

Abstract
Background and Objectives
Pathogenic variations in fused in sarcoma (FUS) are among the most common genetic causes of amyotrophic lateral sclerosis (ALS) worldwide. They are supposedly characterized by a homogeneous pure motor phenotype with early-onset and short disease duration. However, a few FUS-mutated cases with a very late disease onset and slow progression have been reported. To analyze genotype-phenotype correlations and identify the prognostic factors in FUS-ALS cases.
Methods
We identified and cross-sectionally analyzed 22 FUS-ALS patient histories from a single-center cohort of 2,615 genetically tested patients and reviewed 289 previously published FUS-ALS cases. Survival analysis was performed by Kaplan-Meier survival curves, followed by the log-rank test and multivariate Cox analysis.
Results
Survival of FUS-ALS is age-dependent: In our cohort, early-onset cases had a rapid disease progression and short survival (p = 0.000003) while the outcome of FUS-mutated patients with mid-to-late onset did not differ from non–FUS-ALS patients (p = 0.437). Meta-analysis of literature data confirmed this trend (p = 0.00003). This survival pattern is not observed in other ALS-related genes in our series. We clustered FUS-ALS patients in 3 phenotypes: (1) axial ALS, with upper cervical and dropped-head onset in mid-to-late adulthood; (2) benign ALS, usually with a late-onset and slow disease progression; and (3) juvenile ALS, often with bulbar onset and preceded by learning disability or mild mental retardation. Those phenotypes arise from different mutations.
Discussion
We observed specific genotype-phenotype correlations of FUS-ALS and identified age at onset as the most critical prognostic factor. Our results demonstrated that FUS mutations underlie a specific subtype of ALS and enable a careful stratification of newly diagnosed FUS-ALS cases for clinical course and potential therapeutic windows. This will be crucial in the light of incoming gene-specific therapy.
Mutations in the fused in sarcoma (FUS) gene were first reported to cause amyotrophic lateral sclerosis (ALS) in 2009.1,2 Over 50 mutations have been linked to the disease since then. Nearly all of these mutations occurred in the nuclear localization signal (NLS) domain encoded by exons 14 and 15. As the name suggests, this domain enables the FUS protein to enter the nucleus, and mutations in the NLS have been shown to disrupt this function, leading to the loss of nuclear protein and an abnormal aggregation in the cytoplasm.3,4 FUS mutation frequency varies across populations, accounting for approximately 3% of familial ALS cases and 0.3% of sporadic ALS cases in European populations, and 6.4%–11.4% and 0.9%–1.5%, respectively, in Asian countries.5 In addition, based on previous reports of FUS mutations in families of African ancestry,6 FUS could possibly be relevant also in African populations. FUS is also the most common mutated gene in juvenile ALS (age at onset younger than 25 years) cases, accounting for up to 30% of such cases in China7,8 and Japan.9
Because FUS mutations could become an actionable disease target with antisense oligonucleotide therapy (ClinicalTrials.gov: NCT04768972),10-12 it is crucial to understand its clinical manifestations and their genomic substrates. Therefore, we analyzed the clinical phenotype of genetically confirmed FUS cases from an Italian tertiary ALS center and investigated for genotype-phenotype correlations specific to FUS in both our cohort and FUS‐ALS cases previously reported in the literature. A comprehensive description of genotype-phenotype patterns will enable stratification of FUS-ALS patients and a more accurate prediction of their clinical course.
Methods
Study Cohort and Data Collection
In this retrospective study, we used blood samples collected from 2615 ALS patients diagnosed according to El Escorial criteria13 that have been seen at the ALS Center in Turin, Italy, from January 1, 2007, to December 31, 2020. Clinical data were prospectively collected at the first visit and during follow-up. Neuropsychological tests were performed as previously reported.14
DNA Analysis
We extracted DNA using standard procedures. All the coding exons and 50 bp of the flanking intron-exon boundaries of SOD1, exon 6 of TARDBP, and exons 14 and 15 of FUS were polymerase chain reaction amplified, sequenced using the Big-Dye Terminator v3.1 sequencing kit (Applied Biosystems Inc., Foster City, CA) and run on an ABIPrism 3130 genetic analyzer. Genetic analysis and whole-genome sequencing were performed as previously reported.15,16 A repeat-primed polymerase chain reaction assay was used to screen for the presence of the GGGGCC hexanucleotide expansion in the first intron of C9ORF72. Whole-genome sequencing was also performed on 849 ALS patients as previously reported16: Those patients were also screened for FUS variants outside of the C-terminal domain.
