

Abstract
Advances in immuno‐oncology have provided a variety of novel therapeutics that harness the innate immune system to identify and destroy neoplastic cells. It is noteworthy that acceptable safety profiles accompany the development of these targeted therapies, which result in efficacious cancer treatment with higher survival rates and lower toxicities. Adoptive cellular therapy (ACT) has shown promising results in inducing sustainable remissions in patients suffering from refractory diseases. Two main types of ACT include engineered Chimeric Antigen Receptor (CAR) T cells and T cell receptor (TCR) T cells. The application of these immuno‐therapies in the last few years has been successful and has demonstrated a safe and rapid treatment regimen for solid and non‐solid tumors. The current review presents an insight into the clinical pharmacology aspects of immuno‐therapies, especially CAR‐T cells. Here, we summarize the current knowledge of TCR and CAR‐T cell immunotherapy with particular focus on the structure of CAR‐T cells, the effects and toxicities associated with these therapies in clinical trials, risk mitigation strategies, dose selection approaches, and cellular kinetics. Finally, the quantitative approaches and modeling techniques used in the development of CAR‐T cell therapies are described.
INTRODUCTION
Over the past 3 decades, our understanding of the role of the human immune system in various cancers has evolved. From these learnings, a host of new modalities were developed to treat cancer using various components of the immune system, now commonly referred to as “immuno‐oncology” or “cancer immunotherapy.” The targets and mechanisms through which these novel therapies operate are numerous, but generally rely on the ability of a modality to target a specific tumor associated antigen, and the ability of the immune system to selectively inhibit or destroy cancer cells expressing those antigens.
Of these immunotherapies, the Chimeric Antigen Receptor T‐cells (CAR‐T cells) are perhaps the most unique. Engineered using a patient's own T‐cells (i.e., autologous or from an allogeneic donor), CAR‐T cells bind specifically to tumor‐associated antigens located on the surface of cancer cells. Upon binding, an immunological synapse is formed between the CAR‐T cell and target cell, which occurs independently of the major histocompatibility complex (MHC) and antigen presentation, and results in the release of perforins and granzymes that trigger caspase dependent and independent apoptotic cell death. 1 , 2 Recent generations of CAR‐T cells can also trigger T cell activation and proliferation after multiple exposures to tumor antigens. 3
CAR‐T CELL DESIGN
CARs are bioengineered receptors that bind to specific tumor antigens and a CAR is the key component of CAR‐T cells. The core structure of a CAR consists of three domains: the extracellular domain, transmembrane domain, and endodomain. The extracellular domain, which is also called antigen‐recognition domain, is a single chain fragment variant composed of heavy‐chain and light‐chain variable regions that are linked by a small polypeptide. The extracellular domain is tethered to the transmembrane domain through a hinge domain that transmits the receptor‐binding signal. The endodomain, also called the intracellular domain, is the core component of most CARs and is mainly composed of the T cell co‐receptor CD3ζ that contains three immunoreceptor tyrosine‐based activation motifs for signal transduction. 4 Over the past 3 decades, the structure of CARs has evolved over five generations based on their structure and endodomain composition. As shown in Figure 1, in the first generation, only one CD3ζ intracellular domain was included in the CAR structure and its poor T cell proliferation and cytotoxicity led to limited in vivo antitumor activity. The second generation added a co‐stimulatory domain to the CD3ζ intracellular domain to enhance T cell proliferation and cytotoxicity. The third generation added one more co‐stimulatory domain to the second generation to further expand T cell proliferation and cytotoxicity of CARs. The fourth generation was also developed based on the second generation, but it added a protein instead of an additional co‐stimulatory domain to the CD3ζ intracellular domain to activate the downstream transcription factor to induce tumor‐killing cytokine production upon CARs activation. The fifth generation is still under development, and it has been proposed to use a gene editing approach to knock out the HLA and TCR genes of T cells obtained from healthy donors to reduce the risk of graft‐versus‐host disease (GVHD) against transplanted CAR‐T cells. 4 , 5
FIGURE 1.
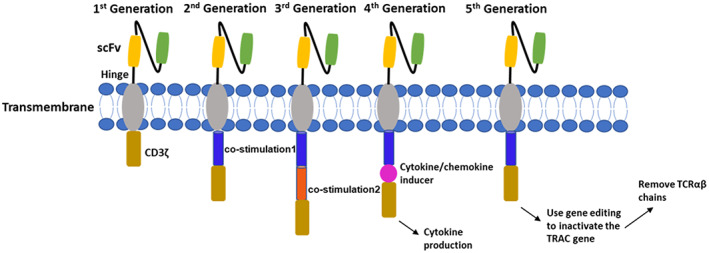
The structures of five generations of CARs.
The success of CAR‐T cells as a novel cancer immunotherapy is reflected in the six drug approvals by the US Food and Drug Administration (FDA) in the past 5 years (Table 1). Notably, each of the six approved CAR‐T cell therapies are indicated for hematologic malignancies, with cluster of differentiation 19 (CD19) and B‐cell maturation antigen (BCMA) comprising the CAR targets. The success of CAR‐T cell therapy in hematologic malignancies can be attributed to several factors. CD19 and BCMA both exhibit desirable qualities of druggable targets. For example, in B‐cell malignancies, CD19 is expressed at normal to high levels, approximating over 80% of malignant cells in acute lymphoblastic leukemia, B‐cell lymphoma, and B‐cell leukemia. 6 Similarly, BCMA has been detected in nearly all multiple myeloma (MM) cell lines (80–100%) and is also more highly expressed in malignant plasma cells versus normal plasma cells. 7 The abundance of the expression of cell surface targets in malignant versus normal cells, combined with the specificity of the CAR, maximizes the potential for clinical efficacy with the inhibition or destruction of the intended target (i.e., malignant) cells. The importance of highly specific tumor‐associated antigens also aid in minimizing the risk of potential on‐target, off‐tumor adverse events (AEs), leading to a more manageable safety profile, relative to traditional chemotherapy. Like other immune modulators, treatment with CAR‐T cell therapy can still result in other serious AEs, such as cytokine release syndrome or neurotoxicity, which can be attributed to the CAR‐T mechanism of action. However, these AEs can often be managed with anti‐IL‐6 agents (such as tocilizumab), corticosteroids, and other supportive care. Finally, as B‐cell targets, CD19‐ and BCMA‐expressing cells exist freely in the systemic circulation, which is where CAR‐T cells are infused back into the patient, once prepared. Thus, the proximity of the newly infused CAR‐T cells and target expressing cells increases the probability for successful CAR binding, formation of the immunological synapse, and downstream tumor cell cytotoxicity. This is unlike targets for solid tumor malignancies, in which several physiological barriers exist that prevent access of the CAR‐T cells to target cells.
TABLE 1.
List of approved CAR‐T therapies in the United States
| Approved CAR‐T brand name (generic name) | Approved indication(s) | Target | Year of first US approval |
|---|---|---|---|
| CARVYKTI (ciltacabtagene autoleucel) | Adult R/R multiple myeloma | BCMA | 2022 |
| ABCEMA (idecabtagene vicleucel) | Adult R/R multiple myeloma | BCMA | 2021 |
| BREYANZI (lisocabtagene maraleucel) | Adult R/R large B‐cell lymphoma | CD19 | 2021 |
| TECARTUS (brexucabtagene autoleucel) | Adult R/R mantle cell lymphoma; adult R/R B‐cell precursor acute lymphoblastic leukemia | CD19 | 2020 |
| YESCARTA (axicabtagene ciloleucel) | Adult R/R large B‐cell lymphoma; adult R/R follicular lymphoma | CD19 | 2017 |
| KYMRIAH (tisagenlecleucel) | Adult R/R large B‐cell lymphoma; pediatric refractory acute lymphoblastic leukemia | CD19 | 2017 |
Abbreviations: BCMA, B‐cell maturation antigen; CD19, cluster of differentiation 19; R/R, relapsed or refractory.
Despite the success of CAR‐T cell therapy in hematologic malignancies, no equivalent successes have been observed for solid tumor malignancies yet. This is mainly attributed to the lack of tumor specific antigens. Unlike liquid tumors, solid tumors do not usually express a unique tumor‐specific antigen and it is common that the same antigen is also expressed on healthy tissues although usually at a lower degree, which leads to higher risk of on‐target, off‐tumor toxicities. In addition, the complicated structure and microenvironment of solid tumors have posed physical challenges for CAR‐T cells to migrate and penetrate tumor tissues, leading to reduced antitumor activities. 8 , 9 A comparison of the top CAR‐T cell therapy targets in hematological malignancy and solid tumors is summarized in Table 2. It demonstrates the approximate number of active CAR‐T cell therapies in each target from 2019 to 2021. 10 As described above, for liquid tumors, CD19 and BCMA are still the dominating targets, whereas HER2, MSLN, GD2, and EGFR are leading targets for solid tumors. It is noteworthy that CAR‐T cell therapies indicated for liver cancer targeting GPC2/GPC3 have witnessed a fast growth during the past 3 years as glypicans are highly expressed in hepatocellular carcinomas.
TABLE 2.
Top targets and approximate number of active CAR‐T cell therapies for hematological malignancies and solid tumors a
| Hematological malignancies | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Target | CD19 | BCMA | CD22 | CD20 | CD123 | CD33 | CD30 | CD38 | CS1 | TAA b |
| Approx. number of active therapies | 542 | 166 | 80 | 64 | 60 | 39 | 38 | 30 | 22 | 2 |
| Solid tumors | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Target | HER2 | MSLN | EGFR | GD2 | GPC2/3 | MUC1 | PSMA | NY‐ESO‐1 | EBV | TAA b |
| Approx. number of active therapies | 59 | 53 | 50 | 49 | 39 | 34 | 28 | 5 | 4 | 5 |
Table was derived from figure 2 in Upadhaya et al. 10 If the actual number is not available in the figure, digitization is used to estimate the number.
Undisclosed tumor‐associate antigen.
CAR‐T CELL MANUFACTURING
As shown in Figure 2, the CAR‐T cell manufacturing process starts from the collection of the patient's T cells by leukapheresis. Once isolated, autologous T cells undergo ex vivo enrichment and activation, followed by the transduction of the CAR cDNA via gene expression vectors, such as retroviral and lentiviral vectors. The genetically engineered T cells are expanded in large scale, washed, concentrated, and cryopreserved, then infused back to the patient for treatment. 11 , 12 , 13 Although protocols for manufacturing clinical grade CAR‐T cells have been well‐established, a number of manufacturing and regulatory challenges, such as processing techniques, quality control mechanisms, logistic developments, and financial liabilities need to be addressed to develop robust CAR‐T cell therapies for the patients. 11 , 14
FIGURE 2.
