

PURPOSE
Waldenstrom Macroglobulinemia (WM) is a rare lymphoma with distinct clinical features, and data from Latin American patients are lacking. Therefore, we aim to investigate the clinical, therapy, and outcome patterns of WM in Latin America.
METHODS
We retrospectively analyzed patients with WM diagnosed between 1991 and 2019 from 24 centers in seven Latin American countries. The study outcomes were overall survival (OS) and progression-free survival (PFS).
RESULTS
We identified 159 cases (median age 67 years, male 62%). Most patients (95%) were symptomatic at diagnosis. The International Prognostic Scoring System for WM (IPSSWM) at diagnosis was available in 141 (89%) patients (high-risk 40%, intermediate-risk 37%, and low-risk 23%). Twenty-seven (17%) patients were tested for MYD88L265P, with 89% (n = 24 of 27) carrying the mutation. First-line and second-line therapies were administered to 142 (89%) and 53 (33%) patients, respectively. Chemoimmunotherapy was the most commonly used first-line (66%) and second-line (45%) approach; only 18 (11%) patients received ibrutinib. With a median follow-up of 69 months, the 5-year OS rate was 81%. In treated patients, the 5-year OS and PFS rates were 78% and 59%, respectively. High-risk IPSSWM at treatment initiation was an independent risk factor for OS (adjusted hazard ratio: 4.73, 95% CI, 1.67 to 13.41, P = .003) and PFS (adjusted hazard ratio: 2.43, 95% CI, 1.31 to 4.50, P = .005).
CONCLUSION
In Latin America, the management of WM is heterogeneous, with limited access to molecular testing and novel agents. However, outcomes were similar to those reported internationally. We validated the IPSSWM score as a prognostic factor for OS and PFS. There is an unmet need to improve access to recommended diagnostic approaches and therapies in Latin America.
INTRODUCTION
Waldenström macroglobulinemia (WM) is an indolent B-cell lymphoma characterized by a monoclonal immunoglobulin M (IgM) paraprotein in the serum and bone marrow infiltration by malignant lymphoplasmacytic cells.1 WM is a rare disease with an estimated incidence of 0.34-0.73 among males and 0.17-0.42 among females per 100,000 persons per year in the United States.2 The incidence of WM in Latin America is unknown. Clinically, WM may present with constitutional symptoms (ie, fever, night sweats, and unintentional weight loss), cytopenias, lymphadenopathy, organomegaly, neuropathy, hemolytic anemia, and hyperviscosity. Consensus guidelines recommend treatment initiation only in symptomatic patients.3,4
CONTEXT
Key Objective
How does Waldenstrom Macroglobulinemia (WM) present in Latin America and what are the most used therapeutic patterns?
Knowledge Generated
In Latin America, the management of WM is heterogeneous, with limited access to molecular testing and novel agents. However, outcomes were similar to those reported internationally. We validated the International Prognostic Scoring System for WM score as a prognostic factor for overall survival and progression-free survival.
Relevance
To our knowledge, this is the first Latin American study to provide real-world data on WM. The International Prognostic Scoring System for WM was validated in our population. There is a need for improvement in diagnostic approaches and access to novel therapies in our region.
The most frequent cytogenetic alteration in WM is the partial loss of the long arm of chromosome 6 (30%-54%), which is associated with a worse prognosis.5 The introduction of whole-genome and next-generation sequencing allowed the detection of somatic activating mutations in the MYD88 and CXCR4 genes, which can be detected in approximately 90% and 40% of WM cases, respectively.6 Several studies have reported the impact of these mutations in the prognosis and therapy responses in WM.7-9
Despite the advent of novel therapeutic strategies, WM remains incurable. A consensus panel has recently updated recommendations for the management of symptomatic WM after the 10th International Workshop for WM.10 However, the treatment approaches used to manage WM outside of clinical trials are heterogeneous. Because data on WM in Latin America are lacking, two Latin American research groups (Grupo de Estudio Latinoamericano de Linfoproliferativos-GELL and Grupo de Estudio Latinoamericano de Mieloma Múltiple-GELAMM) conducted a retrospective review of patients with WM from the region to better understand the clinical, therapy, and outcome patterns of Latin American patients with WM.
METHODS
Patients
We conducted an international, multicenter, retrospective cohort study of patients diagnosed with WM in 24 centers from seven Latin American countries between January 1991 and December 2019 (Data Supplement). We reviewed the medical records of individuals age ≥ 18 years diagnosed using the criteria of the eighth IWWM.11 The Institutional Review Board of each participating center approved this study.
Clinical Data and Risk Stratification
The demographic and clinical data collected at diagnosis included age, sex, presence of lymphadenopathy, splenomegaly, hepatomegaly, bone marrow infiltration, peripheral neuropathy, and hyperviscosity symptoms (not explained otherwise, epistaxis and headaches). Laboratory data included complete blood cell count, serum creatinine, serum β2 microglobulin, serum albumin, serum lactate dehydrogenase, serum ferritin, IgM, IgG, IgA, and the presence of a monoclonal protein in serum protein electrophoresis and immunofixation (SPEP/IFX). When available, results from cytogenetic and mutational analyses for MYD88 and CXCR4 mutations were collected. The International Prognostic Scoring System for WM (IPSSWM) was used for risk stratification.12 We also categorized patients according to the type of treating institution (ie, public v private hospitals).
