

Abstract
Biomineralization requires the controlled movement of ions across cell barriers to reach the sites of crystal growth. Mineral precipitation occurs in aqueous phases as fluids become supersaturated with specific ionic compositions. In the biological world, biomineralization is dominated by the presence of calcium (Ca2+) in crystal lattices. Ca2+ channels are intrinsic modulators of this process, facilitating the availability of Ca2+ within cells in a tightly regulated manner in time and space. Unequivocally, the most mineralized tissue produced by vertebrates, past and present, is dental enamel. With some of the longest carbonated hydroxyapatite (Hap) crystals known, dental enamel formation is fully coordinated by specialized epithelial cells of ectodermal origin known as ameloblasts. These cells form enamel in two main developmental stages: a) secretory; and b) maturation. The secretory stage is marked by volumetric growth of the tissue with limited mineralization, and the opposite is found in the maturation stage, as enamel crystals expand in width concomitant with increased ion transport. Disruptions in the formation and/or mineralization stages result, in most cases, in permanent alterations in the crystal assembly. This introduces weaknesses in the material properties affecting enamel’s hardness and durability, thus limiting its efficacy as a biting, chewing tool and increasing the possibility of pathology. Here, we briefly review enamel development and discuss key properties of ameloblasts and their Ca2+-handling machinery, and how alterations in this toolkit result in enamelopathies.
Keywords: Enamel, Ameloblasts, Ca2+ signaling, Channelopathy
1. Introduction
The evolutionary history of most mammals and their ancestors can be garnered from the analysis of their fossilized dental remains. Tooth size, shape, and enamel microstructure tracks evolution in detail because dental enamel, the outer covering of teeth, is the hardest and most mineralized tissue of vertebrates, and can withstand the trials of time reasonably unchanged. This is not to suggest that enamel formation is a passive event, but the contrary. Dental enamel formation is a remarkable example of cell-tissue interactions encompassing a number of transformations in cytoskeletal elements concomitant with functional differences [1,2]. Ameloblasts are ectoderm-derived epithelial cells responsible for orchestrating the formation and mineralization of dental enamel. Their organized assembly of enamel matrix proteins and interaction with the crystal phase occur extracellularly with limited control by the enamel cells [3]. The geometry of the crystals is guided by the assembly of proteins, many of which are unique to the ameloblasts’ protein synthesis machinery [4]. The mineral phase that constitutes the fully mineralized tooth enamel is composed of calcium Hydroxyapatite (Hap) organized into some of the largest known Hap crystallites in the biological world [3] (Fig. 1).
Fig. 1.

Enamel pattern in mice: Rodent enamel is commonly used as a model to study mammalian enamel development. This SEM micrograph shows enamel prisms (arrowed) oriented in different directions, which is characteristic of rodent enamel but less so in human enamel. Within the prisms, single crystals can be identified. Scale bar = 5 μm.
The biogenesis of enamel (amelogenesis) is, for simplicity, commonly reduced to two main developmental stages: a) secretory; and b) maturation. Secretory ameloblasts develop a distal secretory process and initiate enamel matrix protein secretion (see below) moving in predetermined outward paths toward the outer enamel [5]. Developmentally, the earliest formed regions of the enamel are the cusps, i.e. the pointy surfaces of a chewing (molar) tooth, and the last developmental phase of enamel occurs at the base. Thus, enamel forms outward and downward. Secretory ameloblasts will transition into a maturation stage cell and, rather than dedicate themselves to synthesizing and secreting enamel matrix proteins, they engage primarily in ion transport. Enamel crystal growth magnifies at this time [6]. It is during the maturation stage that many Ca2+ handling proteins and Ca2+-associated functions are upregulated [2] in line with the increased mineral uptake and crystal growth [7]. Ca2+ is a key and necessary component of enamel. Therefore, disruptions in Ca2+ transport may cause the enamel to mineralize poorly, or improperly, resulting in enamelopathies. Here, we review the effects of altered calcium homeostasis on enamel disease. To better understand these effects, we first provide an overview of the ultrastructure of enamel cells, emphasizing the characteristics of Ca2+ handling organelles in these cells.
2. Enamel organ and matrix proteins
Secretory ameloblasts are post-mitotic, tall columnar, and heavily polarized cells. The cell skeleton may extend ~70 μm in height (5 μm diameter), with the nucleus found at the basal pole (away from the enamel). Mitochondria densely occupy the narrow infranuclear cytoplasm (Fig. 2A & 2B), whereas extensive endoplasmic reticulum (ER) networks populate the large infranuclear space. Abundant Golgi saccules are also present [8]. One of the most conspicuous features of secretory ameloblasts is the presence of an apical (facing the enamel) cellular extension known as the Tomes’ process. Many cellular bodies fuse to the Tomes’ process to exocytose and endocytose proteins (Fig. 2B). Cell-cell connections (desmosomes, tight junctions) are found both apically and distally, maintaining narrow intercellular spaces [8]. This limits the movement of molecules between cells in any direction [6]. The protein synthesis apparatus is committed to the production of a key enamel-specific protein known as amelogenin, which constitutes about 90% of all secreted products [9]. Amelogenin is required for the ordering, or alignment, of enamel crystallites in the extracellular space [3]. Other “non-amelogenin” products including enamelin and ameloblastin, are also involved in the normal development of enamel [10]. Mutations in these enamel-specific genes result in a broad range of clinical pathologies known as amelogenesis imperfecta (AI), defined by abnormal mineralization or deficient growth of the enamel layer [11].
Fig. 2.
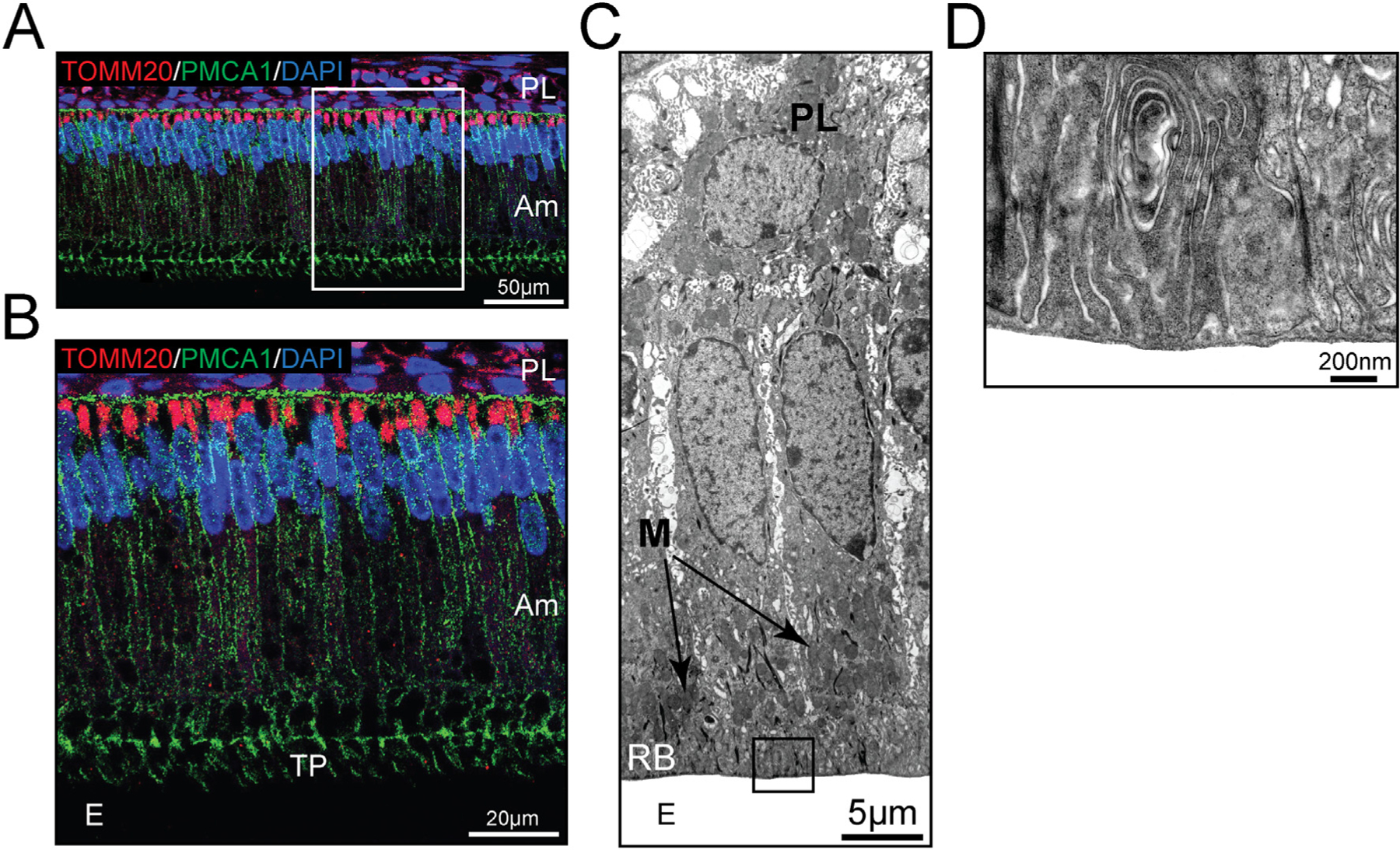
Mitochondria localization in secretory and maturation stage ameloblasts. (A) The mitochondrial outer membrane marker TOMM20 (red) was used to visualize the dominant infranuclear localization of mitochondria in secretory stage ameloblasts. The plasma membrane of ameloblast cells was highlighted by immunofluorescence staining of PMCA (green). Cell nuclei are stained with DAPI (blue). A close-up is shown in (B), which also highlights the Tomes’ process (TP), a cell extension at the apical (distal) end near the enamel (E). (C) TEM image of maturation stage ameloblasts with mitochondria (M) clustering in areas just above the ruffled-border (RB) and below the nucleus. (D) A close-up of the ruffled-border showing the deep infoldings characteristic of this apical membrane structure. Am = ameloblasts. PL = papillary cell layer.