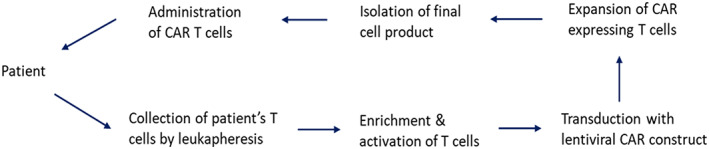
Major steps in CAR‐T cell manufacturing process.
PHARMACOKINETICS OF CAR‐T CELLS
The clinical exposure‐time relationships of cell‐based therapies are commonly summarized under the term “cellular kinetics.” Due to their “living” nature, CAR‐T cells exhibit unique pharmacokinetic (PK) characteristics and traditional PK concepts and properties that apply to small and large molecules, like distribution, metabolism, and excretion, are not directly applicable to CAR‐T cells. These cells do not undergo typical metabolism or clearance pathways. After infusion, they distribute and expand rapidly, and can persist up to multiple years as their concentration levels (typically assessed by number of cells per unit volume or copy number) decrease slowly over time. 15 The cellular kinetic profile of CAR‐T cells is generally characterized by the two terms “cellular expansion” and “cellular persistence” and is shown in Figure 3. The expansion phase describes the rapid exponential expansion of the cells and includes the maximum level of detectable CAR‐T cells in the body following infusion (maximum plasma concentration [C max]), the time to reach C max, and the area under the concentration‐time curve from time zero to time t (AUC0–t ), where t is usually a timepoint during the initial expansion phase, for example 28 days after the infusion of CAR‐T cells (e.g., AUC0–28 days). The persistence phase, consisting of terminal half‐life (t 1/2), the last measurable concentration and the time of the last measurable concentration, describes the perseverance of the CAR‐T cells in the body over time.
FIGURE 3.
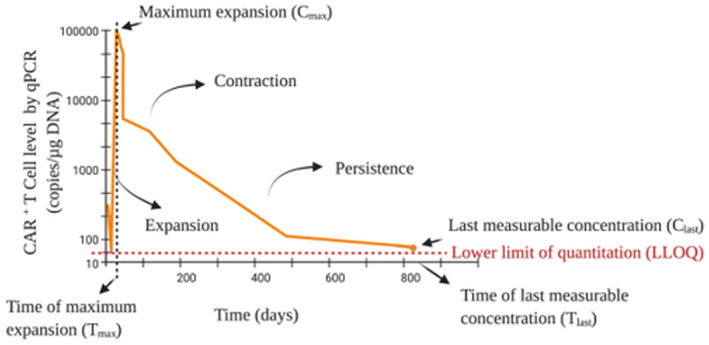
A representative concentration‐time profile for a CAR‐T therapy. Image adapted from figure 1b in Mueller et al. 15 to depict key cellular kinetic parameters, including cellular expansion and persistence. qPCR, quantitative polymerase chain reaction.
Evidently, the investigation of cellular kinetics and the impacting factors help in understanding the safety and efficacy of CAR‐T cells and provide essential information for the selection of the optimal dose range and regimen. 16 Major factors that influence the therapeutic outcome and duration of response following CAR‐T cell infusion are proliferation and persistence of the cells, although exceptions exist. 17 Therefore, detection and monitoring of the circulating CAR‐T cells is important. 18 The two bioanalytical approaches that are most commonly used to characterize the cellular kinetics and quantify the level of persisting CAR‐T cells are quantitative polymerase chain reaction (qPCR) sequencing and flow cytometry. 19
Flow cytometry has been used extensively in various stages of drug development and CAR‐T cell therapy. 19 It is used for quantification of antigen‐bearing cells in patients' samples, assessment of cell viability and functionality, evaluation of CAR expression on cell surface, monitoring therapeutic efficacy, and cell persistence in patients and others. However, there are some challenges associated with flow cytometry for CAR‐T cell characterization and identification as limited availability of certain cell surface CAR molecule antigens often requires the generation of in‐house reagents. 20 , 21
QPCR is the other method that is used commonly to quantify CAR‐T cell levels by utilizing transgene specific primers. In qPCR assays, concentrations are inferred from the level of transgene DNA and CAR‐T cells levels are typically reported as transgene copy per microgram of genomic DNA or in vector copies per ml of blood sample. QPCR allows for the quantification of CAR‐T cells in different fluids and tissues, for example, peripheral blood, bone marrow, and additional tissues if necessary and is generally highly sensitive. 15 , 22
SAFETY CONCERNS FOR CAR‐T CELL THERAPIES
Immuno‐oncology therapies utilize the body's immune system to destroy malignant tissues. Because these therapies are becoming more effective, widespread, and intriguing, optimal management of their toxicities and side effects is crucial. The two most common concerning toxicities for CAR‐T cell therapies are central nervous system toxicities (neurotoxicities) and cytokine release syndrome (CRS). Neurotoxicity symptoms can include confusion, hallucination, delirium, tremor, encephalopathy, cerebral edema, seizure, and others. CRS is caused by increased levels of circulating cytokines that are released from activated and proliferated CAR‐T cells, which disrupt the balance between pro‐inflammatory and anti‐inflammatory cytokines. Both frequently accompany therapeutic effects, with CRS observed in 42% to 94% of patients 23 , 24 and neurotoxicity observed in 18% to 87% of patients, 23 , 25 and can offset the efficacy of the treatment and restrict its widespread application in the clinical setting. 26 , 27 Although some degree of cytokine secretion represents T cell activation and efficacy of treatment, severe CRS, and neurotoxicity after infusion of CAR‐T cells can be lethal and management of the side effects is still challenging for physicians. Understanding the pathophysiology of these toxicities enables researchers to better control AEs and identify predictive biomarkers that can help forecasting the incidence of severe CRS and/or neurotoxicity in patients. 28
CRS is the primary concerning AE associated with CAR‐T cells that largely appears within the first days following CAR‐T cell administration. Upon tumor cell recognition and CAR binding, pro‐inflammatory cytokines, such as TNF‐alpha, IFN‐gamma, and IL‐6, are released from T cells. The excessive concentrations of cytokines result in various clinical symptoms ranging from mild, such as high fever and hypotension, to severe and life‐threatening side effects, such as organ insufficiency which require intensive care. Among life‐threatening complications of CRS, cardiac dysfunction with rapid onset is of particular concern but is often reversible. The presentation of symptoms in CRS varies depending on the type of CAR‐T cell product.
Because CRS occurs because of increased inflammatory cytokines level, excessive circulating cytokines have been suggested to be used as biomarkers to assess the severity and incidence of this syndrome. 29 However, the application of this approach has limitations as frequent cytokine measurements are challenging from a logistical point of view and patients with cancer often have elevated cytokine levels at baseline. 30 Additionally, the degree of cytokine elevation may not be associated with the severity of symptoms, as some patients show clinical symptoms without significant increases in cytokine levels. 31
The second most concerning AE after CAR‐T cell administration is neurotoxicity. Neurotoxic effects were observed both during CRS and days to weeks after resolution of CRS (delayed neurotoxicity). 32 , 33 It is assumed to be caused by high levels of pro‐inflammatory cytokines in the brain which were detectable in the cerebrospinal fluid of some patients. Engineered T cells can pass through the blood–brain barrier (BBB) and stimulate the production of cytokines in the brain. 34
A clinical trial in 133 patients with chronic lymphocytic leukemia (CLL), Non‐Hodgkin's lymphoma (NHL), or acute lymphoblastic leukemia (ALL) who received a CD19 CAR‐T cell therapy following lymphodepletion showed that 40% of patients had one or more neurologic AEs. Five percent of patients developed greater than or equal to grade 4 neurotoxicity. Almost all patients (91%) with any neurologic AEs also had CRS. CRS was observed in 70% of patients and typically occurred before the onset of neurotoxicity. Additionally, the severity of neurotoxicity correlated with severity and early onset of CRS. 35 , 36
In the phase I trial (ZUMA‐1) of axicabtagene ciloleucel in patients with refractory large B‐cell lymphoma, all patients showed at least one adverse reaction and 52% of patients experienced serious adverse reactions. CRS occurrence was almost universal and was observed in 94% of patients with grade 3 or higher CRS observed in 13% of patients. Neurotoxicity, including grade 3 or higher, were reported in 87% and 28% of patients, respectively. 23 In 25% of patients, CRS symptoms lasted more than 2 weeks, however, most of the symptoms resolved with supportive care and treatment. 37
In the phase I‐IIa clinical trial of tisagenlecleucel in children and young adults with relapsed/refractory (R/R) B‐ALL (ELIANA study), 95% of the patients experienced AEs. Seventy‐three percent of these patients had grade 3 or 4 AEs. CRS, including severe or life‐threatening reactions, occurred in 79% of patients. 32 Neurological AEs occurred in 72% of the patients, including 13% with grade 3 or higher toxicities. Most of these events happened during the CRS or after its resolution.
In a phase II clinical trial (KarMMa) of idecabtagene vicleucel in patients with R/R MM (RRMM), 18% of patients developed neurotoxicity, 3% of whom had grade 3. No grade 4 or 5 neurotoxicity was observed, and treatment‐associated neurotoxicity resolved in 92% of patients. Moreover, 84% of patients developed CRS mostly with lower grades, whereas 5% of the patients had grade 3 or higher events. The incidence of both AEs increased with an increasing number of infused CAR‐T cells. 25
Lisocabtagene maraleucel is an autologous CD19‐directed CAR‐T cell therapy that is indicated for the treatment of patients with R/R large B‐cell lymphoma. In the pivotal TRANSCEND NHL 001 clinical trial, in which patients received a flat dose of CAR‐positive viable T cells, CRS occurred in 42% of patients, out of which 2% experienced grade 3 or higher CRS. However, CRS resolved in 98% of the patients within a median duration of 5 days. Fatal or life‐threatening neurological toxicities also occurred following treatment with lisocabtagene maraleucel in 30% of patients, including grade 3 or higher AEs in 10% of them. 24
CRS and neurological toxicities have also been reported following administration of brexucabtagene autoleucel, another autologous CD19‐directed CAR‐T cell therapy. These genetically modified T cells have been approved for the treatment of adult patients with R/R mantle cell lymphoma (MCL) and ALL. Following the treatment, CRS occurred in 91% and 89% of patients, including grade 3 or higher CRS in 15% and 24% in patients with MCL and ALL, respectively. Similarly, neurological events occurred in 63% of patients with MCL and 60% of patients with ALL. Moreover, grade 3 or higher neurological toxicities were reported in 31% and 25% of patients with MCL and ALL, respectively. 38 , 39 , 40
Risk factors that contribute to neurotoxicity and CRS include a high dose of infused CAR‐T cells, high tumor burden, intensive lymphodepletion therapy, and other factors that increase CAR‐T cell numbers in vivo. 41 , 42 , 43 , 44 Some patient‐related factors, such as pre‐existing thrombocytopenia or endothelial activation, may further increase the risk of toxicities. 35 Screening for these risk factors is important to put necessary countermeasures into place and to provide supportive care to manage the toxicities.
