

Abstract
Background:
GM1 gangliosidosis is an autosomal recessive lysosomal storage disease caused by biallelic mutations in the GLB1 gene. Neurodegeneration, hypotonia, visceromegaly, macular cherry-red spots, skeletal dysplasia, and coarse and dysmorphic face are the major clinical features.
Aims:
To evaluate the demographic and clinical data of patients with GM1 gangliosidosis in a single center.
Study Design:
A retrospective clinical study.
Methods:
This study included patients followed at Hacettepe University İhsan Doğramacı Children’s Hospital Pediatric Metabolism Unit with the diagnosis of GM1 gangliosidosis between 1988 and 2021. Hospital records of the patients were reviewed for demographic, clinical, and laboratory findings.
Results:
Fourteen patients were included in the study and 10 (71.4%) were male. The age at onset of clinical symptoms was between 0 and 5 months, and the median time to diagnosis after the first symptom was 4.3 (0-13) months. Motor delay (54%) was the most common initial symptom. The median follow-up period was 14.8 (0.4-92.2) months. Twelve patients (85.7%) died, and all deaths occurred before the age of 24 months. The median survival was 21.3 (95% confidence interval, 15.5-24.9) months. Higher leukocyte beta-galactosidase activity correlated with later age at onset (ρ = 0.575), later age at diagnosis (ρ = 0.618), and longer diagnostic delay (ρ = 0.702) (ρ < 0.05).
Conclusion:
Median survival in patients with GM1 gangliosidosis is less than 24 months. Beta-galactosidase enzyme activity may be associated with clinical onset and time of diagnosis in these patients.
INTRODUCTION
GM1 gangliosidosis is an autosomal recessive lysosomal storage disease characterized by the accumulation of gangliosides, oligosaccharides, keratan sulfate, and their derivatives.1 It results from biallelic pathogenic variants in the GLB1 gene in the chromosome 3p21.33 locus that results in insufficient activity of the lysosomal enzyme β-galactosidase. Although the disease is characterized by the pathological accumulation of GM1 gangliosides in the lysosomes of visceral and connective tissue cells, the accumulation is most prominent in neurons and therefore in the brain.2,3 In addition, keratan sulfate, a glycosaminoglycan, accumulates in the liver and is excreted in the urine.4 The estimated incidence of GM1 gangliosidosis is 1 in 100,000-200,000 live births.1
The clinical findings of the infantile form (type 1) of the disease may be recognized even in the neonatal period by edema (non-immune hydrops fetalis), hepatosplenomegaly, and skin rashes (angiokeratomas). Patients with the infantile form usually present with developmental delay accompanied by tonic-clonic seizures and progressive psychomotor retardation in the first 6 months of life. Clinical signs are variable and include neurodegeneration, hypotonia, visceromegaly, macular cherry-red spots (in 50% of patients), skeletal dysplasia (dysostosis multiplex similar to mucopolysaccharidoses, including anterior beaking of vertebrae, enlargement of the sella turcica and calvarium, etc.), and coarse and dysmorphic facial features, including low-set ears, frontal bossing, depressed nasal bridge, and long philtrum.5 Cardiac involvement (hypertrophic cardiomyopathy) has been reported in patients with certain genotypes.6 Patients usually experience vision and hearing loss around the age of 1 year, have severe neurological disorders accompanied by decerebration rigidity, and often die around the age of 3-4 years.7,8,9,10 While the signs of developmental delay, regression, and involvement of peripheral tissues are prominent in infancy, peripheral findings are more obscure later in life, and the diagnosis of isolated neurological disease may be difficult.11 This study aimed to describe the clinical and biochemical features of patients with GM1 gangliosidosis diagnosed at a single center and determine possible factors associated with clinical presentation.
MATERIALS AND METHODS
Study design and participants
This single center retrospective cross-sectional study was conducted at Hacettepe University İhsan Doğramacı Children’s Hospital Pediatric Metabolism Unit. The study was approved by Hacettepe University Ethics Committee (no. 2021/21-21) on December 21, 2021. Patients followed up at our center with the diagnosis of GM1 gangliosidosis between 1988 and 2021 were included. Hospital records of the patients were reviewed for demographic, clinical, and laboratory findings. Fourteen patients diagnosed with GM1 gangliosidosis were included in the study.
