

Abstract
EDP‐305 is a farnesoid X receptor (FXR) agonist that selectively activates FXR and is a potential treatment for patients with nonalcoholic steatohepatitis (NASH) with liver fibrosis. Results from preclinical studies indicate that CYP3A4 is the primary enzyme involved in EDP‐305 metabolism and that EDP‐305 has low potential to inhibit or induce cytochrome (CYP) isoenzymes and drug transporters. Four studies were conducted in healthy volunteers to evaluate the drug–drug interaction (DDI) potential of EDP‐305 co‐administered with drugs known to be substrates for drug metabolizing enzymes or transporters, and to assess the effect of inhibitors and inducers of CYP3A4 on EDP‐305. Results suggest caution when substrates of CYP3A4 are administered concomitantly with EDP‐305. A potential for increased exposure is apparent when CYP1A2 substrates with a narrow therapeutic index are administered with EDP‐305. In contrast, substrates of drug transporters can be administered concomitantly with EDP‐305 with a low potential for interactions. Coadministration of EDP‐305 and a combined OC had no relevant effects on plasma concentrations of the combined OC. Co‐administration of EDP‐305 with strong or moderate inhibitors and inducers of CYP3A4 is not recommended. These results indicate low overall likelihood of interaction of EDP‐305 and other substrates through CYP mediated interactions. The interaction potential of EDP‐305 with drug transporters was low and of unlikely clinical significance. The EDP‐305 DDI profile allows for convenient administration in patients with NASH and other patient populations with comorbidities, with minimal dose modification of concomitant medications.
Study Highlights
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Drug metabolizing enzymes and transporters are implicated in clinically significant drug interactions. An important aspect for a new chemical entity is to establish the drug interaction potential early in the development process.
WHAT QUESTION DID THIS STUDY ADDRESS?
EDP‐305 is a farnesoid X receptor agonist that is a potential treatment for nonalcoholic steatohepatitis and liver fibrosis. Based on preclinical studies, EDP‐305 had a low potential to inhibit cytochrome (CYP) isoenzymes. Four studies evaluated the potential of EDP‐305 with other drugs known to be substrates for drug metabolizing enzymes or transporters.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
These results indicate caution with co‐administration of EDP‐305 with drugs that are CYP3A4 and CYP1A2 substrates because of potentially clinically significant effects, and co‐administration of EDP‐305 with strong or moderate inhibitors and inducers of CYP3A4 is not recommended. The drug–drug interaction potential of EDP‐305 with transporters or other agents was low and of unlikely clinical significance.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
The results from these four studies with EDP‐305 support the rationale for thorough investigation of potential clinically relevant drug interactions with new chemical entities.
INTRODUCTION
EDP‐305 is a farnesoid X receptor (FXR) agonist that selectively activates FXR and is being developed as a potential treatment for patients with nonalcoholic steatohepatitis (NASH) and liver fibrosis. Preclinical studies indicated that EDP‐305 potently regulates the expression of FXR target genes in vivo and is hepatoprotective in multiple rodent models of liver injury and NASH. 1 , 2 , 3 In phase I studies, EDP‐305 was well‐tolerated in healthy subjects at single doses up to 80 mg and in subjects with or without presumed nonalcoholic fatty liver disease (NAFLD) at multiple daily doses up to 20 mg for 14 days. 4 In a 12‐week, phase II study that evaluated the safety, tolerability, and efficacy of EDP‐305 in 132 non‐cirrhotic patients with fibrotic NASH, treatment with EDP‐305 reduced hepatic enzyme levels and liver fat with a tolerability profile that was consistent with other FXR agonists. 5
Results from preclinical studies indicate that EDP‐305 has a low potential to inhibit cytochrome (CYP) 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4 isoenzymes or to induce CYP1A2, 2B6, and 3A4 in standard hepatic microsome studies. However, incubation in liver microsomes (hepatocytes) in the presence of EDP‐305 resulted in a concentration‐dependent downregulation of messenger RNA (mRNA) for CYP1A2 and CYP3A4, consistent with similar pharmacodynamic effects seen with other FXR agonists. 6 , 7 The latter effects on mRNA for CYP1A2 and CYP3A4 did not translate into effects on enzyme levels in standard in vitro assays but warranted further investigation in the clinic.
Additional in vitro studies suggest the potential for EDP‐305 to inhibit organic anion‐transporting polypeptide (OATP)1B1 and OATP1B3 with a low potential to inhibit bile salt export protein (BSEP), multidrug resistance‐associated protein 2 (MRP2), P‐glycoprotein (P‐gp), and breast cancer resistance protein (BCRP) transporters. EDP‐305 is unlikely to be a BCRP substrate but has potential to be a P‐gp substrate (data on file).
In clinical studies, EDP‐305 geometric mean terminal half‐life (t 1/2) values were similar across all dose levels for fasted and fed subjects and ranged from 11.12 to 14.98 h, following single doses, and 9.86 to 21.2 h following multiple doses. 4 In a radiolabeled mass balance study, fecal excretion was the predominant route of elimination for EDP‐305, with observed mean recovery of total radioactivity of 78.7% in feces and 1.42% in urine (unchanged drug). EDP‐305 was not detected in urine, indicating no urinary clearance of EDP‐305 (data on file; Enanta Pharmaceuticals).
After single doses of EDP‐305 (ranging from 1 to 80 mg), the increase in mean peak and systemic exposures in fasted subjects was slightly greater than dose‐proportional. EDP‐305 exposures generally appeared to increase with multiple dosing (with doses ranging from 0.5 to 20 mg), with little accumulation at doses <10 mg (accumulation index of 1.13–1.74) and moderate to high accumulation at doses greater than or equal to 10 mg (accumulation index of 2.21–5.50) in healthy subjects and subjects with presumptive NAFLD. Based on visual inspection of the trough concentrations, steady‐state pharmacokinetics (PKs) appeared to be reached by day 9 across all doses.
Because of the potential for clinically relevant drug–drug interactions (DDIs) with various drug metabolizing enzymes or drug transporters, a series of phase I clinical studies were conducted in healthy volunteers to evaluate the potential for DDIs with EDP‐305. These DDI studies were conducted at clinically relevant doses, and their results were applied to completed studies in patients with NASH. 5
The DDI potential of EDP‐305 was evaluated at doses of 2.5 (tablet), 5 (tablet) or 10 mg (suspension), and these dose levels are in line with EDP‐305 doses that were administered in patients with NASH. 5 Patients with NASH are reported to have a different metabolic profile than healthy subjects, and plasma exposures in patients with NASH are usually higher than healthy subjects. 8 In the case of EDP‐305, the exposures in patients with NASH are approximately twofold higher than healthy subjects at the same dose/formulation (data on file). A relative bioavailability assessment indicated that EDP‐305 tablet has approximately twofold higher exposures compared to the suspension (data on file). Results from phase I studies in healthy subjects and subjects with presumptive NAFLD 4 and a phase II study in subjects with NASH 5 suggest that the optimal therapeutic dosage range for EDP‐305 is 1–2 mg (tablet) once daily. Thus, these DDI studies were conducted at clinically relevant doses.