Other AEs that are triggered by the release of cytokines and excessive immune activation have been reported in the literature as well. Tumor lysis syndrome or macrophage activation syndrome/hemophagocytic lymphohistiocytosis, for example, can occur concomitantly and proper management of the symptoms becomes crucial to improve clinical outcomes. 45
CRS RISK MITIGATION STRATEGIES
Strategies to attenuate CAR‐T cell‐induced AEs are either preventive and aim to decrease the incidence of severe toxicities like reducing the disease burden with chemotherapy before treatment start, or symptomatic with the aim of minimizing severity and incidence of the toxicities after their occurrence. Overall, the objective of risk mitigation approaches is to decrease the incidence and severity of undesirable immune responses while maintaining the therapeutic effect. 46
A common strategy in mitigating CRS is targeting soluble or membrane‐bound IL‐6 receptors through administration of tocilizumab (anti IL‐6 receptor mAb) which prevents binding of IL‐6 to its receptors. 27 Tocilizumab has been identified as a successful treatment for severe and life‐threatening CRS and does not interfere with the expansion of CAR‐T cells. 47 , 48 Generally, tocilizumab is tolerated well by the patients but, in some cases, it may cause neutropenia and elevate liver enzymes, such as transaminases. Due to recent shortages of tocilizumab, driven by the FDA emergency use authorization granted in June 2021 for tocilizumab use for coronavirus disease 2019 (COVID‐19), coupled with inclusion in the World Health Organization (WHO) guidelines, which saw US demand spiking by greater than 400% in August 2021 compared to pre‐COVID levels, 49 other IL‐6 targeting agents, such as siltuximab or even IL‐1R antagonists like anakinra, are currently being explored as alternatives to tocilizumab.
The downside of tocilizumab is its ineffectiveness of preventing neurotoxicity because it cannot pass the BBB. Therefore, corticosteroids, which suppress the inflammatory system, are typically used to manage cerebral CRS instead. They are also considered the second‐line treatment option in the management of CRS. Although corticosteroids may help to manage CAR‐T cell‐related toxicities, they can also block T cell activation and hamper clinical efficacy of immunotherapy. Therefore, prophylactic use of corticosteroids is usually not recommended and systemic corticosteroid treatment should be limited only to toxicities that are unresponsive or refractory to IL‐6 targeted therapy. 50 , 51
In addition to high‐dose corticosteroids and cytokine inhibitors, premedication with acetaminophen and diphenhydramine, or another H1‐antihistamine, 30 to 60 min prior to treatment with CAR‐T cell therapies is usually recommended to minimize the risk of CRS and infusion‐related reactions. Other factors that can reduce the risk of CRS are a lower number of infused CAR‐T cells, switchable CAR‐T cells, or administration of bispecific T cell engagers as a treatment alternative. 52 , 53
PHARMACOKINETICS/PHARMACODYNAMICS OF CAR‐T CELLS
Dose‐exposure relationships
Generally, there is a lack of an apparent relationship and lack of a consistent trend between the number of infused CAR‐T cells and their subsequent exposure. For instance, no apparent relationship has been found between the dose of tisagenlecleucel (CD19‐directed CAR‐T immunotherapy) and exposure across wide ranges of adjusted doses studied. 15 , 22 Furthermore, in a phase I clinical trial of idecabtagene vicleucel (BCMA‐directed CAR‐T therapy), expansion of CAR‐T cells was detected, and peak blood concentration of all studied dose levels overlapped. 54 However, in the phase II clinical trial of idecabtagene vicleucel, a trend of higher exposures with increasing dose was observed, although interindividual variability was relatively high. 25 This observation has not been observed with anti‐CD19 CAR‐T cell therapies yet.
Potential reasons for the lack of apparent dose‐exposure relationships are not fully understood at this time, but numerous intrinsic and extrinsic factors have been shown to impact the dose‐exposure‐response relationships for both safety and efficacy. Compared to more traditional therapies, which typically show a monotonic dose‐exposure relationship, CAR‐T cells are “living biologic therapies” that expand during the manufacturing process and after administration to the patient. This unique characteristic allows for the potential of a single dose curative paradigm but also results in dose‐exposure‐response relationships for both efficacy and safety that are impacted by numerous factors. Those factors include variability in the starting material itself (for autologous CAR‐T cell therapies the patient's own T‐cells are infused that can vary in T‐cell fitness from patient to patient), differences in conditioning factors when preparing the patient for CAR‐T cell infusion (e.g., choice of the lymphodepletion conditioning regimen), and, last, individual patient‐factors that come into play after CAR‐T cells are infused. Among individual patient characteristics, tumor burden is known to have a more significant impact on the dose‐exposure‐response relationship, with higher tumor burden subjects showing higher expansion and persistence, as well as experiencing higher grade CRS. 15 , 22 , 55 Among conditioning factors preparing the patient for CAR‐T cell infusion, the choice of the lymphodepletion regimen has been shown to have an impact on the dose‐exposure‐response relationship. For example, the addition of fludarabine to the lymphodepletion regimen has shown to improve both cell expansion and persistence as well as the efficacy by increasing disease‐free survival. 55 Additionally, certain manufacturing aspects have been found to impact the dose‐exposure relationship. For example, in a study with a limited number of patients, an apparent dose‐exposure relationship was observed when the ratio of CD4:CD8 was well‐controlled during the manufacturing process, although lisocabtagene maraleucel (liso‐cel), a CAR T whose CD4:CD8 ratio is also well‐controlled, did not show an apparent dose‐exposure relationship in a relatively large study with 344 patients. 24
Exposure‐response relationships for efficacy and safety
For the initial response of CAR‐T cell therapy, presence and expansion of functioning CAR‐engineered T cells is necessary, highlighting the importance of cellular kinetics and functionality of CAR transduced T cells for their efficacy. It has also been shown that maintenance of response and evolution of response at later times are associated with functional CAR‐T cell persistence, however, exceptions have been reported in NHL. 17 Poor or insufficient expansion of CAR‐T cells and short persistence in vivo limits the clinical application of these therapies and may create minimal residual malignancy that results in an apparent complete response (CR) or a partial response. 23 , 56 In addition, some patient‐related factors, such as tumor cell location, environment, and demographics, as well as functional capacity of individual's T cells might not be captured by cellular kinetic assays but can affect exposure‐response relationships. 57 Further research is warranted to enable the integration of these factors into the assessments which results in prediction of treatment response. 15
Based on the clinical trials of tisagenlecleucel (ELIANA and ENSIGN), responding patients showed almost twofold higher expansion in peripheral blood compared to nonresponding individuals and persistence could be measured over 2 years following CAR‐T cell therapy. Nonresponding patients demonstrated delayed expansion and shorter persistence. 22 Likewise, higher cell expansion of idecabtagene vicleucel was associated with response and persistence up to 1 year. 54
Patients with higher exposure and longer persistence show greater response and toxicity. In a phase I clinical trial of CD19‐specific CAR‐T cell therapy in patients with relapsed B‐cell ALL, the peak CAR‐T cell expansion values in vivo have been determined to be the best predictors of short‐term AEs and therapeutic responses. However, in the assessment of long‐term outcomes, disease burden at baseline has been found to be the best predictor of survival and remission period. Patients with low tumor burden showed longer survival compared to high disease burden patients because inflammatory cytokines associated with high tumor burden may affect CAR‐T cell expansion and efficacy. 33 , 58 It has also been reported that the peak blood concentration values are significantly higher in patients with grades 3 or 4 neurotoxicity compared to patients with lower grades. However, CAR‐T cell expansion values were not significantly different among patients with various CRS grades.