Many secretory cells will transition into a maturation stage ameloblast, showing reduced height but maintaining their polarity, and losing the secretory Tomes’ process. They develop deep membrane infoldings or ruffled distal border, which plays an important role in ion transport [6] (Fig. 2C & 2D). These ruffled ameloblasts have less conspicuous ER tubules and contain mitochondria both at the infra- and supranuclear regions with large clusters near the ruffled end [12] (Fig. 2C, Fig. 3). Distal cell-cell tight junctions (TJ) constrain the movement of molecules and ions between cells toward the enamel [6]. Unusually in mammalian cell biology, these ruffled cells will lose and reform these distal infoldings, periodically adopting a smooth distal pole [13] (Fig. 3). These smooth-ended cells, which are a minority of all maturation stage cells, show increased intercellular spaces and distally placed mitochondria. There is only limited knowledge of their role in amelogenesis. It has been suggested that the smooth ameloblast phase facilitates the diffusion of organics from the enamel into the interstitial fluid. They have also been associated with stabilizing pH in the enamel compartment from the mildly acidic pH observed during the ruffled-ended stage [6,14]. An important feature of all maturation stage ameloblasts is their connection with adjacent (basally) cells of the papillary layer (PL). The PL cells contain many mitochondria and likely form a functional syncytium with the ameloblasts [15] (Fig. 2C, Fig. 3). Maturation stage cells orchestrate the bulk of ion transport necessary for the massive expansion of enamel crystals [1,6,15].
Fig. 3.
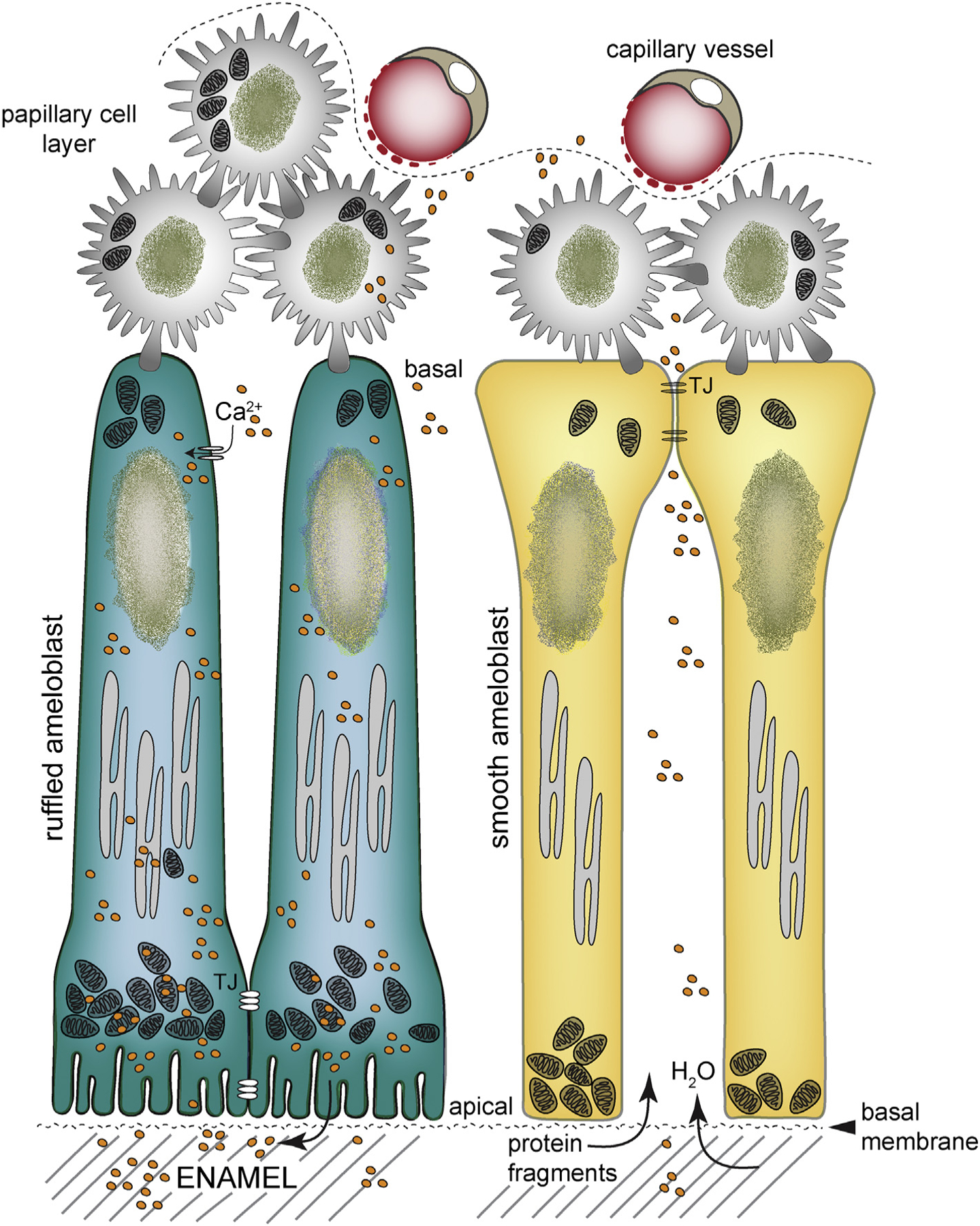
Schematic of ruffled- and smooth-ended ameloblasts: Maturation stage ameloblasts have two main morphologies. In the ruffled-ended ameloblasts (RA), mitochondria localize to the infra- and supranuclear cytoplasm clustering between the nucleus and the ruffled-border. In smooth-ended ameloblasts (SA), mitochondria are found near the apical pole. RA have tight junctions (TJ) apically limiting the flow of ions and molecules between cells. RA is the predominant morphology (70% of cells in the maturation stage) and will cycle into SA and vice versa after a few hours several times per day. Ca2+ uptake is modulated by SOCE channels. Ca2+ ions move across the RA to reach the ruffled-border, but in the SA phase ions can move between cells. In the SA morphology, leaky TJ are found at the basal pole but do not have junctions distally. Water and protein fragments can diffuse into the wider intercellular spaces of SA. The cyclic modulation of RA to SA is poorly understood, but is required for proper enamel mineralization. Papillary cells attach at the basal pole of ameloblasts and are in close proximity to blood vessels. A basal membrane attaches the cells to the mineralizing matrix. Modified from ref. [15].
Given the presence of many junctional complexes communicating adjacent secretory as well as maturation stage ameloblasts, the general consensus is that ameloblasts are the active modulators of transepithelial transport. They do so in a highly organized fashion, with little contribution from paracellular transport [16–18]. This notion is reinforced by the presence of a basal membrane serving as anchorage between maturation ameloblasts and the crystals, adding to the limitations in the passage of interstitial fluid to the enamel (Fig. 3) [19]. It is also intuitive to suggest that the highly structured nature of the biomineralization process of enamel necessitates a delicate balance controlling ion fluxes to coordinate their incorporation into the forming crystals to regulate its stoichiometry. Thus, any alterations that occur either in the secretory (protein synthesis) or maturation (mineralization) stages can upset the delicate balance of enamel crystal assembly.
