

Abstract
Protein arginine methyltransferases (PRMTs) are important therapeutic targets, playing a crucial role in the regulation of many cellular processes and being linked to many diseases. Yet, there is still much to be understood regarding their functions and the biological pathways in which they are involved, as well as on the structural requirements that could drive the development of selective modulators of PRMT activity. Here we report a deconstruction–reconstruction approach that, starting from a series of type I PRMT inhibitors previously identified by us, allowed for the identification of potent and selective inhibitors of PRMT4, which regardless of the low cell permeability show an evident reduction of arginine methylation levels in MCF7 cells and a marked reduction of proliferation. We also report crystal structures with various PRMTs supporting the observed specificity and selectivity.
Introduction
The post-translational methylation of the guanidinium group of arginine residues of histone and nonhistone proteins by protein arginine methyltransferases (PRMTs) plays a fundamental role in many key cellular functions, including gene regulation, signal transduction, RNA processing, and DNA repair.1−7 On the other hand, the aberrant expression of PRMTs or the dysregulation of PRMT activity is associated with several diseases, including many types of cancer.1,3,5,8−11
On the basis of their methylation products, monomethylarginine (Rme1),12 asymmetric dimethylarginine (Rme2a), or symmetric dimethylarginine (Rme2s),1,8 the nine PRMT isoforms identified in human genome to date are classified into three subfamilies:13 type I PRMTs (PRMT1, PRMT2, PRMT3, PRMT4, PRMT6, and PRMT8), catalyzing mono- and asymmetric dimethylation, type II PRMTs (PRMT5 and PRMT9), catalyzing mono- and symmetric dimethylation, and PRMT7, the sole member of type III, which only catalyzes the formation of Rme1.14 Arginine methylation and PRMTs have been associated with a variety of diseases, including cancer and neurological and inflammatory diseases.13
Also, viral proteins from several viruses are methylated by PRMTs,15−19 including SARS-CoV-2 nucleocapsid (N) protein, the methylation of which at residues R95 and R177 is crucial for viral replication.20 Indeed, over the past 15 years the medicinal chemistry community has paid a growing attention to PRMTs,11,13,21−26 in particular to PRMT5 (with a few inhibitors in clinical trials)21,27−33 and to type I enzymes (both pan-type I22,34−36 and selective24,37−45).
In our early studies in the field,46 we developed a series of type I PRMT inhibitors starting from 7,7′-(carbonylbis(azanediyl))bis(4-hydroxynaphthalene-2-sulfonic acid) 1 (AMI-1, Chart 1). In particular, the isosteric bis-4-hydroxy-2-naphthoic acid 2 (EML108, Chart 1) was able to prevent arginine methylation of cellular proteins in whole-cell assays, with activities comparable to AMI-1 (or even better than it). Moreover, compound 2 and its derivatives were found to be selective for arginine methyltransferases and essentially inactive against the lysine methyltransferase SET7/9.47
Chart 1. Structure of the Cofactor SAM, the Byproduct SAH, and Representative Inhibitors of PRMTsa.

a Similar moieties (see text) are depicted in the same blue or red color.
On the basis of molecular modeling studies (docking and binding mode analysis, confirmed by structure-based 3-D QSAR models),46,48 we found that such inhibitors, as well AMI-1, bind PRMT1 (at that time chosen as representative of type I enzymes) between the S-adenosine-l-methionine (SAM) cofactor and substrate arginine binding sites without entirely occupying them. In particular, the binding site of the Arg guanidine group as well as both the adenosine and the methionine ends of the SAM binding pocket seems to be largely unoccupied (Figure 1). This is consistent with previously reported kinetics experiments.49 Therefore, a further decoration of the scaffold of compound 2 aimed to better occupy these pockets could, in principle, result in a gain of affinity.
Figure 1.
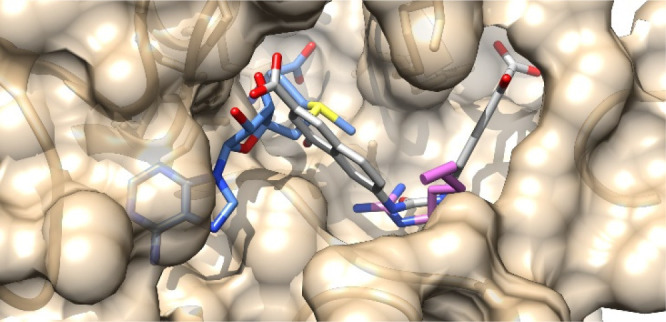
Predicted binding mode of 2 (EML108, in sticks, carbon atoms in gray) into PRMT1 (tan) catalytic site (PDB ID: 1OR8).46−48 SAM is depicted in cornflower blue, histone Arg in orchid (for clarity, only the side chain is shown).
Soon after our studies, Epizyme reported on the development of compound 3 (EPZ004777, Chart 1), a potent and selective inhibitor of lysine methyltransferase DOT1L based on the chemical structures of SAM and the corresponding product (S-adenosylhomocysteine, SAH) of DOT1L catalysis reaction as well as on its mechanism.50 We were intrigued by the structural similarity between the 4′-alkylphenylurea in this compound and the N-naphthylurea moiety of compound 2. Although showing a remarkable selectivity against other histone methyltransferases, compound 3 was confirmed to inhibit also PRMT5 and PRMT7 (with IC50 values of 0.52 and 7.5 μM, respectively).51 Similarly, the amidic derivatives 4a and 4b (Chart 1) of the nonspecific SAM-dependent enzyme inhibitors sinefungin and 6′-homosinefungin were recently reported as PRMT inhibitors, with IC50 values against PRMT4 of 43 nM and 1.9 μM, respectively.52 Again, just like the 4′-alkylphenylurea in compound 3, the N-phenethylamide portion in compounds 4a and 4b resembles the naphthylurea (in red in Chart 1) and mimics the lysine substrate covalently linked to a SAM-like moiety (in blue in Chart 1). It is noteworthy that 4b inhibits modestly (IC50 = 1.9 μM) but selectively PRMT4 with no appreciable activity on other PRMTs. On the other hand, 4a, which differs from 4b only by the methylene group bridging the C5 position of the hexanamide with the benzylamine nitrogen, shows a dramatic increase of potency against PRMT4 (IC50 of 43 nM) but maintains a definite activity against type II and III PRMTs. This supports the hypothesis that the distance between the pharmacophoric moieties that are correlated with PRMTs inhibition plays a key role in potency and selectivity.
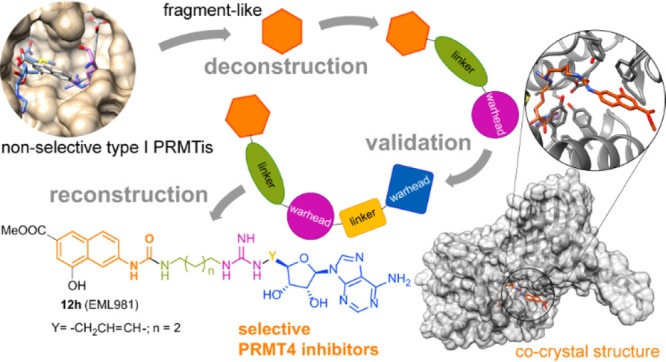
Based on these considerations and pursuing our interest in the identification of potent and selective PRMT inhibitors,7,44,46,47,53−59 we resolved to reduce the structure of 2 to a single N-naphthylurea moiety and to grow it into a more complex structure incorporating warheads able to better bind the above-mentioned available pockets. The derivatives resulting from this design concept (Figure 2) were screened using in vitro biochemical assays to study their potency and selectivity in the inhibition of the various PRMTs. Herein, we report the design and synthesis of such compounds and the identification of 12h (EML981) as a potent and selective inhibitor of PRMT4/CARM1. We also report cocrystallization studies supporting the observed specificity and selectivity of compound 12h.
Figure 2.

Flowchart of our design strategy.
Results and Discussion
Design Strategy
Our previous molecular modeling studies46,48 performed on compound 2 and its derivatives had suggested a binding mode between the cofactor and substrate binding pockets, without fully occupying them. In particular, one of the two 4-hydroxy-2-naphthoic moieties partially occupies the substrate binding site without establishing interactions with the two conserved glutamate residues of the so-called “double-E loop” critical for chelating and orienting the Arg guanidine group.60 On the other hand, the binding of the second 4-hydroxy-2-naphthoic moiety seems to leave the SAM binding pocket largely unoccupied (Figure 1). Therefore, we decided to base our design strategy, schematically depicted in Figure 2, on a “deconstruction–reconstruction” approach that has gained traction in recent years.61−63 The concept underlying this approach is simple: since traditional fragment-based drug discovery (FBDD) combines fragments into a final molecule,64 it is typically possible to deconstruct a known ligand to obtain a relatively smaller fragment library.65,66 Therefore, we deconstructed our previously identified PRMTs inhibitors and decided to extend the resulting naphthylamide fragment using a growing approach.67
We then designed a series of derivatives (Figure 3) incorporating the 4-hydroxy-2-naphthoic group bridged by an amide or urea group with an arginine surrogate (arginine mimetic moiety). The effect of the introduction of a methionine was also explored. The compounds (5–11, Table 1) were synthesized and, at first, analyzed for known classes of assay interference compounds.68 All derivatives were not recognized as PAINS according to the SwissADME web tool (http://www.swissadme.ch),69 the Free ADME-Tox Filtering Tool (FAF-Drugs4) program (http://fafdrugs4.mti.univ-paris-diderot.fr/),70 and the “False Positive Remover” software (http://www.cbligand.org/PAINS/)71 nor as aggregators according to the software “Aggregator Advisor” (http://advisor.bkslab.org/).72
Figure 3.

Compounds designed for the first reconstruction step.
Table 1. Inhibitory Activities of Compounds 5–11 against hPRMT1.

AlphaLISA was used for both fixed dose and IC50 determinations against human recombinant PRMT1 (0.9 nM, final concentration). Histone H4 (1–21) peptide, biotinylated (100 nM, final concentration), and SAM (2 μM, final concentration) were used as substrate and cofactor, respectively.
Compounds were tested at a 100 μM fixed concentration.
Enzyme residual activity percentage calculated with respect to DMSO.
Compounds were tested in 10-concentration IC50 mode with threefold serial dilutions starting at 100 μM. Data were analyzed with GraphPad Prism software (version 6.0) for IC50 curve fitting.
Ligand efficiency (LE) calculated from IC50 as a surrogate for KD.
ND, not determined.
Then, we resolved to determine their effect on the catalytic activity of human recombinant PRMT1, chosen as representative of class I PRMTs. To this aim, we used an in-house peptide-based AlphaLISA assay measuring the levels of H4R3me, developed by us (see the Experimental Section) because the commercially available homogeneous assay kit (BPS, #52054) did not work properly in our hands and gave an unacceptably narrow assay window. All the compounds were tested at a fixed concentration of 100 μM using 2 (EML108) as the reference compound. Then, the compounds that displayed a greater than 85% inhibition (residual enzyme activity <15%) were selected, and the corresponding IC50 values were determined (Table 1).
Overall, the results show that one of the two 4-hydroxy-2-naphthoic groups could be efficiently replaced by an arginine-mimetic group moiety. This result is consistent with our initial assumption that our PRMTs inhibitors bind PRMT1 between the SAM and arginine binding sites without fully occupying them.
Structure–activity relationship (SAR) analysis of tested compounds suggested that, in general, ester derivatives are better than the corresponding acids (for example, compare inhibiting activities of compounds 5a, 5b, 5d, and 5e with those of 6a–6d, respectively) and urea derivatives are more active (compare 7a–7d with 5a–5d, respectively) or comparable (7e vs 5e) to their amide counterparts. The presence of a single methyl on one of the terminal nitrogen atoms of the guanidine group also improves the activity in the amide series (compare 5d and 5e with 5a and 5b, respectively), whereas this is less evident in the urea series (compare 7d and 7e with 7a and 7b, respectively) and in carboxylic acids with respect to ester derivatives. On the contrary, the introduction of a second methyl group on the same nitrogen atom leads to a decrease of inhibiting activity (compare 5f with 5d). When both terminal nitrogens are substituted and blocked into an imidazoline ring (as in the case of compounds 5g and 6f), the inhibiting activity is even lower. Similarly, in the urea series the replacement of the guanidine group with an imidazole leads to the loss of activity (compare 7f with 7a). Regarding the effect of the methylene linker length, in the amide series the activity seems to increase with the number of carbon atoms of the linker (see, for example, the activities of compounds 5a–5c and 7a–7c) at least for ester derivatives, whereas for the corresponding acids the effect is less evident (6a vs 6b) or opposite (6c vs 6d). On the other hand, an “odd/even effect”73−75 seems to occur in the urea series, where an even number of carbon atoms in the alkyl spacer (as in the case of 7b and 7e, n = 2, total carbon atoms in the linker = 4) is less favorable than an odd number (as in the case of 7a and 7d—n = 1, total carbon atoms in the linker = 3—or 7c—n = 3, total carbon atoms in the linker = 5).
The introduction of an α-amino group (as in derivative 11c) does not improve the activity of the compound. On the contrary, the introduction at the same position of a p-methoxyphenylurea resulted in compounds with improved inhibiting capability, even if compared to the α-unsubstituted compounds (compare 11e with 11c and 5a). Last, the removal of the guanidine group is detrimental for the inhibiting activity and cannot be compensated by the sole presence of a p-methoxyphenylurea in the α position relative to the amide carbonyl group. In fact, both the methionine and glycine derivatives 9c, 9d, and 10c are inactive or scarcely active. Noteworthy, the effects appear to be additive. In fact, the ester derivative of the urea series, with three carbon atoms in the linker and a methyl group on the terminal nitrogen of the guanidine, namely compound 7d, is the most active and the most efficient ligand among the tested compounds, showing a submicromolar IC50 value (0.4 μM) and a ligand efficiency76 value (LE) of 0.33.
Therefore, on the basis of the structure–activity relationships, we resolved to select the scaffold of the latter compound for the second step of the construction of our multisubstrate ligands. Considering the effect of the linker length on the activity against PRMT1 and also with the aim to explore a possible difference among various PRMTs, first we designed and synthesized three derivatives in which the linker between the urea and the guanidine groups included three, four, or five carbon atoms and the adenosine moiety was connected to a guanidine ω-nitrogen through a methylene group (Figure 4, derivatives 12a, 12b, and 12c, respectively).
Figure 4.

Compounds designed for the second fragment-growing step.
As in the case of derivatives 5–11, we first determined the effect of compounds 12a–12c on the catalytic activity of human recombinant PRMT1, using our AlphaScreen assay. As expected, the introduction of the adenosine moiety increased the inhibiting activity against PRMT1 as all the derivatives showed IC50 values in the submicromolar range (Table 2, values in parentheses). Prompted by these results, we next used a secondary screening approach to profile the activity of the compounds against a panel of four PRMTs (PRMT1, PRMT3, PRMT4, and PRMT5) using a radioisotope-based filter-binding assay. If compared to the results obtained from the AlphaScreen assay, in the more sensitive radioisotope-based assay compounds 12a–12c were found to be less potent inhibitors of PRMT1 activity, but the IC50 values were still in the micromolar range (Table 2). Noteworthy, they were appreciably more active against PRMT4. We also noticed that the distance between the methyl 4-hydroxy-2-naphthoate moiety and the arginine-mimetic group significantly affects the inhibition of PRMT4 activity, whereas it has no significant effect on the activity of PRMT5 and only a moderate effect on PRMT1 and PRMT3. In fact, compound 12c exhibits a submicromolar activity (IC50 = 0.42 μM) only against PRMT4, with a selectivity for the latter ranging from 20-fold (over PRMT1; Table 2) to 122-fold (over PRMT5; Table 2). Based on these outcomes, we speculated that more potent and selective PRMT4 inhibitors could be generated by modulating the distance between the three pharmacophoric moieties. Therefore, we synthesized a second set of derivatives (compounds 12d–12h, Figure 4) in which the distance between the 4-hydroxy-2-naphthoate moiety and the guanidine group (compound 12d and 12e) or the distance between the guanidine group and the adenosine moiety (compounds 12f–12h) was further increased. Again, all compounds 12a–12h were analyzed for known classes of assay interference compounds68 and were not recognized as PAINS nor as aggregators. The compounds were then tested against a wider panel of PRMTs (including also PRMT6, PRMT7, and PRMT8) using the same radioisotope-based filter-binding assay. The results are reported in Table 2 and summarized as heatmaps in Figure 5. As expected, increasing the linker length between the 4-hydroxy-2-naphthoate and the guanidine group up to a total of four or five carbon atoms yielded a gain in the inhibitory activity against PRMT4, although moderate (6.2-fold for 12d and 2.4-fold for 12e). However, a similar gain was observed also against PRMT1, PRMT5, and PRMT6 (e.g., 9.8-fold and 3.2-fold against PRMT1 and 19.8-fold and 15.2-fold against PRMT5, for 12d and 12e, respectively), with a consequent reduction of selectivity. A lesser effect was observed against PRMT8, and a slight decrease of inhibiting activity was observed against PRMT7. On the other hand, we were pleased to find out that compounds 12f–12h, featuring an increased distance between the guanidine group and the adenosine moiety, showed a remarkable and selective increase in potency against PRMT4, with a consistent gain in selectivity over six other human PRMTs. In particular, compound 12h (EML981) showed a 668-fold gain in potency over its shorter counterpart 12b and 261- to 1266-fold selectivity over the other PRMTs.
Table 2. Inhibitory Activities of Compounds 12a–12h against Various PRMTs.
| IC50 (μM)a,b |
selectivity index for PRMT4 vs other
PRMTsc |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| no. | PRMT1 | PRMT3 | PRMT4 | PRMT5 | PRMT6 | PRMT7 | PRMT8 | PRMT1 | PRMT3 | PRMT5 | PRMT6 | PRMT7 | PRMT8 | LEd |
| 12a | 32.27 (0.5)e | 57.19 | 13.84 | 52.13 | 72.77 | 0.32 | 8.29 | 2 | 4 | 4 | 5 | 0.02 | 1 | 0.14 |
| 12b | 11.67 (0.43)e | 43.41 | 2.14 | 76.7 | >100 | 0.55 | 18.68 | 5 | 20 | 36 | >47 | 0.26 | 9 | 0.18 |
| 12c | 8.46 (0.3)e | 21.26 | 0.42 | 51.41 | >100 | 0.22 | 5.87 | 20 | 51 | 122 | >238 | 0.52 | 14 | 0.19 |
| 12d | 0.86 | 12.2 | 0.068 | 2.6 | 47 | 0.41 | 3.82 | 13 | 179 | 38 | 691 | 6 | 56 | 0.21 |
| 12e | 2.6 | 12.4 | 0.176 | 3.38 | 35.6 | 0.631 | 5.84 | 15 | 70 | 19 | 202 | 4 | 33 | 0.20 |
| 12f | 4.86 | 10.5 | 0.0097 | 0.115 | 22.7 | 0.226 | 6 | 501 | 1082 | 12 | 2340 | 23 | 619 | 0.24 |
| 12g | 1.80 | 13.3 | 0.0084 | 0.778 | 4.34 | 4.68 | 1.41 | 214 | 1583 | 93 | 517 | 557 | 168 | 0.24 |
| 12h | 0.835 | 4.05 | 0.0032 | 1.46 | 1.75 | 1.68 | 1.95 | 261 | 1266 | 456 | 547 | 525 | 609 | 0.25 |
Compounds were tested in 10-concentration IC50 mode with threefold serial dilutions starting at 100 μM. Data were analyzed with GraphPad Prism software (version 6.0) for IC50 curve fitting.
Unless differently indicated, the values were obtained in a radioisotope-based filter assay, using 5 μM histone H4 (for PRMT1, PRMT3, and PRMT8), histone H3 (for PRMT4), histone H2A (for PRMT5), or GST-GAR (for PRMT6 and PRMT7) as the substrate and S-adenosyl-l-[methyl-3H]methionine (1 μM) as methyl donor.
Selectivity index for PRMT4 over the specified PRMT, calculated as the ratio between the IC50 against the specified PRMT and the IC50 against PRMT4 and rounded to the nearest integer.
Ligand efficiency (LE) for PRMT4 calculated from IC50 as a surrogate for KD.
Obtained in the AlphaLISA assay, using human recombinant PRMT1 (0.9 nM, final concentration). Histone H4 (1–21) peptide, biotinylated (100 nM, final concentration), and SAM (2 μM, final concentration) were used as the substrate and cofactor, respectively.
Figure 5.

Inhibitory activities of compounds 12a–12h: the heatmaps depict the IC50 values (nM) for compounds 12a–12h (top panel) and the selectivity index (fold) for PRMT4 over the specified PRMT (bottom).
The selectivity of compound 12h was further assessed against a panel of eight lysine methyltransferases (KMTs), including the SET-domain-containing proteins ASH1L/KMT2H, EZH2/KMT6, MLL1/KMT2A, SET7/9/KMT7, SETD8/KMT5A, SUV39H2/KMT1B, and SUV420H1/KMT5B and the non-SET-domain-containing DOT1L/KMT4.77 To this aim, the inhibition of 12h toward these selected enzymes was assessed at two different concentrations (1 and 10 μM, respectively, >300 and >3000 fold higher than the IC50 value against PRMT4) using SAH,78−80 chaetocin (for ASH1L),81 or ryuvidine (for SETD8)82 as reference compounds. Noteworthy, we found that none of the enzymes was inhibited by 12h even at the higher tested concentration (Figure S2 and Table S1, Supporting Information).
SPR-Based Studies of Binding to PRMT4
First identified as a transcriptional regulator,83 PRMT4, also known as coactivator-associated arginine methyltransferase 1 (CARM1), is a type I enzyme that regulates gene expression by numerous mechanisms. It positively regulates transcription by methylating H3R17 and H3R26,84,85 methylates steroid receptor coactivators including SRC3 and CBP/p300, and can directly act as a transcriptional coactivator of nuclear receptors.86,87 PRMT4 also methylates a variety of other targets, including splicing factors such as CA150,91 to regulate the exon skipping, and RNA-binding proteins (e.g., PABP1, HuR, and HuD),88−90 to modify their ability to bind to the transcription-related proteins. PRMT4 also regulates the looping of enhancers and promoters by methylating MED12, which is a component of the mediator.92
It has been demonstrated that PRMT4 is overexpressed in various cell lines of hematologic cancers and solid tumors, such as leukemia,93,94 breast,95 prostate,96 liver,97 and colorectal cancers.98,99 Moreover, the enzyme is overexpressed in ischemic hearts and hypoxic cardiomyocytes, and it has been suggested that PRMT4 has an essential role in myocardial infarction and cardiomyocyte apoptosis.100 Other emerging functions of PRMT4 include autophagy, metabolism, early development, pre-mRNA splicing and export, and localization to paraspeckles.101 Also, it was recently found that in lymphomas that carry mutation in p300/CBP, PRMT4 loss or inhibition is a vulnerability.102 Therefore, PRMT4 is considered an appealing therapeutic target for anticancer drug development, and in fact a few inhibitors have been developed with different degrees of potency and selectivity.24,41,52,103
To further characterize the effect of compounds 12a–12h on PRMT4, we resolved to evaluate their direct binding to the target protein using Surface Plasmon Resonance (SPR). To this aim, human PRMT4 (full length) was covalently immobilized on a sensor chip surface using an amine-coupling approach, and the three compounds were injected at different concentrations over the protein surface. To reduce false positives from detergent-sensitive, nonspecific aggregation-based binding, detergents (0.05% Tween-20) were added to the running buffer in all experiments. A specific and strong binding interaction was demonstrated between PRMT4 and each compound, with equilibrium dissociation constant (KD) values in the nanomolar range for the most active derivatives 12f–12h (Figure 6 and Table 3). Compound 12f interacts with PRMT4 with higher affinity (KD = 25.2 nM; Table 3) compared to 12g (KD = 51.7 nM) and 12h (KD = 75.9 nM). As shown by the sensorgrams depicted in Figure 6 and Figure S1 (Supporting Information), compounds 12f–12h dissociate from the protein slower than compounds 12a–12e. Nonetheless, the in vitro residence time (τR)104 values are quite similar (Table 3), in particular for compounds 12b, 12c, 12e, 12g, and 12h, and cannot account alone for the higher potency of compounds 12g and 12h. On the contrary, the association rate constants (Kon) certainly contribute to the affinity for the target, being significantly higher for compounds 12f–12h than for compounds 12a–12e (Table 3).
Figure 6.

Sensorgrams obtained from the SPR interaction analysis of compounds 12a and 12f–12h (panels a–d, respectively) binding to immobilized PRMT4. Each compound was injected at different concentrations (from 3 to 0.05 μM for 12a and from 640 to 10 nM for 12f–12h) with an association and a dissociation time of 90 s, with a flow rate of 30 μL/min. The equilibrium dissociation constants (KD) were derived from the ratio between kinetic dissociation (koff) and association (kon) constants.
Table 3. Affinity and Kinetic Parameters Derived from SPR Experiments.
| compound | KD (nM) | kon (1/Ms) | koff (1/s) | τR (s) |
|---|---|---|---|---|
| 12a | 663.4 | 1.67 × 104 | 0.011 | 90.9 |
| 12b | 2300 | 6.52 × 103 | 0.015 | 66.7 |
| 12c | 359.2 | 3.49 × 104 | 0.013 | 76.9 |
| 12d | 2800 | 2.70 × 105 | 0.760 | 1.31 |
| 12e | 603.4 | 2.58 × 104 | 0.015 | 66.7 |
| 12f | 25.2 | 2.70 × 106 | 0.068 | 14.7 |
| 12g | 51.7 | 0.25 × 106 | 0.013 | 76.9 |
| 12h | 75.9 | 0.21 × 106 | 0.016 | 62.5 |
| SAM | 9.6 | 2.90 × 105 | 0.003 | 333.3 |
Structural Studies
Then we resolved to study and compare the binding modes of compounds 12a–12h with different PRMTs. In particular, we resolved to compare compounds 12a–12c (less potent and selective PRMT4 inhibitors in the series 12, upper part of the heatmaps in Figure 5) and compounds 12f–12h (most potent and selective PRMT4 inhibitors of the series, lower part of the heatmaps in Figure 5) in cocrystallization studies performed using five PRMTs, namely isolated domains of mmPRMT4 (Mus musculus PRMT4, residues 130–487 or 140–497), full length and truncated Rattus norvegicus PRMT1, full length Mus musculus PRMT2, mmPRMT6 (Mus musculus PRMT6, residues 34–378), and full length Mus musculus PRMT7.
Unfortunately, no cocrystals were obtained with PRMT1, PRMT2, or PRMT7. On the contrary, we were able to cocrystallize the compounds in the complex with both PRMT4 and PRMT6. Crystallization, data collection, and structure refinement are fully described in the Experimental Section.
In the case of the complexes with PRMT4, all structures were solved and refined (depending on crystals, resolution ranged from 2.1 to 2.4 Å at ESRF or SOLEIL synchrotron beamlines) in the space group P21212 and contain one copy of the PRMT4 tetramer in the asymmetric unit, as previously described.106 In all cases, each PRMT4 monomer binds one molecule of the ligand (Figure S2, Supporting Information). Crystallographic statistics are summarized in Table 4. The electron density maps obtained in the cocrystallization studies with compounds 12a–12c and 12f–12h reveal the conformation of each compound on all four monomers of the asymmetric unit (Figure 7). All six cocrystallized compounds (12a–12c and 12f–12h) adopt an overall similar conformation in the complex with PRMT4, revealing two common anchoring platforms, the methyl 4-hydroxy-2-naphthoate and the adenosine moieties, each one occupying the same binding site on PRMT4 regardless of the different linker lengths (Figure 8a and b). On the contrary, distinctive conformations are observed for the linker in each compound (see below for details).
Table 4. X-ray Data Collection and Refinement Statistics for PRMT4 Complexes with Compounds 12a–12c and 12f–12h.
| ligand | 12a (EML734) | 12b (EML709) | 12c (EML736) | 12f (EML980) | 12g (EML982) | 12h (EML981) |
| PDB ID | 7PV6 | 7PPY | 7PPQ | 7PU8 | 7PUQ | 7PUC |
| data processing | ||||||
| wavelength (Å) | 0.968 | 0.979 | 0.979 | 0.980 | 0.980 | 0.980 |
| resolution range (Å)a | 48.40–2.40 (2.46–2.40) | 42.53–2.42 (2.49–2.42) | 45.80–2.10 (2.13–2.10) | 48.07–2.19 (2.23–2.19) | 48.01–2.09 (2.12–2.09) | 46.06–2.19 (2.23–2.19) |
| space group | P21212 | P21212 | P21212 | P21212 | P21212 | P21212 |
| unit cell | 75.0 99.5 208.5 90 90 90 | 74.7 98.8 207.0 90 90 90 | 74.8 98.5 206.9 90 90 90 | 75.2 98.8 208.4 90 90 90 | 75.1 98.7 207.8 90 90 90 | 75.3 98.9 208.2 90 90 90 |
| total reflections | 424165 (30752) | 249037 (11755) | 382899 (10013) | 1073363 (40161) | 1237125 (48566) | 1072170 (46596) |
| unique reflections | 61974 (4383) | 58136 (3623) | 89784 (3974) | 80189 (3738) | 92014 (4053) | 80457 (3971) |
| multiplicity | 6.8 (7.0) | 4.3 (3.2) | 4.3 (2.5) | 13.4 (10.7) | 13.4 (12.0) | 13.3 (11.7) |
| completeness (%) | 99.7 (96.8) | 98.3 (80.0) | 99.1 (86.7) | 98.7 (81.5) | 99.5 (90.2) | 99.2 (87.0) |
| mean ⟨I/σI⟩b | 11.3 (1.1) | 8.8 (0.9) | 9.2 (0.9) | 14.4 (1.3) | 17.8 (1.8) | 11.1 (1.1) |
| resolution limit for ⟨I/σI⟩ > 2c | 2.63 | 2.69 | 2.33 | 2.24 | 2.13 | 2.39 |
| Wilson B-factor | 50.5 | 46.1 | 38.4 | 46.2 | 42.5 | 45.3 |
| Rmeas | 0.140 (2.177) | 0.131 (1.441) | 0.101 (1.282) | 0.104 (1.678) | 0.089 (1.387) | 0.164 (2.950) |
| CC1/2 | 0.999 (0.547) | 0.998 (0.441) | 0.998 (0.416) | 0.999 (0.543) | 1.000 (0.831) | 0.999 (0.511) |
| refinement | ||||||
| resolution range | 46.17–2.40 (2.49–2.40) | 42.53–2.42 (2.51–2.42) | 45.80–2.10 (2.17–2.10) | 46.09–2.19 (2.27–2.19) | 48.01–2.09 (2.16–2.09) | 46.06–2.19 (2.27–2.19) |
| Rwork | 0.191 (0.318) | 0.205 (0.339) | 0.194 (0.323) | 0.194 (0.322) | 0.181 (0.264) | 0.202 (0.351) |
| Rfree | 0.237 (0.338) | 0.235 (0.359) | 0.228 (0.351) | 0.229 (0.344) | 0.217 (0.304) | 0.230 (0.364) |
| number of non-hydrogen atoms | 11684 | 11303 | 11827 | 11483 | 11659 | 11408 |
| macromolecules | 11282 | 10971 | 11001 | 11000 | 11001 | 10993 |
| ligands | 455 | 436 | 398 | 368 | 410 | 342 |
| solvent | 165 | 104 | 614 | 283 | 442 | 223 |
| validation | ||||||
| RMS(bonds) | 0.006 | 0.010 | 0.004 | 0.006 | 0.005 | 0.009 |
| RMS(angles) | 0.75 | 1.32 | 0.60 | 0.79 | 0.78 | 1.21 |
| Ramachandran favored (%) | 97.08 | 96.47 | 96.70 | 97.14 | 96.77 | 97.58 |
| Ramachandran outliers (%) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| rotamer outliers (%) | 0.41 | 0.84 | 0.08 | 0.67 | 0.42 | 0.42 |
| average B-factor | 55.96 | 49.51 | 45.06 | 52.70 | 48.59 | 52.36 |
| macromolecules | 55.77 | 49.31 | 44.75 | 52.68 | 48.00 | 51.59 |
| ligands | 69.79 | 60.51 | 57.20 | 58.28 | 63.01 | 85.17 |
| solvent | 49.58 | 46.85 | 46.45 | 49.52 | 48.59 | 61.75 |
| Clashscore | 4.24 | 3.72 | 2.95 | 3.22 | 6.35 | 2.64 |
Values in parentheses correspond to the highest-resolution shell.
See ref (105) for crystallographic definitions.
The resolution limits for ⟨I/σI⟩ > 2 are reported.
Figure 7.

Electron density (2Fobs – Fcalc) weighted maps. Compound 12b (a) and compound 12h (b) bound to subunit B of mmPRMT4 (PDB IDs: 7PV6 and 7PUC). PRMT4 is represented as a gray cartoon, and compounds are represented as cornflower blue sticks. Maps are represented as a mesh, with the contouring level set to 1σ. For clarity, N-terminal helices (residues 135–155) of PRMT4 are not shown. E258 and E267 belonging to the double-E loop and H415 of the THW loop are also displayed as sticks.
Figure 8.