Diagnosis of GM1 Gangliosidosis
The diagnosis was based on the blood leukocyte enzyme activity analyse. The enzyme activity of 10 patients was determined in our center, and the enzyme activity of the other three patients was determined in different centers by the same enzyme purification and fluorimetric methods. The enzyme activity of one patient was low, but it was not noted as a numerical data. Therefore, the enzyme level of this patient was not included in the statistical analysis. Patients with leukocyte beta-galactosidase enzyme levels below 50 nmol/h/mg protein level or biallelic pathogenic mutations in the GLB1 gene were diagnosed with GM1 gangliosidosis.
Statistical Analysis
IBM SPSS Statistics for Windows, version 22.0 (IBM Corp., Armonk, NY, USA) was used for statistical analysis. In the descriptive statistics section, categorical variables were presented as numbers and percentages, and numerous variable as mean ± standard deviation and median (minimum-maximum value). The Mann-Whitney U test was used for comparison analyses between the two groups. The relationships between some numerical variables and beta-galactosidase enzyme activity were evaluated by Spearman correlation analysis. The overall survival time and rates of the patients were calculated using Kaplan-Meier analysis. The differences between survival times according to some factors were examined with the log rank test. The significance level was accepted as p < 0.05.
RESULTS
Fourteen patients, all from different families, were diagnosed with GM1 gangliosidosis at our clinic during the study period, 10 (71.4%) of whom were male. The age at onset of clinical symptoms was between 0 and 5 months, and the median age at diagnosis was 6 (0-16) months. The median time to diagnosis after the first symptom was 4.3 (0-13) months. The median follow-up period was 14.8 (0.4-92.2) months. During follow-up, 12 (85.7%) patients died. Parental consanguinity was present in eight patients, and two patients had another family member diagnosed with GM1 gangliosidosis (Table 1).
Table 1. Patient Characteristics.
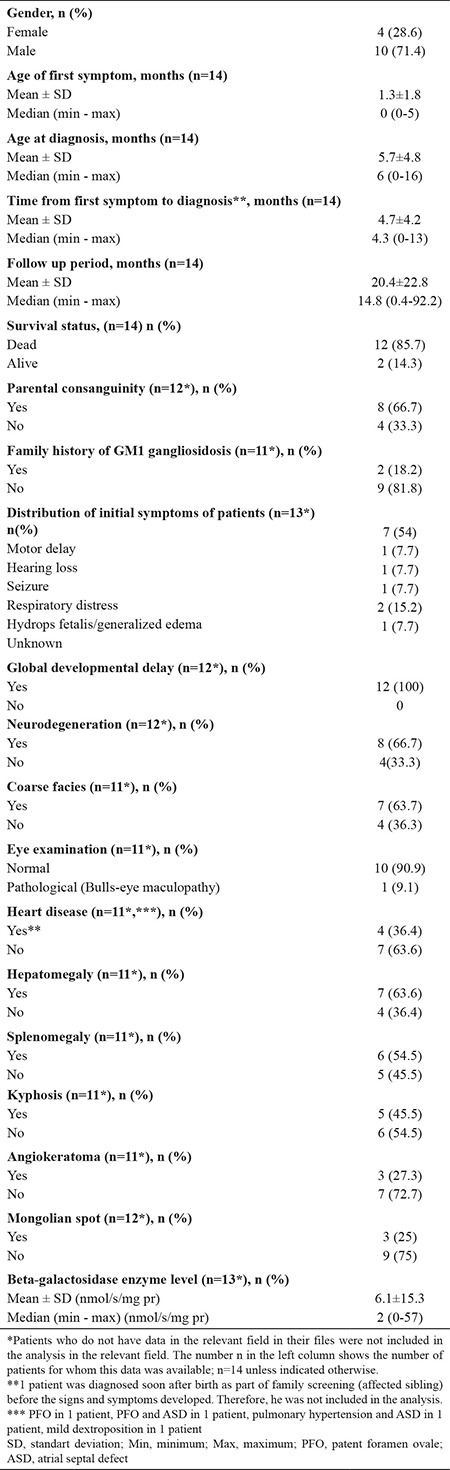
Motor delay (54%) was the most common initial symptom. One patient diagnosed presymptomatically in the newborn period (see text) was not included in this analysis (N = 13).
Clinical findings are also summarized in Table 1. Motor delay was the initial symptom in 54% of the patients. Eye examination was abnormal only in one patient (bulls-eye maculopathy), and cherry-red spot, cardiomyopathy, or scoliosis was not observed in any of the patients. GLB1 (NM_001317040.1) gene sequencing was performed in two patients, in whom homozygous mutations were detected, one by whole-exome sequencing (c.134C > T; p.Pro45Leu) and the other by targeted Sanger sequencing (c.176G > A; p.Arg59His).