3. General effects of systemic calcium changes in enamel
Ca2+ is the fifth most abundant element in the human body weighing about 1000 g, most of which (99%) is found in bone as Hap [20]. Bone Ca2+ serves not only as a stiffening material in the skeleton, but also as the main Ca2+ reservoir in the body. Ca2+ metabolism is mediated by renal reabsorption, intestinal absorption, and bone turnover which are dynamically regulated by parathyroid hormone (PTH), 1,25-dihydroxyvitamin D [1,25(OH)2D], and Ca2+ itself [20]. Bone tissue is a dynamic system that is constantly being remodeled (bone deposition and bone resorption) so that short-term Ca2+ deficiencies can be resolved over time. Ca2+ is released from bone by the activity of PTH, causing its levels to rise in plasma where it is closely monitored, being removed by calcitonin stimulation when levels are too high and then stored in bone. Although enamel is more highly mineralized than bone containing increased levels of Ca2+, this trapped Ca2+ within the enamel crystal lattice cannot be “released” dynamically into the system as it does in bone since enamel does not remodel. However, there has been interest in determining whether disruptions in systemic calcium levels (hypo-hypercalcemia), via PTH or vitamin D (and its receptor VDR), have lasting effects in enamel.
Bonucci and co-workers reported the effects of hypocalcemia in young rats induced by low dietary Ca2+ in nursing mothers [21]. This resulted in hypomineralized enamel in the pups. Researchers also found that this defect could be corrected following a 10-day period of added Ca2+ in the diet. Hypocalcemia was also reported to affect the enamel cells, resulting in the formation of cyst-like structures in the ameloblasts layer and abnormal localization of enamel proteins [22]. In a surgical study, thyroidectomy did not affect enamel in rats but hypocalcemia induced by thyro-parathyroidectomy resulted in severely affected enamel [23]. These data link hypocalcemia with enamel deficiencies, as expected.
In addition, the role of vitamin D has been recognized in enamel [24] among other reasons, as it appears to regulate enamel gene expression [25]. The role of vitamin D in enamel is also supported by reports that deficiency of vitamin D during pregnancy results in increased early childhood caries [26]. This is validated by a study that reported disruptions in the formation of enamel as well as disrupted calbindin expression in vitamin D deficient rats [27]. VDR−/− mice also showed enamel abnormalities although the effects of loss of function of VDR were complex in the context of induced dietary hypocalcemia or in its absence [25]. Other studies analyzed the enamel phenotype of Rickets patients, a condition associated with vitamin D-deficiency, identifying increased caries and tooth decay in this group [25]. However, other vitamin D-deficiency related diseases, including Alzheimer’s disease (AD) and Parkinson’s disease, have not been associated with enamel abnormalities to date. The recently proposed role of vitamin D by Berridge [28] as a regulator of intracellular signaling pathways and redox may be of interest in the future of enamel biology as Ca2+ signaling and redox appear to be tightly associated with enamel mineralization.
Additional effects in enamel linked to systemic dysregulation in Ca2+ homeostasis were recently highlighted in patients with nephrocalcinosis [29]. This disease results from an increase in the Ca2+ content of the kidneys due to increased urinary levels of calcium, phosphate, and oxalate, and leads to progressive renal damage. Nephrocalcinosis patients were reported to show AI and identified mutations in the FAM20A gene associated with this phenotype [30]. FAM20A is a kinase expressed in secretory stage ameloblasts and loss of function mutations prevent phosphorylation of proteins in the secretory pathway causing pathology [30].
4. Channels involved in Ca2+ dysregulation in enamel
As stated above, the transcellular movement of Ca2+ is favored over a paracellular route [16], a model that would thus require Ca2+ channels, pumps, and exchangers as mediators of Ca2+ uptake and extrusion [17].
4.1. Store-operated Ca2+entry
The bulk of Ca2+ uptake in enamel cells is regulated by store-operated Ca2+ entry (SOCE) channel [16,31]. The SOCE archetype is the Ca2+ release activated Ca2+ (CRAC) channel formed by two protein families, the ER-based single pass membrane protein STIM1 (stromal interaction molecule) and its homologue STIM2, as well as the plasma membrane bound Orai proteins [32–34]. STIM1 and STIM2 are Ca2+ sensors and have a luminal EF hand Ca2+ binding domain. Upon a reduction of ER Ca2+ concentration and its detachment from the EF hand, STIM1 proteins undergo conformational changes leading to physical interactions with the Orai proteins, allowing sustained Ca2+ entry [35,36]. There are three mammalian Orai proteins, Orai1–3, all of which function as membrane channel pores [37,38]. Despite the similar roles of STIM1 and STIM2 as Ca2+ sensors and of all three Orai proteins as functional membrane pores, only mutations in STIM1 and ORAI1 genes are known to cause disease [39]. These so called CRAC channelopathies include an ensemble of dysfunctions, the chief being immunodeficiency among the loss-of-function mutations, and result in both AI and a form of ectodermal dysplasia [39,40]. In support of the role of CRAC channels in enamel formation, pharmacological approaches using a number of Orai inhibitors (Synta-66, BTP-2, 2-APB) have demonstrated a significant reduction in Ca2+ influx in thapsigargin stimulated cells [31]. More recently, a double conditional deletion of Stim1 and Stim2 genes in mice reported severe enamel hypomineralization in this mouse model with a gross dental phenotype resembling human AI phenotype [41].
4.2. Cav1.2 channels
The L-type voltage-gated Cav1 (Cav1.2) channel encodes an alpha-1 subunit of a voltage-dependent calcium channel. Loss-of-function mutations in the encoding gene (CACNA1C) result in Brugada syndrome, producing short QT intervals potentially resulting in sudden cardiac death [42]. Gain-of-function mutations in Cav1.2 result in sustained inward Ca2+ currents due to nearly complete loss of voltage-dependent channel inactivation and intracellular Ca2+ overload in multiple cell types, causing a multiorgan dysfunction known as Timothy syndrome [42]. Timothy syndrome patients present with dental abnormalities, including smaller teeth and abnormal enamel [43], likely due to dysregulation of the membrane potential of the enamel cells and the resulting Ca2+ overload, but this has not been studied to date. Yet, it is noteworthy that in vascular smooth muscle cells, STIM1 interacts with Cav1.2 at localized ER/plasma membrane junctions that also contain Orai1 channels, thus controlling Cav1.2 Ca2+ signals [44]. This interaction is not known in enamel cells.
4.3. TRP channels
Transient receptor potential (TRP) channels have recently come to light in association with enamel. The nearly ubiquitous, melastatin-like transient receptor potential cation channel (TRPM7) is an essential permeable ion channel regulating Mg2+ homeostasis [45]. While homozygous deletion of the kinase domain in the c-terminus of TRPM7 is embryonic lethal [45], heterozygous mice are viable for the study of TRPM7 in enamel. These mice showed severely hypomineralized enamel and skeletal abnormalities with lower Ca2+ and higher Mg2+ content in the enamel [46]. Interestingly, recent evidence by Penner and colleagues has linked TRMP7 with SOCE, and while TRPM7 itself is not a store-operated ion channel, its activity via the kinase domain modulates STIM1, thereby participating in Ca2+ homeostasis [47].
5. Ca2+ buffers and pumps
Intracellular Ca2+ buffers have been reported in enamel cells including parvalbumin, calretinin, calmodulin, and calcineurin [reviewed in [16,48]] but, to date, no enamel deficiencies have been associated with mutations in any of these genes. In addition, ATP-dependent Ca2+ pumps, including sarco/endoplasmic reticulum Ca2+ ATP-ases (SERCA) and plasma membrane Ca2+ ATP-ases (PMCA), have been recently revisited as the role of Ca2+ in both signaling and transport in enamel has been under scrutiny [16,49]. Mutations in SERCA2, the predominant isoform in enamel cells [50,51], have not been associated with enamel disease. PMCA1 and PMCA4 are the predominant isoforms found in enamel cells notably showing a basolateral localization [49]. These pumps were originally considered to play a major role in Ca2+ transport in enamel [52], but no mutations have been identified in the PMCA gene coding family (ATP2B1–4) that would result in enamel abnormalities. Thus, they may instead play a minor role in maintenance of Ca2+ homeostasis without being essential components.
6. Calcium exchangers
Both NCX and NCKX are expressed in enamel cells. These bidirectional electrogenic transporters move Na+ in exchange for Ca2+ (and K+ in the case of NCKX). The NCX family (encoded by SLC8A gene) contains six members, but only NCX1 and NCX3 are clearly expressed in enamel cells as showing an apical or apico/lateral distribution [53]. Electrophysiological studies of whole cell recordings of primary enamel cells as well as of the use of pharmacological inhibitors supported the functionality of NCXs in enamel cells [53]. However, no dental abnormalities reported to date have been associated with SLC8A gene mutations.