Structures of mmPRMT4 in complex with compounds 12a–12c and 12f–12h (PDB IDs: 7PV6, 7PPY, 7PPQ, 7PU8, 7PUQ, and 7PUC, respectively). (a) Superimposition (done on protein backbones) of compounds (12a, 12b, 12c, 12f, 12g, and 12h) bound to subunit B of mmPRMT4. Each PRMT4 subunit is represented as a cartoon (shades of gray, lime, cyan, marine, yellow, gray, and pink ribbons), and compounds are represented as sticks (in lime, yellow, cyan, cornflower blue, sea blue, and pink, respectively). (b) Close-up view of bound compound conformations. (c) Binding interactions of compound 12a (lime sticks) with mmPRMT4 monomer B (ribbon). (d) Binding interactions of compound 12h (pink sticks) with mmPRMT4 monomer B (ribbon). Hydrogen bonds are shown as dashed lines. For clarity, N-terminal helices (residues 135–165) of PRMT4 are not shown.
As expected, the adenosine moiety occupies the SAM binding site, and as previously described,106 the main interactions are with Y150, E215, E244, and M269 (Figure 8c and d).
On the other hand, the methyl 4-hydroxy-2-naphthoate moiety partially occupies the peptide substrate binding site on PRMT4 and mainly interacts with Y262, P473, F475, and Y477 on one side and F153 on the other side (Figure 8c and d). Interestingly, superimposition of the conformation of 12h cocrystallized with PRMT4 with the conformations of the PABP1 peptide transition state mimics recently developed by us44 allowed us to assess that the moiety occupies position −1 to −4 of the peptide substrate binding site, −1 being the position of the amino acid located at the N-terminal side of the arginine to be methylated (Figure S3, Supporting Information). Despite slight modifications observed among monomers inside a given tetramer, the crystal structures of compounds 12a–12c and 12f–12h revealed that the conformations of the protein side chains are identical for all compounds.
Surrounded by such a “frozen” binding site, the linker of each compound adopts a unique and distinctive conformation stabilized by interactions with the protein platform in a region mapped on one side by catalytic residues M260, E258, H415, and W416 and on the other side by F153, Y154, and E267 (Figure 8c and d and Figures S4 and S5, Supporting Information). Depending on the length and the nature of the linker, strong hydrogens bonds are established with such frozen sites, and the number and the strength of the binding interactions of each compound appear to be correlated with the affinity and, consequently, with the IC50 value. In fact, longer compounds 12f–12h showed IC50 values in the low nM ranges compared to the μM range observed for shorter compounds 12a–12c.
In the case of compound 12h, the best inhibitor herein reported, the guanidine group lies between the catalytic glutamate residues (E258 and E267) of the double-E loop in the binding site of the guanidine moiety of the peptide transition state, and the N5 atom establishes two hydrogen bonds with the oxygen atoms of the main chain of E258 and M260 (Figure 8d and Figure S10, Supporting Information).
As revealed by the crystal structures, at least a two-carbon atom long linker between the adenosine moiety and the guanidine group is required to bring the latter within the catalytic clamp formed by E258 and E267, with a three-carbon atom linker (as in compounds 12g and 12h) being even better. If the linker is shorter (as in the case of compounds 12a–12c), the two guanidine-stabilizing hydrogen bonds established with the PRMT4 main chain are lost, and this may account for a weaker affinity compared to derivatives 12f–12h. This observation is in agreement with an improvement in inhibition capability observed for compounds 12f–12h compared to compounds 12a–12c (see Table 2 above). In addition, the trans conformation imposed by the double bond in 12h is less constrained than the one adopted by 12g (also featuring a three-carbon atom linker between the adenosine moiety and the guanidine group) and, therefore, more favorable.
Regarding the linker between the naphthylurea moiety and the guanidine group, a length increase from three carbon atoms (n = 1, 12a) to four or five (n = 2, 12b, and n = 3, 12c, respectively) brings additional van der Waals interaction with W416 and Y262, thus substantiating an increase in affinity and a corresponding decrease in IC50 values.
In the case of the complexes with PRMT6, all structures were solved and refined (depending on crystals, resolution ranging from 1.65 to 2.3 Å) in the space group P21 with one copy of the PRMT6 dimer in the asymmetric unit as previously described.107 Structure determinations and refinements revealed that only compounds 12a, 12c, and 12f are visible in the active site of each monomer of the PRMT6 dimer.
For the complexes with the other compounds, a molecule of SAH (constitutively contained in the purified Mus musculus PRMT6) was observed in the active site. Crystallographic statistics are summarized in Table 5. In all the structures of the complexes with PRMT6, the electron density for the compound is always better in one monomer. Compound 12a is the only one for which a complete electron density is visible in one monomer of the PRMT6 structure (Figure 9), whereas for 12c and 12f the electron density becomes fragmented after the guanidine group and the density for the methyl-4-hydroxy-2-naphthoate moiety is weak. Moreover, for compound 12f, the active site of one monomer is occupied by both the compound and SAH.
Table 5. X-ray Data Collection and Refinement Statistics for PRMT6 Complexes with Compounds 12a, 12b, and 12f.
| ligand | 12a (EML734) | 12c (EML736) | 12f (EML980) |
| PDB ID | 7NUD | 7NUE | 7P2R |
| data processing | |||
| wavelength (Å) | 1.54178 | 1.54178 | 1.54178 |
| resolution range (Å)a | 45.08–1.65 (1.68–1.65) | 45.26–2.00 (2.05–2.00) | 45.34–2.30 (2.39–2.30) |
| space group | P21 | P21 | P21 |
| unit cell (Å, deg) | 41.8 118.1 72.0 90 104.3 90 | 41.8 118.5 71.9 90 103.1 90 | 41.8 118.7 72.1 90 102.8 90 |
| total reflections | 1494775 (29889) | 174449 (10045) | 119380 (8711) |
| unique reflections | 80845 (4071) | 45652 (3074) | 30045 (2880) |
| multiplicity | 18.5 (7.3) | 3.8 (3.3) | 4.0 (3.0) |
| completeness (%) | 99.6 (99.8) | 98.7 (91.4) | 98.7 (90.6) |
| ⟨I/σI⟩b | 14.9 (1.1) | 14.4 (2.0) | 7.2 (2.0) |
| resolution limit for ⟨I/σI⟩ > 2c | 1.72 | 2.00 | 2.30 |
| Wilson B-factor (Å2) | 21.3 | 27.9 | 35.2 |
| Rmeas | 0.123 (2.215) | 0.112 (0.737) | 0.130 (0.597) |
| CC1/2 | 0.999 (0.479) | 0.996 (0.679) | 0.989 (0.780) |
| refinement | |||
| resolution range (Å) | 37.46–1.65 (1.71–1.65) | 38.53–2.0 (2–2.0) | 45.34–2.3 (2–2.3) |
| % Rwork | 0.1961 (0.2903) | 0.1957 (0.2659) | 0.1990 (0.2649) |
| % Rfree | 0.2135 (0.3114) | 0.2434 (0.3227) | 0.2685 (0.3706) |
| number of non H atoms | 5375 | 5341 | 5396 |
| protein | 5148 | 5126 | 5266 |
| ligands | 152 | 92 | 92 |
| water | 139 | 123 | 38 |
| validation | |||
| RMS(bonds) | 0.008 | 0.008 | 0.008 |
| RMS(angles) | 1.04 | 0.99 | 1.03 |
| Ramachandran favored (%) | 98.61 | 97.83 | 98.18 |
| Ramachandran outliers (%) | 0.00 | 0.15 | 0.00 |
| rotamer outliers (%) | 0.55 | 0.55 | 0.90 |
| average B-factor (Å2) | 30.87 | 33.39 | 40.92 |
| protein | 30.69 | 33.02 | 40.59 |
| ligands | 43.30 | 58.06 | 62.86 |
| water | 29.82 | 30.33 | 32.21 |
| Clashscore | 3.49 | 4.08 | 3.32 |
Values in parentheses correspond to the highest-resolution shell.
See ref (105) for crystallographic definitions.
The resolution limits for ⟨I/σI⟩ > 2 are reported.
Figure 9.

(a) Electron density (2Fobs – Fcalc) weighted maps of compound 12a (represented as cornflower blue sticks) bound to subunit B of mmPRMT6 (represented as a gray cartoon; PDB ID: 7NUD). Maps are represented as a mesh, with the contouring level set to 1σ. For clarity, N-terminal helices (residues 42–56) of mmPRMT6 are not shown. (b) Superimposition (done on protein backbones) of the conformations of compound 12a (yellow or cornflower blue sticks, respectively) when bound to subunit B of mmPRMT4 (represented as a dark gray cartoon) and when bound to mmPRMT6 (represented as a light gray cartoon). For clarity, N-terminal helices of PRMT4 and PRMT6 are not shown.
The crystal structure revealed an unusual distorted U-shaped conformation adopted by compound 12a (and probably also 12c and 12f) in the binding to PRMT6, with a folding at the level of the guanidine group (Figure 9 and Figure S11, Supporting Information). Whereas the adenosine moiety lies into its canonic binding pocket, the guanidine group of the compounds is unable to reach the PRMT6 double-E loop clamp because the linker with the sugar is too short, even for the longer compound 12f. Instead of binding in the arginine substrate pocket as observed with PRMT4 (Figures S2 and S8–S10, Supporting Information), the methyl-4 hydroxy-2-naphthoate moiety is sandwiched between the adenosine and the side chains of Y50 and Y51 residues of the α helix motif I (Figure S12, Supporting Information). Hence, the binding of compounds 12a, 12c, and 12f affects the proper folding of the PRMT6 alpha-X helix containing the motif I (Y50-Y51-X-X-Y54).
If compared to the structures obtained in the presence of SAH, the binding site of the complex of PRMT6 with compound 12a is larger due to a displacement of the α helix and the flipping of the side chains of Y50 and Y51 residues to give room for the compound naphthoate moiety. As a consequence, the E267 residue of the double-E loop is not coordinated anymore by residues Y50 and Y54 of motif 1 and adopts a different conformation (Figure S11, Supporting Information).
Assessment of Functional Potency and Cell Toxicity in Various Cell Lines
As mentioned above, this study was designed as a proof of concept of our approach to probe the structural differences among the various PRMTs and to develop potent and selective inhibitors. Therefore, we were not surprised to find that, at this stage, compounds 12f–12h showed poor apparent permeability in a parallel artificial membrane permeability assay (PAMPA), using the reference drugs propranolol (highly permeable) and the furosemide (poorly permeable) as positive and negative controls, respectively (see the Supporting Information). Nonetheless, we resolve to investigate if, regardless of the low cell permeability, the compounds were able to affect the activity of PRMT4 in a cellular context. First, we assessed cell toxicity in human embryonic kidney HEK293T cells. To this aim, we incubated the cell line with different concentrations (10, 50, and 100 μM) of each compound and assessed cell viability after 24 and 72 h using the MTT assay. We observed that none among the tested compound was able to reduce the number of metabolically active cells in comparison with the vehicle, at all the tested concentrations (up to 100 μM, Figure 10a). Even after 72 h of treatment cell viability remained above 80% (Figure 10b).
Figure 10.

Cellular effects of compounds 12f–12h. (a, b) The viability of HEK293T cells was assessed by measuring the mitochondrial-dependent reduction of MTT to formazan, with respect to DMSO, after treatment with compounds 12f–12h at three different concentrations (10, 50, and 100 μM) for (a) 24 h and (b) 72 h. Data are reported as the mean ± SD of four independent experiments. (c, d) Western blot analyses were performed (a) on lysates from HEK293T cells after treatment with compounds 12f–12h at 10 and 50 μM for 24 h and (d) on lysates from MCF7 cells after treatment with compound 12h at 10 and 50 μM for 4 and 8 days. Methylation was detected by immunoblotting with a pan-PRMT4 substrate antibody (PRMT4sub; see the main text).102 Total histone H3 (c) or actin (d) was used to check for equal loading. The cell-permeable PRMT4 inhibitor TP064 (10 μM) was used as a reference compound. (e, f) Relative proliferation of (e) HEK293T and (f) MCF7 cells with different concentration of 12h for different time points. The medium was changed at day 4. All the data points represent the relative viability normalized to day 0. The error bars represent the standard deviation of three biological replicates performed at each time point.
We next investigated the functional potency of compounds 12f–12h in reducing the cellular level of arginine methylation catalyzed by PRMT4. To this aim, HEK293T cells were incubated for 24 h with the three compounds (at 10 and 50 μM) or with compound TP-064 (10 μM)24 and used as a positive control, and total cell lysates were then immunoblotted with a pan-PRMT4 substrate antibody (PRMT4sub), originally raised against the R388 site of Nuclear Factor 1 B-type (NFIB-Me) but also capable of recognizing many PRMT4 substrates.102,108 As shown in Figure 10c, the results confirmed the proof of concept concerning the design of these derivatives. In fact, although with a lower effect compared to the cytopermeable TP-064, the compounds, in particular 12h, are able to reduce the activity of PRMT4 in a concentration-dependent way. Noteworthy, TP-064 concentrations higher than 10 μM led to cell death. Based on these findings, we resolved to investigate the effects of compound 12h also on the MCF7 breast cancer cell line, the proliferation of which was previously found to be dependent on PRMT4.109,110 MCF7 cells were incubated with 12h (at 10 and 50 μM) or with the control TP-064 (10 μM) for 4 and 8 days used as positive control, and total cell lysates were then immunoblotted with the pan-PRMT4 substrate antibody PRMT4sub. As shown in Figure 10d, the arginine methylation levels in MCF7 cells are profoundly affected by TP064 but also by the significantly less permeable 12h, in a concentration- and time-dependent way. More importantly, both 10 μM TP064 and 50 μM 12h markedly decreased the proliferation of MCF7 cells, whereas no effect was observed in HEK293T cells (Figure 10e and f).
Conclusions
The pivotal role played by PRMT-mediated arginine methylation in the regulation of many cellular processes and the implications in the genesis of various diseases have attracted growing interest toward PRMTs as potential therapeutic targets. Yet, even if a few clinical-grade small-molecule inhibitors have been identified for these proteins,11 there is still much to be understood about their functions and the biological pathways in which they are involved, as well as on the structural requirements that could drive the development of selective modulators of PRMT activity.11
In this work, starting from a series of type I PRMT inhibitors previously identified by us and with molecular modeling studies of their binding mode,46,47 we deconstructed such ligands into a 4-hydroxy-2-naphthoic fragment and then applied a step-by-step growing approach, in which the fragment was bridged by an amide or urea group with an arginine mimetic or methionine moiety. As a primary screening, we gauged the effect of the synthesized compounds (derivatives 5–11) on the catalytic activity of human recombinant PRMT1, chosen as representative of class I PRMTs, using a purpose-developed AlphaLISA assay. The compounds showing an inhibition greater than 85% were selected, and the corresponding IC50 values were determined. The structure–activity relationships identified a scaffold (namely compound 7d) featuring both naphthylurea and methylguanidine groups as the best candidate for the further growing step. Then, another series of compounds (12a–12h) were designed and synthesized, introducing also an adenosine moiety and exploring the distance between the three groups.
A radioisotope-based filter-binding assay was used as a secondary screening to profile the activity of the compounds against type I PRMT1, PRMT3, PRMT4, PRMT6, and PRMT8, type II PRMT5, and type III PRMT7. The overall length of the compounds and, even more, the length of the two linkers resulted to be crucial for the inhibitory activity especially against PRMT4 and, at a minor extent, against PRMT1. In fact, derivative 12h featuring a four-carbon atom linker between the 4-hydroxy-2-naphthoate and the guanidine group and a three-carbon atom linker between the latter and the adenosine resulted to be the most potent inhibitor against PRMT4 (IC50 = 3 nM), with a 261- to 1266-fold selectivity over the other PRMTs. Noteworthy, 12h was found to be selective also and further assessed against a panel of eight KMTs, including the SET-domain-containing proteins ASH1L/KMT2H, EZH2/KMT6, MLL1/KMT2A, SET7/9/KMT7, SETD8/KMT5A, SUV39H2/KMT1B, and SUV420H1/KMT5B and the non-SET-domain-containing DOT1L/KMT4, showing no inhibiting effect against these enzymes even at the higher tested concentration (>3000 fold higher than the IC50 value against PRMT4). SPR studies confirmed a specific and strong binding interaction between PRMT4 and 12h with a KD value in the nanomolar range (KD = 75.9 nM; τR = 62.5 s), and crystallographic studies showed that the three-carbon atom long linker between the adenosine moiety and the guanidine group brings the latter within the catalytic clamp formed by E258 and E267 of the double-E loop, allowing the establishing of stabilizing hydrogen bonds between the guanidine N5 atom and the main chain oxygen atoms of E258 and M260. The trans conformation imposed by the double bond in 12h impedes the formation of more constrained rotamers. On the other hand, the methyl 4-hydroxy-2-naphthoate moiety partially occupies the peptide substrate binding site on PRMT4 and mainly interacts with Y262, P473, F475, and Y477 on one side and F153 on the other side.
Whereas a similar trend was observed also against PRMT1 and PRMT5, the effect of the linker length on the inhibiting activity was less significant (if any) in the case of PRMT3, PRMT8, and, particularly, PRMT6. In the case of the latter, cocrystallization studies revealed that the compounds adopt an odd distorted U-shaped conformation, where the guanidine group is unable to reach the PRMT6 double-E loop clamp and the methyl-4 hydroxy-2-naphthoate moiety does not bind to the arginine substrate pocket, but it is sandwiched between the adenosine and the side chains of Y50 and Y51 residues of α helix motif I. Interestingly, in the case of PRMT7 the shorter compound 12a shows a certain selective inhibition (IC50 = 0.3 μM; selectivity from 26-fold to 231-fold) compared to other tested PRMTs, in agreement with the restrictive and narrow active site for PRMT7.111
Surprisingly, regardless of the low cell permeability, 12h is able to affect the activity of PRMT4 in a cellular context, showing an evident reduction of arginine methylation levels in MCF7 cells and a marked reduction of proliferation.
In conclusion, this study confirmed the feasibility of our deconstruction–reconstruction approach to achieve potency and selectivity against a specific PRMT isoform starting from nonselective PRMT inhibitors. Although nonoptimized for cell permeability, the identified PRMT4 inhibitor 12h (EML981) is able to reduce the activity of PRMT4 in a concentration-dependent way and can be used for further development of cell-active PRMT4 inhibitors. Also, the approach is versatile and can be applied to identify selective inhibitors of other PRMTs.
Chemistry
The synthetic protocol for the preparation of compounds 5–8 is depicted in Schemes 1 and 2. The 4-acetoxy-6-nitro-2-naphthoate 13, prepared as previously reported by us,47 was straightforwardly transformed in the 4-hydroxy-6-nitro-2-naphthoate 14,112 through treatment with piperidine in dichloromethane (DCM; Scheme 1). Protection of the hydroxyl group with di-tert-butyl dicarbonate (Boc2O) in the presence of triethylamine (TEA) and N,N-dimethyl-4-aminopyridine (DMAP) yielded the intermediate 15 which was then reduced with zinc dust in acetic acid to give the corresponding arylamine 16. The reaction of the latter with the proper Boc-protected aminoalkanoic acid, in the presence of N,N′-dicyclohexylcarbodiimide (DCC) and DMAP, furnished amides 17a–17c. After deprotection with trifluoroacetic acid (TFA) in DCM, the resulting amines 18a–18c were reacted with Boc-protected S-methylisothiourea or N,S-dimethylisothiourea to yield the protected guanidines 19a–19c and 20a and 20b. Removal of the tert-butoxycarbonyl group under acidic conditions gave ester derivatives 5a–5e, from which the corresponding carboxylic acids 6a–6d were obtained by hydrolysis with lithium hydroxide aqueous solution. A similar synthetic pathway was followed to prepare ureido derivatives 7a–7e and 8a (Scheme 1). Briefly, naphthylamine 21, prepared from the same key building block 13 as previously reported by us,47 was reacted with phenyl chloroformate to yield phenyl carbamate 22. The reaction of the latter with the proper mono-Boc-protected alkyldiamine, followed by treatment with piperidine in DCM, furnished derivatives 23a–23e. After trifluoroacetic acid deprotection, the corresponding amines 24a–24e were reacted with Boc-protected S-methylisothiourea or N,S-dimethylisothiourea to yield compounds 25a–25c, 26a, and 26b. Finally, deprotection gave esters 7a–7e. The carboxylic acid derivative 8a was obtained by hydrolysis of ester 7b. As depicted in Scheme 2, the amine 18a was also reacted with 1,1,2-trimethylisothiourea to give the substituted guanidine derivative 5f or with 2-methylthio-2-imidazoline hydroiodide to yield the imidazoline derivative 5g. The carboxylic acids 6e and 6f were obtained from 5f and 5g, respectively, by hydrolysis with lithium hydroxide aqueous solution. Reaction of phenyl carbamate 22 with 3-imidazolylpropan-1-amine followed by treatment with piperidine in DCM furnished ester derivative 7f. Finally, the carboxylic acid 8b was obtained by basic hydrolysis.
Scheme 1. Synthesis of Derivatives 5a–5e, 6a–6d, 7a–7e, and 8a.
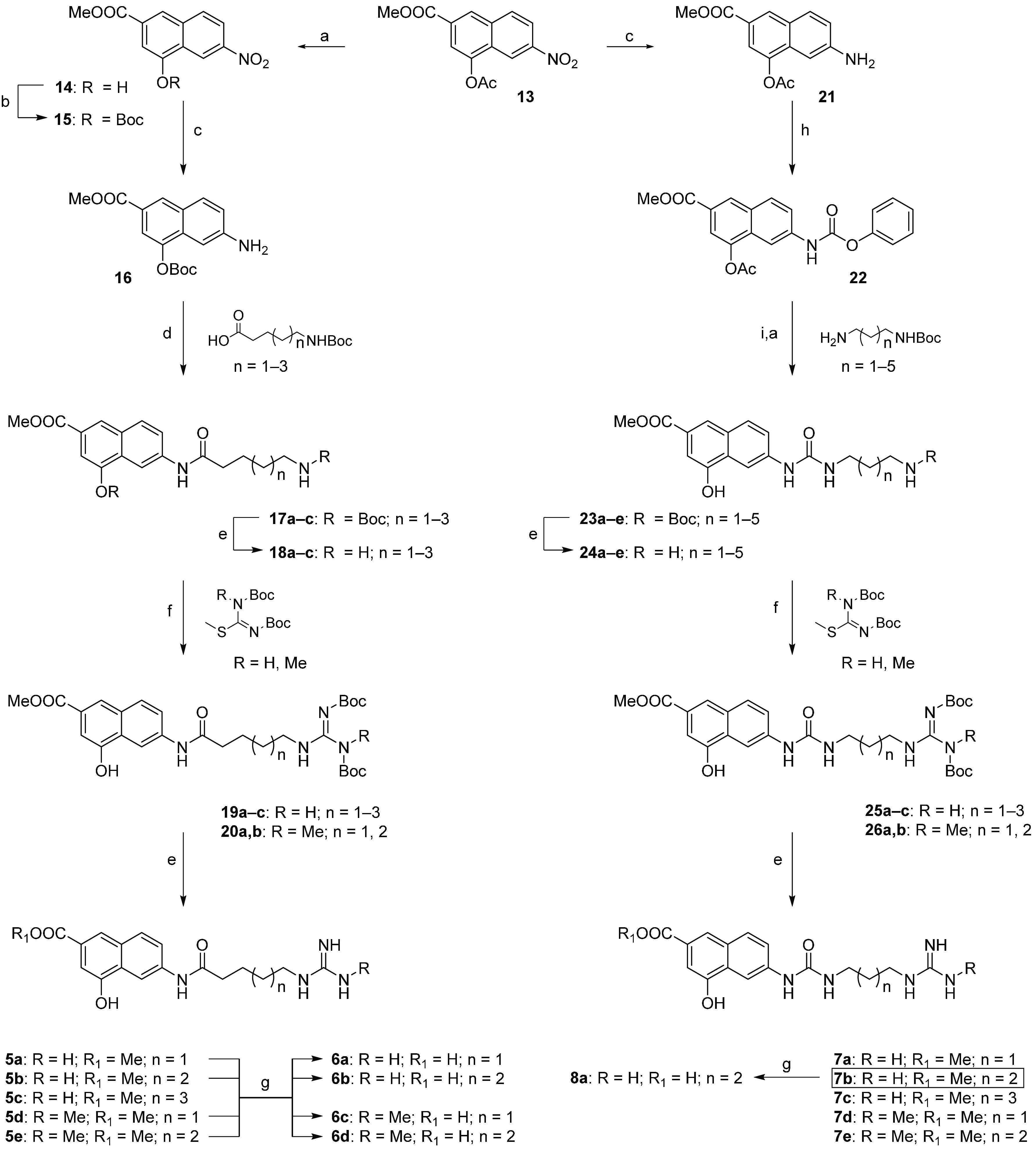
Reagents and conditions: (a) piperidine, DCM, room temperature (r.t.), 30 min. (93–99%); (b) Boc2O, TEA, DMAP, DCM, r.t., 12 h (70%); (c) Zn, AcOH, r.t., 1 h (98–99%; (d) DCC, DMAP, DCM, r.t., 8–12 h (80–88%); (e) DCM/TFA, 9:1, r.t., 1 h (60–99%); (f) TEA, DMAP, DMF, r.t., 24 h (30–85%); (g) LiOH, MeOH/H2O, r.t., 48 h (80–92%); (h) phenyl chloroformate, TEA, EtOAc, r.t., 12 h (70%); (i) TEA, DMF, r.t., 24 h (80–87%).
Scheme 2. Synthesis of Derivatives 5f, 5g, 6e, 6f, 7f, and 8b.

Reagents and conditions: (a) TEA, DMAP, DMF, r.t., 24 h (57–67%); (b) LiOH, MeOH/H2O, r.t., 48 h (62–89%); (c) TEA, DMAP, DMF, r.t., 24 h; (d) piperidine, DCM, room temperature (r.t.), 30 min (71%, over two steps).
Derivatives 9–11 were in turn prepared (Scheme 3) starting from the O-Boc-aminohydroxynaphthoate 16 that was reacted with the orthogonally protected N-Fmoc-l-methionine, N-Boc-glycine, or Nα-Fmoc-Nω-Pbf-l-arginine in the presence of DCC and DMAP to give the corresponding amides 9a, 10a, or 11a. After deprotection with piperidine and/or TFA in DCM, the corresponding compounds 9c, 10b, and 11b were reacted with 4-methoxyphenyl isocyanate in the presence of TEA to obtain ureas 9d, 10c, and 11d. Derivatives 11c and 11e were obtained from 11b and 11d, respectively, after the removal of Pbf protection.
Scheme 3. Synthesis of Derivatives 9–11.

Reagents and conditions: (a) proper N-protected amino acid, DCC, DMAP, DCM, r.t., 8–12 h (79–81%); (b) piperidine, DCM, 30 min (72–99%); (c) DCM/TFA, 9:1, r.t., 1 h (68–74%); (d) 4-methoxyphenyl isocyanate, TEA, THF, r.t., 4 h (79–82%); (e) DCM/TFA, 1:9, r.t., 24 h (68–77%).
Compounds 12a–12h were prepared as outlined in Scheme 4. Adenosine derivatives 27a–27d,44,113−115 obtained according to a slight modification of previously reported procedures116,117 (Scheme 5), were reacted with N,N′-di-Boc-thiourea in the presence of trifluoroacetic anhydride (TFAA) and sodium hydride118 to give thioureas 28a–28d. A coupling reaction with amino derivatives 24a–24e in the presence of EDC hydrochloride as an activating agent yielded derivatives 29–32 which were deprotected with TFA to obtain the target compounds 12a–12h.
Scheme 4. Synthesis of Derivatives 12a–12h.

Reagents and conditions: (a) NaH 60% mineral oil, TFAA, dry THF, r.t., 16 h (33–53%); (b) EDC hydrochloride, TEA, DCM, r.t., 18 h (72–81%); (c) DCM/TFA, 1:1, r.t., 2 h (74–80%).
Scheme 5. Synthesis of Derivatives 27a–27d.

