

Abstract
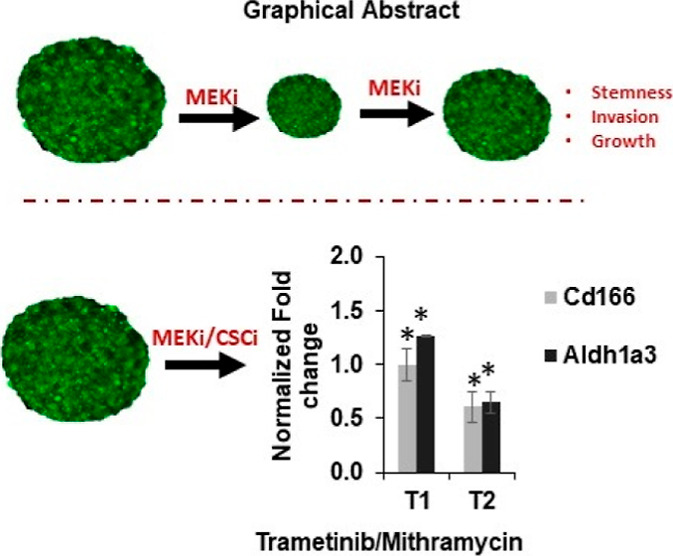
Drug resistance is a leading cause for the failure of cancer treatments. Plasticity of cancer cells to acquire stem cell-like properties enables them to escape drug toxicity through different adaptive mechanisms. Eliminating cancer stem cells (CSCs) can potentially improve treatment outcomes for patients. To determine the role of CSCs in resistance of colorectal cancer cells to targeted therapies and identify treatment strategies, we treated spheroids of BRAFmut and KRASmut colorectal cancer cells with inhibitors of the mitogen-activated protein kinase pathway and studied resistance mechanisms through gene and protein expression analyses. We found that treatments activated several oncogenic pathways and expression of CSC markers CD166 and ALDH1A3. We identified a specific combination treatment using trametinib and mithramycin A to simultaneously inhibit the CSC phenotype and activities of several pathways in cancer cells. This study demonstrates the feasibility of therapeutic targeting of CSCs as a strategy to block tumorigenic activities of cancer cells.
Keywords: drug resistance, tumor spheroids, colorectal cancer, combination treatments, cancer stem cells, invasion
Drug resistance is a major barrier against success of cancer treatments in both primary tumors and metastases, the latter of which accounts for about 90% of mortality among cancer patients.1,2 Colorectal cancer is the third most diagnosed cancer and the second leading cause of cancer-related deaths in the world.3 Over 50% of colorectal cancers have mutations in mitogen-activated protein kinase (MAPK) or phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR pathways,4−6 making them attractive targets for therapies. Although inhibitors of the MAPK pathway such as those targeting RAF, MEK, or ERK were shown to suppress growth of colorectal tumors in vivo,7,8 cancer cells often adapt to these drugs through several mechanisms. A primary mechanism of resistance is activation of compensatory oncogenic signaling pathways. Compensatory signaling is an adaptive response under drug pressure and allows cancer cells to use redundant signaling pathways to avoid drug toxicity and retain their tumorigenic activities. For example, colorectal cancer cells develop resistance to single-agent inhibition of the MAPK pathway by activating PI3K/Akt, JAK/STAT, or Wnt pathways.4,5,9 Motivated by this feedback signaling, several clinical trials used combinations of inhibitors of MAPK and PI3K/Akt pathways. Although exhibiting promising antitumor effects, these trials were largely unsuccessful due to excessive toxicity to patients.10,11
Gain of a stem cell-like trait is considered a major mechanism that allows cancer cells to escape therapies that either directly target the cell cycle or interfere with signaling pathways important for cell division.12 Cancer stem cells (CSCs) present a major therapeutic challenge due to their plasticity. In the absence of treatment pressure, CSCs shift toward a proliferative state and revert to a quiescent stem cell state in the presence of drugs, allowing survival during chemotherapy.13 Despite significant progress in cancer research and treatments, eliminating CSCs remains an unmet clinical need. Several key signaling pathways, such as Wnt, PI3K/Akt, TGF, and JAK/STAT, as well as transcriptional regulators including Oct4, Nanog, and Myc, are commonly activated in various CSCs to regulate their self-renewal and differentiation states.14,15 This broad signaling activity makes therapeutic targeting of CSCs very difficult. Several studies have performed large-scale screening of chemical libraries to identify potential drugs that reduce or eliminate CSC populations from tumors.16,17 Because of their pivotal functions in cancer, efforts are underway to develop or re-purpose drugs against CSCs to improve cancer therapies.
To investigate the role of CSCs in resistance of KRASmut and BRAFmut colorectal cancer cells to targeted therapies, we used a 3D spheroid model in a cyclic treatment regimen that mimics how patients commonly receive chemotherapy.18 Cells treated with inhibitors of the MAPK pathway activated multiple compensatory pathways and gained CSC phenotypes. Using a rationale combination treatment approach, we evaluated the effectiveness of different drug combinations against MAPK and the activated compensatory pathways. However, this approach was ineffective against CSCs and invasiveness of cancer cells. We found that simultaneously targeting the MAPK pathway using trametinib and CSCs using mithramycin A prevented CSC phenotypes and feedback signaling of cancer cells and their matrix invasion. Overall, our approach to mechanistically study the role of CSCs in resistance of colorectal cancer cells to cyclic targeted therapies enabled the design and testing of various drug combinations to identify a treatment that blocked CSCs, reduced signaling of oncogenic pathways, and prevented matrix invasion of cancer cells.
Results
Formation of Tumor Spheroids and Modeling Drug Resistance
The aqueous dextran (DEX) phase nanodrop containing cancer cells stably remained immiscible from the polyethylene glycol (PEG) phase solution, allowing cells to aggregate into a spheroid while ensuring free diffusion of nutrients from the immersion phase (Figure 1a).19−21 After compact spheroids formed, we maintained them in the complete growth medium. This micropatterning approach generates spheroids that are ∼30% more compact than those from the standard ultralow attachment plate method.22 With a density of 1.5 × 104 HCT116 cells, a variation of <5.5% from an average diameter of 468 μm resulted.
Figure 1.
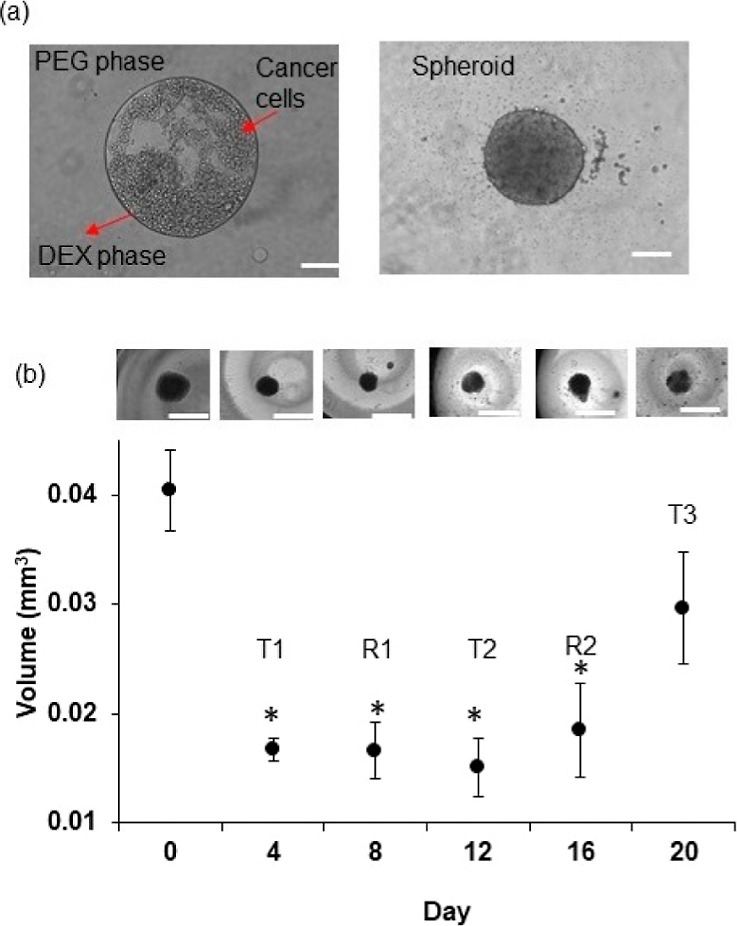
Formation and drug resistance of tumor spheroids. (a) 0.3 μL drop of the aqueous DEX phase containing HCT116 cancer cells is immersed in the aqueous PEG phase. The drop settles to the well bottom and confines the cancer cells to form a spheroid. (b) Size of HCT116 spheroids during cyclic treatments (T1, T2, and T3) with 10 nM trametinib and recovery phases in between (R1 and R2). Inset images from left to right represent spheroids on days 0, 4, 8, 12, 16, and 20. Error bars represent standard errors from the mean (n = 8). The scale bar is 200 μm. * denotes p < 0.01.