NCKX4 is a sodium/potassium/calcium exchanger (encoded by SLC24A4) that operates at a 4Na+: 1Ca2+ +1 K+ stoichiometry [54]. NCKX4 was first identified in rat enamel cells by a genome-wide screening identifying Slc24a4 transcript as highly upregulated in maturation stage enamel cells [2]. Immunohistochemistry showed absence of NCKX4 in secretory cells, but demonstrated a strong apical localization in maturation stage ameloblasts suggesting a direct role in Ca2+ extrusion in these cells [55]. Subsequent studies supported this role for NCKX4 with the identification of hypomaturation and AI in consanguineous patients with null and missense mutations in the SLC24A4 gene [56]. Slc24a4−/− mice also presented with abnormal enamel reminiscent of the human AI phenotype [56]. Later studies identified a novel homozygous [57] and a missense mutation in SLC24A4 causing hypomaturation and AI [58]. In patients with SLC24A4 mutations, AI is manifested as loss of enamel, increased attrition, and heavy-brownish discoloration, all indicative of hypomineralization. More recently, the sequencing of candidate genes in a consanguineous family presenting with AI similar to that described previously (22, 23 [58]) identified full and partial deletions in the last three exons of the SLC24A4 gene caused by homologous recombination [59]. Besides the human and murine genetic studies, Samoyed dogs, the large herding-sized dog of the Samoyedic people of Siberia, were also reported to have a mutation in the SCL24A4 gene associated with AI [60]. These genetic data strongly support the role of NCKX4 in Ca2+ homeostasis in enamel cells and point to its natural role as a key mediator in Ca2+extrusion.
More recently, a functional assessment of SCL24A4 mutations associated with AI and their impact on cytosolic Ca2+ levels were performed in HEK293 cells in the reverse operating mode of NCKX4 [61]. Mutant HEK293 cells showed decreased cytosolic Ca2+ (measured with Fluo-4FF), further strengthening the role of NCKX4 in Ca2+ transport in enamel [61]. Its role in enamel appears to be impacted by its localization in the ruffled-border. In conditions where Ca2+ homeostasis is disrupted [41] or in situations of other physiological insults such as fluorosis [62], NCKX4 localization is altered with strong signals found in the cytosol. This alteration is associated, in at least one instance, with the loss of the ruffled-border [41]. This suggests that this apical membrane feature is necessary for NCKX4 to function as a Ca2+ exchanger and to enable Ca2+ extrusion. This is supported by the loss of NCKX4 expression in smooth-ended ameloblasts [16].
In addition to its now accepted role in AI, NCKX4 has been linked to olfactory responses [63], melanocortin-dependent satiety [64], and, most recently, to vision, with NCKX4 shaping the cone photoresponse [65].
7. The role of intracellular organelles
Several studies have defined the cellular localization of ER tubules and mitochondria in enamel cells at each developmental stage [reviewed in [15]] although only limited knowledge of their putative functions (e.g. their contribution to Ca2+ signaling) in enamel formation is available. The expression of inositol 1,4,5-triphosphate receptors (IP3R) in enamel cells was originally described by Hubbard [66] and later by our group, identifying the expression of all three IP3R isoforms [31]. Ryanodine receptors (RyR) were also reported in ameloblasts, but their expression was much less than that of IP3R [31]. Our ongoing investigation of the effects of physiological agonists in enamel cells (unpublished) lends support to the dominant role of IP3R, rather than RyR, in enamel. Despite the relevance of ER Ca2+ signaling in cell biology, to date there are no known dental defects associated with mutations in IP3R. Yet, there is growing interest in enamel biology by a number of laboratories, including our own, on one of the key ER functions, the unfolded protein response (UPR) [41,67–69]. UPR has been linked to disruptions in enamel formation, but the mechanism affecting enamel is different. Two recent reports have described mutations in AMEL and ENAM that appear to induce UPR. The murine p.Tyr64His mutation in the AMEL gene caused UPR-induced apoptosis resulting in AI [69], whereas the ENAMEMp.L31R mutation resulted in UPR and disruptions in the secretory pathway of ameloblasts, ultimately leading to AI [67]. Our work showed that UPR was linked to dysregulation of Ca2+ homeostasis but did not have a clear effect on the expression of enamel genes [41].
Mitochondrial Ca2+ dynamics and the role of mitochondrial-associated functions such as cell metabolism and cell redox are all poorly known in enamel cells. To date, there are no known mitochondrial genes that are recognized as impacting enamel formation when mutated. This is likely to be a misrepresentation, however, given that alterations in enamel are not always severe or obvious to dental practitioners and primary physicians. In fact, as shown in Fig. 4, enamel cells show high Ca2+ buffering capacity by mitochondria, suggesting a role in Ca2+ signaling.
Fig. 4.
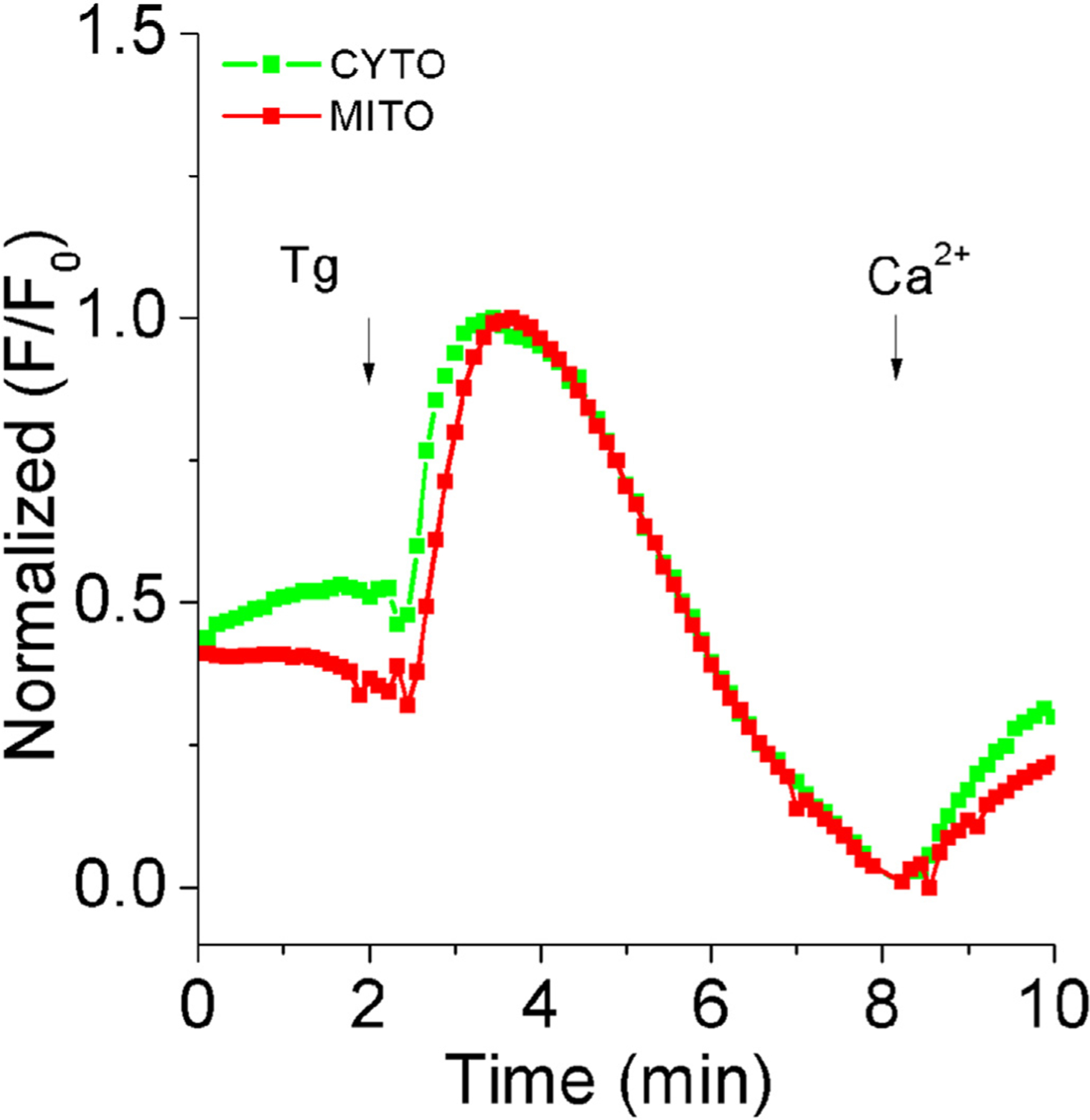
Mitochondrial and cytoplasmic Ca2+ recordings in the enamel cell line LS8 cells. Live-cell imaging using epifluorescence inverted microscopy of cultured LS8 cells loaded with cytoplasmic (Fura 2-AM) and mitochondrial (Rhod-2) fluorescent probes. Plot shows increase in cytosolic and mitochondrial Ca2+ after addition of thaspigargin (1.5 μM) in 0 Ca2+ and following 2mM Ca2+ restitution.