Similarly, no relationship was observed between the dose of viable tisagenlecleucel CAR‐T cells and probability of severe CRS and neurologic events in children and young adults with R/R B‐ALL. 22 However, in responding patients, higher expansion was associated with occurrence of higher grades CRS. In the case of idecabtagene vicleucel, higher expansion values were also associated with higher grades of CRS. 54 Although a phase I clinical trial of axicabtagene ciloleucel CAR‐T cell therapy in large B‐cell lymphoma (ZUMA‐1) showed that expansion was associated with response and peak expansion was correlated with high grade neurological events, but not with high grade CRS. 23 , 58
In the clinical studies of tisagenlecleucel for R/R B‐ALL, pediatric patients with higher tumor burden showed increased expansion which was associated with higher CRS grades and neurological events. Treatment of CRS with low‐dose corticosteroids or tocilizumab did not inhibit in vivo persistence and expansion of CAR‐T cells. 15 Patients with high tumor burden treated with lisocabtagene maraleucel also showed higher incidence of CRS and neurological events. 24
Impact of pathophysiological‐demographic factors on CAR‐T cell exposure
Because cellular kinetics correlate with therapeutic effect and patient response, determining the influential factors on cellular kinetics is important. Interindividual and intertrial variabilities as well as product‐specific factors in CAR‐T cell therapy can affect the therapeutic efficacy. Although prespecified clinical covariates could not clearly predict the efficacy of CAR‐T cells, model‐based analysis of these covariates plays an important role in identifying biological determinants of CAR‐T cells kinetics. 59
Clinical trial data from axicabtagene ciloleucel (ZUMA‐1) showed that key covariates, such as disease stage, age, and administration of tocilizumab or glucocorticoids, did not affect the response rate. Moreover, biological covariates and product characteristics, such as expression of CD19 and T‐cell phenotypes, did not influence the response rate either. 23
Locke et al. applied logistic regression analysis and investigated the impact of covariates on response rates in patients with DLBC receiving axicabtagene ciloleucel. They found that despite similar peak CAR‐T cell concentration levels, patients with higher tumor burden showed lower response rates compared to patients with lower disease burden. 58 However, in a model‐based analysis by Liu et al. that included CAR‐T cell therapy directed against MM and CLL, no correlation between patient response and tumor burden was observed. 59
Stein et al. have investigated the effect of tocilizumab, corticosteroids, and several other covariates on the cellular expansion of tisagenlecleucel CAR‐T cells using data from phase II clinical trials. 60 According to the developed population PK model, none of the studied covariates showed a statistically significant effect on peak plasma concentrations of tisagenlecleucel. Furthermore, they demonstrated that administration of corticosteroids or tocilizumab did not affect the cellular expansion rate. However, Mueller et al. observed that baseline tumor burden and severity of CRS were correlated with tisagenlecleucel cell expansion. 15 , 60 They also denoted that pre‐existing or post‐treatment immunogenicity did not have any clinically meaningful effect on the expansion or persistence of tisagenlecleucel. 61
In the phase Ib/II clinical trial of ciltacabtagene autoleucel (CARTITUDE‐1), a BCMA CAR‐T cell therapy, neurotoxicity was reported in 21% of patients with R/R MM following a single low‐dose infusion. The occurrence was associated with high tumor burden, high CAR‐T cell expansion, and CAR‐T cell persistence. 62
Ogasawara et al. assessed the relationship between in vivo cellular expansion and efficacy or safety after adjusting for key baseline characteristics of lisocabtagene maraleucel in the TRANSCEND trial. 63 As reported earlier, higher cellular expansion was associated with a higher overall response and CR rate as well as higher occurrence of CRS and neurological events in patients with R/R large B cell lymphoma. After accounting for age and tumor burden, the two factors that were confounding the relationship between efficacy and cell expansion, the association became even stronger. Moreover, cellular expansion was lower after repeated dosing. 63 Ogasawara et al. also evaluated the effect of other covariates on cellular expansion, including baseline intrinsic factors like disease characteristics and drug co‐administration. They showed that the covariate associations were smaller than the residual between‐subject variability in the population and concluded that tested covariates did not have a meaningful impact on lisocabtagene maraleucel kinetics. 64
Following the phase II clinical trial of brexucabtagene autoleucel (ZUMA‐2), in which a single dose of CD19‐directed CAR positive cells was administered to patients with R/R MCL, Wang et al. evaluated the relationship between cellular expansion and objective response and incidence of AEs. Higher expansion was associated with deeper response and higher peak levels among responding patients. Moreover, there was no association between expansion and baseline tumor burden. Additionally, for patients with CRS and neurological events, expansion was greater in the patients with grade 3 or higher AEs. 65
Exposure of CAR‐T cells may also be affected by the conditioning therapy or lymphodepletion that patients receive. It has been shown that the addition of fludarabine to cyclophosphamide might affect the clinical outcomes by altering CAR‐T cell expansion and persistence in patients with NHL. 55 , 66 However, other studies show that the expansion and persistence of CAR‐T cells was not affected by the type of lymphodepletion regimen. 22
OTHER IMPORTANT FACTORS
Immunogenicity
CAR‐T cell therapy can induce both cellular and humoral immune responses especially when they contain murine components or non‐human monoclonal antibodies in their structure which might result in treatment failure or reduced efficacy. Therefore, one of the important elements in CAR‐T cell therapy is to characterize the immunogenicity. 67 , 68 In the clinical trial of idecabtagene vicleucel in patients with MM (KarMMa), antidrug antibody (ADAs) was detected in 44% of subjects 6 months after infusion and increased up to 65% at month 12. However, the presence of ADAs did not significantly affect the efficacy, safety, and PKs of idecabtagene vicleucel. 25
After infusion of tisagenlecleucel in patients with R/R B‐cell ALL, immunogenicity was detected in 36.7% of patients that had increased levels of antimurine CAR19 (mCAR19) antibodies in their serum. However, the study groups showed similar exposure, duration of remission and clinical response, and treatment‐induced anti‐mCAR19 antibodies did not have any effect on persistence, expansion, efficacy, and safety of tisagenlecleucel. 15 , 22
Likewise, in the clinical trial of axicabtagene ciloleucel in patients with lymphoma, there was no evidence of kinetic, safety, and efficacy alteration in any of the subjects with positive anti‐product antibody. Like tisagenlecleucel and axicabtagene ciloleucel, brexucabtagene autoleucel has the same mouse‐derived single‐chain variable fragment in its structure. However, infusion of the CD19‐directed CAR‐T cells did not result in worse clinical responses despite the presence of pre‐existing antibodies or treatment‐induced increases in ADA. 69
In a comprehensive review paper summarizing immunogenicity of CAR‐T cells, Wagner et al. described the current clinical evidence of immune responses against CAR‐T cells and suggested ways to decrease the risk of anti‐CAR antibody production and immunogenicity. Construction of CAR‐T cells with humanized components, mutation of the epitopes within the CAR spacer to prevent innate immune cell activation and intensified lymphodepletion before CAR‐T cell therapy highlight approaches that mitigate inherent CAR immunogenicity. 69 , 70 , 71
FIRST‐IN‐HUMAN DOSE STUDY DESIGNS FOR CAR‐T CELLS
First‐in‐human dose selection (Typical starting doses of CAR‐T cells in liquid and solid tumors)
One of the key steps in developing new biopharmaceutical products is the determination of a safe starting dose for clinical trials. Ideally, the starting dose in humans should be low enough to avoid major toxicities but should be high enough to avoid prolonged dose escalation that can expose too many patients to subtherapeutic dose levels and lead to unnecessarily lengthy clinical studies. Data from preclinical toxicology studies helps to establish a safe initial dose for first‐in‐human (FIH) trials and to determine potential toxic or AEs and their severity. It has been recommended to perform preclinical studies in different species and select the initial dose according to the most sensitive group. 72 The European Medicine Agency (EMA) and the FDA have issued various guidance documents to help to determine the FIH dose. The conventional way for determining a safe starting dose for cytotoxic compounds in humans is to use 10% of LD10 (lethal dose to one tenth of studied mice). However, if significant toxicities are observed in toxicological studies in non‐rodent species, the initial dose is recommended to be one sixth to one third of the lowest toxic dose in the more sensitive species. 72 However, protein or cell‐based drugs have shifted the paradigm in drug development because they have more unpredictable safety and efficacy profiles in vivo compared to small‐molecule drugs and it may be difficult to establish an initial starting dose based on the considerations used for small‐molecule drugs. Therefore, available animal or in vitro data and previous clinical experience with the CAR‐T cell product or related products should be leveraged to justify the clinical starting dose. 73
Historically, FIH dose selection strategies were based on drug toxicities, but the focus of immune‐oncology drug development has shifted to approaches that identify minimum effective doses rather than maximum tolerated doses. 74 Immuno‐oncology agents are administered within an extended time frame and might result in delayed AEs that happen outside the dose limiting toxicity (DLT) window of the study, thus determination of proper doses based only on the cycle 1 data performed frequently in the past are inadequate. Several published reports have used the strategy of extending the DLT evaluation period to resolve this limitation. 75 , 76 , 77
In comparison to cytotoxic chemotherapy agents, immuno‐oncology treatments do not directly affect tumor cells and their cytotoxic effects are mediated indirectly by immune cells, such as natural killer cells or T‐cells. Thus, higher concentrations of these therapies are not necessarily associated with better efficacy or more toxicity.
In the case of CAR‐T cell therapy, the total number of infused CAR‐engineered viable T cells has been determined as the dose, which can also be adjusted based on the patients' body weight or body surface area. Moreover, unlike the traditional therapeutic agents with homogeneous characteristics, CAR‐T cells are heterogeneous, and it is likely that only a subgroup of cells induce therapeutic or toxic effects. In addition, the cellular kinetics of T cells and their maximum expansion, which occurs within roughly 1 week after infusion, makes it difficult to apply some risk mitigation approaches, such as administration of a test dose to patients. Therefore, selection of the starting dose should be based on the current knowledge about the target and the related potential toxicities, toxicological parameters, and exposure‐response relationships. 74
OPTIMAL FIH STUDY DESIGNS FOR CAR‐T CELLS
The landscape of drug development in oncology has changed drastically with the introduction of immune‐based cancer therapies, such as monoclonal antibodies and CAR‐T cells, hence traditional methods of clinical trial design have to be modified accordingly. 74 Clinical trial design for cell‐based immunotherapies is challenging and more complex due to their significantly different toxicity and PK profiles compared to conventional anticancer drugs. In addition, there is a need to identify new approaches of response assessment and to identify optimal regimens for combinational therapies. 78 Moreover, variabilities in genetic engineering and manufacturing of cellular products might affect the efficacy and safety of these therapies and therefore dose‐finding trials are not solely driven by toxicity considerations.
FIH trials of oncology agents are generally performed in patients with advanced cancers that are resistant or refractory to current anticancer drugs, as compared to most other therapeutic areas in which FIH studies are conducted in healthy individuals. Therefore, oncology trials should offer a relatively high benefit to risk ratio for patients. The main goal of FIH oncology studies are typically assessments of safety and tolerability, PK and pharmacodynamic (PD) properties, antitumor efficacy, DLTs, and identifying appropriate doses and regimens for phase II clinical trials. It is important to complete these studies as effectively and quickly as possible while avoiding major toxicities and extensive dose escalations. 79
Unlike the obvious importance of phase I clinical trials, the validity of obtained toxicity and recommended phase II dose (RP2D) data is sometimes ambiguous due to the low number of patients enrolled in these trials. 80 However, following the completion of dose‐escalation cohorts in phase I trials, expansion cohorts can be incorporated which enroll additional subjects at the RP2D to increase the quantity and quality of data attained from phase I clinical trials. 81 Depending on disease type or molecular characteristics of the investigational agent, it is now a common practice that phase I immuno‐oncology trials include several dose expansion cohorts and it has been shown that the implementation of expansion cohorts can increase the probability of success in phase II trials. 82
Expedited approvals for oncology agents have resulted in numerous clinical trials investigating immunotherapies. For example, the expedited approval of axicabtagene ciloleucel by the FDA was based on a single‐arm phase I and II clinical trial and the efficacy was evaluated based on the overall response rate (ORR). In addition, brexucabtagene autoleucel was approved in adults with R/R MCL under the accelerated approval pathway. 83 One disadvantage of expedited approval compared to conventional approvals, which are based on phase III clinical studies, is the lack of a control group. Moreover, some of the most meaningful clinical trial end points, such as progression‐free survival and overall survival (OS), can usually not be determined. Therefore, continued approval typically requires verification of clinical benefit in a confirmatory trial.
Locke et al. 84 (axicabtagene ciloleucel) and Bishop et al. 85 (tisagenlecleucel) provide examples of phase III clinical trials (ZUMA‐7 and BELINDA, respectively) that have been conducted with CAR‐T therapies. The results of the ZUMA‐7 study lead to the successful filing of a supplemental Biologics License Application in the second‐line setting in adults with R/R large B‐cell lymphoma recently. 86
Because efforts are ongoing to increase the application of T cell‐based therapies, ideal trial designs that integrate multiple end points are crucial. 87 , 88 The optimum trial designs may eventually result in the selection of an effective therapeutic dose. Apart from clinical trial design, there is also an unmet need to harmonize product generation and efficacy and safety testing all of which could accelerate drug development and benefit patients with cancer.