Literature Review
We searched the PubMed database for the terms “FUS,” “ALS,” and “FUS MUTATION.” Only studies that reported clinical data and the detailed mutation status were included.
Statistical Analysis
As clinical data of FUS carriers have not been uniformly collected and reported in the literature, patients from our cohort and the published case series were first analyzed separately and then confronted to validate the observations.
The association between age at disease onset and a mutation was evaluated with analysis of variance with the Tukey post hoc test. Kaplan-Meier survival curves followed by the log-rank test were used to evaluate the survival of different groups from disease onset. Univariate Cox regression was applied to derive unadjusted hazard ratios (HRs) for death/tracheostomy. Multivariate Cox regression models were used to estimate the covariate-adjusted risk of death/tracheostomy (from onset) and to account for the age at onset—mutations interaction. As the role of variants outside of the C-terminal (NLS) domain of FUS is unclear and they are not consistently assessed during genetic screening, they were treated separately (Supplementary Results). All analyses were performed using R studio (version 3.6.0).
Standard Protocol Approvals, Registrations, and Patient Consents
All patients participating in the study gave written informed consent. Ethical approval was obtained from the medical ethical review board of the A.O.U Città della Salute e della Scienza di Torino, Italy.
Data Availability
Data are available from the authors upon reasonable request by interested researchers.
Results
Case Series
We evaluated the FUS gene in 2,615 Italian ALS patients (1,450 [55.4%] males, median age at onset = 65.8 years, interquartile range [IQR] 57.9–71.8, 293 [11.2%] familial ALS) collected over 14 years.
Among those, 233 (8.9%) patients carried the C9orf72 repeated expansion, 90 (3.4%) pathogenic mutations in SOD1, and 55 (2.1%) in TARDBP. We identified 22 ALS cases carrying a pathogenic FUS mutation (12 [54.5%] males, median diagnostic delay [months] = 7, IQR 4–11, 2 [0.9%] bulbar onset), representing 0.84% of the present series (0.45% of sporadic ALS cases and 6.1% of ALS cases with a family history of the disease in this Italian cohort). Notably, this frequency is higher than in our recent genome-wide mutational screening on ∼1,000 cases from an Italian population-based cohort (3/957),15 likely reflecting a referral bias. Five of these 22 ALS cases have been previously described.15,17-20 Variants outside of the C-terminal domain are reported in eTable 1 (links.lww.com/NXG/A537).
Age at Onset
The mean age at onset was 45.7 (SD 16.4, range 11–74) years (Figure 1), which was significantly younger than the rest of our cohort (difference in mean age at onset: −20.25 years, p = 5.56 × 10−16; CI −20.25 to −25.07). Of note, when comparing the major ALS mutations (C9orf72, SOD1, TARDBP, and FUS), FUS was characterized by the earliest age at disease onset (Figure 1).
Figure 1. Cumulative Distribution Plot of Age at Onset in Our Cohort.
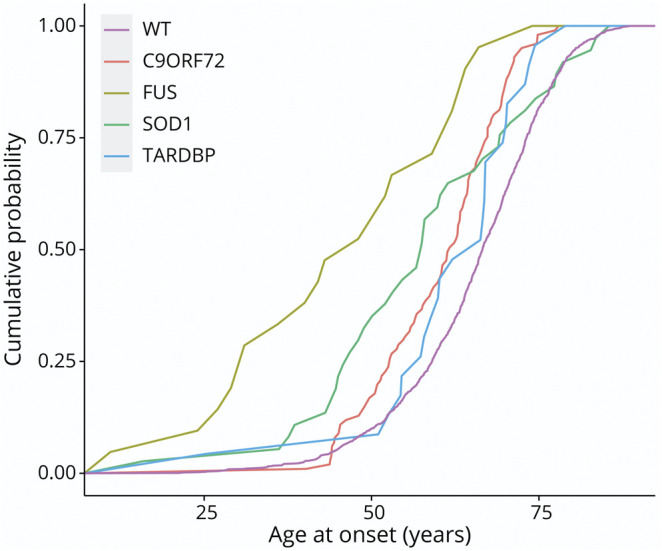
Wild type (WT) median age at onset 68.8 years (IQR 61.7–74.8), C9orf72 median age at onset 62.9 years (IQR 54.0–67.8), SOD1 median age at onset 66.6 years (IQR 46.8–77.23), TARDBP median age at onset 66.6 years (58.8–72.9), and FUS median age at onset 48.00 years (IQR 31.0–62.0). FUS = fused in sarcoma.
