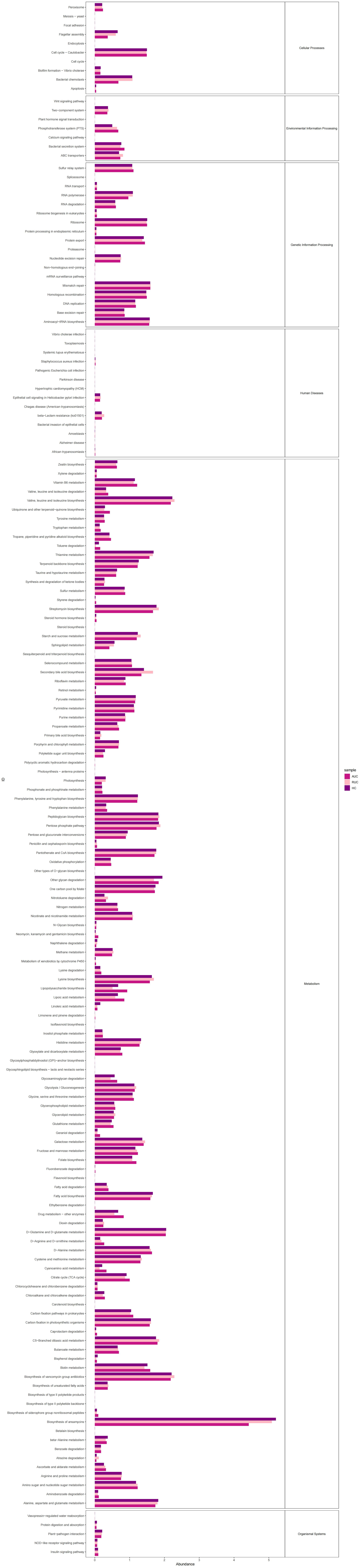
We regarded the microbial functional signatures by implementing PICRUSt 2.0 in the three groups. Differentially abundant metabolic pathways were detected at KEGG level 3 ( Supplementary Figure 4 ). Compared with the healthy control subjects, UC patients with active disease exhibited lower expression of genes involved in bacterial chemotaxis, flagellar assembly, pantothenate and coenzyme A biosynthesis, thiamine metabolism, and biosynthesis of ansamycins. However, this group demonstrated higher expression of genes involved in geraniol degradation, fatty acid degradation, ascorbate and aldarate metabolism, valine, leucine and isoleucine degradation, tyrosine metabolism, and propanoate metabolism. UC patients with active disease, compared with those in remission, also demonstrated a higher abundance of geraniol degradation, lipopolysaccharide biosynthesis, toluene degradation, valine, leucine and isoleucine degradation, ubiquinone and other terpenoid-quinone biosynthesis, vitamin B6 metabolism, and lipoic acid metabolism. However, UC patients in remission had a higher abundance of bacterial chemotaxis, methane metabolism, biosynthesis of ansamycins, valine, leucine and isoleucine biosynthesis, lysine biosynthesis, and thiamine metabolism than patients with active disease. Notably, the expression levels of lipopolysaccharide biosynthesis and toluene degradation pathways were significantly lower in UC patients in remission than in healthy control subjects.
Discussion
In this study, we analyzed the results of high-throughput sequencing of the 16S rRNA V3–V4 variable region of the intestinal flora of UC patients with active disease or in remission and in a healthy population. Significant differences in the diversity and composition of the intestinal flora were identified among the three groups. In particular, alpha diversity in the gut microbiome, evaluated by the observed species, Chao1, and the Shannon Diversity Index, which was significantly lower in UC patients than in healthy control subjects, reflecting the fact that the richness and evenness (i.e. relative density) of bacteria were lower in UC patients than in control subjects. Among UC patients, alpha diversity was lower in those with active disease than in those in remission, but the difference was not statistically significant. Similarly, beta diversity of gut microbiota significantly differed among the three groups. However, the dendrogram based on unweighted UniFrac distance using UPGMA method clustering revealed no clear differences among the three groups. Our findings are analogous to those of previous studies (Ma et al., 2018; Xu et al., 2021). Earlier studies revealed that a reduction in the diversity of fecal microbiota is the most consistent indicator of IBD, and the microbiome of UC patients is more similar to that of healthy control subjects than to that of individuals with CD (Sankarasubramanian et al., 2020).