METHODS
Each study was conducted in accordance with the principles of the Declaration of Helsinki and in compliance with the International Conference on Harmonization Guideline for Good Clinical Practice. The protocol and amendments and the informed consent documents were approved by independent ethics committees/institutional review boards (Midlands IRB, Overland Park, KS). All subjects provided written informed consent prior to participation in any study procedures.
Subjects
For all studies, eligible subjects were between 18 and 55 years (18–45 years in study 008) with a body mass index (BMI) of 18–30 kg/m2 (18–32 kg/m2 in study 008). Subjects were healthy based on a medical evaluation; female subjects were of non‐childbearing potential (except in study 008), and male subjects agreed to use effective contraception and refrain from sperm donation until 90 days after the last dose of study drug. Subjects refrained from caffeine use from 72 h prior to the study until discharge from the clinic. Detailed exclusion criteria are provided in the Data S1.
Study design
All four studies were open‐label, single center studies conducted in healthy subjects. Each study consisted of a screening period that was up to 28 days, an inpatient treatment period, and a follow up at 6 or 7 days after discharge from the study site. Below is an overview of DDI studies (Table 1), with individual study details below.
- EDP‐305 as perpetrator
- Effect of EDP‐305 on CYP3A4 (DDI with midazolam, study 004)
- Effect of EDP‐305 on CYP1A2 (DDI with caffeine, study 004)
- Effect of EDP‐305 on OATP1B1/B3 and BCRP (DDI with rosuvastatin, study 004)
- Effect of EDP‐305 on oral contraceptives (study 008)
- EDP‐305 as victim
- Effect of strong CYP3A4 inhibitor on EDP‐305 (DDI with itraconazole, study 005)
- Effect of strong CYP3A4 inducer on EDP‐305 (DDI with rifampin, study 005)
- Effect of moderate CYP3A4 inhibitor on EDP‐305 (DDI with fluconazole, study 007)
- Effect of selective P‐gp inhibitor on EDP‐305 (DDI with quinidine, study 007)
TABLE 1.
Summary of drug interaction studies conducted with EDP‐305
| Study | EDP‐305 | Probe substrate | |||
|---|---|---|---|---|---|
| Dose/Formulation | Days | Substrate/Dose | Days | CYP or transporter | |
| EDP‐305 as perpetrator | |||||
| 1 | 10 mg/suspension | 5 to 15 |
Midazolam 2 mg Caffeine 200 mg Rosuvastatin 20 mg |
1 and 12 1 and 12 2 and 13 |
CYP3A4 CYP1A2 BRCP/OATP1B1/OATP1B3 |
| 2 | 2.5 mg/tablet | 11 to 21 of cycle 3 | Ethinyl estradiol/norgestimate | 1 to 21 of cycles 1, 2, 3 | CYP3A4 |
| EDP‐305 as victim | |||||
| 3 | 5 mg/tablet |
1 and 14 1 and 8 |
Fluconazole 400 mg QD Quinidine 300 mg BID |
5 to 18 5 to 12 |
CYP3A4 P‐gp |
| 4 | 10 mg/suspension | 1 and 14 |
Itraconazole 200 mg q.d. Rifampin 600 mg q.d. |
5 to 18 5 to 16 |
CYP3A4 CYP3A4 |
Study 004 (clinicaltrials.gov: NCT03187496)
Subjects were confined to a clinical pharmacology research unit during the study, were discharged on day 17, and returned for a follow‐up visit on day 22 (flow chart in Material S1). Subjects abstained from caffeine starting 72 h before day 1 and continuing through discharge from the clinic. Single oral doses of midazolam 2 mg and caffeine 200 mg were administered on days 1 and 12; on days 2 and 13, a single oral dose of rosuvastatin 20 mg was administered; on days 5 to 15, a daily oral dose of EDP‐305 was administered (10 mg). Caffeine, midazolam, and rosuvastatin were provided as commercially available formulations. EDP‐305 was administered as an oral suspension containing 10 mg/30 ml in the morning after an overnight fasting. Plasma samples were collected on days 1 and 2 and days 12 and 13 to determine concentrations of midazolam and caffeine and their metabolites. Plasma samples were collected on days 2 through 5 and days 13 through 16 to determine concentrations of rosuvastatin and its metabolites. Plasma samples were collected predose on days 5, 8, 10, 11, 12, 13, 14, 15, 16, and 17 to determine trough EDP‐305 concentrations. Subjects were discharged on day 17 with a follow‐up visit on day 22.
Study 005 (clinicaltrials.gov: NCT03213145)
Oral itraconazole 200 mg (2 × 100 mg capsules) was administered once daily on days 5–18 in part 1; oral rifampin 600 mg (2 × 300 mg capsules) was administered once daily on days 5–16 in part 2. Oral EDP‐305 10 mg (10 mg/30 ml suspension) was administered once daily on days 1 and 14 in both part 1 and part 2 (flow chart in Material S1). Rifampin and EDP‐305 were administered after a 10 h overnight fast that continued for at least 4 h postdose. Itraconazole was administered while fasted when co‐administered with EDP‐305, but in a fed state when administered alone. This was done due to the food effect observed on EDP‐305 (enhanced exposure, data on file), to avoid confounding that effect with metabolic inhibition. Otherwise, itraconazole was administered in the fed state per package insert. Plasma samples to determine concentrations of EDP‐305 and its metabolites were collected predose and at 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, and 12 h postdose on days 1 and 14 of part 1 and part 2 and before the morning dose of itraconazole at 24, 48, 72, 96, and 120 h after the dose of EDP‐305 on days 15 through 19 in part 1. In part 2 (enrolling subjects different from part 1), plasma samples were collected before the morning dose of rifampin at 24, 48, and 72 h after the dose of EDP‐305 on days 15 through 17. Subjects were discharged on day 17 or day 19 with a follow up visit 6–7 days later.
Study 007 (clinicaltrials.gov: NCT03610945)
In part 1, oral fluconazole 400 mg (2 × 200 mg tablets) was administered once daily on days 5 through 18, and a single oral dose of EDP‐305 5 mg (tablet) was administered on days 1 and 14. In part 2 (enrolling subjects different from part 1), an oral dose of quinidine 300 mg (tablet) was administered twice daily on days 5 through 12, and a single oral dose of EDP‐305 5 mg (tablet) was administered on days 1 and 8 (flow chart in Material S1). All doses were administered after a 10 h overnight fast and for at least 4 h postdose. Plasma samples to measure concentrations of EDP‐305 and its metabolites were collected predose and at 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 12, 24, 48, and 72 h postdose on days 1 and 14 in part 1 and on days 1 and 8 in part 2. In addition, plasma samples were collected at 24, 48, 72, 96, and 120 h after the dose of EDP‐305 on days 15 through 19 in part 1 and on days 9 to 13 in part 2. Subjects were discharged from the inpatient unit on day 19 in part 1 and day 13 in part 2 with a follow‐up visit 6–7 days later.