As mentioned previously, efficacy of CAR‐T cell therapy may not increase following administration of higher doses, therefore traditional dose‐finding approaches that only leverage toxicity data are not appropriate for dose determination. Because efficacy and toxicity of CAR‐T cells can be evaluated simultaneously, Li et al. have proposed a toxicity and efficacy probability interval design for dose finding trials of CAR‐T cells. This approach integrates both efficacy and safety outcomes to determine the optimum dose. This approach is simple, flexible, and transparent because investigators can specify the decision rules before starting the trial. 89
Quantitative approaches to guide the applications of model‐informed drug development to CAR‐T cell therapy
Model‐informed drug development (MIDD) is the application of a wide range of quantitative approaches to inform decision making throughout the drug development process. 90 Recent advances in CAR‐T cell therapy have generated abundant mathematical models 90 , 91 , 92 designed to address different aspects of CAR‐T cell therapy including T‐cell activation, 93 , 94 T cell–tumor cell interactions, 95 and T‐cell/cytokine interactions, 93 , 96 among others. Despite the growing number of available CAR‐T cell therapy modeling approaches, their application in drug development is not yet mature. This is mainly due to the fact that CAR‐T cell therapies exhibit complex mechanisms of action and atypical exposure‐response properties that are not comparable to the traditional PK/PD of small molecule/biologic therapies leading to the term “cellular kinetics” instead of PKs.
So far, the use of noncompartmental analyses is the straightforward approach to quantify dose‐exposure‐response relationships 68 , 97 with the caveat that these approaches are lacking mechanistic interactions between tumor and CAR‐T cell. Stein et al. 60 developed a population cellular kinetic model for CAR‐T cell applied to tisagenlecleucel. Their model has been recently modified by Can Liu et al. 59 to retrospectively characterize CAR‐T cell kinetics in more diverse patients. Chaudhury et al. 98 provided an excellent review of cellular kinetic‐PD models of CAR‐T cells. Most of the cellular kinetic models developed by Stein et al., 60 their derivatives, 59 and those reviewed by Chaudhury et al. 98 are empirical and do not consider the interactions between CAR‐T cells and normal T cells, nor pay much attention to the complex relationships among target engagement, tumor burden, and the dynamics of native immune systems.
Hardiansyah et al. 97 developed a quantitative system pharmacological model of CAR‐T cell kinetics to quantify the complex relationships between CAR‐T cell doses, disease burden, and proinflammatory cytokines. The expansion of CAR‐T cells and elimination of B‐cells was found to be more correlated with disease burden than the administered CAR‐T cell doses. Although the model developed by Hardiansyah et al. was calibrated with only a few subjects, it demonstrated a clear value of using a mechanism‐based approach to capture CAR‐T cell kinetics (i.e., time‐dependent expansion, rapid contraction, and prolonged persistence).
Singh et al. 99 developed a multiscale, mechanistic PK/PD model to quantitatively describe the CAR‐T cell activities in in vitro and in vivo preclinical models using literature data. Their model has the potential to be used to evaluate both the in vitro in vivo correlation and efficacy/safety. Kimmel et al. 100 quantified the roles of T‐cell competition and stochastic extinction events in CAR‐T cell therapy. The outcome of their modeling suggested that variability among patient and disease factors are a major factor impacting the timing and probability of cures. 95
Barros et al. 90 developed a population mathematical model to describe tumor response to CAR‐T cell immunotherapy in immunodeficient mouse models, encompassing interactions between a non‐solid tumor and CAR‐T cells (effector and long‐term memory). Their model accounted for several phenomena, such as tumor‐induced immunosuppression, memory pool formation, and conversion of memory into effector CAR‐T cells in the presence of new tumor cells. Their analysis suggested that the effectiveness of therapy mostly depends on some specific parameters, such as the differentiation of effector to memory CAR‐T cells, CAR‐T cytotoxic capacity, tumor growth rate, and tumor‐induced immunosuppression. However, it is worth noting that their model is based on an immunodeficient mouse model and hence does not consider toxicity effect of CAR‐T cell immunotherapy (e.g., CRS). Table 3 demonstrates the summary of applied models for CAR‐T cell therapy.
TABLE 3.
Summary of applied models for CAR‐T cell therapy
| Articles | Type of models | Model application |
|---|---|---|
| Stein et al. 2019 59 | Population model for cellular kinetics | Assess the impact of tocilizumab therapy on the kinetics of in vivo tisagenlecleucel expansion |
| Liu et al. 2021 58 | Population model for cellular kinetics using seven clinical trials | Analyze systematic of the factors influencing CAR‐T therapy in humans |
| Singh et al. 2020 99 | Multiscale, mechanistic cellular kinetic‐pharmacodynamic model using a preclinical literature data | Analyze the CAR‐T cell activities in in vitro and in vivo preclinical models |
| Mueller et al. 2018 22 | Noncompartmental analysis quantifying dose‐exposure‐response relationship | Characterize the clinical pharmacology of tisagenlecleucel in B‐cell acute lymphoblastic leukemia |
| Awasthi et al. 2020 68 | Noncompartmental analysis quantifying dose‐exposure‐response relationship | Characterize the Tisagenlecleucel cellular kinetics, dose, and immunogenicity |
| Hardiansyah et al. 2019 97 | Quantitative system pharmacology model for CAR‐T cell | To quantify the relationships among CAR‐T doses, disease burden, and proinflammatory cytokines in human and to gain relevant insights into the determinant of clinical toxicity/efficacy in development of CAR‐T therapy |
| Kimmel et al. 2021 100 | Mathematical model quantifying the roles of T‐cell competition and stochastic extinction events in CAR‐T cell therapy | Assess T‐cell and CAR‐T cell dynamic and the resulting tumor cell dynamics |
| Barros et al. 2021 90 | Mathematical model characterizing CAR‐T immunotherapy in preclinical models | To describe tumor response to CAR‐T cell immunotherapy in immunodeficient mouse models |
| Yiu et al. 2021 94 | Mathematical model characterizing the dynamics of a cytokine storm | To study the effects of inhibiting individual cytokines, to predict the effects of different TGN1412 infusion rates, and to reveal effects of empirical uncertainty |
| Sahoo et al. 2020 91 | Modeling of cellular kinetics ‐pharmacodynamic | To quantitatively study the factors that contribute to the efficacy of CAR T‐cell therapy in solid tumors |
Role of clinical pharmacology and translational sciences to enable FIH dose selection and identifying optimal doses for clinical studies and Project Optimus
Given the challenges of CAR‐T therapies in regards to a general lack of dose/exposure‐response relationships, the lack of easy translatability of animal models to humans, the complex mechanism of action, including the interactions between tumor cells and immune cells, more unpredictable safety and efficacy profiles compared to more traditional therapies, the increasing relevance of combination therapies and the associated challenges with identifying optimal regimens, it is fairly obvious to assume that clinical pharmacology and translational sciences approaches will become more important and widely used to address some of these challenges. Some of the issues mentioned above are not unique to CAR‐T therapies but rather apply to the whole field of oncology drug development. The FDA has recognized this and recently started a new initiative named “Project Optimus,” 101 which aims to reform the dose optimization and dose selection approaches in oncology drug development through evaluations of a range of doses in trials early in development to select dose or doses that maximizes not only the efficacy of a drug but the safety and tolerability as well. Analysis of these data and pivotal dose selection will rely heavily on quantitative approaches.
ALLOGENIC CAR‐T CELL THERAPIES – THE FUTURE OF CAR‐T CELL THERAPIES
ALLOGENE phase I safety and efficacy data
Autologous T cells have been commonly used to manufacture CAR products. However, they have several limitations, such as lengthy production time, high production cost, manufacturing shortfalls, and dependence on functional fitness of patient T cells. 102 To overcome these limitations, allogenic CAR‐T cell therapy was developed. Different from autologous CAR‐T cell therapy, T cells for allogenic CAR‐T therapy are harvested from healthy donors. These T cells are then engineered to express CARs to recognize certain cell‐surface proteins that are expressed in hematologic or solid tumors. By leveraging gene editing technology, the risks of GVHD and allogeneic rejection can be greatly reduced. 103
CD19 is an attractive target for CAR‐T cell therapies due to its ubiquitous B‐cell surface expression. This antigen is only expressed on B cells, thereby preventing significant non‐hematological toxicity while demonstrating manageable on‐target toxicity. 104 Allogene Therapeutics' ALLO‐501/501A are anti‐CD19 allogenic CAR‐T cell products indicated for the treatment of R/R NHL. Both products have disrupted TCRα gene to reduce GVHD risk, and edited CD52 gene to permit use of ALLO‐647 (a humanized anti‐CD52 mAb) for host T cell depletion. 105 , 106 Whereas ALLO‐501 contains a rituximab recognition domain, ALLO‐501A eliminates this domain to allow for the use in a broader patient population, including those patients with NHL with recent rituximab exposure. The phase I data from ALLO‐501 ALPHA trial in NHL demonstrated an ORR of 75% and CR rate of 50% in CAR‐T cell naive patients. A 6‐month CR rate of 36% in CAR‐T cell naive patients with large B‐cell lymphoma was also observed following a single ALLO‐501 infusion, which was similar to the pivotal trials of autologous CAR‐T cell therapies (29%–40%). The treatment was also well‐tolerated with no DLTs or GVHD and manageable CRS. Meanwhile, in the ALLO‐501A ALPHA2 trial in NHL, ALLO‐501A demonstrated comparable efficacy to ALLO‐501. 107 However, following a recent report on the presence of a chromosomal abnormality in a patient from the ALPHA2 trial, the FDA issued a clinical hold on all of the company's AlloCAR T clinical trials. Further investigation on this single patient showed that the chromosomal abnormality was unrelated to TALEN gene editing or Allogene's manufacturing process and had no clinical significance. The clinical hold was removed thereafter. 108 , 109
B‐cell maturation antigen (BCMA) is a transmembrane glycoprotein belonging to the tumor necrosis factor (TNF) receptor superfamily that plays an important role in regulating B‐cell proliferation and survival. 110 It is universally and increasingly expressed on malignant plasma cells of patients with MM—thus became one of the most promising target antigens in MM. 111 ALLO‐715 is the first anti‐BCMA allogenic CAR‐T cell product indicated for the treatment of RRMM. In the ongoing phase I UNIVERSAL study in adults with RRMM who have received greater than or equal to three prior lines of therapy, patients received one of two lymphodepletion (LD) regimens followed by ALLO‐715 at one of four dose levels (DL1 = 40 × 10 6 cells, DL2 = 160 × 106 cells, DL3 = 320 × 106 cells, DL4 = 480 × 106 cells) in a 3 + 3 dose escalation design. Both LD regimens include the use of ALLO‐647, an anti‐CD52 monoclonal antibody, for selective and transitory host lymphodepletion. As of the October 14, 2021, data cutoff, higher cellular doses have demonstrated increased response rates with greater cellular expansion. Efficacy data of the DL3 cohort have been extensively discussed as this dose level was selected for dose expansion. In this cohort, a 71% ORR was observed with 46% patients achieving a stringent CR, a CR, or a very good partial response. Among these 46% patients, 92% were minimal residual disease negative. The median duration of response is 8.3 months, with nine patients maintaining ongoing response at the time of the data cutoff. No DLTs or GVHD had been reported as of the data cutoff date. Manageable CRS was reported in 53% patients with grade 1 and grade 2 episodes. 54% infection, 70% grade 3+ neutropenia, and 14% low‐grade reversive neurotoxicity were also observed as of the data cutoff. Overall, these data suggested that ALLO‐715 and ALLO‐647 have a tolerable safety profile and higher cellular doses (DL3/DL4) yield clinical meaningful efficacy. 112 , 113 , 114
TCR‐T CELL THERAPY
Both CAR and TCR‐therapies genetically engineer patient's own T cells to kill cancer cells. However, the mechanism of actions of these two therapies are different. CAR‐T cells utilize antibody fragment to bind specific antigens on the surface of cancer cells, whereas TCRs use heterodimers composed of two different transmembrane polypeptide chains (α and β) to recognize polypeptide fragments presented by MHC molecules. 5 Compared with CAR‐T cells, which can target only cell surface antigens, TCR‐T cells can recognize any antigen fragment that can be displayed by MHC molecules, and it has shown greater potential against solid tumors than CAR‐T cells. 5 , 115
TCR‐T cell therapy clinical data
In January 2022, the FDA approved Immunocore's TCR therapy (KIMMTRAK) for the treatment of unresectable or metastatic uveal melanoma (mUM). It was the first regulatory approval for a TCR therapeutic, and bispecific T cell engager for solid tumors. 116 The approval was based on their phase III IMCgp100‐202 randomized pivotal trial evaluating OS of KIMMTRAK compared to the control group (pembrolizumab, ipilimumab, or dacarbazine) in patients with previously untreated mUM, and data showed that KIMMTRAK demonstrated statistically and clinically meaningful OS benefit, with a hazard ratio of 0.51 in favor of treatment group and a median OS of 21.7 months. 117 The result was not only a milestone for mUM, but also cancer immunotherapies in general, as it was the first pivotal study to show clear superiority over PD1/PD‐L1 inhibitors in solid tumors.