Higher leukocyte beta-galactosidase activities correlated with later age at onset, later age at diagnosis, and longer diagnostic delay (Table 2). Similarly, the enzyme activity was significantly lower in patients diagnosed before 6 months of age (p = 0.042) (Table 3).
Table 2. Variables Related to Leukocyte Beta-galactosidase Enzyme Activity (N=13) *.

Table 3. Association of Beta Galactosidase Enzyme Activity with Selected Patient Characteristics (N=13)*.
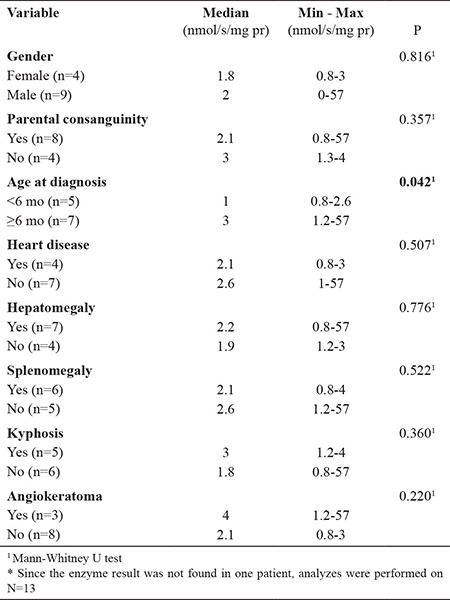
When the beta-galactosidase activity was compared between the groups according to sex, parental consanguinity, presence of heart disease, hepatomegaly, splenomegaly, kyphosis and angiokeratoma, no significant differences were found (p = 0.816, p = 0.317, p = 0.507, p = 0.776, p = 0.522, p = 0.360 and p = 0.220, respectively) (Table 3).
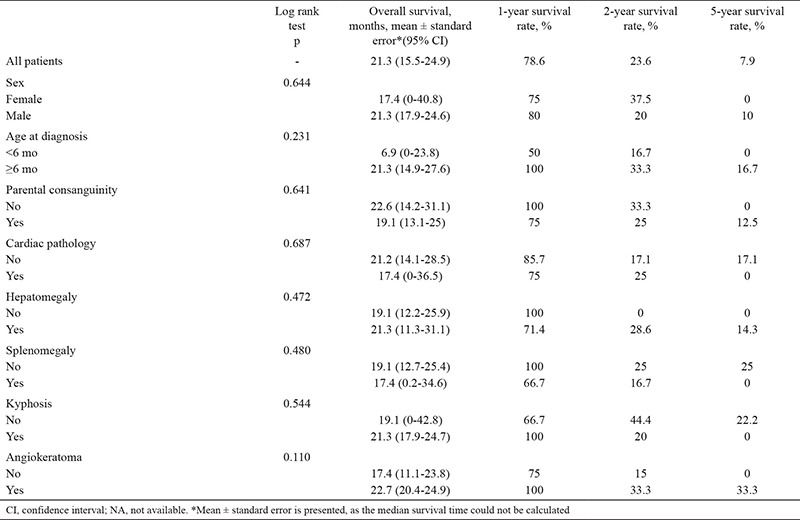
In this study, the median survival of patients with GM1 gangliosidosis was 21.3 (95% confidence interval: 15.5-24.9) months. The 1-, 2-, and 5-year survival rates were 78.6%, 23.6%, and 7.9%, respectively (Figure 1). Survival did not differ significantly according to sex, parental consanguinity, age at diagnosis, presence of heart disease, hepatomegaly, splenomegaly, kyphosis, or angiokeratoma (log rank test, p > 0.05) (Table 4).
Figure 1.
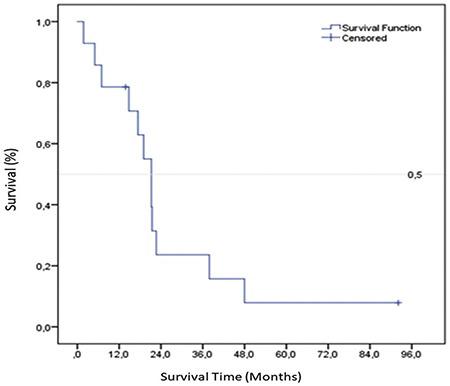
Kaplan-Meier plot of the overall survival rate
Table 4. Evaluation of Survival Rates and Log Rank Test Results (n = 13).
