

Abstract
The coronavirus disease 2019 (COVID-19) is a pandemic that has severely posed substantial health challenges and claimed millions of lives. Though vaccines have been produced to stem the spread of this disease, the death rate remains high since drugs used for treatment have therapeutic challenges. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus that causes the disease, has a slew of potential therapeutic targets. Among them is the furin protease, which has a cleavage site on the virus’s spike protein. The cleavage site facilitates the entry of the virus into human cells via cell–cell fusion. This critical involvement of furin in the disease pathogenicity has made it a viable therapeutic strategy against the virus. This study employs the consensus docking approach using HYBRID and AutoDock Vina to virtually screen a pre-filtered library of 3942 natural product compounds of African origin against the human furin protease (PDB: 4RYD). Twenty of these compounds were selected as hits after meeting molecular docking cut-off of − 7 kcal.mol−1, pose alignment inspection, and having favorable furin-ligand interactions. An area under the curve (AUC) value of 0.72 was computed from the receiver operator characteristic (ROC) curve, and Boltzmann-enhanced discrimination of the ROC curve (BEDROC) value of 0.65 showed that AutoDock Vina was a reasonable tool for selecting actives for this target. Seven of these hits were proposed as potential leads having had bonding interactions with catalytic triad residues Ser368, His194, and Asp153, and other essential residues in the active site with plausible binding free energies between − 189 and − 95 kJ/mol from the Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) calculations as well as favorable ADME/Tox properties. The molecules were also predicted as antiviral, anti-inflammatory, membrane permeability inhibitors, RNA synthesis inhibitors, cytoprotective, and hepatoprotective with probable activity (Pa) above 0.5 and probable inactivity values below 0.1. Some of them also have anti-influenza activity. Influenza virus has many similarities with SARS-CoV-2 in their mode of entry into human cells as both are facilitated by the furin protease. Pinobanksin 3-(E)-caffeate, one of the potential leads is a propolis compound. Propolis compounds have shown inhibitory effects against ACE2, TMPRSS2, and PAK1 signaling pathways of SARS-CoV-2 in previous studies. Likewise, quercitrin is structurally similar to isoquercetin, which is currently in clinical trials as possible medication for COVID-19.
Supplementary Information
The online version contains supplementary material available at 10.1007/s11224-022-02056-1.
Keywords: SARS-CoV-2, COVID-19, Furin protease, Natural products, MM-PBSA, Consensus docking
Introduction
Coronaviruses (CoVs) are a family of viruses that cause several diseases in humans. These diseases range from common cold infections to severe acute respiratory syndromes [1, 2]. In recent times, coronaviruses have caused major outbreaks in the world. Among these are the 2003 severe acute respiratory syndrome coronavirus (SARS-CoV) outbreak in China, the 2012 Middle East respiratory syndrome coronavirus (MERS-CoV) outbreak in Saudi Arabia [3], and recently, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) outbreak that emerged from the Wuhan province of China in December 2019 [4]. According to recent studies, SARS-CoV-2 is a new strain of the virus and is responsible for the coronavirus disease 2019 (COVID-19) [5]. The fast spreading rate of the virus across the world prompted the World Health Organization (WHO) to declare the outbreak as a pandemic [6]. As of January 2022, case numbers above 300 million were reported worldwide with over 5.5 million deaths. When infected with the disease, symptoms such as dry cough, sore throat, fever, and tiredness are commonly experienced. Ageusia, anosmia, and runny nose are also common symptoms of the disease [7]. In more severe cases, patients bear serious symptoms such as difficulty in breathing, chest pains, among others [8]. In some instances, oxygen is administered through ventilators to augment patient’s survival [9, 10]. The severity of this disease has instigated the demand to identify new antiviral drug candidates or repurpose existing drugs for its treatment. Currently, a number of vaccines have been rolled out in the quest to provide immunity against the virus; however, availability and acceptance have become major issues as people continually get infected every day [11]. Thus, effective therapy is required, necessitating the use of medications as an additional therapeutic option in the fight against the disease.