Survival Analysis
FUS mutations are associated with a poor prognosis (difference in median survival: 7.5 months, log-rank p-value = 0.009). Unexpectedly, we found strong age dependence: The interaction of FUS mutation and patients' age is a relevant predictor of patient survival (p = 0.0001). FUS patients with older ages at onset (older than 45.7 years) did not differ in disease duration from the rest of the cohort (p = 0.437 [95% CI 0.65–2.65]), whereas FUS cases with younger age at onset (younger than 45.7 years) had dramatically shorter disease durations (HR = 5.92 [2.57–13.62], p = 0.000003) (Figure 2A). To correct for possible confounders, we performed a multivariate analysis with the relevant covariates (including the type of mutation and site of disease onset), obtaining the same results (eResults, links.lww.com/NXG/A537).
Figure 2. Survival Analyses of FUS Carriers.

(A) Survival analysis of early-onset FUS carriers (age at onset younger than 46 years, left) and mid-to-late onset FUS (older than 46 years, right) vs nonmutated ALS cases. Early-onset FUS median survival 82.7 weeks (IQR 47.9–95.9) and non-FUS early-onset ALS median survival 204.7 weeks (IQR 126.1–274.0); log-rank p-value = 0.000006. Mid-to-late onset FUS median survival 126.1 (IQR 86.7–195.6) and non-FUS mid-to-late onset ALS median survival 121.9 (78.1–195.6); log-rank p-value = 0.2 (B) Survival analysis of early-onset FUS carriers (age at onset <46 years) vs mid-to-late onset FUS (>46 years). ALS = amyotrophic lateral sclerosis; FUS = fused in sarcoma.
Clinical Features
Clinical onset was most commonly in the upper cervical spinal cord, with neck and shoulder girdle weakness as the initial symptoms (n = 10, 45.5% of the FUS mutation carriers). Lower motor neuron (LMN) involvement was clinically evident in all cases while upper motor neuron signs were detected in 81.8% (18/22) of cases: The remaining cases (4/22, 18.2%) had a pure lower motor neuron syndrome (progressive muscular atrophy). None of the patients had evidence of severe cognitive impairment on extensive neuropsychological testing, except for our 2 young cases who manifested mild cognitive impairment and a learning disability. Three cases had a history of postural tremors that predated the onset of motor neuron symptoms (Table 1).
Table 1.
Summary of Genetic and Clinical Data of Patients Carrying FUS Mutations in Our Cohort

Genotype-Phenotype Correlations
The analysis of the clinical phenotypes of the 22 patients revealed 3 genotype groups that showed markedly different clinical features. Carriers of a truncating mutation or a missense variant in the last 2 amino acids of the NLS domain presented with the most aggressive phenotypes, and they were the only FUS carriers in our cohort with a bulbar onset. We also observed an expanded phenotypic spectrum in this group that included abnormal cognitive functions, namely mild intellectual or learning disabilities preceding the onset of motor symptoms. In contrast, carriers of a missense variant in the 517 and 519 residues had the mildest disease of all FUS carriers, with slowly progressing flail leg phenotypes. Finally, other missense variants in the C-terminal domain usually manifested with a proximal upper limb involvement with predominant LMN phenotype: The disease severity associated with these mutations was more variable than the other 2 groups and largely correlated with age at disease onset.
Literature Review
We collected 289 additional published FUS‐ALS cases for whom clinical and genetic information was available: Nonsynonymous mutations affected the C-terminal domain in 253 of them (87.5%). Missense mutations constitute most (75.1%, 190/253) of those cases (eTable 2, links.lww.com/NXG/A537).
Age at Onset
Among published FUS cases the mean age of onset was 37.0 years (SD 15.9, range 11–80), some 8 years younger than observed in our Italian cohort. The observed difference may mirror the different distribution of the amino acid change in the 2 cohorts but can also be related to publication bias. Furthermore, it could not be ruled out that FUS had been prevalently screened in early-onset ALS cases.
