

Abstract
Remdesivir is an adenosine analogue that has a cyano substitution in the C1’ position of the ribosyl moiety and a modified base structure to stabilize the linkage of the base to the C1’ atom with its strong electron-withdrawing cyano group. Within the replication-transcription complex (RTC) of SARS-CoV-2, the RNA-dependent RNA polymerase nsp12 selects remdesivir monophosphate (RMP) over adenosine monophosphate (AMP) for nucleotide incorporation but noticeably slows primer extension after the added RMP of the RNA duplex product is translocated by three basepairs. Cryo-EM structures have been determined for the RTC with RMP at the nucleotide-insertion (i) site or at the i+1, i+2, or the i+3 sites after product translocation to provide a structural basis for a delayed-inhibition mechanism by remdesivir. In this study, we applied molecular dynamics (MD) simulations to extend the resolution of structures to the measurable maximum that is intrinsically limited by MD properties of these complexes. Our MD simulations provide (i) a structural basis for nucleotide selectivity of the incoming substrates of remdesivir triphosphate over adenosine triphosphate and of ribonucleotide over deoxyribonucleotide, (ii) new detailed information on hydrogen atoms involved in H-bonding interactions between the enzyme and remdesivir, and (iii) direct information on the catalytically active complex that is not easily captured by experimental methods. Our improved resolution of interatomic interactions at the nucleotide-binding pocket between remedesivir and the polymerase could help to design a new class of anti-SARS-CoV2 inhibitors.
Keywords: Molecular Dynamics (MD) simulations, variance of atomic motions, the highest theoretical resolution, MD-derived electrostatic potential maps, MD-ESP, MD-ED, PDF, nsp12 RNA polymerase
Graphical Abstract
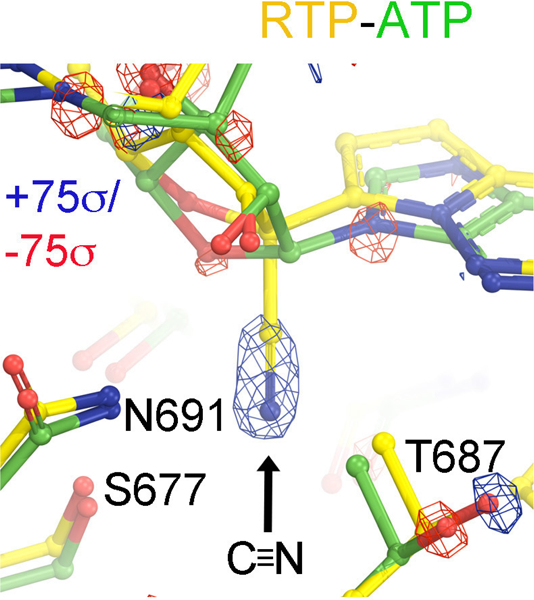
Difference ESP maps between MD-derived RTP and ATP complexes reveal the largest features corresponding to an insertion of the cyano group at the C1’ position, which is accompanied by a subtle displacement of the T687 sidechain.
INTRODUCTION
Remdesivir (RDV) is an adenosine analogue with a cyano group substitution at the C1’ position of the nucleotide base.1, 2 It has been used as a viral suppressor for treatment of SARS-CoV-2 infections although it shows only modest efficacy.3–5 Nonetheless, there is great interest in understanding its binding mode and reaction mechanism. RDV-triphosphate (RTP) is a substrate of the RNA polymerase (pol) but not an inhibitor because it can be efficiently added to the growing RNA chain by the nsp12 RNA polymerase of SARS-CoV-2. Incorporation of RDV into the RNA occurs only after the pro-drug RDV has been converted by the host cell into RTP.6–10 RDV does not act as a chain terminator because it has a 3’-hydroxyl group, thereby allowing the primer to be efficiently extended by the RNA pol after RDV-monophosphate (RMP) incorporation.6–10 The rate of in vitro primer extension after RMP incorporation depends on the location of the added RMP and the concentration of the next incoming nucleotide triphosphate (NTP). When the added RMP in the RNA duplex product was translocated at the i+3 site, an accumulation of this product was observed in the presence of a low concentration of incoming NTP, but not at their physiological concentrations.6 Thus, the reduced primer extension observed in vitro could also imply the reduced viral replication and transcription in vivo is a mechanism known as delayed inhibition.6 It could also permit exoribonuclease to remove some of added RMPs so that AMPs can be reinserted. This extra step of RMP insertion-excision could effectively slow down viral RNA synthesis.
Evidence exists that an exoribonuclease associated with both SARS-CoV and SARS-CoV-2 viruses could reduce a modest fraction of RDV inhibition,11, 12 suggesting that a significant fraction of added RDV nucleotides were not removed and thus have become part of the viral mRNAs. Nevertheless, RMP-containing viral RNAs are likely non-functional and thus are partially responsible for suppression of viral reproduction.13 In fact, the incorporation of RDV and its effects on viral RNA production in the SARS-CoV-2 infected cell lines is highly strand dependent.12 Therefore, a new class of anti-SARS-CoV-2 inhibitors could be rationally designed, including the formation of dead-end inhibitory ternary complexes at the pol active site, translocation-inhibited complexes, or exoribonuclease-inhibited complexes. These designs all require higher-resolution structural information than are available from the current cryo-EM structures.
In this study, we supplement high-resolution structural information using molecular dynamics (MD) simulations,14 starting with medium- and low-resolution cryo-EM structures. Particularly, high-resolution structural information on the binding site of RTP can facilitate the rational design of effective drugs for targeting the viral RNA polymerase. Our structural analysis can also address many fundamental questions, including whether substitution of the RDV 3’-OH group by 3’-NH2 or other functional groups could make the inhibitor a non-extendable chain terminator. If so, the modified RDV could terminate the RNA-synthesis complex after being incorporated. While MD simulations provide specific information about both kinetics and thermodynamics of enzymes in solution, we limit our analysis in this study to thermodynamics of the pol complexes, i.e., the equilibrated structures in the MD simulation trajectories. Here, we focus on the analysis of the catalytically active complex of SARS-CoV-2 nsp12 pol in the presence of the essential Mg2+ ions and the incoming RTP, or ATP base-paired with a template U nucleotide. These simulations provide unique and valuable structural information about the active complex that would be challenging to obtain from X-ray diffraction or cryo-EM methods. Our structural analysis suggests a specific mechanism for incorporation of RMP or AMP by the nsp12 RNA pol of SARS-CoV-2. Our approach differs from recent MD simulations on remdesivir in different positions within the RNA duplex product that relied on free energy calculation in which the proposed remdesivir destabilizes the enzyme-RNA complex.15 How those results correlate with experimental data remains unclear.