Peroxisomes are also known to participate in Ca2+ homeostasis in cells [70]. Although their role in enamel is limited, recent data have shed light on the expression of a number of proteins associated with peroxisomal function during the early stages of amelogenesis [71]. It is also reported that patients with abnormal peroxisome biogenesis present with facial dysmorphology and abnormal enamel [71]. More recently, mild peroxisomal dysfunction caused by hypomorphic mutations in the PEX1 and PEX6 genes caused Heimler syndrome – characterized by sensorineural hearing loss and nail dysplasia, and resulted in AI [72]. These data suggest that maintenance of peroxisomal function is important in enamel development although it is unclear whether a direct role in peroxisomal-mediated Ca2+ signaling can be established.
8. Stim1/2-deficient ameloblasts
Stim1/2-deficient mice were recently developed in collaboration with the laboratory of Stefan Feske, as reported [73], allowing us to investigate the effects of Ca2+ dysregulation in cells and in the enamel. The enamel of these mice were severely hypomineralized, but there were also a number of striking changes in the enamel-forming cells [41]. The ameloblasts of Stim1/2-deficient mice, for example, showed a high number of multinucleated and/or short-bodied cells. Multinucleation has been documented in wild type cells as an intriguing feature of ameloblasts observed in a minority of both secretory and maturation stage cells [74]. The interest in this feature resides in the notion that all ameloblasts are considered to be post-mitotic cells and, thus, this should not be expected. Yet, as cells age, the number of multinucleate ameloblasts increases, a process that has been linked to the fusion of two or, in some cases, more mononucleate cells [74]. While multinucleate ameloblasts are uncommon, our analysis of ameloblasts of Stim1/2-deficient mice showed higher numbers of multinucleated/shorter cells (Fig. 5B & 5C). The reasons for these alterations in the ameloblasts of these mice are unknown, but disrupted Ca2+ homeostasis likely played a role.
Fig. 5.
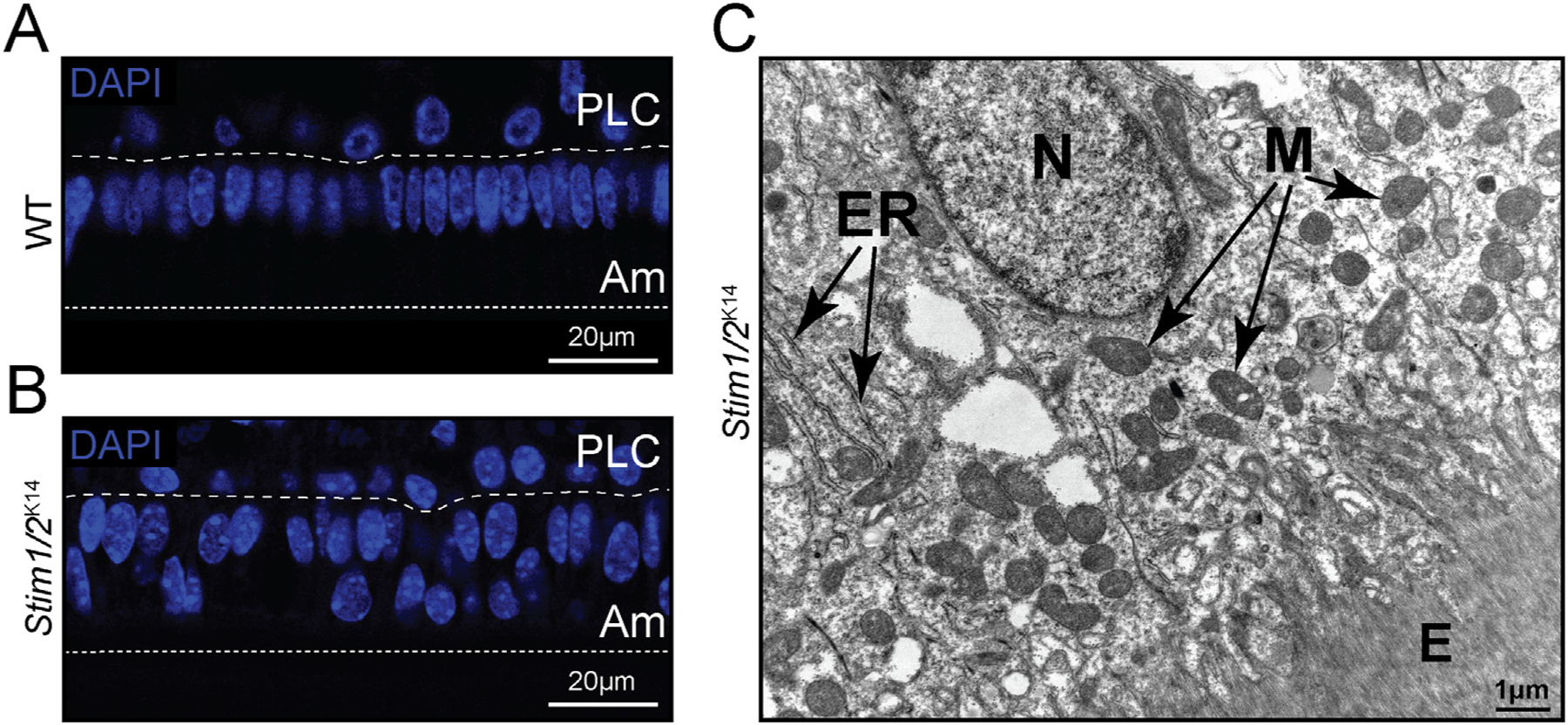
Stim1/2-deficient mice show short/multinucleated ameloblasts. (A–B) Maturation stage ameloblasts of Stim1/2-deficient mice show high numbers of multinucleated and short ameloblasts (B) compared to control mice (A) as visualized by DAPI staining (blue). (C) TEM image of Stim1/2-deficient maturation stage ameloblast showing a nucleus abnormally localized at the apical pole as are the mitochondria. ER = Endoplasmic reticulum. M = mitochondria. E = enamel space.
Closer inspection of the ameloblasts of Stim1/2-deficient mice by transmission electron microscopy (TEM) revealed that maturation ameloblasts did not develop a typical ruffled-border [41] (Fig. 6A). These ameloblasts also showed displaced mitochondria and in many of them the cristae had been disrupted by non-electron dense, multilayered bodies resembling mitophagic structures [41] (Fig. 6B).
Fig. 6.