Therapy Approaches and Response Criteria
The therapy approaches used during the first or subsequent lines were divided as follows: monotherapy (ie, rituximab, chlorambucil, fludarabine, cyclophosphamide, ibrutinib, and thalidomide), chemoimmunotherapy (rituximab plus chemotherapy), and others. The overall response rate (ORR) was defined as the sum of minor response (≥ 25% but < 50% reduction in serum IgM levels), partial response (PR, ≥ 50% but < 90% reduction), very good partial response (VGPR, ≥ 90% reduction or normalization of serum IgM with a persistent monoclonal spike in SPEP/IFX), and complete response (normalization of serum IgM and SPEP/IFX and absence of bone marrow and extramedullary disease).13 The major response rate (MRR) was defined as the sum of PR, VGPR, and complete response.
Outcomes and Statistical Analysis
We used descriptive statistics to summarize all variables. The primary study outcome was overall survival (OS), defined as the time between diagnosis and death from any cause, loss to follow-up, or end of the study (November 2020). The secondary outcome was progression-free survival (PFS) after frontline, defined as the time from the date of treatment initiation until first relapse, loss to follow-up, death from any cause, or end of the study, whichever occurs first. Differences between groups were identified using the chi-square or Fisher's exact test, as appropriate.
Survival probabilities were estimated using the Kaplan-Meier method, and the difference between groups was computed using the log-rank test. We estimated proportional hazard ratios (HRs) for OS and PFS with a 95% CI using univariate and multivariate Cox proportional hazard regression models. Two models were constructed to assess the confounding effect of different predictors in the primary and secondary outcomes. The first model included the IPSSWM score at treatment initiation and rituximab use. The second model included the previous variables plus sex and the type of treating institution. In the regression analysis, patients with low-risk and intermediate-risk IPSSWM scores were grouped into a low-intermediate risk category given the low count in the low-risk group. The variables in our models were selected on the basis of priori assumptions of their public health and clinical relevance regarding survival outcomes rather than their significance in the univariate analysis.12 The hazard assumption was tested by plotting Schoenfeld residuals. All feasible two-way interactions between the included variables in both OS and PFS models were assessed in Cox regression analyses. We did not find any significant interaction. Sensitivity analyses were performed in patients with a follow-up of at least 5 years (n = 105). The 95% CI of the response rates was estimated using the Clopper-Pearson method. Outcomes with a P value < .05 were considered statistically significant. We used the R software for analysis.
RESULTS
Demographic and Clinical Features
A total of 159 patients with WM were identified. Table 1 summarizes the demographic and clinical features of the patients. At diagnosis, the median age was 67 (range, 24-89) years with a male predominance (62%). Fifty-two percent of the patients were treated in private centers, and 48% in public centers. Most patients (n = 151, 95%) were symptomatic at diagnosis with symptomatic adenopathy (28%), symptomatic splenomegaly (25%), and hyperviscosity symptoms (20%) as the most common symptoms. The IPSSWM score was available in 141 patients, of whom 40%, 37%, and 23% were classified as high-risk, intermediate-risk, and low-risk disease, respectively. Most patients had anemia (hemoglobin ≤ 11.5 g/dL; 71%) at the time of diagnosis. Serum IgM levels > 7 g/dL and bone marrow involvement of ≥ 50% were seen in 22% of patients each.
TABLE 1.
Demographic and Clinical Features and Outcomes of Latin American Patients With Waldenstrom Macroglobulinemia
Overall, molecular testing was performed in 21% (n = 23 of 159) of the patients (27% in private and 16% in public centers, P = .146). MYD88L265P testing was performed in 17% (n = 27 of 159), del17p in 6% (n = 9 of 159), and CXCR4 in 1% (n = 1 of 159). For those in whom genetic testing was performed, 89% (n = 24 of 27) had MYD88L265P mutation, none had del17p mutation, and the only patient tested for CXCR4 mutation had MYD88L265P and CXCR4 mutations (Data Supplement).
Therapy Approaches and Responses
Figure 1 shows the therapy patterns during the three lines of therapy.
FIG 1.