Survival Analysis
The age at onset, survival correlation, we identified in our FUS cases was also observed across FUS-ALS cases from the literature review: When dividing patients based on the median age at onset, patients with a later age at onset (age at onset older than 45.7 years) had a considerably (p = 0.00003) longer disease duration (median survival 29.0 months, IQR 20.0–48.0) than younger patients (median survival 16.0 months, IQR 11.0–27.0, Figure 2B). Younger age remained the most important predictor of a poorer prognosis when mutation type and site of disease onset were included in the model (p = 0.00102, OR 1.55, CI 1.19–2.01) (eResults, links.lww.com/NXG/A537). However, further analysis of the literature data revealed that survival varies dramatically among different missense mutations, with mutation occurring in the 510 amino acid having a more prolonged survival. Of interest, survival was also different for substituting amino acid in the same residue. Among the mutations in position 521, a significantly longer median survival is observed in the p. R521H variant (p = 0.0007) (eResults).
Clinical Features
Because of the different nature of the 2 cohorts, we could not compare the same clinical features across the 2 cohorts. However, spinal disease onset was reported in most of the FUS-mutated patients (209/253, 82.6%). It should be noted that among literature cases with a juvenile-onset (younger than 25 years), almost half of patients (47.4%, 18/38) had either a mild intellectual disability or a learning impairment.
Genotype-Phenotypes Correlation
The 3 genotype-phenotype clusters from our cohort were also identified from literature cases. Truncating mutations and missense variants in the last amino acids had young-onset (younger than 30 years) and a severe disease course (survival <1 year). Bulbar onset was more frequent in this mutation group (36%, compared with 10.5% of other C-terminal mutation carriers). Among literature cases, almost half of patients with a juvenile-onset (47.4%, 18/38) had a mild intellectual retardation or a learning disability, and 33.3% (4/12) had upper limbs tremor. Conversely, some mutations (the p.Q519E and to a less extent, the p.K510R and p.R521H variants) had a slower progressing disease with prolonged survival. The remaining majority of FUS mutations typically presented with symmetric weakness of proximal limb and neck flexor/extensors consistent with the classical adult ALS-FUS phenotype.
Phenotype Clusters of FUS Mutations
Based on the joint data of our series and previously published ALS-FUS cases, we identified 3 genotype-phenotype clusters (Figure 3):
Axial ALS-FUS. This is the classic ALS-FUS phenotype (40.7%, 114/280), with neck or proximal upper limbs weakness as the first symptom. The presence of dropped-head in the early stages of the disease should be considered highly suspicious for FUS mutations despite age at disease onset. Cognitive impairment is rare in this group. This clinical course was observed chiefly with missense variants in the NLS and is the typical presentation of substitutions occurring at the 514 and 521 amino acids. However, it is worth noting that based on literature data adult-onset FUS-ALS could also present with unspecified weakness in lower limbs (13.2%, 37/280). Bulbar onset is less frequent (18/280) and usually reflects a disruptive mutation.
Benign ALS-FUS. Specific missense variants in the NLS (p.K510R, p.H517Y, p.Q519E, and p.R521H) lead to a milder disease course, with onset in late-adult life, slow progression, and prolonged survival. This is the rarest (7.9%, 22/280) FUS-related phenotype.
Juvenile ALS-FUS. ALS-FUS in childhood or early adulthood may manifest with bulbar, proximal upper limb, or distal limbs onset and rapid progression to respiratory failure. Intellectual disability or learning impairment are standard features of young-onset ALS and may precede motor neuron symptoms by many years. Coexistent tremor is also possible. This aggressive ALS phenotype is caused by either a truncating mutation or missense variants in the last amino acidic residues (p.P525L and p.Y526L). About one quarter (24.6%, 69/280) of FUS cases have this juvenile aggressive phenotype.
Figure 3. Patterns of Motor and Nonmotor Involvement in FUS-ALS and Genotype-Phenotype Correlation of FUS Mutations.

FUS = fused in sarcoma; NLS = nuclear localization signal.
Of interest the aforementioned phenotypes were not observed in ALS patients who carried FUS variants outside the C-terminal (eResults, links.lww.com/NXG/A537).