MATERIALS AND COMPUTATIONAL METHODS
The structure of the nsp12 RNA pol (7bv2) was retrieved from the Protein Data Bank (PDB) as a starting point of our MD simulations.7 The bound RMP (F86) and pyrophosphate were removed and the 3’ end of the primer chain was truncated so the active site was left open. Triphosphate versions of adenosine, remdesivir, and galidesivir (GLD) were placed at the active site with the triphosphate tail coordinated to two Mg2+ ions via non-bonding interactions. Mg2+ ions were also coordinated with formal bonds to D618, Y619, D760, and D761 using the Metal Center Parameters Builder (MCPB) from AmberTools.16, 17 A weak restraint (10 kcal/mol/Å2 centered at 2.1 Å) was added to constrain the distance between Mg2+ ions and the deprotonated primer-terminal O3’ (ptO3’), accounting for the bond between those two atoms as MCPB does not currently support metal bonds to nucleic acid chains. MCPB was also used to create parameters for the two Zn2+ ions bound to three cysteines and a histidine. Parameters for the triphosphate molecules were generated with Antechamber.17
The system was immersed in a water box and neutralized with Na+ ions. All MD simulations were performed using the Amber force field and NAMD.17, 18 Equilibration was performed in three steps, starting with equilibration of the solvent, then both solvent and sidechains, and finally the complete system. Each equilibration step consisted of minimization, gradual heating to 310 K and thermalization. Once the system was stabilized at 310 K at the end of the thermalization step, a production run of 100 ns was performed. For the ATP-containing MD simulations, the coordinates were sampled at intervals of 20 ps per frame. For the RTP- and GLD-TP-containing MD trajectories, coordinates were sampled at 40 ps per frame.
Individual frames of MD trajectories were aligned using the program VMD, and saved as individual PDB files.19 Electron scattering factors for neutral atoms were assigned for calculation of ESP maps and X-ray scattering factors for the calculations of ED maps. All maps were combined using the CCP4 suite (the programs sfall, fft, and mapsig for adding one map per frame at a time).20 The unit cell box for the ATP-containing structure was a cube with edge length of 120 Å. The grid number was set to 360 (the grid size is 0.333 Å), and the initial ΔB = 8 Å2 was used to minimize the Fourier series termination effect at the nominal resolution of 1.0 Å for calculations of both structure factors and maps. The pre-equilibrated 7bv2 model was used as a starting model to fit the MD-ESP (or MD-ED) maps.7 The models were rebuilt using the graphics program Coot, following residual maps and refined using both Phenix and Refmac5 with standard iterations.21–23 All figures were made using PyMol.24
RESULTS AND DISCUSSION
MD-derived electrostatic potential maps versus cryo-EM maps
Our analysis is based on the thermal distribution of atomic configurations sampled by MD simulations, providing (i) electron density (ED) maps computed with X-ray atomic scattering factors, (ii) electrostatic potential (ESP) maps obtained with electron atomic scattering factors (Fig. 1), and (iii) probability density functions (PDFs) of atomic coordinates. Therefore, the MD trajectories provide both the variance of atomic distributions (i.e., atomic B-factors) and the average atomic positions at thermal equilibrium (i.e., equilibrated structures). When there is a single conformation for a protein residue or nucleic acid base, the individual atomic coordinate distributions can be described to a good approximation by 3-dimensional Gaussian functions. When there are multiple conformations, the distributions are described by a superposition of multiple overlapping Gaussian functions. In this study, electron scattering factors of neutral atoms were used. Partial charges of ionizable atoms, highlighting differences between experimental cryo-EM maps and MD-simulated ESP maps, properly account for the actual partial atomic charges.
Figure 1.
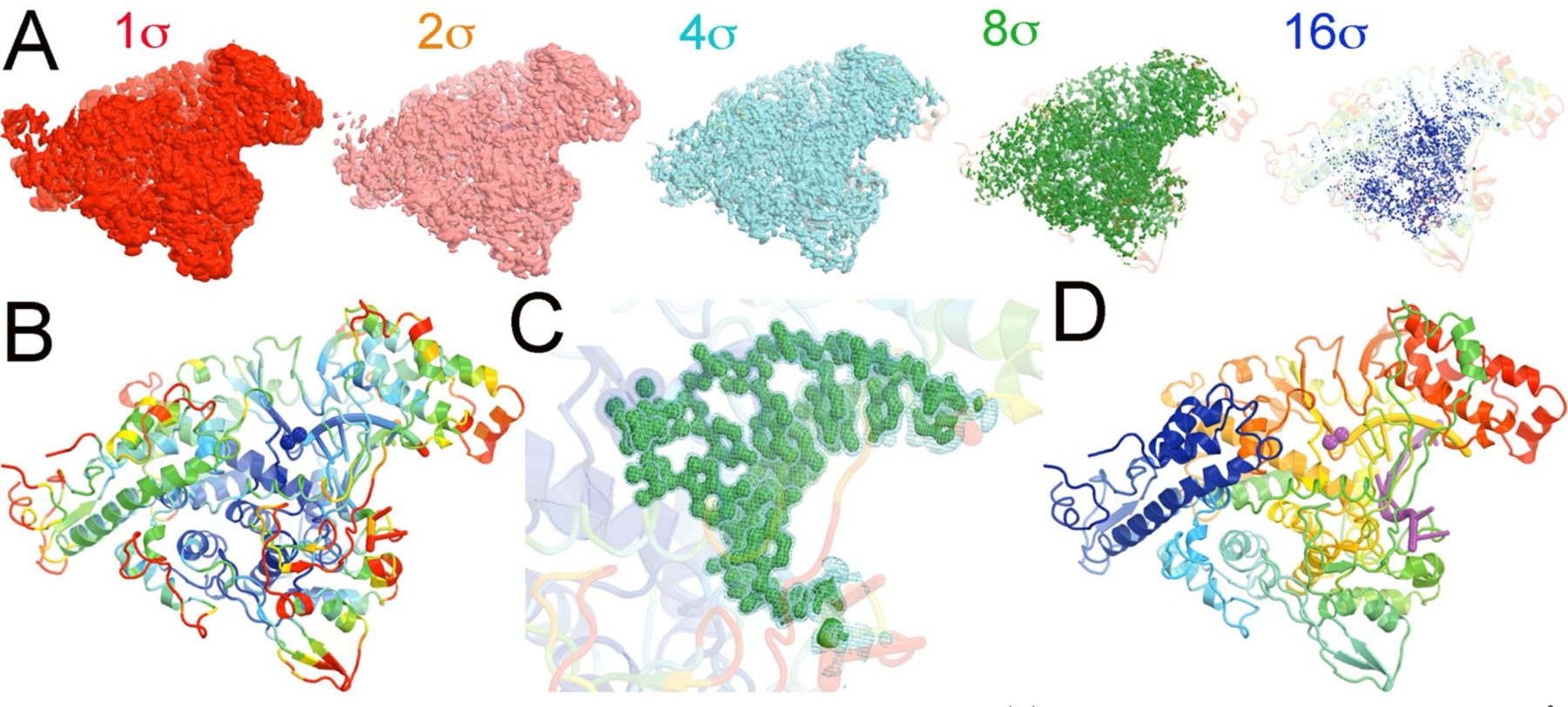
Overall comparison of MD-ESP maps with the experimental Cryo-EM maps. (A) MD-ESP map with added blurring ΔB = 8 Å2 contoured at +1 (red), +2 (salmon), +4 (cyan), +8 (green), or +16σ (blue) superimposed on the cartoon drawing of the refined atomic model colored by rainbow spectrum of atomic B-factors refined in transparency. (B) The refined atomic model colored by rainbow spectrum of atomic B-factors (blue-white-red for small-medium-large B-factors, respectively). (C) A close-up view of the MD-ESP map for the primer/template RNA duplex plus ATP contoured at +4σ (cyan mesh) and +8σ (green isosurface). (D) The refined atomic model colored by rainbow spectrum of residue sequence numbers.