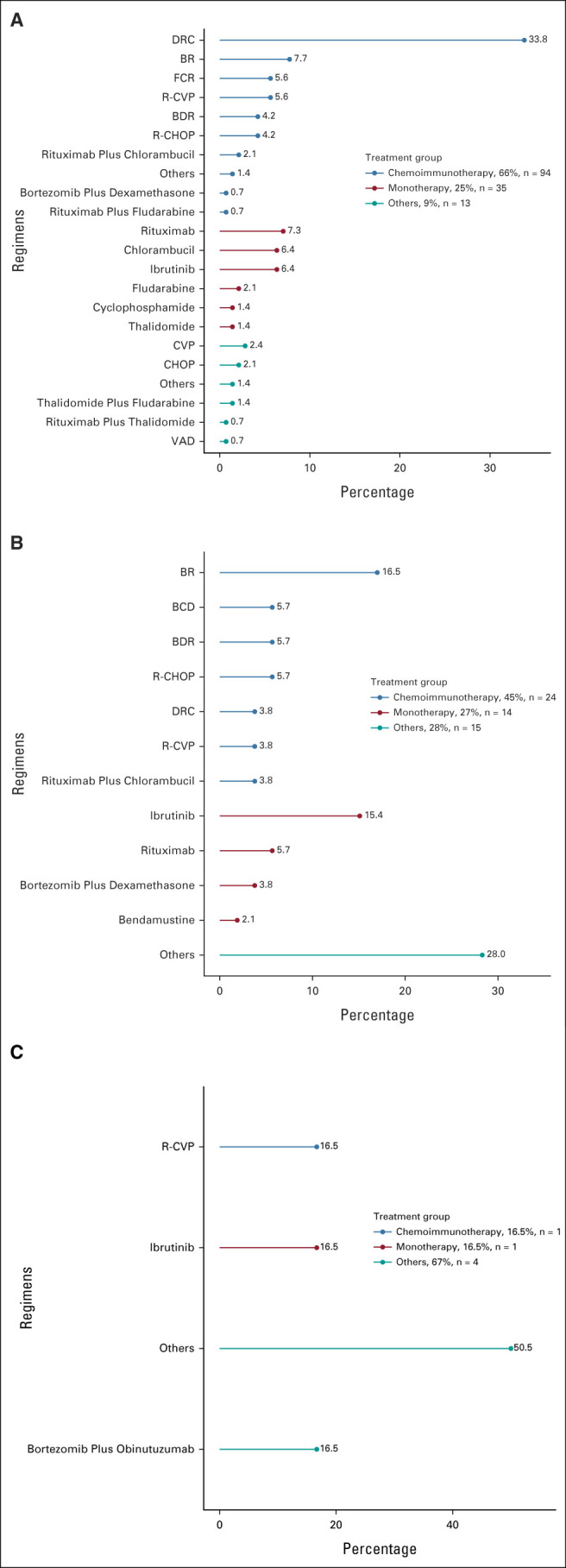
Therapy approaches used during (A) first-line (n = 142), (B) second-line (n = 53), and (C) third-line (n = 6) settings. BCD, bortezomib-cyclophosphamide-dexamethasone; BDR, bortezomib-dexamethasone-rituximab; BR, bendamustine-rituximab; CHOP, cyclophosphamide, doxorubicin, vincristine, and prednisone; CVP, cyclophosphamide, vincristine, and prednisone; DRC, dexamethasone, rituximab, and cyclophosphamide; FCR, fludarabine-cyclophosphamide-rituximab; R-CHOP, rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone; R-CVP, rituximab plus cyclophosphamide, vincristine, and prednisone; VAD, vincristine-doxorubicin-dexamethasone.
First-line therapy was administered to 142 (89%) patients. Chemoimmunotherapy (n = 94 of 142, 66%) was the most frequently used first-line therapy, followed by monotherapy (n = 35 of 142, 25%). Combination of dexamethasone, rituximab, and cyclophosphamide (DRC) was the most frequently used regimen (34%, n = 48 of 142). First-line therapy was heterogeneous among countries. Chemoimmunotherapy was more commonly used in Chile (80%), Uruguay (80%), and Argentina (74%) than Peru (58%) or Mexico (46%). Monotherapy use ranged from 7% in Chile to 40% in Mexico (Data Supplement). Neither patient's age (P = .63) nor IPSSWM risk score (P = .94) influenced the clinician's decision on the first-line therapy approach used. However, chemoimmunotherapy was more frequently administered at private institutions (74% v 58%, P = .03), with an upward trend from 16% in the 1991-2010 period to 83% in the 2011-2020 period (P < .001; Data Supplement). The median time to first-line therapy initiation was 35 days (range 0-15 years). Second-line therapy was administered to 53 patients; chemoimmunotherapy (45%) was the most commonly used approach. Bendamustine plus rituximab (17%) and monotherapy with ibrutinib (15%) were the most frequently used regimens. Six patients received third-line therapy, and one patient received fourth-line therapy. Maintenance rituximab was given to 13 (n = 13 of 137, 10%) patients and only after first-line therapy. Ibrutinib was used in 18 (11%) patients anytime during the disease process (14% in private and 12% in public centers, P = .946). Intrathecal chemotherapy was given to five (n = 5 of 142, 4%) patients. Four patients underwent autologous stem-cell transplantation (ASCT) after failing first-line therapy. All but one received a melphalan-based conditioning regimen and remained disease-free (PFS 24, 48, and 60 months, respectively). The fourth patient did not have enough data to be analyzed.
Table 2 summarizes the clinical features and responses of patients who received therapy. Most patients had an intermediate-risk (41%) or high-risk (43%) IPSSWM score at therapy initiation. The ORR to first-line therapy was 86% (95% CI, 79 to 92), and the MRR was 85% (95% CI, 77 to 91). No differences were seen in ORR (83%, 95% CI, 70 to 93 v 88%, 95% CI, 79 to 94; P = .62) and MRR (81%, 95% CI, 67 to 91 v 87%, 95% CI, 77 to 94; P = .56) between patients managed at private versus public centers, respectively. The ORR and MRR in patients who received the first-line chemoimmunotherapy regimen was 86% (n = 107 of 124; 95% CI, 59 to 77) and 84% (n = 105 of 124; 95% CI, 60 to 78), respectively (Data Supplement).
TABLE 2.