CoVs share a lot of similarities in their genome organization [12]. Recent studies have shown that SARS-CoV-2 uses similar host cell receptors as SARS-CoV to enter human cells. This is attributable to the similarity in sequence of the spike proteins of both viruses. The entry of CoVs into host cells is facilitated by host-cell membrane fusion that involves several processes of receptor binding and proteolytic cleavage of the spike (S) protein. The spike (S) protein consists of two subunits namely S1 and S2 that are responsible for the attachment and entry of the virus into the host cell. While S1 is involved in the receptor binding on host cell surfaces, S2 facilitates the whole fusion mechanism. During the S protein proteolytic activity, several host cell proteases like furin, cathepsin B, trypsin, elastase, plasmin, and cell surface transmembrane protease/serine (TMPRSS) cleave the S protein to facilitate the viral entry [13]. Inhibition of these proteases has been suggested as plausible therapeutic targets for viral infections and can decrease viral infectivity [13]. Fusion between the virus and host cells occurs either via cytoplasmic or endosomal membrane fusion [14]. The first important step for target cell entry is the interaction of the virus spike protein with angiotensin-converting enzyme 2 (ACE-2) [15]. Alongside ACE2, the virus also employs the enzyme TMPRSS2 for priming [16].
Though the virus enters cells using these important receptors, emphasis has been placed on furin protease cleavage sites found on the virus spike protein for its role in facilitating its entry into host cells [17]. Furin belongs to a family of serine secretory proteases known as proprotein convertases (PCs) [18]. PCs are responsible for the regulation of majority of biological processes by activating precursor forms of a wide range of receptors, hormones, and cell surface protein [19]. In viral disease processes, furin and other PCs activate cell surface glycoproteins in the pathogenicity of several family of viruses including CoVs, paramyxoviruses, herpesviruses, togaviruses, bornaviruses, flaviviruses, bunyaviruses, filoviruses, orthomyxoviruses, retroviruses, and pneumoviruses, facilitating their entry into target cells [20–22]. Other pathogenic examples include furin activation of precursor proteins by influenza virus—a virus that has been shown to have several similarities with SARS-CoV virus, the Ebola virus, distemper virus, and many more [19]. These and other roles played by furin proteases make them important in the viral maturation process, SARS-CoV-2 pathogenesis, and viral transmission in humans. CRISPR-Cas9 knockout of furin has been reported to significantly reduce the production of infectious SARS-CoV-2 virus [23]. In ferrets, SARS-CoV-2 virus which lacks the furin cleavage site (FCS) was observed to have low transmission to other animals as compared to the wild-type virus [24]. Also, very low frequencies were observed for SARS-CoV-2 mutants that had FCS deletions in human tissues [24]. The spike FCS has a total of ten amino acid residues, of which the 682RRARSVAS689 region is highly conserved [25] and the 681PRRA684 region has been reported to be unique to SARS-CoV-2 [26]. Inhibiting the furin protease has been shown to prevent SARS-CoV-2 binding to the human furin protease, thereby suppressing viral production [22].
Drug discovery experiments in cell culture and animal models targeting the inhibition of furin protease as therapeutic intervention for specific diseases have been promising. In 2015, some novel furin inhibitors were tested via cell culture experiments against influenza virus, anthrax, and diphtheria toxins. Their findings showed that, in the presence of these inhibitors, the spread of the avian influenza viruses, H5N1 and H7N1, was strongly inhibited [19]. Anthrax and diphtheria toxins which are not viruses but depend on furin for their propagation showed signs of protective effect in the presence of the inhibitors [19].