The RNA pol active site represents the most rigid part of this enzyme complex, with the smallest distribution variance of atomic coordinates as revealed by MD-derived ESP maps. Figure 1 shows the overall ESP map derived from 5,000 frames sampled from 100-ns MD trajectories (MD-ESP) and its comparison to the closely related experimental emd-30120 map.7 This map was generated after filtering the MD-ESP map to comparable resolution and resampling onto the experimental grids.7 The MD-ESP map was averaged over 20 ps per frame and superimposed on the equilibrium positions of the atomic model that was refined against the MD-ESP map.
The ensemble of configurations of the RNA pol complex sampled by MD simulations represents the highest achievable resolution of an error-free image reconstruction. The variance of the distribution resulting from thermal atomic motion determines the atomic B-factor and defines the local resolution. The larger the variance (and the B-factors), the broader the atomic ESP peak distribution, and the lower the amplitude of the atomic ESP. When the MD-ESP map is contoured at +1, +2, +4, +8, or +16σ in the isosurface ESP representation, the region that is visible in the highest contour level of +16σ represents the highest local resolution, while the regions that are only visible in the lowest contour level of +1σ represents the lowest local resolution (Fig. 1A). The pol active site is the region with highest resolution, while the peripheral loops on the surface of the complex correspond to the regions with the lowest local resolution (Fig. 1B). Clearly, the distribution of local resolution in MD-ESP maps is fully consistent with the distribution of refined atomic B-factors in the complex at thermal equilibrium.
We filtered the resolution of MD-ESP map by adding blurring B-factors (ΔB = 28 Å2) to compare the MD-derived ESP maps with Cryo-EM maps, and we resampled the MD-ESP onto the grids of the Cryo-EM emd-30210 map (to ~ 2.5 Å) as reported for the 7bv2 structure (Fig. 1).7 The comparison shows that the two maps are remarkably similar, except for the orientation of the carboxylates of D760 and D761 (Fig. 2). However, modeling the orientation of carboxylate sidechains at medium resolution can be challenging, particularly when the carboxylate groups are bound to divalent metal ions.25–28 Furthermore, we note that the experimental map corresponds to the enzyme-RNA product complex with incorporated RMP and bound pyrophosphate whereas our MD-ESP map corresponds to the ATP substrate complex. Nevertheless, these differences do not appreciably affect the structure of the nsp12 RNA pol except for the triphosphate moiety-binding pocket.7
Figure 2.
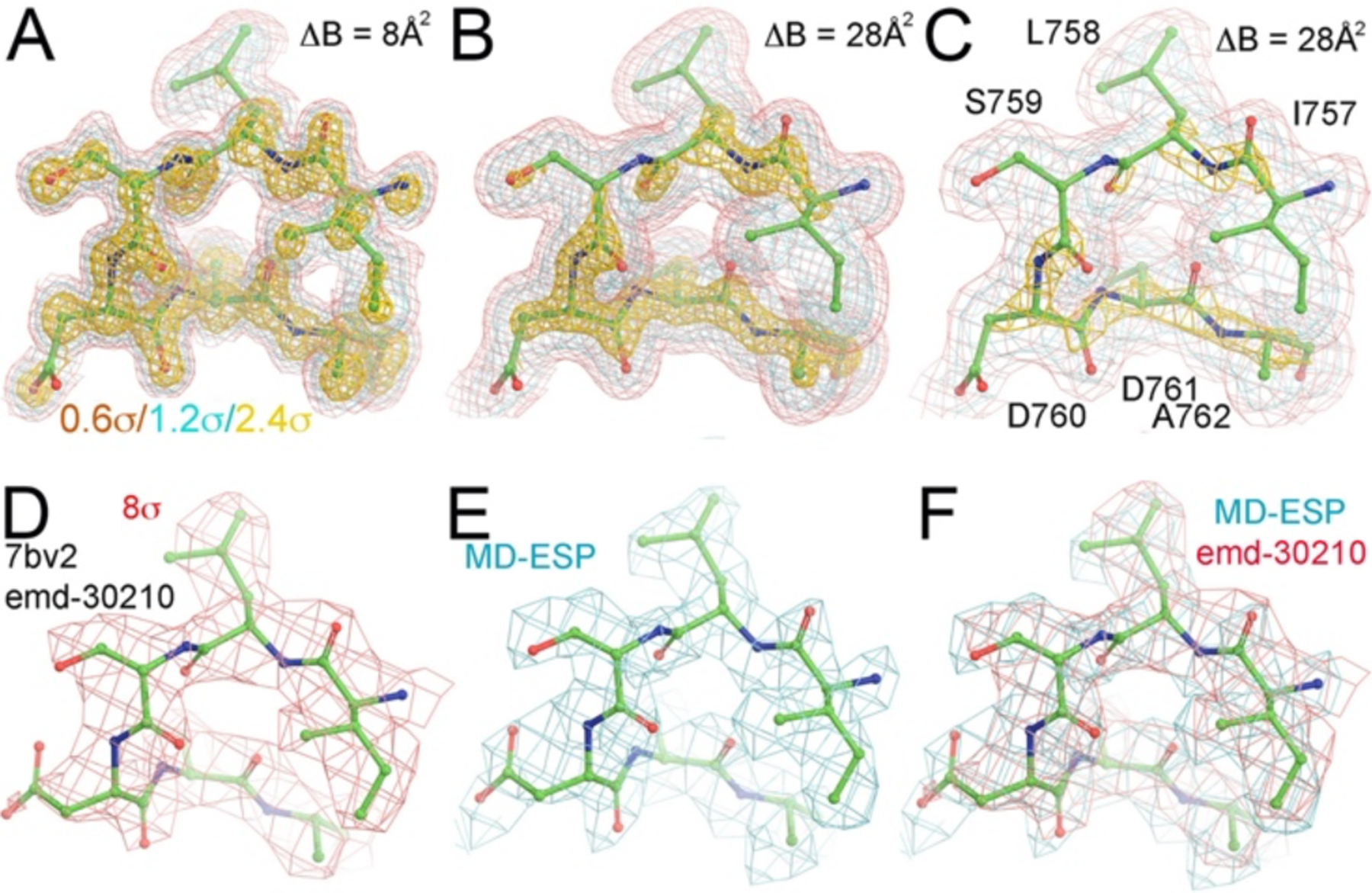
Detailed comparison of MD-ESP maps with the experimental Cryo-EM maps. (A) A close-up view of the MD-ESP map for a catalytic loop (ΔB = 8 Å2) contoured at +0.6σ (salmon), +1.2σ (cyan), and +2.4σ (gold). B) The same catalytic loop of the MD-ESP map as (A) but with ΔB = 28 Å2. (C) The same as (B) but with coarse grid numbers corresponding to about 2.0 Å resolution. (D) The experimental emd-30120 map contoured +8σ. (E) Resampled MD-ESP map on the experimental map grids. (F) Superimposition of MD-ESP maps with the emd-30210 map and reported atomic model.
