

Abstract
Organ infiltration by donor T cells is critical to the development of acute graft-versus-host disease (aGVHD) in recipients after allogeneic hematopoietic stem cell transplant (allo-HCT). However, deconvoluting the transcriptional programs of newly recruited donor T cells from those of tissue-resident T cells in aGVHD target organs remains a challenge. Here, we combined the Serial Intravascular Staining technique with single-cell RNA-seq to dissect the tightly connected processes by which donor T cells initially infiltrate tissues and then establish a pathogenic tissue-residency program in a rhesus macaque allo-HCT model that develops aGVHD. Our results enabled creation of a spatiotemporal map of the transcriptional programs controlling donor CD8+ T cell infiltration into the primary aGVHD target organ, the gastrointestinal (GI) tract. We identified the large and small intestines as the only two sites demonstrating allo-specific, rather than lymphodepletion-driven, T cell infiltration. GI-infiltrating donor CD8+ T cells demonstrated a highly activated, cytotoxic phenotype while simultaneously developing a canonical tissue-resident memory T cell (TRM) transcriptional signature driven by IL-15/IL-21 signaling. We discovered expression of a cluster of genes directly associated with tissue invasiveness, including those encoding adhesion molecules (ITGB2), specific chemokines (CCL3, CCL4L1) and chemokine receptors (CD74), as well as multiple cytoskeletal proteins. This tissue invasion transcriptional signature was validated by its ability to discriminate the CD8+ T cell transcriptome of patients with GI aGVHD from those of GVHD-free patients. These results provide insights into the mechanisms controlling tissue occupancy of target organs by pathogenic donor CD8+ TRM cells during aGVHD in primate transplant recipients.
One sentence summary:
Flow cytometric and transcriptomic analyses reveal coordinated tissue-infiltration and tissue-residence of donor T cells that drive gastrointestinal GVHD.
INTRODUCTION:
T cell infiltration into secondary lymphoid organs and non-lymphoid tissues is central to T cell function and occurs during homeostatic tissue surveillance (1, 2), as well as during T cell-driven immunopathology (including auto- (3, 4) and allo-immune disorders (5, 6)) and T cell-mediated anti-tumor immune attack (7, 8). One of the best-studied clinical instances of T cell infiltration occurs during acute graft-versus-host disease (aGVHD), wherein donor T cells become activated, tissue-infiltrating, and highly cytotoxic after hematopoietic stem cell transplant (HCT) (9). During aGVHD, donor T cells first populate secondary lymphoid organs, where they undergo allo-antigen priming, and then home towards and infiltrate non-lymphoid GVHD-target organs (10-12). Upon infiltration, these cells induce the immunologic and clinical manifestations of aGVHD, including wide-spread organ damage (13). In mouse models, it has been demonstrated that donor T cells acquire tissue resident memory (TRM) features (including expression of CD103) during aGVHD (14-16). However, central questions remain concerning how these pathogenic TRM are distinguished from protective TRM, how donor TRM become activated and cause tissue damage, and perhaps most important, the mechanisms controlling the act of T cell invasion into target organs prior to their evolution into TRM. Determining the drivers of this dynamic process represents a critical unmet need, with broad relevance for alloimmunity, as well as for autoimmune diseases and immuno-oncology.
One of the major barriers to understanding the control of T cell infiltration into target tissues has been the inherent difficulty in capturing the key variable of time in this process. Time is a critical variable, given that T cells are actively homing towards and infiltrating into target organs, and are concordantly evolving a highly pathogenic transcriptional program. Without understanding the time-dependency of T cell infiltration, we cannot fully comprehend key drivers and their dynamics. Here, we have been able to deconvolute time as a variable during T cell infiltration and tissue destruction during primate aGVHD by applying Serial Intravascular Staining (SIVS) and pairing this with multiparameter flow cytometric and single-cell transcriptomic analyses. We performed these experiments using the non-human primate (NHP) aGVHD model (17-20), to best replicate the complex dynamics and molecular drivers of clinical tissue infiltration and disease in an out-bred immunologically experienced transplant recipient. This model takes advantage of the close functional similarity of the immune systems in NHP and humans, such that insights can be most rapidly translated to the clinic. SIVS allowed us to differentially label T cells longitudinally in individual animals during the process of active tissue infiltration, in order to ascertain the time-, tissue- and donor-dependence of T cell-mediated immune pathology during aGVHD.
RESULTS
Applying SIVS to a non-human primate aGVHD model
We built a NHP model of allogeneic HCT (allo-HCT) and aGVHD, in which a donor graft is transplanted into MHC-haploidentical recipient rhesus macaques pre-conditioned with myeloablative total body irradiation (17, 19). For the experiments described herein, transplant recipients received no post-transplant immunosuppression, which enabled interrogation of the natural history of aGVHD (Figure 1A, Table S1). Two control cohorts, encompassing autologous HCT (auto-HCT) recipients and untransplanted healthy controls (HC), were also analyzed (Figure 1A). In accordance with our previous data (17, 19), allo-HCT without immunosuppression resulted in early-onset lethal aGVHD, with skin and GI tract-predominant clinical disease (Figure 1B-D), coinciding with donor T cell engraftment (Figure 1E). Despite histopathologic evidence of lymphocytic infiltration into the liver (Figure S1A), allo-HCT recipients did not develop overt clinical signs of liver dysfunction/damage, with bilirubin, ALT, AST, GGT and alkaline phosphatase remaining relatively stable at the time of terminal analysis (Figure S1B). Thus, lethal GI aGVHD dominated the clinical outcomes in these experiments with transplant recipients reaching humane euthanasia endpoints prior to developing overt clinical liver disease. In contrast, auto-HCT controls survived long-term without aGVHD (Figure 1C). aGVHD clinical activity began on ~day +5 and peaked on day +7-8 (Figure 1B), similar to the kinetics of mouse alloimmune T cell trafficking to GVHD-target organs (13, 15). We performed a series of in vivo studies on day +8 post-transplant, to determine the mechanisms driving tissue infiltration during severe aGVHD.
Figure 1. Spatiotemporal compartmentalization of T cells in tissues during aGVHD.

(A-B) The experimental animals received autologous or allogeneic HCT as shown in (A) and clinical aGVHD scores for skin (B, left panel), gastrointestinal tract (B, middle panel) and liver (B, right panel) were calculated for GVHD (n=11 animals) and auto-HCT (n=9 animals) experimental NHP cohorts. *p<0.05, using two-way ANOVA with multiple comparison Holm-Sidak post-test. (C) Overall survival of NHP recipients following allo- and auto-HCT without immunosuppression is presented. Studies with pre-set experimental endpoints were censored. **p<0.01 using log-rank (Mantel-Cox) test. (D) aGVHD histopathology scores were measured for indicated organs from healthy control (HC) animals or HCT recipients on day +8 after allo- or auto-HCT without immunosuppression. *p<0.05, using multiple comparison Holm-Sidak test. (E) Donor chimerism in whole blood or FACS-purified peripheral blood T cells from allo-HCT recipient animals was quantified by microsatellite analysis. (F) Experimental schema and representative flow cytometry plot from HC lung that illustrate the SIVS method using fluorescent αCD45-antibodies to discern cellular compartmentalization and to track cellular migration into tissues are shown. (G) Relative distribution of CD8+ T cells among the intravascular compartment (Compartment-1; green dots), the recent infiltrating cell compartment (Compartment-2; red dots) and the tissue-localized compartment (Compartment-3; gray dots) in the indicated organs from healthy control animals (HC; n=4 animals), auto-HCT recipients (auto-HCT; n=4 animals) and allo-HCT recipients (GVHD; n=4 animals) on day +8 post-transplant. The green, and gray triangles and the red diamond at the bottom of the figure illustrate the pattern of the three compartments in the tissues examined. pLN, peripheral Lymph Nodes; medLN, mediastinal Lymph Nodes; mesLN, mesenteric Lymph Nodes. (H) Measurement of donor chimerism in the colon of an allo-HCT recipient animal, based on discordant expression of MHC-I allele MAMU-A001, in the three different compartments. (I) Donor CD8+ T cell chimerism in different compartments across different organs on day +8 following MAMU-A001-mismatched MHC-haploidentical allo-HCT (n=3 animals). The gray triangle at the bottom of the figure illustrates the pattern of donor chimerism in the tissues examined.
To dissect the spatiotemporal compartmentalization in aGVHD, we used a method to study T cell trafficking, known as ‘Serial Intravascular Staining’ (SIVS, described in the companion manuscript (21)). SIVS takes advantage of direct intravenous injection of sub-saturating doses of αCD45 antibodies, conjugated to different fluorescent tags, which label leukocytes in the systemic circulation at the time of injection and at different time-points prior to analysis (6 hours and 5 minutes, respectively). This strategy allowed us to differentiate 3 spatiotemporal compartments in multiple tissues (Figure 1F). The first compartment was identified by injecting a green-fluorescent AlexaFluor488 (AF488)-tagged αCD45 antibody 5 minutes prior to euthanasia, which restricted αCD45-AF488 labeling to the intravascular compartment (‘IVas+’ or ‘Compartment-1’) (Figure 1F and Figure S2). Compartments-2 and −3 were identified using a second αCD45 antibody, conjugated with the far red-fluorescent AlexaFluor647 (AF647), injected 6 hours prior to αCD45-AF488 injection. When far red-fluorescent αCD45-AF647 stained cells migrated out of the intravascular space into tissues, they retained their fluorescent tag, and thereby could be distinguished from cells that were in the intravascular space at the time of necropsy. These cells were identified as red (CD45-AF647+), but not green (CD45-AF488−), and are referred to as ‘Compartment-2’ or ‘recent infiltrating’ cells (Figure 1F and Figure S2). These two fluorescently-tagged cell populations could be further distinguished from a third population of cells, which remained negative for both the green and the red CD45 labels. These CD45-AF647−CD45-AF488− cells were present in the tissues before the 6-hour CD45-AF647 injection, and were therefore protected from αCD45 staining, and are referred to herein as ‘Compartment-3’ or ‘tissue-localized’ cells (Figure 1F and Figure S2).
Using the SIVS technique, we were able to deconvolute the organ-specific spatiotemporal compartmentalization of both CD4+ and CD8+ T cells after transplant (gated as shown in Figure S2, T cell subset definitions are provided in Table S2). Given the central role that CD8+ T cells play in aGVHD-mediated organ destruction (22, 23), we concentrated our discussion on the trafficking of these cells. Results from CD4+ T cells are shown in Supplementary Figures. We identified three different classes of organs/tissues based on their relative balance of Compartment-1, −2 or −3 CD8+ T cells (Figure 1G). We did not purify T cells from the skin in this study, and so we limited our discussion to visceral organs and tissues. Liver and lungs contained predominantly the intravascular, Compartment-1 T cells. However, unlike lung IVas+ cells, liver Compartment-1 CD8+ T cell displayed a TRM phenotype (Figure S3A, B), consistent with previous work documenting the localization of liver resident lymphocytes within a specific intra-sinusoidal niche (24, 25). Notably, during aGVHD, these liver Compartment-1 CD8+ T cells expressed the proliferation marker Ki67 as well as high proportions of cells expressing Granzyme B, suggesting an activated status (Figure S3C, D). Bone marrow and spleen contained predominantly the recently infiltrating, Compartment-2 cells (as well as a large proportion of Compartment-1 cells). Lymph nodes, kidneys, small intestine (with T cells isolated from the jejunum) and large intestine contained predominantly tissue-localized Compartment-3 CD8+ T cells (Figure 1G). Similar organ-specific compartmentalization was observed for CD4+ T cells (Figure S4A). Notably, these distributions did not result in statistically significant changes between the three experimental conditions (HC, auto-HCT and aGVHD) suggesting that spatiotemporal T cell compartmentalization may be a fundamental characteristic of organ immune structure that is not impacted by either homeostatic or allo-activated T cell trafficking on the time scales studied.