At the phylum level, the profile of gut microbiota appeared to be dominated by Firmicutes, Bacteroidetes, and Proteobacteria, which is consistent with the findings of previous studies (Forbes et al., 2016; Rehman et al., 2016), confirming the reliability of our samples and analytical methods. The largest group of bacteria in the gut, members of the phylum Firmicutes, ferment dietary fiber to short-chain fatty acids (SCFAs), such as butyrate, which are used as energy sources and are regulators of inflammation (Tremaroli and Bäckhed, 2012). In addition, Firmicutes species, together with members of phylum Bacteroidetes, are involved in the regulation of lipid and bile acid metabolism and in the maintenance of energy balance in the host (Pan et al., 2018). There is substantial evidence to support an increase in Proteobacteria (especially adherent invasive E. coli) in IBD patients (Knox et al., 2019). In our study, UC patients with active disease had significantly higher levels of Proteobacteria than UC patients in the remission and healthy control subjects, and this suggests that Proteobacteria abundance may be related to the severity of inflammation. Several other studies investigating the microbiota in UC patients using mucosal biopsy have also reached similar conclusions (Lucke et al., 2006; Chen et al., 2014; Ahmed et al., 2016; Ma et al., 2018).
At the genus level, we found that abundance levels of many bacteria were altered in UC patients, which was reflected in the LEfSe results. Blautia and Lachnoclostridium were significantly elevated in UC patients in remission, whereas Rothia, Actinomyces, Pediococcus, and TM7x could be considered biomarkers in patients with active disease. Blautia is an anaerobic bacterium with probiotic characteristics that promotes the production of SCFAs, prevents inflammation, and maintains intestinal homeostasis (Liu et al., 2021), and has been found to be significantly reduced in colorectal cancer patients (Chénard et al., 2020) and in animal models (Zhang et al., 2020). In addition, previous research has found that Lachnoclostridium in stool could serve as a biomarker for non-invasive diagnosis of colorectal adenoma and cancer (Liang et al., 2020). Rothia dentocariosa is a conditioned pathogen found mainly in the human oral cavity and is known to cause local periodontal infection, endocarditis, septicemia, peritoneal dialysis-related peritonitis, and pneumonia (Yeung et al., 2014). Our findings reveal the need to be vigilant in monitoring the pathogenicity of this commensal bacterium in patients with active UC and its potential to cause clinically significant infection. TM7x is the first obligate epibiont parasite discovered within the domain of Bacteria that requires other bacteria (Actinomyces odontolyticus strain XH001) as a host (Ibrahim et al., 2021). Actinomyces species are opportunistic pathogens mostly inhabiting the oral cavity and upper respiratory tract, and overgrowth of this genus could cause inflammation. Therefore, UC patients need to pay attention to oral hygiene and ensure timely treatment of oral diseases. The term ‘gum–gut axis’ was first proposed by Kevin and Ajay (Byrd and Gulati, 2021) to describe the relationship between the microbiota of the oral cavity and the gastrointestinal tract. Other studies have since reported the presence of many oral microbes in the intestinal tissues of IBD patients, such as Aggregatibacter, Campylobacter, Enterobacteria, Fusobacterium, Gemella, Neisseria, Pasteurella, Peptostreptococcus, and Streptococcus (Gevers et al., 2014; Yachida et al., 2019; Schmidt et al., 2019). We observed the colonization of distant microbial niches by oral microbes in UC patients with acute disease, which can complement the knowledge and understanding of the gum–gut axis.
Previous research has shown that, compared with healthy individuals, the gut microbiota of UC patients and Bifidobacterium species, and a lower abundance of Faecalibacterium prausnitzii, as well as a lower concentration of organic acids (Sartor and Wu, 2017; Knox et al., 2019). Sulfate-reducing bacteria, whose metabolites (e.g. hydrogen sulfide) can block the use of butyrate by colonocytes (Smith et al., 2005), have also been found to be increased in abundance in patients with IBD (Verma et al., 2010). Lloyd-Price et al. (Lloyd-Price et al., 2019) previously demonstrated a characteristic increase in facultative anaerobes at the expense of obligate anaerobes in the gut microbiota of UC patients, and these previous findings were also reflected in our results. Subsequent studies on the IBD microbiome should involve specific strains that can provide greater insight into correlated changes in the microbiome.