Experimentally, it is difficult to capture the catalytically active complex that represents a minor population in the reaction mixture without using specific methods to enrich its population. To experimentally probe the catalytically relevant complex(es), it would be necessary to stall the RNA synthesis by using, for example, transition-state analogue inhibitors, a 3’-deoxy non-extendable primer, a non-hydrolyzable nucleotide analogue (such as α,β-amide substituted ATP or AMPNPP), or catalytically inactive divalent metal ions (such as Ca2+), as reported for RB69 DNA pol and human pol β (and to a lesser extent, for other DNA pols)29–38. Another method is reliant on certain crystal lattice constraints to trap the pyrophosphate product and a third divalent metal ion.39, 40 However, one must be aware of the fact that any of these methodologies could have artifacts and are prone to misinterpretation.41, 42
Binding of ATP, RTP and Galidesivir-triphosphate
Figure 3 shows the MD-derived electron density (MD-ED) maps for the pol active site of SARS-CoV-2, containing the nascent basepair between a template nucleotide U and each of the incoming substrates: ATP, RTP or Galidesivir (GLD)-triphosphate (GLD-TP). RTP has a cyano substitution at the C1’ position while GLD-TP has an amine substitution at O4’ (Fig. 3B, 3C). Upon RTP binding, the cyano-binding pocket expands only slightly as seen in the outward displacement of the sidechain of T687 (Fig. 3G). The equilibrium conformation of RTP differs from the binding mode of ATP and GLD-TP in that it is missing an intra-molecular hydrogen bond (HB) between its O3’ and the β-phosphate group (Fig. 3A–3C). In the ATP-bound complex, a cluster of positively charged amino acid residues form a well-defined binding pocket for the triphosphate moiety of ATP and exhibit well-ordered MD-ED features (Fig. 3D–3F), of which four directly interact with the triphosphate moiety (Fig. 4). The binding mode observed suggests that ATP adopts a single conformation at the binding site. The same positive residues also bind the triphosphate moiety of RTP and GLD-TP. However, the triphosphate groups of RTP and GLD-TP exhibit multiple conformations, so their MD-ED features are only approximately defined when compared to the ATP complex. Here, we describe only the dominant conformations even though the RTP and GLD-TP exhibit multiple conformations. In the ATP-binding pocket, ordered water molecules occupy the pocket where the substituted cyano-group of RTP occupies it in the RTP complex (Fig. 5)
Figure 3.
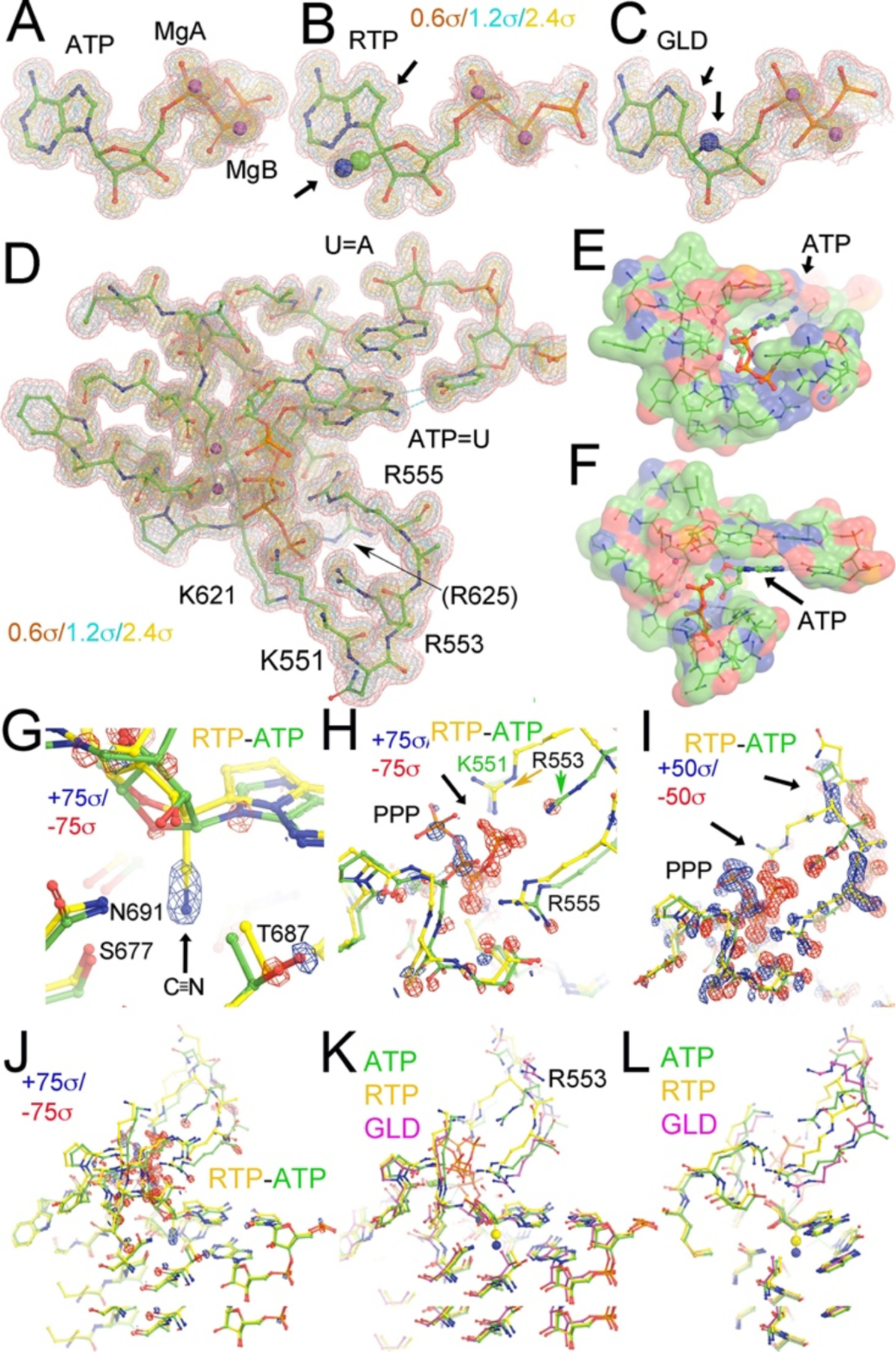
MD-ED maps for ATP-, RTP- or GLD-TP-containing complex and their differences. Maps for ATP (A), RTP (B), and GLD-TP (C) were contoured at +0.6σ (salmon), +1.2σ (cyan), and +2.4σ (gold). Substituted groups are in large CPK model as indicated by black arrows. (D) The pol active site including the nascent base pair and primer-terminal base pair. (E, F). Surface representation of the ATP-binding pocket colored by charges (negative, red, positive, blue, and neutral, green). (G, H, I, J) Four different views of the RTP minus ATP difference map at the highest contouring levels (+75σ, blue/−75σ, red, G. H, and J) show the added cyano substitution in RTP relative to ATP (G, it should be noted that only relative values are meaningful because MD-ED maps are contoured relative to standard deviation of the entire maps and the absolute value of their standard deviation is highly dependent on the size of an arbitrary box used for MD simulations and also on sampling resolution unlike crystallographic ED maps), the displaced triphosphate moiety (H), and at the second highest contouring levels (I, +50σ, blue/−50σ, red), and (J) zoom-out view. Arrows indicate displacements of the triphosphate tail and its interacting loop. (K, L) Two views of comparison of ATP (green), RTP (gold), GLD-TP (magenta) bound structures.
Figure 4.