At the time of writing, two patients are still alive. The first is an 18-month-old girl born at term to consanguineous parents (first cousins). Axial hypotonia was noted at the age of 5 months, followed by loss of motor skills and onset of limb hypertonia and brisk deep tendon reflexes. She was diagnosed at the age of 5 months. Beta-galactosidase activity was 1.6 nmol/h/mg protein (normal: >50). She is currently being followed up with motor losses and swallowing dysfunction. The result of the genetic study of the patient is awaited.
The second surviving patient is a 7-year-old boy who presented with developmental delay and refractory epilepsy. His medical history revealed refractory seizures since at the age of 30 months, and he had begun to fall frequently and walk unsteadily since the age of 36 months. Although he can form sentences of 3-4 words, follow commands, and climb stairs, and get dressed and undressed without support, at the age of 7, he needed support climbing stairs and could not dress properly by himself. Physical examination is significant for scrotal angiokeratomas, wide-based gait, limb hypertonia, hyperactive deep tendon reflexes, ataxia, dysmetria, intentional tremor (more prominently in the right hand), and instantaneous backward tone loss in the whole body. Leukocyte beta-galactosidase enzyme activity level was 10.31 nmol/h/mg/protein (reference: 107.3 ± 35.8).
DISCUSSION
The infantile form of GM1 gangliosidosis is a severe clinical entity known to be associated with infant mortality in most patients. However, juvenile and adult-onset subtypes have also been described. In our single center study, information was presented on 14 patients with GM1 gangliosidosis, two of whom are still alive. Although the number of patients is quite high for such a rare disease from a single center, the number may be considered insufficient for statistical analyses.
Turkey has quite a high rate of consanguineous marriage (24%).12 Therefore, autosomal recessive diseases are expected to be seen more frequently.13 In the study by Ozkara and Topçu14, evaluating 300 patients with sphingolipidosis in Turkey, seven patients with GM1 gangliosidosis had been diagnosed between 1997 and 2002, and the incidence of the disease was reported as 1/185,714. Case reports from different centers in our country have been published, with central nervous system involvement as the prominent finding, in accordance with other published studies.15,16 Mongolian spot was reported in one case. In our case series, three patients had Mongolian spot.16
Published literature on GM1 gangliosidosis consists mostly of case reports and case series similar to ours; however, Lang et al.’s7 meta-analysis published in 2020 is noteworthy in that it includes 44 studies and 154 patients, which indicates that delays in crawling, standing, and walking (motor milestones) are among the clinically distinctive features of the disease. In our study, the first finding of the patients was motor delay, with a rate of 50%, and this result is compatible with the literature.7,17,18 In the published literature, significant systemic and neurological symptoms generally appeared before the age of 18 months.7 In this published cohort, symptom onset was usually before the age of 3 months, and first hospital admission and diagnosis were before the age of 9 months. Specifically, 6-18 months of interval denotes a significant period for neurodevelopmental regression and multi-organ involvement. In our study, the median age at diagnosis was 6 months, which indicates that most diagnoses could be made before significant neurodegeneration. Many early motor milestones, such as head control, transferring objects, and sitting independently, are delayed and the attained motor functions may be lost by neurodegeneration; thus, monitoring the neurodevelopmental stages is important in the follow-up of healthy children.
In our study, the enzyme level at diagnosis was significantly lower in patients diagnosed before 6 months of age (p = 0.042). Our study showed that the lower the beta-galactosidase enzyme activity, the younger the age at first symptom and the age at diagnosis. The enzyme level did not change with other demographic and clinical data. When the factors affecting survival are evaluated in the literature, chest physiotherapy and feeding with a tube preventing aspiration pneumonia come to the fore. However, no study has presented the enzyme level at diagnosis and disease course.7,19 To the best of our knowledge, this is the first case series to suggest an association between the beta-galactosidase enzyme level and the onset of symptoms and timing of diagnosis in GM1 gangliosidosis.
In our study, the mean follow-up period was 20.4 months, with a median of 14.8 months. All patients who died before 24 months of life. In the literature, most of the deaths have been reported between 12 and 24 months.1,7,19 This result is compatible with the literature.