Study 008 (clinicaltrials.gov: NCT03783897)
This was a crossover design study, where the same subjects underwent a lead‐in period, part 1, and part 2 (flow chart in Material S1). Subjects self‐administered the study OC (35 μg ethinyl estradiol [EE] and 250 μg norgestimate [NGM]) during a 29‐day lead‐in period. In part 1, the study consisted of an outpatient visit on day 14 (±2), an inpatient stay on days 20–23, and an outpatient visit on day 28 (−3). In part 2 (the same subjects as in part 1), the study consisted of an inpatient stay on days 10–23 and an outpatient visit on day 28 (−3). In both parts 1 and 2, subjects received a tablet of the active OC on days 1 through 21 and an inert tablet on days 22 through 28. In part 2, an oral dose of 2.5 mg EDP‐305 (tablet) was co‐administered with the OC on days 11–21 at approximately the same time each day. EDP‐305 was taken with an overnight fast of at least 10 h and for a minimum of 4 h postdose. In part 1, plasma samples for measuring NGMN (NGM active metabolite), levonorgestrel (LNG, NGM active metabolite), and EE concentrations were collected predose on days 14 and 20, and on day 21 predose and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, and 24 h (day 22) postdose. In part 2, plasma samples for assessment of NGM, LNG, and EE concentrations were collected predose on days 14 and 20, and on day 21 predose and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, and 24 h (day 22) postdose. Plasma samples to measure concentrations of EDP‐305 and its metabolites were collected predose on days 14 and 20, and on day 21 predose and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, and 24 h (day 22) postdose. Subjects returned to the inpatient unit for a follow up visit 7 days (+2 days) after their last dose.
Analytical methods
Concentrations of EDP‐305 and its metabolites (EP‐022571, EP‐022572, and EP‐022679) in human plasma were quantified by high performance liquid chromatography (HPLC) with tandem mass spectrometric (LC–MS/MS) detection with an assay range of 0.0300–30.0 ng/ml (004, 005: Medpace Bioanalytical Laboratory, Cincinnati, OH, 007, 008: Covance Laboratories, Salt Lake City, UT). Plasma concentrations of midazolam, caffeine, and rosuvastatin and their respective metabolites (1′‐hydroxymidazolam, 4′‐hydroxymidazolam, paraxanthine, and N‐desmethyl‐rosuvastatin) were analyzed using validated methodology (Covance Central Laboratory, Indianapolis, IN). Plasma concentrations of EE, NGM, and LNG were determined using a validated analytical procedure (Covance Laboratories). Details for assay methodology are provided in the Data S1.
Pharmacokinetics
Generally, all available data was used per analyte for PK parameter determination, per standard practice. The predose sample result was included in parameter estimation, if the value was quantifiable. PK parameters included area under the plasma concentration–time curve, from time 0 to the last measurable non‐zero concentration calculated by the linear up/log down method (AUC0‐t); area under the plasma concentration–time curve from time 0 extrapolated to infinity (AUC0‐inf); maximum plasma concentration (Cmax); apparent t 1/2; time to reach Cmax (Tmax); apparent terminal elimination rate constant (lambda z); apparent total clearance from plasma (CL/F); apparent volume of distribution (V d); predose plasma concentration (Ctrough); metabolite‐to‐parent plasma AUC0‐inf ratio (AUCR0‐inf).
Plasma PK parameters were estimated using noncompartmental methods with Phoenix WinNonlin version 6.3 or version 8.1 (Pharsight Corporation, Mountain View, CA). Actual sampling times were used and values below the limit of quantitation were considered missing. PK parameters by treatment were provided using descriptive statistics (n, mean, SD, median, Q1, Q3, min, max, GM, and % coefficient of variation [%CV]).
Statistical analysis
The effect of concomitant administration of EDP‐305 on a substrate or effect of a probe on EDP‐305 was assessed using the ratio and 90% confidence intervals (CIs) of the geometric mean of the plasma PK parameters: Cmax, AUC0‐t, AUC0‐inf, and AUCR0‐inf. A linear mixed model with a fixed effect for treatment and a random effect for subject was used for natural log‐transformed Cmax, AUC0‐t, AUC0‐inf, and AUCR0‐inf parameters. Geometric least‐squares means were provided for each treatment. In all comparisons, substrate administered alone was used as the reference. No adjustments were made for multiplicity.
In addition, for study 008, the pooled estimate (across all treatments) of within‐subject CV was calculated. This procedure was equivalent to Schuirmann’s 2 one‐sided tests at the 0.05 level of significance. A statistical analysis using Wilcoxon signed‐rank test was conducted to compare the Tmax of the test treatment to the Tmax of the reference treatment on PK profile day 21 for EE, NGM, and LNG analytes. For each PK parameter and treatment, median was calculated. Hodges‐Lehmann estimates of the median of the differences and the related 90% CIs were calculated for comparison between test and reference treatments.
RESULTS
Baseline characteristics of subjects from each study are shown in Table 2. Characteristics were typical of a population of healthy subjects. Subjects were predominantly men, except for the OC study (008), not Hispanic or Latino, and approximately equally divided between White and Black or African American.
TABLE 2.
Baseline characteristics of subjects in each study
| Study 004 midazolam caffeine rosuvastatin | Study 008 oral contraceptive | Study 007 fluconazole quinidine | Study 005 itraconazole rifampin | ||
|---|---|---|---|---|---|
| N = 24 | N = 43 | Part 1 N = 24 | Part 2 N = 24 | N = 48 | |
| Age, years a | 36.9 ± 9.9 | 33.0 ± 6.7 | 37.2 ± 11.4 | 26.4 ± 8.2 | 34.5 ± 8.2 |
| Female, n (%) | 2 (8.3) | 43 (100) | 2 (8.3) | 2 (8.3) | 5 (10.4) |
| Race, n (%) | |||||
| White | 11 (45.8) | 24 (55.8) | 22 (91.6) | 20 (83.3) | 20 (41.7) |
| Black or African American | 11 (45.8) | 19 (44.2) | 1 (4.2) | 2 (8.3) | 24 (50.0) |
| Other | 2 (8.4) | 0 | 1 (4.2) | 2 (8.3) | 4 (8.3) |
| Not Hispanic or Latino, n (%) | 23 (95.8) | 36 (83.7) | 21 (87.5) | 21 (87.5) | 42 (87.5) |
| Weight, kg a | 79.7 ± 10.0 | 71.4 ± 10.8 | 80.5 ± 10.4 | 75.0 ± 10.8 | 82.6 ± 10.4 |
| Body mass index, kg/m2 a | 25.3 ± 2.7 | 25.9 ± 3.8 | 25.3 ± 2.7 | 23.6 ± 3.1 | 26.5 ± 2.6 |
Mean ± standard deviation.