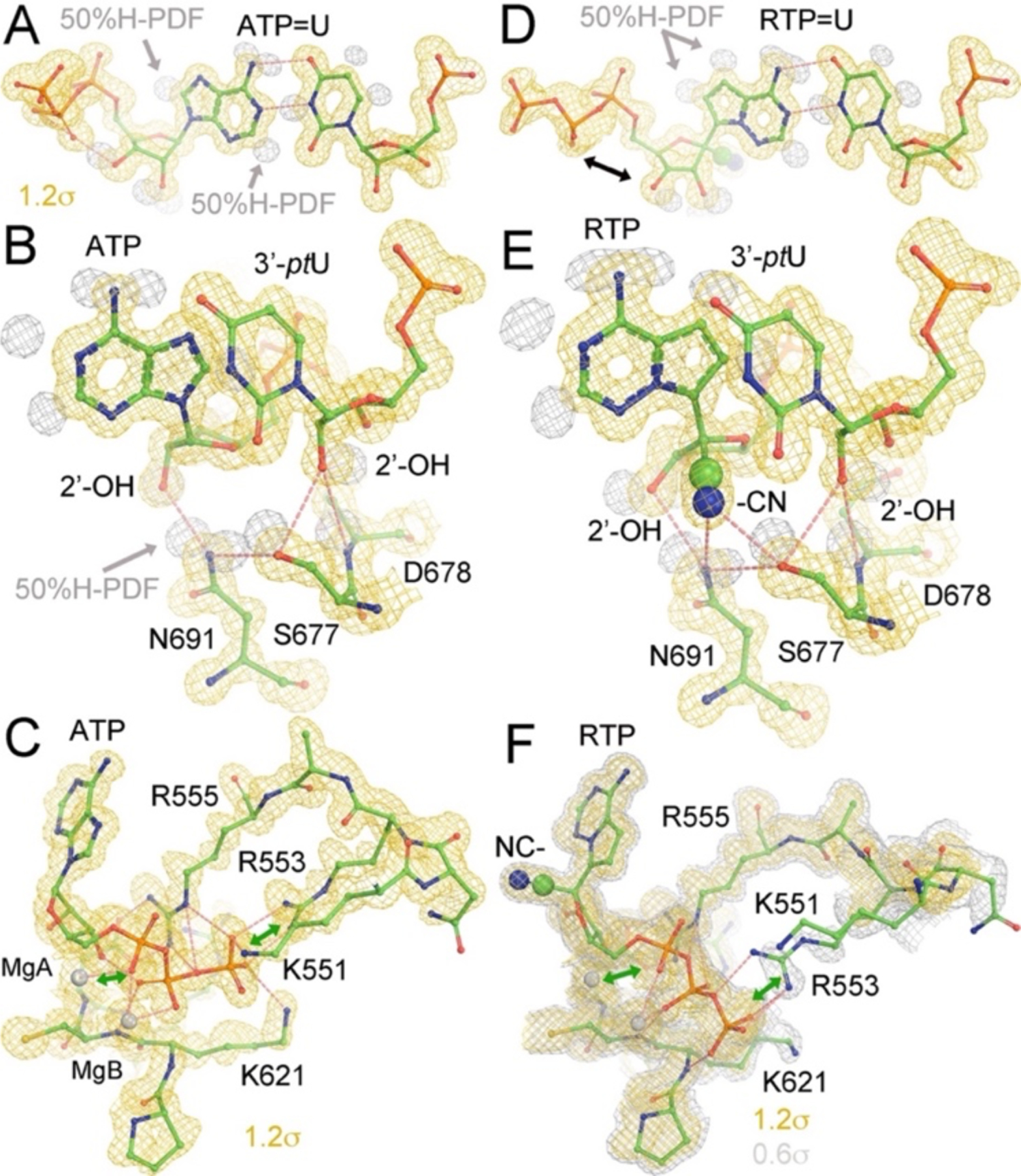
HB patterns for recognition of 2’-OH and cyano groups in (A-C) ATP and (D-F) RTP complexes, respectively. MD-ED maps are contoured at a low (+0.6σ, silver, in F) and a high levels (+1.2σ, gold, all panels). H atom probability density function (PDF) maps are contoured at about 50% probability.
Figure 5.
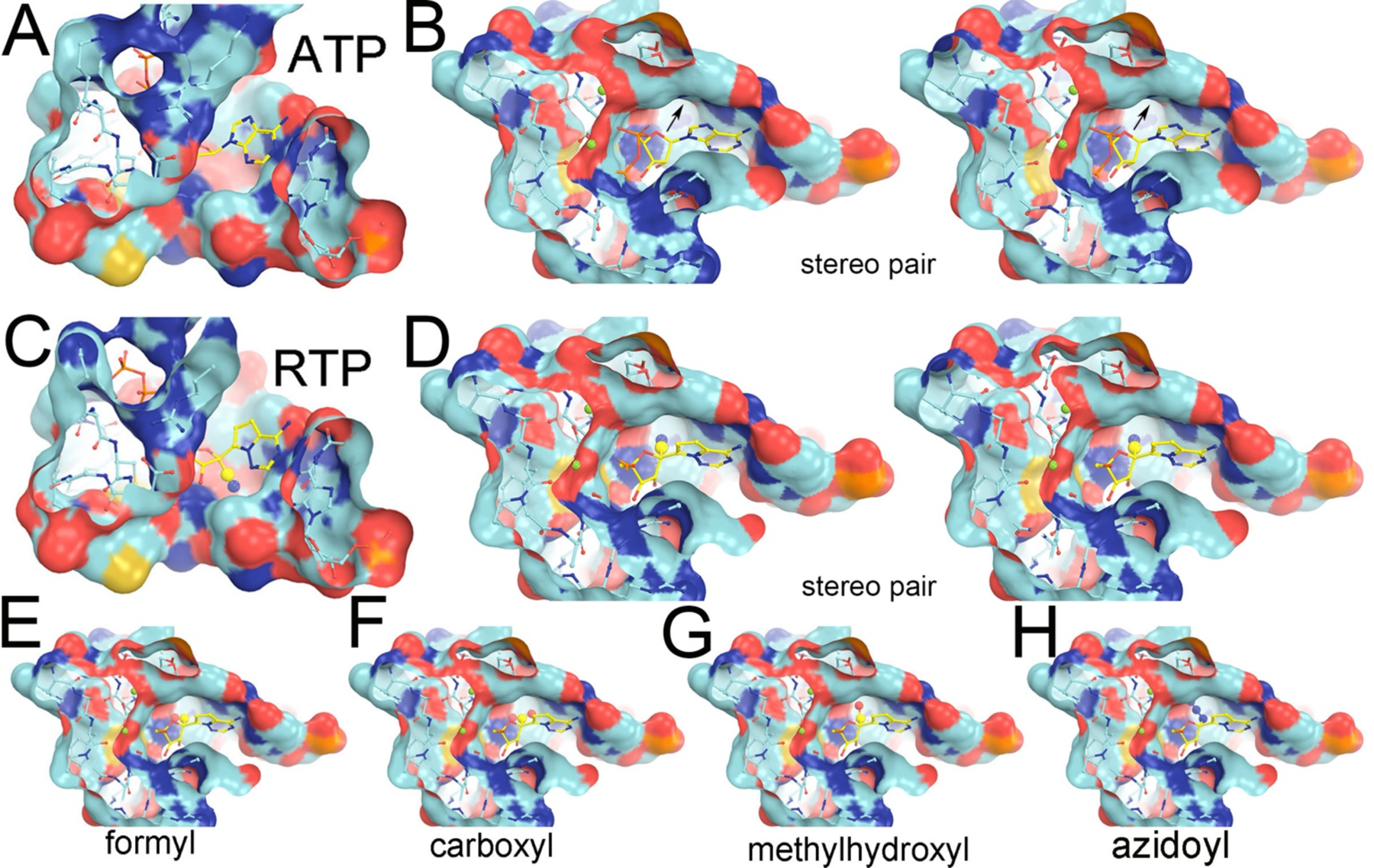
The ATP-binding pocket and the pre-existing binding pocket for the cyano group of RTP and hypothetical models for new drug design. (A, B) Two views of the ATP-bound complex show the pre-existing pocket underneath the C1’ atom where the cyano group is substituted in the RTP complex. (B) In stereodiagram. Arrows indicate a pre-existing pocket in the pol. (C, D) The same two views of the RTP-bound complex. (D) In stereodiagram. (E-H) With hypothetical formal, carboxyl, methylhydroxyl, or azidoyl substitution at C1’. Solvent-accessible surface is colored by negative charge (red), positive charge (blue), uncharged (cyan) atoms, and sulfur atom (gold).