Discussion
We have described the clinical features of an Italian cohort of ALS patients carrying FUS mutations and compared our findings with the published literature. Overall, clinical features agreed with those previously reported in smaller series. Although the FUS-related phenotype was initially thought to be homogenous, characterized by early onset and a relatively short disease duration,21 it has become increasingly clear that some FUS‐ALS patients also develop the disease at an older age.22 Truncating mutations and some missense variants (most notably, the p.P525L) were consistently associated with an earlier onset and an aggressive form of the disease.23,24 This phenotypic variability is more pronounced across our cohort; we detected an unexpectedly high rate (13.6%) of slow-progressing FUS cases. We speculate that the lower frequency (4.3%) of this phenotype observed in the literature arises from the FUS screening primarily performed in young-onset ALS.
We found that the prognosis in FUS-ALS strongly correlated with age at disease onset. More specifically, only individuals with a younger age at onset had an aggressive disease with a poor prognosis, and the survival of late-adulthood FUS cases did not differ from the other sporadic ALS patients. This age-survival relationship is FUS-specific25 and contrasts with other ALS cases and ALS-related mutations, where earlier ages at onset are usually associated with a milder phenotype and longer disease durations.
Most ALS-FUS cases manifest with a dropped-head or proximal upper limbs weakness; bulbar onset is more typical of juvenile ALS-FUS and the mutations associated with it (truncating mutation or missense variants in the last amino acidic residues). It is worth noting that juvenile FUS patients had a worse prognosis than adult onset FUS regardless of the site of symptoms onset and despite predominant axial or neck weakness being known features associated with rapid progression to respiratory failure.26
Conversely, few FUS missense mutations (p.K510R, p.H517Y, p.Q519E, and p.R521H) had a slowly progressive disease with either a flail leg or a pure lower motor neuron (progressive muscular atrophy) presentation.
We additionally report that the juvenile, more aggressive ALS-FUS had a coexistent extra-motor involvement (learning and intellectual disabilities or tremor) that appears years before the onset of motor symptoms.
Although the lack of biological data prevents us from understanding the molecular basis of those genotype-phenotype clusters, it could be postulated that the different severity of the 3 phenotypes mirrors the pathologic changes associated with different FUS mutations.27,28 However, the FUS mutation is not exclusively responsible for the phenotype, as a marked phenotypic variability exists among patients carrying the same variant and often belonging to the same pedigree. For example, the parent of our juvenile-onset p.R495* case carried the same mutation but was asymptomatic at age 47 years.
Our results have several clinical implications, especially in the light of targeted therapies under development for FUS.12,29 The age-dependent effect of FUS mutations on survival yields a primary clinical value in both genetic counseling and trial design. Indeed, other than dramatically different prognoses, FUS-related subtypes are likely to have different therapeutic windows of opportunity before disease spreading occurs. The different ages of onset of FUS-ALS subtypes should also be accounted for appropriate timing of potential presymptomatic treatments. In addition, extra-motor features of FUS mutations should be routinely investigated and integrated into clinical practice and research settings to optimize genetic counseling and trial outcomes.
The description we provided here of FUS-related phenotypes is also relevant to interpreting FUS mutations outside the NLS. Because non-NLS FUS variants showed diverse and less specific phenotypes, it is likely that they are not pathogenic or had a different contribution to motor neuron degeneration.
Finally, the highly specific phenotype and natural history of patients with FUS mutations suggest that FUS-ALS is a unique subtype of motor neuron disorder. This gene-specific feature deserves further inquiry given the close similarity of FUS with other ALS-related proteins such as TARDBP. Nevertheless, our findings have important implications regarding the pathogenic mechanism of FUS-related neurodegeneration and require a more detailed investigation.
Acknowledgment
This work was supported by the Italian Ministry of Health (Ministero della Salute, Ricerca Sanitaria Finalizzata, grant RF-2016-02362405), the Progetti di Rilevante Interesse Nazionale program of the Ministry of Education, University and Research (grant 2017SNW5MB), the European Commission's Health Seventh Framework Programme (FP7/2007–2013 under grant agreement 259867), and the Joint Programme–Neurodegenerative Disease Research (Strength, ALS-Care and Brain-Mend projects), granted by Italian Ministry of Education, University and Research. This study was performed under the Department of Excellence grant of the Italian Ministry of Education, University and Research to the “Rita Levi Montalcini” Department of Neuroscience, University of Torino, Italy. This work was also supported by the Intramural Research Program of the NIH, National Institute on Aging (Z01-AG000949-02) and by the National Institute of Neurological Disorders and Stroke (1ZIANS003154). The sponsor organizations had no role in data collection or analysis and did not participate in writing or approving the manuscript. The Authors thank the Laboratory of Neurogenetics (NIH) staff for their collegial support and technical assistance.