In addition to Immunocore, some biotechnology companies have also initiated the clinical development of their TCR‐T cell products. For example, Adaptimmune's specific peptide enhanced affinity receptor (SPEAR) T‐cell therapy platform engineered patient's own T cells to enhance the binding affinity of natural TCR cells to recognize and bind to specific cancer peptides. The company has ongoing clinical trials for three SPEAR T‐cells including Afami‐cel (formerly ADP‐A2M4), ADP‐A2M4CD8, and ADP‐A2AFP in multiple solid tumor indications. The latest clinical data of Afami‐cel showed that patients with synovial sarcoma, whose tumors were HLA‐A*02 serotype and expressed the MAGEA4 peptide, had durable responses. The ORR per independent review was 34% (36% in patients with synovial sarcoma, and 25% for patients with myxoid round‐cell liposarcoma) and the disease control rate was 85%. The therapy was well‐tolerated and has a favorable benefit–risk profile in general with only one patient (2%) experiencing grade 3 CRS and 66% patients experiencing grade 1 or grade 2 CRS. 107 , 118
Immatics presented their clinical data on dose escalation from their TCR‐T programs (ACTengine) IMA201, IMA202, and IMA203. Patients who were heavily pretreated but failed all previous therapies, entered the study with recurrent and/or refractory solid tumors, including non‐small cell lung cancer, head and neck cancer, melanoma, synovial sarcoma, and others. At the first and second dose level (below one billion transduced cells), nine out of 10 evaluable subjects showed antitumor activity with disease control and tumor shrinkage was observed in eight patients, including one partial response. All product candidates demonstrated a manageable safety and tolerability profile with transient and manageable treatment‐emergent adverse events. Although one patient receiving IMA203 at the second dose level experienced a DLT, it was transient and fully resolved within 48 h. 119
Besides solid tumors, TCR‐T therapy has also been utilized in the liquid tumor space. Medigene reported their preliminary efficacy and immune monitoring data from the phase I part of the phase I/II clinical trial for MDG1011, a TCR‐T therapy directed against the tumor antigen PReferentially expressed Antigen in MElanoma (PRAME) indicated for the treatment of advanced‐stage blood cancers. Nine patients received MDG1011 as a single intravenous infusion at dose levels of 0.5, 1 or 5 million TCR‐transduced T cells per kg body weight. One patient with AML treated at the lowest dose experienced complete remission at week 4 after treatment; one patient with multilineage myelodysplastic syndrome and myeloproliferative neoplasm treated at the highest dose, remained without apparent progression to secondary AML 3 and 6 months after MDG1011 administration; two patients with AML, treated at the intermediate and highest dose, respectively, experienced transient grade 1 or 2 CRS within 3 days of drug administration, indicating in vivo biological activity. The drug appears safe and well‐tolerated with no immune effector cell‐associated neurotoxicity syndrome or DLTs. 120
CONCLUSIONS
The clinical success of immunotherapy, which is due to the understanding of cancer immunobiology and effective application of this knowledge to eliminate cancerous cells, has changed the field of cancer therapy. Currently, immunotherapy is considered as one of the most important and effective approaches of cancer care. The success of genetically modified CAR‐T cells has created substantial excitement among Biotech companies and researchers. In this regard, the scientific communities have produced tremendous efforts in developing abundant quantitative approaches for CAR‐T cells that can assist the safety and efficacy evaluation of CAR‐T cell products. As discussed above, approaches that enable quantifying the correlation between CAR‐T cell mechanism of action and clinical outcome could be an attractive platform addressing most of the components of MIDD (e.g., increase the probability of regulatory success, guiding personalized doses for specific patient populations, etc.). However, developing such models is currently challenging due to the lack of clinical data. Availability of more clinical data and experiences derived from multiple CAR‐T cell therapies could facilitate development of quantitative approaches.
CONFLICT OF INTEREST
Johannes Kast, Di Zhou, Marc R. Yago, Po‐Wei Chen, Malidi Ahamadi, Sandeep Dutta, and Vijay V. Upreti are employees of and stockholders in Amgen Inc. Saeideh Nozohouri is a previous employee of Amgen Inc.
Kast J, Nozohouri S, Zhou D, et al. Recent advances and clinical pharmacology aspects of Chimeric Antigen Receptor (CAR) T‐cellular therapy development. Clin Transl Sci. 2022;15:2057‐2074. doi: 10.1111/cts.13349
Funding information
No funding was received for this work
REFERENCES
- 1. Cullen SP, Martin SJ. Mechanisms of granule‐dependent killing. Cell Death Differ. 2008;15:251‐262. [DOI] [PubMed] [Google Scholar]
- 2. de Saint Basile G, Menasche G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol. 2010;10:568‐579. [DOI] [PubMed] [Google Scholar]
- 3. Singh AK, McGuirk JP. CAR T cells: continuation in a revolution of immunotherapy. Lancet Oncol. 2020;21:e168‐e178. [DOI] [PubMed] [Google Scholar]
- 4. Tokarew N, Ogonek J, Endres S, von Bergwelt‐Baildon M, Kobold S. Teaching an old dog new tricks: next‐generation CAR T cells. Br J Cancer. 2019;120:26‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhao L, Cao YJ. Engineered T cell therapy for cancer in the clinic. Front Immunol. 2019;10:2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang K, Wei G, Liu D. CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Exp Hematol Oncol. 2012;1:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yu B, Jiang T, Liu D. BCMA‐targeted immunotherapy for multiple myeloma. J Hematol Oncol. 2020;13:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marofi F, Motavalli R, Safonov VA, et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther. 2021;12:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. 2020;17:147‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Upadhaya S, Yu JX, Shah M, Correa D, Partridge T, Campbell J. The clinical pipeline for cancer cell therapies. Nat Rev Drug Discov. 2021;20:503‐504. [DOI] [PubMed] [Google Scholar]
- 11. Hernandez‐Lopez A, Tellez‐Gonzalez MA, Mondragon‐Teran P, Meneses‐Acosta A. Chimeric antigen receptor‐T cells: a pharmaceutical scope. Front Pharmacol. 2021;12:720692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hucks G, Rheingold SR. The journey to CAR T cell therapy: the pediatric and young adult experience with relapsed or refractory B‐ALL. Blood Cancer J. 2019;9:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Levine BL, Miskin J, Wonnacott K, Keir C. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev. 2017;4:92‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang X, Riviere I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolytics. 2016;3:16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mueller KT, Maude SL, Porter DL, et al. Cellular kinetics of CTL019 in relapsed/refractory B‐cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood. 2017;130:2317‐2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grigor EJM, Fergusson D, Kekre N, et al. Risks and benefits of chimeric antigen receptor T‐cell (CAR‐T) therapy in cancer: a systematic review and meta‐analysis. Transfus Med Rev. 2019;33:98‐110. [DOI] [PubMed] [Google Scholar]
- 17. Gupta A, Gill S. CAR‐T cell persistence in the treatment of leukemia and lymphoma. Leuk Lymphoma. 2021;62:2587‐2599. [DOI] [PubMed] [Google Scholar]
- 18. Demaret J, Varlet P, Trauet J, et al. Monitoring CAR T‐cells using flow cytometry. Cytometry B Clin Cytom. 2021;100:218‐224. [DOI] [PubMed] [Google Scholar]
- 19. Kakkanaiah VN, Lang KR, Bennett PK. Flow cytometry in cell‐based pharmacokinetics or cellular kinetics in adoptive cell therapy. Bioanalysis. 2018;10:1457‐1459. [DOI] [PubMed] [Google Scholar]
- 20. Litwin V, Marder P. Flow Cytometry in Drug Discovery and Development. John Wiley & Sons; 2011. [Google Scholar]
- 21. De Oliveira SN, Wang J, Ryan C, et al. A CD19/fc fusion protein for detection of anti‐CD19 chimeric antigen receptors. J Transl Med. 2013;11:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mueller KT, Waldron E, Grupp SA, et al. Clinical pharmacology of Tisagenlecleucel in B‐cell acute lymphoblastic leukemia. Clin Cancer Res. 2018;24:6175‐6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T‐cell therapy in refractory large B‐cell lymphoma. N Engl J Med. 2017;377:2531‐2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abramson JS, Palomba ML, Gordon LI, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B‐cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396:839‐852. [DOI] [PubMed] [Google Scholar]
- 25. Munshi NC, Anderson LD Jr, Shah N, et al. Idecabtagene Vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384:705‐716. [DOI] [PubMed] [Google Scholar]
- 26. Wang Z, Guo Y, Han W. Current status and perspectives of chimeric antigen receptor modified T cells for cancer treatment. Protein Cell. 2017;8:896‐925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Neelapu SS, Tummala S, Kebriaei P, et al. Chimeric antigen receptor T‐cell therapy – assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15:47‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Z, Han W. Biomarkers of cytokine release syndrome and neurotoxicity related to CAR‐T cell therapy. Biomark Res. 2018;6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Teachey DT, Lacey SF, Shaw PA, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T‐cell therapy for acute lymphoblastic leukemia. Cancer Discov. 2016;6:664‐679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19‐28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Klinger M, Brandl C, Zugmaier G, et al. Immunopharmacologic response of patients with B‐lineage acute lymphoblastic leukemia to continuous infusion of T cell‐engaging CD19/CD3‐bispecific BiTE antibody blinatumomab. Blood. 2012;119:6226‐6233. [DOI] [PubMed] [Google Scholar]