The RTP-minus-ATP MD-ED difference map, when comparing RTP to ATP binding, shows that the largest positive feature corresponds to the cyano substitution of RTP at the C1’ position (Fig. 3G). The largest negative difference corresponds to the triphosphate moiety, due to the different conformation of the nucleotide triphosphate in the two structures (Fig. 3G). Associated with these structural differences, there are many other MD-ED difference features due to the repositioning of five positively charged sidechains of the key amino acid residues and the corresponding main chain backbone, suggesting correlated motions and different conformations of the triphosphate moiety for the three incoming NTPs. Therefore, the MD-ED difference Fourier analysis is very sensitive to subtle structural changes.
The conformation of the triphosphate moiety of GLD-TP resembles that of ATP. However, the MD-ED features of the triphosphate group of RTP and GLD-TP are less well defined than those of ATP (Figs. 3, 4). In all three cases, the nucleobase and α-phosphate group exhibit distinct MD-ED features (Fig. 3) as well as distinct dynamics in response to the different incoming nucleoside triphosphates. In addition, we have repeated all of our MD simulations, starting with the 6xez coordinates,43 which largely reproduced the overall structural features reported in this study. However, these replicas also showed that there were more than two alternate conformations of the triphosphate-binding loops for substrates described here.
H-bonding interactions of RTP within the polymerase complex
MD simulations allows us to analyze detailed H-bond (HB) networks that are difficult to determine by simple inspection of medium- and low-resolution experimental maps. HB analysis provides valuable insights into the structural and functional roles of the HB network and shows that it involves the sidechain of S677, the 2’ and 3’ hydroxyl groups, and pol backbone. The 2’-OH groups of both ATP and the 3’ primer-terminal nucleotide U (ptU) are recognized by the pol via HB interactions (Fig. 4). The N691 sidechain and the backbone amide of D678 provide selectivity against 2’-deoxy nucleotides, ensuring that this pol only accommodates RNA and discriminates against DNA (Fig. 4B, 4E). We note that the HB between N691 and S667 can also keep these two nucleotides adjacent to each other, which is essential for the RNA polymerization reaction.
ATP and RTP form two nearly identical base pairing HBs with the template nucleotide U (Fig. 4). Recognition of the 2’-OH group of RTP and ptU in the RTP complex is nearly identical to that of the ATP complex and is not affected by differences in the MD-ED maps due to the orientations of the 2’-OH and 3’-OH groups in RTP and ATP. The cyano group exhibits an HB interaction with the sidechain of S677 and a polar interaction with the sidechain of N691 (Fig. 4E). These interactions increase the binding affinity of RTP in the pol-substrate complex. The same base pairing interaction between GLD-TP and the template is observed between ATP and the template U.
DNA pols use a “sugar-gate” residue to stereochemically prevent rNTPs with 2’-OH groups from binding to the dNTP-binding pocket.44, 45 RNA pols have a specific interacting residue(s) to establish the HB that recognizes the 2’-OH group, enabling them to select rNTPs over dNTPs.46–48 However, the detailed HB pattern responsible for introducing selectivity for the substrate has remained unknown due to the limited resolution of cryo-EM maps of the RTC of SARS-CoV-2.7, 49–51 Here, we find evidence that the sidechain of N691 recognizes the 2’-OH group of the incoming ATP (and other NTP substrates). The orientation of the N691 sidechain is constrained by a HB between the sidechains of N691 and S677. In addition, the pre-existing pocket underneath the C1’ atom of ATP is filled by ordered water molecules, providing a strong cooperative effect for recognition of the RTP substrate as observed in our MD simulations. The HB network also assists in the recognition of the 2’-OH of the 3’ primer-terminal nucleotide by the backbone amide of D678 (Fig. 4B, 4E).
The pre-existing cyano-binding pocket is also observed in the ATP complex although filled with water molecules and replaced by the hydrophilic cyano group of remdesivir in the RTP complex (Fig. 5A–5D). The size and hydrophilic nature of the pocket also makes it suitable for other types of polar moieties such as formyl, carboxyl, methylhydroxyl, keto, or amide groups that could substitute for the cyano group of C1’ (Fig. 5E–5H). Also, with modest puckering of the nucleotide sugar, the pocket could also fit an azidoyl group (Fig. 5H). Additional MD simulations with these new compounds are ongoing.
Our analysis provides new insights on specific interactions responsible for binding of nucleotide analogues that could mimic ATP and outperform remdesivir. Therefore, our structural insight is expected to stimulate further studies of modified nucleotides with new functional groups designed for high affinity binding at the nascent base pair-binding pocket.
Relationship between MD simulations and biophysical studies
MD simulations and simulated annealing have been the standard procedures of model refinement for both X-ray crystallography and NMR for many decades because they can rapidly find the equilibrated structure present in crystals or in solution starting with only moderately accurate coordinates.52, 53 Slow-cooling procedures are commonly used for finding a single equilibrated structure. We provide an alternative in this study. The equilibrated structures derived from MD-ESP or MD-ED maps can also be used for interpretation of corresponding medium- and low-resolution cryo-EM ESP maps and crystallographic ED maps. We present evidence that MD simulations can complement high-resolution structural information for these structures because they provide MD-ESP and MD-ED maps that are highly comparable with the experimental data. Figure 1 shows a striking similarity between the MD-ESP and cryo-EM map when they are compared locally. However, during a global comparison, we discovered that the magnification used for image reconstruction was often only approximate and required additional correction for voxel rescaling between a global comparison of MD-ESP and cryo-EM maps to reliably be made.
MD simulations are powerful theoretical single-molecule techniques and should be comparable to experimental single-molecule spectroscopy. Because single-molecule kinetic behaviors are fully reproduced by the kinetic behaviors of bulk solution assembly complexes (i.e., Michaelis-Menten kinetics for complex enzymatic reactions),54 one expects that MD simulations should reproduce nearly all macroscopically measurable kinetic and thermodynamic properties of any enzyme, even though this study focuses on only on the equilibrated structures for the purpose of drug discovery. At the single-molecule level, enzymes fluctuate stochastically, and this information should be present in the MD simulation trajectories, alongside information for the kinetic rates of conversion between any pair of specific functional states.55–58 With assistance of MD simulations between two closely functional states, one may be able to deconstruct such states from experimental maps when they are inadvertently merged into a single state. Therefore, this structure is just an example but it opens up a new avenue for powerful MD simulations-based rationale for drug design.
Supplementary Material
Funding Sources
This work was supported by the NIH grant GM106121 (V.S.B.) and a generous allocation of high-performance computing time from NERSC.