KRAS and PIK3CA gene mutations in HCT116 cells activate oncogenic MAPK and PI3K/Akt/mTOR pathways.23,24 We previously showed that targeting the MAPK pathway blocks HCT116 cell proliferation in spheroid cultures.25 To mimic cyclic chemotherapy, we treated HCT116 spheroids with the LD50 concentration of trametinib (MEK inhibitor), using a treatment/recovery regimen.18 This approach helped model how cancer cells spared by the treatment develop adaptive resistance. Although trametinib potently inhibited cell proliferation during the initial treatment phase (T1) and reduced the size of spheroids 2.5-fold, cells became less responsive to the subsequent treatments (Figure 1b). At the end of the cyclic treatments, spheroids treated with trametinib were 1.80-fold larger than those at the end of T1. Our drug resistance model reliably emulated several in vivo studies where treating tumor xenografts with MEK1/2 inhibitors did not reduce the tumor size.26
Mechanism of Drug Resistance
Motivated by the role of CSCs in tumor heterogeneity and chemoresistance,14,27,28 we quantified the expression of several CSC and pluripotency genes following each cycle of treatment and recovery. First, we evaluated the expression of these genes following cyclic treatments of HCT116 and HT29 spheroids with trametinib, selumetinib, and PD0325901 (Figure S1). Most of the genes were significantly upregulated in both cell lines especially after T1 and T2, including CD166 and ALDH1A3, which are prominent CSC markers of colorectal cancer.29,30 Therefore, we focused on these two markers in our studies. The expression of CD166 and ALDH1A3 significantly increased in spheroids of both cell lines, with trametinib and PD0325901 after both T1 and T2 and with selumetinib after T2 (Figures 2a,b and S1). The expression levels were generally higher during T1 and T2 phases and often reduced during recovery phases. To evaluate this effect at a functional level, we quantified colony-forming capacity of drug-treated cells and found that both HCT116 and HT29 cells had significantly greater colony formation following treatments with the MEK inhibitors (Figures 2c,d and S2). Moreover, the efficiency of colony formation was almost always higher after T2 than T1 with both cell lines when compared to nontreated control samples, with the exception of PD0325901-treated HCT116 cells. Overall, resistance of cancer cells to MEK inhibition in cyclic treatments was consistent with the gain of a CSC phenotype.
Figure 2.

MEK inhibition promotes stemness in colorectal cancer cells. The heatmaps show the normalized fold change values of prominent CSC gene markers after treating (a) HCT116 and (b) HT29 spheroids with 5 nM trametinib, 100 nM PD0325901, or 100 nM selumetinib. The fold change values are relative to the control spheroids at 48 h (n = 2 independent experiments, two replicates/experiment). (c,d) Colony-forming efficiency of HCT116 and HT29 cells obtained from spheroids following cyclic treatments T1 and T2. A threshold of 50 μm colony diameter was used. Error bars represent standard errors from the mean (n = 8). ** denotes p < 0.01 compared to the control.
Signaling Mechanisms of Adaptive Resistance of Colorectal Cancer Cells
Next, we investigated molecular mechanisms that enable colorectal cancer cells to adapt to MEK inhibition. We previously showed that MEK inhibition of BRAFmut HT29 spheroids activates the PI3K/Akt pathway.18 First, we validated this finding in KRASmut HCT116 cells and found that trametinib treatment significantly activates the PI3K/Akt pathway (Figures 3a and S3). All three gene markers, that is, PIK3CA, PIK3CB, and Akt, showed increased expression following T1 and T2. Increased activity of this pathway also occurred following treatments with the other two MEK inhibitors (Figure S3). Then, we asked whether MEK inhibition may also result in compensatory activation of JAK/STAT or Wnt pathways. Our focus on these two pathways was due to their roles in resistance to MEK inhibitors, invasion, and stemness of colorectal tumors.5,31 We used both single-agent trametinib treatment and its combination with dactolisib that we previously showed to generate antiproliferative effects in colorectal cancer cells.25 Our analysis showed that treatments activated both pathways. STAT1 and STAT6 from the JAK/STAT pathway and AXIN1, AXIN2, and β-catenin 1 from the Wnt pathway showed significantly elevated expression (Figure 3b,c). Interestingly, the expression of Wnt pathway markers was higher following the combination treatment than trametinib treatment alone. We also validated this finding with HT29 cells (Figure S4).
Figure 3.

Mechanisms of adaptive resistance to MEK inhibitors. mRNA expression of gene markers of the (a) PI3K/Akt pathway, (b) STAT pathway, and (c) WNT pathway in HCT116 spheroids following cyclic treatments T1 and T2 shown under the x-axis of each graph. Fold change values are relative to nontreated spheroids. Error bars represent standard errors from the mean (n = 2 independent experiments, two replicates/experiment). * denotes p < 0.05 when compared to nontreated spheroids.
Drug-Resistant Cells Display an Invasive Phenotype
Local invasion is a key process leading to metastasis.32 Several kinase pathways including those activated following MEK inhibition may promote tumor cell invasion.33,34 To study invasive behavior of cancer cells that develop resistance to MEK inhibition and effects of combined targeting of MAPK and PI3K/Akt pathways, we treated HCT116 spheroids with trametinib, PD0325901, or selumetinib for one round or two rounds, recovered them from the treatments, and embedded them in a collagen hydrogel. To account for size differences of drug-treated and control spheroids, we quantified the invasion area relative to the spheroid area. Pretreated spheroids showed significantly higher matrix invasion than the vehicle control spheroids (Figure 4a). Additionally, spheroids from T2 were significantly more invasive than those from T1. To validate this finding, we quantified gene expression of three prominent EMT markers (Figure 4b). Among them, E47 expression significantly increased from T1 and T2. Interestingly, the expression levels decreased during recovery phases, suggesting adaptive behavior of cancer cells.
Figure 4.

Cyclic treatments with MEK inhibitors promote invasive and CSC phenotypes. (a) HCT116 spheroids pretreated with trametinib, PD0325901, or selumetinib using T1 and T2 regimens and embedded in a human type I collagen to evaluate cell invasion for 5 days. Quantified invasion area/spheroid area for vehicle control and pretreated spheroids are shown. Error bars represent standard errors (n = 5). (b) mRNA expression of EMT markers following cyclic trametinib treatments (T1 and T2) and recovery (R1 and R2). (c) Representative images of collagen invasion of HCT116 cells from spheroids pretreated with combinations of MEK and PI3K/Akt inhibitors for two rounds, and the quantified results are shown. (d) mRNA expression of CSC markers in HCT116 spheroids following the combination treatments for two rounds (T1 and T2) is shown. *p < 0.05 and **p < 0.01. In images of panels (a,c), red lines denote the border of invading cells, and the scale bar is 200 μm. Error bars represent standard errors from the mean (n = 2 independent experiments, two replicates/experiment).
Next, we combined trametinib with two different PI3K/Akt inhibitors (dactolisib or VS-5584) to evaluate the effect of this strategy against cancer cell invasion. Despite pretreatments with these drug combinations, significant cell invasion from spheroids still occurred (Figure 4c). Spheroids treated for two rounds showed even greater cell invasion of up to fivefold to eightfold than the control, untreated spheroids. Our subsequent molecular analysis showed that HCT116 spheroids pretreated with these combinations had significant upregulated CD166 and ALDH1A3 genes (Figure 4d), which we also observed in HT29 cells (Figure S5). Overall, these results indicate that despite the benefit of simultaneously inhibiting MAPK and PI3K/Akt pathways to block cell proliferation, activation of pathways that promote cancer cell invasion and stemness renders the treatments ineffective.