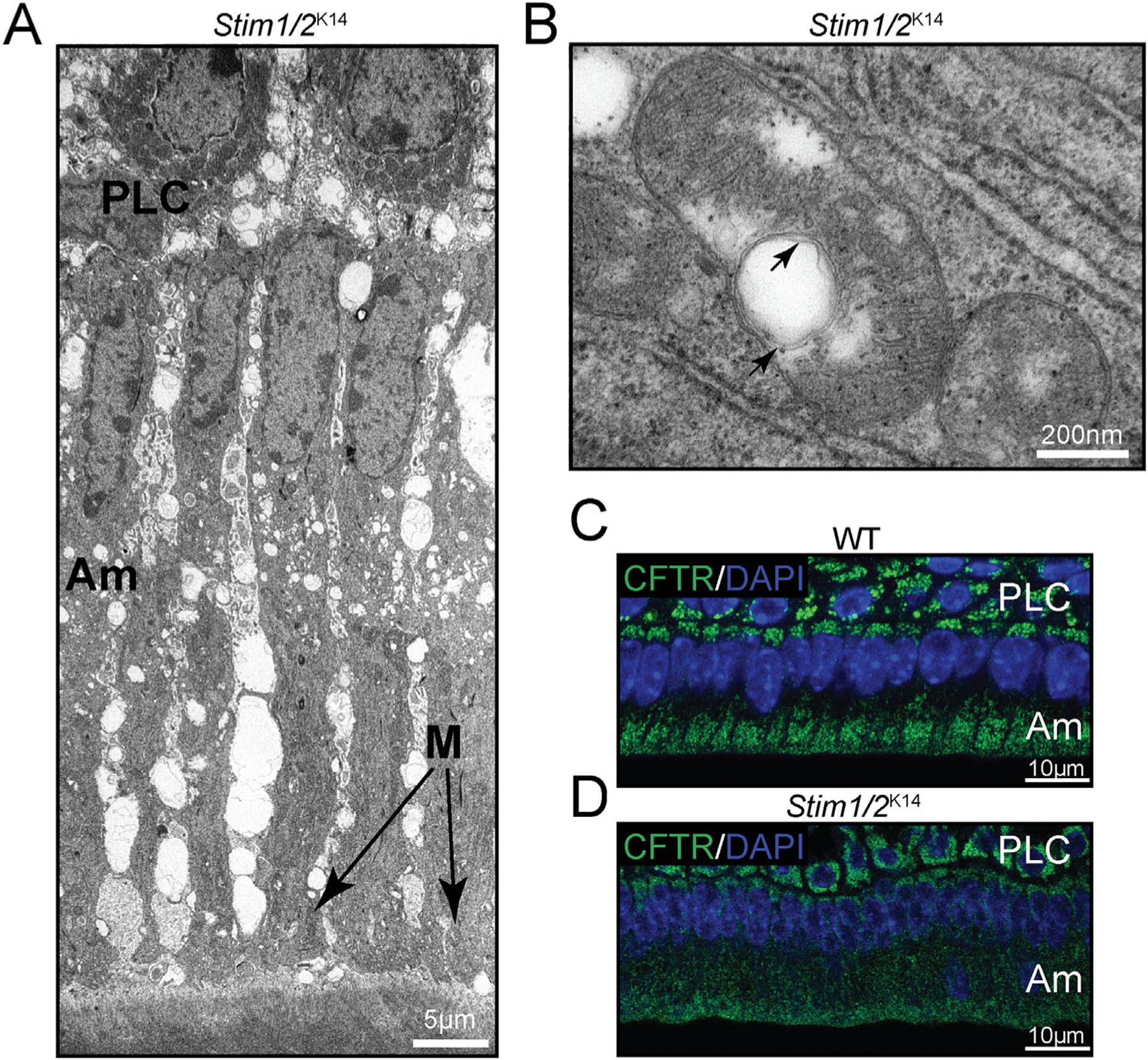
Stim1/2-deficient ameloblasts. (A) TEM micrograph of Stim1/2-deficient maturation stage ameloblasts (Am) showing arrested development of the ruffled-border, increased intercellular space, and apically placed mitochondria. These characteristics are atypical of ruffled-ended ameloblasts but define smooth-ended ameloblasts. Stim1/2-deficient ameloblasts analyzed in different positions of the lower incisors all showed a smooth-ended morphology, suggesting that Ca2+ signals are important for the formation of the ruffled-border. (B) Mitochondria of Stim1/2-deficient mice commonly showed white (non-electron dense) bodies with multiple outer membranes in their matrix. (C–D) The cystic fibrosis transmembrane conductance regulator (CFTR) protein is important in chloride transport during enamel maturation. CFTR is a key target of S-glutathionylation. In WT ameloblasts, CFTR is localized at the apical side of the ameloblasts as shown by the immunofluorescence analysis of WT ameloblasts (C), but in Stim1/2-deficient ameloblasts, CFTR is mislocalized (D), suggesting that it was not S-glutathionylated. DAPI staining (blue) was used to indicate cell nuclei.
Bioinformatic analysis of RNAseq data of enamel cells of these mice, however, revealed an unexpected link to the glutathione system. It was shown that the glutathione metabolism pathway was one of the most enriched in enamel organ cells [41]. We confirmed this by real-time PCR showing downregulation of glutathione S-transferases, which are important enzymes in protein S-glutathionylation [75]. Our hypothesis that the glutathione system was altered in Stim1/2-deficient mice was supported by the lack of actin expression in the distal (apical) pole of the cells but not in the wild type cells. Actin is one of the best known targets of S-glutathionylation [76] and dysregulation results in altered cell cytoskeleton [77]. Thus, we surmised that disruptions in Ca2+ homeostasis associated with abnormal SOCE affected the glutathione system and, in turn, impacted the ability of actin to support the formation of a ruffled-border [41].
Another known target of S-glutathionylation is the cystic fibrosis transmembrane conductance regulator (CFTR). CFTR is an ATP-dependent chloride channel that controls salt and water transport across epithelial tissues. CFTR mutations principally cause exocrine pancreatic destruction and airway disease through infection and inflammation, also producing intestinal obstruction and neuropathy [21]. This anion channel has been shown to be important in enamel mineralization despite the very low level of chloride present in the enamel crystals (~0.0065 mol/g) [78–81]. CFTR function is altered by oxidized forms of glutathione which, at the single channel level, inhibited the opening rates of individual CFTR channels, likely via a poorly conserved cysteine (cys-1344) found in a region predicted to participate in ATP-dependent channel opening [82]. The lower chloride concentration in the enamel of Stim1/2-deficient mice suggested that CFTR may have been impacted by abnormal glutathione. Fig. 6C and D show the localization of CFTR at the apical pole of wild type maturation stage ameloblasts but not in Stim1/2-deficient mice, where CFTR signals were weak and found across the cell cytosol. These data are in line with the role of S-glutathionylation in CFTR function, leading us to consider that glutathione metabolism plays an unexpected role in mediating ion transport in enamel cells. This potentially raises another issue; the subcellular localization of NCKX4 in ameloblasts of Stim1/2-deficient mice was abnormal as reported [41]. While we originally considered the possibility that its mislocalization was associated with the lack of a ruffled-border in the ameloblasts, it could also be hypothesized that NCKX4 is a target of S-glutathionylation. The 622 amino acids sequence of NCKX4 contains 11 cysteine residues, although currently there are no published data on S-glutathionylation of NCKX4. Other proteins such as STIM1, with fewer cysteines in a larger protein sequence than NCKX4, are s-glutathionylated [83].
9. Non-Ca2+ enamelopathies
It should be noted that the altered function of a number of proteins involved in transepithelial transport in enamel cells also result in enamelopathies [1]. Some of these, such as sodium/bicarbonate cotransporters (NBCE1) and anion exchangers (AE2), are involved in acid-base balance [84], highlighting the role of pH balance in enamel [14]. There is, however, a growing list of transporters that affect enamel [85], including poorly known proteins like the sodium-coupled citrate NaCT. This protein modulates cellular uptake of citrate and results in AI associated with the rare autosomal Kohlschütter–Tönz syndrome (KTZS), otherwise characterized by epilepsy and intellectual disability [86]. Studies of KTZS emphasize the disparity of systemic effects on enamel, but also reiterate how sensitive biomineralization is to a vast array of conditions.
10. Summary and outlook
Understanding the role of Ca2+ signaling and transport in enamelopathies presents new challenges when compared to other systems. Biomineralizing systems have two check points: a) the cell; and b) its output, i.e. the formation of Hap crystals. Alterations in cell kinetics can impact the latter dramatically, leading to a record of these defects in the enamel crystals. This record becomes permanently registered in enamel as it does not remodel. So sensitive are enamel cells that even periods of high fever or the dramatic moment of birth produce alterations in enamel formation that will remain registered in enamel for life [87]. The assembly of the crystals via interaction with matrix proteins and other ions occurs in the extracellular space and while this is beautifully controlled by the ameloblasts, once materials exit the cells there is limited direct control of the biochemical interactions that take place. However, ameloblasts are clearly the directors of the assemble and while some progress has been made to define their Ca2+ toolkit, it is now apparent that a panoply of intracellular Ca2+ signaling processes are yet to be elucidated, including mitochondrial Ca2+ signaling, the redox environment, and how these processes interact to form Hap crystals. Data described above highlight the relevance of Ca2+ signaling molecules in enamel disease. The severity of these pathologies is remarkable in that it has elucidated two main links to disease to date: a) Ca2+ uptake and Ca2+ extrusion are essential components of healthy enamel; whereas b) the intracellular Ca2+ machinery is seemingly less critical based on currently available data.
Acknowledgements
This work was funded by the National Institute of Dental and Craniofacial Research grants DE022799 and DE025639 to RSL.
Abbreviations:
- Hap
hydroxyapatite
- SOCE
store-operated Ca2+ entry
- RA
ruffled-ended ameloblast
- SA
smooth-ended ameloblast
- AI
amelogenesis imperfecta
Footnotes
Transparency document
The http://dx.doi.org/10.1016/j.bbamcr.2018.04.013 associated this article can be found, in online version.