Several types of synthetic inhibitors have been designed for furin protease since the human and mouse protein crystal structures were solved. Majority of these inhibitors are either peptide-based or non-peptide based derivatives such as nona-d-Arg-amide (D9R) and 2,5-dideoxystreptamine [27, 28]. However, only a few of these inhibitors have entered human clinical trials. Diminazene, an antiparasitic drug was identified to be a potent furin inhibitor via structure-based virtual screening and in vitro enzyme-based assay with an IC50 of 5.42 ± 0.11 μM [29]. Another study reported an IC50 of 13.2 μM for diaminazene against the furin protease [30]. It has also been reported that furin inhibitors, decanoyl-RVKR-chloromethylketone (CMK), and naphthofluorescein showed antiviral activity against the SARS-CoV-2 virus in VeroE6 cells by blocking viral entry and suppressing viral RNA transcription [22].
Few studies have targeted the inhibition of the enzyme using natural product sources [31]. Natural products and their associated moieties have historically been great sources of therapeutic agents [32]. Due to the resistance most viruses have towards antiviral therapy, there is a growing interest in natural products as one of the best resource for finding new chemically diverse leads that can be used to develop therapeutically new antiviral agents [33]. Natural products are rich sources of diverse chemical compounds from which drugs can be isolated [34]. Compared to synthetic drugs, drugs from natural sources have lower side effects, are less expensive, and are mostly less toxic [35]. In view of this, the exploration of its chemical space for scaffold purposes has been an essential part of drug discovery.
In this study, we aimed to identify putative antiviral inhibitors of furin protease using natural product-derived compounds of African origin as potential therapeutic agents for COVID-19 disease. We carried out consensus docking using pre-filtered libraries of African natural products against the binding site of the protein. The binding mechanisms, active site residue interactions, and binding free energies of the potential leads were evaluated using molecular dynamics simulations and the Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) calculations. The biological activity and pharmacological profiles of the compounds were predicted [36–38].
Methods
The study employed consensus docking approach by using OEDocking HYBRID (version 3.5.0.4) [39] and AutoDock Vina [40] for the molecular docking studies of furin protease and natural product-derived compounds from African sources (Fig. 1). The compounds with binding energies of − 7 kcal.mol−1 or less from both docking studies were considered [41]. The best poses of each compound from the two docking applications were compared and those with RMSD better than 2 Å were considered for downstream analysis (Fig. 1) [42]. The molecular interactions between the furin protease and the selected ligands were investigated using PyMOL (v 2.0.6) and Ligplot + [43]. Molecular dynamics simulations and MM-PBSA computations were also performed to support the selection of the potential lead compounds (Fig. 1).
Fig. 1.

A graphical representation of the step-by-step approach employed in this study. The work involved screening of natural product library against furin protease using molecular docking, molecular dynamics (MD) simulations, elucidation of molecular interactions, and prediction of biological activity. ANPDB, African Natural Products Database; MM-PBSA, Molecular Mechanics Poisson-Boltzmann Surface Area; PASS, Prediction of Activity Spectra for Substances; ADMET, absorption, distribution, metabolism, excretion and toxicity
Database preparation
A library of 7391 natural product compounds were obtained from three databases comprising Northern African Natural Products Database (NANPDB) [44], East African Natural Products Database (EANPDB) [45], and AfroDb database [34], a catalog of ZINC15 [46]. NANPBD and EANPDB have recently been merged to form the African Natural Products Database (ANPDB) [45].
The compounds were then filtered using FILTER (v4.0.0.4, OpenEye Scientific Software, Inc., Santa Fe, NM, USA; www.eyesopen.com) to eliminate all compounds with undesirable properties from the library especially non-drug-likeness, non-lead-likeness, and toxicity. FILTER is a molecular filtering application whose algorithm works based on physical property calculations and functional group knowledge. Its default drug/lead-like parameters were used for the filtration. After filtering, a total of 3942 compounds passed and were used for the virtual screening.
Protein structure retrieval and preparation
Crystal structure of the human furin protease was retrieved from the protein databank (PDB ID: 4RYD). The structure was solved using X-ray diffraction at a resolution of 2.15 Å [19]. The hexameric protein structure of 4RYD is composed of six similar chains namely A, B, C, D, E, and F, each having 482 amino acids sequence length [19, 47]. As a result of the similarity, chain A was selected for this study. Within the active site of the crystal structure was the bound ligand para-guanidinomethyl-Phac-R-Tle-R-Amba (MI-1148). The bound ligand and ions such as calcium and sodium found within the structure were removed using PyMOL (v 2.0.6).