Combination Treatments to Suppress CSC and Invasion
Due to the role of the Wnt pathway in cancer stemness and invasion and elevated activity of this pathway in drug-resistant cells in our studies, we asked whether targeting this pathway would inhibit stemness in colorectal cancer cells. We selected PRI-724, a small molecular inhibitor of the Wnt/β-catenin signaling pathway, and niclosamide, an FDA-approved salicylamide.35,36 Our analysis of HCT116 spheroids treated with a combination of trametinib and PRI-724 or niclosamide showed increased expression of CD166 and ALDH1A3 genes (Figure 5a). In particular, ALDH1A3 expression significantly increased by approximately sixfold. The combination treatments were also ineffective against collagen invasion of HCT116 cells from spheroids. Compared to the vehicle control condition, treated spheroids had an increased invasion of ∼7–10-fold (Figure 5b). In addition, targeting STAT6 using AS151799 after combination treatments of spheroids with MEK and PI3K/Akt inhibitors significantly increased expression levels of CD166 and ALDH1A3 genes (Figure 5c) despite blocking collagen invasion of cancer cells (Figure 5d).
Figure 5.

Targeting Wnt or STAT pathways enriches CSCs. (a) mRNA expression of CSC markers and (b) collagen invasion of HCT116 spheroids following combination treatments using 5 nM trametinib with 1 μM PRI-724 or niclosamide. Error bars represent standard errors from the mean (n = 5). (c) mRNA expression of CSC markers and (d) collagen invasion of HCT116 spheroids treated sequentially with a STAT inhibitor (AS151799) following a combination treatment with MEK and Akt inhibitors. The fold change values are relative to non-treated spheroids. * denotes p < 0.05, and **denotes p < 0.01 compared to nontreated spheroids. The scale bar is 200 μm. Error bars represent standard errors from the mean (n = 2 independent experiments, two replicates/experiment) for qPCR.
Mithramycin A, an FDA-approved drug, was recently shown to inhibit stemness of cancer cells.37 Thus, we asked whether mithramycin combined with trametinib would block the CSC state and invasiveness of colorectal cancer spheroids. First, we conducted a matrix format combination treatment and computed the fraction of cells affected and combination index values, which indicate synergy levels of drug combinations (Figure 6a,b). The treatments significantly reduced cell proliferation and showed high efficacy and synergy at low drug concentrations. Based on this analysis and visual inspection of morphology of spheroids to avoid highly toxic concentrations (Figure S6), we selected a 2.5 nM trametinib/2.5 nM mithramycin combination to cyclically treat HCT116 spheroids and analyze CSC genes. This approach effectively suppressed CSC markers CD166 and ALDH1A3 (Figure 6c). Importantly, the expression levels further reduced from T1 to T2, suggesting that the combination treatment prevented adaptive responses of cancer cells and stemness.
Figure 6.

Therapeutic targeting of CSCs in colorectal tumor spheroids. Heatmap plots in (a,b) represent fraction of cells affected (Fa) by single-agent and combination treatments (trametinib and mithramycin) and the respective combination index values corresponding to the combined concentrations of the two compounds. (c) Gene expression analysis of HCT116 tumor spheroids following cyclic treatments with trametinib/mithramycin combination for (c) CSC genes, (d) STAT pathway (Stat6), Wnt pathway (axin1 and β-catenin), EMT (E47 and Zeb1), and (e) PI3K/Akt pathway. All fold change values are relative to nontreated tumor samples. * denotes p < 0.05 compared to nontreated spheroids. Error bars represent standard errors from the mean (n = 2 independent experiments, two replicates/experiment).
Because treating colorectal cancer spheroids with MEK inhibitors led to activation of several pathways and EMT markers, we asked to what extent this combination could inhibit these diverse events. Our gene expression analysis following T1 and T2 showed that mithramycin/trametinib combination downregulated STAT6, a marker for the STAT pathway, AXIN1 and beta catenin, markers for the Wnt pathway, and E47 and ZEB1, markers of EMT (Figure 6d). This treatment also reduced gene expression of the PI3K/Akt pathway, which is a common bypass mechanism in many cancers including colorectal cancer (Figure 6e). We observed that the p-Akt level increased after T1 but reduced following T2, suggesting that the inhibition offered by the combination treatment is sustained (Figure S7). To ensure that these inhibitory effects resulted from the combination treatment, we also performed single-agent mithramycin treatment of HCT116 spheroids for two rounds (T1 and T2). Mithramycin alone was insufficient against activation of markers of CSC, STAT, WNT, and PI3K pathways (Figure S8). However, the treatment reduced the expression of EMT markers (Figure S8). Finally, we showed that the combination treatment effectively blocks collagen invasion of HCT116 cells from spheroids. Unlike trametinib-treated spheroids that showed up to sixfold increased matrix invasion than nontreated spheroids, mithramycin-treated and combination-treated spheroids showed no invasion (Figure 7). In addition, the size of combination-treated spheroids significantly reduced. These results suggest that mithramycin prevents invasiveness, consistent with the inhibitory effect of mithramycin against mRNA expression of EMT markers (Figure S8), while trametinib blocks proliferation and produces cytotoxicity.
Figure 7.
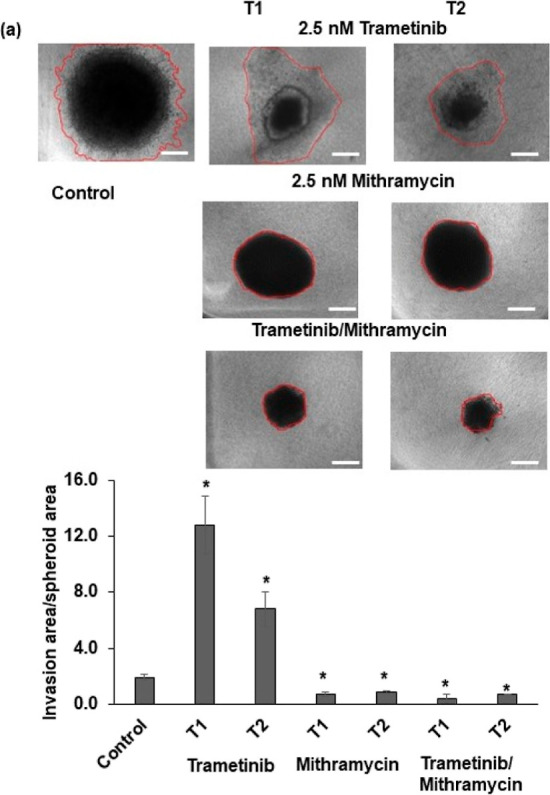
Targeting CSCs inhibits matrix invasion of colorectal tumor spheroids. Representative images of collagen invasion of the vehicle control and pretreated HCT116 spheroids. Quantified invasion area/spheroid area for the vehicle control and pretreated spheroids from T1 and T2 treatments, each normalized to the size of its respective spheroid from day 0. Error bars represent standard errors (n = 5). The scale bar is 200 μm. * denotes p < 0.05.
Overall, our studies using molecular and functional analysis of spheroid cultures of colorectal cancer cells demonstrated the role of CSCs in resistance to MEK inhibition and the potential of a combination treatment using mithramycin and trametinib in blocking different compensatory signaling pathways and CSCs to maintain drug sensitivity of cancer cells.
Discussion
Our study emphasizes a need for a combinatorial approach targeting CSCs and a constitutively active signaling pathway in KRASmut and BRAFmut colorectal cancer to prevent acquired drug resistance. We showed that single-agent inhibition of the MAPK pathway in colorectal tumor spheroids led to resistance by feedback activation of PI3K/Akt, JAK/STAT, and WNT pathways and gain of stemness marked by CD166 and ALDH1A3. This is consistent with the role of these CSC markers in colorectal carcinoma and correlation with a poor prognosis for patients.9,38,39 Simultaneous targeting of CSCs using mithramycin and MAPK pathway blocked stemness, feedback signaling, and EMT and invasiveness of cancer cells.