- 32. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B‐cell lymphoblastic leukemia. N Engl J Med. 2018;378:439‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Park JH, Rivière I, Gonen M, et al. Long‐term follow‐up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378:449‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hu Y, Sun J, Wu Z, et al. Predominant cerebral cytokine release syndrome in CD19‐directed chimeric antigen receptor‐modified T cell therapy. J Hematol Oncol. 2016;9:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gust J, Hay KA, Hanafi LA, et al. Endothelial activation and blood‐brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR‐T cells. Cancer Discov. 2017;7:1404‐1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hay KA, Hanafi L‐A, Li D, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor–modified T‐cell therapy. Blood. 2017;130:2295‐2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bouchkouj N, Kasamon YL, de Claro RA, et al. FDA approval summary: Axicabtagene Ciloleucel for relapsed or refractory large B‐cell lymphoma. Clin Cancer Res. 2019;25:1702‐1708. [DOI] [PubMed] [Google Scholar]
- 38. Anderson MK, Torosyan A, Halford Z. Brexucabtagene Autoleucel: a novel chimeric antigen receptor T‐cell therapy for the treatment of mantle cell lymphoma. Ann Pharmacother. 2022;56:609‐619. [DOI] [PubMed] [Google Scholar]
- 39. Mian A, Hill BT. Brexucabtagene autoleucel for the treatment of relapsed/refractory mantle cell lymphoma. Expert Opin Biol Ther. 2021;21:435‐441. [DOI] [PubMed] [Google Scholar]
- 40. Shah BD, Ghobadi A, Oluwole OO, et al. KTE‐X19 for relapsed or refractory adult B‐cell acute lymphoblastic leukaemia: phase 2 results of the single‐arm, open‐label, multicentre ZUMA‐3 study. Lancet. 2021;398:491‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rouce RH, Heslop HE. Forecasting cytokine storms with new predictive biomarkers. Cancer Discov. 2016;6:579‐580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gardner RA, Finney O, Annesley C, et al. Intent‐to‐treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129:3322‐3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xu Y, Zhang M, Ramos CA, et al. Closely related T‐memory stem cells correlate with in vivo expansion of CAR.CD19‐T cells and are preserved by IL‐7 and IL‐15. Blood. 2014;123:3750‐3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee DW, Kochenderfer JN, Stetler‐Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose‐escalation trial. Lancet. 2015;385:517‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Teachey DT, Rheingold SR, Maude SL, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine‐directed therapy. Blood. 2013;121:5154‐5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Van De Vyver AJ, Marrer‐Berger E, Wang K, Lehr T, Walz AC. Cytokine release syndrome by T‐cell‐redirecting therapies: can we predict and modulate patient risk? Clin Cancer Res. 2021;27:6083‐6094. [DOI] [PubMed] [Google Scholar]
- 47. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127:3321‐3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 results of ZUMA‐1: a multicenter study of KTE‐C19 anti‐CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25:285‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Genentech . Update on actemra® (tocilizumab) supply in the U.S, 2022. https://www.gene.com/media/statements/ps_081621. Accessed May 19, 2022.
- 50. Kotch C, Barrett D, Teachey DT. Tocilizumab for the treatment of chimeric antigen receptor T cell‐induced cytokine release syndrome. Expert Rev Clin Immunol. 2019;15:813‐822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kauer J, Hörner S, Osburg L, et al. Tocilizumab, but not dexamethasone, prevents CRS without affecting antitumor activity of bispecific antibodies. J Immunother Cancer. 2020;8:e000621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. De Benedetti F, Brunner HI, Ruperto N, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367:2385‐2395. [DOI] [PubMed] [Google Scholar]
- 53. Han D, Xu Z, Zhuang Y, Ye Z, Qian Q. Current progress in CAR‐T cell therapy for hematological malignancies. J Cancer. 2021;12:326‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Raje N, Berdeja J, Lin Y, et al. Anti‐BCMA CAR T‐cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019;380:1726‐1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR‐T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126:2123‐2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Poorebrahim M, Melief J, Pico de Coaña Y, L. Wickström S, Cid‐Arregui A, Kiessling R. Counteracting CAR T cell dysfunction. Oncogene. 2021;40:421‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Locke FL, Rossi JM, Neelapu SS, et al. Tumor burden, inflammation, and product attributes determine outcomes of axicabtagene ciloleucel in large B‐cell lymphoma. Blood Adv. 2020;4:4898‐4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu C, Ayyar VS, Zheng X, et al. Model‐based cellular kinetic analysis of chimeric antigen receptor‐T cells in humans. Clin Pharmacol Ther. 2021;109:716‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stein AM, Grupp SA, Levine JE, et al. Tisagenlecleucel model‐based cellular kinetic analysis of chimeric antigen receptor‐T cells. CPT Pharmacometrics Syst Pharmacol. 2019;8:285‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Thudium Mueller K, Grupp SA, Maude SL, et al. Tisagenlecleucel immunogenicity in relapsed/refractory acute lymphoblastic leukemia and diffuse large B‐cell lymphoma. Blood Adv. 2021;5:4980‐4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Berdeja JG, Madduri D, Usmani SZ, et al. Ciltacabtagene autoleucel, a B‐cell maturation antigen‐directed chimeric antigen receptor T‐cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE‐1): a phase 1b/2 open‐label study. Lancet. 2021;398:314‐324. [DOI] [PubMed] [Google Scholar]
- 63. Ogasawara K, Lymp J, Mack T, et al. In vivo cellular expansion of Lisocabtagene Maraleucel and association with efficacy and safety in relapsed/refractory large B‐cell lymphoma. Clin Pharmacol Ther. 2022;112:81‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ogasawara K, Dodds M, Mack T, Lymp J, Dell’Aringa J, Smith J. Population cellular kinetics of Lisocabtagene Maraleucel, an autologous CD19‐directed chimeric antigen receptor T‐cell product, in patients with relapsed/refractory large B‐cell lymphoma. Clin Pharmacokinet. 2021;60:1621‐1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang M, Munoz J, Goy A, et al. KTE‐X19 CAR T‐cell therapy in relapsed or refractory mantle‐cell lymphoma. N Engl J Med. 2020;382:1331‐1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Turtle CJ, Hanafi L‐A, Berger C, et al. Immunotherapy of non‐Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19‐specific chimeric antigen receptor‐modified T cells. Sci Transl Med. 2016;8:355ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jensen MC, Popplewell L, Cooper LJ, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19‐specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245‐1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Awasthi R, Pacaud L, Waldron E, et al. Tisagenlecleucel cellular kinetics, dose, and immunogenicity in relation to clinical factors in relapsed/refractory DLBCL. Blood Adv. 2020;4:560‐572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wagner DL, Fritsche E, Pulsipher MA, et al. Immunogenicity of CAR T cells in cancer therapy. Nat Rev Clin Oncol. 2021;18:379‐393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jonnalagadda M, Mardiros A, Urak R, et al. Chimeric antigen receptors with mutated IgG4 fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol Ther. 2015;23:757‐768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Shum T, Kruse RL, Rooney CM. Strategies for enhancing adoptive T‐cell immunotherapy against solid tumors using engineered cytokine signaling and other modalities. Expert Opin Biol Ther. 2018;18:653‐664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Le Tourneau C, Stathis A, Vidal L, Moore MJ, Siu LL. Choice of starting dose for molecularly targeted agents evaluated in first‐in‐human phase I cancer clinical trials. J Clin Oncol. 2010;28:1401‐1407. [DOI] [PubMed] [Google Scholar]
- 73. US Food and Drug Administration . Considerations for the design of early‐phase clinical trials of cellular and gene therapy products – guidance for industry, 2015. https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/considerations‐design‐early‐phase‐clinical‐trials‐cellular‐and‐gene‐therapy‐products. Accessed May 19, 2022.
- 74. Wages NA, Chiuzan C, Panageas KS. Design considerations for early‐phase clinical trials of immune‐oncology agents. J Immunother Cancer. 2018;6:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med. 2012;366:2443‐2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Brahmer JR, Tykodi SS, Chow LQM, et al. Safety and activity of anti‐PD‐L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455‐2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chakiba C, Grellety T, Bellera C, Italiano A. Encouraging trends in modern phase 1 oncology trials. N Engl J Med. 2018;378:2242‐2243. [DOI] [PubMed] [Google Scholar]
- 79. Mittapalli RK, Yin D, Beaupre D, Palaparthy R. Starting dose selection and dose escalation for oncology small molecule first‐in‐patient trials: learnings from a survey of FDA‐approved drugs. Cancer Chemother Pharmacol. 2021;87:23‐30. [DOI] [PubMed] [Google Scholar]
- 80. Manji A, Brana I, Amir E, et al. Evolution of clinical trial design in early drug development: systematic review of expansion cohort use in single‐agent phase I cancer trials. J Clin Oncol. 2013;31:4260‐4267. [DOI] [PubMed] [Google Scholar]
- 81. Ezzalfani M, Audrey D, Caroline M, et al. The role of the expansion cohort in phase I trials in oncology: guidelines of the phase I HUB. Bull Cancer. 2015;102:73‐82. [DOI] [PubMed] [Google Scholar]
- 82. Bugano DDG, Hess K, Jardim DLF, et al. Use of expansion cohorts in phase I trials and probability of success in phase II for 381 anticancer drugs. Clin Cancer Res. 2017;23:4020‐4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. US Food and Drug Administration . FDA approves brexucabtagene autoleucel for relapsed or refractory mantle cell lymphoma, 2020. https://www.fda.gov/drugs/resources‐information‐approved‐drugs/fda‐approves‐brexucabtagene‐autoleucel‐relapsed‐or‐refractory‐mantle‐cell‐lymphoma. Accessed May 19, 2022.