Reagents and conditions: (a) acetone, HClO4 70%, r.t., 5 h (81%); (b) NaN3, DPPA, DBU, 15-crown-5, dioxane, 2 h (86%); (c) H2, Pd/C 10%, MeOH, r.t., 5 h (99%); (d) α-hydroxyisobutyronitrile, DEAD, PPh3, dry THF, 0–20 °C, 24 h, (89%); (e) Boc2O, NaBH4, NiCl2·6H2O, dry MeOH, 0 °C, 2 h (80%); (f) DCM/TFA 95:5, t.a, 8 h, (65%); (g) o-iodoxybenzoic acid (IBX), CH3CH2O2CCH=P(C6H5)3, DMSO, 20 °C, 72 h, (70%); (h) DIBAL-H, DCM, −78 °C, 2 h, (98%); (i) phthalimide, DEAD, PPH3, dry THF, 20 °C, 16 h, (70%); (l) NH2NH2·H2O, MeOH, 0–25 °C, 16 h, (90%); (m) H2, Pd/C 10%, AcOEt, 20 °C, 18 h (99%).
Experimental Section
Chemistry
General Directions
All chemicals, purchased from Merck KGaA and Fluorochem Ltd., were of the highest purity. All solvents were reagent grade and, when necessary, were purified and dried by standard methods. All reactions requiring anhydrous conditions were conducted under a positive atmosphere of nitrogen in oven-dried glassware. Standard syringe techniques were used for anhydrous addition of liquids. Reactions were routinely monitored by TLC performed on aluminum-backed silica gel plates (Merck KGaA, Alufolien Kieselgel 60 F254) with spots visualized by UV light (λ = 254, 365 nm) or using a KMnO4 alkaline solution. Solvents were removed using a rotary evaporator operating at a reduced pressure of ∼10 Torr. Organic solutions were dried over anhydrous Na2SO4. Chromatographic purification was done on an automated flash-chromatography system (Isolera Dalton 2000, Biotage) using cartridges packed with KP-SIL, 60 Å (40–63 μm particle size). All microwave-assisted reaction were conducted in a CEM Discover SP microwave synthesizer equipped with a vertically focused IR temperature sensor.
Analytical high-performance liquid chromatography (HPLC) was performed on a Shimadzu SPD 20A UV/vis detector (λ = 220 and 254 nm) using a C-18 column Phenomenex Synergi Fusion-RP 80A (75 × 4.60 mm; 4 μm) at 25 °C using a mobile phase A (water + 0.1% TFA) and B (ACN + 0.1% TFA) at a flow rate of 1 mL/min. Preparative HPLC was performed using a Shimadzu Prominence LC-20AP with the UV detector set to 220 and 254 nm. Samples were injected onto a Phenomenex Synergi Fusion-RP 80A (150 × 21 mm; 4 mm) C-18 column at room temperature. Mobile phases of A (water + 0.1% TFA) and B (ACN + 0.1% TFA) were used with a flow rate of 20 mL/min.
1H spectra were recorded at 400 MHz on a Bruker Ascend 400 spectrometer while 13C NMR spectra were obtained by distortionless enhancement by polarization transfer quaternary (DEPTQ) spectroscopy on the same spectrometer. Chemical shifts are reported in δ (ppm) relative to the internal reference tetramethylsilane (TMS). Low-resolution mass spectra were recorded on a Finnigan LCQ DECA TermoQuest mass spectrometer in electrospray positive and negative ionization modes (ESI-MS). High-resolution mass spectra were recorded on a ThermoFisher Scientific Orbitrap XL mass spectrometer in electrospray positive ionization modes (ESI-MS). All tested compounds possessed a purity of at least 95% established by HPLC unless otherwise noted.
Methyl 6-(5-Guanidinopentanamido)-4-hydroxy-2-naphthoate (5a)
Compound 19a (280 mg, 0.501 mmol) was dissolved in 10 mL of a solution of DCM/TFA (9:1), and the mixture was stirred for 48 h. The solvent was evaporated, and the resulting solid was washed with CHCl3 to give the TFA salt of compound 5a as a brown solid (201 mg, 85%). 1H NMR (400 MHz, DMSO-d6) δ 10.45 (s, 1H, exchangeable with D2O), 10.24 (s, 1H, exchangeable with D2O), 8.54 (s, 1H), 8.01–7.95 (m, 2H), 7.72 (d, J = 8.7 Hz, 1H), 7.49 (br t, 1H, exchangeable with D2O), 7.34 (s, 1H), 7.21–6.75 (m, 3H, exchangeable with D2O), 3.88 (s, 3H), 3.18–3.12 (m, 2H), 2.42 (t, J = 7.2 Hz, 2H), 1.68–1.53 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 171.96, 167.02, 156.91, 153.20, 138.43, 130.18, 130.14, 127.79, 126.06, 121.49, 121.09, 110.05, 106.93, 40.90, 40.42, 36.21, 28.35, 22.48. HRMS (ESI): m/z [M + H]+ calcd for C18H22N4O4 + H+: 359.1714. Found: 359.1705.
Methyl 6-(6-Guanidinohexanamido)-4-hydroxy-2-naphthoate (5b)
The TFA salt of compound 5b was obtained as a pale brown solid (254 mg, 81%) starting from compound 19b (370 mg, 0.646 mmol) following the procedure described for 5a. 1H NMR (400 MHz, DMSO-d6) δ 10.43 (s, 1H, exchangeable with D2O), 10.20 (s, 1H, exchangeable with D2O), 8.54 (s, 1H), 8.00 (s, 1H), 7.97 (d, J = 8.5 Hz, 1H), 7.72 (d, J = 8.5 Hz, 1H), 7.50 (br t, 1H, exchangeable with D2O), 7.33 (s, 1H), 7.22–6.83 (m, 3H, exchangeable with D2O), 3.87 (s, 3H), 3.12–3.10 (m, 2H), 2.39 (t, J = 6.9 Hz, 2H), 1.67–1.63 (m, 2H), 1.53–1.49 (m, 2H), 1.37–1.33 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 171.94, 167.05, 157.18, 153.45, 138.77, 130.27, 130.19, 128.00, 126.10, 121.55, 121.13, 110.05, 107.10, 52.51, 41.13, 36.82, 28.84, 26.23, 25.13. HRMS (ESI): m/z [M + H]+ calcd for C19H24N4O4 + H+: 373.1870. Found: 373.1863.
Methyl 6-(7-Guanidinoheptanamido)-4-hydroxy-2-naphthoate (5c)
The TFA salt of compound 5c was obtained as a pale brown solid (76 mg, 90%) starting from 19c (100 mg, 0.170 mmol) following the procedure described for 5a. 1H NMR (400 MHz, DMSO-d6) δ 10.45 (s, 1H, exchangeable with D2O), 10.20 (s, 1H, exchangeable with D2O), 8.54 (s, 1H), 8.01 (s, 1H), 7.95 (d, J = 8.9 Hz, 1H) 7.71 (d, J = 8.9 Hz, 1H), 7.55 (br t, 1H, exchangeable with D2O), 7.33 (s, 1H), 7.23–6.77 (m, 3H, exchangeable with D2O), 3.87 (s, 3H), 3.12–3.07 (m, 2H), 2.38 (t, J = 7.3 Hz, 2H), 1.65–1.62 (m, 2H), 1.51–1.47 (m, 2H), 1.35–1.33 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 172.04, 167.05, 157.22, 153.45, 138.79, 130.26, 130.19, 128.01, 126.09, 121.55, 121.15, 110.05, 107.09, 52.50, 41.21, 36.88, 28.81, 28.75, 26.39, 25.44. HRMS (ESI): m/z [M + H]+ calcd for C20H26N4O4 + H+: 387.2027. Found: 387.2017.
Methyl 4-Hydroxy-6-(5-(3-methylguanidino)pentanamido)-2-naphthoate (5d)
The TFA salt of compound 5d was obtained as a brown solid (204 mg, 86%) starting from 20a (280 mg, 0.489 mmol) following the procedure described for 5a. 1H NMR (400 MHz, DMSO-d6) δ 10.47 (s, 1H, exchangeable with D2O), 10.28 (s, 1H, exchangeable with D2O), 8.55 (s, 1H), 8.00 (s, 1H) 7.96 (d, J = 8.9 Hz, 1H), 7.72 (d, J = 8.7 Hz, 1H), 7.54 (br t, 1H, exchangeable with D2O), 7.45 (s, 1H, exchangeable with D2O), 7.36–7.34 (m, 3H, 2H exchangeable with D2O), 3.87 (s, 3H), 3.18–3.14 (m, 2H), 2.75–2.74 (m, 3H), 2.44–2.40 (m, 2H), 1.68–1.53 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 171.85, 167.09, 156.80, 153.47, 138.73, 130.29, 130.20, 128.01, 126.12, 121.53, 121.15, 110.10, 107.11, 52.51, 41.13, 36.42, 28.64, 28.39, 22.67. HRMS (ESI): m/z [M + H]+ calcd for C19H24N4O4 + H+: 373.1870. Found: 373.1873.
Methyl 4-Hydroxy-6-(6-(3-methylguanidino)hexanamido)-2-naphthoate (5e)
The TFA salt of compound 5e was obtained as a pale brown solid (279 mg, 80%) starting from 20b (410 g, 0.699 mmol) following the procedure described for 5a. 1H NMR (400 MHz, DMSO-d6) δ 10.48 (s, 1H, exchangeable with D2O), 10.24 (s, 1H, exchangeable with D2O), 8.55 (s, 1H), 8.01 (s, 1H), 7.96 (d, J = 8.5 Hz, 1H), 7.72 (d, J = 8.5 Hz, 1H), 7.47 (t, J = 5.4 Hz, 1H, exchangeable with D2O), 7.40 (s, 1H, exchangeable with D2O), 7.34–7.32 (m, 3H, 2H exchangeable with D2O), 3.87 (s, 3H), 3.13–3.09 (m, 2H), 2.73 (s, 3H), 2.39 (t, J = 7.3 Hz, 2H), 1.66–1.63 (m, 2H), 1.53–1.50 (m, 2H), 1.36–1.33 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 171.96, 167.05, 156.73, 153.45, 138.77, 130.25, 130.20, 128.00, 126.10, 121.55, 121.12, 110.05, 107.10, 52.51, 41.23, 36.82, 28.86, 28.40, 26.22, 25.14. HRMS (ESI): m/z [M + H]+ calcd for C20H26N4O4 + H+: 387.2027. Found: 387.2020.
Methyl 6-(5-(3,3-Dimethylguanidino)pentanamido)-4-hydroxy-2-naphthoate (5f)
To a stirred solution of 18a (200 mg, 0.464 mmol) and DMAP (6 mg, 0.046 mmol) in dry DMF (2.5 mL), TEA (65 μL, 0.464 mmol) and 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea (228 mg, 0.929 mmol) were added. The resulting mixture was stirred at room temperature for 24 h. The solution was diluted with AcOEt (30 mL) and brine (10 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography (gradient Hex/AcOEt 80:20 to 20:80) to give 5f as a white solid (268 mg, 67%). 1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H, exchangeable with D2O), 10.24 (s, 1H, exchangeable with D2O), 8.55 (d, J = 2.1 Hz, 1H), 8.01 (s, 1H), 7.96 (d, J = 8.9 Hz, 1H), 7.71 (dd, J = 8.9, 2.1 Hz, 1H), 7.42 (s, 1H, exchangeable with D2O), 7.37 (t, 1H, exchangeable with D2O), 7.36–7.34 (m, 1H), 3.87 (s, 3H), 3.23–3.18 (m, 2H), 2.96 (s, 6H), 2.42 (t, J = 7.0 Hz, 2H), 1.68–1.60 (m, 2H), 1.60–1.57 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.87, 167.05, 156.20, 153.46, 138.72, 130.30, 130.22, 127.99, 126.14, 121.55, 121.13, 110.09, 107.11, 52.51, 42.13, 38.60, 36.47, 28.67, 22.63. HRMS (ESI): m/z [M + H]+ calcd for C20H26N4O4 + H+: 387.2027. Found: 387.2019.
Methyl 6-(5-((4,5-Dihydro-1H-imidazol-2-yl)amino)pentanamido)-4-hydroxy-2-naphthoate (5g)
Compound 5g was obtained as a white product (136 mg, 61%) by the reaction of compound 18a (250 mg, 0.58 mmol) with 2-methylthio-2-imidazoline hydroiodide (283 mg, 1.16 mmol) following the procedure described for 5f. 1H NMR (400 MHz, DMSO-d6) δ 10.45 (s, 1H, exchangeable with D2O), 10.23 (s, 1H, exchangeable with D2O), 8.54 (d, J = 2.1 Hz, 1H), 8.30 (br t, 1H, exchangeable with D2O), 8.00 (s, 1H), 7.95 (d, J = 8.9 Hz, 1H), 7.70 (dd, J = 8.9, 2.1 Hz, 1H), 7.33 (s, 1H), 4.27–3.97 (m, 4H), 3.87 (s, 3H), 3.19–3.14 (m, 2H), 2.41 (t, J = 7.1 Hz, 2H), 1.66–1.60 (m, 2H), 1.60–1.51 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 171.79, 167.04, 159.87, 153.45, 138.70, 130.30, 130.22, 127.99, 126.14, 121.55, 121.13, 110.09, 107.11, 52.51, 42.92, 42.49, 36.39, 28.85, 22.62. HRMS (ESI): m/z [M + H]+ calcd for C20H24N4O4 + H+: 385.1870. Found: 385.1863.
6-(5-Guanidinopentanamido)-4-hydroxy-2-naphthoic Acid (6a)
To a stirred solution of 5a (100 mg, 0.211 mmol) in 4 mL of MeOH, an aqueous solution (1 mL) of LiOH (15 mg, 0.633 mmol) was added. The reaction mixture was stirred at room temperature for 48–72 h (TLC analysis). The resulting mixture was acidified with 1 N HCl and then concentrated under vacuum. The crude product was purified by reversed-phase high-performance liquid chromatography (RP-HPLC) to afford 6a as the TFA salt (89 mg, 92%). 1H NMR (400 MHz, DMSO-d6) δ 12.81 (s, 1H, exchangeable with D2O), 10.38 (s, 1H, exchangeable with D2O), 10.23 (s, 1H, exchangeable with D2O), 8.54 (s, 1H), 7.99–7.93 (m, 2H), 7.70 (d, J = 8.6 Hz, 1H), 7.53 (br t, 1H, exchangeable with D2O), 7.34 (s, 1H), 7.22–6.75 (m, 3H, exchangeable with D2O), 3.19–3.10 (m, 2H), 2.43 (t, J = 7.4 Hz, 2H), 1.69–1.53 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 171.26, 167.64, 156.71, 152.78, 137.95, 129.86, 129.61, 127.30, 121.07, 120.48, 109.65, 107.07, 40.54, 35.91, 28.13, 22.17. HRMS (ESI): m/z [M + H]+ calcd for C17H20N4O4 + H+: 345.1557. Found: 345.1549.
6-(6-Guanidinohexanamido)-4-hydroxy-2-naphthoic Acid (6b)
The TFA salt of compound 6b was obtained as a white solid (149 mg, 77%) starting from 5b (200 mg, 0.411 mmol) following the procedure described for 6a. 1H NMR (400 MHz, DMSO-d6) δ 12.79 (s, 1H, exchangeable with D2O), 10.38 (s, 1H, exchangeable with D2O), 10.21 (s, 1H, exchangeable with D2O), 8.54 (s, 1H), 7.97–7.92 (m, 2H), 7.69 (d, J = 8.0 Hz, 1H), 7.59 (br t 1H, exchangeable with D2O), 7.33 (s, 1H), 7.17–6.77 (m, 3H, exchangeable with D2O), 3.12–3.09 (m, 2H), 2.39 (t, J = 7.1 Hz, 2H),1.67–1.64 (m, 2H), 1.53–1.50 (m, 2H), 1.37–1.34 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 172.08, 168.13, 157.06, 153.24, 138.35, 130.33, 130.13, 127.72, 127.26, 121.61, 120.95, 110.14, 107.51, 41.13, 36.74, 28.76, 26.18, 25.13. HRMS (ESI): m/z [M + H]+ calcd for C18H22N4O4 + H+: 359.1714. Found: 359.1707.
4-Hydroxy-6-(5-(3-methylguanidino)pentanamido)-2-naphthoic Acid (6c)
The TFA salt of compound 6c was obtained as a pale brown solid (84 mg, 88%) starting from 5d (100 mg, 0.204 mmol) following the procedure described for 6a. 1H NMR (400 MHz, DMSO-d6) δ 12.79 (s, 1H, exchangeable with D2O), 10.37 (s, 1H, exchangeable with D2O), 10.22 (s, 1H, exchangeable with D2O), 8.54 (s, 1H), 7.99–7.93 (m, 2H), 7.70 (d, J = 8.5 Hz, 1H), 7.39–7.30 (m, 5H, 4H exchangeable with D2O), 3.19–3.14 (m, 2H), 2.76 (s, 3H), 2.45–2.40 (m 2H), 1.70–1.53 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 171.87, 168.10, 138.28, 130.35, 130.16, 127.75, 127.27, 121.63, 121.05, 110.17, 107.46, 41.02, 36.34, 28.55, 28.23, 22.63. HRMS (ESI): m/z [M + H]+ calcd for C18H22N4O4 + H+: 359.1714. Found: 359.1709.
4-Hydroxy-6-(6-(3-methylguanidino)hexanamido)-2-naphthoic Acid (6d)
The TFA salt of compound 6d was obtained as a white solid (155 mg, 80%) starting from 5e (200 mg, 0.399 mmol) following the procedure described for 6a. 1H NMR (400 MHz, DMSO-d6) δ 12.75 (s, 1H, exchangeable with D2O), 10.37 (s, 1H, exchangeable with D2O), 10.26 (s, 1H, exchangeable with D2O), 8.55 (s, 1H), 7.97–7.92 (m, 2H), 7.72 (d, J = 7.9 Hz, 1H), 7.50 (br t, 1H,, exchangeable with D2O) 7.50–7.34 (m, 4H, 3H exchangeable with D2O), 3.15–3.10 (m, 2H), 2.74 (s, 3H), 2.40 (t, J = 7.4 Hz, 2H), 1.68–1.64 (m, 2H), 1.55–1.51 (m, 2H), 1.39–1.36 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 171.97, 168.15, 156.89, 153.29, 138.55, 130.31, 130.03, 127.82, 127.29, 121.54, 121.01, 110.11, 107.56, 41.20, 36.82, 28.84, 28.38, 26.21, 25.17. HRMS (ESI): m/z [M + H]+ calcd for C19H24N4O4 + H+: 373.1870. Found: 373.1862.
6-(5-(3,3-Dimethylguanidino)pentanamido)-4-hydroxy-2-naphthoic Acid (6e)
Compound 6e was obtained as a pale brown solid (74 mg, 62%) starting from 5f (95 mg, 0.245 mmol) following the procedure described for 6a. 1H NMR (400 MHz, DMSO-d6) δ 10.37 (s, 1H, exchangeable with D2O), 10.21 (s, 1H, exchangeable with D2O), 8.53 (d, J = 2.1 Hz, 1H), 7.97 (s, 1H), 7.93 (d, J = 8.9 Hz, 1H), 7.70 (dd, J = 8.9, 2.2 Hz, 1H), 7.41 (s, 1H, exchangeable with D2O), 7.37 (br t, 1H, exchangeable with D2O), 7.33 (s, 1H), 3.23–3.18 (m, 2H), 2.42 (t, J = 7.0 Hz, 2H), 1.70–1.63 (m, 2H), 1.61–1.53 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.81, 168.13, 156.20, 153.27, 138.45, 130.35, 130.10, 127.80, 127.35, 121.57, 120.97, 110.14, 107.56, 42.13, 38.60, 36.47, 28.67, 22.65. HRMS (ESI): m/z [M + H]+ calcd for C19H24N4O4 + H+: 373.1870. Found: 373.1865.
6-(5-((4,5-Dihydro-1H-imidazol-2-yl)amino)pentanamido)-4-hydroxy-2-naphthoic Acid (6f)
Compound 6f was obtained as a pale brown solid (84 mg, 79%) starting from 5g (85 mg, 0.221 mmol) following the procedure described for 6a. 1H NMR (400 MHz, DMSO-d6) δ 12.78 (s, 1H, exchangeable with D2O), 10.37 (s, 1H, exchangeable with D2O), 10.21 (s, 1H, exchangeable with D2O), 8.53 (d, J = 2.1 Hz, 1H), 8.31 (br t, 1H, exchangeable with D2O), 7.97 (s, 1H), 7.93 (d, J = 8.9 Hz, 1H), 7.70 (dd, J = 8.9, 2.1 Hz, 1H), 7.33 (s, 1H), 3.67–3.54 (m, 4H), 3.20–3.15 (m, 2H), 2.41 (t, J = 7.1 Hz, 2H), 1.69–1.59 (m, 2H), 1.59–1.52 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 171.74, 168.13, 159.87, 153.27, 138.44, 130.35, 130.10, 127.80, 127.35, 121.56, 120.97, 110.15, 107.56, 42.92, 42.49, 36.39, 28.85, 22.63. HRMS (ESI): m/z [M + H]+ calcd for C19H22N4O4 + H+: 371.1714. Found: 371.1706.
Methyl 6-(3-(3-Guanidinopropyl)ureido)-4-hydroxy-2-naphthoate (7a)
The TFA salt of compound 7a (36 mg, 99%) was obtained starting from 25a (43 mg, 0.077 mmol) following the procedure described for 5a. 1H NMR (400 MHz, DMSO-d6) δ 10.26 (s, 1H, exchangeable with D2O), 8.94 (s, 1H, exchangeable with D2O), 8.24 (d, J = 2.2 Hz, 1H), 7.93 (s, 1H), 7.85 (d, J = 9.0 Hz, 1H), 7.54–7.47 (m, 2H, 1H exchangeable with D2O), 7.26 (s, 1H), 7.07 (br s, 3H, exchangeable with D2O), 6.40 (br t, 1H, exchangeable with D2O), 3.82 (s, 3H), 3.16–3.11 (m, 4H), 1.70–1.58 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 166.62, 156.75, 155.32, 152.54, 139.72, 129.70, 128.77, 127.91, 124.63, 121.23, 120.16, 107.22, 106.46, 51.92, 38.44, 36.45, 29.39. HRMS (ESI): m/z [M + H]+ calcd for C17H21N5O4 + H+: 360.1666. Found: 360.1659.
Methyl 6-(3-(4-Guanidinobutyl)ureido)-4-hydroxy-2-naphthoate (7b)
The TFA salt of compound 7b was obtained as a yellow solid (39 mg, 97%) starting from compound 25b (49 mg, 0.082 mmol) following the procedure described for 5a. 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H, exchangeable with D2O), 8.96 (s, 1H, exchangeable with D2O), 8.28 (d, J = 2.2 Hz, 1H), 7.96 (s, 1H), 7.88 (d, J = 8.9 Hz, 1H), 7.59–7.52 (m, 2H, 1H exchangeable with D2O), 7.29 (s, 1H), 7.06 (br s, 3H, exchangeable with D2O), 6.44 (br t, 1H, exchangeable with D2O), 3.85 (s, 3H), 3.23–3.03 (m, 4H), 1.60–1.39 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 166.63, 156.67, 155.18, 152.52, 139.79, 129.70, 128.72, 127.93, 124.58, 121.23, 120.10, 107.08, 106.45, 51.92, 27.04, 26.00. HRMS (ESI): m/z [M + H]+ calcd for C18H23N5O4 + H+: 374.1823. Found: 374.1816.
Methyl 6-(3-(5-Guanidinopentyl)ureido)-4-hydroxy-2-naphthoate (7c)
The TFA salt of compound 7c was obtained as a yellow solid (77 mg, 90%) starting from compound 25c (100 mg, 0.170 mmol) following the procedure described for 5a. 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H, exchangeable with D2O), 8.91 (s, 1H, exchangeable with D2O), 8.28 (d, J = 2.2 Hz, 1H), 7.97 (s, 1H), 7.88 (d, J = 8.9 Hz, 1H), 7.56–7.52 (m, 2H, 1H, exchangeable with D2O), 7.30 (s, 1H), 7.08 (br s, 3H, exchangeable with D2O), 6.36 (br t, 1H, exchangeable with D2O), 3.86 (s, 3H), 3.15–3.08 (m, 4H), 1.53–1.45 (m, 4H), 1.36–1.31 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 167.13, 157.19, 155.61, 153.02, 140.33, 130.19, 129.20, 128.44, 125.05, 121.74, 120.58, 107.52, 106.95, 52.41, 41.21, 29.88, 28.67, 23.94. HRMS (ESI): m/z [M + H]+ calcd for C19H25N5O4 + H+: 388.1979. Found: 388.1969.
Methyl 4-Hydroxy-6-(3-(3-(3-methylguanidino)propyl)ureido)-2-naphthoate (7d)
The TFA salt of compound 7d was obtained as a yellow solid (39 mg, 97%) starting from compound 26a (47 mg, 0.082 mmol) following the procedure described for 5a. 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H, exchangeable with D2O), 8.95 (s, 1H, exchangeable with D2O), 8.27 (d, J = 2.2 Hz, 1H), 7.97 (s, 1H), 7.89 (d, J = 8.9 Hz, 1H), 7.57 (dd, J = 8.9, 2.2 Hz, 1H), 7.42–7.30 (m, 4H, 3H exchangeable with D2O), 6.39 (br t, 1H, exchangeable with D2O), 3.86 (s, 3H), 3.20–3.15 (m, 4H), 2.76 (d, J = 4.7 Hz, 3H), 1.72–1.65 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.12, 156.76, 155.82, 153.03, 140.19, 130.21, 129.28, 128.40, 125.15, 121.73, 120.66, 107.74, 106.96, 52.43, 36.97, 29.89, 28.44. HRMS (ESI): m/z [M + H]+ calcd for C18H23N5O4 + H+: 374.1823. Found: 374.1816.
Methyl 4-Hydroxy-6-(3-(4-(3-methylguanidino)butyl)ureido)-2-naphthoate (7e)
The TFA salt of compound 7e was obtained as a yellow solid (136 mg, 92%) starting from compound 26b (125 mg, 0.250 mmol) following the procedure described for 5a. 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H, exchangeable with D2O), 8.94 (s, 1H, exchangeable with D2O), 8.28 (d, J = 2.1 Hz, 1H), 7.97 (s, 1H), 7.89 (d, J = 8.8 Hz, 1H), 7.55 (dd, J = 8.9, 2.2 Hz, 1H), 7.44–7.21 (m, 4H, 3H exchangeable with D2O), 6.41 (br t, 1H, exchangeable with D2O), 3.86 (s, 3H), 3.26–3.08 (m, 4H), 2.74 (d, J = 4.5 Hz, 3H), 1.60–1.42 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 166.63, 156.23, 155.21, 152.53, 139.81, 129.69, 128.72, 127.94, 124.58, 121.23, 120.11, 107.09, 106.45, 51.91, 40.60, 38.59, 27.91, 27.04, 26.01. HRMS (ESI): m/z [M + H]+ calcd for C19H25N5O4 + H+: 388.1979. Found: 388.1972.
Methyl 6-(3-(3-(1H-Imidazol-1-yl)propyl)ureido)-4-hydroxy-2-naphthoate (7f)
To a stirring solution of methyl 4-acetoxy-6-((phenoxycarbonyl)amino)-2-naphthoate 22 (400 mg, 1.05 mmol) in dry DMF (4 mL), a solution of 3-(1H-imidazol-1-yl)propan-1-amine (263 mg, 2.11 mmol) and TEA (293 μL, 2.10 mmol) in dry DMF (4 mL) was added, and the reaction was stirred at room temperature for 2 h. NaHCO3 saturated solution was added (25 mL), and the resulting mixture was extracted with AcOEt (3 × 25 mL). The combined organic layers were washed with NaHCO3 saturated solution (2 × 10 mL) and brine (10 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a light brown solid that was purified by flash chromatography (gradient AcOEt/MeOH 100:0 to 90:10) yielding a mixture of acetylated and deacetylated compounds. Therefore, the solid was suspended in DCM (15 mL), and piperidine (290 μL) was added. After 1 h, the solvent was removed, and the crude product was diluted with AcOEt (50 mL). The organic layer was washed with 1 N HCl (3× 20 mL) and brine (30 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure, yielding pure 7f as a white solid (274 mg, 71%). 1H NMR (400 MHz, DMSO-d6) δ 10.32 (s, 1H, exchangeable with D2O), 8.87 (s, 1H, exchangeable with D2O), 8.27 (d, J = 2.2 Hz, 1H), 8.00–7.97 (m, 1H), 7.89 (d, J = 8.9 Hz, 1H), 7.67 (s, 1H), 7.56 (dd, J = 8.9, 2.2 Hz, 1H), 7.30 (d, J = 1.6 Hz, 1H), 7.22 (s, 1H), 6.91 (s, 1H), 6.36 (br t, 1H, exchangeable with D2O), 4.02 (t, J = 6.9 Hz, 2H), 3.86 (s, 3H), 3.12–3.07 (m, 2H), 1.94–1.87 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.12, 155.65, 153.03, 140.20, 137.75, 130.19, 129.25, 128.87, 128.40, 125.11, 121.72, 120.65, 119.80, 107.70, 106.95, 52.41, 44.16, 36.92, 31.85. HRMS (ESI): m/z [M + H]+ calcd for C19H20N4O4 + H+: 369.1557. Found: 369.1550.
6-(3-(4-Guanidinobutyl)ureido)-4-hydroxy-2-naphthoic Acid (8a)
The TFA salt of compound 8a was obtained as a yellow solid (45 mg, 92%) starting from compound 7b (50 mg, 0.102 mmol) following the procedure described for 6a. 1H NMR (400 MHz, DMSO-d6) δ 10.23 (s, 1H, exchangeable with D2O), 8.94 (s, 1H, exchangeable with D2O), 8.27 (d, J = 2.2 Hz, 1H), 7.93 (s, 1H), 7.86 (d, J = 8.9 Hz, 1H), 7.61–7.52 (m, 2H, 1H exchangeable with D2O), 7.29 (s, 1H), 7.25–6.63 (br s, 3H, exchangeable with D2O), 6.44 (br t, 1H, exchangeable with D2O), 3.23–3.05 (m, 4H), 1.68–1.39 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 167.73, 156.66, 155.21, 152.35, 139.50, 129.59, 128.79, 127.73, 125.80, 121.26, 119.95, 107.16, 106.92, 27.06, 26.00. HRMS (ESI): m/z [M + H]+ calcd for C17H21N5O4 + H+: 360.1666. Found: 360.1658.
6-(3-(3-(1H-Imidazol-1-yl)propyl)ureido)-4-hydroxy-2-naphthoic Acid (8b)
Compound 8b was obtained as a yellow solid (62 mg, 89%) starting from compound 7f (72 mg, 0.195 mmol) following the procedure described for 6a. 1H NMR (400 MHz, DMSO-d6) δ 12.69 (s, 1H, exchangeable with D2O), 10.22 (s, 1H, exchangeable with D2O), 9.13 (s, 1H, exchangeable with D2O), 9.02 (s, 1H), 8.27 (d, J = 2.2 Hz, 1H), 7.94 (s, 1H), 7.89–7.83 (m, 2H), 7.70 (s, 1H), 7.56 (dd, J = 8.9, 2.2 Hz, 1H), 7.30 (s, 1H), 6.53 (br t, 1H, exchangeable with D2O), 4.26 (t, J = 7.0 Hz, 2H), 3.18–3.13 (m, 2H), 2.05–1.98 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.21, 155.84, 152.87, 139.90, 135.99, 130.06, 129.35, 128.20, 126.36, 122.47, 121.74, 120.65, 120.54, 107.86, 107.42, 46.92, 36.51, 30.94. HRMS (ESI): m/z [M + H]+ calcd for C18H18N4O4 + H+: 355.1401. Found: 355.1394.
Methyl (S)-6-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-(methylthio)butanamido)-4-((tert-butoxycarbonyl)oxy)-2-naphthoate (9a)
Under nitrogen atmosphere 16 (692 mg, 2.18 mmol), (((9H-fluoren-9-yl)methoxy)carbonyl)-l-methionine (969 mg, 2.61 mmol), and DMAP (27 mg, 0.218 mmol) were dissolved in dry DCM (8 mL), and then DCC (536 mg, 2.60 mmol) was added. The reaction mixture was stirred until the disappearance of the starting material (TLC analysis, 8–12 h). The resulting suspension was filtered, and the filtrate was evaporated under reduced pressure. The crude material was purified by flash chromatography (gradient Hex/AcOEt 80:20 to 20:80) yielding 9a as a pale yellow solid (1.18 g, 81%). 1H NMR (400 MHz, DMSO-d6) δ 10.57 (s, 1H, exchangeable with D2O), 8.51 (s, 1H, exchangeable with D2O), 8.47–8.41 (m, 1H), 8.19 (d, J = 9.0 Hz, 1H), 7.92–7.88 (m, 2H), 7.86–7.72 (m, 5H), 7.45–7.39 (m, 2H), 7.36–7.31 (m, 2H), 4.35–4.28 (m, 3H), 4.27–4.22 (m, 1H), 3.92 (s, 3H), 3.32–3.28 (m, 2H), 2.08 (s, 3H), 2.05–1.92 (m, 2H), 1.52 (s, 9H). MS (ESI) m/z: 671 (M + H)+.
Methyl (S)-6-(2-Amino-4-(methylthio)butanamido)-4-hydroxy-2-naphthoate (9c)
To a solution of 9a (1.07 g, 1.60 mmol) in 20 mL of DCM, piperidine (1 mL) was added. After 30 min, the solvent was removed, and the crude product was diluted with AcOEt (40 mL). The organic layer was washed with 1 N HCl (3 × 10 mL) and brine (30 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude material 9b was dissolved in 10 mL of a solution of DCM/TFA (9:1), and the mixture was stirred for 1 h. The solvent was evaporated, and the resulting solid was washed with CHCl3 to give the pure TFA salt of compound 9c as a light brown solid (503 mg, 68%). 1H NMR (400 MHz, DMSO-d6) δ 10.43 (s, 2H, exchangeable with D2O), 8.58 (s, 1H), 8.04–8.01 (m, 1H), 7.97 (d, J = 8.8 Hz, 1H), 7.77 (dd, J = 8.8, 2.2 Hz, 1H), 7.34 (d, J = 2.2 Hz, 1H), 3.87 (s, 3H), 3.49–3.46 (m, 1H), 3.00–2.97 (m, 1H), 2.64–2.55 (m, 2H), 2.06 (s, 3H), 2.00–1.93 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 174.11, 166.55, 153.00, 137.81, 129.93, 127.46, 125.73, 121.05, 120.83, 109.84, 106.64, 54.72, 52.02, 34.26, 29.89, 14.62. HRMS (ESI): m/z [M + H]+ calcd for C17H20N2O4S + H+: 349.1217. Found: 349.1211.
Methyl (S)-4-Hydroxy-6-(2-(3-(4-methoxyphenyl)ureido)-4-(methylthio)butanamido)-2-naphthoate (9d)