We previously showed that single-agent MEK inhibition transiently downregulates pERK and significantly shrinks the size of BRAFmut colorectal tumor spheroids but results in feedback Akt activation.18,25 In this study, we demonstrated that blocking MEK in KRASmut colorectal cancer cells also activates Akt (Figures 3a and S3),40 suggesting that compensatory PI3K/Akt signaling is a common resistance mechanism in consensus molecular subtype II of colorectal cancer.41,42 Moreover, we found significant activation of STAT1 and STAT6 kinases following MEK inhibition or combined MEK/Akt inhibition (Figure 3b). Other studies had also shown activation of STAT3 in colorectal cancer cells following treatments with MAPK inhibitors.5,43 Additionally, activation of the WNT signaling pathway (Figure 3c) was consistent with previous reports.9,44
Our study highlights the difficulty of blocking feedback activation of multiple signaling pathways using dual combination treatments in colorectal cancer cells. Clinically, this introduces a major challenge when selecting effective drug combinations.45 Although we previously showed that combinations of MEK and Akt inhibitors prevent growth of colorectal tumor spheroids by long-term blocking of feedback Akt signaling,25 we found that this approach is unsuccessful in blocking gain of stemness and invasiveness of cancer cells and leads to activation of other pathways. We demonstrated that an additional treatment with a STAT inhibitor blocked invasiveness of cancer cells but was ineffective against activation of CSC markers. Altogether, our results suggest that dual inhibition of MAPK and PI3K/Akt pathways does not offer significant benefits against tumorigenic activities of colorectal cancer cells. We also note that combined inhibition of these two pathways generated excessive toxicity to patients in several clinical trials,10 due to the importance of these pathways in proliferation and survival of cells in normal tissues. Motivated by the vital role of the Wnt pathway for the function of CSCs especially in colorectal cancer,46 we evaluated whether targeting of Wnt may block stemness of cancer cells. However, this approach also proved insufficient.
In our studies, cancer cells developed adaptive responses to different dual-targeted therapies and gained stem cell-like properties. Such compensatory mechanisms emerge from the redundancy and complex connectivity of signaling in colorectal cancer cells.47 Thus, we hypothesized that direct targeting of CSCs may bypass these mechanisms. We selected mithramycin A, an FDA-approved antibiotic that has been used in clinical trials for different cancers. Although it has been suggested that anticancer effects of mithramycin result from its inhibition of specificity protein 1 (Sp1),48 a recent study also showed its efficacy against CSCs.37 Our strategy to use a combination of mithramycin and trametinib generated significant benefits by suppressing the expression of CD166 and ALDH1A3 CSC markers, simultaneously downregulating PI3K/Akt, STAT, and Wnt signaling pathways and blocking matrix invasion and proliferation of colorectal tumor spheroids (Figures 6 and 7). Our results support the hypothesis that CSCs confer drug resistance in cancer and targeting them would improve treatment outcomes by blocking drug resistance.
Spheroid and organoid cultures of cancer cells have been leveraged to study mechanisms of drug resistance in different cancers.49−52 For example, a 3D model of colorectal cancer composed of cells derived from a primary tumor reproduced resistance to 5-FU treatment in the animal model,53 and an organoid culture model developed under an air-liquid interface contained CSCs and exhibited resistance to anticancer drugs.54,55 Over the past several years, we have developed tumor spheroid models of colorectal cancers for mechanistic studies of drug resistance and to test and identify effective treatment strategies.56 Here, we leveraged our established 3D tumor spheroid model in a cyclic treatment regimen to demonstrate that colorectal cancer cells develop adaptive resistance to single-agent inhibition of MEK through feedback signaling and gain of stemness. We found that a combination treatment approach targeting MEK and CSCs effectively blocks adaptive resistance of cancer cells by suppressing tumor stemness. Due to the role of tumor stroma in tumorigenesis and drug responses of cancer cells,57,58 incorporating key components of the stroma including fibroblasts and immune cells in our model and using patient-derived tumor cells and validation in animal experiments are critical to increase the translational potential of this approach. We note that using combinations of cancer drugs may generate excessive toxicities to normal tissues.59 Beyond investigating efficacy of drug combinations against cancer cells, it is critical to study their potential toxic effects. While we recently used a 3D model of normal cells to study toxicities of combinations of MAPK and PI3K/Akt inhibitors to normal bone marrow and colon cells,60 more complex models such as organs-on-a-chip are developed to address this important question.61
Materials and Methods
Cell Culture and Spheroid Formation
McCoy’s 5A medium was used to culture HT29 and HCT116 colorectal cancer cells (ATCC). The medium was supplemented with 10% fetal bovine serum (FBS) (Sigma), 1% antibiotic (Life Technologies), and 1% glutamine (Life Technologies). Cells were dissociated using 0.25% trypsin (Life Technologies) from 80 to 90% confluent monolayer cultures in tissue culture flasks. Trypsin was neutralized using the complete growth medium. The cell suspension was centrifuged down at 1000 rpm for 5 min. After removing the supernatant, cells were suspended in 1 mL of the culture medium and counted using a hemocytometer prior to spheroid formation.
A polymeric aqueous two-phase system was used to form colorectal tumor spheroids.21,62 Bio-ultra-PEG with a molecular weight of 35 kDa (Sigma) and DEX with a molecular weight of 500 kDa (Pharmacosmos) were dissolved in the complete growth medium to obtain final stock solutions of 5% (w/v) PEG and 12.8% (w/v) DEX. A round-bottom ultralow attachment 384-well plate (Corning) was used as a “destination plate”. Each well of this plate was loaded with 30 μL of the aqueous PEG phase medium. A suspension of 1 × 108 cells/mL was prepared, and 100 μL of the suspension was thoroughly mixed with 100 μL of the 12.8% (w/v) aqueous DEX phase medium. This mixing reduced DEX concentration to 6.4% (w/v) and adjusted the density of cells to 5 × 107 cells/mL. Each well from one column of a flat-bottom 384-well plate (Corning), which was used as a “source plate”, was filled with 25 μL of the resulting cell suspension in the DEX phase. A robotic liquid handler (Bravo SRT) (Agilent Technologies) was used to aspirate 0.3 μL of the suspension containing 1.5 × 104 cells from each well and dispense it into each well of the destination plate containing the aqueous PEG phase. The destination plate was incubated for 48 h to allow cells in each well to aggregate into a compact spheroid.
Cyclic Drug Treatments of Spheroids
Trametinib, PD0325901, selumetinib, dactolisib, VS-5584, AS151799, PRI-724, and niclosamide were purchased from Selleckchem. Mithramycin A was purchased from Sigma. Stock solutions of these compounds were prepared according to the manufacturers’ instructions. Stock solutions were stored at −80 °C. The compounds were tested against tumor spheroids according to our published protocol.40,63 Concentrations of 5 nM trametinib, 100 nM PD0325901, and 100 nM selumetinib were selected from dose-dependent tests against HT29 and HCT116 spheroids and used for cyclic treatments. Spheroids were subjected to two rounds of treatment (T1 and T2) each for 4 days and followed by recovery phases (R1 and R2) each for 4 days.18 Prior to the start of the experiments and at the end of each cycle of treatment and recovery, phase images of spheroids were captured using an inverted microscope (Axio Observer, Zeiss) equipped with a high-resolution camera (AxioCam MRM, Zeiss). The images were analyzed using ImageJ software (NIH) to measure the diameter of spheroids. The volume of spheroids was approximated from the diameter data assuming a spherical shape.64
Combination Drug Treatments of Spheroids
For combination treatments with MEK and AKT inhibitors, 5 nM trametinib was used with 200 nM dactolisib or 1 μM VS-5584. To further evaluate the effect of STAT6 inhibition, this was followed by a treatment with 10 μM AS15179. For combination treatments with MEK and WNT inhibitors, 5 nM trametinib was used with 1 μM niclosamide or 1 μM PRI-724. Combination treatments with MEK and CSC inhibitors were carried out in a 5 × 5 matrix format. Solutions of 4X concentrations of trametinib (1X, 10X, 50X, 100X, and 1000X) and mithramycin (1X, 5X, 10X, 50X, and 100X) were prepared. From these preparations, 15 μL of trametinib solution and 15 μL of mithramycin solution were added to each well containing a spheroid in 30 μL of cell culture medium to dilute each compound fourfold. Treatments were carrid out for 4 days, and viability of cells was quantified using a Prestoblue assay. The fraction of cells affected by each treatment was calculated as (1-viability). A synergy analysis was performed,63,65 resulting in 25 combination indices (CI) for the combined concentrations. In addition, images of spheroids were captured to observe the morphology of the spheroids after drug treatments.