References
- [1].Lacruz RS, Enamel: molecular identity of its transepithelial ion transport system, Cell Calcium 65 (2017) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lacruz RS, et al. , Identification of novel candidate genes involved in mineralization of dental enamel by genome-wide transcript profiling, J. Cell. Physiol 227 (5) (2012) 2264–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].F. AG, et al. , Enamel biomineralization: the assembly and disaseembly of protein extracellular organic matrix, in: Teaford MF, Meredith Smith M, Ferguson M (Eds.), Development, Function and Evolution of Teeth, 2000. [Google Scholar]
- [4].Paine ML, Snead ML, Tooth developmental biology: disruptions to enamel-matrix assembly and its impact on biomineralization, Orthod. Craniofac. Res 8 (4) (2005) 239–251. [DOI] [PubMed] [Google Scholar]
- [5].Boyde A, Microstructure of enamel, CIBA Found. Symp 205 (1997) 18–27 (Discussion 27–31). [DOI] [PubMed] [Google Scholar]
- [6].Smith CE, Cellular and chemical events during enamel maturation, Crit. Rev. Oral Biol. Med 9 (2) (1998) 128–161. [DOI] [PubMed] [Google Scholar]
- [7].Smith CE, et al. , Mineral acquisition rates in developing enamel on maxillary and mandibular incisors of rats and mice: implications to extracellular acid loading as apatite crystals mature, J. Bone Miner. Res 20 (2) (2005) 240–249. [DOI] [PubMed] [Google Scholar]
- [8].Sasaki T, et al. , Cell biology of tooth enamel formation. Functional electron microscopic monographs, Monogr. Oral Sci 14 (1990) 1–199. [PubMed] [Google Scholar]
- [9].Termine JD, et al. , Properties of dissociatively extracted fetal tooth matrix proteins. I. Principal molecular species in developing bovine enamel, J. Biol. Chem 255 (20) (1980) 9760–9768. [PubMed] [Google Scholar]
- [10].Bartlett JD, et al. , 3. Protein-protein interactions of the developing enamel matrix, Curr. Top. Dev. Biol 74 (2006) 57–115. [DOI] [PubMed] [Google Scholar]
- [11].Witkop CJ Jr., Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: problems in classification, J. Oral Pathol 17 (9–10) (1988) 547–553. [DOI] [PubMed] [Google Scholar]
- [12].Smith CE, Nanci A, Overview of morphological changes in enamel organ cells associated with major events in amelogenesis, Int. J. Dev. Biol 39 (1) (1995) 153–161. [PubMed] [Google Scholar]
- [13].Smith CE, McKee MD, Nanci A, Cyclic induction and rapid movement of sequential waves of new smooth-ended ameloblast modulation bands in rat incisors as visualized by polychrome fluorescent labeling and GBHA-staining of maturing enamel, Adv. Dent. Res 1 (2) (1987) 162–175. [DOI] [PubMed] [Google Scholar]
- [14].Lacruz RS, et al. , Regulation of pH during amelogenesis, Calcif. Tissue Int 86 (2) (2010) 91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sasaki T, Cell Biology of Tooth Enamel Formation, Vol. 14 (1990). [PubMed] [Google Scholar]
- [16].Nurbaeva MK, et al. , Ca2+ transport and signalling in enamel cells, J. Physiol 595 (10) (2016) 3015–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hubbard MJ, Calcium transport across the dental enamel epithelium, Crit. Rev. Oral Biol. Med 11 (4) (2000) 437–466. [DOI] [PubMed] [Google Scholar]
- [18].Nanci A, Fortin M, Ghitescu L, Endocytotic functions of ameloblasts and odontoblasts: immunocytochemical and tracer studies on the uptake of plasma proteins, Anat. Rec 245 (2) (1996) 219–234. [DOI] [PubMed] [Google Scholar]
- [19].Nanci A, Zalzal S, Kogaya Y, Cytochemical characterization of basement membranes in the enamel organ of the rat incisor, Histochemistry 99 (4) (1993) 321–331. [DOI] [PubMed] [Google Scholar]
- [20].Peacock M, Calcium metabolism in health and disease, Clin. J. Am. Soc. Nephrol 5 (Suppl. 1) (2010) S23–30. [DOI] [PubMed] [Google Scholar]
- [21].Bonucci E, et al. , Morphological studies of hypomineralized enamel of rat pups on calcium-deficient diet, and of its changes after return to normal diet, Anat. Rec 239 (4) (1994) 379–395. [DOI] [PubMed] [Google Scholar]
- [22].Nanci A, et al. , Morphological and immunocytochemical analyses on the effects of diet-induced hypocalcemia on enamel maturation in the rat incisor, J. Histochem. Cytochem 48 (8) (2000) 1043–1058. [DOI] [PubMed] [Google Scholar]
- [23].Acevedo AC, et al. , Morphological study of amelogenesis in the rat lower incisor after thyro-parathyroidectomy, parathyroidectomy and thyroidectomy, Cell Tissue Res 283 (1) (1996) 151–157. [DOI] [PubMed] [Google Scholar]
- [24].Berdal A, et al. , Cell- and stage-specific expression of vitamin D receptor and calbindin genes in rat incisor: regulation by 1,25-dihydroxyvitamin D3, Dev. Biol 155 (1) (1993) 172–179. [DOI] [PubMed] [Google Scholar]
- [25].Descroix V, et al. , Physiopathology of dental rickets in vitamin D receptor-ablated mice, J. Dent. Res 89 (12) (2010) 1427–1432. [DOI] [PubMed] [Google Scholar]
- [26].Gyll J, et al. , Vitamin D status and dental caries in healthy Swedish children, Nutr. J 17 (1) (2018) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Berdal A, et al. , Calbindins D-9kDa and −28kDa and enamel secretion in vitamin D-deficient and control rats, Connect. Tissue Res 22 (1–4) (1989) 165–171. [PubMed] [Google Scholar]
- [28].Berridge MJ, Vitamin D deficiency accelerates ageing and age-related diseases: a novel hypothesis, J. Physiol 595 (22) (2017) 6825–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yamaguti PM, et al. , Amelogenesis imperfecta in familial hypomagnesaemia and hypercalciuria with nephrocalcinosis caused by CLDN19 gene mutations, J. Med. Genet 54 (1) (2017) 26–37. [DOI] [PubMed] [Google Scholar]
- [30].Wang SK, et al. , FAM20A mutations associated with enamel renal syndrome, J. Dent. Res 93 (1) (2014) 42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nurbaeva MK, et al. , Dental enamel cells express functional SOCE channels, Sci. Rep 5 (2015) 15803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Vig M, et al. , CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry, Science 312 (5777) (2006) 1220–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Feske S, et al. , A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function, Nature 441 (7090) (2006) 179–185. [DOI] [PubMed] [Google Scholar]
- [34].Prakriya M, et al. , Orai1 is an essential pore subunit of the CRAC channel, Nature 443 (7108) (2006) 230–233. [DOI] [PubMed] [Google Scholar]
- [35].Prakriya M, Lewis RS, Store-operated calcium channels, Physiol. Rev 95 (4) (2015) 1383–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Smyth JT, et al. , Activation and regulation of store-operated calcium entry, J. Cell. Mol. Med 14 (10) (2010) 2337–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hoth M, Niemeyer BA, The neglected CRAC proteins: Orai2, Orai3, and STIM2, Curr. Top. Membr 71 (2013) 237–271. [DOI] [PubMed] [Google Scholar]
- [38].Trebak M, Putney JW Jr., ORAI Calcium Channels, Physiology (Bethesda) 32 (4) (2017) 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lacruz RS, Feske S, Diseases caused by mutations in ORAI1 and STIM1, Ann. N. Y. Acad. Sci 1356 (1) (2015) 45–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lian J, et al. , ORAI1 mutations abolishing store-operated Ca(2+) entry cause anhidrotic ectodermal dysplasia with immunodeficiency, J. Allergy Clin. Immunol (2017), 10.1016/j.jaci.2017.10.031 [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- [41].Eckstein M, et al. , Store-operated Ca2+ entry controls ameloblast cell function and enamel development, JCI Insight 2 (6) (2017) e91166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Splawski I, et al. , Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism, Cell 119 (1) (2004) 19–31. [DOI] [PubMed] [Google Scholar]
- [43].Papineau SD, Wilson S, Dentition abnormalities in a Timothy syndrome patient with a novel genetic mutation: a case report, Pediatr. Dent 36 (3) (2014) 245–249. [PubMed] [Google Scholar]