A solution of 4-methoxyphenyl isocyanate (77 mg, 0.519 mmol) in dry THF (6 mL) was added dropwise to a solution of 9c (200 mg, 0.432 mmol) and TEA (126 μL, 0.950 mmol) in dry THF (20 mL) under N2 atmosphere. The reaction was stirred at room temperature for 6 h. The precipitate was collected by filtration and rinsed with water and diethyl ether successively. Purification by crystallization (EtOH) gave the title compound as a pale yellow solid (169 mg, 79%). 1H NMR (400 MHz, DMSO-d6) δ 10.55–10.45 (m, 2H, exchangeable with D2O), 8.55 (d, J = 2.2 Hz, 1H), 8.47 (s, 1H, exchangeable with D2O), 8.05–8.01 (m, 1H), 7.99 (d, J = 8.9 Hz, 1H), 7.76 (dd, J = 8.9, 2.2 Hz, 1H), 7.34 (s, 1H), 7.30 (d, J = 9.0 Hz, 2H), 6.82 (d, J = 9.0 Hz, 2H), 6.50 (br d, 1H exchangeable with D2O), 4.53–4.48 (m, 1H), 3.87 (s, 3H), 3.69 (s, 3H), 2.57–2.53 (m, 2H), 2.08 (s, 3H), 2.08–2.02 (m, 1H), 2.02–1.87 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 171.26, 166.53, 155.03, 154.06, 153.01, 137.77, 133.26, 130.01, 129.81, 127.41, 125.85, 121.05, 120.82, 119.33, 113.93, 110.16, 106.68, 55.12, 53.15, 52.03, 32.84, 29.52, 14.74. HRMS (ESI): m/z [M + H]+ calcd for C25H27N3O6S + H+: 498.1693. Found: 498.1687.
Methyl 6-(2-((tert-Butoxycarbonyl)amino)acetamido)-4-((tert-butoxycarbonyl)oxy)-2-naphthoate (10a)
Compound 10a was obtained as a pale yellow solid (1.58 g, 79%) by the reaction of amine 16 (1.34 g, 4.22 mmol) and (tert-butoxycarbonyl)glycine (888 mg, 5.07 mmol) following the procedure described for 9a. 1H NMR (400 MHz, CDCl3) δ 8.56 (br s, 1H, exchangeable with D2O), 8.47 (s, 1H), 8.27 (s, 1H), 7.94 (d, J = 8.9 Hz, 1H), 7.85 (d, J = 1.5 Hz, 1H), 7.14–7.09 (m, 1H), 5.27 (br s, 1H, exchangeable with D2O), 4.00 (s, 3H), 3.69 (d, J = 6.2 Hz, 2H), 1.53–1.48 (m, 18H). MS (ESI) m/z: 475 (M + H)+.
Methyl 6-(2-Aminoacetamido)-4-hydroxy-2-naphthoate (10b)
Compound 10b was obtained as a light brown solid (434 mg, 74%) by the reaction of 10a (716 mg, 1.51 mmol) following the procedure described for 5a. 1H NMR (400 MHz, DMSO-d6) δ 10.42 (s, 1H, exchangeable with D2O), 10.28 (s, 1H, exchangeable with D2O), 8.54 (s, 1H), 8.03–7.87 (m, 2H), 7.81–7.61 (m, 4H, 3H exchangeable with D2O), 7.32 (s, 1H), 3.92–3.86 (m, 2H), 3.86 (s, 3H). MS (ESI) m/z: 275 (M + H)+.
Methyl 4-Hydroxy-6-(2-(3-(4-methoxyphenyl)ureido)acetamido)-2-naphthoate (10c)
Compound 10c was obtained as a white solid (250 mg, 81%) by the reaction of amine 10b (283 mg, 0.731 mmol) and 4-methoxyphenyl isocyanate (129 mg, 0.871 mmol) following the procedure described for 9d. 1H NMR (400 MHz, DMSO-d6) δ 10.56 (s, 1H, exchangeable with D2O), 10.33 (s, 1H, exchangeable with D2O), 8.63 (s, 1H, exchangeable with D2O), 8.51 (s, 1H), 8.04–7.95 (m, 2H), 7.70 (dd, J = 8.9, 2.2 Hz, 1H), 7.32–7.28 (m, 3H), 6.82 (d, J = 9.0 Hz, 2H), 6.36 (br t, 1H, exchangeable with D2O), 3.97 (d, J = 5.6 Hz, 2H), 3.86 (s, 3H), 3.68 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 169.33, 166.84, 155.84, 154.32, 153.16, 138.05, 133.50, 130.12, 130.0986, 129.79, 127.51, 125.71, 121.02, 120.64, 119.32, 113.88, 109.75, 106.67, 55.12, 52.01, 21.66. HRMS (ESI): m/z [M + H]+ calcd for C22H21N3O6 + H+: 424.1503. Found: 424.1500.
Methyl (S)-6-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-5-(3-((2,2,4,6,7-pentamethyl-2,3-dihydrobenzofuran-5-yl)sulfonyl)guanidino)pentanamido)-4-((tert-butoxycarbonyl)oxy)-2-naphthoate (11a)
Compound 11a was obtained as a pale yellow solid (1.50 g, 79%) by the reaction of Nα-(((9H-fluoren-9-yl)methoxy)carbonyl)-Nω-((2,2,4,6,7-pentamethyl-2,3-dihydrobenzofuran-5-yl)sulfonyl)-l-arginine (1.55 g, 2.40 mmol) with amine 16 (634 mg, 2.00 mmol) following the procedure described for 9a. 1H NMR (400 MHz, DMSO-d6) δ 10.55 (s, 1H, exchangeable with D2O), 8.51 (d, J = 1.8 Hz, 1H), 8.50–8.43 (m, 1H), 8.22–8.16 (m, 1H), 7.93–7.87 (m, 2H), 7.87–7.84 (m, 2H), 7.82–7.72 (m, 3H), 7.45–7.39 (m, 1H), 7.39–7.30 (m, 2H), 6.72 (br s, 2H, exchangeable with D2O), 6.38 (br s, 2H, exchangeable with D2O), 6.28 (s, 1H), 4.37–4.27 (m, 1H), 4.27–4.19 (m, 1H), 3.92 (s, 3H), 3.15–3.05 (m, 2H), 2.95–2.84 (m, 2H), 2.48–2.45 (m, 3H), 2.42–2.38 (m, 4H), 1.98–1.92 (m, 4H), 1.55 (s, 9H), 1.52 (s, 3H), 1.37 (s, 3H), 1.36 (s, 3H). MS (ESI) m/z: 948 (M + H)+.
Methyl (S)-6-(2-Amino-5-(3-((2,2,4,6,7-pentamethyl-2,3-dihydrobenzofuran-5-yl)sulfonyl)guanidino)pentanamido)-4-((tert-butoxycarbonyl)oxy)-2-naphthoate (11b)
To a solution of 11a (948 mg, 1.00 mmol) in 20 mL of DCM, piperidine (1 mL) was added. After 30 min, the solvent was removed, and the crude product was diluted with AcOEt (40 mL). The organic layer was washed with 1 N HCl (3 × 10 mL) and brine (30 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure yielding pure 11b as a light yellow solid (522 mg, 72%). 1H NMR (400 MHz, DMSO-d6) δ 10.31 (s, 1H, exchangeable with D2O), 8.57 (d, J = 2.2 Hz, 1H), 8.01 (s, 1H), 7.97 (d, J = 8.9 Hz, 1H), 7.76 (dd, J = 8.8, 2.2 Hz, 1H), 7.34 (d, J = 1.8 Hz, 1H), 6.75 (br s, 2H, exchangeable with D2O), 6.39 (br s, 2H, exchangeable with D2O), 3.87 (s, 3H), 3.10–3.03 (m, 3H), 2.90–2.85 (m, 4H), 2.48 (s, 6H), 2.46 (s, 3H), 2.39 (s, 3H), 1.96 (s, 3H), 1.37 (s, 9H). MS (ESI) m/z: 726 (M + H)+.
Methyl (S)-6-(2-Amino-5-guanidinopentanamido)-4-hydroxy-2-naphthoate (11c)
Compound 11b (100 mg, 0.137 mmol) was dissolved in a solution of DCM/TFA 1:9, and then a drop of water was added. The resulting mixture was stirred at room temperature for 24 h. The solvent was evaporated, and the resulting solid was washed with CHCl3 to afford the TFA salt as a white solid (45 mg, 68%). 1H NMR (400 MHz, DMSO-d6) δ 10.95 (s, 1H, exchangeable with D2O), 8.55 (d, J = 2.0 Hz, 1H), 8.45–8.35 (m, 3H, exchangeable with D2O), 8.04–7.99 (m, 1H), 7.92–7.89 (m, 1H), 7.74 (dd, J = 8.7, 2.4 Hz, 1H), 7.46–7.22 (m, 4H, 1H, exchangeable with D2O), 3.87 (s, 3H), 3.19–3.14 (m, 2H), 1.91–1.86 (m, 2H), 1.63–1.57 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.05, 166.99, 157.38, 153.63, 137.38, 130.83, 130.55, 127.80, 126.79, 121.45, 121.09, 111.11, 107.36, 53.32, 52.53, 40.61, 28.87, 24.74. HRMS (ESI): m/z [M + H]+ calcd for C18H23N5O4 + H+: 374.1823. Found: 374.1821.
Methyl (S)-4-((tert-Butoxycarbonyl)oxy)-6-(2-(3-(4-methoxyphenyl)ureido)-5-(3-((2,2,4,6,7-pentamethyl-2,3-dihydrobenzofuran-5-yl)sulfonyl)guanidino)pentanamido)-2-naphthoate (11d)
Compound 11d was obtained as a pale yellow solid (239 mg, 82%) by the reaction of 4-methoxyphenyl isocyanate (61 mg, 0.413 mmol) with amine 11b (250 mg, 0.334 mmol) following the procedure described for 9d. 1H NMR (400 MHz, DMSO-d6) δ 10.58–10.41 (m, 2H, exchangeable with D2O), 8.55 (d, J = 2.1 Hz, 1H), 8.50 (s, 1H, exchangeable with D2O), 8.04–8.01 (m, 1H), 7.98 (d, J = 8.9 Hz, 1H), 7.75 (dd, J = 8.9, 2.2 Hz, 1H), 7.34 (s, 1H), 7.29 (d, J = 9.0 Hz, 2H), 6.82 (d, J = 9.0 Hz, 2H), 6.74 (brs, 2H, exchangeable with D2O), 6.43 (br d, 1H, exchangeable with D2O), 6.35 (s, 2H, exchangeable with D2O), 4.45–4.41 (m, 1H), 3.87 (s, 3H), 3.69 (s, 3H), 3.15–3.06 (m, 2H), 2.82 (s, 3H), 2.44 (s, 3H), 2.37 (s, 3H), 1.93 (s, 3H), 1.82–1.68 (m, 2H), 1.69–1.58 (m, 2H), 1.49–1.38 (m, 2H), 1.35 (s, 9H). MS (ESI) m/z: 875 (M + H)+.
Methyl (S)-6-(5-Guanidino-2-(3-(4-methoxyphenyl)ureido)pentanamido)-4-hydroxy-2-naphthoate (11e)
Compound 11e was obtained as a pale yellow solid (74 mg, 77%) starting from 11d (131 mg, 0.150 mmol) following the procedure described for 11c. 1H NMR (400 MHz, DMSO-d6) δ 10.50–10.47 (m, 2H, exchangeable with D2O), 8.56 (d, J = 3.5 Hz, 1H), 8.02 (s, 1H), 8.00 (d, J = 8.9 Hz, 1H), 7.76 (dd, J = 8.9, 2.2 Hz, 1H), 7.53–7.48 (m, 1H), 7.35–7.34 (m, 1H), 7.30 (d, J = 9.1 Hz 2H), 7.05 (br s, 3H, exchangeable with D2O), 6.84 (d, J = 9.1 Hz, 2H), 6.52–6.49 (m, 1H, exchangeable with D2O), 4.52–4.45 (m, 1H), 3.88 (s, 3H), 3.69 (s, 3H), 3.19–3.11 (m, 2H), 1.85–1.65 (m, 2H), 1.59–1.45 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.44, 166.51, 156.61, 154.98, 154.07, 153.01, 137.67, 133.26, 130.04, 129.88, 127.40, 125.91, 121.06, 120.72, 119.30, 113.95, 110.12, 106.72, 55.13, 53.12, 52.05, 40.44, 30.51, 25.07. HRMS (ESI): m/z [M + H]+ calcd for C26H30N6O6 + H+: 523.2300. Found: 523.2293.
Methyl 6-(3-(3-(3-(((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)guanidino)propyl)ureido)-4-hydroxy-2-naphthoate (12a)
Compound 29a (100 mg, 0.118 mmol) was dissolved in a solution of DCM/TFA 1:1, and then a drop of water was added. The resulting mixture was stirred at room temperature for 1 h. The solvent was evaporated, and the resulting solid was washed with CHCl3 to afford the TFA salt 12a as a white solid (64 mg, 76%). 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H, exchangeable with D2O), 8.95 (s, 1H, exchangeable with D2O), 8.45 (s, 1H), 8.27–8.21 (m, 2H), 7.98 (s, 1H), 7.88 (d, J = 8.9 Hz, 1H), 7.86 (br s, 2H, exchangeable with D2O), 7.56 (dd, J = 9.0, 2.2 Hz, 1H), 7.49–7.42 (m, 4H, exchangeable with D2O), 7.30 (s, 1H), 6.39 (br t, 1H, exchangeable with D2O), 5.95 (d, J = 5.7 Hz, 1H), 4.72 (t, J = 5.4 Hz, 1H), 4.18–4.16 (m, 1H), 4.06–4.01 (m, 1H), 3.87 (s, 3H), 3.18–3.12 (m, 4H), 1.70–1.60 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 166.63, 155.88, 155.31, 152.53, 149.03, 140.76, 139.69, 129.72, 128.79, 127.90, 124.66, 121.25, 120.17, 119.22, 107.25, 106.47, 87.82, 82.34, 72.71, 71.07, 51.94, 43.17, 36.47, 29.32. HRMS (ESI): m/z [M + H]+ calcd for C27H32N10O7 + H+: 609.2528. Found: 609.2520.
Methyl 6-(3-(4-(3-(((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)guanidino)butyl)ureido)-4-hydroxy-2-naphthoate (12b)
Compound 12b was obtained as a white solid (33 mg, 75%) starting from compound 29b (52 mg, 0.060 mmol) following the procedure described for 12a. 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H, exchangeable with D2O), 8.90 (s, 1H, exchangeable with D2O), 8.42 (s, 1H), 8.28–8.21(m, 2H), 7.97 (s, 1H), 7.88 (d, J = 8.9 Hz, 1H), 7.78 (br s, 2H, exchangeable with D2O), 7.54 (dd, J = 8.9, 2.2 Hz, 1H), 7.42–7.33 (m, 4H, exchangeable with D2O), 7.30 (s, 1H), 6.34 (br t, 1H, exchangeable with D2O), 5.93 (d, J = 5.7 Hz, 1H), 4.71 (t, J = 5.7 Hz, 1H), 4.17–4.15 (m, 1H), 4.06–3.98 (m, 1H), 3.86 (s, 3H), 3.60–3.48 (m, 2H), 3.17–3.05 (m, 4H), 1.51–1.45 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 166.62, 155.80, 155.14, 152.51, 151.14, 149.05, 140.59, 139.77, 129.70, 128.72, 127.91, 121.23, 120.09, 119.22, 107.08, 106.45, 87.79, 82.34, 72.64, 71.05, 51.92, 43.12, 26.99, 25.88. HRMS (ESI): m/z [M + H]+ calcd for C28H34N10O7 + H+: 623.2685. Found: 623.2689.
Methyl 6-(3-(5-(3-(((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)guanidino)pentyl)ureido)-4-hydroxy-2-naphthoate (12c)
Compound 12c was obtained as a pale yellow solid (63 mg, 78%) starting from compound 29c (95 mg, 0.108 mmol) following the procedure described for 12a. 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H, exchangeable with D2O), 8.83 (s, 1H, exchangeable with D2O), 8.38 (s, 1H), 8.25 (d, J = 2.2 Hz, 1H), 8.20 (s,1H), 7.96 (s, 1H), 7.88 (d, J = 8.9 Hz, 1H), 7.59 (s, 2H, exchangeable with D2O), 7.53 (dd, J = 8.9, 2.2 Hz, 1H), 7.37–7.32 (m, 4H, exchangeable with D2O), 7.29 (s, 1H), 6.25 (br t, 1H, exchangeable with D2O), 5.92 (d, J = 5.7 Hz, 1H), 4.71 (t, J = 5.4 Hz, 1H), 4.16–4.14 (m, 1H), 4.03–3.99 (m, 1H), 3.85 (s, 3H), 3.10–3.05 (m, 4H), 1.47–1.40 (m, 4H), 1.28–1.24 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.13, 156.32, 155.59, 153.01, 149.59, 141.00, 140.29, 130.21, 129.21, 128.42, 125.08, 121.75, 120.59, 119.77, 107.54, 106.95, 88.33, 82.87, 73.10, 71.54, 52.42, 43.62, 41.49, 29.88, 28.58, 23.91. HRMS (ESI): m/z [M + H]+ calcd for C29H36N10O7 + H+: 637.2841. Found: 637.2843.
Methyl 6-(3-(6-(3-(((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)guanidino)hexyl)ureido)-4-hydroxy-2-naphthoate (12d)
Compound 12d was obtained as a pale yellow solid (0.065 g, 74%) starting from compound 29d (92 mg, 0.103 mmol) following the procedure described for 12a. 1H NMR (400 MHz, DMSO-d6) δ 10.27 (s, 1H, exchangeable with D2O), 8.86 (s, 1H, exchangeable with D2O), 8.41 (s, 1H), 8.25 (d, J = 2.2 Hz, 1H), 8.24 (s,1H), 7.46 (s, 1H), 7.87 (d, J = 8.9 Hz, 1H), 7.80 (s, 2H, exchangeable with D2O), 7.53 (dd, J = 8.9, 2.2 Hz, 1H), 7.42–7.35 (m, 4H), 7.29 (s, 1H), 6.30 (br t, 1H, exchangeable with D2O), 5.92 (d, J = 5.7 Hz, 1H), 4.69 (t, J = 5.4 Hz, 1H), 4.18–4.14 (m, 1H), 4.03–3.99 (m, 1H), 3.85 (s, 3H), 3.58–3.45 (m, 2H), 3.12–3.03 (m, 4H), 1.48–1.34 (m, 4H), 1.26–1.18 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 166.63, 155.83, 155.08, 152.51, 151.04, 149.03, 140.63, 139.383, 129.69, 128.70, 127.94, 124.55, 121.24, 120.08, 119.23, 107.01, 106.45, 87.81, 82.42, 72.66, 71.02, 51.92, 43.10, 40.98, 29.65, 28.32, 25.95, 25.76. HRMS (ESI): m/z [M + H]+ calcd for C30H38N10O7 + H+: 651.2998. Found: 651.2997.
Methyl 6-(3-(7-(3-(((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)guanidino)heptyl)ureido)-4-hydroxy-2-naphthoate (12e)
Compound 12e was obtained as a pale yellow solid (58 mg, 75%) starting from compound 29e (0.100 g, 0.110 mmol) following the procedure described for 12a. 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H, exchangeable with D2O), 8.85 (s, 1H, exchangeable with D2O), 8.42 (s, 1H), 8.27 (d, J = 2.1 Hz, 1H), 8.24 (s,1H), 7.97 (s, 1H), 7.88 (d, J = 8.9 Hz, 1H), 7.80 (s, 2H, exchangeable with D2O), 7.54 (dd, J = 8.9, 2.1 Hz, 1H), 7.42–7.26 (m, 5H, 4H exchangeable with D2O), 6.29 (br t, 1H, exchangeable with D2O), 5.93 (d, J = 5.7 Hz, 1H), 4.70 (t, J = 5.4 Hz, 1H), 4.17–4.14 (m, 1H), 4.03–4.01 (m, 1H), 3.86 (s, 3H), 3.56–3.50 (m, 2H), 3.13–3.03 (m, 4H), 1.44–1.40 (m, 4H), 1.29–1.24 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 167.13, 156.32, 155.57, 153.01, 151.60, 149.54, 141.11, 140.32, 130.20, 129.20, 128.44, 125.05, 121.74, 120.57, 119.74, 107.50, 106.95, 88.31, 82.92, 73.16, 71.52, 52.41, 43.60, 41.47, 30.18, 28.81, 26.78, 26.49. HRMS (ESI): m/z [M + H]+ calcd for C31H40N10O7 + H+: 665.3154. Found: 665.3148.
Methyl 6-(3-(4-(3-(2-((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)ethyl)guanidino)butyl)ureido)-4-hydroxy-2-naphthoate (12f)
Compound 12f was obtained as a pale yellow solid (85 mg, 80%) starting from compound 30 (110 mg, 0.141 mmol) following the procedure described for 12a. 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H, exchangeable with D2O), 8.90 (s, 1H, exchangeable with D2O), 8.41 (s, 1H), 8.27 (d, J = 2.2 Hz, 1H), 8.22 (s, 1H), 7.97 (s, 1H), 7.88 (d, J = 8.9 Hz, 1H), 7.66 (br s, 2H, exchangeable with D2O), 7.55 (dd, J = 8.9, 2.2 Hz, 1H), 7.34–7.24 (m, 5H, 4H, exchangeable with D2O), 6.36 (br t, 1H, exchangeable with D2O), 5.90 (d, J = 5.0 Hz, 1H), 4.69 (t, J = 5.4 Hz, 1H), 4.13–4.11 (m, 1H), 3.94–3.89 (m, 1H), 3.86 (s, 3H), 3.23–3.18 (m, 2H), 3.16–3.09 (m, 4H), 1.95–1.86 (m, 2H), 1.50–1.46 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 167.12, 156.00, 155.66, 153.01, 149.65, 140.96, 140.28, 130.20, 129.22, 128.42, 125.09, 121.74, 120.60, 119.68, 107.59, 106.95, 88.33, 81.64, 73.77, 73.40, 52.42, 46.24, 41.15, 38.54, 32.85, 27.52, 26.47. HRMS (ESI): m/z [M + H]+ calcd for C29H36N10O7 + H+: 637.2841. Found: 637.2838.
Methyl 6-(3-(4-(3-(3-((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)propyl)guanidino)butyl)ureido)-4-hydroxy-2-naphthoate (12g)
Compound 12g was obtained as a white solid (71 mg, 78%) starting from compound 31 (95 mg, 0.120 mmol) following the procedure described for 12a. 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H, exchangeable with D2O), 8.90 (s, 1H, exchangeable with D2O), 8.38 (s, 1H), 8.27 (d, J = 2.1 Hz, 1H), 8.23 (s, 1H), 7.97 (s, 1H), 7.88 (d, J = 8.9 Hz, 1H), 7.70 (s, 2H, exchangeable with D2O), 7.55 (dd, J = 8.9, 2.1 Hz, 1H), 7.34–7.28 (m, 5H, 4H exchangeable with D2O), 6.36 (br t, 1H, exchangeable with D2O), 5.87 (d, J = 5.0 Hz, 1H), 4.64 (t, J = 5.4 Hz, 1H), 4.08–4.06 (m, 1H), 3.86 (s, 3H), 3.16–3.12 (m, 6H), 1.70–1.48 (m, 8H). 13C NMR (101 MHz, DMSO-d6) δ 167.13, 155.95, 155.67, 153.01, 149.63, 140.93, 140.27, 130.21, 129.23, 128.42, 125.09, 121.74, 120.60, 107.60, 106.96, 88.32, 83.63, 73.72, 73.53, 52.42, 41.21, 41.13, 39.09, 30.52, 27.54, 26.49, 25.55. HRMS (ESI): m/z [M + H]+ calcd for C30H38N10O7 + H+: 651.2998. Found: 651.2996.
Methyl 6-(3-(4-(3-((E)-3-((2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)allyl)guanidino)butyl)ureido)-4-hydroxy-2-naphthoate (12h)
Compound 12h was obtained as a white solid (63 mg, 78%) starting from compound 32 (84 mg, 0.106 mmol) following the procedure described for 12a. 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H, exchangeable with D2O), 8.92 (s, 1H, exchangeable with D2O), 8.39 (s, 1H), 8.27 (d, J = 2.1 Hz, 1H), 8.24 (s, 1H), 7.97 (s, 1H), 7.88 (d, J = 9.0 Hz, 1H), 7.72 (s, 2H, exchangeable with D2O), 7.57–7.49 (m, 2H, 1H exchangeable with D2O), 7.43 (br t, 1H, exchangeable with D2O), 7.35 (s, 2H, exchangeable with D2O), 7.30 (s, 1H), 6.37 (br t, 1H, exchangeable with D2O), 5.93 (d, J = 5.0 Hz, 1H), 5.89–5.86 (m, 1H), 4.69 (t, J = 5.4 Hz, 1H), 4.38–4.36 (m, 1H), 4.15–4.12 (m, 1H), 3.84 (s, 3H), 3.84–3.81 (m, 2H), 3.17–3.13 (m, 4H), 1.55–1.42 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 167.13, 155.94, 155.68, 153.01, 149.63, 140.97, 140.27, 130.48, 130.21, 129.23, 128.38, 125.09, 121.74, 120.60, 88.38, 84.21, 74.44, 73.28, 54.07, 52.42, 44.66, 42.31, 41.21, 27.52, 26.47. HRMS (ESI): m/z [M + H]+ calcd for C30H36N10O7 + H+: 649.2841. Found: 649.2829.
Methyl 4-Hydroxy-6-nitro-2-naphthoate (14)
Compound 14 was obtained as a pale yellow solid (1.53 g, 99%) starting from compound 13 (1.80 g, 6.22 mmol) following the procedure described for 11b. 1H NMR (400 MHz, DMSO-d6) δ 8.99 (s, 1H, exchangeable with D2O), 8.38–8.06 (m, 4H), 7.50 (s, 1H), 3.91 (s, 3H). MS (ESI) m/z: 248 (M + H)+.
Methyl 4-((tert-Butoxycarbonyl)oxy)-6-nitro-2-naphthoate (15)
To a stirred suspension of 14 (1.53 g, 6.22 mmol) in dry DCM (8 mL), DMAP (75 mg, 0.613 mmol), TEA (0.95 mL, 6.80 mmol), and Boc2O (1.75 g, 8.04 mmol) were added. The resulting mixture was stirred at room temperature for 12 h. Then, the solvent was removed at reduced pressure, and the crude residue was purified by flash chromatography (gradient Hex/AcOEt 80:20 to 40:60) yielding 15 as a white solid (1.51 g, 70%). 1H NMR (400 MHz, CDCl3) δ 8.98 (d, J = 2.0 Hz, 1H), 8.57 (s, 1H), 8.34 (dd, J = 9.0, 2.0 Hz, 1H), 8.11 (m, 2H), 4.01 (s, 3H), 1.64 (s, 9H). MS (ESI) m/z: 348 (M + H)+.
Methyl 6-Amino-4-((tert-butoxycarbonyl)oxy)-2-naphthoate (16)
To a solution of 15 (1.51 g, 4.35 mmol) in acetic acid (50 mL), Zn dust (2.83 g, 43.50 mmol) was added. The resulting mixture was stirred for 1 h at room temperature, filtered, and concentrated in vacuo. The acid residue was dissolved in saturated aqueous solution of NaHCO3 (50 mL) and extracted with AcOEt (3 × 50 mL). The organic layer was washed with brine (50 mL), dried (Na2SO4), and filtered. Vacuum evaporation of the solvent gave the title compound 16 (1.35 g, 99%) as a pale yellow solid which was directly used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 7.78 (s, 1H), 7.73 (d, J = 8.7 Hz, 1H), 6.98 (s, 1H), 6.95 (d, J = 8.7 Hz, 1H), 4.15 (br s, 2H, exchangeable with D2O), 3.92 (s, 3H), 1.58 (s, 9H). MS (ESI) m/z: 318 (M + H)+.
Methyl 6-(5-((tert-Butoxycarbonyl)amino)pentanamido)-4-((tert-butoxycarbonyl)oxy)-2-naphthoate (17a)
Compound 17a was obtained as a pale yellow solid (1.80 g, 83%) starting from compound 16 (1.34 g, 4.22 mmol) and 5-tert-butoxycarbonylaminovaleric acid (1.10 g, 5.07 mmol) following the procedure described for 9a. 1H NMR (400 MHz, DMSO-d6) δ 10.34 (s, 1H, exchangeable with D2O), 8.48 (s, 1H), 8.43 (s, 1H), 8.16 (d, J = 9.0 Hz, 1H), 7.83–7.70 (m, 2H), 6.80 (br t, 1H, exchangeable with D2O), 3.90 (s, 3H), 3.02–2.89 (m, 2H), 2.38 (t, J = 7.2 Hz, 2H), 1.63–1.50 (m, 11H), 1.49–1.28 (m, 11H). MS (ESI) m/z: 517 (M + H)+.
Methyl 6-(6-((tert-Butoxycarbonyl)amino)hexanamido)-4-((tert-butoxycarbonyl)oxy)-2-naphthoate (17b)
Compound 17b was obtained as a pale yellow solid (950 mg, 80%) by the reaction of 6-tert-butoxycarbonylaminohexanoic acid (633 mg, 2.74 mmol) with amine 16 (725 mg, 2.28 mmol) following the procedure described for 9a. 1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H, exchangeable with D2O), 8.49 (s, 1H), 8.42 (s, 1H), 8.16 (d, J = 8.9 Hz, 1H), 7.78–7.73 (m, 2H), 6.81 (br t, 1H, exchangeable with D2O), 3.91 (s, 3H), 3.01–2.88 (m, 2H), 2.38 (m, 2H), 1.75–1.50 (m, 11H), 1.44–1.20 (m, 13H). MS (ESI) m/z: 531 (M+H)+.
Methyl 6-(7-((tert-Butoxycarbonyl)amino)heptanamido)-4-((tert-butoxycarbonyl)oxy)-2-naphthoate (17c)
Compound 17c was obtained as a pale yellow solid (1.52 g, 88%) by the reaction of 7-tert-butoxycarbonylaminoheptanoic acid (928 mg, 3.78 mmol) with amine 16 (1.00 g, 3.15 mmol) following the procedure described for 9a. 1H NMR (400 MHz, DMSO-d6) δ 10.35 (s, 1H, exchangeable with D2O), 8.49 (s, 1H), 8.42 (s, 1H), 8.16 (d, J = 8.9 Hz, 1H), 7.83–7.70 (m, 2H), 6.81 (br t, 1H, exchangeable with D2O), 3.91 (s, 3H), 3.01–2.88 (m, 2H), 2.39 (t, J = 7.4 Hz, 2H), 1.75–1.50 (m, 11H), 1.44–1.20 (m, 15H). MS (ESI) m/z: 545 (M+H)+.
Methyl 6-(5-Aminopentanamido)-4-hydroxy-2-naphthoate (18a)
The TFA salt of compound 18a was obtained as a pale brown solid (870 mg, 71%) starting from compound 17a (1.47 g, 2.85 mmol) following the procedure described for 5a. 1H NMR (400 MHz, DMSO-d6) δ 10.44 (s, 1H, exchangeable with D2O), 10.26 (s, 1H, exchangeable with D2O), 8.54 (s, 1H), 8.05–7.89 (m, 2H), 7.79–7.64 (m, 4H, 3H exchangeable with D2O), 7.33 (s, 1H), 3.86 (s, 3H), 2.90–2.76 (m, 2H), 2.41 (t, J = 7.0 Hz, 2H), 1.80–1.56 (m, 4H). MS (ESI) m/z: 317 (M + H)+.
Methyl 6-(6-Aminohexanamido)-4-hydroxy-2-naphthoate (18b)
The TFA salt of compound 18b was obtained as a brown solid (507 mg, 70%) starting from compound 17b (868 mg, 1.63 mmol) following the procedure described for 5a. 1H NMR (400 MHz, DMSO-d6) δ 10.44 (s, 1H, exchangeable with D2O), 10.26 (s, 1H, exchangeable with D2O), 8.56 (s, 1H), 8.05–7.95 (m, 2H), 7.73–7.66 (m, 4H, 3H exchangeable with D2O), 7.34 (s, 1H), 3.86 (s, 3H), 2.80–2.82 (m, 2H), 2.41 (t, J = 7.2 Hz, 2H), 1.70–1.56 (m, 4H), 1.40–1.37 (m, 2H). MS (ESI) m/z: 331 (M + H)+.
Methyl 6-(7-Aminoheptanamido)-4-hydroxy-2-naphthoate (18c)
The TFA salt of compound 18c was obtained as a yellow solid (503 mg, 60%) starting from compound 17c (1.00 g, 1.83 mmol) following the procedure described for 5a. 1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H, exchangeable with D2O), 10.26 (s, 1H, exchangeable with D2O), 8.55 (s, 1H), 8.05–7.96 (m, 2H), 7.73–7.66 (m, 4H, 3H exchangeable with D2O), 7.34 (s, 1H), 3.86 (s, 3H), 2.79–2.75 (m, 2H), 2.41 (t, J = 7.0 Hz, 2H), 1.65–1.52 (m, 4H), 1.40–1.27 (m, 4H). MS (ESI) m/z: 345 (M + H)+.
Methyl 6-(5-(2,3-Bis(tert-butoxycarbonyl)guanidino)pentanamido)-4-hydroxy-2-naphthoate (19a)
Compound 19a was obtained as a pale yellow solid (301 mg, 58%) by the reaction of 18a (400 mg, 0.929 mmol) and 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea (540 mg, 1.86 mmol) following the procedure described for 5f. 1H NMR (400 MHz, DMSO-d6) δ 11.50 (s, 1H, exchangeable with D2O), 10.42 (s, 1H, exchangeable with D2O), 10.20 (s, 1H, exchangeable with D2O), 8.53 (s, 1H), 8.32 (br t, 1H, exchangeable with D2O), 7.99 (s, 1H), 7.94 (d, J = 8.8 Hz, 1H), 7.70 (d, J = 8.8 Hz, 1H), 7.32 (s, 1H), 3.86 (s, 3H), 2.40 (m, 2H), 1.73–1.51 (m, 6H), 1.47 (s, 9H), 1.38 (s, 9H). MS (ESI) m/z: 559 (M + H)+.
Methyl 6-(6-(2,3-bis(tert-Butoxycarbonyl)guanidino)hexanamido)-4-hydroxy-2-naphthoate (19b)
Compound 19b was obtained as a yellow product (422 mg, 76%) by the reaction of compound 18b (433 mg, 0.974 mmol) with 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea (565 mg, 1.94 mmol) following the procedure described for 5f. 1H NMR (400 MHz, DMSO-d6) δ 11.49 (s, 1H, exchangeable with D2O), 10.40 (s, 1H, exchangeable with D2O), 10.16 (s, 1H, exchangeable with D2O), 8.53 (s, 1H), 8.32 (br t, 1H, exchangeable with D2O), 7.99 (s, 1H), 7.94 (d, J = 8.8 Hz, 1H), 7.70 (d, J = 8.8 Hz, 1H), 7.32 (s, 1H), 3.86 (s, 3H), 3.30–3.22 (m, 2H), 2.40 (t, J = 7.4 Hz, 2H), 1.73–1.51 (m, 6H), 1.47 (s, 9H), 1.38 (s, 9H). MS (ESI) m/z: 573 (M + H)+.
Methyl 6-(7-(2,3-bis(tert-Butoxycarbonyl)guanidino)heptanamido)-4-hydroxy-2-naphthoate (19c)
Compound 19c was obtained as a yellow product (226 mg, 85%) by the reaction of compound 18c (250 mg, 0.545 mmol) with 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea (317 mg, 1.09 mmol) following the procedure described for 5f. 1H NMR (400 MHz, DMSO-d6) δ 11.50 (s, 1H, exchangeable with D2O), 10.42 (s, 1H, exchangeable with D2O), 10.20 (s, 1H, exchangeable with D2O), 8.53 (s, 1H), 8.32 (br t, 1H, exchangeable with D2O), 7.99 (s, 1H), 7.94 (d, J = 8.8 Hz, 1H), 7.70 (d, J = 8.8 Hz, 1H), 7.32 (s, 1H), 3.86 (s, 3H), 2.40 (m, 2H), 1.72–1.55 (m, 2H), 1.53–1.44 (m, 11 H), 1.38–1.33 (m, 13 H). MS (ESI) m/z: 587 (M + H)+.
Methyl 6-(5-(1,2-Bis(tert-butoxycarbonyl)-3-methylguanidino)pentanamido)-4-hydroxy-2-naphthoate (20a)
Compound 20a was obtained as a yellow product (280 mg, 53%) by the reaction of compound 18a (400 mg, 0.929 mmol) with N,N′-bis(tert-butoxycarbonyl)-N,S-dimethylisothiourea (566 mg, 1.86 mmol) following the procedure described for 5f. 1H NMR (400 MHz, DMSO-d6) δ 10.44 (s, 1H, exchangeable with D2O), 10.20 (s, 1H, exchangeable with D2O), 8.53 (s, 1H), 8.11–7.82 (m, 3H, 1H exchangeable with D2O), 7.70 (d, J = 9.1 Hz, 1H), 7.32 (s, 1H), 3.86 (s, 3H), 3.17–3.11 (m, 2H), 2.87 (s, 3H), 2.39–2.31 (m, 2H), 1.74–1.48 (m, 4H), 1.36 (m, 18H). MS (ESI) m/z: 573 (M + H)+.
Methyl 6-(6-(2,3-Bis(tert-Butoxycarbonyl)-3-methylguanidino)hexanamido)-4-hydroxy-2-naphthoate (20b)
Compound 20b was obtained as a yellow product (456 mg, 72%) by the reaction of compound 18b (480 mg, 1.08 mmol) with N,N′-bis(tert-butoxycarbonyl)-N,S-dimethylisothiourea (657 mg, 2.16 mmol) following the procedure described for 5f. 1H NMR (400 MHz, DMSO-d6) δ 10.44 (s, 1H, exchangeable with D2O), 10.20 (s, 1H, exchangeable with D2O), 8.53 (s, 1H), 8.00 (s, 1H), 7.95 (d, J = 8.9 Hz, 1H), 7.71 (dd, J = 8.9 Hz, 1H), 7.32 (s, 1H), 3.86 (s, 3H), 3.15–3.11 (m, 2H), 2.87 (s, 3H), 2.38 (t, J = 7.4 Hz, 2H), 1.74–1.48 (m, 6H), 1.38 (s, 9H), 1.36 (s, 9H). MS (ESI) m/z: 587 (M + H)+.
Methyl 4-Acetoxy-6-amino-2-naphthoate (21)