Demographic and Clinical Features and Outcomes of Latin American Patients With Waldenstrom Macroglobulinemia According to the Treating Institution

In the subset of patients with MYD88L265P mutation who received therapy (chemoimmunotherapy n = 15 and ibrutinib n = 5), the ORR, MRR, and VGPR rates were 100%, 95%, and 15%, respectively (Data Supplement). The ORR in the five patients treated with ibrutinib was 100%, of whom four (80%) achieved PR and one (20%) attained a VGPR. The one patient with CXCR4 mutation received ibrutinib and attained a PR.
Disease Outcomes
With a median follow-up of 69 months (95% CI, 59 to 90), the overall 5-year OS was 81% (95% CI, 76 to 89) and the median OS was not reached. In patients who received therapy, the 5-year OS and PFS rates were 78% (95% CI, 70 to 86) and 59% (95% CI, 50 to 69), respectively, with median survival times not reached for both outcomes. Worse 5-year OS (P = .003) and PFS (P = .004) rates were seen in patients with a high-risk IPSSWM score (OS: 64%; PFS: 35%) compared with patients with intermediate-risk (OS: 88%; PFS: 59%) and low-risk (OS: 100%; PFS: 82%) diseases (Figs 2 and 3). Males had worse 5-year OS (75% v 94%, P = .006) and PFS (50% v 72%, P = .004) rates than females (Figs 2 and 3). The use of rituximab did not influence the survival estimates in the study periods of 2001-2010 (OS, P = .16; PFS, P = .86), 2011-2020 (OS, P = .73; PFS, P = .73), and 2001-2020 (OS, P = .32; PFS, P = .69; Data Supplement).
FIG 2.
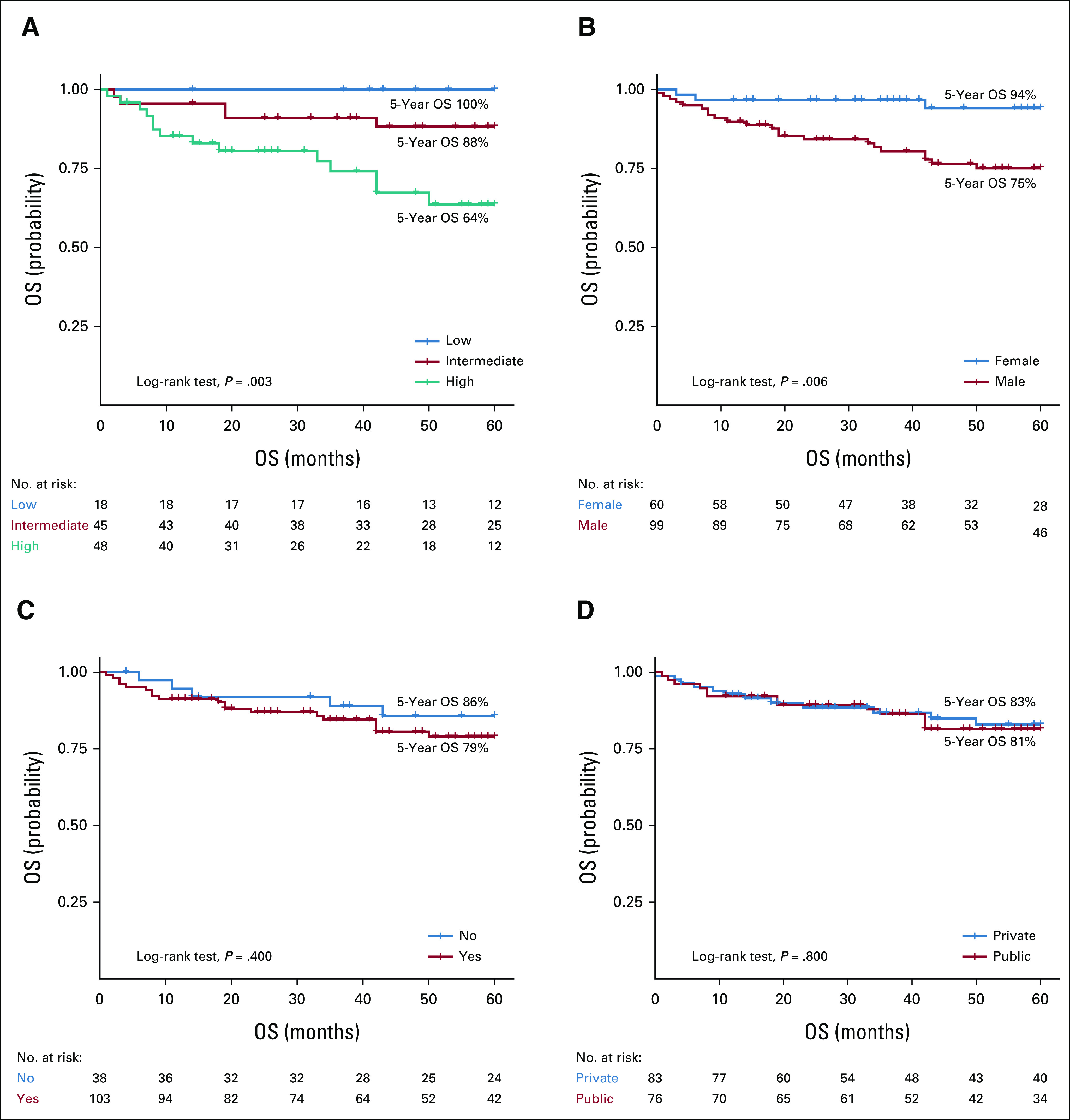
OS of patients with Waldenstrom Macroglobulinemia according to (A) treatment IPSSWM score, (B) sex, (C) rituximab use, and (D) type of treating institution. IPSSWM, International Prognostic Scoring System for WM; OS, overall survival.