Pharmacokinetics
EDP‐305 as perpetrator
In study 004, mean midazolam plasma concentrations over time generally were higher when co‐administered with EDP‐305 (Figure 1). When EDP‐305 was co‐administered with the CYP3A4 probe substrate, midazolam, a 1.25‐fold increase in the Cmax, a 1.53‐fold increase in the AUC0‐t, and a 1.55‐fold increase in the AUC0‑∞ of midazolam was observed (Table 3). The increase in midazolam AUC was less than twofold, indicating that EDP‑305 is a weak inhibitor of CYP3A4. EDP‐305 co‐administered with midazolam resulted in a delayed Tmax (↑98%), lower V d/F (↓14%), lower CL/F (↓36%), and longer t 1/2 (↑34%) for midazolam. Based on the magnitude of this effect, caution should be exercised when substrates of CYP3A4 are administered concomitantly with EDP‐305.
FIGURE 1.
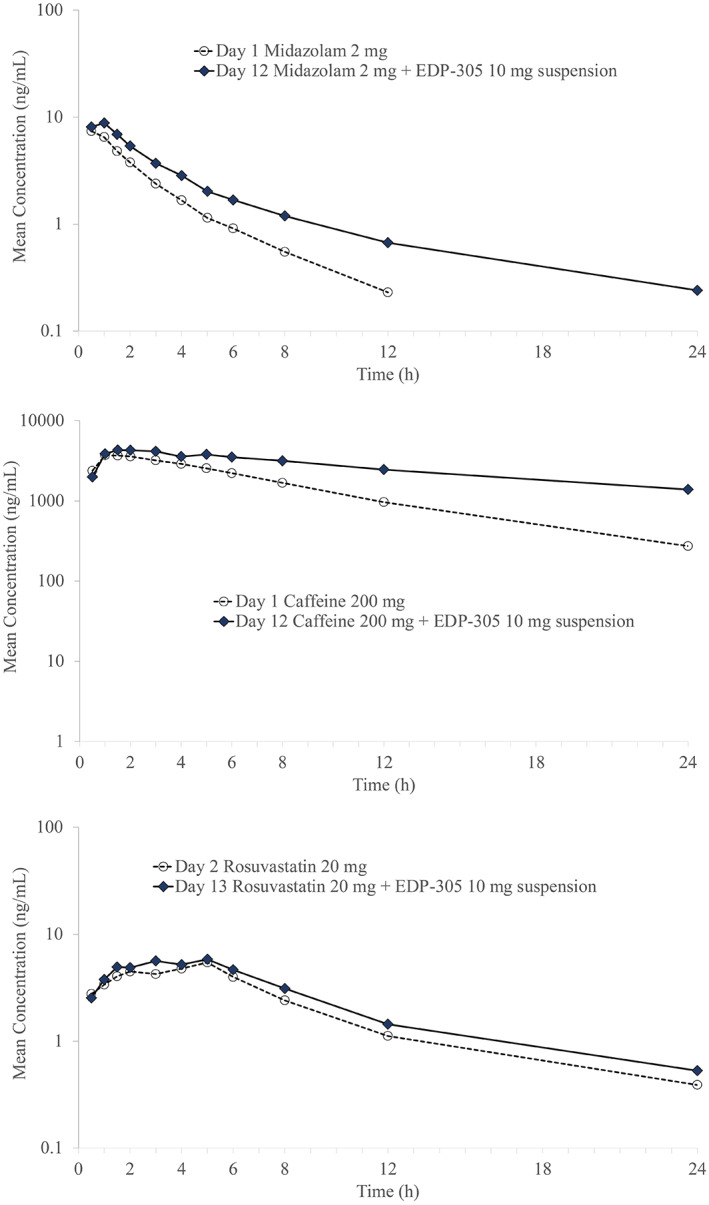
Mean plasma concentration vs. time curves for midazolam (top), caffeine (middle), and rosuvastatin (bottom) when administered along or combined with EDP‐305.
TABLE 3.
Statistical comparison of pharmacokinetic exposure parameters after co‐administration of EDP‐305 with different probes
| Treatment | N | Probe + EDP‐305 test | N | Probe alone reference | Geo LSMean ratio (90% CI) |
|---|---|---|---|---|---|
| Geo LSMean | Geo LSMean | ||||
| EDP‐305 as perpetrator | |||||
| Study 004 effect on midazolam | |||||
| Cmax, ng/ml | 24 | 9.41 | 24 | 7.51 | 1.25 (1.11, 1.41) |
| AUC0,inf, h ng/ml | 24 | 32.22 | 24 | 20.73 | 1.55 (1.32, 1.83) |
| Study 004 effect on caffeine | |||||
| Cmax, ng/ml | 24 | 4718 | 24 | 4191 | 1.13 (1.04, 1.21) |
| AUC0,inf, h ng/ml | 15 | 61,746 | 23 | 31,655 | 1.95 (1.61, 2.36) |
| Study 004 effect on rosuvastatin | |||||
| Cmax, ng/ml | 24 | 5.59 | 24 | 5.04 | 1.11 (0.94, 1.31) |
| AUC0,inf, h ng/ml | 22 | 61.19 | 22 | 49.49 | 1.24 (1.03, 1.48) |
| Study 008 effect on ethinyl estradiol | |||||
| Cmax, pg/ml | 31 | 148 | 35 | 137 | 1.08 (0.99, 1.19) |
| AUC0,t, h pg/ml | 31 | 1350 | 35 | 1250 | 1.08 (1.00, 1.16) |
| Study 008 effect on norelgestromin | |||||
| Cmax, pg/ml | 31 | 2470 | 35 | 2640 | 0.94 (0.86, 1.02) |
| AUC0,t, h pg/ml | 31 | 25,600 | 35 | 25,900 | 0.99 (0.92, 1.05) |
| Study 008 effect on levonorgestrel | |||||
| Cmax, pg/ml | 31 | 3850 | 35 | 2990 | 1.29 (1.21, 1.38) |
| AUC0,t, h pg/ml | 31 | 74,800 | 35 | 57,200 | 1.31 (1.23, 1.39) |
| Probe + EDP‐305 test | N | EDP‐305 alone reference | |||
|---|---|---|---|---|---|
| EDP‐305 as victim | |||||
| Study 005 itraconazole effect | |||||
| Cmax, ng/ml | 21 | 50.48 | 24 | 31.25 | 1.62 (1.28, 2.04) |
| AUC0,inf, h ng/ml | 18 | 1891.47 | 24 | 360.31 | 5.25 (4.26, 6.48) |
| Study 005 rifampin effect | |||||
| Cmax, ng/ml | 21 | 27.02 | 24 | 29.62 | 0.91 0.77, 1.08) |
| AUC0,inf, h ng/ml | 19 | 137.92 | 24 | 393.59 | 0.35 (0.30, 0.41) |
| Study 007 fluconazole effect | |||||
| Cmax, ng/ml | 24 | 57.71 | 24 | 32.73 | 1.76 (1.56, 1.99) |
| AUC0,inf, h ng/ml | 24 | 1537.60 | 24 | 408.40 | 3.77 (3.40, 4.17) |
| Study 007 quinidine effect | |||||
| Cmax, ng/ml | 24 | 31.78 | 24 | 31.64 | 1.00 (0.92, 1.10) |
| AUC0,inf, h ng/ml | 24 | 367.17 | 24 | 353.24 | 1.04 (0.95, 1.14) |
Abbreviations: AUC0,inf, area under the concentration curve from time 0 to infinity; CI, 90% confidence interval; Cmax, peak plasma concentration; Geo LSMean, geometric least square mean.