Combining SIVS and measurement of donor chimerism to identify T cells infiltrating GVHD target organs
While the Compartment-3-predominant organs (lymph nodes, kidney, and GI tract) all appeared similar through SIVS, by specifically tracking donor versus host T cell identity during SIVS, we discovered a critical distinction between these tissues after allo-HCT. This distinction was based on the balance between donor and host T cells in these tissues (Figure 1H), reflecting the pace of replacement of recipient T cells by donor cells after allo-HCT. These studies revealed that donor-origin Compartment-3 CD8+ and CD4+ T cells predominated in the lymph nodes on day +8 (Figure 1I and Figure S4B), consistent with rapid entry of transplanted T cells and replacement of host cells early after transplant. In contrast, a substantial fraction of host-origin T cells remained in the tissue-localized Compartment-3 cells of the large intestine, small intestine, kidney, and bone marrow (Figure 1I and Figure S4B) during the period of active infiltration of donor T cells into these tissues. Regulatory CD4+ T cells (TREG) represent an additional radiation-resistant population, with host TREG previously demonstrated to contribute to the reconstitution of the TREG pool following lethal irradiation (26, 27). To determine the relative contribution of host TREG to post-transplant reconstitution, we compared conventional CD4+ T cell (CD4+ TCONV) and CD4+ TREG donor chimerism in allo-HCT recipients after MAMU-A001-mismatched transplant. We found that, in the large intestine, a higher ratio of donor-versus-recipient TREG were present on day +8 compared to donor-versus-recipient CD4+ TCONV. This correlated with higher expression of the tissue-residence marker CD69 in TREG versus Tconv (Figure S4C-E). The greater donor TREG chimerism in the GI tract may be due to the counter-regulatory signaling that occurs during aGVHD (18), potentially driving these cells into the GI tract during disease.
Together, the data suggest a tenacious resident CD8+ T cell niche in the GI tract, kidney, and bone marrow, in which host T cells persist after transplant, even surviving lethal irradiation (28), and further link the timing of donor T cell infiltration into these tissues with the onset of clinical disease. Importantly, while a high proportion of host CD8+ T cells remained in Compartment-3 in the intestines, kidney and bone marrow, Compartment-2 was dominated by donor T cells in these same tissues, (with some variability observed in the bone marrow compared to other tissues, Figure 1I and Figure S4B). This dichotomy suggests that our analysis was able to catch donor T cells in the act of organ infiltration. This is particularly relevant for donor T cells infiltrating the intestines, since this infiltration was associated with lethal GI aGVHD (Figure 1B, C). This provided a strategy to interrogate the protein and gene expression patterns that mark these cells and to control their infiltration into aGVHD target tissues.
To rigorously distinguish pathogenic T cell infiltration during aGVHD from the physiologic T cell trafficking that occurs during homeostatic reconstitution, we employed a control: applying SIVS after autologous HCT. While our studies in allo-HCT identified multiple organs undergoing T cell infiltration after transplant, homeostatic expansion occurs alongside allo-activated T cell expansion during allo-HCT, given the exposure of recipients to pre-transplant lymphodepleting irradiation. Comparing the extent of tissue infiltration by Compartment-2 cells in allo-HCT versus auto-HCT controls (both analyzed on day +8 post-transplant) allowed us to deconvolute the role that homeostatic reconstitution played in T cell tissue infiltration. To complete this analysis, the extent of tissue migration was determined by calculating the percentage of tissue-infiltrating Compartment-2 T cells compared to total extravascular (IVas−, Compartment-2 + Compartment-3) cells (representative flow cytometry from the colon shown in Figure 2A). This analysis demonstrated that for many tissues (including peripheral and visceral lymph nodes, spleen, bone marrow, liver, lungs and kidneys), the amount of post-transplant CD8+ (Figure 2B-D) and CD4+ (Figure 2B-D, Figure S5A-E) T cell migration on day +8 was not different after allo-HCT compared to post-auto-HCT conditions, suggesting that most of the T cell trafficking was in response to lymphodepletion, which abolished competition for a niche in these organs. Indeed, when corrected using the auto-HCT controls, only two sites exhibited statistically significant (p<0.05) differential CD8+ T cell infiltration during aGVHD: the large and small intestines (Figure 2D). Only the large intestine was identified as a site of GVHD-specific CD4+ T cell infiltration (Figure S5E). This result is especially notable given that the GI tract has been repeatedly demonstrated in the NHP aGVHD model as key site of clinical and histopathologic disease (17, 19).
Figure 2. Dynamics of CD8+ T cell trafficking into GVHD-target organs following HCT in NHP.

(A) Mononuclear cells were isolated from different organs from HC animals (n=4) or from recipient animals on day +8 following auto-HCT (n=4 animals) or MAMU-A001-mismatched allo-HCT (n=3 animals). A representative gating tree depicts SIVS-based measurement of donor and host CD8+ T cell migration in MAMU-A001-mismatched allo-HCT transplantation. (B-D) Plots, depicting relative migration of donor and host CD8+ T cells from MAMU-A001-mismatched allo-HCT recipients, normalized to the migration of CD8+ T cells in untransplanted HC animals (B); relative migration of CD8+ T cells from autologous HCT recipients, normalized to the migration of CD8+ T cells in untransplanted HC (C) and relative migration of donor and host CD8+ T cells from MAMU-A001-mismatched allo-HCT recipients, normalized to the migration of CD8+ T cells following auto-HCT (D). pLN – peripheral Lymph Nodes, medLN – mediastinal Lymph Nodes, mesLN – mesenteric Lymph Nodes. *p<0.05 using t-test with Holm-Sidak multiple comparison correction.
Defining the characteristics of actively infiltrating T cells
The major impact that day +8 GI T cell infiltration had on clinical aGVHD was further underscored by the positive correlation between the extent of CD8+ T cell migration into the large and (to a lesser extent) small intestine and the severity of aGVHD pathology in individual transplant recipients – a correlation that was not observed in any other organ (Figure 3A). We therefore determined the prominent characteristics of donor cells infiltrating the GI tract, in order to understand the drivers of lethal aGVHD. As shown in Figures 3B-H, flow cytometry identified a number of unique attributes of intestinal tissue-infiltrating Compartment-2 CD8+ T cells. This included the observation that, while they were rigorously demonstrated to be extra-vascular and tissue-infiltrating by their lack of staining by the green-fluorescent IVas+ CD45 antibody, these intestinal Compartment-2 CD8+ T cells did not yet co-express the canonical TRM CD69+CD103+ phenotype (Figure 3B), consistent with their migratory phenotype. A proportion of these Compartment-2 cells did, however, express CD69 (Figure S6A, B), previously demonstrated to be up-regulated early in the process of TRM differentiation, prior to up-regulation of CD103 expression (29).
Figure 3. Migration of donor CD8+ T cells to the large and small intestine correlates with the severity of tissue pathology during aGVHD in NHP.
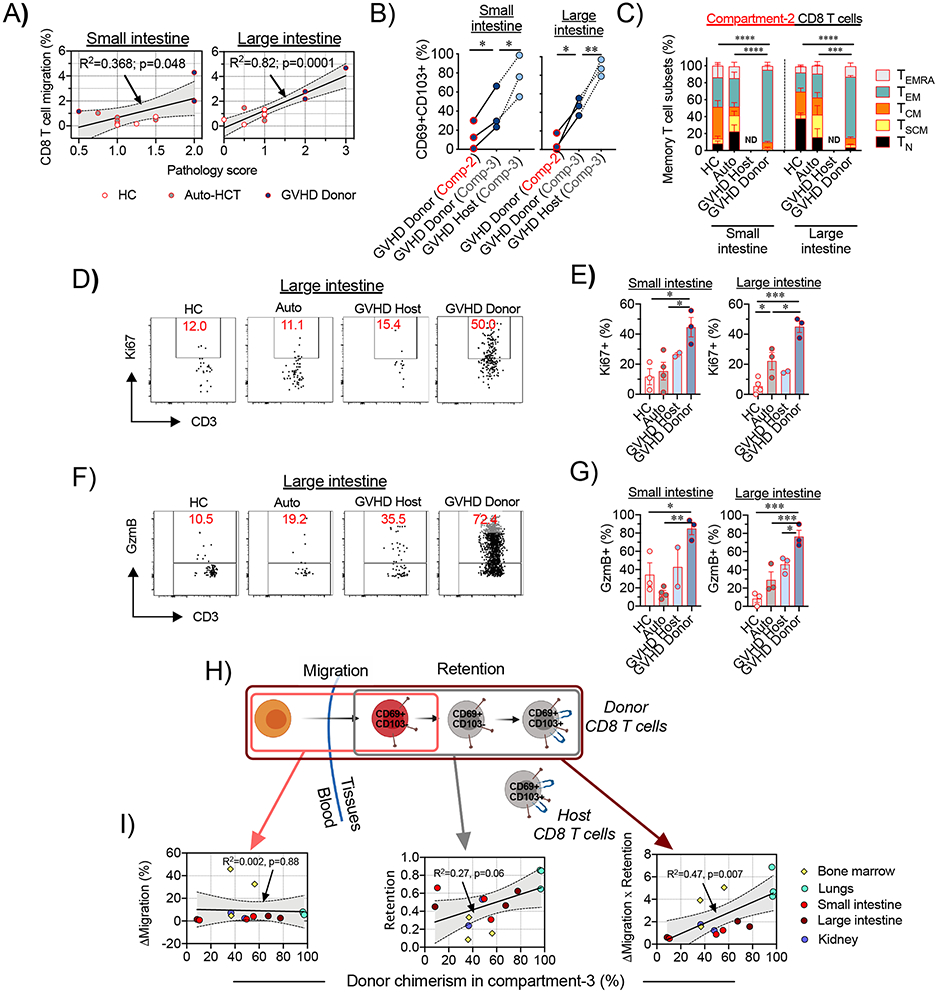
(A) Correlations between the migration of CD8+ T cells and GVHD histopathology scores in the small intestine (jejunum; left panel) and large intestine (colon; right panel) are shown. Lines with tinted areas indicate the 95% CI using linear regression. (B) Percentage of donor or host CD69+CD103+ CD8+ T cells were measured within the recent infiltrating compartment (Compartment-2) and the tissue-localized compartment (Comparment-3). Lines connect corresponding values that were obtained from the same animal. *p<0.05, **p<0.01 using paired t-test. Comp-2 = Compartment-2; Comp-3= Compartment-3. (C) Recent infiltrating (Compartment-2) CD8+ T cells in the small and large intestines from healthy control (HC) animals (n=4 for small intestine and n=3 for large intestine), auto-HCT cohort on day +8 (Auto, n=4 animals) and host (GVHD Host) and donor (GVHD Donor) cells following MAMU-A001-mismatched allo-HCT on day +8 (n=3 animals) were stained for CCR7, CD45RA and CD95 expression and the distributions between memory subsets are shown. ND – no data. (D-E) Representative flow cytometry plots (D) and summary data (E) depict Ki67 expression in recent infiltrating (Compartment-2) CD8+ T cells in the small and large intestine in different experimental cohorts. (F-G) Representative flow cytometry plots (F) and the summary data (G) depict Granzyme B (GzmB) expression in recent infiltrating (Compartment-2) CD8+ T cells in the small and large intestine in different experimental cohorts. For E and G: *p <0.05, **p<0.01, ***p<0.001 using one-way ANOVA with Holm-Sidak multiple comparison post-test. (H) Schematic representation of donor T cell immigration into GVHD-target non-lymphoid tissues and gradual acquisition of the CD69+CD103+ TRM phenotype. (I) Correlations between donor chimerism in tissue-localized CD8+ T cells (Compartment-3) and the difference between the extent of donor and host CD8+ T cell migration (ΔMigration, left panel), extent of acquisition of the CD69+CD103+ TRM phenotype in the immigrated donor CD8+ T cells (Retention, middle panel) and the difference between the extent of donor and host CD8+ T cell migration, adjusted to the extent of acquisition of the CD69+CD103+ TRM phenotype (Migration X Retention), across different non-lymphoid organs and tissues are shown. The lines with the tinted areas indicate the 95% CI using linear regression.