FIG 3.
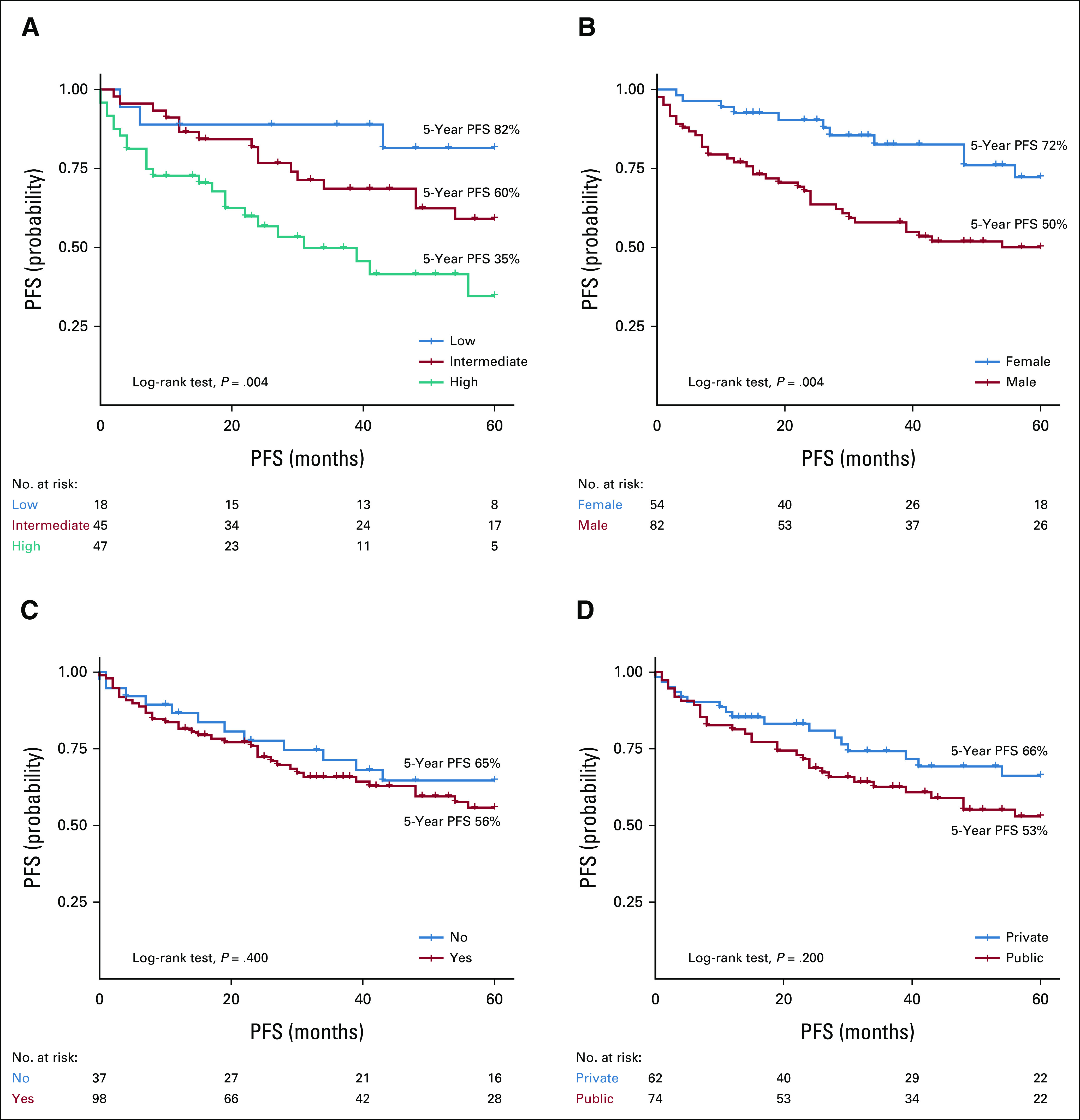
PFS of patients with Waldenstrom Macroglobulinemia according to (A) treatment IPSSWM score, (B) sex, (C) rituximab use, and (D) type of treating institution. IPSSWM, International Prognostic Scoring System for WM; PFS, progression-free survival.