Glossary
- ALS
amyotrophic lateral sclerosis
- FUS
fused in sarcoma
- HR
hazard ratio
- IQR
interquartile range
- LMN
lower motor neuron
- NLS
nuclear localization signal
Appendix. Authors
Contributor Information
Giorgia Brodini, Email: giorgia.brodini@edu.unito.it.
Giovanni De Marco, Email: giovanni.demarco@unito.it.
Federico Casale, Email: federico.casale@unito.it.
Giuseppe Fuda, Email: giuseppe.fuda@unito.it.
Paolina Salamone, Email: paolina.salamone@unito.it.
Maura Brunetti, Email: maura.brunetti@unito.it.
Luca Sbaiz, Email: lsbaiz@cittadellasalute.to.it.
Salvatore Gallone, Email: salvatore.gallone@unito.it.
Alessandro Bombaci, Email: alessandro.bombaci@unito.it.
Rosario Vasta, Email: rosario.vasta@unito.it.
Umberto Manera, Email: umberto.manera@unito.it.
Antonio Canosa, Email: antonio.canosa@unito.it.
Cristina Moglia, Email: cristina.moglia@unito.it.
Andrea Calvo, Email: PhDandrea.calvo@unito.it.
Bryan J. Traynor, Email: traynorb@mail.nih.gov.
Adriano Chio, Email: adriano.chio@unito.it.
Study Funding
Italian Ministry of Health (Ricerca Sanitaria Finalizzata, grant RF-2016-02362405), Italian Ministry of Education, University and Research (Progetti di Rilevante Interesse Nazionale program, grant 2017SNW5MB), European Commission's Health Seventh Framework Programme (FP7/2007–2013 under grant agreement 259867), Italian Ministry of Education, University and Research, Joint Programme–Neurodegenerative Disease Research (Strength, ALS-Care and Brain-Mend projects), National Institute on Aging, Intramural Research Program of the NIH (Z01-AG000949-02), NINDS (1ZIANS003154).
Disclosure
A. Calvo has received research grant from Cytokinetics; B. J. Traynor holds European, Canadian, and American patents on the clinical testing and therapeutic intervention for the hexanucleotide repeat expansion of C9orf72; A. Chio serves on scientific advisory boards for Mitsubishi Tanabe, Roche, and Cytokinetics and has received a research grant from Italfarmaco. The remaining authors report no disclosures relevant to the manuscript. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Kwiatkowski TJ, Bosco DA, LeClerc AL, et al. . Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205-1208. [DOI] [PubMed] [Google Scholar]
- 2.Vance C, Rogelj B, Hortobágyi T, et al. . Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shelkovnikova TA, Peters OM, Deykin AV, et al. . Fused in sarcoma (FUS) protein lacking nuclear localization signal (NLS) and major RNA binding motifs triggers proteinopathy and severe motor phenotype in transgenic mice. J Biol Chem. 2013;288(35):25266-25274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gal J, Zhang J, Kwinter DM, et al. . Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol Aging. 2011;32(12):2323.e27-2323.e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2017;88(7):540-549. [DOI] [PubMed] [Google Scholar]
- 6.Gromicho M, Oliveira Santos M, Pinto A, Pronto-Laborinho A, De Carvalho M. Young-onset rapidly progressive ALS associated with heterozygous FUS mutation. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(5-6):451-453. [DOI] [PubMed] [Google Scholar]
- 7.Liu ZJ, Lin HX, Liu GL, et al. . The investigation of genetic and clinical features in Chinese patients with juvenile amyotrophic lateral sclerosis. Clin Genet. 2017;92(3):267-273. [DOI] [PubMed] [Google Scholar]
- 8.Zou ZY, Liu MS, Li XG, Cui LY. Mutations in FUS are the most frequent genetic cause in juvenile sporadic ALS patients of Chinese origin. Amyotroph Lateral Scler Frontotemporal Degener. 2016;17(3-4):249-252. [DOI] [PubMed] [Google Scholar]
- 9.Chen L. FUS mutation is probably the most common pathogenic gene for JALS, especially sporadic JALS. Revue Neurol. 2021;177(4):333-340. [DOI] [PubMed] [Google Scholar]
- 10.Amado DA, Davidson BL. Gene therapy for ALS: a review. Mol Ther. 2021;29(12):3345-3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ito D. Promise of nucleic acid therapeutics for amyotrophic lateral sclerosis. Ann Neurol. 2022;91(1):13-20. [DOI] [PubMed] [Google Scholar]