The rapid evolution of donor CD8+ T cells towards the canonical TRM phenotype in situ was evident, as donor CD8+ T cells acquired the TRM-associated CD69+CD103+ dual positive phenotype during their transition from Compartment-2 to Compartment-3, with the highest proportion of CD69+CD103+ co-expression in residual host CD8+ T cells in the intestine and other non-lymphoid tissues (Figure 3B, Figure S6C-D), confirming their TRM phenotype. Compartment-3 CD4+ T cells did not express CD103, (Figure S7), consistent with previous work documenting low expression of CD103 on CD4+ TRM cells (30).These data suggest that during intestinal aGVHD, donor-derived CD8+ TRM cells differentiate in situ from migratory precursors, likely in response to microenvironmental instructive signals (as has been demonstrated in other models (1, 31-33), including intestinal transplantation (6, 34)). Taken together, these results document the rapid acquisition of a TRM phenotype (within 8 days after transplant) as donor CD8+ T cells infiltrate and then occupy the GI tract.
Pathogenic hallmarks of actively infiltrating and TRM donor CD8+ T cells
Flow cytometric analysis demonstrated that a large proportion of intestinal tissue-infiltrating Compartment-2 CD8+ T cells were skewed towards a CCR7−CD45RA− effector-memory (TEM) phenotype compared to both HC and auto-HCT controls, while other CD8+ T cell subpopulations (including naïve, memory stem cells, central memory and effector memory-RA (subpopulation gating in Table S2 and Figure S2) were not overrepresented in Compartment-2 (Figure 3C). These intestinal Compartment-2 CD8+ TEM cells were highly proliferative and cytotoxic during aGVHD, as measured by their increased expression of Ki67 (Figures 3D-E) and Granzyme B (Figures 3F-G) compared to untransplanted and auto-HCT controls, or to host Compartment-2 cells, consistent with the observed clinical pathogenicity that accompanied donor T cell tissue infiltration. Up to 44.4% of donor-derived Compartment-2 CD8+ T cells demonstrated dual expression of Granzyme B/Perforin and up to 23.8% expressed Granzyme B/CD107a, confirming their high cytotoxic potential (Figure S6E-F). Notably, in the tissue-localized Compartment-3, CD8+ T cells from healthy and auto-HCT controls, as well as the pathogenic donor T cells during aGVHD, also exhibited a prominent CD45RA−CCR7− TEM phenotype (Figure S6G). However, only the Compartment-3 donor cells retained high expression of Ki67 and Granzyme B, (along with frequent co-expression of Perforin, and CD107a), documenting their distinction from homeostatic Compompartment-3 cells and consistent with their pathogenicity (Figure S6E, F, H, I).
Tissue T-cell chimerism is dependent on acquisition of TRM characteristics by CD8+ donor T cells
It has previously been demonstrated that the expression of CD103 is critical for donor CD8+ T cell pathogenicity in the GI tract, as it enables their retention in proximity to the epithelial layer, where these cells mediate their cytotoxic functions (14, 16, 35). However, whether the acquisition of TRM characteristics is also a foundational requirement for donor T cells to anchor in recipient tissues has previously not been a tractable question. By combining SIVS with donor chimerism measurements, we were able to directly address this issue. Our data demonstrate that the acquisition of the CD69+CD103+ phenotype was critical for establishing long-term residence in non-lymphoid tissues, as both the extent of donor CD8+ T cell migration into tissues and their rate of conversion into TRM cells (calculations described in detail in Methods) together, but not separately, defined tissue donor CD8+ T cell chimerism (Figure 3H-I).
Determining the transcriptomes of pathogenic donor TRM CD8+ T cells during aGVHD
SIVS allowed us to rigorously identify tissue-associated CD8+ T cells in healthy controls and after transplant. We next combined SIVS with single-cell RNA-Sequencing (scRNA-seq) to identify the transcriptional programs associated with the infiltration and pathogenicity of donor-derived tissue-associated CD8+ T cells during aGVHD. Because the data shown in Figure 2 and Figure S3 demonstrate that the only organs experiencing allo-specific T cell infiltration at day +8 after transplant were the small and large intestines, and because the large intestine demonstrated the most robust correlation between the influx of donor CD8+ T cells and tissue pathology (Figure 3A), we confined our scRNA-seq analysis to the large intestine. Prior to performing scRNA-seq, we flow cytometrically sorted viable, tissue-associated CD45+ cells from the large intestine of healthy control animals (n = 4) and from the aGVHD cohort (n = 4) by gating on IVas− (non-Compartment-1) cells (Table S3). Auto-HCT recipients had too few T cells in the GI tract on day 8 post-transplant to reliably purify by cell sorting, and thus were not included in this analysis. Where possible, donor and host cells were flow cytometrically purified based on disparate expression of MAMU-A001 (in two of the four aGVHD animals analyzed, Table S1). In the two remaining allo-HCT recipients, we used computational methods to discern donor and host cells based on sex-mismatch between the transplant pairs, using Y-chromosome-derived transcripts (see Methods for details). Using this methodology, we successfully reconstructed transcriptomes from 21,490 single cells, followed by the identification of 14,185 T cells, based on their expression of canonical T cell genes (Figure S8). To identify 4,715 CD8+ T cells, we used the annotation tool VISION to score each cell using a CD8+ vs CD4+ signature (36) and retained high-scoring clusters.
To systematically determine the features that differentiated pathogenic donor CD8+ TRM in the GI tract from physiologic host or HC CD8+ TRM, we used VISION to score a TRM-signature from Milner et al., 2017 (37) in all donor, host, and HC CD8+ T cells, thereby identifying the continuum of TRM transcriptional programing at a single-cell level in all GI-associated CD8+ T cells (Figure 4A-B, Figure S9A). We chose the Milner TRM signature because it identifies the key genetic regulators of TRM differentiation from multiple tissues regardless of a cell’s activation status, thus providing the most accurate measure of TRM transcriptional features. As shown in Figure 4A-B, and in agreement with flow cytometric analysis (Figure 3B), donor-derived GI CD8+ T cells had a lower mean TRM score compared to either host- or HC-derived cells (p<0.001 using the Wilcoxon test), consistent with there being a higher proportion of actively infiltrating (rather than established TRM) donor cells during GI aGVHD. Given that the Milner signature was derived from studies in mice, we also compared the NHP donor, host and HC CD8+ T cells to two human-derived GI-tract-specific tissue-resident T cell signatures (Zhang_CD8_C05_CD6 and Zhang_CD8_C06_CD160) (38), which confirmed the higher TRM differentiation status of large intestine host and HC CD8+ T cells compared to donor CD8+ T cells (Figure S9). Notably, the murine Milner signature was highly correlated with both of the human-derived intestinal TRM cell signatures (Figure S9B). A third human-derived, but not GI-specific, TRM signature, identified by Kumar et al (39), also showed significant correlation with the Milner TRM signature (p<0.0001), but this non-GI-derived signature did not discriminate intestinal donor CD8+ T cells from residual host CD8+ T cells and CD8+ T cells from HC large intestines (Figure S9C-E). This observation may have been expected, given previous work that has demonstrated that the transcriptomic profiles of TRM cells display considerable interorgan variation (40).
Figure 4. Transcriptomic profile of donor-derived CD8+ TRM cells in the large intestine during aGVHD in NHP.

(A) CD8+ T cell census (n=4,715 cells), colored by experimental cohort, was clustered in Uniform Manifold Approximation and Projection (UMAP) space. (B) Enrichment scores for the TRM-signature from Milner et al, (2017) (37)) were calculated for CD8+ T cells from healthy control (HC) animals, and host and donor CD8+ T cells on day +8 following allo-HCT. Horizontal lines represent the 25th and 75th percentiles for enrichment scores of the healthy control cohort, which were used as cut off values to determine TRM-low and TRM-high cells. (C) Differentially expressed (DE) genes were identified between donor CD8+ TRM-high cells and their counterparts from healthy control (HC) animals (HC; top panel) and host CD8+ T cells (bottom panel). (D) Pathway analysis was performed using Ingenuity Pathway Analysis on differentially expressed genes between donor CD8+ TRM-high cells, and their counterparts from healthy controls and host CD8+ T cells. Signaling pathways with positive enrichment scores and p<0.05 using a t-test with the Benjamini-Hochberg correction are depicted. (E) CD8+ T cell census (n=4,715 cells), colored by the enrichment score for the GVHD signature from Ichiba et al, (2003) (52), was clustered in UMAP space. (F) Enrichment scores for the aGVHD signature (from Ichiba et al, (2003) (52)) were calculated for healthy controls (HC), host, and donor CD8+ TRM-high cells. (G) Predicted up-stream regulators were determined using Ingenuity Pathway Analysis tool performed on DE genes between donor CD8+ TRM-high cells and their counterparts from healthy controls (left panel) and host CD8+ T cells (right panel). (H) Expression of IL-21R in Compartment-2 and Compartment-3 donor and host CD8+ T cells from the large intestines of MAMU-A001-mismatched allo-HCT recipients on day +8 (n=3). *p<0.05 using paired t-test. (I) Spleen cells, isolated from healthy control (HC) animals or animals with aGVHD on day +8 after allo-HCT, were incubated with the indicated cytokines for 48 hours. Then, percentage of CD69+CD103+ CD8+ T cells was measured by flow cytometry. *p<0.05, using one-way paired ANOVA and Holm-Sidak multiple comparison post-test.