Compound 21 was obtained as a pale yellow solid (2.69 g, 98%) starting from 13 (3.00 g, 10.37 mmol) following the procedure described for 16. 1H NMR (400 MHz, DMSO-d6) δ 8.28 (s, 1H), 7.86 (d, J = 9 Hz, 1 H), 7.54 (s, 1 H), 7.06 (d, J = 9 Hz, 1 H), 6.79 (s, 1H), 6.04 (s, 2 H, exchangeable with D2O), 3.87 (s, 3 H), 2.42 (s, 3H). MS (ESI) m/z: 260 (M + H)+.
Methyl 4-Acetoxy-6-((phenoxycarbonyl)amino)-2-naphthoate (22)
Compound 21 (2.68 g, 10.33 mmol) was dissolved in AcOEt, and then TEA (1.58 mL, 11.37 mmol) was added. The resulting mixture was cooled at 0 °C, and phenyl chloroformate (1.94 mL, 15.50 mmol) was added dropwise. The yellow suspension obtained was allowed to warm to room temperature and stirred for 16 h. The reaction was then diluted with AcOEt (60 mL), washed with water (3 × 20 mL), 1N HCl (3× 20 mL), NaHCO3 saturated solution (3 × 20 mL), and brine (30 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting solid was crystallized from AcOEt to give the title compound 22 as a pale yellow solid (2.74 g, 70%).
1H NMR (400 MHz, DMSO-d6) δ 10.74 (s, 1H, exchangeable with D2O), 8.50 (s, 1H), 8.23–8.14 (m, 2H), 7.79–7.72 (m, 2H), 7.49–7.43 (m, 2H), 7.33–7.24 (m, 3H), 3.91 (s, 3H), 2.41 (s, 3H). MS (ESI) m/z: 380 (M + H)+.
Methyl 6-(3-(3-((tert-Butoxycarbonyl)amino)propyl)ureido)-4-hydroxy-2-naphthoate (23a)
To a stirring solution of methyl 4-acetoxy-6-((phenoxycarbonyl)amino)-2-naphthoate 22 (856 mg, 2.26 mmol) in dry DMF (7 mL), a solution of tert-butyl (3-aminopropyl)carbamate (786 mg, 4.51 mmol) and TEA (629 μL, 4.51 mmol) in dry DMF (7 mL) was added, and the reaction was stirred at room temperature for 2 h. NaHCO3 saturated solution was added (25 mL), and the resulting mixture was extracted with AcOEt (3 × 25 mL). The combined organic layers were washed with NaHCO3 saturated solution (2 × 10 mL) and brine (10 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a light brown solid that was purified by flash chromatography (gradient AcOEt/MeOH 100:0 to 90:10) yielding a mixture of acetylated and deacetylated compounds. Therefore, the solid was suspended in DCM (30 mL), and piperidine (580 μL) was added. After 1 h, the solvent was removed, and the crude product was diluted with AcOEt (50 mL). The organic layer was washed with 1 N HCl (3× 20 mL) and brine (30 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure, yielding pure 23a as an orange solid (754 mg, 80%). 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H, exchangeable with D2O), 8.91 (s, 1H, exchangeable with D2O), 8.27 (d, J = 2.1 Hz, 1H), 7.97 (s, 1H), 7.89 (d, J = 8.9 Hz, 1H), 7.55 (dd, J = 8.9, 2.1 Hz, 1H), 7.30 (s, 1H), 6.85 (t, J = 5.0 Hz, 1H, exchangeable with D2O), 6.25 (t, J = 5.7 Hz, 1H, exchangeable with D2O), 3.87 (s, 3H), 3.14–3.08 (m, 2H), 3.01–2.94 (m, 2H), 1.59–1.50 (m, 2H), 1.40 (s, 9H). MS (ESI) m/z: 418 (M + H)+.
Methyl 6-(3-(4-((tert-Butoxycarbonyl)amino)butyl)ureido)-4-hydroxy-2-naphthoate (23b)
Compound 23b was obtained as a yellow solid (828 mg, 85%) by the reaction of 22 (856 mg, 2.26 mmol) with tert-butyl (4-aminobutyl)carbamate (849 mg, 4.51 mmol) following the procedure described for 23a. 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H, exchangeable with D2O), 8.80 (s, 1H, exchangeable with D2O), 8.26 (d, J = 2.1 Hz, 1H), 7.97 (s, 1H), 7.90 (d, J = 8.9 Hz, 1H), 7.54 (dd, J = 8.9, 2.1 Hz, 1H), 7.29 (s, 1H), 6.82 (t, J = 5.0 Hz, 1H, exchangeable with D2O), 6.22 (t, J = 5.7 Hz, 1H, exchangeable with D2O), 3.85 (s, 3H), 3.17–3.03 (m, 2H), 3.02–2.86 (m, 2H), 1.53–1.29 (m, 13H). MS (ESI) m/z: 432 (M + H)+.
Methyl 6-(3-(5-((tert-Butoxycarbonyl)amino)pentyl)ureido)-4-hydroxy-2-naphthoate (23c)
Compound 23c was obtained as a yellow solid (855 mg, 85%) by the reaction of 22 (856 mg, 2.26 mmol) with tert-butyl (5-aminopentyl)carbamate (911 mg, 4.51 mmol) following the procedure described for 23a. 1H NMR (400 MHz, DMSO-d6) δ 10.31 (s, 1H, exchangeable with D2O), 8.83 (s, 1H, exchangeable with D2O), 8.27 (d, J = 2.3 Hz 1H), 7.96 (s, 1H), 7.88 (d, J = 8.7 Hz, 1H), 7.52 (dd, J = 8.7, 2.3 Hz, 1H), 7.30 (s, 1H), 6.81 (t, J = 4.8 Hz, 1H, exchangeable with D2O), 6.25 (t, J = 5.1 Hz, 1H, exchangeable with D2O), 3.85 (s, 3H), 3.17–3.03 (m, 2H), 3.02–2.86 (m, 2H), 1.53–1.29 (m, 15H). MS (ESI) m/z: 446 (M + H)+.
Methyl 6-(3-(6-((tert-Butoxycarbonyl)amino)hexyl)ureido)-4-hydroxy-2-naphthoate (23d)
Compound 23d was obtained as a yellow solid (882 mg, 85%) by the reaction of 22 (856 mg, 2.26 mmol) with tert-butyl (6-aminohexyl)carbamate (974 mg, 4.51 mmol) following the procedure described for 23a. 1H NMR (400 MHz, DMSO-d6) δ 10.32 (s, 1H, exchangeable with D2O), 8.84 (s, 1H, exchangeable with D2O), 8.28 (d, J = 2.4 Hz 1H), 7.96 (s, 1H), 7.87 (d, J = 8.5 Hz, 1H), 7.52 (dd, J = 8.5, 2.4 Hz, 1H), 7.28 (s, 1H), 6.80 (t, J = 5.1 Hz, 1H, exchangeable with D2O), 6.24 (t, J = 5.0 Hz, 1H, exchangeable with D2O), 3.85 (s, 3H), 3.19–3.05 (m, 2H), 3.03–2.89 (m, 2H), 1.58–1.26 (m, 17H). MS (ESI) m/z: 460 (M + H)+.
Methyl 6-(3-(7-((tert-Butoxycarbonyl)amino)heptyl)ureido)-4-hydroxy-2-naphthoate (23e)
Compound 23e was obtained as a yellow solid (931 mg, 87%) by the reaction of 22 (856 mg, 2.26 mmol) with tert-butyl (7-aminoheptyl)carbamate (1.04 g, 4.51 mmol) following the procedure described for 23a. 1H NMR (400 MHz, DMSO-d6) δ 10.27 (s, 1H, exchangeable with D2O), 8.82 (s, 1H, exchangeable with D2O), 8.27 (d, J = 2.1 Hz, 1H), 7.97 (s, 1H), 7.88 (d, J = 8.9 Hz, 1H), 7.53 (dd, J = 8.9, 2.2 Hz, 1H), 7.30 (s, 1H), 6.81 (t, J = 5.0 Hz, 1H, exchangeable with D2O), 6.26 (t, J = 5.2 Hz, 1H, exchangeable with D2O), 3.86 (s, 3H), 3.16–3.05 (m, 4H), 1.51–1.42 (m, 4H), 1.53–1.30 (m, 15H). MS (ESI) m/z: 474 (M + H)+.
Methyl 6-(3-(3-Aminopropyl)ureido)-4-hydroxy-2-naphthoate (24a)
The TFA salt of compound 24a was obtained as a white solid (578 mg, 80%) starting from 23a (700 mg, 1.67 mmol) following the procedure described for 5a. 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H, exchangeable with D2O), 9.02 (s, 1H, exchangeable with D2O), 8.28 (d, J = 2.1 Hz, 1H), 7.99–7.94 (m, 1H), 7.89 (d, J = 8.9 Hz, 1H), 7.69 (br s, 3H, exchangeable with D2O), 7.55 (dd, J = 8.9, 2.1 Hz, 1H), 7.30 (s, 1H), 6.49 (br t, 1H, exchangeable with D2O), 3.86 (s, 3H), 3.29–3.10 (m, 2H), 2.99–2.79 (m, 2H), 1.85–1.58 (m, 2H). MS (ESI) m/z: 318 (M + H)+.
Methyl 6-(3-(4-Aminobutyl)ureido)-4-hydroxy-2-naphthoate (24b)
The TFA salt of compound 24b was obtained as a white solid (679 mg, 82%) starting from 23b (800 mg, 1.86 mmol) following the procedure described for 5a. 1H NMR (400 MHz, DMSO-d6) δ 10.31 (s, 1H, exchangeable with D2O), 8.99 (s, 1H, exchangeable with D2O), 8.30 (d, J = 2.1 Hz, 1H), 7.98 (s, 1H), 7.86 (d, J = 8.9 Hz, 1H), 7.71 (br s, 3H, exchangeable with D2O) 7.56 (dd, J = 8.9, 2.1 Hz, 1H), 7.31 (s, 1H), 6.45 (br t, 1H, exchangeable with D2O), 3.85 (s, 3H), 3.16–3.08 (m, 2H), 2.83–2.74 (m, 2H), 1.66–1.43 (m, 4H). MS (ESI) m/z: 332 (M + H)+.
Methyl 6-(3-(5-Aminopentyl)ureido)-4-hydroxy-2-naphthoate (24c)
The TFA salt of compound 24c was obtained as a white solid (728 mg, 83%) starting from 23c (850 mg, 1.91 mmol) following the procedure described for 5a. 1H NMR (400 MHz, DMSO-d6) δ 10.34 (s, 1H, exchangeable with D2O), 8.95 (s, 1H, exchangeable with D2O), 8.26 (d, J = 2.1 Hz, 1H), 7.97 (s, 1H), 7.89 (d, J = 8.9 Hz, 1H), 7.67 (br s, 3H, exchangeable with D2O), 7.54 (dd, J = 8.9, 2.1 Hz, 1H), 7.29 (s, 1H), 6.85 (br t, 1H, exchangeable with D2O), 3.86 (s, 3H), 3.11 (m, 2H), 2.85–2.73 (m, 2H), 1.63–1.42 (m, 4H), 1.39–1.27 (m, 2H). MS (ESI) m/z: 346 (M + H)+.
Methyl 6-(3-(6-Aminohexyl)ureido)-4-hydroxy-2-naphthoate (24d)
The TFA salt of compound 24d was obtained as a white solid (745 mg, 85%) starting from compound 23d (850 mg, 1.85 mmol) following the procedure described for 5a. 1H NMR (400 MHz, DMSO-d6) δ 10.34 (s, 1H, exchangeable with D2O), 8.92 (s, 1H, exchangeable with D2O), 8.28 (d, J = 2.1 Hz, 1H), 7.99–7.94 (m, 1H), 7.88 (d, J = 8.9 Hz, 1H), 7.68 (br s, 3H, exchangeable with D2O), 7.54 (dd, J = 8.9, 2.1 Hz, 1H), 7.29 (s, 1H), 6.37 (br t, 1H, exchangeable with D2O), 3.86 (s, 3H), 3.16–3.09 (m, 2H), 2.83–2.75 (m, 2H), 1.59–1.46 (m, 6H), 1.39–1.34 (m, 2H). MS (ESI) m/z: 360 (M + H)+.
Methyl 6-(3-(7-Aminoheptyl)ureido)-4-hydroxy-2-naphthoate (24e)
The TFA salt of compound 24e was obtained as a white solid (750 mg, 81%) starting from compound 23e (900 mg, 1.90 mmol) following the procedure described for 5a. 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H, exchangeable with D2O), 8.82 (s, 1H, exchangeable with D2O), 8.27 (d, J = 2.2 Hz, 1H), 7.98–7.94 (m, 1H), 7.85 (d, J = 8.8 Hz, 1H), 7.68 (br s, 3H, exchangeable with D2O), 7.53 (dd, J = 8.8, 2.2 Hz, 1H), 7.30 (s, 1H), 6.26 (br t, 1H, exchangeable with D2O), 3.86 (s, 3H), 3.16–3.05 (m, 4H), 1.51–1.42 (m, 6H), 1.34–1.30 (m, 4H). MS (ESI) m/z: 374 (M + H)+.
Methyl 6-(3-(3-(2,3-Bis(tert-butoxycarbonyl)guanidino)propyl)ureido)-4-hydroxy-2-naphthoate (25a)
Compound 25a was obtained as a white solid (62 mg, 31%) by the reaction of 24a (154 mg, 0.357 mmol) with 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea (207 mg, 0.714 mmol) following the procedure described for 5f. 1H NMR (400 MHz, DMSO-d6) δ 11.50 (s, 1H, exchangeable with D2O), 10.30 (s, 1H, exchangeable with D2O), 8.86 (s, 1H, exchangeable with D2O), 8.35 (br t, 1H, exchangeable with D2O), 8.26 (d, J = 2.2 Hz, 1H), 7.96 (s, 1H), 7.87 (d, J = 8.9 Hz, 1H), 7.54 (dd, J = 8.9, 2.2 Hz, 1H), 7.28 (s, 1H), 6.37 (br t, 1H, exchangeable with D2O), 3.85 (s, 3H), 3.35–3.23 (m, 2H) 3.19–3.06 (m, 2H), 1.58–1.49 (m, 11H), 1.39 (s, 9H). MS (ESI) m/z: 560 (M + H)+.
Methyl 6-(3-(4-(2,3-Bis(tert-butoxycarbonyl)guanidino)butyl)ureido)-4-hydroxy-2-naphthoate (25b)
Compound 25b was obtained as a white solid (59 mg, 30%) by the reaction of 24b (155 mg, 0.348 mmol) with 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea (202 mg, 0.696 mmol) following the procedure described for 5f. 1H NMR (400 MHz, DMSO-d6) δ 11.52 (s, 1H, exchangeable with D2O), 10.35 (s, 1H, exchangeable with D2O), 8.89 (s, 1H, exchangeable with D2O), 8.41–8.20 (m, 2H, 1H exchangeable with D2O), 7.96 (s, 1H), 7.85 (d, J = 8.7 Hz, 1H), 7.58 (dd, J = 8.7, 2.1 Hz, 1H), 7.30 (s, 1H), 6.31 (br t, 1H, exchangeable with D2O), 3.86 (s, 3H), 3.37–3.22 (m, 2H), 3.20–2.98 (m, 2H), 1.56–1.47 (m, 13H), 1.38 (s, 9H). MS (ESI) m/z (%): 574 (M + H)+.
Methyl 6-(3-(5-(2,3-Bis(tert-butoxycarbonyl)guanidino)pentyl)ureido)-4-hydroxy-2-naphthoate (25c)
Compound 25c was obtained as a yellow solid (128 mg, 40%) by the reaction of 24c (250 mg, 0.544 mmol) with 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea (316 mg, 1.09 mmol) following the procedure described for 5f. 1H NMR (400 MHz, DMSO-d6) δ 11.51 (s, 1H, exchangeable with D2O), 10.39 (s, 1H, exchangeable with D2O), 9.01 (s, 1H, exchangeable with D2O), 8.33–8.24 (m, 2H, 1H exchangeable with D2O), 7.95 (s, 1H), 7.88 (d, J = 8.9 Hz, 1H), 7.53 (dd, J = 8.9, 2.2 Hz, 1H), 7.28 (s, 1H), 6.55 (br t, 1H, exchangeable with D2O), 3.85 (s, 3H), 3.35–3.23 (m, 2H) 3.14–3.06 (m, 2H), 1.55–1.44 (m, 15H), 1.38 (s, 9H). MS (ESI) m/z (%): 588 (M + H)+.
Methyl 6-(3-(3-(2,3-Bis(tert-butoxycarbonyl)-3-methylguanidino)propyl)ureido)-4-hydroxy-2-naphthoate (26a)
Compound 26a was obtained as a white solid (64 mg, 32%) by the reaction of 24a (150 mg, 0.348 mmol) with N,N′-bis(tert-butoxycarbonyl)-N,S-dimethylisothiourea (212 mg, 0.696 mmol) following the procedure described for 5f. 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H, exchangeable with D2O), 8.88 (s, 1H, exchangeable with D2O), 8.26 (d, J = 2.2 Hz, 1H), 7.96 (s, 1H), 7.88 (d, J = 8.9 Hz, 1H), 7.54 (dd, J = 8.9, 2.2 Hz, 1H), 7.28 (s, 1H), 6.34 (br t, 1H, exchangeable with D2O), 3.86 (s, 3H), 3.20–3.12 (m, 4H), 2.89 (s, 3H), 1.74–1.57 (m, 2H), 1.39 (s, 9H) 1.37 (s, 9H). MS (ESI) m/z: 574 (M + H)+.
Methyl 6-(3-(4-(2,3-Bis(tert-butoxycarbonyl)-3-methylguanidino)butyl)ureido)-4-hydroxy-2-naphthoate (26b)
Compound 26b was obtained as a white solid (126 mg, 40%) by the reaction of 24b (240 mg, 0.539 mmol) with N,N′-bis(tert-butoxycarbonyl)-N,S-dimethylisothiourea (328 mg, 1.08 mmol) following the procedure described for 5f. 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H, exchangeable with D2O), 8.82 (s, 1H, exchangeable with D2O), 8.26 (d, J = 2.2 Hz, 1H), 7.96 (s, 1H), 7.88 (d, J = 8.9 Hz, 1H), 7.54 (dd, J = 8.9, 2.2 Hz, 1H), 7.28 (s, 1H), 6.27 (br t, 1H, exchangeable with D2O), 3.86 (s, 3H), 3.16–3.12 (m, 4H), 2.89 (s, 3H), 1.74–1.57 (m, 4H), 1.39 (s, 9H) 1.37 (s, 9H). MS (ESI) m/z: 588 (M + H)+.
9-((4R,6R)-6-(Aminomethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-amine (27a)
Pd/C (5 wt % on activated carbon, 0.1 equiv) was added to a solution of 34 (1.00 g, 3.01 mmol) in MeOH (100 mL), and the reaction was stirred under H2 (1 atm, balloon) for 16 h. The reaction mixture was filtered and concentrated to give the title compound 27a, used in the next step without further purification. 1H NMR (400 MHz, DMSO-d6) δ 8.37 (s, 1H), 8.16 (s, 1H), 7.32 (br s, 2H), 6.08 (d, J = 3.2 Hz, 1H), 5.45 (dd, J = 6.4, 3.3 Hz, 1H), 4.98 (dd, J = 6.4, 2.7 Hz, 1H), 4.09 (td, J = 5.8, 2.6 Hz, 1H), 2.70 (m, 2H), 1.54 (s, 3H), 1.33 (s, 3H). MS (ESI) m/z: 307 (M + H)+.
9-((4R,6R)-6-(2-Aminoethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-amine (27b)
36 (0.25 g, 0.60 mmol) was dissolved in 10 mL of a solution of DCM/TFA (95:5), and the mixture was stirred at room temperature. After 24 h, the solvent was removed under vacuum, and the crude was resuspended in a solution of citric acid 10%. The aqueous solution was washed with DCM (3 × 25 mL), basified with 2 N NaOH until pH 12, and then extracted with CHCl3 (3 × 25 mL). The collected organic phases were dried (Na2SO4), filtered, and concentrated under vacuum to give 27b as a white solid (0.30 g, 65%). 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 7.89 (s, 1H), 6.03 (d, J = 2.4 Hz, 1H), 5.70 (br s, 2H), 5.48 (dd, J = 6.5, 2.4 Hz, 1H), 4.90 (dd, J = 6.5, 4.1 Hz, 1H), 4.33–4.21 (m, 1H), 2.86–2.72 (m, 2H), 1.90–1.78 (m, 2H), 1.61 (s, 3H), 1.39 (s, 3H). MS (ESI) m/z: 321 (M + H)+.
9-((4R,6R)-6-((E)-3-Aminoprop-1-en-1-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-amine (27c)
To a stirred solution of compound 39 (0.40 g, 0.86 mmol) in MeOH (2 mL), a solution of hydrazine monohydrate (0.87 g, 1.73 mmol) was added, and the mixture was stirred at room temperature for 16 h. Then, the solvent was evaporated, and the crude reside was resuspended in a solution of 10% citric acid. The aqueous solution was washed with DCM (3 × 25 mL), basified with 2 N NaOH until pH 12, and then extracted with CHCl3 (3 × 25 mL). The collected organic phases were dried (Na2SO4), filtered, and concentrated under vacuum to give the amine 27c as a white solid (0.26 g, 90%). 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 7.87 (s, 1H), 6.08 (d, J = 2.0 Hz, 1H), 5.87 (br s, 2H), 5.82–5.79 (m, 1H), 5.75–5.65 (m, 1H), 5.53 (dd, J = 6.3, 2.0 Hz, 1H), 4.99 (dd, J = 6.4, 3.5 Hz, 1H), 4.71–4.66 (m, 1H), 3.23–3.20 (m, 2H), 1.62 (s, 3H), 1.39 (s, 3H). MS (ESI) m/z: 333 (M + H)+.
9-((4R,6R)-6-(3-aminopropyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-amine (27d)
Compound 27d was obtained as a white solid (0.35 g, 99%) starting from 27c (0.35 g, 1.05 mmol) following the procedure described for 27a. 1H NMR (400 MHz, CDCl3) δ 8.37 (s, 1H), 7.89 (s, 1H), 6.06 (d, J = 2.4 Hz, 1H), 5.72 (br s, 2H), 5.48 (dd, J = 6.5, 2.4 Hz, 1H), 4.95 (dd, J = 6.5, 4.1 Hz, 1H), 4.35–4.24 (m, 1H), 2.86–2.72 (m, 2H), 1.97–1.65 (m, 4H), 1.61 (s, 3H), 1.39 (s, 3H). MS (ESI) m/z: 335 (M + H)+.
tert-Butyl (9-((3aR,4R,6R,6aR)-6-((3-tert-Butyloxythioureido)methyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-yl)carbamate (28a)
To a cooled solution of N,N′-bis-tert-butoxycarbonylthiourea (1.00 g, 3.62 mmol) dissolved in dry THF, NaH (60% dispersion in mineral oil, 173 mg, 4.34 mmol) was added, and the resulting mixture was stirred at the same temperature for 1 h. Then, TFAA (603 μL, 4.34 mmol) was added, and the stirring continued for an additional 1 h at 0 °C; afterward, amine 27a (1.61 g, 3.96 mmol) was added, and the resulting suspension was stirred for 16 h. The crude was diluted with brine (50 mL), and the aqueous phase was extracted with AcOEt (3 × 25 mL). The collected organic phases were washed with brine, dried (Na2SO4), and concentrated under vacuum, yielding a residue that was purified by column chromatography (gradient Hex/AcOEt 80:20 to 40:50 + 10% MeOH) to give 28a as a white solid (716 mg, 35%). 1H NMR (400 MHz, CDCl3) δ 9.80 (t, J = 4.3 Hz, 1H, exchangeable with D2O), 8.76 (s, 1H, exchangeable with D2O), 8.07 (s, 1H, exchangeable with D2O), 7.92 (s, 1H), 7.84 (s, 1H), 6.14 (d, J = 2.5 Hz, 1H), 5.42 (dd, J = 6.4, 2.5 Hz, 1H), 5.09 (dd, J = 6.4, 3.7 Hz, 1H), 4.54–4.50 (m, 1H), 4.18–3.94 (m, 2H), 1.62 (s, 3H), 1.57 (s, 9H), 1.45 (s, 9H), 1.39 (s, 3H). MS (ESI) m/z: 566 (M + H)+.
tert-Butyl (9-((3aR,4R,6R,6aR)-6-((3-tert-Butyloxythioureido)ethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-yl)carbamate (28b)
Compound 28b was obtained as a white solid (198 mg, 53%) starting from 27b (350 mg, 1.09 mmol) following the procedure described for 28a. 1H NMR (400 MHz, CDCl3) δ 9.77 (br s, 1H, exchangeable with D2O), 8.34 (s, 1H), 8.14 (br s, 1H, exchangeable with D2O), 7.88 (s, 1H), 6.04 (d, J = 2.5 Hz, 1H), 5.89 (br s, 2H, exchangeable with D2O), 5.47 (dd, J = 6.6, 2.5 Hz, 1H), 4.97 (dd, J = 6.6, 4.0 Hz, 1H), 4.29–4.24 (m, 1H), 3.90–3.80 (m, 1H), 3.69–3.59 (m, 1H), 2.17–2.10 (m, 2H), 1.60 (s, 3H), 1.38 (s, 12H). MS (ESI) m/z: 480 (M + H)+.
tert-Butyl (9-((3aR,4R,6R,6aR)-6-((3-tert-Butyloxythioureido)propyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-yl)carbamate (28c)
Compound 28c was obtained as a white solid (191 mg, 35%) starting from 27c (250 mg, 0.75 mmol) following the procedure described for 28a. 1H NMR (400 MHz, CDCl3) δ 9.77 (br s, 1H, exchangeable with D2O), 8.34 (s, 1H), 8.14 (br s, 1H, exchangeable with D2O), 7.88 (s, 1H), 6.03 (d, J = 2.5 Hz, 1H), 5.89 (br s, 2H, exchangeable with D2O), 5.45 (dd, J = 6.6, 2.5 Hz, 1H), 4.92 (dd, J = 6.6, 4.0 Hz, 1H), 4.29–4.24 (m, 1H), 3.70–3.58 (m, 2H), 1.82–1.74 (m, 2H), 1.70–1.64 (m, 2H), 1.61 (s, 3H), 1.47 (s, 9H), 1.38 (s, 3H). MS (ESI) m/z: 494 (M + H)+.
tert-Butyl (9-((3aR,4R,6R,6aR)-6-((3-tert-Butyloxythioureido)propyl-1-en)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-yl)carbamate (28d)
Compound 28d was obtained as a white solid (121 mg, 33%) starting from 27d (250 mg, 0.75 mmol) following the procedure described for 28a. 1H NMR (400 MHz, CHCl3) δ 9.70 (br s, 1H exchangeable with D2O), 8.37 (s, 1H), 8.01 (br s, 1H, exchangeable with D2O), 7.88 (s, 1H), 6.09 (d, J = 2.1 Hz, 1H), 5.85–5.79 (m, 2H), 5.75 (br s, 2H, exchangeable with D2O), 5.51 (dd, J = 6.3, 2.0 Hz, 1H), 5.03 (dd, J = 6.4, 3.5 Hz, 1H), 4.73–4.66 (m, 1H), 4.23–4.19 (m, 2H), 1.62 (s, 3H), 1.48 (s, 9H), 1.39 (s, 3H). MS (ESI) m/z: 492 (M + H)+.
Methyl 6-(3-(3-((E)-2-(tert-Butoxycarbonyl)-3-(((3aR,4R,6R,6aR)-6-(6-((tert-butoxycarbonyl)amino)-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)guanidino)propyl)ureido)-4-hydroxy-2-naphthoate (29a)
To a stirred suspension of 28a (100 mg, 0.177 mmol) and 24a (152 mg, 0.354 mmol) in dry DCM, EDC hydrochloride (69 mg, 0.354 mmol) and TEA (74 μL, 0.531 mmol) were added at room temperature. The resulting mixture was stirred for 18 h, then the solvent was evaporated under reduced pressure, and the resulting oil was taken up with water. The aqueous phase was extracted with AcOEt (3 × 25 mL), and the collected organic phases were washed with brine, dried (Na2SO4), and concentrated under reduced pressure. The crude material was purified by flash chromatography (gradient DCM/MeOH 95:5 to 90:10) yielding 29a as white solid (114 mg, 75%). 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H, exchangeable with D2O), 10.18 (s, 1H, exchangeable with D2O), 8.83 (s, 1H, exchangeable with D2O), 8.66–8.62 (m, 2H), 8.24 (d, J = 2.2 Hz, 1H), 7.95 (s, 1H), 7.87 (d, J = 8.9 Hz, 1H), 7.54 (dd, J = 8.9, 2.2 Hz, 1H), 7.28 (s, 1H), 6.32 (br t, 1H, exchangeable with D2O), 6.25 (s, 1H), 5.51–5.46 (m, 1H), 5.08–5.02 (m, 1H), 4.34–4.29 (m, 1H), 3.85 (s, 3H), 3.45–3.39 (m, 2H), 3.20–3.08 (m, 4H), 1.70–1.60 (m, 2H), 1.54 (s, 3H), 1.46 (s, 9H), 1.35 (s, 9H), 1.32 (s, 3H). MS (ESI) m/z: 849 (M + H)+.
Methyl 6-(3-(4-((E)-2-(tert-Butoxycarbonyl)-3-(((3aR,4R,6R,6aR)-6-(6-((tert-butoxycarbonyl)amino)-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)guanidino)butyl)ureido)-4-hydroxy-2-naphthoate (29b)
Compound 29b was obtained as a white solid (37 mg, 72%) by the reaction of 28a (52 mg, 0.060 mmol) with compound 24b (53 mg, 0.120 mmol) following the procedure described for 29a. 1H NMR (400 MHz, DMSO-d6) δ 10.27 (s, 1H, exchangeable with D2O), 10.15 (s, 1H, exchangeable with D2O), 8.86 (s, 1H, exchangeable with D2O), 8.69–8.58 (m, 2H), 8.26 (d, J = 2.2 Hz, 1H), 7.96 (s, 1H), 7.87 (d, J = 8.9 Hz, 1H), 7.55 (dd, J = 8.9, 2.2 Hz, 1H), 7.29 (s, 1H), 6.31 (br t, 1H, exchangeable with D2O), 6.24 (s, 1H), 5.53–5.45 (m, 1H), 5.12–4.94 (m, 1H), 4.39–4.28 (m, 1H), 3.85 (s, 3H), 3.44–3. 38 (m, 2H), 3.17–3.07 (m, 4H), 1.58–1.51 (m, 4H), 1.49–1.46 (m, 12H), 1.37 (s, 9H), 1.30 (s, 3H). MS (ESI) m/z: 863 (M + H)+.
Methyl 6-(3-(5-((E)-2-(tert-Butoxycarbonyl)-3-(((3aR,4R,6R,6aR)-6-(6-((tert-butoxycarbonyl)amino)-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)guanidino)pentyl)ureido)-4-hydroxy-2-naphthoate (29c)
Compound 29c was obtained as a white solid (197 mg, 77%) by the reaction of compound 28a (165 mg, 0.292 mmol) with compound 24c (268 mg, 0.584 mmol) following the procedure described for 29a. 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H, exchangeable with D2O), 10.15 (s, 1H, exchangeable with D2O), 8.86 (s, 1H, exchangeable with D2O), 8.64–8.59 (m, 2H), 8.26 (d, J = 2.2 Hz, 1H), 7.96 (s, 1H), 7.88 (d, J = 8.9 Hz, 1H), 7.55 (dd, J = 8.9, 2.2 Hz, 1H), 7.29 (s, 1H), 6.29 (br t, 1H, exchangeable with D2O), 6.24 (s, 1H), 5.52–5.46 (m, 1H), 5.13–5.02 (m, 1H), 4.36–4.31 (m, 1H), 3.86 (s, 3H), 3.45–3.39 (m, 2H), 3.21–3.11 (m, 4H), 1.55 (s, 3H), 1.48 (s, 9H), 1.40–1.17 (m, 18H). MS (ESI) m/z: 878 (M + H)+.
Methyl 6-(3-(6-((E)-2-(tert-Butoxycarbonyl)-3-(((3aR,4R,6R,6aR)-6-(6-((tert-butoxycarbonyl)amino)-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)guanidino)hexyl)ureido)-4-hydroxy-2-naphthoate (29d)
Compound 29d was obtained as a white solid (195 mg, 73%) by the reaction of 28a (170 mg, 0.300 mmol) with compound 24d (284 mg, 0.600 mmol) following the procedure described for 29a. 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H, exchangeable with D2O), 10.16 (s, 1H, exchangeable with D2O), 8.85 (s, 1H, exchangeable with D2O), 8.60–8.54 (m, 2H), 8.27 (d, J = 2.3 Hz, 1H), 7.98 (s, 1H), 7.89 (d, J = 8.9 Hz, 1H), 7.61 (dd, J = 8.9, 2.3 Hz, 1H), 7.33 (s, 1H), 6.34 (br t, 1H, exchangeable with D2O), 6.29 (s, 1H), 5.55–5.49 (m, 1H), 5.13–5.05 (m, 1H), 4.34–4.28 (m, 1H), 3.86 (s, 3H), 3.47–3.41 (m, 2H), 3.26–3.13 (m, 4H), 2.31–2.23 (m, 4H), 1.59 (s, 3H), 1.48–1.43 (m, 13H), 1.39- 1.32 (m, 12H). MS (ESI) m/z: 892 (M + H)+.
Methyl 6-(3-(7-((E)-2-(tert-Butoxycarbonyl)-3-(((3aR,4R,6R,6aR)-6-(6-((tert-butoxycarbonyl)amino)-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)guanidino)heptyl)ureido)-4-hydroxy-2-naphthoate (29e)
Compound 29e was obtained as a white solid (201 mg, 72%) by the reaction of compound 28a (175 mg, 0.309 mmol) with compound 24e (301 mg, 0.618 mmol) following the procedure described for 29a. 1H NMR (400 MHz, DMSO-d6) δ 10.31 (s, 1H, exchangeable with D2O), 10.25 (s, 1H, exchangeable with D2O), 8.79 (s, 1H, exchangeable with D2O), 8.63–8.58 (m, 2H), 8.25 (d, J = 1.7 Hz, 1H), 7.96 (s, 1H), 7.89 (d, J = 8.9 Hz, 1H), 7.51 (dd, J = 8.9, 1.7 Hz, 1H), 7.31 (s, 1H), 6.33 (br t, exchangeable with D2O), 6.21 (s, 1H), 5.52–5.49 (m, 1H), 5.08–5.04 (m, 1H), 4.37–4.30 (m, 1H), 3.81 (s, 3H), 3.40–3.27 (m, 2H), 3.19–3.09 (m, 4H), 1.54 (s, 3H), 1.51–1.42 (m, 13H), 1.36 (s, 9H), 1.29–1.17 (m, 9H). MS (ESI) m/z: 906 (M + H)+.
Methyl 6-(3-(4-((E)-3-(2-((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)ethyl)-2-(tert-butoxycarbonyl)guanidino)butyl)ureido)-4-hydroxy-2-naphthoate (30)
Compound 30 was obtained as a white solid (129 mg, 80%) by the reaction of compound 28b (100 mg, 0.208 mmol) with compound 24b (185 mg, 0.416 mmol) following the procedure described for 29a. 1H NMR (400 MHz, DMSO-d6) δ 10.26 (s, 1H, exchangeable with D2O), 8.80 (s, 1H, exchangeable with D2O), 8.34–8.30 (m, 2H), 8.25 (d, J = 2.1 Hz, 1H), 8.19–8.13 (m, 1H), 7.96 (s, 1H), 7.87 (d, J = 8.9 Hz, 1H), 7.54 (dd, J = 8.9, 2.1 Hz, 1H), 7.36–7.25 (m, 3H, 2H exchangeable with D2O), 6.27 (br t, 1H, exchangeable with D2O), 6.11 (s, 1H), 5.47–5.34 (m, 1H), 5.01–4.89 (m, 1H), 4.17–4.05 (m, 1H), 3.86 (s, 3H), 3.20–3.09 (m, 6H), 1.99–1.73 (m, 2H), 1.53 (s, 3H), 1.49–1.42 (m, 4H), 1.35 (s, 9H), 1.31 (s, 3H). MS (ESI) m/z: 778 (M + H)+.
Methyl 6-(3-(4-((E)-3-(3-((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)propyl)-2-(tert-butoxycarbonyl)guanidino)butyl)ureido)-4-hydroxy-2-naphthoate (31)
Compound 31 was obtained as a white solid (155 mg, 79%) starting from compound 28c (123 mg, 0.249 mmol) and compound 24b (222 mg, 0.498 mmol) following the procedure described for 29a. 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H, exchangeable with D2O), 8.85 (s, 1H, exchangeable with D2O), 8.29–8.21 (m, 2H), 8.26 (d, J = 2.2 Hz, 1H), 7.92 (s, 1H), 7.85 (d, J = 8.9 Hz, 1H), 7.56 (dd, J = 8.9, 2.2 Hz, 1H), 7.30–7.22 (m, 3H, 2H exchangeable with D2O), 6.27 (br t, 1H, exchangeable with D2O), 6.07 (s, 1H), 5.47–5.44 (m, 1H), 4.89–4.77 (m, 1H), 4.11–4.02 (m, 1H), 3.86 (s, 3H), 3.46–3.37 (m, 2H), 3.17–3.10 (m, 4H), 1.67–1.58 (m, 2H), 1.57–1.44 (m, 9H), 1.37 (s, 9H), 1.30 (s, 3H). MS (ESI) m/z: 791 (M + H)+.