In the univariate analysis, a high-risk IPSSWM at treatment initiation was a predictor of mortality (HR, 4.76, 95% CI, 1.71 to 13.28) and PFS (HR, 2.56, 95% CI, 1.39 to 4.70). After adjusting for sex, type of treating institution, and rituximab use, a high-risk IPSSWM score remained an independent risk factor for OS (adjusted hazard ratio [aHR], 4.73, 95% CI, 1.67 to 13.41, P = .003) and PFS (aHR, 2.43, 95% CI, 1.31 to 4.50, P = .005; Table 3). In addition, male sex was associated with mortality (aHR, 3.75, 95% CI, 1.08 to 13.04, P = .038) and PFS (aHR, 2.64, 95% CI, 1.29 to 5.40, P = .008; Table 3). This outcome was not modified by the IPSSWM score (P for interaction = .998), treatment period (P for interaction = .743), or treating institution (P for interaction = .904). In the sensitivity analysis, a high-risk IPSSWM was a predictor for mortality (aHR, 6.24, 95% CI, 2.19 to 17.82, P = .001) and PFS (aHR, 3.77, 95% CI, 1.87 to 7.62, P < .001; Data Supplement).
TABLE 3.
Univariate and Multivariate Cox Regression Analyses of OS and PFS Predictors in Latin American Patients With Waldenstrom Macroglobulinemia

After 5 years from diagnosis, 38 (26%) patients had relapsed and 29 (20%) had died. Cause of death was reported in 19 cases; the most common were infection (n = 7) and disease progression/transformation to aggressive non-Hodgkin lymphoma (n = 5).
DISCUSSION
Herein, we describe the clinical features, therapy approaches, and survival outcomes of patients with WM encountered in 24 centers from seven Latin American countries over the past three decades. We found that treatment patterns are heterogeneous with limited access to molecular testing and novel agents. Although chemoimmunotherapy was the most commonly used approach, there was heterogeneity in the choice of treatments across the countries. Nonetheless, the survival outcomes were comparable with those reported in high-income countries. In addition, the use of rituximab did not seem to have an impact on patient outcomes. To the best of our knowledge, this is the first study that provides real-world data on WM in Latin America.
In our cohort, the median age at diagnosis was 67 years, with a slight male predominance. These features were similar to those reported in previous studies.14 Less than 5% of our patients were asymptomatic during the initial presentation. This number is lower compared with historical data of 25%.1-3 In this study, the most common indications for therapy were organomegaly, anemia, and hyperviscosity symptoms, with a median time to therapy initiation of 35 days. Although the reasons why our patients required early therapy are out of the scope of this work, we believe that delayed access to specialized cancer care by a saturated health care system might have contributed to the observed outcome. Indeed, a late diagnosis has been associated with a more aggressive course of the disease and a worse prognosis.15,16
The MYD88L265P mutation has been reported in more than 90% of patients with WM, allowing in some cases the distinction from other IgM-secreting malignancies.7 The CXCR4 mutation has been associated with a more aggressive course of the disease and resistance to ibrutinib.17 In our study, most patients did not undergo molecular testing because of the lack of molecular laboratories in many of the participating institutions and its high cost. In addition, given that ibrutinib is not readily available in most Latin American countries, cytogenetics or mutational analysis does not influence clinicians' choice of therapy. Despite these caveats, ibrutinib therapy was associated with a high response rate in our patients, similar to reported clinical trials.18
There are few randomized trials comparing different regimens in WM. Thus, treatment recommendations follow retrospective studies and phase II trials. Real-world data from the United States showed that between 2009 and 2013, single-agent rituximab (45%) and purine analogs (15%) were the most frequently used frontline approaches.19 In Europe, monotherapy (eg, chlorambucil, rituximab, and fludarabine) was the most frequent frontline approach (43%), whereas chemoimmunotherapy (predominantly rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone) was used in 36% of patients.14 In our study, chemoimmunotherapy was the most common approach in the first-line and second-line setting, often with dexamethasone, rituximab, and cyclophosphamide and bendamustine-rituximab, respectively. Bortezomib or ibrutinib was given to < 10% of our patients. In Latin America, both drugs require either institutional approval for reimbursement or the cost is borne by the patient, which might explain the low rate of use of these agents. We did not find differences in responses or outcomes between patients managed at private versus public institutions, which is not surprising considering that access to therapies is mostly similar in both settings.
The role of ASCT has been explored in small series in the relapsed or refractory setting. The largest European experience using ASCT in WM reported a 5-year PFS of 49% and an OS of 69%, with a nonrelapse mortality of 4% at 1 year.20 A more recent review by Gertz et al21 showed that this strategy is effective but currently underutilized, possibly because of the high rate of toxicity and not being a curative strategy. In our study, four patients underwent ASCT after failing frontline therapy. Thus, although ASCT is available in the region, it is rarely used for WM.
In this study, both the median OS and PFS were not reached. At a 5-year follow-up, 81% of patients were alive and survival rates in those treated were 78% and 59% for OS and PFS, respectively. These results are similar to previous real-world reports.14,19 In addition, we were able to validate the IPSSWM score in Latin American patients. We could not validate the revised IPSSWM scoring system because of inconsistency in lactate dehydrogenase reporting.22 In our cohort, the use of rituximab did not seem to have an impact on survival. Clinical trials have shown improved responses and delayed disease progression with rituximab-based therapies.23,24 However, neither randomized controlled studies nor observational studies have demonstrated improvement in OS.14,25-29 A recent study using the US SEER database identified a lower mortality risk in patients receiving rituximab-based regimens, but no difference in OS when comparing rituximab monotherapy versus combination with chemotherapy.30 Although we cannot entirely explain our finding, residual confounding such as patient comorbidities, delayed diagnosis, and delayed access to therapy might have influenced the observed outcome. Therefore, we recommend that current therapy guidelines on the use of rituximab in WM should be followed.