To further determine what differentiated pathogenic donor CD8+ T cells from both host and HC CD8+ TRM, we first defined a TRM-high cutoff score by identifying the upper TRM score quartile from HC CD8+ T cells (Figure 4B) and then applied this score cutoff to both the donor and host CD8+ T cells, to identify TRM-high subpopulations in each. This allowed us to probe the transcriptional differentiators between pathogenic donor TRM-high cells and both host and HC TRM controls through differential expression (DE) calculations, followed by pathway enrichment analysis and classification of up-stream regulators. This pipeline distinguished the following features of pathogenic CD8+ TRM cells: they develop rapidly (within 8 days) after allo-HCT, and they consist of pathogenic donor cells that simultaneously enact TRM transcriptional programming in addition to upregulation of key T cell activation programs. These programs include upregulation of specific cellular metabolic pathways (including oxidative phosphorylation and mitochondrial respiration, which have been demonstrated to be central to both TRM development (41-43) and aGVHD pathogenesis (44, 45)), co-stimulatory signaling pathways (including aGVHD-promoting ICOS- (46, 47), IL-2- (48, 49) and OX40- (20, 50) signaling pathways) and cytotoxicity pathways (51)) compared to their physiologic counterparts (Figure 4C-D, Tables S4-S5). We also identified a higher enrichment score for the aGVHD pathogenicity signature adopted from Ichiba et al (52) in donor TRM-high cells in comparison with host and HC TRM-high cells (Figure 4E-F), further substantiating the pathogenicity of the donor TRM. Of note, our analysis of the up-stream regulators that control the activation pathways in donor TRM-high CD8+ T cells revealed involvement of IL-15 and IL-21 cytokine signaling (Figure 4G, Table S6), suggestive of a central role for these cytokines in TRM differentiation (particularly during lymphopenic conditions) (29, 53), as well as their pathogenic role during aGVHD (54-58). We further confirmed that both IL-15 and IL-21 could induce maturation towards a CD69+CD103+ TRM phenotype in NHP splenocytes in vitro. We found that IL-15 induced TRM differentiation in splenocytes isolated from both HC animals and from those with aGVHD, whereas IL-21 promoted TRM differentiation only in aGVHD-derived cells (Figure 4H). Given our observation of the specificity of IL-21 in the differentiation of CD8+ TRM during aGVHD, we further investigated this biological pathway. IL-21R was expressed on both Compartment-2 and Compartment-3 donor CD8+ T cells in the large intestine during aGVHD, with higher expression on the surface of recently infiltrating Compartment-2 cells. In contrast, expression of IL-21R on residual host cells was low (Figure 4I). Of note, both IL-21R+ and IL-21R− donor CD8+ T cells in Compartments 2- and 3- demonstrated similar expression of Ki67, Perforin and Granzyme B, suggesting that, as previously reported in mice (54, 58), expression of IL-21R does not differentiate between proliferating/cytotoxic and more quiescent cells. We also measured the expression of IL21 itself and found a proportion of donor cells in the large intestine expressing this gene (8.69%) following allo-HCT, thus providing a local source of this cytokine during GI aGVHD (Figure S10D-F). Together, these data identify multiple transcriptional pathways and up-stream regulators that are strongly associated with aGVHD-mediated tissue destruction, providing the first direct link between TRM differentiation and donor T cell pathogenesis in primate aGVHD.
Identifying transcriptional control of actively infiltrating donor CD8+ T cells
In addition to identifying the drivers of pathogenic CD8+ TRM, this study also provided an opportunity to interrogate the transcriptional hallmarks of actively tissue-infiltrating pathogenic CD8+ T cells at unprecedented specificity. To accomplish this, based on the SIVS results (Figure 3B) which demonstrated that Compartment-2 actively infiltrating cells display a migratory, rather than TRM-like, phenotype, we first identified a TRM-low cut-off score (by identifying the lower Milner TRM score quartile from HC CD8+ T cells, Figure 4C), and then applied this score cut-off to both donor and host CD8+ T cells. This allowed us to isolate donor cells that were tissue associated, given that they were first sorted as non-intravascular (Compartment-1-negative) prior to scRNA-seq, and simultaneously did not yet display features of TRM. We then defined the transcriptional differentiators between pathogenic donor TRM-low cells and both host and HC TRM-low controls by applying differential expression calculations (Figure 5A, Tables S7-S9). Similar to their donor TRM-high counterparts, donor CD8+ TRM-low cells demonstrated high enrichment scores for the aGVHD pathogenicity signature from Ichiba et al (52) (Figure S10G). In agreement with flow cytometric analysis (Figure 3F-G and Figure S6E-F), transcriptome analysis demonstrated that these cells co-expressed genes encoding multiple cytotoxic mediators (Figure S10H), also consistent with their pathogenic status. Of note, a direct comparison of donor CD8+ TRM-low cells with their TRM-high counterparts revealed that while the TRM-low cells had similar expression of the cytotoxicity-related transcripts GZMA, GZMB, and PRF1, they demonstrated lower expression of the genes encoding TNFRSF1B (59), ID2 (60), IFNG (61) and RGS1 (62) (Figure S10I and Table S10), which have been demonstrated to play major roles in the pathogenicity of both GI GVHD and T cell-driven colitis. Moreover, and consistent with the role for IL21 in TRM differentiation described above (Figure 4H), our analysis of predicted up-stream regulators of TRM-high versus TRM-Low CD8+ T cells confirmed activation of IL-21 signaling in TRM-High > TRM-Low (Table S11). Together, these data suggest that while Compartment-2 and Compartment-3 CD8+ T cells both display pathogenic characteristics, Compartment 3 cells express additional genes associated with auto- and allo-immunity, likely enabling them to perpetuate intestinal inflammation during GI GVHD.
Figure 5. Transcriptomic profile of donor tissue-infiltrating CD8+ T cells in the large intestine during aGVHD in NHP.

(A) Differentially expressed (DE) genes were identified between donor CD8+ TRM-low cells and their counterparts from healthy control animals (HC; left panel) and host CD8+ T cells (right panel). (B) Venn diagram illustrating the identification of the donor TRM-low-specific gene signature. (C-D) Pathway analysis was performed using Ingenuity Pathway Analysis on the donor TRM-low-specific gene signature. Signaling pathways with p<0.05 using a t-test with the Benjamini-Hochberg correction (C) and the predicted top 25 up-stream regulators (D) are depicted. (E) Functional annotation of migration-related genes from donor TRM-low-specific gene signature. (F) GSEA plot depict the enrichment of two TRM-low gene signatures in patients with grade 3-4 aGVHD (n=4) compared to patients with grade 0-2 aGVHD (n=38). The curve labeled “TRM-Low Donor vs Host and HC UP” encompasses genes over-represented in donor TRM-low CD8+ T cells in comparison with both host and HC TRM-low counterparts – blue line). The curve labeled “Donor TRM-Low specific signature UP” encompasses genes over-represented exclusively in the donor TRM-low CD8+ T cell comparison (but not present in the comparison of donor TRM-high CD8+ T cells versus both host and HC TRM-high counterparts) – red line). NES – normalized enrichment score.
Finally, we focused on the TRM-low-specific transcriptome by identifying those differentially expressed genes that were unique to the TRM-low donor versus control comparisons (and not identified in the TRM-high versus control comparisons). This analysis distinguished 53 transcripts (46 up-regulated genes and 7 down-regulated genes) that were specifically dysregulated in donor CD8+ TRM-low cells, as well as their associated pathways and up-stream regulators (Figure 5B-D, Tables S12-S14). Regulatory network analysis again identified IL-15, as well as type I and type II Interferons (and their corresponding signals) as key up-stream regulators controlling the transcriptional programming of these donor tissue-infiltrating T cells (Figure 5D, Table S14). Pertinent to the goal of defining the drivers of T cell infiltration, we identified a subset of genes that have been shown to be critical for cellular adhesion, migration and tissue infiltration in other clinical scenarios. These included chemokines, as well as regulators of their secretion and receptors (CCL3 (63), CCL4L1 (64), GZMM (65), CD74 (66)), surface adhesion receptors and regulators of their expression (LTB (67) and ITGB2 (68)); and cytoskeleton components and regulators (ACTB (69), ACTG1 (69), CAP1 (70), COTL1 (71), ISG15 (72), LSP1 (73), PFN1 (74)) (Figure 5E). These data thus provide the first transcriptional map of this unique subpopulation of actively infiltrating donor CD8+ T cells, nominating a new class of molecules that enable pathologic CD8+ T cell tissue invasion during GVHD.
Validating the clinical relevance of the NHP tissue-infiltration transcriptional signature
To validate that the genes and pathways identified through the NHP experiments were relevant to human disease, we compared our NHP transcriptional signatures to a CD8+ T cell signature generated from patients with aGVHD. The human aGVHD signature was constructed from peripheral blood CD8+ T cells that were sorted on days +21 and +28 post-transplant from 42 patients receiving an unrelated-donor HCT and standard calcineurin inhibitor/methotrexate aGVHD prophylaxis (Table S15). Twenty-eight of these patients developed Grade 2-4 aGVHD (with 27 of 28 having either upper or lower GI disease). All patients with Grade 3-4 aGVHD demonstrated lower GI disease (Table S15). The resulting transcriptomes were dichotomized between patients who developed severe Grades 3-4 aGVHD versus those with Grades 0-2 aGVHD (Figure 5F) or those with moderate-severe Grades 2-4 aGVHD versus those with Grades 0-1 aGVHD (Figure S10L). These analyses demonstrated significant overlap (q=0.02) between the migratory donor CD8+ TRM-low signatures and the human CD8+ T cell aGVHD signature (Figure 5F, Figure S10L, Table S16). These results provide strong evidence for the relevance of the NHP data to human aGVHD, and they demonstrate the ability to probe the peripheral blood for signatures relevant to the migrating pathogenic cells that ultimately infiltrate the GI tract during clinical aGVHD. Thus, while the peripheral blood did not contain the complete pathologic signature derived from GI-tract infiltrating T cells (Table S16), the peripheral blood signature complemented the organ-specific results, which successfully dichotomized patients with moderate-to-severe clinical disease.
Discussion
Here, the application of SIVS plus scRNA-seq to NHP aGVHD has enabled the interrogation of tissue-localized T cell immunity at an unprecedented depth of detail in an outbred, pathogen-exposed, immunologically-experienced system. By applying serial infusions of differentially-labeled anti-CD45 antibodies (‘SIVS’, (21)), we have been able to include time as a variable in our analysis, facilitating the detection of three spatiotemporally-defined T cell compartments: (1) T cells that were intravascular at the time of analysis; (2) T cells that were intravascular six hours prior to analysis, and subsequently infiltrated into target tissues; and, (3) T cells that were tissue-associated for at least six hours prior to analysis and did not acquire either of the two CD45 labels. By overlaying our ability to distinguish donor versus host cells after allo-HCT onto these three compartments, we have developed a highly sensitive and specific paradigm to identify pathogenic donor CD8+ T cells, either in the act of organ infiltration or in the act of tissue destruction during GI aGVHD.