Methyl 6-(3-(4-((E)-3-((E)-3-((3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)allyl)-2-(tert-butoxycarbonyl)guanidino)butyl)ureido)-4-hydroxy-2-naphthoate (32)
Compound 32 was obtained as a white solid (143 mg, 81%) by the reaction of compound 28d (110 mg, 0.224 mmol) with compound 24b (199 mg, 0.448 mmol) following the procedure described for 29a. 1H NMR (400 MHz, DMSO-d6) δ 10.27 (s, 1H, exchangeable with D2O), 8.84 (s, 1H, exchangeable with D2O), 8.31–8.23 (m, 2H), 8.16 (d, J = 1.9 Hz, 1H), 7.96 (s, 1H), 7.87 (d, J = 8.9 Hz, 1H), 7.54 (dd, J = 8.9, 1.9 Hz, 1H), 7.33–7.25 (m, 3H, 2H exchangeable with D2O), 6.30 (br t, exchangeable with D2O), 6.15 (s, 1H), 5.76–5.70 (m, 2H), 5.43 (m, 1H), 4.95 (m, 1H), 4.60–4.55 (m, 1H), 3.85 (s, 3H), 3.78–3.73 (m, 2H), 3.15–3.10 (m, 4H), 1.55 (s, 3H), 1.47–1.38 (m, 4H), 1.36 (s, 9H), 1.31 (s, 3H). MS (ESI) m/z: 789 (M + H)+.
((4R,6R)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methanol (33)
To a suspension of adenosine (2.00 g, 7.50 mmol) in dry acetone (150 mL), 0.75 mL of HClO4 was added dropwise. After stirring overnight, the clear solution was treated with a saturated solution of NaHCO3 until the pH was 7. The solvent was concentrated under vacuum, and the product was extracted with AcOEt (3 × 25 mL). The collected organic phases were washed with brine (30 mL), dried (Na2SO4), filtered, and concentrated under vacuum, yielding compound 33 as a white solid (1.86 g, 81%). 1H NMR (400 MHz, DMSO-d6) δ 8.16 (s, 1H), 8.00 (s, 1H), 6.48 (br s, 2H), 6.09 (s, 1H), 5.25 (s, 1H), 5.01 (s, 1H), 4.47 (s, 1H), 4.24 (m, 2H), 1.53 (s, 3H), 1.30 (s, 3H). MS (ESI) m/z: 308 (M + H)+.
9-((4R,6R)-6-(Azidomethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-amine (34)
To a solution of 33 (1.00 g, 3.25 mmol) in dry 1,4-dioxane (10 mL), DPPA (1.48 mL, 6.50 mmol) and DBU (1.50 mL, 9.80 mmol) were added dropwise, and the reaction mixture was stirred at room temperature. After 24 h, sodium azide (1.06 g, 16.3 mmol) and 15-crown-5 (0.06 mL, 0.33 mmol) were added, and the resulting mixture was refluxed for 2 h. The mixture was warmed up to room temperature, filtered, and concentrated under vacuum. The residue was taken up with CHCl3 (50 mL), washed with water (50 mL) and brine (50 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The crude brown oil was purified by flash column chromatography (gradient DCM/AcOEt/MeOH 80:20:0 to 20:80:10), yielding the azide 34 as a yellow solid (0.93 g, 86%). 1H NMR (400 MHz, DMSO-d6) δ 8.35 (s, 1H), 8.18 (s, 1H), 7.34 (br s, 2H), 6.21 (d, J = 3.2 Hz, 1H), 5.52 (dd, J = 6.4, 3.3 Hz, 1H), 5.01 (dd, J = 6.4, 2.7 Hz, 1H), 4.31 (td, J = 5.8, 2.6 Hz, 1H), 3.68–3.48 (m, 2H), 1.55 (s, 3H), 1.34 (s, 3H). MS (ESI) m/z: 333 (M + H)+.
2-((4R,6R)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)acetonitrile (35)
To a solution of 33 (0.76 g, 2.47 mmol) and PPh3 (1.62 g, 6.17 mmol) in THF (25 mL), acetone cyanohydrin (0.57 mL, 6.20 mmol) was added. Within 5 min, DEAD (40% in toluene, 2.79 mL, 6.10 mmol) was added at 0 °C. The solution was stirred at the same temperature for 10 min and then warmed to room temperature. After 16 h, the solvent was evaporated, and the crude residue was purified by flash column chromatography (gradient AcOEt/MeOH 99:1 to 80:20), yielding the title compound (0.70 g, 89%) as a brown oil. 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 7.88 (s, 1H), 6.09 (s, 1H), 5.60 (br s, 2H), 5.48 (dd, J = 6.4, 2.0 Hz, 1H), 5.13 (dd, J = 6.3, 3.1 Hz, 1H), 4.56–4.50 (m, 1H), 3.03–2.86 (m, 2H), 1.62 (s, 3H), 1.40 (s, 3H). MS (ESI) m/z: 317 (M + H)+.
tert-Butyl (2-((4R,6R)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)ethyl)carbamate (36)
To a stirred solution of 35 (0.50 g, 1.58 mmol) in MeOH (20 mL), di-tert-butyl dicarbonate (1.03 g, 4.74 mmol) and nickel(II) chloride hexahydrate (0.04 g, 0.16 mmol) were added. The solution was cooled at 0 °C, and sodium borohydride (0.42 g, 11.1 mmol) was added portionwise; the black suspension was stirred at room temperature for 4 h. Then, diethylenetriamine (0.17 mL, 1.58 mmol) was added, and the resulting mixture was stirred for 24 h. The solvent was removed under vacuum, and the crude was extracted with AcOEt (3 × 50 mL). The collected organic phases were washed with water (50 mL) and brine (50 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The crude product was purified by flash column chromatography (gradient DCM/MeOH 99:1 to 90:10), yielding the protected amine 36 as a pale yellow solid (0.53 g, 80%). 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 1H), 7.87 (s, 1H), 5.99 (d, J = 2.6 Hz, 1H), 5.60 (br s, 2H), 5.48 (dd, J = 6.6, 2.7 Hz, 1H), 5.03 (s, 1H), 4.91 (dd, J = 6.6, 4.0 Hz, 1H), 4.28–4.18 (m, 1H), 3.30–3.11 (m, 1H), 1.97–1.87 (m, 2H), 1.60 (s, 3H), 1.41 (s, 9H), 1.38 (s, 3H). MS (ESI) m/z: 421 (M + H)+.
Ethyl (E)-3-((4R,6R)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethylperhydrofuro[3,4-d][1,3]dioxol-4-yl)acrylate (37)
To a solution of 33 (1.50 g, 4.88 mmol) in 15 mL of DMSO, (ethoxycarbonylmethylene)triphenylphosphorane (4.25 g, 12.2 mmol) and o-iodoxybenzoic acid (IBX) (1.37 g, 12.2 mmol) were added. The mixture was stirred at room temperature for 72 h. Water (25 mL) was added, and the mixture was extracted with AcOEt (3 × 50 mL). The combined organic phases were dried (Na2SO4), filtered, and concentrated under vacuum to obtain a crude residue which was purified by column chromatography (AcOEt/MeOH 95:5) to give the title compound as an orange solid (1.28 g, 70%). 1H NMR (400 MHz, CDCl3) δ 8.33 (s, 1H), 7.87 (s, 1H), 6.96 (dd, J = 15.7, 5.5 Hz, 1H), 6.13 (d, J = 1.9 Hz, 1H), 5.81 (dd, J = 15.6, 1.7 Hz, 1H), 5.61 (br s, 2H), 5.56 (dd, J = 6.2, 1.9 Hz, 1H), 5.14 (dd, J = 6.3, 3.5 Hz, 1H), 4.84–4.77 (m, 1H), 4.12 (q, J = 7.1 Hz, 2H), 1.63 (s, 3H), 1.40 (s, 3H), 1.23 (t, J = 7.1 Hz, 3H). MS (ESI) m/z: 376 (M + H)+.
(E)-3-((4R,6R)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)prop-2-en-1-ol (38)
To a solution of 37 (1.00 g, 2.65 mmol) in dry DCM (10 mL), DIBAL-H (1 M in THF, 20 mL, 20.0 mmol) was added dropwise at −78 °C, and the mixture was stirred for 2 h. Then, the reaction was quenched with MeOH (8 mL) and warmed to room temperature. A saturated solution of Rochelle salt (40 mL) was added, and the resulting suspension was stirred overnight; then the aqueous phase was extracted with AcOEt (3 × 25 mL). The collected organic phases were washed with brine (30 mL), dried (Na2SO4), and filtered. Evaporation under vacuum of the solvent gave the title compound 38 (0.86 g, 98%) as a white yellow solid, which was directly used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 1H), 7.87 (s, 1H), 6.10 (d, J = 2.0 Hz, 1H), 5.87–5.84 (m, 1H), 5.58–5.53 (m, 3H), 5.02 (dd, J = 6.3, 3.4 Hz, 1H), 4.73–4.71 (m, 1H), 4.09–4.06 (m, 2H), 1.63 (s, 3H), 1.40 (s, 3H). MS (ESI) m/z: 334 (M + H)+.
2-((E)-3-((4R,6R)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4 yl)allyl)isoindoline-1,3-dione (39)
To a stirred suspension of 38 (0.48 g, 1.44 mmol), phthalimide (0.21 g, 1.44 mmol), and Ph3P (0.38 g, 1.44 mmol) in THF (10 mL), DEAD (40% in toluene, 0.66 mL, 1.44 mmol) was added dropwise. After stirring for 1.5 h at room temperature, a colorless solid started to precipitate. Stirring was continued for 1 h, after which the mixture was cooled to 0 °C and filtered. The residue was washed with Et2O and dried in vacuum to give 39 (0.47 g, 70%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.24 (s, 1H), 7.86–7.83 (m, 3H), 7.74–7.71 (m, 2H), 6.07 (d, J = 2.0 Hz, 1H), 5.87–5.80 (m, 1H), 5.77–5.68 (m, 1H), 5.60 (br s, 2H), 5.48 (dd, J = 6.3, 2.0 Hz, 1H), 4.98 (dd, J = 6.3, 3.3 Hz, 1H), 4.71–4.65 (m, 1H), 4.21–4.18 (m, 2H), 1.59 (s, 3H), 1.37 (s, 3H). MS (ESI) m/z: 463 (M + H)+.
AlphaLISA PRMT1 Activity Assay
PRMT1 activity assays were performed by taking advantage of the AlphaLISA homogeneous proximity assay.
The assays were performed in white opaque OptiPlate-384 (PerkinElmer, #6007299) at 22 °C in a final volume of 25 μL, using the following assay buffer: 30 mM Tris–HCl pH 8.0, 1 mM DTT, 0.01% BSA, 0.01% Tween-20.
In each well, 3 μL of human recombinant PRMT1 (BPS BioScience, #51041) (final concentration, 0.9 nM) was first incubated for 30 min with 2.5 μL of each compound (dissolved in DMSO and diluted in assay buffer to obtain 1% DMSO); then, 4.5 μL of a mixture of histone H4 (1–21) peptide biotinylated (AnaSpec, # 62555) (final concentration, 100 nM) and SAM (Sigma, #A7007) (final concentration, 2 μM) was added. The reaction was incubated for 60 min, and then it was stopped by the addition of 5 μL of antimethyl-histone H4 arginine 3 (H4R3me) AlphaLISA acceptor beads (PerkinElmer, #AL150) (final concentration, 20 μg/mL) diluted in Epigenetic Buffer 1X (PerkinElmer, #AL008). After an incubation of 60 min, 10 μL of streptavidin donor beads (PerkinElmer, #6760002) diluted in Epigenetic Buffer 1X was added in each well (final concentration, 20 μg/mL). After an incubation of 30 min, signals were read in Alpha mode with a PerkinElmer EnSight multimode microplate reader (excitation at 680 nm and emission at 615 nm).
For each incubation step, the OptiPlate was sealed with protective foil to prevent evaporation and contamination. Donor and acceptor beads were added in subdued light.
The 100% activity (positive control) was reached using the vehicle (DMSO) while the 0% activity (negative control) was obtained without the protein. Data were analyzed using Excel software. Values obtained for each compound are mean ± SD determined for three separate experiments.
Radioisotope-Based IC50 Profiling against PRMTs
The effects of compounds 12a–12h on the catalytic activity of PRMT1, PRMT3, PRMT4, PRMT5, PRMT6, PRMT7, and PRMT8 were determined with a HotSpot PRMT activity assay by Reaction Biology Corporation (Malvern, PA) according to the company’s standard operating procedure.119,120 Briefly, the full-length human recombinant proteins PRMT1 (residues 2–371, C-terminus; with an N-terminal GST-tag; MW = 68.3 kDa; Genbank Accession # NM_001536) or PRMT3 (residues 2–531, C-terminus; with an N-terminal His-tag; MW = 62.0 kDa; Genbank Accession # NM_005788), or PRMT4 (residues 2–608, C-terminus; with an N-terminal GST-tag; MW = 91.7 kDa; Genbank Accession # NM_199141), or PRMT5/MEP50 complex121,122 (residues PRMT5 2–637, C-terminus, and MEP50 2–342, C-terminus; with an N-terminal FLAG-tag, PRMT5, or His-tag, MEP50; MW = 73.7/39.9 kDa; Genbank Accession #NM_006109, #NM_006109), or PRMT6 (residues 2–375, C-terminus; with an N-terminal GST-tag; MW = 67.8 kDa; Genbank Accession # NM_018137), or PRMT7 (residues 2–692, C-terminus; with an N-terminal His-tag; MW = 81.7 kDa; Genbank Accession # NM_019023), or ΔN(1–60)-PRMT8123 (residues 61–394, C-terminus; with C- and N-terminal His-tags; MW = 43.2 kDa; Genbank Accession # NM_019854) were added to a solution of the proper substrate (histone H4 for PRMT1, PRMT3, and PRMT8; histone H3.3 for PRMT4; histone H2A for PRMT5/MEP50; GST-GAR for PRMT6 and PRMT7; final concentration 5 μM) in freshly prepared reaction buffer (50 mM Tris-HCl (pH 8.5), 5 mM MgCl2, 50 mM NaCl, 1 mM DTT, 1 mM PMSF, 1% DMSO) and gently mixed. The proper solution of compounds 12a–12h in DMSO was delivered into the PRMT reaction mixture by using an Acoustic Technology instrument (Echo 550, LabCyte Inc. Sunnyvale, CA) in the nanoliter range and incubated for 20 min at room temperature. Then, 3H-SAM (final concentration 1 μM) was delivered into the reaction mixture to initiate the reaction. After incubation for 60 min at 30 °C, the reaction mixture was delivered to filter paper for detection (as assessed by scintillation). Data were analyzed using Excel and GraphPad Prism 6.0 software (GraphPad Software Inc., San Diego, CA) for IC50 curve fits using sigmoidal dose vs response-variable slope (four parameters) equations.
Selectivity Assay against KMTs
The effects of compound 12h on the catalytic activity of ASH1L/KMT2H, EZH2/KMT6, MLL1/KMT2A, SET7/9/KMT7, SETD8/KMT5A, SUV39H2/KMT1B, SUV420H1/KMT5B, and DOT1L/KMT4 were determined with a HotSpot KMT activity assay by the Reaction Biology Corporation (Malvern, PA) according to the company’s standard operating procedure.119,120 Briefly, the human recombinant ASH1L (residues 2046–2330, with an N-terminal His-tag; MW = 35.4 kDa; Genbank Accession # NM_018489), or the human recombinant EZH2-containing five-member polycomb repressive complex 2 (including EZH2 residues 2–746, AEBP2 2–517, EED 2–441, RbAp48 2–425, SUZ12 2–739; all full-length; with N-terminal Flag-tag on EED and N-terminal His-tag on all others; MW = 333.8 kDa; Genbank Accession # NM_001203247, NM_001114176, NM_003797, NM_005610, NM_015355), or the human recombinant MLL1 complex (including MLL1 residues 3745–3969, C-terminus, WDR5 22–334, C-terminus, RbBP5 1–538, C-terminus, ASH2L 2–534, C-terminus, DPY-30 1–99, C-terminus; N-terminal His-tag on all subunits; MW = 212.0 kDa; Genbank Accession # NM_005933, NM_017588, NM_005057, NM_001105214, NM_0325742), or the human recombinant SET7/9 (residues 2–366, C-terminus; with a N-terminal GST-tag and a C-terminal His-tag; MW = 68.5 kDa; Genbank Accession # NM_030648), or the human recombinant SETD8 (residues 190–352, C-terminus; N-terminal His-tag; MW = 22.0 kDa; Genbank Accession # NM_020382), or the human recombinant SUV39H2 (residues 46–410, C-terminus; N-terminal fusion protein with a C-terminal His-tag; MW = 98.8 kDa; GenBank Accession No. NM_001193424), or the human recombinant SUV420H1-tv2 (residues 2–393; C-terminus; N-terminal GST-tag; MW = 73.2 kDa; GenBank Accession No. NM_016028), or the human recombinant DOT1L (residues 1–416; N-terminal GST-tag; MW = 80.0 kDa; Genbank Accession # NM_032482) were added to a solution of the proper substrate (oligonucleosomes for ASH1L, MLL1 complex, SETD8, SUV420H1 and DOT1L, final concentration 0.05 mg/mL; core histone for EZH2 complex and SET7/9, final concentration 0.05 mg/mL; histone H3 for SUV39H2, final concentration 5 μM) in freshly prepared reaction buffer (50 mM Tris-HCl (pH 8.5), 5 mM MgCl2, 50 mM NaCl, 1 mM DTT, 1 mM PMSF, 1% DMSO) and gently mixed. The proper solution (1 or 10 μM fixed concentrations) of compound 12h in DMSO was delivered into the KMT reaction mixture by using Acoustic Technology (Echo 550, LabCyte Inc. Sunnyvale, CA) in nanoliter range and incubated for 20 min at room temperature. Then, 3H-SAM (final concentration 1 μM) was delivered into the reaction mixture to initiate the reaction. After incubation for 60 min at 30 °C, the reaction mixture was delivered to filter-paper for detection (as assessed by scintillation). SAH,78−80 chaetocin (for ASH1L),81 or ryuvidine (for SETD8)82 were used as reference compounds and tested in 10-dose IC50 mode with threefold serial dilution starting at 100 μM. No inhibitor control (DMSO) was considered as 100% enzyme activity. Data were analyzed using Excel and GraphPad Prism 6.0 software (GraphPad Software Inc., San Diego, CA). Values obtained for each compound are mean ± SD determined for two separate experiments.
PRMT4 SPR Experiments
SPR experiments were performed on a Biacore T200 biosensor (Cytiva). PBS buffer (phosphate-buffered saline, pH 7.4) supplemented with 0.05% Tween-20 was used as the running buffer. Full-length recombinant PRMT4 (Active motif, # 81107) was diluted at the concentration of 50 μg/mL in 10 mM sodium acetate, pH 4.5, and then immobilized on a Series S Sensor Chip CM5 at a flow rate of 10 μL/min by using standard amine-coupling protocols to obtain densities of 4.6 kRU. One flow cell was left empty for background subtractions. Compounds 12f–12h were diluted in PBS supplemented with 0.05% Tween-20, keeping a final 2% DMSO concentration. Each compound was tested in 12 serial dilutions, prepared starting from 640 to 10 nM. Binding experiments were performed at 25 °C by using a flow rate of 30 μL/min, with 90 s monitoring of association and 90 s monitoring of dissociation. Regeneration of the surfaces was performed, when necessary, by a 10 s injection of 5 mM NaOH. The sensorgrams obtained at the 12 concentrations of each compound were first corrected taking advantage of the solvent correction performed by the instrument (correction range from 1.5% to 2.8% DMSO), and then they were double referenced. The corrected sensorgrams for each compound were fitted simultaneously using a 1:1 Langmuir model to obtain equilibrium dissociation constants (KD) and kinetic dissociation (koff) and association (kon) constants.
PAINS Analysis
Compounds 5–12 were analyzed for known classes of assay interference compounds.68 All derivatives were not recognized as PAINS according to the SwissADME web tool (http://www.swissadme.ch),69 the Free ADME-Tox Filtering Tool (FAF-Drugs4) program (http://fafdrugs4.mti.univ-paris-diderot.fr/),70 and the “False Positive Remover” software (http://www.cbligand.org/PAINS/)71 nor as aggregators according to the software “Aggregator Advisor” (http://advisor.bkslab.org/).72
Crystallography
PRMT4 Cloning, Expression, and Purification
The Mus musculus PRMT4 gene sequence corresponding to the PRMT core (residues 130 to 487 or 497, mmPRMT4130–487 or mmPRMT4130–497, respectively) were amplified by PCR from the original GST-PRMT4 construct. The sequences were cloned in the pDONR207 (Invitrogen) vector using a BP reaction (Gateway Cloning, Life Technologies). The positive clones were confirmed by sequencing (GATC). The sequences were subcloned in a pDEST20 vector using an LR reaction. The resulting recombinant proteins harbor an amino-terminal glutathione S-transferase (GST) tag followed by a Tobacco etch virus (TEV) protease cleavage site. DH10Bac competent cells containing the baculovirus genome were transformed with the pDEST20-PRMT4 plasmids and plated onto LB agar media containing 15 μg mL–1 tetracycline, 7 μg mL–1 gentamicin, 50 μg mL–1 kanamycin, 25 μg mL–1 X-Gal, and 40 μg mL–1 IPTG. Bacmid DNA purified from recombination-positive white colonies was transfected into Sf9 cells using the Lipofectin reagent (Invitrogen). Viruses were harvested 10 days after transfection. Sf9 cells were grown at 300 K in suspension culture in Grace medium (Gibco) using Bellco spinner flasks. Sf9 cell culture (1 L, at 0.8 × 106 cells mL–1) was infected with recombinant GST-mmPRMT4 viruses with an infection multiplicity of 1. Cells were harvested 48 h postinfection. Cell lysis was performed by sonication in 50 mL of buffer A [50 mM Tris-HCl pH 8.0, 250 mM NaCl, 5% glycerol, 5 mM TCEP, 0.01% NP40 and antiproteases (Roche, cOmplete, EDTA-free)], and cellular debris was sedimented by centrifugation of the lysate at 40 000g for 30 min. The supernatant was incubated overnight at 277 K with 2 mL of glutathione Sepharose resin (GE Healthcare). After a short centrifugation, the supernatants were discarded, and the beads were poured in an Econo-column (Bio-Rad). After two wash steps with 10 mL of buffer A, 2 mL of buffer A supplemented with in-house produced TEV protease was applied to the columns, and digestion was performed for 4 h at 303 K with gentle mixing. The digest was concentrated with an Amicon Ultra 10K (Millipore), loaded on a gel-filtration column (HiLoadTM 16/60 SuperdexTM S200, GE Healthcare), and eluted at 1 mL min–1 with buffer B (20 mM Tris-HCl pH 7.5, 50 mM NaCl, 1 mM TCEP) using an ÄKTA Purifier device (GE Healthcare). Fractions containing mmPRMT4 were pooled and concentrated to 5 mg mL–1.
PRMT4 Crystallization, Data Collection, and Structure Determination
The inihibitors were solubilized in water before addition to the protein solution (2–3 mg mL–1) at the final concentration of 0.5–2 mM. A vapor diffusion method utilizing hanging drop trays (VDX plate, Hampton Research) with a 0.5 mL reservoir was used for crystallization. Typically, 2 μL of protein–ligand solution was added to 1 μL of a well solution consisting of (1) 100 mM Tris-HCl pH 8.0, 10% (v/v) PEG 2000 MME for cocrystallization with 12a; (2) 100 mM Tris-HCl pH 8.0, 10% (v/v) PEG 2000 MME and 100 mM NaCl for cocrystallization with 12b and 12c; (3) 100 mM Tris-HCl pH 8.5, 20% (v/v) PEG 3350 and 200 mM ammonium sulfate for cocrystallization with 12f and 12h; and (4) 100 mM Tris-HCl pH 8.5, 20% (v/v) PEG 3350 and 100 mM NaCl for cocrystallization with 12g. Crystals grew in a few days at 293 K. Crystals were flash-frozen in liquid nitrogen after a brief transfer to 5 μL of reservoir solution containing 15% (v/v) PEG 400 as a cryoprotectant and were stored in liquid nitrogen.
The diffraction data sets were collected using SOLEIL PROXIMA1 and PROXIMA2 and ESRF ID30-B beamlines, on a Pilatus 6M, EIGER 9M, and EIGER 6 M (Dectris) detector, respectively, and processed with XDS124 and HKL-2000.125 The crystals belonged to the P21212 space group with four monomers of mmPRMT4 in the asymmetric unit. The structures were solved by molecular replacement using a mmPRMT4 structure as a probe.106 Model building and refinement were carried out using Coot,126 PHENIX,127−129 and BUSTER.130 TLS refinement with 20 groups per polypeptide chain was used. All other crystallographic calculations were carried out with the CCP4 package.131 A summary of the data statistics for the solved and structures is provided in Table 4. The atomic coordinates and experimental data (PDB ID XXXX) have been deposited in the Protein Data Bank. Structure figures were generated with PyMol (http://www.pymol.org). Initial 3D structures of compounds 12a–12c and 12f–12h were generated using the Marvin suite, Marvin version 5.2.3_1, ChemAxon (https://www.chemaxon.com). Stereochemical restraint dictionaries for all compounds have been produced using the grade Web Server (http://grade.globalphasing.org).
PRMT6 Cloning, Expression, and Purification
The Mus musculus PRMT6 gene sequence (UNP Q6NZB1) corresponding to the residues 34–378 of the protein was cloned in the pnEA-vHis or pnEA/vHis vector (TEV protease cleavable hexahistidine tag at the amino- or carboxy-terminus of the recombinant protein, respectively)132 between the NdeI and XhoI restriction sites and used to transform Escherichia coli BL21 (DE3) cells. The protein also contains an F315L point mutation, reported for the entry as a natural variant. The construction in the pnEA/vHis plasmid also contains a C53S point mutation to prevent a disufide bond formation with the C232 in the second monomer of the homodimer. The protein expression was induced for 4 h at 310 K, with 0.1 mM IPTG in LB medium. The cells were harvested by centrifugation and resuspended in 10 mL per pellet g of buffer A (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 7 mM β-ME, 20 mM imidazole) supplemented with 1 mM PMSF. After sonication, the lysate was centrifuged at 40 000 × g for 25 min at 4 °C. The supernatant was mixed with 0.5 mL of NiNTA superflow resin (Qiagen) per liter of culture, incubated for 1 h at 4 °C, and then loaded on an Econo column (Bio-Rad). After the column was washed with 10 column volumes (c.v.) of buffer A, the bound protein was eluted with 5 c.v. of buffer A supplemented with 500 mM imidazole. The fractions containing the recombinant protein were detected by a Bradford assay, pooled, and supplemented with TEV protease. The sample was then dialyzed in a 10 000 MWCO dialysis membrane (Spectrum) against 1 L of buffer B (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 7 mM β-ME, and 1 mM EDTA]) for 16 h at 4 °C. The dialyzed sample was loaded onto a HiLoad 16/60 Superdex 200 prep grade column (GE Healthcare) and equilibrated in buffer C (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 5 mM TCEP). The protein was finally concentrated on an Amicon Ultra-4 10 000 MWCO filter (Millipore) to a 6 mg mL–1 final concentration. Samples were either directly used for crystallization or flash-frozen as 25 μL aliquots in liquid nitrogen and stored at −80 °C.
PRMT6 Crystallization, Data Collection, and Structure Determination
EML ligands were solubilized in water before addition to the protein solution (6 mg mL–1) at the final concentration of 1 mM. A vapor diffusion method utilizing hanging drop trays (VDX plate, Hampton Research) with a 1 mL reservoir was used for crystallization. Typically, 1 μL of protein–ligand solution was added to 1 μL of well solution consisting of (1) 100 mM Tris-HCl pH 7.5, 200 mM NaNO3, and 20% (v/v) PEG 3350 for the cocrystallization with 12a; (2) 100 mM Tris-HCl pH 8.0, 200 mM HCOONa, and 19% PEG 3350 for the cocrystallization with 12c; and (3) 100 mM Tris-HCl pH 7.5, 200 mM HCOONa, and 19% PEG 3350 for the cocrystallization with 12f. Crystals grew in a week at 293 K. Crystals were flash-frozen in liquid nitrogen after a brief transfer to 5 μL of reservoir solution containing 15% (v/v) PEG 400 as a cryoprotectant and were stored in liquid nitrogen.
Data sets were all collected on an in-house RIGAKU FR-X diffraction source, using a PILATUS 4 M detector (Dectris), and processed with XDS. Three structures of mmPRMT6 have been solved and refined. The crystals belong to the P21 space group with two mmPRMT6 molecules in the asymmetric unit. The structures were solved by molecular replacement using previously solved mmPRMT6 structures as the search model (PDB ID 4C03).107 Iterative cycles of model building and refinement were carried out using Coot, PHENIX, and Buster. A summary of the data statistics for the two solved and refined structures is provided in Table 5. The atomic coordinates and experimental data (PDB ID 7NUD, 7NUE, and 7P2R) have been deposited in the Protein Data Bank.
Parallel Artificial Membrane Permeability Assay (PAMPA)