ABBREVIATIONS
- RdRp
RNA-dependent RNA polymerase
- pol
polymerase
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- P/T
primer/template
- RDV
remdesivior
- RMP
remdesivir monophosphate
- RTP
remdesivir triphosphate
- AMP
adenosine monophosphate
- ATP
adenosine triphosphate
- GLD
galidesivir
- GLD-TP
galidesivir triphosphate
- MD
molecular dynamics
- MD-ESP
MD-derived electrostatic potential maps
- MD-ED
MD-derived electron density maps
probability density function of atomic distribution
- HB
hydrogen bond or hydrogen bonding
Footnotes
Competing interest statement
The authors declare that there is no competing interest in this study.
Supporting Information
Containing the refined PDB coordinates of the equilibrated ATP-bound complex against MD-derived ESP maps.
REFERENCES
- [1].Seley-Radtke KL, and Yates MK (2018) The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold, Antiviral Res 154, 66–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yates MK, and Seley-Radtke KL (2019) The evolution of antiviral nucleoside analogues: A review for chemists and non-chemists. Part II: Complex modifications to the nucleoside scaffold, Antiviral Res 162, 5–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wang Y, Zhang D, and Du G (2020) Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial (vol 395, 1569, 2020), Lancet 395, 1694–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Eastman RT, Roth JS, Brimacombe KR, Simeonov A, Shen M, Patnaik S, and Hall MD (2020) Remdesivir: A review of its discovery and development leading to emergency use authorization for treatment of COVID-19, ACS Cent Sci 6, 672–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Grein J, Ohmagari N, Shin D, Diaz G, Asperges E, Castagna A, Feldt T, Green G, Green ML, Lescure FX, Nicastri E, Oda R, Yo K, Quiros-Roldan E, Studemeister A, Redinski J, Ahmed S, Bernett J, Chelliah D, Chen D, Chihara S, Cohen SH, Cunningham J, D’Arminio Monforte A, Ismail S, Kato H, Lapadula G, L’Her E, Maeno T, Majumder S, Massari M, Mora-Rillo M, Mutoh Y, Nguyen D, Verweij E, Zoufaly A, Osinusi AO, DeZure A, Zhao Y, Zhong L, Chokkalingam A, Elboudwarej E, Telep L, Timbs L, Henne I, Sellers S, Cao H, Tan SK, Winterbourne L, Desai P, Mera R, Gaggar A, Myers RP, Brainard DM, Childs R, and Flanigan T (2020) Compassionate use of remdesivir for patients with severe Covid-19, N Engl J Med 382, 2327–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gordon CJ, Tchesnokov EP, Woolner E, Perry JK, Feng JY, Porter DP, and Gotte M (2020) Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency, J Biol Chem 295, 6785–6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yin W, Mao C, Luan X, Shen DD, Shen Q, Su H, Wang X, Zhou F, Zhao W, Gao M, Chang S, Xie YC, Tian G, Jiang HW, Tao SC, Shen J, Jiang Y, Jiang H, Xu Y, Zhang S, Zhang Y, and Xu HE (2020) Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir, Science 368, 1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tchesnokov EP, Feng JY, Porter DP, and Gotte M (2019) Mechanism of inhibition of Ebola virus RNA-dependent RNA polymerase by remdesivir, Viruses 11, E326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z, Lu G, Qiao C, Hu Y, Yuen KY, Wang Q, Zhou H, Yan J, and Qi J (2020) Structural and functional basis of SARS-CoV-2 entry by using human ACE2, Cell 181, 894–904 e899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dangerfield TL, Huang NZ, and Johnson KA (2020) Remdesivir is effective in combating COVID-19 because it is a better substrate than ATP for the viral RNA-dependent RNA polymerase, iScience 23, 101849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Agostini ML, Andres EL, Sims AC, Graham RL, Sheahan TP, Lu X, Smith EC, Case JB, Feng JY, Jordan R, Ray AS, Cihlar T, Siegel D, Mackman RL, Clarke MO, Baric RS, and Denison MR (2018) Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease, mBio 9, e00221–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhao Y, Sun J, Li Y, Li Z, Xie Y, Feng R, Zhao J, and Hu Y (2021) The strand-biased transcription of SARS-CoV-2 and unbalanced inhibition by remdesivir, iScience 24, 102857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang J, Reiss K, Shi Y, Lolis E, Lisi GP, and Batista VS (2021) Mechanism of inhibition of the reproduction of SARS-CoV-2 and Ebola Viruses by remdesivir, Biochemistry 60, 1869–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang J, Shi Y, Reiss K, Allen B, Maschietto F, Lolis E, Konigsberg WH, Lisi GP, and Batista VS (2022) Insights into binding of single-stranded viral RNA template to the replication-transcription complex of SARS-CoV-2 for the priming reaction from molecular dynamics simulations, Biochemistry 61, 424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bylehn F, Menendez CA, Perez-Lemus GR, Alvarado W, and de Pablo JJ (2021) Modeling the binding mechanism of remdesivir, favilavir, and ribavirin to SARS-CoV-2 RNA-dependent RNA polymerase, ACS Cent Sci 7, 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li P, and Merz KM Jr. (2016) MCPB.py: A python based metal center prameter builder, J Chem Inf Model 56, 599–604. [DOI] [PubMed] [Google Scholar]
- [17].Case DA, Cerutti DS, Cheatham TE III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Greene D, Homeyer N, Izadi S, Kovalenko A, Lee TS, LeGrand S, Li P, Lin C, Liu J, Luchko T, Luo R, Mermelstein D, Merz KM, Monard G, Nguyen H, Omelyan I, Onufriev A, Pan F, Qi R, Roe DR, Roitberg A, Sagui C, Simmerling CL, Botello-Smith WM, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Xiao L, York DM, and Kollman PA (2017) AMBER 2017, University of California, San Francisco. [Google Scholar]
- [18].Phillips JC, Hardy DJ, Maia JDC, Stone JE, Ribeiro JV, Bernardi RC, Buch R, Fiorin G, Henin J, Jiang W, McGreevy R, Melo MCR, Radak BK, Skeel RD, Singharoy A, Wang Y, Roux B, Aksimentiev A, Luthey-Schulten Z, Kale LV, Schulten K, Chipot C, and Tajkhorshid E (2020) Scalable molecular dynamics on CPU and GPU architectures with NAMD, J Chem Phys 153, 044130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Humphrey W, Dalke A, and Schulten K (1996) VMD: visual molecular dynamics, J Mol Graph 14, 33–38, 27–38. [DOI] [PubMed] [Google Scholar]
- [20].Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, and Wilson KS (2011) Overview of the CCP4 suite and current developments, Acta Crystallogr D Biol Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Emsley P, and Cowtan K (2004) Coot: model-building tools for molecular graphics, Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- [22].Murshudov GN, Vagin AA, and Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method, Acta Crystallogr D Biol Crystallogr 53, 240–255. [DOI] [PubMed] [Google Scholar]
- [23].Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution, Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Delano WL Pymol Schrodinger, Inc., http://pymol.org/, software downloaded on March, 2020. [Google Scholar]