Our observations provide strong support for the rapid evolution of a pathogenic TRM transcriptional program in donor CD8+ T cells after allo-HCT, representing a striking counterpart to recent observations documenting the longevity of donor-origin TRM in transplanted lung and intestinal allografts (5, 6, 34). Our results demonstrate that within eight days after allo-HCT, donor T cells infiltrate target organs and rapidly exhibit protein and transcriptional TRM hallmarks. However, despite clear expression of a TRM program, when compared to either host or HC GI-tract CD8+ T cells, the donor CD8+ TRM are clearly not homeostatic. They upregulate multiple transcriptional networks driving T cell activation, including evidence of both metabolic reprograming toward mitochondrial respiration and massively upregulated cytotoxicity pathways. Our results also identify IL-15 and IL-21 as key up-stream regulators, which together coordinate the transcriptional programming responsible for the pathogenic donor CD8+ TRM differentiation. These two cytokines have been shown to drive aGVHD in mouse models (54-57) and IL-15 signaling is required for CD8+ TRM generation in multiple barrier tissues (29, 76). Compared to IL15, the role of IL-21 in the generation of TRM cells is far less established (53), and in this study, we provide new evidence that IL-21 can drive TRM differentiation from circulatory precursors during aGVHD, but not under homeostatic conditions. Notably, the demonstration of redundant T cell activation programs may provide an explanation for the well-documented difficulty of reversing ongoing clinical aGVHD: pathogenic GI-tract TRM are not only physically sequestered from therapeutic agents (1, 2), they are also potentially protected from monomorphic treatment approaches, given the non-overlapping pathways that they activate.
This study also enabled the identification of the transcriptional network controlling donor CD8+ T cells ‘caught in the act’ of tissue infiltration. These rare cells have previously been intractable to detailed examination, because of their small numbers and their fleeting nature. Through the combination of SIVS and scRNA-seq, we were able to identify and deeply interrogate this critical cell population. By enabling flow-sorting of GI-tract T cells that were Compartment-1 (iVas)-negative, SIVS allowed us to rigorously exclude intravascular cells from our analysis. While Compartment-2 cells were too rare in the GI tract for sorting, the fact that these cells were rigorously demonstrated to be non-TRM by flow cytometry allowed us to transcriptionally identify them by their TRM-low signature. Transcriptional analysis subsequently identified central features of these actively infiltrating cells. While these include several ‘usual suspects’ in aGVHD, comprising co-stimulatory/co-inhibitory pathways (79), the immunoproteosome (80) and several targetable metabolic pathways (44, 45, 81), one of the most important findings was the transcriptional program of adhesion/extravasation/migration that these actively infiltrating cells initiated. These represent distinct pathways compared to donor CD8+ T cells that had already enacted TRM programming, as well as from both host and HC Compartment-2 cells. The identification of tissue infiltration-associated gene networks may have relevance to other immune phenomena: These include solid organ transplant rejection and auto-immune diseases, which both require infiltration and long-term tissue persistence of pathogenic cells (82). In addition, the discovery of a CD8+ T cell infiltration gene network may be used to improve the ability of anti-tumor T cells to more successfully infiltrate their targets and survive in the tumor microenvironment, given the recently-discovered mechanistic connections between tumor-infiltrating lymphocytes and TRM biology (7, 8).
This study has the following limitations: First, we studied the non-prophylaxed ‘natural history’ of aGVHD, and mechanisms of infiltration and pathogenic T cell residency may be altered by immunoprophylaxis. Second, this study was limited to two anti-CD45 infusions, six hours apart, thus constraining our ability to discriminate shorter- versus- longer-lived tissue-resident cells. Additional CD45-antibody infusions will enable more precise dissection of the pace/character of pathogenic tissue residency. Finally, the scRNA-seq experiments focused only on the GI tract. Mechanisms controlling aGVHD are expected to be at least partially organ-specific; thus, further work with other target tissues will add important insights.
In summary, by combining SIVS with scRNA-seq we have interrogated pathogenic donor CD8+ T cells in the act of infiltrating and causing tissue destruction during GI aGVHD. Our results document a transcriptional network defining T cell infiltration during this disease and validate the clinical relevance of this network. They nominate a new class of targets by which to control the act of T cell infiltration, which constitutes a necessary prelude to T cell-mediated organ damage.
Materials and Methods
Study Design.
This was a prospective study in rhesus macaques designed to determine the biological role of T cell migration and to identify the molecular signature of pathogenic tissue-infiltrating T cells in aGVHD-induced tissue immunopathology. Several cohorts of transplant recipients (assigned in an unblinded, non-randomized manner) were studied: (1) autologous transplants (abbreviated as ‘Auto-HCT’, n = 9 for clinical analysis and n=4 for immunological analysis); (2) allogeneic transplants with no GVHD prophylaxis (abbreviated as ‘allo-HCT’ and ‘aGVHD’, n = 11 for clinical analysis and n = 4, for immunological analysis, including 3 MAMU-A001-mismatched transplantations); (3) a control cohort of healthy, immunologically naïve macaques (abbreviated as ‘HC’, n= 4 for immunological analysis). Auto- and allo-HCT recipients used for SIVS experiments were euthanized at pre-determined experimental endpoint on day +8 and were censored from survival analysis. Animal demographic parameters, transplant characteristics, and doses of αCD45 antibodies, administered in vivo are shown in Table S1. Sample size calculations were not performed for the present study, given existing data from multiple previous transplant cohorts published by our group (17-20), documenting the substantial differences in median GVHD-free and overall survival after MHC haplo-identical allogeneic HCT versus autologous transplantation in NHP. Blinding was performed on all pathologic analysis and on the initial analysis of flow cytometry data, as well as on transcriptomic sample handling and data processing.
NHP Ethics Statement.
This study was conducted in strict accordance with USDA regulations and the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. It was approved by the Biomere Inc and the University of Washington Institutional Animal Care and Use Committees.
Human studies ethics statement.
The patients described in this study were enrolled in clinical trial #NCT01743131 that was conducted according to the principles set forth in the Declaration of Helsinki and that was approved by participating center IRBs. Written informed consent was received from all participants. Hematopoietic Cell Transplantations (HCT) in NHP. We used our previously described strategy for HCT in rhesus macaques (17-19), described in detail in Supplementary Materials and Methods.
αCD45 infusions.
Purified αCD45 (clone MB4-6D6) was purchased from Miltenyi Biosciences. Antibodies were conjugated in Dr. Roederer’s laboratory to AlexaFluor dyes. Conjugated antibodies were diluted in sterile Normal Saline Solution, and 5 ml were slowly infused over 5 minutes to animals via a central catheter placed in the femoral vein (auto-HCT and allo-HCT cohorts) or to sedated animals via a peripheral catheter placed in saphenous vein (HC cohort). To track lymphocyte migration, αCD45 antibodies conjugated with AlexaFluor647 (αCD45-AF647) were administered 6 hours before euthanasia/tissue collection at a dose of 30 mcg/kg. To discriminate tissue-residing cells from cells remaining in the vasculature, αCD45 antibodies, conjugated with AlexaFluor488 (αCD45-AF488), were administered 5 minutes before euthanasia/tissue collection at a dose of 30 or 60 mcg/kg (Figure 1, Figure S2; Table S1). Blood samples were drawn from the catheter before and 5 minutes after each αCD45 antibody injection to ensure that all blood leukocytes were uniformly labeled with the injected antibodies.
Necropsy and tissue processing.
Tissues from euthanized animals were processed after perfusion with PBS using enzymatic digestion and mechanic separation as described in the Supplementary Materials and Methods.
Immune Analysis.
Blinding was performed on all pathologic analysis. Flow cytometry data were collected using BD FACS LSRFortessa and analyzed using FloJo 10 in an unblinded manner. The gating strategy and the assessment of cellular migration using intravascular aCD45 labeling are described in Figure S2, Table S2 and Supplementary Materials and Methods.
Transcriptomic analysis.
Single cell libraries from sorted samples were generated using the 10X Chromium 3’ platform, and then sequenced using a NovaSeq S2 (Illumina). Single-cell RNA-seq reads were aligned to a transcriptome based on the Mmul 10 Macaca mulatta reference genome with Ensembl's (v98) annotations and the analysis was performed in an unblinded manner using Seurat v3.1, VISION, clusterProfiler and IPA as described in the Supplementary Materials and Methods. Bulk RNA-Seq from human samples were trimmed using Trimmomatic v.39 (83) and aligned with kallisto v.0.46.1 (84). Analysis was performed using DESeq2 (85) and clusterProfiler(86).
Statistical analysis
Both paired and unpaired Student’s t-test were used for comparing two groups, where appropriate. One-way ANOVA with Holm-Sidak multiple comparison post-test or two-way ANOVA analysis with Holm-Sidak multiple comparison post-test were used for comparing multiple groups, where appropriate. Groups were considered significantly different when p<0.05.
Supplementary Material
Table S1. Transplant characteristics
Table S2. List of antibodies used, flow cytometry panels and flow-based definitions of lymphocyte populations.
Table S3. scRNA-seq sample information and QC metrics.
Table S4. Differentially expressed genes between donor, host and HC CD8+ T cells with high enrichment scores for the TRM signature.
Table S5. Pathway analysis of differentially expressed genes between donor, host and HC CD8+ T cells with high enrichment scores for the TRM signature.
Table S6. Predicted activated up-stream regulators in donor CD8+ T cells with high enrichment scores for the TRM signature in comparison with their HC and host counterparts.
Table S7. Differentially expressed genes between donor, host and HC CD8+ T cells with low enrichment scores for the TRM signature.
Table S8. Pathway analysis of differentially expressed genes between donor, host and HC CD8+ T cells with low enrichment scores for the TRM signature.
Table S9. Predicted activated up-stream regulators in donor CD8+ T cells with low enrichment scores for the TRM signature in comparison with their HC and host counterparts.
Table S10. Differentially expressed genes between donor CD8+ T cells with low and high enrichment scores for the TRM signature.
Table S11. Predicted activated up-stream regulators in donor CD8+ T cells with low enrichment scores compared to cells with high enrichment scores for the TRM signature.
Table S12. Gene signature of donor CD8+ T cells with low enrichment scores for the TRM signature, defined as shown in Figure 5B.
Table S13. Pathway analysis on the gene signature of donor CD8+ T cells with low enrichment scores for the TRM signature.
Table S14. Predicted activated up-stream regulators based on the gene signature of donor CD8+ T cells with low enrichment scores for the TRM signature.
Table S15. Clinical characteristics of patients from the ABA2 clinical trial (NCT01743131), included in the current study.
Table S16. Results of GSEA analysis comparing patients from the ABA2 clinical trial (NCT01743131) with Grades 2-4 and 3-4 aGVHD versus patients with Grades 0-1 and 0-2 aGVHD, respectively.
Table S17. scRNA-seq-derived gene sets used for GSEA analysis.
Data File S1. Primary data
Figure S1. Liver function tests in recipients following autologous or allogeneic HCT in NHP.