A donor solution (0.2 mM) was prepared by diluting 20 mM dimethyl sulfoxide (DMSO) compound stock solution using phosphate buffer (pH 7.4, 0.01 M). Filters were coated with 5 μL of a 1% (w/v) dodecane solution of l-α-phosphatidylcholine. Donor solution (150 μL) was added to each well of the filter plate. To each well of the acceptor plate, 300 μL of solution (5% DMSO in phosphate buffer) was added. Selected compounds were tested in triplicate; propranolol and furosemide were used as positive and negative controls. The sandwich was incubated for 24 h at room temperature under gentle shaking. After the incubation time, the sandwich plates were separated, and 250 μL of the acceptor plate was transferred to a UV quartz microtiter plate and measured by UV spectroscopy, using a Multiskan GO microplate spectrophotometer (Thermo Fisher Scientific) at 250–500 nm at a step of 5 nm. Reference solutions (250 μL) were prepared diluting the sample stock solutions to the same concentration as that with no membrane barrier. The apparent permeability value Papp is determined from the ratio r of the absorbance of the compound found in the acceptor chamber divided by the theoretical equilibrium absorbance (determined independently), the Faller modification133 of the Sugano equation:134
In this equation, VR is the volume of the acceptor compartment (0.3 cm3), VD is the donor volume (0.15 cm3), A is the accessible filter area (0.24 cm2), and t is the incubation time in seconds.
Cell Viability Assay
The HEK293T cell line was cultured in DMEM (Euroclone) supplemented with 10% (v/v) fetal bovine serum (Euroclone), 100 U/mL penicillin, 100 μg/mL streptomycin (Euroclone), and 2 mM l-glutamine (Euroclone) at 37 °C in a 5% CO2 atmosphere. Cell viability was measured via a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. A total of 200 μL of cells seeded in 96-well microtiter plates (5 × 104 cells/mL) was exposed for 24 or 72 h to different concentrations of compounds 12f–12h (10, 50, and 100 μM) in media containing 0.2% DMSO. The mitochondrial-dependent reduction of MTT to formazan was used to assess cell viability. Live cells reduce yellow MTT to purple formazan. The resulting formazan was solubilized in DMSO, and the absorbance was measured at 550 nm and corrected for 620 nm background. Experiments were performed in quadruplicate and all values are expressed as the percentage of the control containing 0.2% DMSO.
Western Blotting Method
HEK293T or MCF7 cells were treated with different compounds and then harvested and lysed at the indicated time, and the cell lysates were applied to Western blot analysis. In brief, cells were harvested and washed three times with cold PBS, and then cell lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.1% SDS, 1% sodium deoxycholate, 5 mM EDTA, supplemented with proteinase inhibitor mixture) was added to obtain the cell lysates. Cell debris was pelleted and discarded, whereas the supernatant was kept. Protein samples were added with SDS loading buffer and boiled for 10 min, followed by SDS-PAGE. Then the proteins were transferred to a PVDF membrane. The membrane was blocked with 5% fat-free milk for 1 h at room temperature and then incubated with primary antibodies (antihistone H3, Abcam, #ab18521; pan-PRMT4 substrate antibody, made in-house) at 4 °C overnight. The blot was then washed three times with PBST and incubated with secondary antibodies for 1 h at room temperature. After being washed three times with PBST, the membrane was incubated with ECL reagent, and the signals were detected on X-ray film.
Anti-PRMT4 Substrate Antibody
The anti-PRMT4 substrate antibody was raised against the R388 site in the transcription factor NFIB. This antibody recognizes NFIB-Me in a PRMT4-dependent manner but also recognizes additional PRMT4 substrates.102,108
Cell Proliferation Assay
HEK293T cells were seeded into 96-well plates at the density of 250 cells/well in 100 μL of medium. MCF7 cells were seeded into 96-well plates at the density of 1000 cells/well in 100 μL of medium. Cell proliferation was analyzed at day 0, day 2, day 4, day 6, and day 8, and the medium was changed at day 4. The proliferation analysis was based on viability as determined by CellTiter-Glo (#G7572, Promega) according to the manufacturer’s instructions. Triple independent repeats were conducted.
Acknowledgments
G.S. is supported by grants from the Italian Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR), Progetti di Ricerca di Interesse Nazionale (PRIN 20152TE5PK), from the University of Salerno (FARB grant), and from the Regione Campania (Italy) grant “Combattere la resistanza tumorale: piattaforma integrata multidisciplinare per un approccio tecnologico innovativo alle oncoterapie—CAMPANIA ONCOTERAPIE” (project no. B61G18000470007). D.R. was supported by an STSM Grant from the COST Action CM1406. S.C. is supported by grants from the Italian Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR), Progetti di Ricerca di Interesse Nazionale (PRIN 2017MT3993). L.B., J.C., V.C., N.M., and N.T-C. are supported by grants from CNRS, Université de Strasbourg, INSERM, Instruct-ERIC, part of the European Strategy Forum on Research Infrastructures (ESFRI) supported by national member subscriptions as well as the French Infrastructure for Integrated Structural Biology (FRISBI) [ANR-10-INSB-005, grant ANR-10-LABX-0030-INRT, a French State fund managed by the Agence Nationale de la Recherche under the frame program Investissements d’Avenir labeled ANR-19-CE11-0010-01 JC and IGBMC], grants from the Association pour la Recherche contre le Cancer (ARC) (ARC 2016, n° PJA 20161204817), and grants from “Ligue d’Alsace contre le Cancer”. We thank Alastair McEwen for help in the structural studies. We thank members of SOLEIL Proxima1 beamlines and the European Synchrotron Radiation Facility–European Molecular Biology Laboratory joint Structural Biology groups for the use of beamline facilities and for assistance during X-ray data collection.
Glossary
Abbreviations Used
- AcOEt
ethyl acetate
- ACN
acetonitrile
- AEBP2
zinc finger protein AEBP2, also known as adipocyte enhancer-binding protein 2
- ALPHA
amplified luminescent proximity homogeneous assay
- ASH1L
absent small and homeotic disks protein 1 homologue
- ASH2L
Set1/Ash2 histone methyltransferase complex subunit ASH2, also known as absent small and homeotic disks protein 2 homologue
- CA150
also known as TCERG1
- CARM1
coactivator-associated arginine methyltransferase 1
- CBP
CREB-binding protein
- CREB
cAMP response element-binding protein
- DEPTQ
distortionless enhancement by polarization transfer quaternary
- DOT1L
DOT1-like (disruptor of telomeric silencing 1-like)
- DPY30
protein dpy-30 homologue, also known as Dpy-30-like protein
- EDC
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- ECL
enhanced chemiluminescence
- EED
polycomb protein EED, also known as embryonic ectoderm development protein
- ELAV
embryonic lethal, abnormal vision, Drosophila
- ELAVL1
ELAV like RNA binding protein 1
- ELAVL4
ELAV like RNA binding protein 4
- EZH2
enhancer of zeste homologue 2
- GAR
glycine- and arginine-rich motif
- HuD
human antigen D, also known as ELAVL4
- HuR
human antigen R, also known as ELAVL1
- MED12
mediator of RNA polymerase II transcription subunit 12
- MLL1
histone-lysine N-methyltransferase 2A, also known as myeloid/lymphoid or mixed-lineage leukemia protein 1
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NFIB-Me
nuclear factor 1 B-type
- p300
E1A-associated protein of 300 kDa
- Papp
apparent permeability
- PABP1
poly(A)-binding protein 1
- Pbf
2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl
- PBST
phosphate-buffered saline with Tween-20
- PMSF
phenylmethylsulfonyl fluoride
- PRMT
protein arginine methyltransferase
- PRMT1
protein arginine methyltransferase 1
- PRMT3
protein arginine methyltransferase 3
- PRMT4
protein arginine methyltransferase 4
- PRMT5
protein arginine methyltransferase 5
- PRMT6
protein arginine methyltransferase 6
- PRMT7
protein arginine methyltransferase 7
- PRMT8
protein arginine methyltransferase 8
- PVDF
pol(vinylidene difluoride)
- RBAP48
histone-binding protein RBBP4, also known as retinoblastoma-binding protein p48
- RBBP5
retinoblastoma-binding protein 5
- SAH
S-5′-adenosyl-l-homocysteine
- SAM
S-adenosyl-l-methionine
- SD
standard deviation
- SRC-3
steroid receptor coactivator-3
- SET
suppressor of variegation 3-9 enhancer-of-zeste trithorax
- SET7/9
SET domain-containing protein 7
- SETD8
SET domain-containing protein 8, also known as PR/SET domain-containing protein 07
- siRNA
small interference ribonucleic acid
- SUV39H2
suppressor of variegation 3-9 homologue 2, also known as Su(var.)3-9 homologue 2
- SUV420H1-TV2
suppressor of variegation 4-20 homologue 1, -transcription variant 2
- SUZ12
polycomb protein SUZ12, also known as suppressor of zeste 12 protein homologue
- TCERG1
transcription elongation regulator 1
- WDR5
WD repeat-containing protein 5
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00252.
Synthesis of intermediates 27a–d, PAMPA assay, additional results from crystallographic studies, and copies of 1H NMR and 13C NMR spectra of all final compounds (PDF)
PDB validation report for 7PPY (PDF)
PDB validation report for 7PV6 (PDF)
PDB validation report for 7PPQ (PDF)
PDB validation report for 7PU8 (PDF)
PDB validation report for 7PUC (PDF)
PDB validation report for 7PUQ (PDF)
PDB validation report for 7NUD (PDF)
PDB validation report for 7NUE (PDF)
PDB validation report for 7P2R (PDF)
Molecular formula strings (CSV)
Accession Codes
The atomic coordinates and structure factors of PRMT4 in complex with 12a–c and 12f–h in space group P21212 (PDB ID: 7PV6, 7PPY, 7PPQ, 7PU8, 7PUQ, 7PUC) and of PRMT6 in complex with 12a, 12c, and 12f in space group P21 (PDB ID: 7NUD, 7NUE, and 7P2R) have been deposited in the Protein Data Bank, and authors will release the atomic coordinates and experimental data upon article publication.
Author Present Address
¶ Center for Drug Discovery and Development-DMPK, Aptuit, an Evotec Company, Via A. Fleming, 4, 37135 Verona, Italy
Author Contributions
∥ The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. G.I. and C.M. contributed equally to this work and are listed in alphabetical order.
The authors declare no competing financial interest.
Special Issue
Published as part of the Journal of Medicinal Chemistry special issue “Epigenetics 2022”.
Supplementary Material
References
- Bedford M. T.; Clarke S. G. Protein Arginine Methylation in Mammals: Who, What, and Why. Mol. Cell 2009, 33, 1–13. 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann J.; Clancy K. W.; Thompson P. R. Chemical Biology of Protein Arginine Modifications in Epigenetic Regulation. Chem. Rev. 2015, 115, 5413–5461. 10.1021/acs.chemrev.5b00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H.; Mundade R.; Lange K. C.; Lu T. Protein Arginine Methylation of Non-Histone Proteins and its Role in Diseases. Cell Cycle 2014, 13, 32–41. 10.4161/cc.27353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrowsmith C. H.; Bountra C.; Fish P. V.; Lee K.; Schapira M. Epigenetic Protein Families: a New Frontier for Drug Discovery. Nat. Rev. Drug Discovery 2012, 11, 384–400. 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- Guccione E.; Richard S. The Regulation, Functions and Clinical Relevance of Arginine Methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 642–657. 10.1038/s41580-019-0155-x. [DOI] [PubMed] [Google Scholar]
- Lee S.-Y.; Vuong T. A.; So H.-K.; Kim H.-J.; Kim Y. B.; Kang J.-S.; Kwon I.; Cho H. PRMT7 Deficiency Causes Dysregulation of the HCN Channels in the CA1 Pyramidal Cells and Impairment of Social Behaviors. Exp. Mol. Med. 2020, 52, 604–614. 10.1038/s12276-020-0417-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai W. C.; Gayatri S.; Reineke L. C.; Sbardella G.; Bedford M. T.; Lloyd R. E. Arginine Demethylation of G3BP1 Promotes Stress Granule Assembly. J. Biol. Chem. 2016, 291, 22671–22685. 10.1074/jbc.M116.739573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Bedford M. T. Protein Arginine Methyltransferases and Cancer. Nat. Rev. Cancer 2013, 13, 37–50. 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- Cha B.; Jho E.-H. Protein Arginine Methyltransferases (PRMTs) as Therapeutic Targets. Expert Opin. Ther. Tar. 2012, 16, 651–664. 10.1517/14728222.2012.688030. [DOI] [PubMed] [Google Scholar]
- Blanc R. S.; Richard S. Arginine Methylation: The Coming of Age. Mol. Cell 2017, 65, 8–24. 10.1016/j.molcel.2016.11.003. [DOI] [PubMed] [Google Scholar]
- Xu J.; Richard S. Cellular Pathways Influenced by Protein Arginine Methylation: Implications for Cancer. Mol. Cell 2021, 81, 4357–4368. 10.1016/j.molcel.2021.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner B. M. Reading Signals on the Nucleosome with a New Nomenclature for Modified Histones. Nat. Struct. Mol. Biol. 2005, 12, 110–112. 10.1038/nsmb0205-110. [DOI] [PubMed] [Google Scholar]
- Wu Q.; Schapira M.; Arrowsmith C. H.; Barsyte-Lovejoy D. Protein Arginine Methylation: From Enigmatic Functions to Therapeutic Targeting. Nat. Rev. Drug Discovery 2021, 20, 509–530. 10.1038/s41573-021-00159-8. [DOI] [PubMed] [Google Scholar]
- Zurita-Lopez C. I.; Sandberg T.; Kelly R.; Clarke S. G. Human Protein Arginine Methyltransferase 7 (PRMT7) Is a Type III Enzyme Forming ω-NG-Monomethylated Arginine Residues*. J. Biol. Chem. 2012, 287, 7859–7870. 10.1074/jbc.M111.336271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger M.-C.; Liang C.; Russell R. S.; Lin R.; Bedford M. T.; Wainberg M. A.; Richard S. Methylation of Tat by PRMT6 Regulates Human Immunodeficiency Virus Type 1 Gene Expression. J. Virol. 2005, 79, 124–131. 10.1128/JVI.79.1.124-131.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kzhyshkowska J.; SchÜTt H.; Liss M.; Kremmer E.; Stauber R.; Wolf H.; Dobner T. Heterogeneous Nuclear Ribonucleoprotein E1B-AP5 is Methylated in Its Arg-Gly-Gly (RGG) Box and Interacts with Human Arginine Methyltransferase HRMT1L1. Biochem. J. 2001, 358, 305–314. 10.1042/bj3580305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho J.; Choi S.; Seong Y. R.; Choi J.; Im D.-S. The Arginine-1493 Residue in QRRGRTGR1493G Motif IV of the Hepatitis C Virus NS3 Helicase Domain Is Essential for NS3 Protein Methylation by the Protein Arginine Methyltransferase 1. J. Virol. 2001, 75, 8031–8044. 10.1128/JVI.75.17.8031-8044.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shire K.; Kapoor P.; Jiang K.; Hing M. N. T.; Sivachandran N.; Nguyen T.; Frappier L. Regulation of the EBNA1 Epstein-Barr Virus Protein by Serine Phosphorylation and Arginine Methylation. J. Virol. 2006, 80, 5261–5272. 10.1128/JVI.02682-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souki S. K.; Gershon P. D.; Sandri-Goldin R. M. Arginine Methylation of the ICP27 RGG Box Regulates ICP27 Export and Is Required for Efficient Herpes Simplex Virus 1 Replication. J. Virol. 2009, 83, 5309–5320. 10.1128/JVI.00238-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai T.; Yu Z.; Wang Z.; Liang C.; Richard S. Arginine Methylation of SARS-Cov-2 Nucleocapsid Protein Regulates RNA Binding, Its Ability to Suppress Stress Granule Formation, and Viral Replication. J. Biol. Chem. 2021, 297, 100821. 10.1016/j.jbc.2021.100821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Penebre E.; Kuplast K. G.; Majer C. R.; Boriack-Sjodin P. A.; Wigle T. J.; Johnston L. D.; Rioux N.; Munchhof M. J.; Jin L.; Jacques S. L.; West K. A.; Lingaraj T.; Stickland K.; Ribich S. A.; Raimondi A.; Scott M. P.; Waters N. J.; Pollock R. M.; Smith J. J.; Barbash O.; Pappalardi M.; Ho T. F.; Nurse K.; Oza K. P.; Gallagher K. T.; Kruger R.; Moyer M. P.; Copeland R. A.; Chesworth R.; Duncan K. W. A Selective Inhibitor of PRMT5 with in Vivo and in Vitro Potency in MCL Models. Nat. Chem. Biol. 2015, 11, 432–437. 10.1038/nchembio.1810. [DOI] [PubMed] [Google Scholar]
- Eram M. S.; Shen Y. D.; Szewczyk M. M.; Wu H.; Senisterra G.; Li F. L.; Butler K. V.; Kaniskan H. U.; Speed B. A.; dela Sena C.; Dong A. P.; Zeng H.; Schapira M.; Brown P. J.; Arrowsmith C. H.; Barsyte-Lovejoy D.; Liu J.; Vedadi M.; Jin J. A Potent, Selective, and Cell-Active Inhibitor of Human Type I Protein Arginine Methyltransferases. ACS Chem. Biol. 2016, 11, 772–781. 10.1021/acschembio.5b00839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoriw A.; Rajapurkar S. R.; O’Brien S.; Gerhart S. V.; Mitchell L. H.; Adams N. D.; Rioux N.; Lingaraj T.; Ribich S. A.; Pappalardi M. B.; Shah N.; Laraio J.; Liu Y.; Butticello M.; Carpenter C. L.; Creasy C.; Korenchuk S.; McCabe M. T.; McHugh C. F.; Nagarajan R.; Wagner C.; Zappacosta F.; Annan R.; Concha N. O.; Thomas R. A.; Hart T. K.; Smith J. J.; Copeland R. A.; Moyer M. P.; Campbell J.; Stickland K.; Mills J.; Jacques-O’Hagan S.; Allain C.; Johnston D.; Raimondi A.; Porter Scott M.; Waters N.; Swinger K.; Boriack-Sjodin A.; Riera T.; Shapiro G.; Chesworth R.; Prinjha R. K.; Kruger R. G.; Barbash O.; Mohammad H. P. Anti-tumor Activity of the Type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell 2019, 36, 100–114. 10.1016/j.ccell.2019.05.014. [DOI] [PubMed] [Google Scholar]
- Nakayama K.; Szewczyk M. M.; Sena C. d.; Wu H.; Dong A.; Zeng H.; Li F.; de Freitas R. F.; Eram M. S.; Schapira M.; Baba Y.; Kunitomo M.; Cary D. R.; Tawada M.; Ohashi A.; Imaeda Y.; Saikatendu K. S.; Grimshaw C. E.; Vedadi M.; Arrowsmith C. H.; Barsyte-Lovejoy D.; Kiba A.; Tomita D.; Brown P. J. TP-064, a Potent and Selective Small Molecule Inhibitor of PRMT4 for Multiple Myeloma. Oncotarget 2018, 9, 18480–18493. 10.18632/oncotarget.24883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szewczyk M. M.; Ishikawa Y.; Organ S.; Sakai N.; Li F.; Halabelian L.; Ackloo S.; Couzens A. L.; Eram M.; Dilworth D.; Fukushi H.; Harding R.; dela Seña C. C.; Sugo T.; Hayashi K.; McLeod D.; Zepeda C.; Aman A.; Sánchez-Osuna M.; Bonneil E.; Takagi S.; Al-Awar R.; Tyers M.; Richard S.; Takizawa M.; Gingras A.-C.; Arrowsmith C. H.; Vedadi M.; Brown P. J.; Nara H.; Barsyte-Lovejoy D. Pharmacological Inhibition of PRMT7 Links Arginine Monomethylation to the Cellular Stress Response. Nat. Commun. 2020, 11, 2396. 10.1038/s41467-020-16271-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominici C.; Sgarioto N.; Yu Z.; Sesma-Sanz L.; Masson J.-Y.; Richard S.; Raynal N. J. M. Synergistic Effects of Type I PRMT and PARP Inhibitors Against Non-Small Cell Lung Cancer Cells. Clin. Epigenetics 2021, 13, 54. 10.1186/s13148-021-01037-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart S. V.; Kellner W. A.; Thompson C.; Pappalardi M. B.; Zhang X.-P.; Montes de Oca R.; Penebre E.; Duncan K.; Boriack-Sjodin A.; Le B.; Majer C.; McCabe M. T.; Carpenter C.; Johnson N.; Kruger R. G.; Barbash O. Activation of the p53-MDM4 Regulatory Axis Defines the Anti-Tumour Response to PRMT5 Inhibition Through Its Role in Regulating Cellular Splicing. Sci. Rep. 2018, 8, 9711. 10.1038/s41598-018-28002-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao H.; Yan X.; Zhu K.; Zhang H. Discovery of Novel PRMT5 Inhibitors by Virtual Screening and Biological Evaluations. Chem. Pharm. Bull. 2019, 67, 382–388. 10.1248/cpb.c18-00980. [DOI] [PubMed] [Google Scholar]
- Duncan K. W.; Rioux N.; Boriack-Sjodin P. A.; Munchhof M. J.; Reiter L. A.; Majer C. R.; Jin L.; Johnston L. D.; Chan-Penebre E.; Kuplast K. G.; Porter Scott M.; Pollock R. M.; Waters N. J.; Smith J. J.; Moyer M. P.; Copeland R. A.; Chesworth R. Structure and Property Guided Design in the Identification of PRMT5 Tool Compound EPZ015666. ACS Med. Chem. Lett. 2016, 7, 162–166. 10.1021/acsmedchemlett.5b00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroz R. V.; Reutershan M. H.; Schneider S. E.; Sloman D.; Lacey B. M.; Swalm B. M.; Yeung C. S.; Gibeau C.; Spellman D. S.; Rankic D. A.; Chen D.; Witter D.; Linn D.; Munsell E.; Feng G.; Xu H.; Hughes J. M. E.; Lim J.; Saurí J.; Geddes K.; Wan M.; Mansueto M. S.; Follmer N. E.; Fier P. S.; Siliphaivanh P.; Daublain P.; Palte R. L.; Hayes R. P.; Lee S.; Kawamura S.; Silverman S.; Sanyal S.; Henderson T. J.; Ye Y.; Gao Y.; Nicholson B.; Machacek M. R. The Discovery of Two Novel Classes of 5,5-Bicyclic Nucleoside-Derived PRMT5 Inhibitors for the Treatment of Cancer. J. Med. Chem. 2021, 64, 3911–3939. 10.1021/acs.jmedchem.0c02083. [DOI] [PubMed] [Google Scholar]
- Smith C. R.; Aranda R.; Bobinski T. P.; Briere D. M.; Burns A. C.; Christensen J. G.; Clarine J.; Engstrom L. D.; Gunn R. J.; Ivetac A.; Jean-Baptiste R.; Ketcham J. M.; Kobayashi M.; Kuehler J.; Kulyk S.; Lawson J. D.; Moya K.; Olson P.; Rahbaek L.; Thomas N. C.; Wang X.; Waters L. M.; Marx M. A. Fragment-Based Discovery of MRTX1719, a Synthetic Lethal Inhibitor of the PRMT5•MTA Complex for the Treatment of MTAP-Deleted Cancers. J. Med. Chem. 2022, 65, 1749–1766. 10.1021/acs.jmedchem.1c01900. [DOI] [PubMed] [Google Scholar]
- Bonday Z. Q.; Cortez G. S.; Grogan M. J.; Antonysamy S.; Weichert K.; Bocchinfuso W. P.; Li F.; Kennedy S.; Li B.; Mader M. M.; Arrowsmith C. H.; Brown P. J.; Eram M. S.; Szewczyk M. M.; Barsyte-Lovejoy D.; Vedadi M.; Guccione E.; Campbell R. M. LLY-283, a Potent and Selective Inhibitor of Arginine Methyltransferase 5, PRMT5, with Antitumor Activity. ACS Med. Chem. Lett. 2018, 9, 612–617. 10.1021/acsmedchemlett.8b00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney D. C.; McMillan B. J.; Ranaghan M. J.; Moroco J. A.; Brousseau M.; Mullin-Bernstein Z.; O’Keefe M.; McCarren P.; Mesleh M. F.; Mulvaney K. M.; Robinson F.; Singh R.; Bajrami B.; Wagner F. F.; Hilgraf R.; Drysdale M. J.; Campbell A. J.; Skepner A.; Timm D. E.; Porter D.; Kaushik V. K.; Sellers W. R.; Ianari A. Discovery of a First-in-Class Inhibitor of the PRMT5–Substrate Adaptor Interaction. J. Med. Chem. 2021, 64, 11148–11168. 10.1021/acs.jmedchem.1c00507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniskan H. U.; Konze K. D.; Jin J. Selective inhibitors of protein methyltransferases. J. Med. Chem. 2015, 58, 1596–1629. 10.1021/jm501234a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Jiang H.; Jin J.; Xie Y.; Chen Z.; Zhang H.; Lian F.; Liu Y.-C.; Zhang C.; Ding H.; Chen S.; Zhang N.; Zhang Y.; Jiang H.; Chen K.; Ye F.; Yao Z.; Luo C. Development of Potent Type I Protein Arginine Methyltransferase (PRMT) Inhibitors of Leukemia Cell Proliferation. J. Med. Chem. 2017, 60, 8888–8905. 10.1021/acs.jmedchem.7b01134. [DOI] [PubMed] [Google Scholar]
- Halby L.; Marechal N.; Pechalrieu D.; Cura V.; Franchini D.-M.; Faux C.; Alby F.; Troffer-Charlier N.; Kudithipudi S.; Jeltsch A.; Aouadi W.; Decroly E.; Guillemot J.-C.; Page P.; Ferroud C.; Bonnefond L.; Guianvarc’h D.; Cavarelli J.; Arimondo P. B. Hijacking DNA methyltransferase transition state analogues to produce chemical scaffolds for PRMT inhibitors. Philos. Trans. R. Soc. London, B,Biol. Sci. 2018, 373, 20170072. 10.1098/rstb.2017.0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan M.; Manku S.; Therrien E.; Nguyen N.; Styhler S.; Robert M.-F.; Goulet A.-C.; Petschner A. J.; Rahil G.; Robert MacLeod A.; Déziel R.; Besterman J. M.; Nguyen H.; Wahhab A. N-Benzyl-1-Heteroaryl-3-(Trifluoromethyl)-1H-Pyrazole-5-Carboxamides as Inhibitors of Co-Activator Associated Arginine Methyltransferase 1 (CARM1). Bioorg. Med. Chem. Lett. 2009, 19, 1218–1223. 10.1016/j.bmcl.2008.12.075. [DOI] [PubMed] [Google Scholar]
- Huynh T.; Chen Z.; Pang S.; Geng J.; Bandiera T.; Bindi S.; Vianello P.; Roletto F.; Thieffine S.; Galvani A.; Vaccaro W.; Poss M. A.; Trainor G. L.; Lorenzi M. L.; Gottardis M.; Jayaraman L.; Purandare A. V. Optimization of Pyrazole Inhibitors of Coactivator Associated Arginine Methyltransferase 1 (CARM1). Bioorg. Med. Chem. Lett. 2009, 19, 2924–2927. 10.1016/j.bmcl.2009.04.075. [DOI] [PubMed] [Google Scholar]
- Purandare A. V.; Chen Z.; Huynh T.; Pang S.; Geng J.; Vaccaro W.; Poss M. A.; Oconnell J.; Nowak K.; Jayaraman L. Pyrazole Inhibitors of Coactivator Associated Arginine Methyltransferase 1 (CARM1). Bioorg. Med. Chem. Lett. 2008, 18, 4438–4441. 10.1016/j.bmcl.2008.06.026. [DOI] [PubMed] [Google Scholar]
- Ferreira de Freitas R.; Eram M. S.; Smil D.; Szewczyk M. M.; Kennedy S.; Brown P. J.; Santhakumar V.; Barsyte-Lovejoy D.; Arrowsmith C. H.; Vedadi M.; Schapira M. Discovery of a Potent and Selective Coactivator Associated Arginine Methyltransferase 1 (CARM1) Inhibitor by Virtual Screening. J. Med. Chem. 2016, 59, 6838–6847. 10.1021/acs.jmedchem.6b00668. [DOI] [PubMed] [Google Scholar]
- Guo Z.; Zhang Z.; Yang H.; Cao D.; Xu X.; Zheng X.; Chen D.; Wang Q.; Li Y.; Li J.; Du Z.; Wang X.; Chen L.; Ding J.; Shen J.; Geng M.; Huang X.; Xiong B. Design and Synthesis of Potent, Selective Inhibitors of Protein Arginine Methyltransferase 4 against Acute Myeloid Leukemia. J. Med. Chem. 2019, 62, 5414–5433. 10.1021/acs.jmedchem.9b00297. [DOI] [PubMed] [Google Scholar]
- Therrien E.; Larouche G.; Manku S.; Allan M.; Nguyen N.; Styhler S.; Robert M.-F.; Goulet A.-C.; Besterman J. M.; Nguyen H.; Wahhab A. 1,2-Diamines as Inhibitors of Co-Activator Associated Arginine Methyltransferase 1 (CARM1). Bioorg. Med. Chem. Lett. 2009, 19, 6725–6732. 10.1016/j.bmcl.2009.09.110. [DOI] [PubMed] [Google Scholar]
- Wan H.; Huynh T.; Pang S.; Geng J.; Vaccaro W.; Poss M. A.; Trainor G. L.; Lorenzi M. V.; Gottardis M.; Jayaraman L.; Purandare A. V. Benzo[d]imidazole Inhibitors of Coactivator Associated Arginine Methyltransferase 1 (CARM1)-Hit to Lead Studies. Bioorg. Med. Chem. Lett. 2009, 19, 5063–5066. 10.1016/j.bmcl.2009.07.040. [DOI] [PubMed] [Google Scholar]