In study 004, mean caffeine plasma concentrations over time generally were higher when co‐administered with EDP‐305. When co‐administered with caffeine, a CYP1A2 probe, EDP‐305 marginally increased caffeine Cmax by 1.13‐fold, AUC0‐t by 1.90‐fold, and AUC0‐∞ by 1.95‐fold (Table 3). The increase in caffeine AUC was less than twofold, indicating that EDP‐305 is a weak inhibitor of CYP1A2. EDP‐305 co‐administered with caffeine resulted in a delayed Tmax (↑50%), lower CL/F (↓45%), and longer t 1/2 (↑64%) but had little effect on the V d/F for caffeine. Based on the magnitude of this effect, CYP1A2 substrates with a narrow therapeutic index may have potential for increased exposure when administered with EDP‐305.
In study 004, mean rosuvastatin plasma concentrations over time generally were higher when co‐administered with EDP‐305. When co‐administered with rosuvastatin, a probe for BCRP and OATP1B1/1B3, EDP‐305 marginally increased the rosuvastatin Cmax by 1.11‐fold, AUC0‐t by 1.24‐fold, and AUC0‐∞ by 1.24‐fold (Table 3). The increase in rosuvastatin AUC was less than 1.25‐fold, suggesting that EDP‐305 is unlikely to be an inhibitor of OATP1B1, OATP1B3, and BCRP transporters. EDP‐305 co‐administration with rosuvastatin had no apparent effect on Tmax and V d/F (↓6%), but resulted in lower CL/F (↓20%), and longer t 1/2 (↑17%) for rosuvastatin. Based on the magnitude of this effect, substrates of BCRP and OATP1B1/1B3 can be administered concomitantly with EDP‐305.
The metabolite‐to‐parent ratio data was available (based on quantifiable concentrations) for both midazolam and rosuvastatin metabolites, 1′‐hydroxymidazolam, and N‐desmethyl‐rosuvastatin, respectively. The metabolite‐to‐parent ratio based on total systemic exposure (AUCR0‐inf) decreased when midazolam was administered with EDP‐305 compared to when it was administered alone. When midazolam was administered with EDP‐305, GM AUCR0‐inf was 20.6% compared to 39.1% when administered alone. The metabolite‐to‐parent ratio based on total systemic exposure (AUCR0‐inf) decreased when rosuvastatin was administered with EDP‐305 compared to when it was administered alone. When rosuvastatin was administered with EDP‐305, GM AUCR0‐inf was 11.2% compared to 16.6% when administered alone.
An exploratory objective of study 004 was to assess trough levels of EDP‐305 and its metabolites in the presence and absence of single doses of midazolam, caffeine, and rosuvastatin. As indicated by generally comparable trough concentrations from day 12 onward, steady‐state concentrations of EDP‐305 and its metabolites were achieved by day 12 in the majority of subjects. Co‐administration of metabolizing enzyme or transporter probes had no apparent effect on the PK of EDP‐305 and its metabolites.
In study 008, mean plasma concentrations of EE, NGM, and LNG were comparable whether the OC was administered alone or combined with EDP‐305 (Figure 2). Peak and systemic exposures (Cmax and AUC0‐τ) of EE and NGM were similar following co‐administration of the combined OC with EDP‐305 or administration of OC alone (Table 3). A slight increase (1.3‐fold) in LNG Cmax and AUC0‐τ was observed following the co‐administration of the OC with EDP‐305 compared to OC alone. Following co‐administration of OC with EDP‐305, the median Tmax of EDP‐305 was 4.0 h and the geometric mean t 1/2 was 9.6 h.
FIGURE 2.
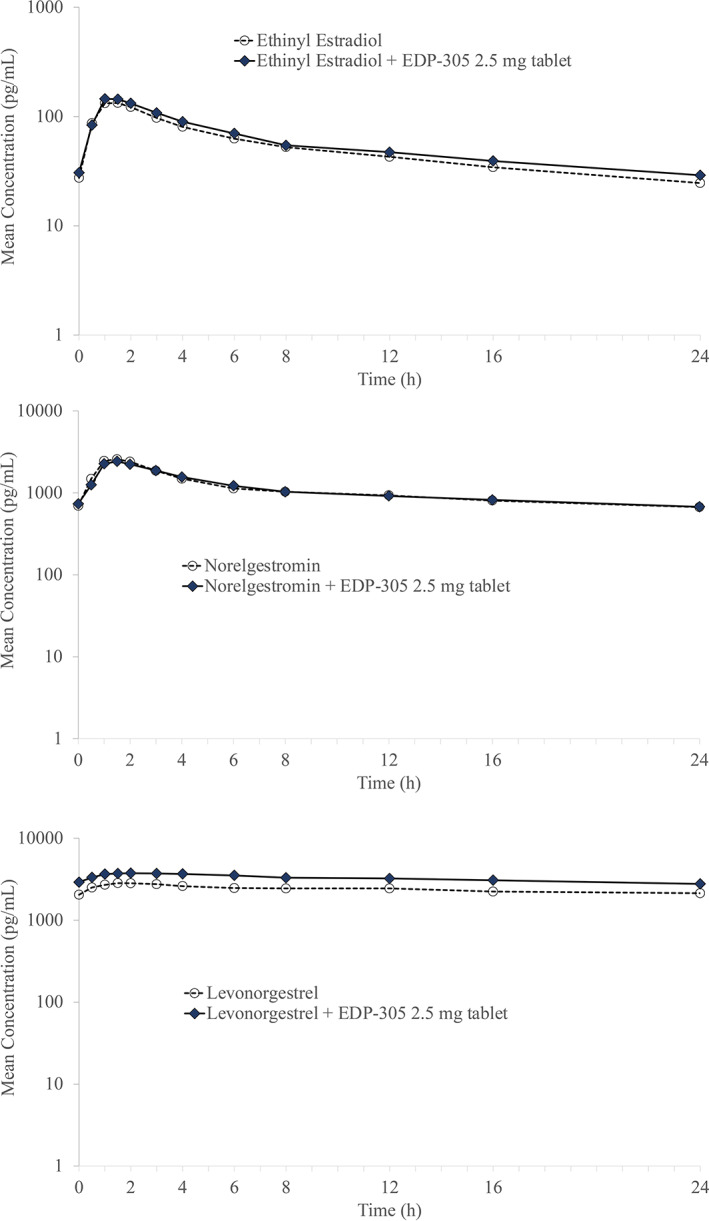
Mean plasma concentration of ethinyl estradiol (top), norelgestromin (middle), and levonorgestrel (bottom) after co‐administration with EDP‐305.