- 12.Korobeynikov VA, Lyashchenko AK, Blanco-Redondo B, Jafar-Nejad P, Shneider NA. Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat Med. 2022;28(1):104-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293-299. [DOI] [PubMed] [Google Scholar]
- 14.Chiò A, Moglia C, Canosa A, et al. . Cognitive impairment across ALS clinical stages in a population-based cohort. Neurology. 2019;93(10):e984-e994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiò A, Calvo A, Mazzini L, et al. . Extensive genetics of ALS: a population-based study in Italy. Neurology. 2012;79(19):1983-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grassano M, Calvo A, Moglia C, et al. . Mutational analysis of known ALS genes in an Italian population-based cohort. Neurology. 2020;96(4):e600-e609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiò A, Restagno G, Brunetti M, et al. . Two Italian kindreds with familial amyotrophic lateral sclerosis due to FUS mutation. Neurobiol Aging. 2009;30(8):1272-1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moglia C, Calvo A, Brunetti M, Chiò A, Grassano M. Broadening the clinical spectrum of FUS mutations: a case with monomelic amyotrophy with a late progression to amyotrophic lateral sclerosis. Neurol Sci. 2021;42(3):1207-1209. [DOI] [PubMed] [Google Scholar]
- 19.Lanteri P, Meola I, Canosa A, et al. . The heterozygous deletion c.1509_1510delAG in exon 14 of FUS causes an aggressive childhood-onset ALS with cognitive impairment. Neurobiol Aging. 2021;103:130.e1-130.e7. [DOI] [PubMed] [Google Scholar]
- 20.Canosa A, Lomartire A, De Marco G, et al. . A novel splice site FUS mutation in a familial ALS case: effects on protein expression. Amyotroph Lateral Scler Frontotemporal Degener. 2021;23(1-2):128-136. [DOI] [PubMed] [Google Scholar]
- 21.Akiyama T, Warita H, Kato M, et al. . Genotype-phenotype relationships in familial amyotrophic lateral sclerosis with FUS/TLS mutations in Japan: FUS/TLS linked FALS in Japan. Muscle Nerve. 2016;54(3):398-404. [DOI] [PubMed] [Google Scholar]
- 22.Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat. 2013;34(6):812-826. [DOI] [PubMed] [Google Scholar]
- 23.Conte A, Lattante S, Zollino M, et al. . P525L FUS mutation is consistently associated with a severe form of juvenile Amyotrophic Lateral Sclerosis. Neuromuscul Disord. 2012;22(1):73-75. [DOI] [PubMed] [Google Scholar]
- 24.Zhou B, Wang H, Cai Y, et al. . FUS P525L mutation causing amyotrophic lateral sclerosis and movement disorders. Brain Behav 2020;10(6):e01625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Naumann M, Peikert K, Günther R, et al. . Phenotypes and malignancy risk of different FUS mutations in genetic amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2019;6(12):2384-2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pinto S, Gromicho M, Swash M, deCarvalho M. Cervical muscle weakness is a marker of respiratory dysfunction in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2020;91(3):323-324. [DOI] [PubMed] [Google Scholar]
- 27.Lenzi J, De Santis R, de Turris V, et al. . ALS mutant FUS proteins are recruited into stress granules in induced Pluripotent Stem Cells (iPSCs) derived motoneurons. Dis Models Mech. 2015;8(7):755-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lim SM, Choi WJ, Oh KW, et al. . Directly converted patient-specific induced neurons mirror the neuropathology of FUS with disrupted nuclear localization in amyotrophic lateral sclerosis. Mol Neurodegener. 2016;11(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arnold C. Tailored treatment for ALS poised to move ahead. Nat Med. May 30, 2019. doi: 10.1038/d41591-019-00013-w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available from the authors upon reasonable request by interested researchers.