Figure S2. Gating tree for flow cytometric identification and characterization of T cells within different spatiotemporal compartments using Serial Intravascular Staining (SIVS) in NHP.
Figure S3. Phenotypic characteristics of liver intravascular Compartment-1 T cells in NHP.
Figure S4. Compartmentalization of CD4+ T cells at steady state and after HCT in NHP.
Figure S5. Migration of CD4+ and CD8+ T cells at steady state and after HCT in NHP.
Figure S6. Phenotypic characterization of Compartment-2 and Compartment-3 CD8+ T cells in NHP.
Figure S7. Phenotypic characteristics of intestinal CD4+ T cells in NHP.
Figure S8. Transcriptomic identification of intestinal CD8+ T cells in NHP.
Figure S9. Transcriptomic identification of intestinal TRM-High and TRM-Low CD8+ T cells from large intestine in NHP.
Figure S10. Immunophenotypic and transcriptomic characteristics of CD8+ T cells from the large intestine.
Figure S11. Threshold testing of TRM-High and TRM-Low CD8+ T cells.
ACKNOWLEDGEMENTS
Funding: This project was funded by the following grants: NIH 2U19 AI051731, NIH 2R01 HL095791, NIH/FDA, R01 FD004099, CURE Childhood (https://curechildhoodcancer.org) to LSK, ASTCT New Investigator Award, CIBMTR/Be the Match Foundation Amy Strelzer Manasevit Research Program to VT, NIH K23HL136900 to BW, NIH R01 HL56067; NIH R37 AI34495 to BRB, Sloan Fellowship in Chemistry, NIH 2R01 HL095791 to AKS, Richard and Susan Smith Family Foundation, the HHMI Damon Runyon Cancer Research Foundation Fellowship (DRG-2274-16), the AGA Research Foundation’s AGA-Takeda Pharmaceuticals Research Scholar Award in IBD – AGA2020-13-01, the HDDC Pilot and Feasibility P30 DK034854, and the Food Allergy Science Initiative to JOM. The authors gratefully acknowledge the veterinary and animal husbandry staff at the Washington National Primate Research Center, and Harry Leung at the Boston Children’s Hospital/Harvard Medical School Microscopy Core.
Footnotes
Competing interests: BW, MQ and AL report personal fees from Bristol Myers Squibb, JTH reports research funding and personal fees from Bristol Myers Squibb, JOM reports compensation for consulting services with Cellarity. BRB serves as an advisor to Kamon Pharmaceuticals, Inc, Five Prime Therapeutics Inc, Regeneron Pharmaceuticals, Magenta Therapeutics and BlueRock Therapeuetics; he reports research support from Fate Therapeutics, RXi Pharmaceuticals, Alpine Immune Sciences, Inc, Abbvie Inc., Leukemia and Lymphoma Society, Childrens' Cancer Research Fund, KidsFirst Fund and is a co-founder of Tmunity. AKS reports compensation for consulting and/or scientific advisory board membership from Merck, Honeycomb Biotechnologies, Cellarity, Repertoire Immune Medicines, Ochre Bio, and Dahlia Biosciences. LSK is on the scientific advisory board for HiFiBio; she reports research funding from Kymab Limited, Magenta Therapeutics, BlueBird Bio, and Regeneron Pharmaceuticals. She reports consulting fees from Equillium, Vertex, Novartis Inc, EMD Serono, Gillead Sciences, Interrius, Takeda Pharmaceuticals and Bristol Myers Squibb. The other authors declare no competing interests.
Data availability:
All data associated with this study are present in the paper or supplementary materials. The NHP scRNA-seq data were deposited in the NCBI’s Gene Expression Omnibus database (GSE142483). The RNA-seq data from the ABA2 clinical trial were deposited in the NCBI’s Gene Expression Omnibus database (GEO157280).
References
- 1.Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, Wang J, Casey KA, Barber DL, Kawamura KS, Fraser KA, Webby RJ, Brinkmann V, Butcher EC, Newell KA, Ahmed R, Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med 207, 553–564 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCully ML, Kouzeli A, Moser B, Peripheral Tissue Chemokines: Homeostatic Control of Immune Surveillance T Cells. Trends Immunol 39, 734–747 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Zundler S, Becker E, Spocinska M, Slawik M, Parga-Vidal L, Stark R, Wiendl M, Atreya R, Rath T, Leppkes M, Hildner K, Lopez-Posadas R, Lukassen S, Ekici AB, Neufert C, Atreya I, van Gisbergen K, Neurath MF, Hobit- and Blimp-1-driven CD4(+) tissue-resident memory T cells control chronic intestinal inflammation. Nat Immunol 20, 288–300 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Park CO, Kupper TS, The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med 21, 688–697 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snyder ME, Finlayson MO, Connors TJ, Dogra P, Senda T, Bush E, Carpenter D, Marboe C, Benvenuto L, Shah L, Robbins H, Hook JL, Sykes M, D'Ovidio F, Bacchetta M, Sonett JR, Lederer DJ, Arcasoy S, Sims PA, Farber DL, Generation and persistence of human tissue-resident memory T cells in lung transplantation. Sci Immunol 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zuber J, Shonts B, Lau SP, Obradovic A, Fu J, Yang S, Lambert M, Coley S, Weiner J, Thome J, DeWolf S, Farber DL, Shen Y, Caillat-Zucman S, Bhagat G, Griesemer A, Martinez M, Kato T, Sykes M, Bidirectional intragraft alloreactivity drives the repopulation of human intestinal allografts and correlates with clinical outcome. Sci Immunol 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amsen D, van Gisbergen K, Hombrink P, van Lier RAW, Tissue-resident memory T cells at the center of immunity to solid tumors. Nat Immunol 19, 538–546 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Park SL, Gebhardt T, Mackay LK, Tissue-Resident Memory T Cells in Cancer Immunosurveillance. Trends Immunol 40, 735–747 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Zeiser R, Socie G, Blazar BR, Pathogenesis of acute graft-versus-host disease: from intestinal microbiota alterations to donor T cell activation. Br J Haematol 175, 191–207 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Anderson BE, Taylor PA, McNiff JM, Jain D, Demetris AJ, Panoskaltsis-Mortari A, Ager A, Blazar BR, Shlomchik WD, Shlomchik MJ, Effects of donor T-cell trafficking and priming site on graft-versus-host disease induction by naive and memory phenotype CD4 T cells. Blood 111, 5242–5251 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reshef R, Luger SM, Hexner EO, Loren AW, Frey NV, Nasta SD, Goldstein SC, Stadtmauer EA, Smith J, Bailey S, Mick R, Heitjan DF, Emerson SG, Hoxie JA, Vonderheide RH, Porter DL, Blockade of lymphocyte chemotaxis in visceral graft-versus-host disease. N Engl J Med 367, 135–145 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wysocki CA, Panoskaltsis-Mortari A, Blazar BR, Serody JS, Leukocyte migration and graft-versus-host disease. Blood 105, 4191–4199 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beilhack A, Schulz S, Baker J, Beilhack GF, Wieland CB, Herman EI, Baker EM, Cao YA, Contag CH, Negrin RS, In vivo analyses of early events in acute graft-versus-host disease reveal sequential infiltration of T-cell subsets. Blood 106, 1113–1122 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El-Asady R, Yuan R, Liu K, Wang D, Gress RE, Lucas PJ, Drachenberg CB, Hadley GA, TGF-(3)-dependent CD103 expression by CD8(+) T cells promotes selective destruction of the host intestinal epithelium during graft-versus-host disease. J Exp Med 201, 1647–1657 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santos ESP, Cire S, Conlan T, Jardine L, Tkacz C, Ferrer IR, Lomas C, Ward S, West H, Dertschnig S, Blobner S, Means TK, Henderson S, Kaplan DH, Collin M, Plagnol V, Bennett CL, Chakraverty R, Peripheral tissues reprogram CD8+ T cells for pathogenicity during graft-versus-host disease. JCI Insight 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Romagnani A, Vettore V, Rezzonico-Jost T, Hampe S, Rottoli E, Nadolni W, Perotti M, Meier MA, Hermanns C, Geiger S, Wennemuth G, Recordati C, Matsushita M, Muehlich S, Proietti M, Chubanov V, Gudermann T, Grassi F, Zierler S, TRPM7 kinase activity is essential for T cell colonization and alloreactivity in the gut. Nat Commun 8, 1917 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furlan SN, Watkins B, Tkachev V, Cooley S, Panoskaltsis-Mortari A, Betz K, Brown M, Hunt DJ, Schell JB, Zeleski K, Yu A, Giver CR, Waller EK, Miller JS, Blazar BR, Kean LS, Systems analysis uncovers inflammatory Th/Tc17-driven modules during acute GVHD in monkey and human T cells. Blood 128, 2568–2579 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furlan SN, Watkins B, Tkachev V, Flynn R, Cooley S, Ramakrishnan S, Singh K, Giver C, Hamby K, Stempora L, Garrett A, Chen J, Betz KM, Ziegler CG, Tharp GK, Bosinger SE, Promislow DE, Miller JS, Waller EK, Blazar BR, Kean LS, Transcriptome analysis of GVHD reveals aurora kinase A as a targetable pathway for disease prevention. Sci Transl Med 7, 315ra191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller WP, Srinivasan S, Panoskaltsis-Mortari A, Singh K, Sen S, Hamby K, Deane T, Stempora L, Beus J, Turner A, Wheeler C, Anderson DC, Sharma P, Garcia A, Strobert E, Elder E, Crocker I, Crenshaw T, Penedo MC, Ward T, Song M, Horan J, Larsen CP, Blazar BR, Kean LS, GVHD after haploidentical transplantation: a novel, MHC-defined rhesus macaque model identifies CD28- CD8+ T cells as a reservoir of breakthrough T-cell proliferation during costimulation blockade and sirolimus-based immunosuppression. Blood 116, 5403–5418 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tkachev V, Furlan SN, Watkins B, Hunt DJ, Zheng HB, Panoskaltsis-Mortari A, Betz K, Brown M, Schell JB, Zeleski K, Yu A, Kirby I, Cooley S, Miller JS, Blazar BR, Casson D, Bland-Ward P, Kean LS, Combined OX40L and mTOR blockade controls effector T cell activation while preserving Treg reconstitution after transplant. Sci Transl Med 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lake Potter HPGE, Tkachev Victor, Fabozzi Giulia, Chassiakos Alexander, Petrovas Costantinos, Darrah Patricia A., Lin Philana L., Foulds Kathryn E., Kean Leslie S., Flynn JoAnne L., Roederer Mario. Sci Transl Med 2021. (in press). [DOI] [PubMed] [Google Scholar]
- 22.Champlin R, Ho W, Gajewski J, Feig S, Burnison M, Holley G, Greenberg P, Lee K, Schmid I, Giorgi J, et al. , Selective depletion of CD8+ T lymphocytes for prevention of graft-versus-host disease after allogeneic bone marrow transplantation. Blood 76, 418–423 (1990). [PubMed] [Google Scholar]
- 23.Schattenfroh NC, Hoffman RA, McCarthy SA, Simmons RL, Phenotypic analysis of donor cells infiltrating the small intestinal epithelium and spleen during graft-versus-host disease. Transplantation 59, 268–273 (1995). [PubMed] [Google Scholar]