We also found that the relative abundances of Sutterella, Haemophilus, and Klebsiella species were significantly higher in UC patients with active disease than in those in remission. Sutterella is known to impair gut antibacterial immune function and increase susceptibility to gut disease (Moon et al., 2015). Enrichment of Sutterella leads to excessive secretion of IgA protease and, thus, a decreased IgA concentration in the intestinal mucosa, which facilitates pathobiont invasion of epithelial cells and intracellular survival (Kaakoush, 2020). Interestingly, the relative abundance of Butyricicoccus, a recognized pathogenic bacterium, was higher in UC patients in remission than in those with active disease. Butyricicoccus is known to prevent cytokine-induced epithelial integrity losses and promote the proliferation of beneficial flora (such as Bifidobacterium and Lactobacillus) (Eeckhaut et al., 2013; Devriese et al., 2017). In addition, we found no significant difference in Faecalibacterium prausnitzii abundance among the three groups, similar to the finding of Brigida et al. (Barberio et al., 2022), but in contrast to the vast majority of previous studies (Sokol et al., 2009; Machiels et al., 2014). This may be related to the ethnic and geographical differences of UC patients, and follow-up of a large number of Asian UC patients will be necessary to explore this issue.
To our knowledge, this is the first trial to report a positive correlation between Roseburia and Lachnospira in gut microbiota, especially in UC patients. Both Roseburia and Lachnospira are butyrate-producing genera, and they play an important role in the control of intestinal inflammatory processes (Wu et al., 2020; Nakamura et al., 2022).
As is widely known, microorganisms have an impact on human immunity, the degradation and absorption of nutrients, and even the metabolism of substances (Martin et al., 2010). To further assess the impact on the host of a change in gut microbiome, we employed PICRUSt2 analysis. Our results suggest that the levels of methane, lipopolysaccharide, geraniol, ansamycins, valine, leucine, and isoleucine, among others, are increased in UC patients with severe inflammation. This finding has only rarely been reported before and might play a certain anti-inflammatory role in the clinical course of disease in UC patients.
It should be noted that our study also had certain limitations. The focus of our research was on dysbiosis in the stool of UC patients, and the perturbation of the structure of the gut microbiota in the biopsy mucosa deserves further research. Furthermore, owing to its cross-sectional design, our study was unable to dynamically observe changes in the intestinal flora of the same UC patient during the recurrence and remission stages. Extensive effort is required to identify changes in the microbiota and their role in the pathogenesis of UC.
Overall, our study revealed that the bacterial community composition of stool differs between UC patients with active disease and those in remission. Our findings will certainly lay the foundation for further research on the function and signaling pathways of gut microbiota in UC. Further comprehensive investigations of microbiomics and metabonomics have great potential to expand our knowledge of the pathogenesis of IBD and to identify potential treatments.
Data availability statement
The name of the repository is NCBI Sequence Read Archive (SRA) database, and accession number is PRJNA866870.
Ethics statement
The studies involving human participants were reviewed and approved by the Medical Ethical Committee of the Beijing Friendship Hospital Affiliated to Capital Medical University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
Conceived and designed the experiments: SZ, HS, and PL. Collection of fecal specimens: SL and LF. Performed the experiments: SZ and MH. Analyzed the data: MH and SZ. Manuscript preparation: MH. Manuscript revisions: SZ, HS, and PL. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work, ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
This work was funded by the National Natural Science Foundation of China (82070575); Beijing Municipal Administration of Hospitals’ Youth Programme (QML20190104, QML20180102); Beijing Nova Program (Z201100006820147); Beijing Municipal Science & Technology Commission (Z181100001718221).
Acknowledgments
We thank Allwegene Company (Beijing) for technical assistance in sequencing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2022.953962/full#supplementary-material
The microbiota basic line of the subjects. (A) Sequence distribution map. The abscissa shows the sequence length gradient, and the ordinate the number of tags. (B) Venn diagram. Different colors represent different samples, and overlapping circles represent OTUs that occur in more than one sample.