- [44].Wang Y, et al. , The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels, Science 330 (6000) (2010) 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ryazanova LV, et al. , TRPM7 is essential for Mg(2+) homeostasis in mammals, Nat. Commun 1 (2010) 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nakano Y, et al. , A critical role of TRPM7 as an Ion Channel protein in mediating the mineralization of the craniofacial hard tissues, Front. Physiol 7 (2016) 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Faouzi M, et al. , The TRPM7 channel kinase regulates store-operated calcium entry, J. Physiol 595 (10) (2017) 3165–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lacruz RS, et al. , Dental enamel formation and implications for oral health and disease, Physiol. Rev 97 (3) (2017) 939–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Robertson SYT, et al. , Multiple calcium export exchangers and pumps are a prominent feature of enamel organ cells, Front. Physiol 8 (2017) 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hubbard MJ, McHugh NJ, Mangum JE, Exclusion of all three calbindins from a calcium-ferry role in rat enamel cells, Eur. J. Oral Sci 119 (Suppl. 1) (2011) 112–119. [DOI] [PubMed] [Google Scholar]
- [51].Franklin IK, Winz RA, Hubbard MJ, Endoplasmic reticulum Ca2+-ATPase pump is up-regulated in calcium-transporting dental enamel cells: a non-housekeeping role for SERCA2b, Biochem. J 358 (Pt 1) (2001) 217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Takano Y, Enamel mineralization and the role of ameloblasts in calcium transport, Connect. Tissue Res 33 (1–3) (1995) 127–137. [DOI] [PubMed] [Google Scholar]
- [53].Okumura R, et al. , Sodium-calcium exchangers in rat ameloblasts, J. Pharmacol. Sci 112 (2) (2010) 223–230. [DOI] [PubMed] [Google Scholar]
- [54].Schnetkamp PP, The SLC24 Na+/Ca2+-K+ exchanger family: vision and beyond, Pflugers Arch 447 (5) (2004) 683–688. [DOI] [PubMed] [Google Scholar]
- [55].Hu P, et al. , Expression of the sodium/calcium/potassium exchanger, NCKX4, in ameloblasts, Cells Tissues Organs 196 (6) (2012) 501–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Parry DA, et al. , Identification of mutations in SLC24A4, encoding a potassium-dependent sodium/calcium exchanger, as a cause of amelogenesis imperfecta, Am. J. Hum. Genet 92 (2) (2013) 307–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wang S, et al. , STIM1 and SLC24A4 are critical for enamel maturation, J. Dent. Res 93 (7 suppl) (2014) 94S–100S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Herzog CR, et al. , Hypomaturation amelogenesis imperfecta caused by a novel SLC24A4 mutation, Oral Surg. Oral Med. Oral Pathol. Oral Radiol 119 (2) (2015) e77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Seymen F, et al. , Exonal deletion of SLC24A4 causes hypomaturation amelogenesis imperfecta, J. Dent. Res 93 (4) (2014) 366–370. [DOI] [PubMed] [Google Scholar]
- [60].Pedersen NC, Shope B, Liu H, An autosomal recessive mutation in SCL24A4 causing enamel hypoplasia in Samoyed and its relationship to breed-wide genetic diversity, Canine Genet. Epidemiol 4 (2017) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Jalloul AH, et al. , A functional study of mutations in K+-dependent Na+-Ca2+ exchangers associated with Amelogenesis imperfecta and non-syndromic oculocutaneous albinism, J. Biol. Chem 291 (25) (2016) 13113–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Bronckers AL, Jalali R, Lytton J, Reduced protein expression of the Na(+)/Ca(2+)+K(+)-exchanger (SLC24A4) in apical plasma membranes of maturation ameloblasts of fluorotic mice, Calcif. Tissue Int 100 (1) (2017) 80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Stephan AB, et al. , The Na(+)/Ca(2+) exchanger NCKX4 governs termination and adaptation of the mammalian olfactory response, Nat. Neurosci 15 (1) (2011) 131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Li XF, Lytton J, An essential role for the K+−dependent Na+/Ca2+−exchanger, NCKX4, in melanocortin-4-receptor-dependent satiety, J. Biol. Chem 289 (37) (2014) 25445–25459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Vinberg F, et al. , The Na(+)/Ca(2+), K(+) exchanger NCKX4 is required for efficient cone-mediated vision, elife 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hubbard MJ, Abundant calcium homeostasis machinery in rat dental enamel cells. Up-regulation of calcium store proteins during enamel mineralization implicates the endoplasmic reticulum in calcium transcytosis, Eur. J. Biochem 239 (3) (1996) 611–623. [DOI] [PubMed] [Google Scholar]
- [67].Brookes SJ, et al. , Amelogenesis imperfecta caused by N-terminal enamelin point mutations in mice and men is driven by endoplasmic reticulum stress, Hum. Mol. Genet 26 (10) (2017) 1863–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Barron MJ, et al. , A mutation in the mouse Amelx tri-tyrosyl domain results in impaired secretion of amelogenin and phenocopies human X-linked amelogenesis imperfecta, Hum. Mol. Genet 19 (7) (2010) 1230–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Brookes SJ, et al. , Endoplasmic reticulum stress in amelogenesis imperfecta and phenotypic rescue using 4-phenylbutyrate, Hum. Mol. Genet 23 (9) (2014) 2468–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lasorsa FM, et al. , Peroxisomes as novel players in cell calcium homeostasis, J. Biol. Chem 283 (22) (2008) 15300–15308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Stelzig I, et al. , Peroxisomes in dental tissues of the mouse, Histochem. Cell Biol 140 (4) (2013) 443–462. [DOI] [PubMed] [Google Scholar]
- [72].Ratbi I, et al. , Heimler syndrome is caused by hypomorphic mutations in the peroxisome-biogenesis genes PEX1 and PEX6, Am. J. Hum. Genet 97 (4) (2015) 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Concepcion AR, et al. , Store-operated Ca2+ entry regulates Ca2+-activated chloride channels and eccrine sweat gland function, J. Clin. Invest 126 (11) (2016) 4303–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Smith CE, Warshawsky H, Multinucleate ameloblasts in the rat incisor, Anat. Rec 188 (4) (1977) 407–415. [DOI] [PubMed] [Google Scholar]
- [75].Tew KD, et al. , The role of glutathione S-transferase P in signaling pathways and S-glutathionylation in cancer, Free Radic. Biol. Med 51 (2) (2011) 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wang J, et al. , Reversible glutathionylation regulates actin polymerization in A431 cells, J. Biol. Chem 276 (51) (2001) 47763–47766. [DOI] [PubMed] [Google Scholar]
- [77].Townsend DM, S-glutathionylation: indicator of cell stress and regulator of the unfolded protein response, Mol. Interv 7 (6) (2007) 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Sui W, Boyd C, Wright JT, Altered pH regulation during enamel development in the cystic fibrosis mouse incisor, J. Dent. Res 82 (5) (2003) 388–392. [DOI] [PubMed] [Google Scholar]
- [79].Chang EH, et al. , Enamel pathology resulting from loss of function in the cystic fibrosis transmembrane conductance regulator in a porcine animal model, Cells Tissues Organs 194 (2–4) (2011) 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Bronckers AL, et al. , Composition of mineralizing incisor enamel in cystic fibrosis transmembrane conductance regulator-deficient mice, Eur. J. Oral Sci 123 (1) (2015) 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Hayashi Y, et al. , Organic-inorganic relationships, and immunohistochemical localization of amelogenins and enamelins in developing enamel, Basic Appl. Histochem 30 (3) (1986) 291–299. [PubMed] [Google Scholar]
- [82].Wang W, et al. , Reversible silencing of CFTR chloride channels by glutathionylation, J. Gen. Physiol 125 (2) (2005) 127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Hawkins BJ, et al. , S-glutathionylation activates STIM1 and alters mitochondrial homeostasis, J. Cell Biol 190 (3) (2010) 391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Lacruz RS, et al. , The sodium bicarbonate cotransporter (NBCe1) is essential for normal development of mouse dentition, J. Biol. Chem 285 (32) (2010) 24432–24438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Wright JT, Carrion IA, Morris C, The molecular basis of hereditary enamel defects in humans, J. Dent. Res 94 (1) (2015) 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Schossig A, et al. , SLC13A5 is the second gene associated with Kohlschutter-Tonz syndrome, J. Med. Genet 54 (1) (2017) 54–62. [DOI] [PubMed] [Google Scholar]
- [87].Goodman A, JC R, Assessment of systemic physiological perturbations from dental enamel hypoplasias and associated histological structures, Yearb. Phys. Anthropol 33 (1990) 59–110. [Google Scholar]