We found a worse survival in males. Previous studies have described that males with WM have a worse survival and higher risk of mortality.19,31 This study did not find an effective modification between males and females by IPSSWM score, treatment period, or type of treating institution. The observed outcome may indicate unmeasured confounders in the WM population not explained by high-risk disease, treatment variation over time, and the demographic variables accounted for in this study. Thus, further research is needed to understand this disparity.
Our study is limited by its retrospective nature. Patients were classified using archived records, laboratory, and pathology reports. The absence of a centralized pathology review in this international study lays a risk for selection bias. Similarly, response assessment was evaluated by each investigator, which is subjected to observer bias. However, our outcomes are consistent with previous studies. Our main strength is this study's multinational scale, which included patients seen at specialized cancer centers in Latin America.
In conclusion, to our knowledge, this is the first study to provide real-world data on WM in Latin America. We found that in Latin America, the management of WM is heterogeneous, with limited access to molecular testing and modern therapies. Despite that, outcomes were similar to those reported internationally. Moreover, we validated the IPSSWM score as a predictor of OS and PFS. We believe that the clinical information gathered in this cohort study should provide invaluable insight and guidance into treating this relatively rare and challenging disease, especially in resource-limited settings. Finally, there is an unmet need to improve access to recommended diagnostic approaches and therapies in our region.
DATA SHARING STATEMENT
The data that support the findings of this study are available from the corresponding author, E.R., upon reasonable request.
AUTHOR CONTRIBUTIONS
Conception and design: Eloísa Riva, Patricio José Duarte, Bryan Valcárcel, Guilherme Fleury-Perini, Efreen Montaño, Henry Idrobo, Brady E. Beltran, Jorge J. Castillo, Luis E. Malpica Castillo
Provision of study materials or patients: Eloísa Riva, Ivan Murrieta, Daniel del Carpio, Camila Peña, Jule Vásquez, Sebastian Yantorno, Sergio Lopresti, Milagros Altamirano, Guillermo J. Ruiz-Arguelles, Elia Zamora Pérez, Henry Idrobo, Humberto Martínez-Cordero
Collection and assembly of data: Eloísa Riva, Patricio José Duarte, Guillermina Remaggi, Ivan Murrieta, Ariel Corzo, Daniel del Carpio, Camila Peña, Jule Vásquez, Virginia Bove, Larissa Teixeira, Guilherme Fleury-Perini, Sebastian Yantorno, César Samánez, Sergio Lopresti, Milagros Altamirano, Luis Villela, Guillermo J. Ruiz-Arguelles, Guillermo J. Ruiz-Delgado, Efreen Montaño, Verónica Verri, Elia Zamora Pérez, Fernando Pérez Jacobo, Henry Idrobo, Humberto Martínez-Cordero, Brady E. Beltran, Jhoanna Ramírez, Luis E. Malpica Castillo
Data analysis and interpretation: Eloísa Riva, Patricio José Duarte, Bryan Valcárcel, Jorge J. Castillo, Luis E. Malpica Castillo
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/go/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
No potential conflicts of interest were reported.
REFERENCES
- 1.García-Sanz R, Montoto S, Torrequebrada A, et al. : Waldenström macroglobulinaemia: Presenting features and outcome in a series with 217 cases. Br J Haematol 115:575-582, 2001 [DOI] [PubMed] [Google Scholar]
- 2.Yin X, Chen L, Fan F, et al. : Trends in incidence and mortality of Waldenström macroglobulinemia: A population-based study. Front Oncol 10:1712, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dimopoulos MA, Kyle RA, Anagnostopoulos A, et al. : Diagnosis and management of Waldenstrom’s macroglobulinemia. J Clin Oncol 23:1564-1577, 2005 [DOI] [PubMed] [Google Scholar]
- 4.Dimopoulos MA, Kastritis E, Owen RG, et al. : Treatment recommendations for patients with Waldenström macroglobulinemia (WM) and related disorders: IWWM-7 consensus. Blood 124:1404-1411, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nguyen-Khac F, Lambert J, Chapiro E, et al. : Chromosomal aberrations and their prognostic value in a series of 174 untreated patients with Waldenström’s macroglobulinemia. Haematologica 98:649-654, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morel P, Duhamel A, Gobbi PG, et al. : MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. Blood 5:2654-2661, 2015 [Google Scholar]
- 7.Treon SP, Xu L, Yang G, et al. : MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med 367:826-833, 2012 [DOI] [PubMed] [Google Scholar]
- 8.Treon SP, Cao Y, Xu L, et al. : Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenström macroglobulinemia. Blood 123:2791-2796, 2014 [DOI] [PubMed] [Google Scholar]
- 9.Castillo JJ, Xu L, Gustine JN, et al. : CXCR4 mutation subtypes impact response and survival outcomes in patients with Waldenström macroglobulinaemia treated with ibrutinib. Br J Haematol 187:356-363, 2019 [DOI] [PubMed] [Google Scholar]