- 24.Fernandez-Ruiz D, Ng WY, Holz LE, Ma JZ, Zaid A, Wong YC, Lau LS, Mollard V, Cozijnsen A, Collins N, Li J, Davey GM, Kato Y, Devi S, Skandari R, Pauley M, Manton JH, Godfrey DI, Braun A, Tay SS, Tan PS, Bowen DG, Koch-Nolte F, Rissiek B, Carbone FR, Crabb BS, Lahoud M, Cockburn IA, Mueller SN, Bertolino P, McFadden GI, Caminschi I, Heath WR, Liver-Resident Memory CD8(+) T Cells Form a Front-Line Defense against Malaria Liver-Stage Infection. Immunity 45, 889–902 (2016). [DOI] [PubMed] [Google Scholar]
- 25.McNamara HA, Cai Y, Wagle MV, Sontani Y, Roots CM, Miosge LA, O'Connor JH, Sutton HJ, Ganusov VV, Heath WR, Bertolino P, Goodnow CG, Parish IA, Enders A, Cockburn IA, Up-regulation of LFA-1 allows liver-resident memory T cells to patrol and remain in the hepatic sinusoids. Sci Immunol 2, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chopra M, Biehl M, Steinfatt T, Brandl A, Kums J, Amich J, Vaeth M, Kuen J, Holtappels R, Podlech J, Mottok A, Kraus S, Jordan-Garrote AL, Bauerlein CA, Brede C, Ribechini E, Fick A, Seher A, Polz J, Ottmuller KJ, Baker J, Nishikii H, Ritz M, Mattenheimer K, Schwinn S, Winter T, Schafer V, Krappmann S, Einsele H, Muller TD, Reddehase MJ, Lutz MB, Mannel DN, Berberich-Siebelt F, Wajant H, Beilhack A, Exogenous TNFR2 activation protects from acute GvHD via host T reg cell expansion. J Exp Med 213, 1881–1900 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komatsu N, Hori S, Full restoration of peripheral Foxp3+ regulatory T cell pool by radioresistant host cells in scurfy bone marrow chimeras. Proc Natl Acad Sci U S A 104, 8959–8964 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arina A, Beckett M, Fernandez C, Zheng W, Pitroda S, Chmura SJ, Luke JJ, Forde M, Hou Y, Burnette B, Mauceri H, Lowy I, Sims T, Khodarev N, Fu YX, Weichselbaum RR, Tumor-reprogrammed resident T cells resist radiation to control tumors. Nat Commun 10, 3959 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, Tscharke DC, Heath WR, Inouye M, Carbone FR, Gebhardt T, The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol 14, 1294–1301 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Beura LK, Fares-Frederickson NJ, Steinert EM, Scott MC, Thompson EA, Fraser KA, Schenkel JM, Vezys V, Masopust D, CD4(+) resident memory T cells dominate immunosurveillance and orchestrate local recall responses. J Exp Med 216, 1214–1229 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bergsbaken T, Bevan MJ, Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8(+) T cells responding to infection. Nat Immunol 16, 406–414 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, Lucas PJ, Artis D, Wherry EJ, Hogquist K, Vezys V, Masopust D, Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol 188, 4866–4875 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang N, Bevan MJ, Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 39, 687–696 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bartolome-Casado R, Landsverk OJB, Chauhan SK, Richter L, Phung D, Greiff V, Risnes LF, Yao Y, Neumann RS, Yaqub S, Oyen O, Horneland R, Aandahl EM, Paulsen V, Sollid LM, Qiao SW, Baekkevold ES, Jahnsen FL, Resident memory CD8 T cells persist for years in human small intestine. J Exp Med, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou S, Ueta H, Xu XD, Shi C, Matsuno K, Predominant donor CD103+CD8+ T cell infiltration into the gut epithelium during acute GvHD: a role of gut lymph nodes. Int Immunol 20, 385–394 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, Mathis D, Benoist C, Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity 27, 786–800 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, Wang D, Getzler AJ, Nguyen T, Crotty S, Wang W, Pipkin ME, Goldrath AW, Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature 552, 253–257 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, Gao R, Kang B, Zhang Q, Huang JY, Konno H, Guo X, Ye Y, Gao S, Wang S, Hu X, Ren X, Shen Z, Ouyang W, Zhang Z, Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 564, 268–272 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, Senda T, Sun X, Ho SH, Lerner H, Friedman AL, Shen Y, Farber DL, Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep 20, 2921–2934 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Behr FM, Chuwonpad A, Stark R, van Gisbergen K, Armed and Ready: Transcriptional Regulation of Tissue-Resident Memory CD8 T Cells. Front Immunol 9, 1770 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borges da Silva H, Beura LK, Wang H, Hanse EA, Gore R, Scott MC, Walsh DA, Block KE, Fonseca R, Yan Y, Hippen KL, Blazar BR, Masopust D, Kelekar A, Vulchanova L, Hogquist KA, Jameson SC, The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8(+) T cells. Nature 559, 264–268 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li C, Zhu B, Son YM, Wang Z, Jiang L, Xiang M, Ye Z, Beckermann KE, Wu Y, Jenkins JW, Siska PJ, Vincent BG, Prakash YS, Peikert T, Edelson BT, Taneja R, Kaplan MH, Rathmell JC, Dong H, Hitosugi T, Sun J, The Transcription Factor Bhlhe40 Programs Mitochondrial Regulation of Resident CD8(+) T Cell Fitness and Functionality. Immunity 51, 491–507 e497 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, Luo C, O'Malley JT, Gehad A, Teague JE, Divito SJ, Fuhlbrigge R, Puigserver P, Krueger JG, Hotamisligil GS, Clark RA, Kupper TS, Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543, 252–256 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Byersdorfer CA, Tkachev V, Opipari AW, Goodell S, Swanson J, Sandquist S, Glick GD, Ferrara JL, Effector T cells require fatty acid metabolism during murine graft-versus-host disease. Blood 122, 3230–3237 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gatza E, Wahl DR, Opipari AW, Sundberg TB, Reddy P, Liu C, Glick GD, Ferrara JL, Manipulating the bioenergetics of alloreactive T cells causes their selective apoptosis and arrests graft-versus-host disease. Sci Transl Med 3, 67ra68 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hubbard VM, Eng JM, Ramirez-Montagut T, Tjoe KH, Muriglan SJ, Kochman AA, Terwey TH, Willis LM, Schiro R, Heller G, Murphy GF, Liu C, Alpdogan O, van den Brink MR, Absence of inducible costimulator on alloreactive T cells reduces graft versus host disease and induces Th2 deviation. Blood 106, 3285–3292 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taylor PA, Panoskaltsis-Mortari A, Freeman GJ, Sharpe AH, Noelle RJ, Rudensky AY, Mak TW, Serody JS, Blazar BR, Targeting of inducible costimulator (ICOS) expressed on alloreactive T cells down-regulates graft-versus-host disease (GVHD) and facilitates engraftment of allogeneic bone marrow (BM). Blood 105, 3372–3380 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP 3rd, Armand P, Cutler C, Ho VT, Treister NS, Bienfang DC, Prasad S, Tzachanis D, Joyce RM, Avigan DE, Antin JH, Ritz J, Soiffer RJ, Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med 365, 2055–2066 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perol L, Martin GH, Maury S, Cohen JL, Piaggio E, Potential limitations of IL-2 administration for the treatment of experimental acute graft-versus-host disease. Immunol Lett 162, 173–184 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Blazar BR, Sharpe AH, Chen AI, Panoskaltsis-Mortari A, Lees C, Akiba H, Yagita H, Killeen N, Taylor PA, Ligation of OX40 (CD134) regulates graft-versus-host disease (GVHD) and graft rejection in allogeneic bone marrow transplant recipients. Blood 101, 3741–3748 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Du W, Cao X, Cytotoxic Pathways in Allogeneic Hematopoietic Cell Transplantation. Front Immunol 9, 2979 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ichiba T, Teshima T, Kuick R, Misek DE, Liu C, Takada Y, Maeda Y, Reddy P, Williams DL, Hanash SM, Ferrara JL, Early changes in gene expression profiles of hepatic GVHD uncovered by oligonucleotide microarrays. Blood 102, 763–771 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Tian Y, Cox MA, Kahan SM, Ingram JT, Bakshi RK, Zajac AJ, A Context-Dependent Role for IL-21 in Modulating the Differentiation, Distribution, and Abundance of Effector and Memory CD8 T Cell Subsets. J Immunol 196, 2153–2166 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hanash AM, Kappel LW, Yim NL, Nejat RA, Goldberg GL, Smith OM, Rao UK, Dykstra L, Na IK, Holland AM, Dudakov JA, Liu C, Murphy GF, Leonard WJ, Heller G, van den Brink MR, Abrogation of donor T-cell IL-21 signaling leads to tissue-specific modulation of immunity and separation of GVHD from GVL. Blood 118, 446–455 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hippen KL, Bucher C, Schirm DK, Bearl AM, Brender T, Mink KA, Waggie KS, Peffault de Latour R, Janin A, Curtsinger JM, Dillon SR, Miller JS, Socie G, Blazar BR, Blocking IL-21 signaling ameliorates xenogeneic GVHD induced by human lymphocytes. Blood 119, 619–628 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blaser BW, Roychowdhury S, Kim DJ, Schwind NR, Bhatt D, Yuan W, Kusewitt DF, Ferketich AK, Caligiuri MA, Guimond M, Donor-derived IL-15 is critical for acute allogeneic graft-versus-host disease. Blood 105, 894–901 (2005). [DOI] [PubMed] [Google Scholar]
- 57.Roychowdhury S, Blaser BW, Freud AG, Katz K, Bhatt D, Ferketich AK, Bergdall V, Kusewitt D, Baiocchi RA, Caligiuri MA, IL-15 but not IL-2 rapidly induces lethal xenogeneic graft-versus-host disease. Blood 106, 2433–2435 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bucher C, Koch L, Vogtenhuber C, Goren E, Munger M, Panoskaltsis-Mortari A, Sivakumar P, Blazar BR, IL-21 blockade reduces graft-versus-host disease mortality by supporting inducible T regulatory cell generation. Blood 114, 5375–5384 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brown GR, Lee EL, Thiele DL, TNF enhances CD4+ T cell alloproliferation, IFN-gamma responses, and intestinal graft-versus-host disease by IL-12-independent mechanisms. J Immunol 170, 5082–5088 (2003). [DOI] [PubMed] [Google Scholar]