The microbiota basic analysis of the subjects. (A) Rarefaction curve. The abscissa shows the number of randomly selected sequencing data, and the ordinate the number of observed OTUs. (B) Shannon–Wiener curve. The abscissa shows the sequencing depth and the ordinate Shannon Diversity Index. (C) Species accumulation curves. The abscissa shows the sample size, and the ordinate the number of OTUs after sampling. (D) Rank abundance curve. The abscissa shows the OTU level, and the ordinate the relative percentage of sequence numbers in OTUs of this level, that is, the number of sequences belonging to this OTU divided by the total number of sequences. AUC, UC patients with active disease; RUC, UC patients in remission; and HC, healthy control subjects.
Network interaction diagram. The size of the dot represents the abundance, the thickness of the line represents the correlation, the color of the dot represents the phylum it belongs to, and the red line represents a positive correlation.
Bar plot of differentially enriched metabolic pathways at KEGG level 3. AUC, UC patients with active disease; RUC, UC patients in remission; and HC, healthy control subjects.
{kind=link}
References
- Ahmed I., Roy B. C., Khan S. A., Septer S., Umar S. (2016). Microbiome, metabolome and inflammatory bowel disease. Microorganisms 4, 20. doi: 10.3390/microorganisms4020020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberio B., Facchin S., Patuzzi I., Ford A. C., Massimi D., Valle G., et al. (2022). A specific microbiota signature is associated to various degrees of ulcerative colitis as assessed by a machine learning approach. Gut Microbes 14, 2028366. doi: 10.1080/19490976.2022.2028366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd K. M., Gulati A. S. (2021). The "Gum-gut" axis in inflammatory bowel diseases: A hypothesis-driven review of associations and advances. Front. Immunol. 12. doi: 10.3389/fimmu.2021.620124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chénard T., Malick M., Dubé J., Massé E. (2020). The influence of blood on the human gut microbiome. BMC Microbiol. 20, 44. doi: 10.1186/s12866-020-01724-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Wang W., Zhou R., Ng S. C., Li J., Huang M., et al. (2014). Characteristics of fecal and mucosa-associated microbiota in Chinese patients with inflammatory bowel disease. Medicine 93, e51. doi: 10.1097/MD.0000000000000051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole J. R., Wang Q., Cardenas E., Fish J., Chai B., Farris R. J., et al. (2009). The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37, D141–D145. doi: 10.1093/nar/gkn879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devriese S., Eeckhaut V., Geirnaert A., Van den Bossche L., Hindryckx P., Van de Wiele T., et al. (2017). Reduced mucosa-associated butyricicoccus activity in patients with ulcerative colitis correlates with aberrant claudin-1 expression. J. Crohns Colitis 11, 229–236. doi: 10.1093/ecco-jcc/jjw142 [DOI] [PubMed] [Google Scholar]
- Edgar R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604 [DOI] [PubMed] [Google Scholar]
- Eeckhaut V., Machiels K., Perrier C., Romero C., Maes S., Flahou B., et al. (2013). Butyricicoccus pullicaecorum in inflammatory bowel disease. Gut 62, 1745–1752. doi: 10.1136/gutjnl-2012-303611 [DOI] [PubMed] [Google Scholar]
- Forbes J. D., Van Domselaar G., Bernstein C. N. (2016). Microbiome survey of the inflamed and noninflamed gut at different compartments within the gastrointestinal tract of inflammatory bowel disease patients. Inflammation Bowel Dis. 22, 817–825. doi: 10.1097/MIB.0000000000000684 [DOI] [PubMed] [Google Scholar]