EDP‐305 metabolites (EP‐022571, EP‐022572, and EP‐022679) were also evaluated. Metabolite EP‐022571 was less abundant than the parent with respective geometric mean AUC0‐τ (MRAUC) ~ 0.892% of the parent. Metabolite EP‐022572 was less abundant than the parent with mean MRAUC ~ 1.74% of the parent. Metabolite EP‐022679 was less abundant than the parent with mean MRAUC of ~6.30% of the parent. The MRAUC appeared to be higher for EP‐022679 (0.0630) compared to EP‑022571 (0.00892) and EP‐022572 (0.0174), indicating that EP‐022679 was the most abundant metabolite of the three metabolites tested in this study.
In addition, co‐administration of the combined OC and EDP‑305 had no effect on serum progesterone levels, and mean progesterone concentration was below 3 μg/L throughout the study.
Based on these assessments, EDP‐305 does not appear to affect the PKs of a combined OC or serum progesterone levels as no relevant changes were observed following co‐administration of the OC with EDP‐305 compared to the OC alone. These results suggest that a combined OC may be considered an acceptable form of contraception for premenopausal female patients receiving EDP‑305 therapy.
EDP‐305 as victim
In study 005, mean plasma itraconazole concentrations, the strong CYP3A4 inhibitor, were markedly higher when coadministered with EDP‐305 compared with itraconazole alone (Figure 3). When EDP‐305 was co‐administered with itraconazole, EDP‐305 peak and systemic exposures increased by ~1.6‐ and 5.3‐fold, respectively (Table 3). Co‐administration of itraconazole with EDP‐305 resulted in a relatively delayed Tmax (2‐fold), lower V d/F (0.47‐fold), lower CL/F (0.81‐fold), and longer t 1/2 (2.79‐fold) for EDP‐305. These PK results are consistent with inhibition of a major metabolic pathway (CYP3A4).
FIGURE 3.
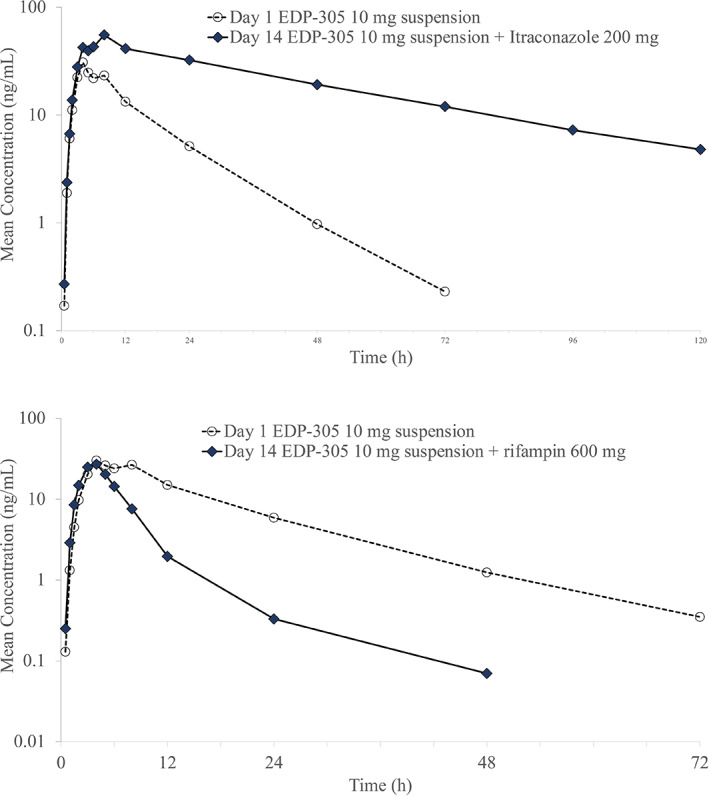
Mean plasma concentrations of EDP‐305 following co‐administration with itraconazole (top) or rifampin (bottom).
Following co‐administration with itraconazole, the peak exposure decreased 0.84‐fold, 0.45‐fold, and 0.63‐fold for metabolites EP‐022571, EP‐022572, and EP‐022679, respectively. The total exposure (AUC0‐t and AUC0‐inf) increased 1.99‐fold to 2.29‐fold in all metabolites except EP‐022571, for which AUC0‐t decreased 0.66‐fold. The t 1/2 could not be calculated for metabolite EP‐022679 and could not be reliably estimated for EP‐002571 due to the limited number of evaluable subjects (n = 5). However, t 1/2 increased 3.88‐fold for EP‐022572.
Furthermore, in study 007, mean plasma EDP‐305 concentrations were markedly higher when co‐administered with. Fluconazole versus EDP‐305 alone (Figure 4). When EDP‐305 was co‐administered with the moderate CYP3A4 inhibitor, fluconazole, EDP‐305 peak and systemic exposures increased by ~1.8‐ and 3.8‐fold, respectively (Table 3). EDP‐305 CL/F and V d/F were decreased, and the t 1/2 increased when EDP‐305 was co‐administered with fluconazole compared to when EDP‐305 was administered alone.
FIGURE 4.
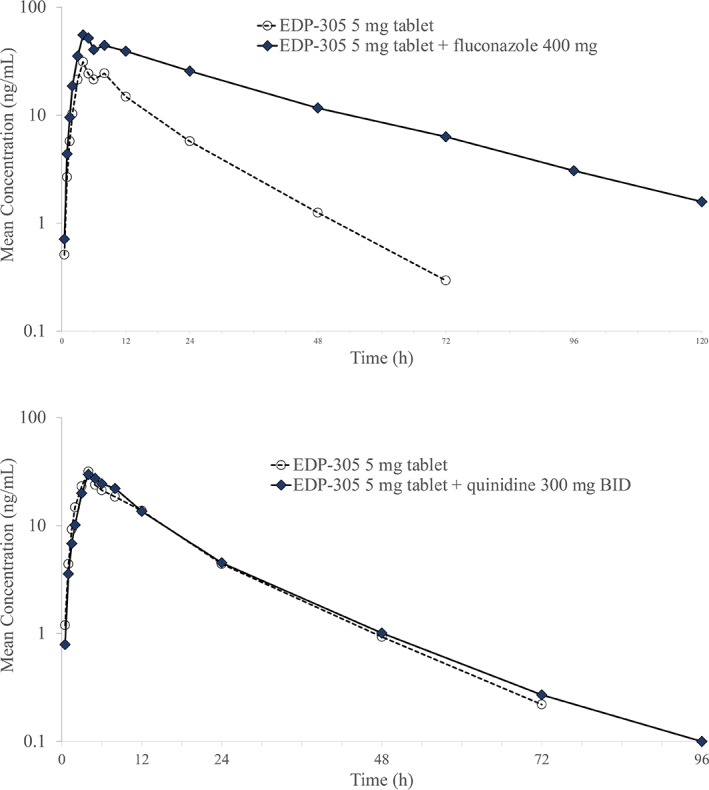
Mean plasma concentrations of EDP‐305 after co‐administration with fluconazole (top) or quinidine (bottom).