- 10.Castillo JJ, Advani RH, Branagan AR, et al. : Consensus treatment recommendations from the tenth international Workshop for Waldenström macroglobulinaemia. Lancet Haematol 7:e827-e837, 2020 [DOI] [PubMed] [Google Scholar]
- 11.Leblond V, Kastritis E, Advani R, et al. : Treatment recommendations from the Eighth International Workshop on Waldenström’s macroglobulinemia. Blood 128:1321-1328, 2016 [DOI] [PubMed] [Google Scholar]
- 12.Morel P, Duhamel A, Gobbi P, et al. : International Prognostic Scoring System for Waldenström macroglobulinemia. Blood 113:4163-4170, 2009 [DOI] [PubMed] [Google Scholar]
- 13.Owen RG, Kyle RA, Stone MJ, et al. : Response assessment in Waldenström macroglobulinaemia: Update from the VIth International Workshop. Br J Haematol 160:171-176, 2013 [DOI] [PubMed] [Google Scholar]
- 14.Buske C, Sadullah S, Kastritis E, et al. : Treatment and outcome patterns in European patients with Waldenström’s macroglobulinaemia: A large, observational, retrospective chart review. Lancet Haematol 5:e299-309, 2018 [DOI] [PubMed] [Google Scholar]
- 15.Gobbi PG, Baldini L, Broglia C, et al. : Prognostic validation of the international classification of immunoglobulin M gammopathies: A survival advantage for patients with immunoglobulin M monoclonal gammopathy of undetermined significance? Clin Cancer Res 11:1786-1790, 2005 [DOI] [PubMed] [Google Scholar]
- 16.Oza A, Rajkumar SV: Waldenstrom macroglobulinemia: Prognosis and management. Blood Cancer J 5:e296-e298, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu L, Tsakmaklis N, Yang G, et al. : Acquired mutations associated with ibrutinib resistance in Waldenström macroglobulinemia. Blood 129:2519-2525, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Treon SP, Tripsas CK, Meid K, et al. : Ibrutinib in previously treated Waldenström’s macroglobulinemia. N Engl J Med 372:1430-1440, 2015 [DOI] [PubMed] [Google Scholar]
- 19.Olszewski AJ, Treon SP, Castillo JJ: Evolution of management and outcomes in Waldenström macroglobulinemia: A population‐based analysis. Oncologist 21:1377-1386, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kyriakou C, Boumendil A, Finel H, et al. : The impact of advanced patient age on mortality after allogeneic hematopoietic cell transplantation for non-Hodgkin’s lymphoma: A retrospective study by the EBMT Lymphoma Working Party. Bone Marrow Transplant 25:86-93. 2019 [DOI] [PubMed] [Google Scholar]
- 21.Gertz MA, Reeder CB, Kyle RA, et al. : Stem cell transplant for Waldenström macroglobulinemia: An underutilized technique. Bone Marrow Transplant 47:1147-1153, 2012 [DOI] [PubMed] [Google Scholar]
- 22.Kastritis E, Morel P, Duhamel A, et al. : A revised International Prognostic Score System for Waldenström’s macroglobulinemia. Leukemia 33:2654-2661, 2019 [DOI] [PubMed] [Google Scholar]
- 23.Treon SP, Ioakimidis L, Soumerai JD, et al. : Primary therapy of Waldenström macroglobulinemia with bortezomib, dexamethasone, and rituximab: WMCTG clinical trial 05-180. J Clin Oncol 27:3830-3835, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dimopoulos MA, Anagnostopoulos A, Kyrtsonis MC, et al. : Primary treatment of Waldenström macroglobulinemia with dexamethasone, rituximab, and cyclophosphamide. J Clin Oncol 25:3344-3349, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Buske C, Hoster E, Dreyling M, et al. : The addition of rituximab to front-line therapy with CHOP (R-CHOP) results in a higher response rate and longer time to treatment failure in patients with lymphoplasmacytic lymphoma: Results of a randomized trial of the German Low-Grade Lymphoma Study Group (GLSG). Leukemia 23:153-161, 2009 [DOI] [PubMed] [Google Scholar]
- 26.Leblond V, Johnson S, Chevret S, et al. : Results of a randomized trial of chlorambucil versus fludarabine for patients with untreated Waldenström macroglobulinemia, marginal zone lymphoma, or lymphoplasmacytic lymphoma. J Clin Oncol 31:301-307, 2013 [DOI] [PubMed] [Google Scholar]
- 27.Rummel MJ, Niederle N, Maschmeyer G, et al. : Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: An open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 381:1203-1210, 2013 [DOI] [PubMed] [Google Scholar]
- 28.Dimopoulos MA, Tedeschi A, Trotman J, et al. : Phase 3 trial of ibrutinib plus rituximab in Waldenström’s macroglobulinemia. N Engl J Med 378:2399-2410, 2018 [DOI] [PubMed] [Google Scholar]
- 29.Tam CS, Opat S, D’Sa S, et al. : A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: The ASPEN study. Blood 136:2038-2050, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olszewski AJ, Chen C, Gutman R, et al. : Comparative outcomes of immunochemotherapy regimens in Waldenström macroglobulinaemia. Br J Haematol 179:106-115, 2017 [DOI] [PubMed] [Google Scholar]
- 31.Kristinsson SY, Eloranta S, Dickman PW, et al. : Patterns of survival in lymphoplasmacytic lymphoma/Waldenström macroglobulinemia: A population-based study of 1,555 patients diagnosed in Sweden from 1980 to 2005. Am J Hematol 88:60-65, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, E.R., upon reasonable request.