- GBD 2017 Inflammatory Bowel Disease Collaborators (2020). The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet Gastroenterol. Hepatol. 5, 17–30. doi: 10.1016/S2468-1253(19)30333-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevers D., Kugathasan S., Denson L. A., Vázquez-Baeza Y., Van, Treuren W., Ren B., et al. (2014). The treatment-naive microbiome in new-onset crohn's disease. Cell Host Microbe 15, 382–392. doi: 10.1016/j.chom.2014.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen J., Gulati A., Sartor R. B. (2010). The role of mucosal immunity and host genetics in defining intestinal commensal bacteria. Curr. Opin. Gastroenterol. 26, 564–571. doi: 10.1097/MOG.0b013e32833f1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X. X., Li Y. H., Yan P. G., Meng X. C., Chen C. Y., Li K. M., et al. (2021). Relationship between clinical features and intestinal microbiota in Chinese patients with ulcerative colitis. World J. Gastroenterol. 27, 4722–4737. doi: 10.3748/wjg.v27.i28.4722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim A., Maatouk M., Rajaonison A., Zgheib R., Haddad G., Bou Khalil J., et al. (2021). Adapted protocol for saccharibacteria cocultivation: Two new members join the club of candidate phyla radiation. Microbiol. Spectr. 9, e0106921. doi: 10.1128/spectrum.01069-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaakoush N. O. (2020). Sutterella species, IgA-degrading bacteria in ulcerative colitis. Trends Microbiol. 28, 519–522. doi: 10.1016/j.tim.2020.02.018 [DOI] [PubMed] [Google Scholar]
- Knox N. C., Forbes J. D., Peterson C. L., Van Domselaar G., Bernstein C. N. (2019). The gut microbiome in inflammatory bowel disease: Lessons learned from other immune-mediated inflammatory diseases. Am. J. Gastroenterol. 114, 1051–1070. doi: 10.14309/ajg.0000000000000305 [DOI] [PubMed] [Google Scholar]
- Kostic A. D., Xavier R. J., Gevers D. (2014). The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterolog 146, 1489–1499. doi: 10.1053/j.gastro.2014.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille M. G., Zaneveld J., Caporaso J. G., McDonald D., Knights D., Reyes J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J. Q., Li T., Nakatsu G., Chen Y. X., Yau T. O., Chu E., et al. (2020). A novel faecal lachnoclostridium marker for the non-invasive diagnosis of colorectal adenoma and cancer. Gut 69, 1248–1257. doi: 10.1136/gutjnl-2019-318532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Mao B., Gu J., Wu J., Cui S., Wang G., et al. (2021). Blautia-a new functional genus with potential probiotic properties? Gut Microbes 13, 1–21. doi: 10.1080/19490976.2021.1875796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Price J., Arze C., Ananthakrishnan A. N., Schirmer M., Avila-Pacheco J., Poon T. W., et al. (2019). Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662. doi: 10.1038/s41586-019-1237-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucke K., Miehlke S., Jacobs E., Schuppler M. (2006). Prevalence of bacteroides and prevotella spp. in ulcerative colitis. J. Med. Microbiol. 55, 617–624. doi: 10.1099/jmm.0.46198-0 [DOI] [PubMed] [Google Scholar]
- Machiels K., Joossens M., Sabino J., De Preter V., Arijs I., Eeckhaut V., et al. (2014). A decrease of the butyrate-producing species roseburia hominis and faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 63, 1275–1283. doi: 10.1136/gutjnl-2013-304833 [DOI] [PubMed] [Google Scholar]
- Malik A., Sharma D., Malireddi R. K. S., Guy C. S., Chang T. C., Olsen S. R., et al. (2018). SYK-CARD9 signaling axis promotes gut fungi-mediated inflammasome activation to restrict colitis and colon cancer. Immunity 49, 515–530. doi: 10.1016/j.immuni.2018.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R., Nauta A. J., Ben Amor K., Knippels L. M., Knol J., Garssen J. (2010). Early life: gut microbiota and immune development in infancy. Benef Microbes 1, 367–382. doi: 10.3920/BM2010.0027 [DOI] [PubMed] [Google Scholar]