None of the patients in our study group received a disease-specific treatment option. Studies have claimed that miglustat may result in clinical improvement via substrate reduction.20,21,22 However, available data are limited. Studies on gene therapy with different methods, especially adenoviral vectors, continue in some centers.23 The finding that the disease does not have a specific treatment or cure option yet reveals the importance of prenatal diagnosis and genetic counseling. Although the possibility of prenatal diagnosis was possible in one of our patients because of his family history and consanguinity, the family did not use this option, and the disease recurred in the subsequent pregnancy, resulting in death from the same disease. This case is an example of the inadequate adoption of genetic counseling and prenatal or preimplantation diagnosis services in Turkey.24,25,26
The study has some limitations. The small number of patients with genetic confirmation of the diagnosis limited the results. Other limitations include missing data, small number of patients, and all enzyme studies, although are performed with the same method, are not conducted in the same center.
Our center registered information on 14 patients with GM1 gangliosidosis, two of whom are still alive. The results of this study suggest that the lower the enzyme level, the younger the age at initial symptoms and the age at diagnosis. Most patients had a severe clinical course, and all deaths occurred before the age of 24 months.
Footnotes
Ethics Committee Approval: The study was approved by Hacettepe University Ethics Committee (no. 2021/21-21) on December 21, 2021.
Data Sharing Statement: Data available on request from the authors. The data that support the findings of this study are available from the corresponding author.
Author Contributions: Concept – H.T.A.; Data Collection or Processing – A.B.K., G.G., K.Ç.; Analysis or Interpretation – Y.Y., H.S.S.;Literature Search – Y.Y., A.B.K., G.G., K.Ç., İ.E., A.D., A.T.; Writing – A.B.K., K.Ç., A.D., H.S.S., A.T.
Conflict of Interest: The authors have no conflicts of interest to declare.
Funding: The financial expenses have covered by the authors in this study.
References
- 1.Brunetti-Pierri N, Scaglia F. GM1 gangliosidosis: review of clinical, molecular, and therapeutic aspects. Mol Genet Metab. 2008;94:391–396. doi: 10.1016/j.ymgme.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 2.Arash-Kaps L, Komlosi K, Seegräber M, et al. The clinical and molecular spectrum of GM1 gangliosidosis. J Pediatr. 2019;215:152–157.e3. doi: 10.1016/j.jpeds.2019.08.016. [DOI] [PubMed] [Google Scholar]
- 3.Tonin R, Caciotti A, Procopio E, et al. Pre-diagnosing and managing patients with GM1 gangliosidosis and related disorders by the evaluation of GM1 ganglioside content. Sci Rep. 2019;9:17684. doi: 10.1038/s41598-019-53995-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lawrence R, Van Vleet JL, Mangini L, et al. Characterization of glycan substrates accumulating in GM1 Gangliosidosis. Mol Genet Metab Rep. 2019;21:100524. doi: 10.1016/j.ymgmr.2019.100524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sperb F, Vairo F, Burin M, Mayer FQ, Matte U, Giugliani R. Genotypic and phenotypic characterization of Brazilian patients with GM1 gangliosidosis. Gene. 2013;512:113–116. doi: 10.1016/j.gene.2012.09.106. [DOI] [PubMed] [Google Scholar]
- 6.Morrone A, Bardelli T, Donati MA, et al. beta-galactosidase gene mutations affecting the lysosomal enzyme and the elastin-binding protein in GM1-gangliosidosis patients with cardiac involvement. Hum Mutat. 2000;15:354–366. doi: 10.1002/(SICI)1098-1004(200004)15:4<354::AID-HUMU8>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 7.Lang FM, Korner P, Harnett M, Karunakara A, Tifft CJ. The natural history of Type 1 infantile GM1 gangliosidosis: A literature-based meta-analysis. Mol Genet Metab. 2020;129:228–235. doi: 10.1016/j.ymgme.2019.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tasso MJ, Martinez-Gutierrez A, Carrascosa C, Vazquez S, Tebar R. GM1- gangliosidosis presenting as nonimmune hydrops fetalis: a case report. J Perinat Med. 1996;24:445–449. doi: 10.1515/jpme.1996.24.5.445. [DOI] [PubMed] [Google Scholar]
- 9.Denis R, Wayenberg JL, Vermeulen M, et al. Hyperphosphatasemia in early diagnosed infantile GM1 gangliosidosis presenting as transient hydrops fetalis. Acta Clin Belg. 1996;51:320–327. doi: 10.1080/22953337.1996.11718526. [DOI] [PubMed] [Google Scholar]
- 10.Sinelli MT, Motta M, Cattarelli D, Cardone ML, Chirico G. Fetal hydrops in GM(1) gangliosidosis: a case report. Acta Paediatr. 2005;94:1847–1849. doi: 10.1111/j.1651-2227.2005.tb01867.x. [DOI] [PubMed] [Google Scholar]
- 11.Lee JS, Choi JM, Lee M, et al. Diagnostic challenge for the rare lysosomal storage disease: Late infantile GM1 gangliosidosis. Brain Dev. 2018;40:383–390. doi: 10.1016/j.braindev.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 12.Bilim N. 2018 Türkiye Nüfus ve Sağlık Araştırması Temel Bulgular . 2019. [Internet] http://www.openaccess.hacettepe.edu.tr:8080/xmlui/bitstream/handle/11655/23356/2018_TNSA_Ozet_Rapor.pdf?sequence=1&isAllowed=y.