Quantitative PCR
Gene expression analysis was carried out after T1, R1, T2, and R2. All fold changes values were expressed relative to that after 24 h, which was used as the control for all four time points. Spheroids were lysed using a total RNA kit (TRK) lysis buffer (Omega Biotek), and the lysate was homogenized by passing it through homogenizer mini columns (Omega Biotek). Total RNA was obtained using an RNA isolation kit (Omega Biotek). After removing DNA using RNase-free DNase (Omega Biotek), purity and concentration of isolated RNA were assessed using optical density (OD) 260/280 spectrophotometry (Synergy H1M, Biotek Instruments). cDNA was synthesized from 1 μg of total RNA using random hexamer primers (Roche). Real-time q-PCR was performed in a LightCycler 480 instrument II using a SYBR Green Master Mix (Roche). After combining 50 ng of cDNA with primers and the SYBR Green Master Mix to a final volume of 15 μL, the reactions were incubated at 95 °C for 5 min followed by 45 cycles of amplification, that is, at 95 °C for 10 s, at 60 °C for 10 s, and at 72 °C for 10 s. The primer sequences for the genes are listed in Table S1. Expression levels of mRNA for different proliferation gene markers were calculated relative to β-actin and hypoxanthine phosphoribosyltransferase (HPRT) using the ΔΔCt method. All the expressions were calculated with respect to control (nontreated) samples. The fold change in mRNA expression was determined according to the 2–ΔΔCt method.66
Invasion Assay
Single-agent treatments with 5 nM trametinib, 100 nM selumetinib, or 100 nM PD0325901 and combination treatments with 5 nM trametinib/200 nM dactolisib or 5 nM trametinib/1 μM VS-5584 were used against HCT116 and HT29 spheroids. After 4 days of treatment, spheroids were suspended in a 2.75 mg/mL human type I collagen solution. Incubation at 37 °C for 120 min resulted in collagen gelation. Invasion of cells from spheroids into collagen was captured using a confocal microscope (Nikon A1) after 5 days. Z-projected images were reconstructed by collapsing the stacks in ImageJ. Cell invasion from each spheroid was quantified as the ratio of invasion pixel area relative to the spheroid pixel area. In a separate experiment, HCT116 spheroids were pretreated for 4 days with a 5 nM trametinib/200 nM dactolisib combination, recovered, embedded in 2.75 mg/ml collagen, and treated with 10 μM of AS1517499 (STAT6 inhibitor). The collagen-embedded spheroids were maintained in drug-free medium to evaluate inhibitory effects of STAT inhibition on matrix invasion of cancer cells. Additionally, HCT116 spheroids were pretreated for 4 days with 2.5 nM trametinib, or 2.5 nM mithramycin, or their combination, recovered, and embedded in 2.75 mg/mL collagen to evaluate collagen invasion of cancer cells.
Clonogenicity Assay
HCT116 and HT29 cells were seeded as single cells in a 1.5% (w/v) methylcellulose gel made using the complete growth medium. Each well of a flat-bottom 384-well plate was loaded with 20 μL of a 1:4 ratio of methylcellulose solution and the growth medium containing 200 cells. Culture medium was added after 2 days of cell seeding in the methylcellulose gel and replaced every 2 days. The cultures were treated with a single dose of MEK inhibitors or a combination of MEK and PI3K inhibitors and maintained for 8–10 days prior to imaging the colonies. The colonies were stained with calcein AM prior to imaging at a 2.5X magnification. An in-house python code in ImageJ was used to automatically detect the colonies in each image and compute the area of the colonies. A threshold colony diameter of 50 μm was used for statistical analysis.67 To compute the colony-forming efficiency, first plating efficiency was computed as plating efficiency = (number of colonies counted/number of cells plated)*100% followed by computing the colony-forming capacity for each condition as colony-forming capacity = [(number of colonies counted/number of cells plated)/plating efficiency]*100%.68
Western Blotting
Western blot analysis with spheroids was performed using our established protocol.40 Primary antibodies purchased from Cell Signaling Technology were phospho-p44/42 MAPK (pErk 1/2), p44/42 MAPK (Erk1/2), phospho-Akt (Ser473), and Akt (pan). Solutions of primary antibodies were prepared at concentrations recommended by the manufacturer. Nitrocellulose membranes were incubated overnight at 4 °C with primary antibody solutions. After several washing steps, membranes were incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h, followed by repeated washing. Detection was carried out using an ECL chemiluminescence detection kit (GE Healthcare) with a FluorChem E imaging system (ProteinSimple).
Statistical Analysis
Student’s t-test was used to evaluate spheroid volume differences between single-agent treatments and vehicle control, to compare the fold change between treated and nontreated samples, to evaluate protein phosphorylation levels from a drug-treated group compared to its respective vehicle control group, to compare colony-forming capacity, and to evaluate matrix invasion of cells from a treated sample and the respective vehicle control. The analysis was performed in Microsoft Excel..
Acknowledgments
This research was supported by a grant CA216413 from National Institutes of Health.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.1c00257.
Fold change in the expression of CSC genes in tumor spheroids during cyclic treatment and recovery with the MEK inhibitor trametinib; representative images of colony formation in HCT116 cells; treating colorectal tumor spheroids with MEK inhibitors activates the PI3K/Akt pathway; fold change in the expression of Wnt pathway genes in HT29 spheroids; treatment-induced stemness in colorectal cancer cells; morphological images of HCT116 spheroids; combined trametinib/mithramycin effect on PI3K/Akt signaling; gene expression of single-agent mithramycin treatment on HCT116 cells; and list and sequences of primers of the genes analyzed for CSC, WNT, STAT, and AKT markers of colorectal cancer cells (PDF)
Author Contributions
Concept and design: A.L. and H.T.; development and methodology: A.L. and H.T.; acquisition, analysis, and interpretation of data: A.L., P.S.T., S.S., P.R.N., and J.H.; writing, review, and/or revision of the manuscript: A.L., G.D.L., and H.T.; study supervision: H.T.
The authors declare no competing financial interest.
Supplementary Material
References
- Housman G.; Byler S.; Heerboth S.; Lapinska K.; Longacre M.; Snyder N.; Sarkar S. Drug resistance in cancer: an overview. Cancers (Basel) 2014, 6, 1769–1792. 10.3390/cancers6031769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holohan C.; Van Schaeybroeck S.; Longley D. B.; Johnston P. G. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- Xi Y.; Xu P. Global colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 2021, 14, 101174. 10.1016/j.tranon.2021.101174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C.; Hobor S.; Bertotti A.; Zecchin D.; Huang S.; Galimi F.; Cottino F.; Prahallad A.; Grernrum W.; Tzani A.; Schlicker A.; Wessels L. F.; Smit E. F.; Thunnissen E.; Halonen P.; Lieftink C.; Beijersbergen R. L.; Di Nicolantonio F.; Bardelli A.; Trusolino L.; Bernards R. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 2014, 7, 86–93. 10.1016/j.celrep.2014.02.045. [DOI] [PubMed] [Google Scholar]