The co‐administration of fluconazole with EDP‐305 resulted in a decrease in EP‐022571 Cmax (55%), AUC0‐t (39%), and AUC0‐inf (34%) relative to EDP‐305 administered alone. Additionally, EP‐022571 t 1/2 increased (1.9‐fold) when EDP‐305 was co‐administered with fluconazole relative to EDP‐305 administered alone, whereas Tmax was similar. EP‐022572. The co‐administration of fluconazole with EDP‐305 resulted in a slight decrease in EP‐022572 Cmax (24%) and an increase in AUC0‐t (1.9‐fold), and AUC0‐inf (1.8‐fold), relative to EDP‐305 administered alone. Additionally, EP‐022572 t 1/2 increased (2.9‐fold) when EDP‐305 was co‐administered with fluconazole relative to EDP‐305 administered alone, whereas Tmax was similar.
The co‐administration of fluconazole with EDP‐305 resulted in an increase in EP‐022679 Cmax (1.5‐fold), AUC0‐t (3.0‐fold), AUC0‐inf (2.2‐fold), delayed Tmax (1.5‐fold longer), and higher t 1/2 (3.3‐fold).
These PK results are also consistent with inhibition of a major metabolic pathway (CYP3A4), similar to study 005.
In study 005, mean plasma EDP‐305 concentrations were markedly lower when co‐administered with rifampin versus EDP‐305 alone (Figure 3). When EDP‐305 was co‐administered with the strong CYP3A4 inducer, rifampin, no apparent effect on EDP‐305 peak exposure was observed, but total systemic exposure decreased by ~65% (Table 3). The co‐administration of rifampin with EDP‐305 had no apparent effect on Tmax but resulted in a relatively higher V d/F (1.79‐fold), higher CL/F (2.77‐fold), and shorter t 1/2 (0.35‐fold) for EDP‐305.
The major metabolites of EDP‐305 generally increased when EDP‐305 was co‐administered with rifampin compared to when EDP‐305 was administered alone. The peak exposure increased 1.58‐fold, 2.77‐fold, and 1.66‐fold for metabolites EP‐022571, EP‐022572, and EP‐022679, respectively. The total exposure (AUC0‐t and AUC0‐inf) increased 1.05‐fold to 1.25‐fold for EP‐022571, and EP‐022572 decreased 0.39‐fold for EP‐022679. The t 1/2 decreased 1.54‐ to 1.79‐fold for all metabolites.
Taken together, the lower exposures of EDP‐305 and the generally increased exposures of the major metabolites of EDP‐305, are consistent with increased metabolism and clearance of the parent drug and indicate that the metabolism of EDP‐305 was induced by the strong CYP3A4 inducer rifampin.
These PK results are consistent with induction of a major metabolic pathway (CYP3A4). Based on these results, co‐administration of EDP‐305 with strong or moderate inducers of CYP3A4 is not recommended.
In study 007, mean plasma quinidine concentrations were comparable when co‐administered with EDP‐305 versus quinidine alone (Figure 4). When EDP‐305 was co‐administered with the selective P‐gp inhibitor, quinidine, no apparent effect on EDP‐305 peak or systemic exposure was observed (Table 3). Co‐administration of quinidine with EDP‐305 had minimal effect on Tmax, V d/F, CL/F, and t 1/2 for EDP‐305. The PK results are consistent with lack of an effect of P‐gp on EDP‐305.
The co‐administration of quinidine with EDP‐305 resulted in a decrease in EP‐022571 Cmax (36%), AUC0‐t (39%), and AUC0‐inf (36%) relative to EDP‐305 administered alone, whereas the Tmax and t 1/2 were not impacted. The PK parameters of EP‐022572 were generally similar when EDP‐305 was administered alone or in combination with quinidine. The co‐administration of quinidine with EDP‐305 resulted in an increase in EP‐022679 Cmax (1.4‐fold), AUC0‐t (1.6‐fold), AUC0‐inf (1.5‐fold), and higher t 1/2 (1.6‐fold), whereas Tmax was not impacted.
Based on these results, no dose adjustments are anticipated when EDP‐305 is co‐administered with quinidine or other P‐gp inhibitors or inducers.
Detailed PK results from each study are provided in the Data S1.
Safety/tolerability
In these four studies, EDP‐305 generally was well‐tolerated. The most common adverse events (AEs) were headache, diarrhea, and pruritus, and most AEs were of mild or moderate severity. No deaths or serious AEs considered related to EDP‐305 were reported. No clinically significant findings were reported from physical examination, electrocardiogram (ECG), or clinical laboratory testing that were considered related to EDP‐305. Additional details on the safety and tolerability findings for each study are found in the Data S1.
DISCUSSION
Clinically significant drug interactions are an important cause of hospitalization and increased healthcare costs in the United States. 9 A wide range of drug metabolizing enzymes as well as drug transporters have been implicated as causes of clinically significant drug interactions. 10 , 11 Thus, an important aspect of drug development for a new chemical entity is to determine the drug interaction potential early in the development process to avoid serious, potentially life‐threatening complications. 10 , 11 The US Food and Drug Administration (FDA) and European regulatory authorities have established guidance for industry for evaluating the drug interaction potential of drugs. 12 , 13 The FDA Guidance provides definitions for the magnitude of an effect on CYP enzymes and OATP1B1/1B3 and BCRP transporters based on the increase of AUC for the probes as weak (≥1.25 but <2‐fold increase), moderate (≥2 but <5‐fold increase), and strong (≥5‐fold increase). 13
Previous work suggests the potential for clinically significant drug interactions with FXR agonists. In human hepatocytes, a potentially clinically significant interaction was identified between FXR agonists and CYP3A4. 14 Studies in healthy subjects with the FXR agonist, obeticholic acid, identified a significant interaction with CYP1A2. 15 Because of the complex nature of NASH, a need exists to evaluate the potential for interactions with drugs being developed for NASH because of the frequent use of concomitant medications. 16
On the basis of results from preclinical studies, four phase I studies were conducted in healthy subjects to characterize the DDI potential of EDP‐305. These studies used guidelines provided by regulatory agencies for the design of studies to explore drug interaction potential of drugs. From studies of EDP‐305 co‐administered with drugs metabolized by CYP3A4, caution should be exercised when substrates of CYP3A4 are administered concomitantly with EDP‐305, and co‐administration of EDP‐305 with strong or moderate inhibitors and inducers of CYP3A4 is not recommended. In addition, CYP1A2 substrates with a narrow therapeutic index may have the potential for increased exposure when co‐administered with EDP‐305; therefore, monitoring drug concentrations of CYP1A2 substrates with a narrow therapeutic index (e.g., tizanidine and theophylline) is recommended. In contrast, substrates of BCRP and OATP1B1/1B3 can be administered concomitantly with EDP‐305 with a low potential for DDIs. Further, no dose adjustments are anticipated when EDP‐305 is co‐administered with quinidine or other P‐gp inhibitors or inducers. Co‐administration of EDP‐305 and a combined OC had no relevant effects on plasma concentrations of the combined OC components, which suggests that a combined OC may be considered an acceptable form of contraception for premenopausal female patients receiving EDP‐305.