- 84. Locke FL, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel as second‐line therapy for large B‐cell lymphoma. N Engl J Med. 2022;386:640‐654. [DOI] [PubMed] [Google Scholar]
- 85. Bishop MR, Dickinson M, Purtill D, et al. Second‐line Tisagenlecleucel or standard Care in Aggressive B‐cell lymphoma. N Engl J Med. 2022;386:629‐639. [DOI] [PubMed] [Google Scholar]
- 86. Gilead . Yescarta® receives U.S. FDA approval as first CAR T‐cell therapy for initial treatment of relapsed or refractory large B‐cell lymphoma (LBCL), 2022. https://www.gilead.com/news‐and‐press/press‐room/press‐releases/2022/4/yescarta‐receives‐us‐fda‐approval‐as‐first‐car‐tcell‐therapy‐for‐initial‐treatment‐of‐relapsed‐or‐refractory‐large‐bcell‐lymphoma‐lbcl. Accessed May 19, 2022.
- 87. Sachs JR, Mayawala K, Gadamsetty S, Kang SP, de Alwis DP. Optimal dosing for targeted therapies in oncology: drug development cases leading by example. Clin Cancer Res. 2016;22:1318‐1324. [DOI] [PubMed] [Google Scholar]
- 88. Kruger SF, Cadilha BL, von Bergwelt‐Baildon M, Endres S, Kobold S. Challenges in clinical trial Design for T Cell‐Based Cancer Immunotherapy. Clin Pharmacol Ther. 2020;107:47‐49. [DOI] [PubMed] [Google Scholar]
- 89. Li DH, Whitmore JB, Guo W, Ji Y. Toxicity and efficacy probability interval Design for Phase I Adoptive Cell Therapy Dose‐Finding Clinical Trials. Clin Cancer Res. 2017;23:13‐20. [DOI] [PubMed] [Google Scholar]
- 90. Barros LRC, Paixão EA, Valli AMP, Naozuka GT, Fassoni AC, Almeida RC. CARTmath—A mathematical model of CAR‐T immunotherapy in preclinical studies of hematological cancers. Cancers. 2021;13:2941‐2962. 10.3390/cancers13122941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sahoo P, Yang X, Abler D, et al. Mathematical deconvolution of CAR T‐cell proliferation and exhaustion from real‐time killing assay data. J R Soc Interface. 2020;17:20190734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Marshall S, Madabushi R, Manolis E, et al. Model‐informed drug discovery and development: current industry good practice and regulatory expectations and future perspectives. CPT Pharmacometrics Syst Pharmacol. 2019;8:87‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Cess CG, Finley SD. Data‐driven analysis of a mechanistic model of CAR T cell signaling predicts effects of cell‐to‐cell heterogeneity. J Theor Biol. 2020;489:110125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yiu HH, Graham AL, Stengel RF. Dynamics of a cytokine storm. PLoS One. 2012;7:e45027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Nukala U, Rodriguez Messan M, Yogurtcu ON, Wang X, Yang H. A systematic review of the efforts and hindrances of modeling and simulation of CAR T‐cell therapy. AAPS J. 2021;23:52. [DOI] [PubMed] [Google Scholar]
- 96. Rohrs JA, Zheng D, Graham NA, Wang P, Finley SD. Computational model of chimeric antigen receptors explains site‐specific phosphorylation kinetics. Biophys J. 2018;115:1116‐1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hardiansyah D, Ng CM. Quantitative systems pharmacology model of chimeric antigen receptor T‐cell therapy. Clin Transl Sci. 2019;12:343‐349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Chaudhury A, Zhu X, Chu L, et al. Chimeric antigen receptor T cell therapies: a review of cellular kinetic‐pharmacodynamic modeling approaches. J Clin Pharmacol. 2020;60(Suppl 1):S147‐S159. [DOI] [PubMed] [Google Scholar]
- 99. Singh AP, Zheng X, Lin‐Schmidt X, et al. Development of a quantitative relationship between CAR‐affinity, antigen abundance, tumor cell depletion and CAR‐T cell expansion using a multiscale systems PK‐PD model. MAbs. 2020;12:1688616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kimmel GJ, Locke FL, Altrock PM. The roles of T cell competition and stochastic extinction events in chimeric antigen receptor T cell therapy. Proc Biol Sci. 2021;288:20210229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. US Food and Drug Administration . Project Optimus – Reforming the dose optimization and dose selection paradigm in oncology, 2022. https://www.fda.gov/about‐fda/oncology‐center‐excellence/project‐optimus. Accessed May 19, 2022.
- 102. Martinez Bedoya D, Dutoit V, Migliorini D. Allogeneic CAR T cells: an alternative to overcome challenges of CAR T cell therapy in glioblastoma. Front Immunol. 2021;12:640082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Allogene Therapeutics . Allogeneic car T therapy—the next revolution in cell therapy, 2022. https://allogene.com/allocar‐t/. Accessed April 20, 2022.
- 104. Orlowski RJ, Porter DL, Frey NV. The promise of chimeric antigen receptor T cells (CARTs) in leukaemia. Br J Haematol. 2017;177:13‐26. [DOI] [PubMed] [Google Scholar]
- 105. Locke FL, Malik S, Tees MT, et al. First‐in‐human data of ALLO‐501A, an allogeneic chimeric antigen receptor (CAR) T‐cell therapy and ALLO‐647 in relapsed/refractory large B‐cell lymphoma (R/R LBCL): ALPHA2 study. J Clin Oncol. 2021;39(Suppl 15):2529. 10.1200/JCO.2021.39.15_suppl.2529 [DOI] [Google Scholar]
- 106. Neelapu SS, Munoz J, Locke FL, et al. First‐in‐human data of ALLO‐501 and ALLO‐647 in relapsed/refractory large B‐cell or follicular lymphoma (R/R LBCL/FL): ALPHA study. J Clin Oncol. 2020;38(Suppl 15):8002. 10.1200/JCO.2020.38.15_suppl.8002 [DOI] [Google Scholar]
- 107. Allogene Therapeutics . Allogene therapeutics CD19 forum, 2021. https://ir.allogene.com/static‐files/7a26a1c0‐55f9‐4474‐a362‐9a49b598c43c. Accessed July 15, 2021.
- 108. Allogene Therapeutics . Allogene therapeutics reports FDA clinical hold of AlloCAR T trials based on a single patient case in ALPHA2 trial, 2021. https://ir.allogene.com/news‐releases/news‐release‐details/allogene‐therapeutics‐reports‐fda‐clinical‐hold‐allocar‐t‐trials. Accessed May 19, 2022.
- 109. Allogene Therapeutics . Allogene therapeutics announces removal of FDA clinical hold across all AlloCAR T™ clinical trials, 2022. https://ir.allogene.com/news‐releases/news‐release‐details/allogene‐therapeutics‐announces‐removal‐fda‐clinical‐hold‐across. Accessed May 19, 2022.
- 110. Cho SF, Anderson KC, Tai YT. Targeting B cell maturation antigen (BCMA) in multiple myeloma: potential uses of BCMA‐based immunotherapy. Front Immunol. 2018;9:1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Seckinger A, Delgado JA, Moser S, et al. Target expression, generation, preclinical activity, and pharmacokinetics of the BCMA‐T cell bispecific antibody EM801 for multiple myeloma treatment. Cancer Cell. 2017;31:396‐410. [DOI] [PubMed] [Google Scholar]
- 112. Mailankody S, Matous JV, Liedtke M, et al. Universal: an Allogeneic first‐in‐human study of the anti‐Bcma ALLO‐715 and the anti‐CD52 ALLO‐647 in relapsed/refractory multiple myeloma. Blood. 2020;136:24‐25.32430494 [Google Scholar]
- 113. Mailankody S, Liedtke M, Sidana S, et al. Universal updated phase 1 data validates the feasibility of Allogeneic anti‐BCMA ALLO‐715 therapy for relapsed/refractory multiple myeloma. Blood. 2021;138:651. [Google Scholar]
- 114. Allogene Therapeutics . Allogene therapeutics reports positive results from phase 1 UNIVERSAL study of single dose ALLO‐715 AlloCAR T™ cell therapy in relapsed/refractory multiple myeloma at the 63rd american society of hematology annual meeting, 2021. <https://ir.allogene.com/news‐releases/news‐release‐details/allogene‐therapeutics‐reports‐positive‐results‐phase‐1‐universal>. Accessed May 19, 2022.
- 115. Zhang J, Wang L. The emerging world of TCR‐T cell trials against cancer: a systematic review. Technol Cancer Res Treat. 2019;18:1533033819831068. 10.1177/1533033819831068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Immunocore . Immunocore announces FDA approval of KIMMTRAK® (tebentafusp‐tebn) for the treatment of unresectable or metastatic uveal melanoma, 2022. <https://ir.immunocore.com/news‐releases/news‐release‐details/immunocore‐announces‐fda‐approval‐kimmtrakr‐tebentafusp‐tebn>. Accessed May 19, 2022.
- 117. Nathan P, Hassel JC, Rutkowski P, et al. Overall survival benefit with Tebentafusp in metastatic uveal melanoma. N Engl J Med. 2021;385:1196‐1206. [DOI] [PubMed] [Google Scholar]
- 118. Adaptimmune . Adaptimmune reports positive results from its pivotal SPEARHEAD‐1 trial in patients with synovial sarcoma and MRCLS at CTOS, 2021. <https://www.adaptimmune.com/investors‐and‐media/news‐center/press‐releases/detail/202/adaptimmune‐reports‐positive‐results‐from‐its‐pivotal>. Accessed May 19, 2022.
- 119. Immatics Biotechnology . Immatics presents data update on dose escalation from ongoing ACTengine® cell therapy programs – press release, 2021. https://investors.immatics.com/news‐releases/news‐release‐details/immatics‐presents‐data‐update‐dose‐escalation‐ongoing‐actenginer. Accessed May 19, 2022.
- 120. Medigene . Medigene reports preliminary efficacy and immune monitoring data of phase i of phase i/ii mdg1011 trial in blood cancers, 2022. https://www.medigene.com/investors‐media/press‐releases/detail/medigene‐reports‐preliminary‐efficacy‐and‐immune‐monitoring‐data‐of‐phase‐i‐of‐phase‐i‐ii‐mdg1011‐trial‐in‐blood‐cancers‐1. Accessed May 19, 2022.