- Van Schaeybroeck S.; Kalimutho M.; Dunne P. D.; Carson R.; Allen W.; Jithesh P. V.; Redmond K. L.; Sasazuki T.; Shirasawa S.; Blayney J.; Michieli P.; Fenning C.; Lenz H. J.; Lawler M.; Longley D. B.; Johnston P. G. ADAM17-dependent c-MET-STAT3 signaling mediates resistance to MEK inhibitors in KRAS mutant colorectal cancer. Cell Rep. 2014, 7, 1940–1955. 10.1016/j.celrep.2014.05.032. [DOI] [PubMed] [Google Scholar]
- Muzny D. M.; Bainbridge M. N.; Chang K.; Dinh H. H.; Drummond J. A.; Fowler G.; Kovar C. L.; Lewis L. R.; Morgan M. B.; Newsham I. F.; Reid J. G.; Santibanez J.; Shinbrot E.; Trevino L. R.; Wu Y.-Q.; Wang M.; Gunaratne P.; Donehower L. A.; Creighton C. J.; Wheeler D. A.; Gibbs R. A.; Lawrence M. S.; Voet D.; Jing R.; Cibulskis K.; Sivachenko A.; Stojanov P.; McKenna A.; Lander E. S.; Gabriel S.; Getz G.; Ding L.; Fulton R. S.; Koboldt D. C.; Wylie T.; Walker J.; Dooling D. J.; Fulton L.; Delehaunty K. D.; Fronick C. C.; Demeter R.; Mardis E. R.; Wilson R. K.; Chu A.; Chun H.-J. E.; Mungall A. J.; Pleasance E.; Gordon Robertson A.; Stoll D.; Balasundaram M.; Birol I.; Butterfield Y. S. N.; Chuah E.; Coope R. J. N.; Dhalla N.; Guin R.; Hirst C.; Hirst M.; Holt R. A.; Lee D.; Li H. I.; Mayo M.; Moore R. A.; Schein J. E.; Slobodan J. R.; Tam A.; Thiessen N.; Varhol R.; Zeng T.; Zhao Y.; Jones S. J. M.; Marra M. A.; Bass A. J.; Ramos A. H.; Saksena G.; Cherniack A. D.; Schumacher S. E.; Tabak B.; Carter S. L.; Pho N. H.; Nguyen H.; Onofrio R. C.; Crenshaw A.; Ardlie K.; Beroukhim R.; Winckler W.; Getz G.; Meyerson M.; Protopopov A.; Zhang J.; Hadjipanayis A.; Lee E.; Xi R.; Yang L.; Ren X.; Zhang H.; Sathiamoorthy N.; Shukla S.; Chen P.-C.; Haseley P.; Xiao Y.; Lee S.; Seidman J.; Chin L.; Park P. J.; Kucherlapati R.; Todd Auman J.; Hoadley K. A.; Du Y.; Wilkerson M. D.; Shi Y.; Liquori C.; Meng S.; Li L.; Turman Y. J.; Topal M. D.; Tan D.; Waring S.; Buda E.; Walsh J.; Jones C. D.; Mieczkowski P. A.; Singh D.; Wu J.; Gulabani A.; Dolina P.; Bodenheimer T.; Hoyle A. P.; Simons J. V.; Soloway M.; Mose L. E.; Jefferys S. R.; Balu S.; O’Connor B. D.; Prins J. F.; Chiang D. Y.; Neil Hayes D.; Perou C. M.; Hinoue T.; Weisenberger D. J.; Maglinte D. T.; Pan F.; Berman B. P.; Van Den Berg D. J.; Shen H.; Triche T. Jr.; Baylin S. B.; Laird P. W.; Getz G.; Noble M.; Voet D.; Saksena G.; Gehlenborg N.; DiCara D.; Zhang J.; Zhang H.; Wu C.-J.; Yingchun Liu S.; Shukla S.; Lawrence M. S.; Zhou L.; Sivachenko A.; Lin P.; Stojanov P.; Jing R.; Park R. W.; Nazaire M.-D.; Robinson J.; Thorvaldsdottir H.; Mesirov J.; Park P. J.; Chin L.; Thorsson V.; Reynolds S. M.; Bernard B.; Kreisberg R.; Lin J.; Iype L.; Bressler R.; Erkkilä T.; Gundapuneni M.; Liu Y.; Norberg A.; Robinson T.; Yang D.; Zhang W.; Shmulevich I.; de Ronde J. J.; Schultz N.; Cerami E.; Ciriello G.; Goldberg A. P.; Gross B.; Jacobsen A.; Gao J.; Kaczkowski B.; Sinha R.; Arman Aksoy B.; Antipin Y.; Reva B.; Shen R.; Taylor B. S.; Chan T. A.; Ladanyi M.; Sander C.; Akbani R.; Zhang N.; Broom B. M.; Casasent T.; Unruh A.; Wakefield C.; Hamilton S. R.; Craig Cason R.; Baggerly K. A.; Weinstein J. N.; Haussler D.; Benz C. C.; Stuart J. M.; Benz S. C.; Zachary Sanborn J.; Vaske C. J.; Zhu J.; Szeto C.; Scott G. K.; Yau C.; Ng S.; Goldstein T.; Ellrott K.; Collisson E.; Cozen A. E.; Zerbino D.; Wilks C.; Craft B.; Spellman P.; Penny R.; Shelton T.; Hatfield M.; Morris S.; Yena P.; Shelton C.; Sherman M.; Paulauskis J.; Gastier-Foster J. M.; Bowen J.; Ramirez N. C.; Black A.; Pyatt R.; Wise L.; White P.; Bertagnolli M.; Brown J.; Chan T. A.; Chu G. C.; Czerwinski C.; Denstman F.; Dhir R.; Dörner A.; Fuchs C. S.; Guillem J. G.; Iacocca M.; Juhl H.; Kaufman A.; Kohl B.; Van Le X.; Mariano M. C.; Medina E. N.; Meyers M.; Nash G. M.; Paty P. B.; Petrelli N.; Rabeno B.; Richards W. G.; Solit D.; Swanson P.; Temple L.; Tepper J. E.; Thorp R.; Vakiani E.; Weiser M. R.; Willis J. E.; Witkin G.; Zeng Z.; Zinner M. J.; Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebolt-Leopold J. S.; Dudley D. T.; Herrera R.; Becelaere K.; Wiland A.; Gowan R. C.; Tecle H.; Barrett S. D.; Bridges A.; Przybranowski S.; Leopold W. R.; Saltiel A. R. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat. Med. 1999, 5, 810–816. 10.1038/10533. [DOI] [PubMed] [Google Scholar]
- Yang H.; Higgins B.; Kolinsky K.; Packman K.; Bradley W. D.; Lee R. J.; Schostack K.; Simcox M. E.; Kopetz S.; Heimbrook D.; Lestini B.; Bollag G.; Su F. Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res. 2012, 72, 779–789. 10.1158/0008-5472.Can-11-2941. [DOI] [PubMed] [Google Scholar]
- Zhan T.; Ambrosi G.; Wandmacher A. M.; Rauscher B.; Betge J.; Rindtorff N.; Häussler R. S.; Hinsenkamp I.; Bamberg L.; Hessling B.; Müller-Decker K.; Erdmann G.; Burgermeister E.; Ebert M. P.; Boutros M. MEK inhibitors activate Wnt signalling and induce stem cell plasticity in colorectal cancer. Nat. Commun. 2019, 10, 2197. 10.1038/s41467-019-09898-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T.; Tolcher A. W.; Papadopoulos K. P.; Beeram M.; Rasco D. W.; Smith L. S.; Gunn S.; Smetzer L.; Mays T. A.; Kaiser B.; Wick M. J.; Alvarez C.; Cavazos A.; Mangold G. L.; Patnaik A. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 2316–2325. 10.1158/1078-0432.Ccr-11-2381. [DOI] [PubMed] [Google Scholar]
- Molina A. M.; Feldman D. R.; Voss M. H.; Ginsberg M. S.; Baum M. S.; Brocks D. R.; Fischer P. M.; Trinos M. J.; Patil S.; Motzer R. J. Phase 1 trial of everolimus plus sunitinib in patients with metastatic renal cell carcinoma. Cancer 2012, 118, 1868–1876. 10.1002/cncr.26429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapnick D. A.; Warner L.; Bernet J.; Rao T.; Liu X. Partners in crime: the TGFβ and MAPK pathways in cancer progression. Cell Biosci. 2011, 1, 42. 10.1186/2045-3701-1-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.; Dong J.; Haiech J.; Kilhoffer M. C.; Zeniou M. Cancer Stem Cell Quiescence and Plasticity as Major Challenges in Cancer Therapy. Stem Cells Int. 2016, 2016, 1740936. 10.1155/2016/1740936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phi L. T. H.; Sari I. N.; Yang Y.-G.; Lee S.-H.; Jun N.; Kim K. S.; Lee Y. K.; Kwon H. Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. 10.1155/2018/5416923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabiraman D. R.; Weinberg R. A. Tackling the cancer stem cells—what challenges do they pose?. Nat. Rev. Drug Discovery 2014, 13, 497–512. 10.1038/nrd4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naujokat C.; Steinhart R. Salinomycin as a drug for targeting human cancer stem cells. J. Biomed. Biotechnol. 2012, 2012, 950658. 10.1155/2012/950658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P. B.; Onder T. T.; Jiang G.; Tao K.; Kuperwasser C.; Weinberg R. A.; Lander E. S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahi Thakuri P.; Luker G. D.; Tavana H. Cyclical Treatment of Colorectal Tumor Spheroids Induces Resistance to MEK Inhibitors. Transl. Oncol. 2019, 12, 404–416. 10.1016/j.tranon.2018.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atefi E.; Joshi R.; Mann J. A. Jr.; Tavana H. Interfacial Tension Effect on Cell Partition in Aqueous Two-Phase Systems. ACS Appl. Mater. Interfaces 2015, 7, 21305–21314. 10.1021/acsami.5b05757. [DOI] [PubMed] [Google Scholar]