- 13.Hiz Kurul S, Oktay Y, Töpf A, et al. High diagnostic rate of trio exome sequencing in consanguineous families with neurogenetic diseases. Brain. 2022;145:1507–1518. doi: 10.1093/brain/awab395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ozkara HA, Topçu M. Sphingolipidoses in Turkey. Brain Dev. 2004;26:363–366. doi: 10.1016/j.braindev.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 15.Kara DÖ, Şahpaz A. Pathological findings of central nervous system, two GM1 gangliosidosis autopsy cases. Turk J Pediatr. 2019;61:953–957. doi: 10.24953/turkjped.2019.06.020. [DOI] [PubMed] [Google Scholar]
- 16.Yüksel D, Öksüz Ş, Senbil N. Extensive mongolian spots associated with GM1 gangliosidosis. Çocuk Sağlığı ve Haslıkları Dergisi. 2007;50:189–192. [Google Scholar]
- 17.Regier, D.S., C.J. Tifft, and C.E. Rothermel, GLB1-Related Disorders, in GeneReviews(®), M.P. Adam, et al., Editors. 1993, University of Washington, Seattle. [Internet] [PubMed]
- 18.Jarnes Utz JR, Kim S, King K, et al. Infantile gangliosidoses: Mapping a timeline of clinical changes. Mol Genet Metab. 2017;121:170–179. doi: 10.1016/j.ymgme.2017.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jarnes Utz JR, Kim S, King K, et al. Infantile gangliosidoses: Mapping a timeline of clinical changes. Mol Genet Metab. 2017;121:170–179. doi: 10.1016/j.ymgme.2017.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deodato F, Procopio E, Rampazzo A, et al. The treatment of juvenile/adult GM1-gangliosidosis with Miglustat may reverse disease progression. Metab Brain Dis. 2017;32:1529–1536. doi: 10.1007/s11011-017-0044-y. [DOI] [PubMed] [Google Scholar]
- 21.Fischetto R, Palladino V, Mancardi MM, et al. Substrate reduction therapy with Miglustat in pediatric patients with GM1 type 2 gangliosidosis delays neurological involvement: A multicenter experience. Mol Genet Genomic Med. 2020;8:e1371. doi: 10.1002/mgg3.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garzone A, Pompilio A, Ciuccarelli F, et al. CPC-086 miglustat off-label in a paediatric formulation for a rare metabolic disease: early infantile GM1 gangliosidosis. Eur J Hosp Pharm. 2013;20:A1–A238. [Google Scholar]
- 23.Nicoli ER, Annunziata I, d’Azzo A, Platt FM, Tifft CJ, Stepien KM. GM1 Gangliosidosis-A Mini-Review. Front Genet. 2021;12:734878. doi: 10.3389/fgene.2021.734878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yakin K, Urman B, Balaban B. Dynamic view of assisted reproduction in Turkey from 1996 to 2020. Reprod BioMed Online. 2022;44:747–754. doi: 10.1016/j.rbmo.2021.12.010. [DOI] [PubMed] [Google Scholar]
- 25.Olgac A, Gulenc N, Kasapkara CS, Kilic M, Senel S, Tumer L. The intersection of two global problems: Refugees and inborn errors of metabolism. Annals of Medical Research. 2021;28:296–302. [Google Scholar]
- 26.Seven M, Paşalak Şİ, Sahin E, Akyuz A. Genetic Literacy of pregnant waonmd en their use of prenatal screening and diagnostic tests in Turke yJ. Genet Couns . 2019;28:578–586. doi: 10.1002/jgc4.1082. [DOI] [PubMed] [Google Scholar]