- Ma H. Q., Yu T. T., Zhao X. J., Zhang Y., Zhang H. J. (2018). Fecal microbial dysbiosis in Chinese patients with inflammatory bowel disease. World J. Gastroenterol. 24, 1464–1477. doi: 10.3748/wjg.v24.i13.1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon C., Baldridge M. T., Wallace M. A., CA D., Burnham, Virgin H. W., et al. (2015). Vertically transmitted faecal IgA levels determine extra-chromosomal phenotypic variation. Nature 521, 90–93. doi: 10.1038/nature14139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M., Maeda K., Yamamoto K., Yamamura T., Sawada T., Ishikawa E., et al. (2022). Preliminary comparison of endoscopic brush and net catheters as the sampling tool to analyze the intestinal mucus in the rectum with ulcerative colitis patients. Digestion 103, 232–243. doi: 10.1159/000521929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakase H., Uchino M., Shinzaki S., Matsuura M., Matsuoka K., Kobayashi T., et al. (2016). Lower neighborhood socioeconomic status associated with reduced diversity of the colonic microbiota in healthy adults. PloS One 11, e0148952. doi: 10.1371/journal.pone.0148952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakase H., Uchino M., Shinzaki S., Matsuura M., Matsuoka K., Kobayashi T., et al. (2021). Evidence-based clinical practice guidelines for inflammatory bowel disease 2020. J. Gastroenterol. 56, 489–526. doi: 10.1007/s00535-021-01784-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng S. C., Shi H. Y., Hamidi N., Underwood F. E., Tang W., Benchimol E. I., et al. (2017). Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet 390, 2769–2778. doi: 10.1016/S0140-6736(17)32448-0 [DOI] [PubMed] [Google Scholar]
- Pan H., Guo R., Zhu J., Wang Q., Ju Y., Xie Y., et al. (2018). A gene catalogue of the sprague-dawley rat gut metagenome. Gigascience 7, giy055. doi: 10.1093/gigascience/giy055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M. N., Dehal P. S., Arkin A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26 (7), 1641–1650. doi: 10.1093/molbev/msp077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman A., Rausch P., Wang J., Skieceviciene J., Kiudelis G., Bhagalia K., et al. (2016). Geographical patterns of the standing and active human gut microbiome in health and IBD. Gut 65, 238–248. doi: 10.1136/gutjnl-2014-308341 [DOI] [PubMed] [Google Scholar]
- Rognes T., Flouri T., Nichols B., Quince C., Mahé F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584. doi: 10.7717/peerj.2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabino J., Vieira-Silva S., Machiels K., Joossens M., Falony G., Ballet V., et al. (2016). Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut 65, 1681–1689. doi: 10.1136/gutjnl-2015-311004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeedi B. J., Liu K. H., Owens J. A., Hunter-Chang S., Camacho M. C., Eboka R. U., et al. (2020). Gut-resident lactobacilli activate hepatic Nrf2 and protect against oxidative liver injury. Cell Metab. 31, 956–968. doi: 10.1016/j.cmet.2020.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankarasubramanian J., Ahmad R., Avuthu N., Singh A. B., Guda C. (2020). Gut microbiota and metabolic specificity in ulcerative colitis and crohn's disease. Front. Med. 7. doi: 10.3389/fmed.2020.606298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartor R. B. (2008). Microbial influences in inflammatory bowel diseases. Gastroenterology 134, 577–594. doi: 10.1053/j.gastro.2007.11.059 [DOI] [PubMed] [Google Scholar]
- Sartor R. B., Wu G. D. (2017). Roles for intestinal bacteria, viruses, and fungi in pathogenesis of inflammatory bowel diseases and therapeutic approaches. Gastroenterology 152, 327–339. doi: 10.1053/j.gastro.2016.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T. S., Hayward M. R., Coelho L. P., Li S. S., Costea P. I., Voigt A. Y., Coelho L. P., et al. (2019). Extensive transmission of microbes along the gastrointestinal tract. Elife 8, e42693. doi: 10.7554/eLife.42693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder K. W., Tremaine W. J., Ilstrup D. M. (1987). Coated oral 5-aminosalicylic acid therapy for mildly to moderately active ulcerative colitis. A randomized study N Engl. J. Med. 317, 1625–1629. doi: 10.1056/NEJM198712243172603 [DOI] [PubMed] [Google Scholar]
- Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. doi: 10.1186/gb-2011-12-6-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith F. M., Coffey J. C., Kell M. R., O'Sullivan M., Redmond H. P., Kirwan W. O. (2005). A characterization of anaerobic colonization and associated mucosal adaptations in the undiseased ileal pouch. Colorectal Dis. 7, 563–570. doi: 10.1111/j.1463-1318.2005.00833.x [DOI] [PubMed] [Google Scholar]