- Lemmo S.; Atefi E.; Luker G. D.; Tavana H. Optimization of Aqueous Biphasic Tumor Spheroid Microtechnology for Anti-Cancer Drug Testing in 3D Culture. Cell. Mol. Bioeng. 2014, 7, 344–354. 10.1007/s12195-014-0349-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atefi E.; Lemmo S.; Fyffe D.; Luker G. D.; Tavana H. High Throughput, Polymeric Aqueous Two-Phase Printing of Tumor Spheroids. Adv. Funct. Mater. 2014, 24, 6509–6515. 10.1002/adfm.201401302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakuri P. S.; Gupta M.; Plaster M.; Tavana H. Quantitative Size-Based Analysis of Tumor Spheroids and Responses to Therapeutics. Assay Drug Dev. Technol. 2019, 17, 140–149. 10.1089/adt.2018.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed D.; Eide P. W.; Eilertsen I. A.; Danielsen S. A.; Eknæs M.; Hektoen M.; Lind G. E.; Lothe R. A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71 10.1038/oncsis.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charitou T.; Srihari S.; Lynn M. A.; Jarboui M.-A.; Fasterius E.; Moldovan M.; Shirasawa S.; Tsunoda T.; Ueffing M.; Xie J.; Xin J.; Wang X.; Proud C. G.; Boldt K.; Al-Khalili Szigyarto C.; Kolch W.; Lynn D. J. Transcriptional and metabolic rewiring of colorectal cancer cells expressing the oncogenic KRASG13D mutation. Br. J. Cancer 2019, 121, 37–50. 10.1038/s41416-019-0477-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakuri P. S.; Gupta M.; Joshi R.; Singh S.; Tavana H. Synergistic Inhibition of Kinase Pathways Overcomes Resistance of Colorectal Cancer Spheroids to Cyclic Targeted Therapies. ACS Pharmacol. Transl. Sci. 2019, 2, 275–284. 10.1021/acsptsci.9b00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich K. P.; Merchant M.; Orr C.; Chan J.; Den Otter D.; Berry L.; Kasman I.; Koeppen H.; Rice K.; Yang N.-Y.; Engst S.; Johnston S.; Friedman L. S.; Belvin M. Intermittent Administration of MEK Inhibitor GDC-0973 plus PI3K Inhibitor GDC-0941 Triggers Robust Apoptosis and Tumor Growth Inhibition. Cancer Res. 2012, 72, 210. 10.1158/0008-5472.CAN-11-1515. [DOI] [PubMed] [Google Scholar]
- Visvader J. E.; Lindeman G. J. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- Dean M.; Fojo T.; Bates S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- Levin T. G.; Powell A. E.; Davies P. S.; Silk A. D.; Dismuke A. D.; Anderson E. C.; Swain J. R.; Wong M. H. Characterization of the intestinal cancer stem cell marker CD166 in the human and mouse gastrointestinal tract. Gastroenterology 2010, 139, 2072–2082. 10.1053/j.gastro.2010.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S.; Yang W.; Zong S.; Li H.; Liu S.; Li W.; Shi Q.; Hou F. Clinicopathological, prognostic and predictive value of CD166 expression in colorectal cancer: a meta-analysis. Oncotarget 2017, 8, 64373–64384. 10.18632/oncotarget.17442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S.; Haase G.; Ben-Ze’ev A. Wnt signaling in cancer stem cells and colon cancer metastasis. F1000Research 2016, 5, 1–11. 10.12688/f1000research.7579.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G.; Li X.; Guo B.; Ke Q.; Dong M.; Li F. PAK5-mediated E47 phosphorylation promotes epithelial–mesenchymal transition and metastasis of colon cancer. Oncogene 2016, 35, 1943–1954. 10.1038/onc.2015.259. [DOI] [PubMed] [Google Scholar]
- Wei Z.-J.; Tao M.-L.; Zhang W.; Han G.-D.; Zhu Z.-C.; Miao Z.-G.; Li J.-Y.; Qiao Z.-B. Up-regulation of microRNA-302a inhibited the proliferation and invasion of colorectal cancer cells by regulation of the MAPK and PI3K/Akt signaling pathways. Int. J. Clin. Exp. Pathol. 2015, 8, 4481–4491. [PMC free article] [PubMed] [Google Scholar]
- Zhou G.; Yang J.; Song P. Correlation of ERK/MAPK signaling pathway with proliferation and apoptosis of colon cancer cells. Oncol. Lett. 2019, 17, 2266–2270. 10.3892/ol.2018.9857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabata R.; Harada K.; Mizutani Y.; Ouchi H.; Yoshimura K.; Sato Y.; Kitao A.; Kimura K.; Kouji H.; Miyashita T.; Tajima H.; Ohta T. Anti-tumor Activity of the Small Molecule Inhibitor PRI-724 Against β-Catenin-activated Hepatocellular Carcinoma. Anticancer Res. 2020, 40, 5211–5219. 10.21873/anticanres.14524. [DOI] [PubMed] [Google Scholar]
- Arend R. C.; Londoño-Joshi A. I.; Samant R. S.; Li Y.; Conner M.; Hidalgo B.; Alvarez R. D.; Landen C. N.; Straughn J. M.; Buchsbaum D. J. Inhibition of Wnt/β-catenin pathway by niclosamide: A therapeutic target for ovarian cancer. Gynecol. Oncol. 2014, 134, 112–120. 10.1016/j.ygyno.2014.04.005. [DOI] [PubMed] [Google Scholar]
- Quarni W.; Dutta R.; Green R.; Katiri S.; Patel B.; Mohapatra S. S.; Mohapatra S. Mithramycin A Inhibits Colorectal Cancer Growth by Targeting Cancer Stem Cells. Sci. Rep. 2019, 9, 15202. 10.1038/s41598-019-50917-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinikova E.; Kozovska Z.; Poturnajova M.; Plava J.; Cierna Z.; Babelova A.; Bohovic R.; Schmidtova S.; Tomas M.; Kucerova L.; Matuskova M. ALDH1A3 upregulation and spontaneous metastasis formation is associated with acquired chemoresistance in colorectal cancer cells. BMC Cancer 2018, 18, 848. 10.1186/s12885-018-4758-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichert W.; Knösel T.; Bellach J.; Dietel M.; Kristiansen G. ALCAM/CD166 is overexpressed in colorectal carcinoma and correlates with shortened patient survival. J. Clin. Pathol. 2004, 57, 1160–1164. 10.1136/jcp.2004.016238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahi Thakuri P.; Lamichhane A.; Singh S.; Gupta M.; Luker G. D.; Tavana H. Modeling Adaptive Resistance of KRAS Mutant Colorectal Cancer to MAPK Pathway Inhibitors with a Three-Dimensional Tumor Model. ACS Pharmacol. Transl. Sci. 2020, 3, 1176–1187. 10.1021/acsptsci.0c00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turke A. B.; Song Y.; Costa C.; Cook R.; Arteaga C. L.; Asara J. M.; Engelman J. A. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012, 72, 3228–3237. 10.1158/0008-5472.Can-11-3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinney J.; Dienstmann R.; Wang X.; de Reyniès A.; Schlicker A.; Soneson C.; Marisa L.; Roepman P.; Nyamundanda G.; Angelino P.; Bot B. M.; Morris J. S.; Simon I. M.; Gerster S.; Fessler E.; De Sousa E Melo F.; Missiaglia E.; Ramay H.; Barras D.; Homicsko K.; Maru D.; Manyam G. C.; Broom B.; Boige V.; Perez-Villamil B.; Laderas T.; Salazar R.; Gray J. W.; Hanahan D.; Tabernero J.; Bernards R.; Friend S. H.; Laurent-Puig P.; Medema J. P.; Sadanandam A.; Wessels L.; Delorenzi M.; Kopetz S.; Vermeulen L.; Tejpar S. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J.; Guo Q.; Xie J.; Jin D.; Zhu Y. Combination of MEK Inhibitor and the JAK2-STAT3 Pathway Inhibition for the Therapy of Colon Cancer. Pathol. Oncol. Res. 2019, 25, 769–775. 10.1007/s12253-019-00592-6. [DOI] [PubMed] [Google Scholar]