- [25].Wang J, and Moore PB (2017) On the interpretation of electron microscopic maps of biological macromolecules, Protein Sci 26, 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang J, Natchiar SK, Moore PB, and Klaholz BP (2021) Identification of Mg(2+) ions next to nucleotides in cryo-EM maps using electrostatic potential maps, Acta Crystallogr D Struct Biol 77, 534–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang J, Liu Z, Frank J, and Moore PB (2018) Identification of ions in experimental electrostatic potential maps, IUCrJ 5, 375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wang J (2017) On the appearance of carboxylates in electrostatic potential maps, Protein Sci 26, 396–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang M, Xia S, Blaha G, Steitz TA, Konigsberg WH, and Wang J (2011) Insights into base selectivity from the 1.8 A resolution structure of an RB69 DNA polymerase ternary complex, Biochemistry 50, 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Xia S, Wang M, Blaha G, Konigsberg WH, and Wang J (2011) Structural insights into complete metal ion coordination from ternary complexes of B family RB69 DNA polymerase, Biochemistry 50, 9114–9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Batra VK, Beard WA, Shock DD, Krahn JM, Pedersen LC, and Wilson SH (2006) Magnesium-induced assembly of a complete DNA polymerase catalytic complex, Structure 14, 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Franklin MC, Wang J, and Steitz TA (2001) Structure of the replicating complex of a pol alpha family DNA polymerase, Cell 105, 657–667. [DOI] [PubMed] [Google Scholar]
- [33].Sawaya MR, Pelletier H, Kumar A, Wilson SH, and Kraut J (1994) Crystal structure of rat DNA polymerase beta: evidence for a common polymerase mechanism, Science 264, 1930–1935. [DOI] [PubMed] [Google Scholar]
- [34].Pelletier H, Sawaya MR, Kumar A, Wilson SH, and Kraut J (1994) Structures of ternary complexes of rat DNA polymerase beta, a DNA template-primer, and ddCTP, Science 264, 1891–1903. [PubMed] [Google Scholar]
- [35].Doublie S, Tabor S, Long AM, Richardson CC, and Ellenberger T (1998) Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution, Nature 391, 251–258. [DOI] [PubMed] [Google Scholar]
- [36].Huang H, Chopra R, Verdine GL, and Harrison SC (1998) Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance, Science 282, 1669–1675. [DOI] [PubMed] [Google Scholar]
- [37].Li Y, Korolev S, and Waksman G (1998) Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation, EMBO J 17, 7514–7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Li Y, Kong Y, Korolev S, and Waksman G (1998) Crystal structures of the Klenow fragment of Thermus aquaticus DNA polymerase I complexed with deoxyribonucleoside triphosphates, Protein Sci 7, 1116–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zamyatkin DF, Parra F, Alonso JM, Harki DA, Peterson BR, Grochulski P, and Ng KK (2008) Structural insights into mechanisms of catalysis and inhibition in Norwalk virus polymerase, J Biol Chem 283, 7705–7712. [DOI] [PubMed] [Google Scholar]
- [40].Zamyatkin DF, Parra F, Machin A, Grochulski P, and Ng KK (2009) Binding of 2’-amino-2’-deoxycytidine-5’-triphosphate to norovirus polymerase induces rearrangement of the active site, J Mol Biol 390, 10–16. [DOI] [PubMed] [Google Scholar]
- [41].Park J, Youn HS, An JY, Lee Y, Eom SH, and Wang J (2021) Structure of New Binary and Ternary DNA Polymerase Complexes From Bacteriophage RB69, Front Mol Biosci 8, 704813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang J, and Konigsberg WH (2022) Two-metal-ion catalysis: Inhibition of DNA polymerase activity by a third divalent metal ion, Front Mol BioSci 9, 824794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chen J, Malone B, Llewellyn E, Grasso M, Shelton PMM, Olinares PDB, Maruthi K, Eng ET, Vatandaslar H, Chait BT, Kapoor TM, Darst SA, and Campbell EA (2020) Structural basis for helicase-polymerase coupling in the SARS-CoV-2 replication-transcription complex, Cell 182, 1560–1573 e1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Brown JA, and Suo Z (2011) Unlocking the sugar “steric gate” of DNA polymerases, Biochemistry 50, 1135–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Joyce CM, and Steitz TA (1994) Function and structure relationships in DNA polymerases, Annu Rev Biochem 63, 777–822. [DOI] [PubMed] [Google Scholar]
- [46].Su Y, Egli M, and Guengerich FP (2016) Mechanism of ribonucleotide incorporation by human DNA polymerase eta, J Biol Chem 291, 3747–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Vaisman A, and Woodgate R (2018) Ribonucleotide discrimination by translesion synthesis DNA polymerases, Crit Rev Biochem Mol Biol 53, 382–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yoon H, and Warshel A (2016) The control of the discrimination between dNTP and rNTP in DNA and RNA polymerase, Proteins 84, 1616–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hillen HS, Kokic G, Farnung L, Dienemann C, Tegunov D, and Cramer P (2020) Structure of replicating SARS-CoV-2 polymerase, Nature 584, 154–156. [DOI] [PubMed] [Google Scholar]
- [50].Gao Y, Yan L, Huang Y, Liu F, Zhao Y, Cao L, Wang T, Sun Q, Ming Z, Zhang L, Ge J, Zheng L, Zhang Y, Wang H, Zhu Y, Zhu C, Hu T, Hua T, Zhang B, Yang X, Li J, Yang H, Liu Z, Xu W, Guddat LW, Wang Q, Lou Z, and Rao Z (2020) Structure of the RNA-dependent RNA polymerase from COVID-19 virus, Science 368, 779–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wang Q, Wu J, Wang H, Gao Y, Liu Q, Mu A, Ji W, Yan L, Zhu Y, Zhu C, Fang X, Yang X, Huang Y, Gao H, Liu F, Ge J, Sun Q, Yang X, Xu W, Liu Z, Yang H, Lou Z, Jiang B, Guddat LW, Gong P, and Rao Z (2020) Structural basis for RNA replication by the SARS-CoV-2 polymerase, Cell 182, 417–428 e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Rice LM, and Brunger AT (1994) Torsion angle dynamics: reduced variable conformational sampling enhances crystallographic structure refinement, Proteins 19, 277–290. [DOI] [PubMed] [Google Scholar]
- [53].Stein EG, Rice LM, and Brunger AT (1997) Torsion-angle molecular dynamics as a new efficient tool for NMR structure calculation, J Magn Reson 124, 154–164. [DOI] [PubMed] [Google Scholar]
- [54].Xie XS (2013) Biochemistry. Enzyme kinetics, past and present, Science 342, 1457–1459. [DOI] [PubMed] [Google Scholar]
- [55].English BP, Min W, van Oijen AM, Lee KT, Luo G, Sun H, Cherayil BJ, Kou SC, and Xie XS (2006) Ever-fluctuating single enzyme molecules: Michaelis-Menten equation revisited, Nat Chem Biol 2, 87–94. [DOI] [PubMed] [Google Scholar]
- [56].Min W, English BP, Luo G, Cherayil BJ, Kou SC, and Xie XS (2005) Fluctuating enzymes: lessons from single-molecule studies, Acc Chem Res 38, 923–931. [DOI] [PubMed] [Google Scholar]
- [57].Xie XS (2002) Single-molecule approach to dispersed kinetics and dynamic disorder: Probing conformational fluctuation and enzymatic dynamics, J Chem Phys 117, 11024–11032. [Google Scholar]
- [58].Yang H, and Xie XS (2002) Statistical approaches for probing single-molecule dynamics photon-by-photon, Chem Phys 284, 423–437. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.