- Sokol H., Seksik P., Furet J. P., Firmesse O., Nion-Larmurier I., Beaugerie L., et al. (2009). Low counts of faecalibacterium prausnitzii in colitis microbiota. Inflammation Bowel Dis. 15, 1183–1189. doi: 10.1002/ibd.20903 [DOI] [PubMed] [Google Scholar]
- Tremaroli V., Bäckhed F. (2012). Functional interactions between the gut microbiota and host metabolism. Nature 489, 242–249. doi: 10.1038/nature11552 [DOI] [PubMed] [Google Scholar]
- Tremelling M., Cummings F., Fisher S. A., Mansfield J., Gwilliam R., Keniry A., et al. (2007). IL23R variation determines susceptibility but not disease phenotype in inflammatory bowel disease. Gastroenterology 132, 1657–1664. doi: 10.1053/j.gastro.2007.02.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R., Verma A. K., Ahuja V., Paul J. (2010). Real-time analysis of mucosal flora in patients with inflammatory bowel disease in India. J. Clin. Microbiol. 48, 4279–4282. doi: 10.1128/JCM.01360-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Pan S., Luo W., Shen Z., Meng X., Xiao M., et al. (2020). Roseburia intestinalis derived flagellin ameliorates colitis by targeting miR 223 3p mediated activation of NLRP3 inflammasome and pyroptosis. Mol. Med. Rep. 22, 2695–2704. doi: 10.3892/mmr.2020.11351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N., Bai X., Cao X., Yue W., Jiang W., Yu Z. (2021). Changes in intestinal microbiota and correlation with TLRs in ulcerative colitis in the coastal area of northern China. Microb. Pathog. 150, 104707. doi: 10.1016/j.micpath.2020.104707 [DOI] [PubMed] [Google Scholar]
- Yachida S., Mizutani S., Shiroma H., Shiba S., Nakajima T., Sakamoto T., et al. (2019). Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 25, 968–976. doi: 10.1038/s41591-019-0458-7 [DOI] [PubMed] [Google Scholar]
- Yeung D. F., Parsa A., Wong J. C., Chatur N., Salh B. (2014). A case of rothia dentocariosa bacteremia in a patient receiving infliximab for ulcerative colitis. Am. J. Gastroenterol. 109, 297–298. doi: 10.1038/ajg.2013.366 [DOI] [PubMed] [Google Scholar]
- Zhang H., Lan M., Cui G., Zhu W. (2020). The influence of caerulomycin a on the intestinal microbiota in SD rats. Mar. Drugs 18, 277. doi: 10.3390/md18050277 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The microbiota basic line of the subjects. (A) Sequence distribution map. The abscissa shows the sequence length gradient, and the ordinate the number of tags. (B) Venn diagram. Different colors represent different samples, and overlapping circles represent OTUs that occur in more than one sample.
The microbiota basic analysis of the subjects. (A) Rarefaction curve. The abscissa shows the number of randomly selected sequencing data, and the ordinate the number of observed OTUs. (B) Shannon–Wiener curve. The abscissa shows the sequencing depth and the ordinate Shannon Diversity Index. (C) Species accumulation curves. The abscissa shows the sample size, and the ordinate the number of OTUs after sampling. (D) Rank abundance curve. The abscissa shows the OTU level, and the ordinate the relative percentage of sequence numbers in OTUs of this level, that is, the number of sequences belonging to this OTU divided by the total number of sequences. AUC, UC patients with active disease; RUC, UC patients in remission; and HC, healthy control subjects.
Network interaction diagram. The size of the dot represents the abundance, the thickness of the line represents the correlation, the color of the dot represents the phylum it belongs to, and the red line represents a positive correlation.
Bar plot of differentially enriched metabolic pathways at KEGG level 3. AUC, UC patients with active disease; RUC, UC patients in remission; and HC, healthy control subjects.
Data Availability Statement
The name of the repository is NCBI Sequence Read Archive (SRA) database, and accession number is PRJNA866870.