Of note, these four studies evaluated the DDI potential of EDP‐305 at doses of 2.5 (tablet), 5 (tablet), or 10 mg (suspension). A relative bioavailability assessment indicated that EDP‐305 tablet has approximately twofold higher exposures compared to the suspension (data on file). Results from phase I studies in healthy subjects and subjects with presumptive NAFLD 4 and a phase II study in subjects with NASH 5 suggest that the optimal therapeutic dosage range for EDP‐305 is 1–2 mg (tablet) once daily. These DDI studies were conducted at clinically relevant doses, and can be applied to future studies in patients with NASH. A phase IIb study in NASH is currently ongoing (NCT04378010).
There are no dose adjustments for EDP‐305 or concomitant medications based on all four study results (co‐administration of EDP‐305 with strong or moderate inhibitors and inducers of CYP3A4 is not recommended, with no dose adjustment a priori). The magnitude of effect on concomitant medications estimated from the DDI studies is expected to be a worst‐case scenario as healthy volunteers may have higher levels of functional enzymes (e.g., CYP3A4) compared to patients with NASH, 8 and thus results from the DDI studies presented here are expected to translate and have been already applied, to studies in patients with NASH. Of note, cirrhotic subjects were excluded from current NASH studies.
In summary, the EDP‐305 DDI profile allows for convenient administration in patients with NASH and other patient populations with comorbidities, with minimal dose modification of concomitant medications.
AUTHOR CONTRIBUTIONS
A.A. and N.A. wrote the manuscript, designed the research, performed the research, and analyzed the data.
CONFLICT OF INTEREST
All authors are employees of Enanta Pharmaceuticals, Inc., Watertown, MA.
Supporting information
Appendix S1
ACKNOWLEDGEMENT
The authors acknowledge the editorial assistance of Richard S. Perry, PharmD, in the preparation of this manuscript, which was supported by Enanta Pharmaceuticals, Inc., Watertown, MA.
Ahmad A, Adda N. Assessment of drug–drug interaction potential with EDP‐305, a farnesoid X receptor agonist, in healthy subjects. Clin Transl Sci. 2022;15:2146‐2158. doi: 10.1111/cts.13348
Funding information
These studies were funded by Enanta Pharmaceuticals, Inc., Watertown, MA.
REFERENCES
- 1. An P, Wei G, Huang P, et al. A novel non‐bile acid FXR agonist EDP‐305 potently suppresses liver injury and fibrosis without worsening of ductular reaction. Liver Int. 2020;40(7):1655‐1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Erstad DJ, Farrar CT, Ghoshal S, et al. Molecular magnetic resonance imaging accurately measures the antifibrotic effect of EDP‐305, a novel farnesoid X receptor agonist. Hepatol Commun. 2018;2(7):821‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li S, Ghoshal S, Sojoodi M, et al. The farnesoid X receptor agonist EDP‐305 reduces interstitial renal fibrosis in a mouse model of unilateral ureteral obstruction. Faseb J. 2019;33(6):7103‐7112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ahmad A, Sanderson K, Dickerson D, Adda N. Phase 1 study of EDP‐305, a novel once‐daily oral farsenoid × receptor agonist, in healthy subjects and those with presumptive nonalcoholic fatty liver disease. Int J Gastroenterol. 2021;5:68‐79. [Google Scholar]
- 5. Ratziu V, Rinella ME, Neuschwander‐Tetri BA, et al. EDP‐305 in patients with NASH: A phase II double‐blind placebo‐controlled dose‐ranging study. J Hepatol. 2021;76:506‐517. [DOI] [PubMed] [Google Scholar]
- 6. Zhang S, Pan X, Jeong H. GW4064, an agonist of farnesoid X receptor, represses CYP3A4 expression in human hepatocytes by inducing small heterodimer partner expression. FASEB J. 2015;43:743‐758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ishida C, Sanoh S, Kotake Y. CYP1A2 downregulation by obeticholic acid: Usefulness as a positive control for the in vitro evaluation of drug:drug interactions. J Pharm Sci. 2019;108:3903‐3910. [DOI] [PubMed] [Google Scholar]
- 8. Jamwal R, de la Monte SM, Ogasawara K, Adusumalli S, Barlock BB, Akhlaghi F. Nonalcoholic fatty liver disease and diabetes are associated with decreased CYP3A4 protein expression and activity in human liver. Mol Pharm. 2018;15(7):2621‐2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carpenter M, Berry H, Pelletier AL. Clinically relevant drug–drug interactions in primary care. Am Fam Physician. 2019;99(9):558‐564. [PubMed] [Google Scholar]
- 10. Peng Y, Cheng Z, Xie F. Evaluation of pharmacokinetic drug–drug interactions: a review of the mechanisms, in vitro and in silico approaches. Metabolites. 2021;11(2):75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yu J, Petrie ID, Levy RH, Ragueneau‐Majlessi I. Mechanisms and clinical significance of pharmacokinetic‐based drug–drug interactions with drugs approved by the U.S. Food and Drug Administration in 2017. Drug Metab Dispos. 2018;47:135‐144. [DOI] [PubMed] [Google Scholar]
- 12. European Medicines Agency Guideline on the Investigation of Drug Interactions . EMA, London, UK2012. Accessed: September 24, 2019 http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf.
- 13. U.S. Food and Drug Administration . Guidance for Industry. In Vitro Drug Interaction Studies—Cytochrome P450 Enzymes‐ and Transporter‐Mediated Drug Interactions Guidance for Industry. Accessed: 3 December 2020 https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/vitro‐drug‐interactionstudies‐cytochrome‐p450‐enzyme‐and‐transporter‐mediated‐drug‐interactions.
- 14. Zhang S, Pan X, Jeong H. GW4064, an agonist of farnesoid X receptor, represses CYP3A4 expression in human hepatocytes by inducing small heterodimer partner expression. Drug Metab Dispos. 2015;43(5):743‐748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Edwards JE, Eliot L, Parkinson A, Karan S, MacConell L. Assessment of pharmacokinetic interactions between obeticholic acid and caffeine, midazolam, warfarin, dextromethorphan, omeprazole, rosuvastatin, and digoxin in phase 1 studies in healthy subjects. Adv Ther. 2017;34(9):2120‐2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dash RP, Babu RJ, Srinivas NR. Non‐alcoholic steatohepatitis (NASH) drug discovery – building a consensus on ADME screening tools and clinical pharmacology strategies to aid candidate development. J Pharm Pharm Sci. 2018;21(1):481‐495. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1