- 60.Masson F, Ghisi M, Groom JR, Kallies A, Seillet C, Johnstone RW, Nutt SL, Belz GT, Id2 represses E2A-mediated activation of IL-10 expression in T cells. Blood 123, 3420–3428 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Takashima S, Martin ML, Jansen SA, Fu Y, Bos J, Chandra D, O'Connor MH, Mertelsmann AM, Vinci P, Kuttiyara J, Devlin SM, Middendorp S, Calafiore M, Egorova A, Kleppe M, Lo Y, Shroyer NF, Cheng EH, Levine RL, Liu C, Kolesnick R, Lindemans CA, Hanash AM, T cell-derived interferon-gamma programs stem cell death in immune-mediated intestinal damage. Sci Immunol 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gibbons DL, Abeler-Dorner L, Raine T, Hwang IY, Jandke A, Wencker M, Deban L, Rudd CE, Irving PM, Kehrl JH, Hayday AC, Cutting Edge: Regulator of G protein signaling-1 selectively regulates gut T cell trafficking and colitic potential. J Immunol 187, 2067–2071 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Castor MG, Rezende B, Resende CB, Alessandri AL, Fagundes CT, Sousa LP, Arantes RM, Souza DG, Silva TA, Proudfoot AE, Teixeira MM, Pinho V, The CCL3/macrophage inflammatory protein-1alpha-binding protein evasin-1 protects from graft-versus-host disease but does not modify graft-versus-leukemia in mice. J Immunol 184, 2646–2654 (2010). [DOI] [PubMed] [Google Scholar]
- 64.Howard OM, Turpin JA, Goldman R, Modi WS, Functional redundancy of the human CCL4 and CCL4L1 chemokine genes. Biochem Biophys Res Commun 320, 927–931 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Baschuk N, Wang N, Watt SV, Halse H, House C, Bird PI, Strugnell R, Trapani JA, Smyth MJ, Andrews DM, NK cell intrinsic regulation of MIP-1alpha by granzyme M. Cell Death Dis 5, e1115 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle-Rowson G, Ghezzi P, Kleemann R, McColl SR, Bucala R, Hickey MJ, Weber C, MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med 13, 587–596 (2007). [DOI] [PubMed] [Google Scholar]
- 67.Brinkman CC, Iwami D, Hritzo MK, Xiong Y, Ahmad S, Simon T, Hippen KL, Blazar BR, Bromberg JS, Treg engage lymphotoxin beta receptor for afferent lymphatic transendothelial migration. Nat Commun 7, 12021 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liang Y, Liu C, Djeu JY, Zhong B, Peters T, Scharffetter-Kochanek K, Anasetti C, Yu XZ, Beta2 integrins separate graft-versus-host disease and graft-versus-leukemia effects. Blood 111, 954–962 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dupre L, Houmadi R, Tang C, Rey-Barroso J, T Lymphocyte Migration: An Action Movie Starring the Actin and Associated Actors. Front Immunol 6, 586 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ono S, The role of cyclase-associated protein in regulating actin filament dynamics - more than a monomer-sequestration factor. J Cell Sci 126, 3249–3258 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim J, Shapiro MJ, Bamidele AO, Gurel P, Thapa P, Higgs HN, Hedin KE, Shapiro VS, Billadeau DD, Coactosin-like 1 antagonizes cofilin to promote lamellipodial protrusion at the immune synapse. PLoS One 9, e85090 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Desai SD, Reed RE, Burks J, Wood LM, Pullikuth AK, Haas AL, Liu LF, Breslin JW, Meiners S, Sankar S, ISG15 disrupts cytoskeletal architecture and promotes motility in human breast cancer cells. Exp Biol Med (Maywood) 237, 38–49 (2012). [DOI] [PubMed] [Google Scholar]
- 73.Hwang SH, Jung SH, Lee S, Choi S, Yoo SA, Park JH, Hwang D, Shim SC, Sabbagh L, Kim KJ, Park SH, Cho CS, Kim BS, Leng L, Montgomery RR, Bucala R, Chung YJ, Kim WU, Leukocyte-specific protein 1 regulates T-cell migration in rheumatoid arthritis. Proc Natl Acad Sci U S A 112, E6535–6543 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schoppmeyer R, Zhao R, Cheng H, Hamed M, Liu C, Zhou X, Schwarz EC, Zhou Y, Knorck A, Schwar G, Ji S, Liu L, Long J, Helms V, Hoth M, Yu X, Qu B, Human profilin 1 is a negative regulator of CTL mediated cell-killing and migration. Eur J Immunol 47, 1562–1572 (2017). [DOI] [PubMed] [Google Scholar]
- 75.Anderson KG, Sung H, Skon CN, Lefrancois L, Deisinger A, Vezys V, Masopust D, Cutting edge: intravascular staining redefines lung CD8 T cell responses. J Immunol 189, 2702–2706 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, Braun A, Masson F, Kallies A, Belz GT, Carbone FR, T-box Transcription Factors Combine with the Cytokines TGF-beta and IL-15 to Control Tissue-Resident Memory T Cell Fate. Immunity 43, 1101–1111 (2015). [DOI] [PubMed] [Google Scholar]
- 77.Clark RA, Watanabe R, Teague JE, Schlapbach C, Tawa MC, Adams N, Dorosario AA, Chaney KS, Cutler CS, Leboeuf NR, Carter JB, Fisher DC, Kupper TS, Skin effector memory T cells do not recirculate and provide immune protection in alemtuzumab-treated CTCL patients. Sci Transl Med 4, 117ra117 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schenkel JM, Fraser KA, Vezys V, Masopust D, Sensing and alarm function of resident memory CD8(+) T cells. Nat Immunol 14, 509–513 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kean LS, Turka LA, Blazar BR, Advances in targeting co-inhibitory and co-stimulatory pathways in transplantation settings: the Yin to the Yang of cancer immunotherapy. Immunol Rev 276, 192–212 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Magenau JM, Reddy P, Proteasome: target for acute and chronic GVHD? Blood 124, 1551–1552 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hippen KL, Aguilar EG, Rhee SY, Bolivar-Wagers S, Blazar BR, Distinct Regulatory and Effector T Cell Metabolic Demands during Graft-Versus-Host Disease. Trends Immunol 41, 77–91 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Beura LK, Rosato PC, Masopust D, Implications of Resident Memory T Cells for Transplantation. Am J Transplant 17, 1167–1175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bolger AM, Lohse M, Usadel B, Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bray NL, Pimentel H, Melsted P, Pachter L, Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
- 85.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yu G, Wang LG, Han Y, He QY, clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Blazar BR, Taylor PA, McElmurry R, Tian L, Panoskaltsis-Mortari A, Lam S, Lees C, Waldschmidt T, Vallera DA, Engraftment of severe combined immune deficient mice receiving allogeneic bone marrow via In utero or postnatal transfer. Blood 92, 3949–3959 (1998). [PubMed] [Google Scholar]
- 88.Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Giron CG, Gil L, Gordon L, Haggerty L, Haskell E, Hourlier T, Izuogu OG, Janacek SH, Juettemann T, To JK, Laird MR, Lavidas I, Liu Z, Loveland JE, Maurel T, McLaren W, Moore B, Mudge J, Murphy DN, Newman V, Nuhn M, Ogeh D, Ong CK, Parker A, Patricio M, Riat HS, Schuilenburg H, Sheppard D, Sparrow H, Taylor K, Thormann A, Vullo A, Walts B, Zadissa A, Frankish A, Hunt SE, Kostadima M, Langridge N, Martin FJ, Muffato M, Perry E, Ruffier M, Staines DM, Trevanion SJ, Aken BL, Cunningham F, Yates A, Flicek P, Ensembl 2018. Nucleic Acids Res 46, D754–D761 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Luecken MD, Theis FJ, Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol 15, e8746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.DeTomaso D, Jones MG, Subramaniam M, Ashuach T, Ye CJ, Yosef N, Functional interpretation of single cell similarity maps. Nat Commun 10, 4376 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Korotkevich G, Sukhov V, Sergushichev A, Fast gene set enrichment analysis. bioRxiv, 060012 (2019). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Transplant characteristics
Table S2. List of antibodies used, flow cytometry panels and flow-based definitions of lymphocyte populations.
Table S3. scRNA-seq sample information and QC metrics.
Table S4. Differentially expressed genes between donor, host and HC CD8+ T cells with high enrichment scores for the TRM signature.
Table S5. Pathway analysis of differentially expressed genes between donor, host and HC CD8+ T cells with high enrichment scores for the TRM signature.
Table S6. Predicted activated up-stream regulators in donor CD8+ T cells with high enrichment scores for the TRM signature in comparison with their HC and host counterparts.
Table S7. Differentially expressed genes between donor, host and HC CD8+ T cells with low enrichment scores for the TRM signature.
Table S8. Pathway analysis of differentially expressed genes between donor, host and HC CD8+ T cells with low enrichment scores for the TRM signature.
Table S9. Predicted activated up-stream regulators in donor CD8+ T cells with low enrichment scores for the TRM signature in comparison with their HC and host counterparts.
Table S10. Differentially expressed genes between donor CD8+ T cells with low and high enrichment scores for the TRM signature.
Table S11. Predicted activated up-stream regulators in donor CD8+ T cells with low enrichment scores compared to cells with high enrichment scores for the TRM signature.
Table S12. Gene signature of donor CD8+ T cells with low enrichment scores for the TRM signature, defined as shown in Figure 5B.
Table S13. Pathway analysis on the gene signature of donor CD8+ T cells with low enrichment scores for the TRM signature.
Table S14. Predicted activated up-stream regulators based on the gene signature of donor CD8+ T cells with low enrichment scores for the TRM signature.
Table S15. Clinical characteristics of patients from the ABA2 clinical trial (NCT01743131), included in the current study.
Table S16. Results of GSEA analysis comparing patients from the ABA2 clinical trial (NCT01743131) with Grades 2-4 and 3-4 aGVHD versus patients with Grades 0-1 and 0-2 aGVHD, respectively.
Table S17. scRNA-seq-derived gene sets used for GSEA analysis.
Data File S1. Primary data
Figure S1. Liver function tests in recipients following autologous or allogeneic HCT in NHP.
Figure S2. Gating tree for flow cytometric identification and characterization of T cells within different spatiotemporal compartments using Serial Intravascular Staining (SIVS) in NHP.
Figure S3. Phenotypic characteristics of liver intravascular Compartment-1 T cells in NHP.
Figure S4. Compartmentalization of CD4+ T cells at steady state and after HCT in NHP.
Figure S5. Migration of CD4+ and CD8+ T cells at steady state and after HCT in NHP.
Figure S6. Phenotypic characterization of Compartment-2 and Compartment-3 CD8+ T cells in NHP.
Figure S7. Phenotypic characteristics of intestinal CD4+ T cells in NHP.
Figure S8. Transcriptomic identification of intestinal CD8+ T cells in NHP.
Figure S9. Transcriptomic identification of intestinal TRM-High and TRM-Low CD8+ T cells from large intestine in NHP.
Figure S10. Immunophenotypic and transcriptomic characteristics of CD8+ T cells from the large intestine.
Figure S11. Threshold testing of TRM-High and TRM-Low CD8+ T cells.
Data Availability Statement
All data associated with this study are present in the paper or supplementary materials. The NHP scRNA-seq data were deposited in the NCBI’s Gene Expression Omnibus database (GSE142483). The RNA-seq data from the ABA2 clinical trial were deposited in the NCBI’s Gene Expression Omnibus database (GEO157280).