- Solberg N. T.; Melheim M.; Strand M. F.; Olsen P. A.; Krauss S. MEK Inhibition Induces Canonical WNT Signaling through YAP in KRAS Mutated HCT-15 Cells, and a Cancer Preventive FOXO3/FOXM1 Ratio in Combination with TNKS Inhibition. Cancers (Basel) 2019, 11, 164. 10.3390/cancers11020164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel A.; Hofheinz R. D.; Kubicka S.; Arnold D. Treatment decisions in metastatic colorectal cancer—Beyond first and second line combination therapies. Cancer Treat. Rev. 2017, 59, 54–60. 10.1016/j.ctrv.2017.04.007. [DOI] [PubMed] [Google Scholar]
- Reya T.; Clevers H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- Mastrogamvraki N.; Zaravinos A. Signatures of co-deregulated genes and their transcriptional regulators in colorectal cancer. npj Syst. Biol. Appl. 2020, 6, 23. 10.1038/s41540-020-00144-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi E.-S.; Nam J.-S.; Jung J.-Y.; Cho N.-P.; Cho S.-D. Modulation of specificity protein 1 by mithramycin A as a novel therapeutic strategy for cervical cancer. Sci. Rep. 2014, 4, 7162. 10.1038/srep07162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S.; Song J. M. 3D bioprinted drug-resistant breast cancer spheroids for quantitative in situ evaluation of drug resistance. Acta Biomater. 2022, 138, 228–239. 10.1016/j.actbio.2021.10.031. [DOI] [PubMed] [Google Scholar]
- Barrera-Rodríguez R.; Fuentes J. M. Multidrug resistance characterization in multicellular tumour spheroids from two human lung cancer cell lines. Cancer Cell Int. 2015, 15, 47. 10.1186/s12935-015-0200-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukai S.; Honma R.; Sakamoto N.; Yamamoto Y.; Pham Q. T.; Harada K.; Takashima T.; Taniyama D.; Asai R.; Fukada K.; Naka K.; Tanabe K.; Ohdan H.; Yasui W. Molecular biological analysis of 5-FU-resistant gastric cancer organoids; KHDRBS3 contributes to the attainment of features of cancer stem cell. Oncogene 2020, 39, 7265–7278. 10.1038/s41388-020-01492-9. [DOI] [PubMed] [Google Scholar]
- Casagrande N.; Borghese C.; Agostini F.; Durante C.; Mazzucato M.; Colombatti A.; Aldinucci D. In Ovarian Cancer Multicellular Spheroids, Platelet Releasate Promotes Growth, Expansion of ALDH+ and CD133+ Cancer Stem Cells, and Protection against the Cytotoxic Effects of Cisplatin, Carboplatin and Paclitaxel. Int. J. Mol. Sci. 2021, 22, 3019. 10.3390/ijms22063019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange C. S.; Djordjevic B.; Brock W. A. The hybrid spheroid clonogenic assay for the intrinsic radio- and chemo-sensitivities of human tumors. Int. J. Radiat. Oncol., Biol., Phys. 1992, 24, 511–517. 10.1016/0360-3016(92)91067-w. [DOI] [PubMed] [Google Scholar]
- Usui T.; Sakurai M.; Enjoji S.; Kawasaki H.; Umata K.; Ohama T.; Fujiwara N.; Yabe R.; Tsuji S.; Yamawaki H.; Hazama S.; Takenouchi H.; Nakajima M.; Tsunedomi R.; Suzuki N.; Nagano H.; Sato K. Establishment of a Novel Model for Anticancer Drug Resistance in Three-Dimensional Primary Culture of Tumor Microenvironment. Stem Cells Int. 2016, 2016, 7053872. 10.1155/2016/7053872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui T.; Sakurai M.; Umata K.; Yamawaki H.; Ohama T.; Sato K. Preparation of Human Primary Colon Tissue-Derived Organoid Using Air Liquid Interface Culture. Curr. Protoc. Toxicol. 2018, 75, 22.6.1–22.6.7. 10.1002/cptx.40. [DOI] [PubMed] [Google Scholar]
- Lamichhane A.; Thakuri P. S.; Rafsanjani Nejad P.; Tavana H. Modeling adaptive drug resistance of colorectal cancer and therapeutic interventions with tumor spheroids. Exp. Biol. Med. 2021, 246, 2372–2380. 10.1177/15353702211014185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S.; Lamichhane A.; Rafsanjani Nejad P.; Heiss J.; Baumann H.; Gudneppanavar R.; Leipzig N. D.; Konopka M.; Luker G. D.; Tavana H. Therapeutic Targeting of Stromal-Tumor HGF-cMET Signaling in an Organotypic Triple Negative Breast Tumor Model. Mol. Cancer Res. 2022, 20, OF1–OF12. 10.1158/1541-7786.Mcr-21-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S.; Ray L. A.; Shahi Thakuri P.; Tran S.; Konopka M. C.; Luker G. D.; Tavana H. Organotypic breast tumor model elucidates dynamic remodeling of tumor microenvironment. Biomaterials 2020, 238, 119853. 10.1016/j.biomaterials.2020.119853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell D.; Alper J.; Ptak K.; Panaro N. J.; Grodzinski P.; Barker A. D. Recent Advances from the National Cancer Institute Alliance for Nanotechnology in Cancer. ACS Nano 2010, 4, 589–594. 10.1021/nn100073g. [DOI] [PubMed] [Google Scholar]
- Rafsanjani Nejad P.; Shahi Thakuri P.; Singh S.; Lamichhane A.; Heiss J.; Tavana H. Toxicity of Combinations of Kinase Pathway Inhibitors to Normal Human Cells in a Three-Dimensional Culture. SLAS Technol. 2021, 26, 255–264. 10.1177/24726303211008858. [DOI] [PubMed] [Google Scholar]
- Si L.; Bai H.; Rodas M.; Cao W.; Oh C. Y.; Jiang A.; Moller R.; Hoagland D.; Oishi K.; Horiuchi S.; Uhl S.; Blanco-Melo D.; Albrecht R. A.; Liu W.-C.; Jordan T.; Nilsson-Payant B. E.; Golynker I.; Frere J.; Logue J.; Haupt R.; McGrath M.; Weston S.; Zhang T.; Plebani R.; Soong M.; Nurani A.; Kim S. M.; Zhu D. Y.; Benam K. H.; Goyal G.; Gilpin S. E.; Prantil-Baun R.; Gygi S. P.; Powers R. K.; Carlson K. E.; Frieman M.; tenOever B. R.; Ingber D. E. A human-airway-on-a-chip for the rapid identification of candidate antiviral therapeutics and prophylactics. Nat. Biomed. Eng. 2021, 5, 815–829. 10.1038/s41551-021-00718-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham S. L.; Atefi E.; Fyffe D.; Tavana H. Robotic production of cancer cell spheroids with an aqueous two-phase system for drug testing. J. Visualized Exp. 2015, 98, e52754 10.3791/52754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahi Thakuri P.; Tavana H. Single and Combination Drug Screening with Aqueous Biphasic Tumor Spheroids. SLAS Discovery 2017, 22, 507–515. 10.1177/2472555217698817. [DOI] [PubMed] [Google Scholar]
- Shahi Thakuri P.; Ham S. L.; Luker G. D.; Tavana H. Multiparametric Analysis of Oncology Drug Screening with Aqueous Two-Phase Tumor Spheroids. Mol. Pharm. 2016, 13, 3724–3735. 10.1021/acs.molpharmaceut.6b00527. [DOI] [PubMed] [Google Scholar]
- Chou T. C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. 10.1158/0008-5472.Can-09-1947. [DOI] [PubMed] [Google Scholar]
- Joshi R.; Thakuri P. S.; Buchanan J. C.; Li J.; Tavana H. Microprinted Stem Cell Niches Reveal Compounding Effect of Colony Size on Stromal Cells-Mediated Neural Differentiation. Adv. Healthcare Mater. 2018, 7, 1700832. 10.1002/adhm.201700832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.-C.; Ingram P. N.; Fouladdel S.; McDermott S. P.; Azizi E.; Wicha M. S.; Yoon E. High-Throughput Single-Cell Derived Sphere Formation for Cancer Stem-Like Cell Identification and Analysis. Sci. Rep. 2016, 6, 27301. 10.1038/srep27301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafehi H.; Orlowski C.; Georgiadis G. T.; Ververis K.; El-Osta A.; Karagiannis T. C. Clonogenic assay: adherent cells. J. Visualized Exp. 2011, 49, 2573. 10.3791/2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.