- van Haren M. J.; Marechal N.; Troffer-Charlier N.; Cianciulli A.; Sbardella G.; Cavarelli J.; Martin N. I. Transition State Mimics Are Valuable Mechanistic Probes for Structural Studies with the Arginine Methyltransferase CARM1. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 3625–3630. 10.1073/pnas.1618401114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland M.; Li A.; Kaghad A.; Panagopoulos D.; Li F.; Szewczyk M.; Smil D.; Scholten C.; Bouché L.; Stellfeld T.; Arrowsmith C. H.; Barsyte D.; Vedadi M.; Hartung I. V.; Steuber H.; Britton R.; Santhakumar V. Rational Design and Synthesis of Selective PRMT4 Inhibitors: A New Chemotype for Development of Cancer Therapeutics. ChemMedChem. 2021, 16, 1116–1125. 10.1002/cmdc.202100018. [DOI] [PubMed] [Google Scholar]
- Ragno R.; Simeoni S.; Castellano S.; Vicidomini C.; Mai A.; Caroli A.; Tramontano A.; Bonaccini C.; Trojer P.; Bauer I.; Brosch G.; Sbardella G. Small Molecule Inhibitors of Histone Arginine Methyltransferases: Homology Modeling, Molecular Docking, Binding Mode Analysis, and Biological Evaluations. J. Med. Chem. 2007, 50, 1241–1253. 10.1021/jm061213n. [DOI] [PubMed] [Google Scholar]
- Castellano S.; Milite C.; Ragno R.; Simeoni S.; Mai A.; Limongelli V.; Novellino E.; Bauer I.; Brosch G.; Spannhoff A.; Cheng D.; Bedford M. T.; Sbardella G. Design, Synthesis and Biological Evaluation of Carboxy Analogues of Arginine Methyltransferase Inhibitor 1 (AMI-1). ChemMedChem. 2010, 5, 398–414. 10.1002/cmdc.200900459. [DOI] [PubMed] [Google Scholar]
- Ragno R.; Mai A.; Simeoni S.; Caroli A.; Valente S.; Perrone A.; Castellano S.; Sbardella G. In Small Molecule Inhibitors of Histone Arginine Methyltransferases: Updated Structure-Based 3-D QSAR Models with Improved Robustness and Predictive Ability; Frontiers in CNS and Oncology Medicinal Chemistry, ACS-EFMC, Siena, Italy, October 7–9, 2007; COMC-010. [Google Scholar]
- Cheng D.; Yadav N.; King R. W.; Swanson M. S.; Weinstein E. J.; Bedford M. T. Small Molecule Regulators of Protein Arginine Methyltransferases. J. Biol. Chem. 2004, 279, 23892–23899. 10.1074/jbc.M401853200. [DOI] [PubMed] [Google Scholar]
- Daigle S. R.; Olhava E. J.; Therkelsen C. A.; Majer C. R.; Sneeringer C. J.; Song J.; Johnston L. D.; Scott M. P.; Smith J. J.; Xiao Y.; Jin L.; Kuntz K. W.; Chesworth R.; Moyer M. P.; Bernt K. M.; Tseng J.-C.; Kung A. L.; Armstrong S. A.; Copeland R. A.; Richon V. M.; Pollock R. M. Selective Killing of Mixed Lineage Leukemia Cells by a Potent Small-Molecule DOT1L Inhibitor. Cancer Cell 2011, 20, 53–65. 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smil D.; Eram M. S.; Li F.; Kennedy S.; Szewczyk M. M.; Brown P. J.; Barsyte-Lovejoy D.; Arrowsmith C. H.; Vedadi M.; Schapira M. Discovery of a Dual PRMT5–PRMT7 Inhibitor. ACS Med. Chem. Lett. 2015, 6, 408–412. 10.1021/ml500467h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X.-C.; Zhang T.; Kim E.-J.; Jiang M.; Wang K.; Wang J.; Chen S.; Zhang N.; Wu H.; Li F.; Dela Seña C. C.; Zeng H.; Vivcharuk V.; Niu X.; Zheng W.; Lee J. P.; Chen Y.; Barsyte D.; Szewczyk M.; Hajian T.; Ibáñez G.; Dong A.; Dombrovski L.; Zhang Z.; Deng H.; Min J.; Arrowsmith C. H.; Mazutis L.; Shi L.; Vedadi M.; Brown P. J.; Xiang J.; Qin L.-X.; Xu W.; Luo M. A Chemical Probe of CARM1 Alters Epigenetic Plasticity Against Breast Cancer Cell Invasion. eLife 2019, 8, e47110. 10.7554/eLife.47110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai A.; Valente S.; Cheng D.; Perrone A.; Ragno R.; Simeoni S.; Sbardella G.; Brosch G.; Nebbioso A.; Conte M.; Altucci L.; Bedford M. T. Synthesis and Biological Validation of Novel Synthetic Histone/Pprotein Methyltransferase Inhibitors. ChemMedChem. 2007, 2, 987–991. 10.1002/cmdc.200700023. [DOI] [PubMed] [Google Scholar]
- Mai A.; Cheng D.; Bedford M. T.; Valente S.; Nebbioso A.; Perrone A.; Brosch G.; Sbardella G.; De Bellis F.; Miceli M.; Altucci L. Epigenetic Multiple Ligands: Mixed Histone/Protein Methyltransferase, Acetyltransferase, and Class III Deacetylase (Sirtuin) Inhibitors. J. Med. Chem. 2008, 51, 2279–2290. 10.1021/jm701595q. [DOI] [PubMed] [Google Scholar]
- Cheng D.; Valente S.; Castellano S.; Sbardella G.; Di Santo R.; Costi R.; Bedford M. T.; Mai A. Novel 3,5-Bis(Bromohydroxybenzylidene)Piperidin-4-ones as Coactivator-Associated Arginine Methyltransferase 1 Inhibitors: Enzyme Selectivity and Cellular Activity. J. Med. Chem. 2011, 54, 4928–4932. 10.1021/jm200453n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano S.; Spannhoff A.; Milite C.; Dal Piaz F.; Cheng D.; Tosco A.; Viviano M.; Yamani A.; Cianciulli A.; Sala M.; Cura V.; Cavarelli J.; Novellino E.; Mai A.; Bedford M. T.; Sbardella G. Identification of Small-Molecule Enhancers of Arginine Methylation Catalyzed by Coactivator-Associated Arginine Methyltransferase 1. J. Med. Chem. 2012, 55, 9875–9890. 10.1021/jm301097p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H.; Wu J.; Bedford M. T.; Sbardella G.; Hoffmann F. M.; Bi K.; Xu W. A TR-FRET-Based Functional Assay for Screening Activators of CARM1. Chembiochem 2013, 14, 827–835. 10.1002/cbic.201300029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franek M.; Legartova S.; Suchankova J.; Milite C.; Castellano S.; Sbardella G.; Kozubek S.; Bartova E. CARM1Modulators Affect Epigenome of Stem Cells and Change Morphology of Nucleoli. Physiol. Res. 2015, 64, 769–782. 10.33549/physiolres.932952. [DOI] [PubMed] [Google Scholar]
- Legartova S.; Sbardella G.; Kozubek S.; Bartova E. Ellagic Acid-Changed Epigenome of Ribosomal Genes and Condensed RPA194-Positive Regions of Nucleoli in Tumour Cells. Folia Biol. 2015, 61, 49–59. [DOI] [PubMed] [Google Scholar]
- Schapira M.; Ferreira de Freitas R. Structural Biology and Chemistry of Protein Arginine Methyltransferases. MedChemComm 2014, 5, 1779–1788. 10.1039/C4MD00269E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Zhou X.; Wang A.; Zheng Y.; Gao Y.; Zhou J. Evolutions in Fragment-Based Drug Design: the Deconstruction–Reconstruction Approach. Drug Discovery Today 2015, 20, 105–113. 10.1016/j.drudis.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallesen J. S.; Narayanan D.; Tran K. T.; Solbak S. M. Ø.; Marseglia G.; Sørensen L. M. E.; Høj L. J.; Munafò F.; Carmona R. M. C.; Garcia A. D.; Desu H. L.; Brambilla R.; Johansen T. N.; Popowicz G. M.; Sattler M.; Gajhede M.; Bach A. Deconstructing Noncovalent Kelch-like ECH-Associated Protein 1 (Keap1) Inhibitors into Fragments to Reconstruct New Potent Compounds. J. Med. Chem. 2021, 64, 4623–4661. 10.1021/acs.jmedchem.0c02094. [DOI] [PubMed] [Google Scholar]
- Jahnke W.; Erlanson D. A.; de Esch I. J. P.; Johnson C. N.; Mortenson P. N.; Ochi Y.; Urushima T. Fragment-to-Lead Medicinal Chemistry Publications in 2019. J. Med. Chem. 2020, 63, 15494–15507. 10.1021/acs.jmedchem.0c01608. [DOI] [PubMed] [Google Scholar]
- Congreve M.; Chessari G.; Tisi D.; Woodhead A. J. Recent Developments in Fragment-Based Drug Discovery. J. Med. Chem. 2008, 51, 3661–3680. 10.1021/jm8000373. [DOI] [PubMed] [Google Scholar]
- Ciulli A.; Williams G.; Smith A. G.; Blundell T. L.; Abell C. Probing Hot Spots at Protein–Ligand Binding Sites: A Fragment-Based Approach Using Biophysical Methods. J. Med. Chem. 2006, 49, 4992–5000. 10.1021/jm060490r. [DOI] [PubMed] [Google Scholar]
- Vieth M.; Siegel M. G.; Higgs R. E.; Watson I. A.; Robertson D. H.; Savin K. A.; Durst G. L.; Hipskind P. A. Characteristic Physical Properties and Structural Fragments of Marketed Oral Drugs. J. Med. Chem. 2004, 47, 224–232. 10.1021/jm030267j. [DOI] [PubMed] [Google Scholar]
- Chevillard F.; Rimmer H.; Betti C.; Pardon E.; Ballet S.; van Hilten N.; Steyaert J.; Diederich W. E.; Kolb P. Binding-Site Compatible Fragment Growing Applied to the Design of β2-Adrenergic Receptor Ligands. J. Med. Chem. 2018, 61, 1118–1129. 10.1021/acs.jmedchem.7b01558. [DOI] [PubMed] [Google Scholar]
- Aldrich C.; Bertozzi C.; Georg G. I.; Kiessling L.; Lindsley C.; Liotta D.; Merz K. M.; Schepartz A.; Wang S. The Ecstasy and Agony of Assay Interference Compounds. J. Med. Chem. 2017, 60, 2165–2168. 10.1021/acs.jmedchem.7b00229. [DOI] [PubMed] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. SwissADME: a Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagorce D.; Sperandio O.; Baell J. B.; Miteva M. A.; Villoutreix B. O. FAF-Drugs3: a Web Server for Compound Property Calculation and Chemical Library Design. Nucleic Acids Res. 2015, 43, W200–W207. 10.1093/nar/gkv353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Irwin J. J.; Duan D.; Torosyan H.; Doak A. K.; Ziebart K. T.; Sterling T.; Tumanian G.; Shoichet B. K. An Aggregation Advisor for Ligand Discovery. J. Med. Chem. 2015, 58, 7076–7087. 10.1021/acs.jmedchem.5b01105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocasoy V.; Dedeoglu B.; Demir-Ordu O.; Aviyente V. Influence of Odd–Even Effect and Intermolecular Interactions in 2D Molecular Layers of Bisamide Organogelators. RSC Adv. 2018, 8, 35195–35204. 10.1039/C8RA06224B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumiyoshi T.; Nishimura K.; Nakano M.; Handa T.; Miwa Y.; Tomioka K. Molecular Assembly of C2-Symmetric Bis-(2S)-2-methyldodecanoylamides of α,ω-Alkylidenediamines into Coiled Coil and Twisted Ribbon Aggregates. J. Am. Chem. Soc. 2003, 125, 12137–12142. 10.1021/ja035085t. [DOI] [PubMed] [Google Scholar]
- Tao F.; Bernasek S. L. Understanding Odd–Even Effects in Organic Self-Assembled Monolayers. Chem. Rev. 2007, 107, 1408–1453. 10.1021/cr050258d. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Keserü G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The Role of Ligand Efficiency Metrics in Drug Discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Feoli A.; Viviano M.; Cipriano A.; Milite C.; Castellano S.; Sbardella G. Lysine Methyltransferase Inhibitors: Where We Are Now. RSC Chem. Biol. 2022, 3, 359. 10.1039/D1CB00196E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchardt R. T.; Huber J. A.; Wu Y. S. Potential Inhibitors of S-Adenosylmethionine-Dependent Methyltransferases. 2. Modification of the Base Portion of S-Adenosylhomocysteine. J. Med. Chem. 1974, 17, 868–873. 10.1021/jm00254a017. [DOI] [PubMed] [Google Scholar]
- Coward J. K.; Slisz E. P. Analogs of S-Adenosylhomocysteine as Potential Inhibitors of Biological Transmethylation. Specificity of the S-Adenosylhomocysteine Binding Site. J. Med. Chem. 1973, 16, 460–463. 10.1021/jm00263a008. [DOI] [PubMed] [Google Scholar]
- Richon V. M.; Johnston D.; Sneeringer C. J.; Jin L.; Majer C. R.; Elliston K.; Jerva L. F.; Scott M. P.; Copeland R. A. Chemogenetic Analysis of Human Protein Methyltransferases. Chem. Biol. Drug Des. 2011, 78, 199–210. 10.1111/j.1747-0285.2011.01135.x. [DOI] [PubMed] [Google Scholar]
- Coussens N. P.; Kales S. C.; Henderson M. J.; Lee O. W.; Horiuchi K. Y.; Wang Y.; Chen Q.; Kuznetsova E.; Wu J.; Chakka S.; Cheff D. M.; Cheng K. C.-C.; Shinn P.; Brimacombe K. R.; Shen M.; Simeonov A.; Lal-Nag M.; Ma H.; Jadhav A.; Hall M. D. High-Throughput Screening with Nucleosome Substrate Identifies Small-Molecule Inhibitors of the Human Histone Lysine Methyltransferase NSD2. J. Biol. Chem. 2018, 293, 13750–13765. 10.1074/jbc.RA118.004274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum G.; Ibáñez G.; Rao X.; Shum D.; Radu C.; Djaballah H.; Rice J. C.; Luo M. Small-Molecule Inhibitors of SETD8 with Cellular Activity. ACS Chem. Biol. 2014, 9, 2471–2478. 10.1021/cb500515r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D.; Ma H.; Hong H.; Koh S. S.; Huang S.-M.; Schurter B. T.; Aswad D. W.; Stallcup M. R. Regulation of Transcription by a Protein Methyltransferase. Science 1999, 284, 2174–2177. 10.1126/science.284.5423.2174. [DOI] [PubMed] [Google Scholar]
- Schurter B. T.; Koh S. S.; Chen D.; Bunick G. J.; Harp J. M.; Hanson B. L.; Henschen-Edman A.; Mackay D. R.; Stallcup M. R.; Aswad D. W. Methylation of Histone H3 by Coactivator-Associated Arginine Methyltransferase 1. Biochemistry 2001, 40, 5747–5756. 10.1021/bi002631b. [DOI] [PubMed] [Google Scholar]
- Selvi B. R.; Batta K.; Kishore A. H.; Mantelingu K.; Varier R. A.; Balasubramanyam K.; Pradhan S. K.; Dasgupta D.; Sriram S.; Agrawal S.; Kundu T. K. Identification of a Novel Inhibitor of Coactivator-associated Arginine Methyltransferase 1 (CARM1)-mediated Methylation of Histone H3 Arg-17. J. Biol. Chem. 2010, 285, 7143–7152. 10.1074/jbc.M109.063933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevillard-Briet M.; Trouche D.; Vandel L. Control of CBP Co-Activating Activity by Arginine Methylation. EMBO J. 2002, 21, 5457–5466. 10.1093/emboj/cdf548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q.; Yi P.; Wong J.; O’Malley B. W. Signaling within a Coactivator Complex: Methylation of SRC-3/AIB1 Is a Molecular Switch for Complex Disassembly. Mol. Cell. Biol. 2006, 26, 7846–7857. 10.1128/MCB.00568-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T.; Mori Y.; Chu D. L.; Koyama Y.; Miyata S.; Tanaka H.; Yachi K.; Kubo T.; Yoshikawa H.; Tohyama M. CARM1 Regulates Proliferation of PC12 Cells by Methylating HuD. Mol. Cell. Biol. 2006, 26, 2273–2285. 10.1128/MCB.26.6.2273-2285.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Bedford M. T. PABP1 Identified as an Arginine Methyltransferase Substrate Using High-Density Protein Arrays. EMBO Rep. 2002, 3, 268–273. 10.1093/embo-reports/kvf052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Park S.; Kilburn B.; Jelinek M. A.; Henschen-Edman A.; Aswad D. W.; Stallcup M. R.; Laird-Offringa I. A. Lipopolysaccharide-induced Methylation of HuR, an mRNA-stabilizing Protein, by CARM1. J. Biol. Chem. 2002, 277, 44623–44630. 10.1074/jbc.M206187200. [DOI] [PubMed] [Google Scholar]
- Cheng D.; Côté J.; Shaaban S.; Bedford M. T. The Arginine Methyltransferase CARM1 Regulates the Coupling of Transcription and mRNA Processing. Mol. Cell 2007, 25, 71–83. 10.1016/j.molcel.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Cheng D.; Vemulapalli V.; Lu Y.; Shen J.; Aoyagi S.; Fry C. J.; Yang Y.; Foulds C. E.; Stossi F.; Treviño L. S.; Mancini M. A.; O’Malley B. W.; Walker C. L.; Boyer T. G.; Bedford M. T. CARM1Methylates MED12 to Regulate Its RNA-Binding Ability. Life Sci. Alliance 2018, 1, e201800117. 10.26508/lsa.201800117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt S. M.; Man N.; Hamard P. J.; Asai T.; Karl D.; Martinez C.; Bilbao D.; Stathias V.; McGrew-Jermacowicz A.; Duffort S.; Tadi M.; Blumenthal E.; Newman S.; Vu L.; Xu Y.; Liu F.; Schurer S. C.; McCabe M. T.; Kruger R. G.; Xu M. J.; Yang F. C.; Tenen D.; Watts J.; Vega F.; Nimer S. D. CARM1 Is Essential for Myeloid Leukemogenesis but Dispensable for Normal Hematopoiesis. Cancer Cell 2018, 33, 1111–1127. 10.1016/j.ccell.2018.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu L. P.; Perna F.; Wang L.; Voza F.; Figueroa M. E.; Tempst P.; Erdjument-Bromage H.; Gao R.; Chen S.; Paietta E.; Deblasio T.; Melnick A.; Liu Y.; Zhao X.; Nimer S. D. PRMT4 Blocks Myeloid Differentiation by Assembling a Methyl-RUNX1-Dependent Repressor Complex. Cell Rep. 2013, 5, 1625–1638. 10.1016/j.celrep.2013.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Dhaheri M.; Wu J.; Skliris G. P.; Li J.; Higashimato K.; Wang Y.; White K. P.; Lambert P.; Zhu Y.; Murphy L.; Xu W. CARM1 Is an Important Determinant of ER-Alpha-Dependent Breast Cancer Cell Differentiation and Proliferation in Breast Cancer Cells. Cancer Res. 2011, 71, 2118–2128. 10.1158/0008-5472.CAN-10-2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H.; Kao C.; Jeng M.-H.; Eble J. N.; Koch M. O.; Gardner T. A.; Zhang S.; Li L.; Pan C.-X.; Hu Z.; MacLennan G. T.; Cheng L. Aberrant Expression of CARM1, a Transcriptional Coactivator of Androgen Receptor, in the Development of Prostate Carcinoma and Androgen-Independent Status. Cancer 2004, 101, 83–89. 10.1002/cncr.20327. [DOI] [PubMed] [Google Scholar]
- Osada S.; Suzuki S.; Yoshimi C.; Matsumoto M.; Shirai T.; Takahashi S.; Imagawa M. Elevated Expression of Coactivator-Associated Arginine Methyltransferase 1 Is Associated with Early Hepatocarcinogenesis. Oncol. Rep. 2013, 30, 1669. 10.3892/or.2013.2651. [DOI] [PubMed] [Google Scholar]
- Kim D.; Lee J.; Cheng D.; Li J.; Carter C.; Richie E.; Bedford M. T. Enzymatic Activity Is Required for the in Vivo Functions of CARM1. J. Biol. Chem. 2010, 285, 1147–1152. 10.1074/jbc.M109.035865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou C.-Y.; LaBonte M. J.; Manegold P. C.; So A. Y.-L.; Ianculescu I.; Gerke D. S.; Yamamoto K. R.; Ladner R. D.; Kahn M.; Kim J. H.; Stallcup M. R. A Coactivator Role of CARM1 in the Dysregulation of β-Catenin Activity in Colorectal Cancer Cell Growth and Gene Expression. Mol. Cancer Res. 2011, 9, 660–670. 10.1158/1541-7786.MCR-10-0223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Ju C.; Hu J.; Huang K.; Yang L. PRMT4 Overexpression Aggravates Cardiac Remodeling Following Myocardial Infarction by Promoting Cardiomyocyte Apoptosis. Biochem. Biophys. Res. Commun. 2019, 520, 645–650. 10.1016/j.bbrc.2019.10.085. [DOI] [PubMed] [Google Scholar]
- Suresh S.; Huard S.; Dubois T. CARM1/PRMT4: Making Its Mark beyond Its Function as a Transcriptional Coactivator. Trends Cell Biol. 2021, 31, 402–417. 10.1016/j.tcb.2020.12.010. [DOI] [PubMed] [Google Scholar]
- Veazey K. J.; Cheng D.; Lin K.; Villarreal O. D.; Gao G.; Perez-Oquendo M.; Van H. T.; Stratton S. A.; Green M.; Xu H.; Lu Y.; Bedford M. T.; Santos M. A. CARM1 Inhibition Reduces Histone Acetyltransferase Activity Causing Synthetic Lethality in CREBBP/EP300-Mutated Lymphomas. Leukemia 2020, 34, 3269–3285. 10.1038/s41375-020-0908-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew A. E.; Moradei O.; Jacques S. L.; Rioux N.; Boriack-Sjodin A. P.; Allain C.; Scott M. P.; Jin L.; Raimondi A.; Handler J. L.; Ott H. M.; Kruger R. G.; McCabe M. T.; Sneeringer C.; Riera T.; Shapiro G.; Waters N. J.; Mitchell L. H.; Duncan K. W.; Moyer M. P.; Copeland R. A.; Smith J.; Chesworth R.; Ribich S. A. Identification of a CARM1 Inhibitor with Potent In Vitro and In Vivo Activity in Preclinical Models of Multiple Myeloma. Sci. Rep. 2017, 7, 17993. 10.1038/s41598-017-18446-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A.; Pompliano D. L.; Meek T. D. Drug–Target Residence Time and Its Implications for Lead Optimization. Nat. Rev. Drug Discovery 2006, 5, 730–739. 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- Karplus P. A.; Diederichs K. Assessing and Maximizing Data Quality in Macromolecular Crystallography. Curr. Opin. Struct. Biol. 2015, 34, 60–68. 10.1016/j.sbi.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troffer-Charlier N.; Cura V.; Hassenboehler P.; Moras D.; Cavarelli J. Functional Insights from Structures of Coactivator-Associated Arginine Methyltransferase 1 Domains. EMBO J. 2007, 26, 4391–4401. 10.1038/sj.emboj.7601855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnefond L.; Stojko J.; Mailliot J.; Troffer-Charlier N.; Cura V.; Wurtz J.-M.; Cianférani S.; Cavarelli J. Functional Insights from High Resolution Structures of Mouse Protein Arginine Methyltransferase 6. J. Struct. Biol. 2015, 191, 175–183. 10.1016/j.jsb.2015.06.017. [DOI] [PubMed] [Google Scholar]
- Cheng D.; Gao G.; Di Lorenzo A.; Jayne S.; Hottiger M. O.; Richard S.; Bedford M. T. Genetic Evidence for Ppartial Redundancy Between the Arginine Methyltransferases CARM1 and PRMT6. J. Biol. Chem. 2020, 295, 17060–17070. 10.1074/jbc.RA120.014704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupien M.; Eeckhoute J.; Meyer C. A.; Krum S. A.; Rhodes D. R.; Liu X. S.; Brown M. Coactivator Function Defines the Active Estrogen Receptor Alpha Cistrome. Mol. Cell. Biol. 2009, 29, 3413–3423. 10.1128/MCB.00020-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng B.-l.; Li W.-j.; Ding J.-c.; He Y.-h.; Ran T.; Xie B.-l.; Wang Z.-r.; Shen H.-f.; Xiao R.-q.; Gao W.-w.; Ye T.-y.; Gao X.; Liu W. A Hypermethylation Strategy Utilized by Enhancer-Bound CARM1 to Promote Estrogen Receptor α-Dependent Transcriptional Activation and Bbreast Carcinogenesis. Theranostics 2020, 10, 3451–3473. 10.7150/thno.39241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain K.; Clarke S. G. PRMT7 as a Unique Member of the Protein Arginine Methyltransferase Family: A Review. Arch. Biochem. Biophys. 2019, 665, 36–45. 10.1016/j.abb.2019.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W. S.; Lee E. J.; Ko I. S.. Preparation of Benzoindazolone Derivatives as NQO1 Activators. WO2020175851, 2020.
- Vrudhula V. M.; Kappler F.; Hampton A. Isozyme-Specific Enzyme Inhibitors. 13. S-[5′(R)-[′N-Triphosphoamino)Methyl]Adenosyl]-L-Homocysteine, a Potent Inhibitor of Rat Methionine Adenosyltransferases. J. Med. Chem. 1987, 30, 888–894. 10.1021/jm00388a024. [DOI] [PubMed] [Google Scholar]
- Wintner E. A.; Conn M. M.; Rebek J. Self-Replicating Molecules: A Second Generation. J. Am. Chem. Soc. 1994, 116, 8877–8884. 10.1021/ja00099a003. [DOI] [Google Scholar]
- Wang J.-F.; Yang X.-D.; Zhang L.-R.; Yang Z.-J.; Zhang L.-H. Synthesis and Biological Activities of 5′-Ethylenic and Acetylenic Modified l-Nucleosides and Isonucleosides. Tetrahedron 2004, 60, 8535–8546. 10.1016/j.tet.2004.06.131. [DOI] [Google Scholar]
- van Haren M.; van Ufford L. Q.; Moret E. E.; Martin N. I. Synthesis and Evaluation of Protein Arginine N-Methyltransferase Inhibitors Designed to Simultaneously Occupy Both Substrate Binding Sites. Org. Biomol. Chem. 2015, 13, 549–560. 10.1039/C4OB01734J. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Richardson S. L.; Mao Y.; Huang R. Design, Synthesis, and Kinetic Analysis of Potent Protein N-Terminal Methyltransferase 1 Inhibitors. Org. Biomol. Chem. 2015, 13, 4149–4154. 10.1039/C5OB00120J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin B.; Liu Z.; Yi M.; Zhang J. An Efficient Method for the Synthesis of Disubstituted Thioureas Via the Reaction of N,N′-di-Boc-Substituted Thiourea with Alkyl and Aryl Aamines Under Mild Conditions. Tetrahedron Lett. 2008, 49, 3687–3690. 10.1016/j.tetlet.2008.03.158. [DOI] [Google Scholar]
- Anastassiadis T.; Deacon S. W.; Devarajan K.; Ma H.; Peterson J. R. Comprehensive Assay of Kinase Catalytic Activity Reveals Features of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2011, 29, 1039–1045. 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi K. Y.; Eason M. M.; Ferry J. J.; Planck J. L.; Walsh C. P.; Smith R. F.; Howitz K. T.; Ma H. Assay Development for Histone Methyltransferases. ASSAY Drug Dev. Technol. 2013, 11, 227–236. 10.1089/adt.2012.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonysamy S.; Bonday Z.; Campbell R. M.; Doyle B.; Druzina Z.; Gheyi T.; Han B.; Jungheim L. N.; Qian Y.; Rauch C.; Russell M.; Sauder J. M.; Wasserman S. R.; Weichert K.; Willard F. S.; Zhang A.; Emtage S. Crystal Structure of the Human PRMT5:MEP50 Complex. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 17960–17965. 10.1073/pnas.1209814109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddershaw A. R.; Stubbs C. J.; Edwardes L. V.; Underwood E.; Hamm G. R.; Davey P. R. J.; Clarkson P. N.; Syson K. Characterization of the Kinetic Mechanism of Human Protein Arginine Methyltransferase 5. Biochemistry 2020, 59, 4775–4786. 10.1021/acs.biochem.0c00554. [DOI] [PubMed] [Google Scholar]
- Sayegh J.; Webb K.; Cheng D.; Bedford M. T.; Clarke S. G. Regulation of Protein Arginine Methyltransferase 8 (PRMT8) Activity by Its N-Terminal Domain. J. Biol. Chem. 2007, 282, 36444–36453. 10.1074/jbc.M704650200. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr. D 2010, 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997, 276, 307–326. 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and Development of Coot. Acta Crystallogr. D 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. D 2010, 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Grosse-Kunstleve R. W.; Hung L. W.; Ioerger T. R.; McCoy A. J.; Moriarty N. W.; Read R. J.; Sacchettini J. C.; Sauter N. K.; Terwilliger T. C. PHENIX: Building New Software for Automated Crystallographic Structure Determination. Acta Crystallogr. D 2002, 58, 1948–1954. 10.1107/S0907444902016657. [DOI] [PubMed] [Google Scholar]
- Liebschner D.; Afonine P. V.; Baker M. L.; Bunkoczi G.; Chen V. B.; Croll T. I.; Hintze B.; Hung L.-W.; Jain S.; McCoy A. J.; Moriarty N. W.; Oeffner R. D.; Poon B. K.; Prisant M. G.; Read R. J.; Richardson J. S.; Richardson D. C.; Sammito M. D.; Sobolev O. V.; Stockwell D. H.; Terwilliger T. C.; Urzhumtsev A. G.; Videau L. L.; Williams C. J.; Adams P. D. Macromolecular Structure Determination Using X-rays, Neutrons and Electrons: Recent Developments in Phenix. Acta Crystallogr. D 2019, 75, 861–877. 10.1107/S2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricogne G.; Blanc E.; Brandl M.; Flensburg C.; Keller P.; Paciorek W.; Roversi P.; Sharff A.; Smart O.; Vonrhein C.. BUSTER, version 2.10. 0; Global Phasing Ltd.: Cambridge, U.K., 2011.
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr. D 2011, 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romier C.; Ben Jelloul M.; Albeck S.; Buchwald G.; Busso D.; Celie P. H. N.; Christodoulou E.; De Marco V.; van Gerwen S.; Knipscheer P.; Lebbink J. H.; Notenboom V.; Poterszman A.; Rochel N.; Cohen S. X.; Unger T.; Sussman J. L.; Moras D.; Sixma T. K.; Perrakis A. Co-Expression of Protein Complexes in Prokaryotic and Eukaryotic Hosts: Experimental Procedures, Database Tracking and Case Studies. Acta Crystallogr. D 2006, 62, 1232–1242. 10.1107/S0907444906031003. [DOI] [PubMed] [Google Scholar]
- Wohnsland F.; Faller B. High-Throughput Permeability pH Profile and High-Throughput Alkane/Water log P with Artificial Membranes. J. Med. Chem. 2001, 44, 923–930. 10.1021/jm001020e. [DOI] [PubMed] [Google Scholar]
- Sugano K.; Hamada H.; Machida M.; Ushio H. High Throughput Prediction of Oral Absorption: Improvement of the Composition of the Lipid Solution Used in Parallel Artificial Membrane Permeation Assay. J. Biomol. Screen. 2001, 6, 189–196. 10.1177/108705710100600309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.