Consensus docking
Consensus docking methodology is an approach that combines multiple docking programs by comparing their top scoring poses. A previous study [48] applied the consensus docking methodology on co-crystallized complexes using AutoDock [49], AutoDock Vina [40], and DOCK6 [50]. A success rate of 82% was observed by using more than one docking tool as compared to individual accuracies of 55%, 64%, and 58%, respectively [48]. Furthermore, because docking tools differ so greatly in their search and scoring algorithms, putting more emphasis on their intersection should compensate for their weaknesses [51–53]. OEDocking HYBRID [39] and AutoDock Vina [40] were combined in this study to predict potential inhibitors of the human furin protease. Only ligands which had binding energies equal to or less than − 7.0 kcal.mol−1 from both OEDocking and AutoDock Vina were selected for further studies since this threshold has been shown to distinguish between putative and non-putative binders of proteins [41]. Although, a more negative binding energy does not imply a better inhibition [54, 55], previous studies have shown that ~ 97.7% of known inhibitors have binding energies of − 7.0 kcal.mol−1 or less [41, 56] and this threshold filters ~ 95% of non-inhibitors. Other studies have also used the − 7.0 kcal.mol−1 threshold to prioritize compounds [57, 58].
Docking validation
HYBRID and AutoDock Vina were both validated before employed in the virtual screening process. A dataset of 52 furin active compounds and 224 decoys were used to assess the performance of the tools in the enrichment of actives for the furin protease. Fifty of the active compounds were retrieved from BindingDB [47] while the remaining two were the co-crystallized ligands obtained from complexes (PDB IDs: 4RYD and 4OMC). All 52 actives were submitted to Database of Useful Decoys (DUD-E) [59] and 224 decoys were obtained for them. For HYBRID, conformers of the compounds were generated using OMEGA [39, 60]. An OEDocking receptor was also created. An OEDocking receptor usually consists of the protein structure, its co-crystallized ligand, and descriptions of the active site. OpenEye Spruce4Docking (OpenEye Scientific Software, Inc., Santa Fe, NM, USA, www.eyesopen.com) utility program was used for generating the OEDocking receptor with default parameters. The OEDocking receptor of the furin protease was generated using the obtained complex (PDB ID: 4RYD) devoid of the other cofactors and water molecules. In other words, the furin protease and the bound ligand para-guanidinomethyl-Phac-R-Tle-R-Amba were used for the OEDocking receptor. The presence of the bound ligand guides the tool to select similar poses for the compounds used in the virtual screening process based on the reference ligand’s shape and 3-dimensional arrangement of its chemical features. HYBRID (version 3.5.0.4) was used to dock the conformer-generated active-decoy dataset into the active site of the produced receptor.
For AutoDock Vina, the set of actives and decoys were initially energy minimized before docking was carried out. The protein’s active site was defined using grid box dimensions set at X = 26.86 Å, Y = 27.66 Å, and Z = 0.38 Å for the center while box size dimensions were set at X = 23.31 Å, Y = 23.79 Å, and Z = 26.44 Å.
The validation of each of the docking protocols was assessed via Screening Explorer by evaluating the ROC curves and Boltzmann-enhanced discrimination of ROC (BEDROC) [61]. Results from this docking validation were used as guide for the subsequent virtual screening of our pre-filtered natural product library against furin in HYBRID and AutoDock Vina.
Virtual screening via OEDocking HYBRID
The first phase of the molecular docking process was performed using the OEDocking HYBRID (version 3.5.0.4) [39]. HYBRID has an improved scoring algorithm with a higher mean area under the curve (AUC) value of 0.78 than its predecessor, FRED (0.75) [39, 60]. Using the pose mode in OMEGA [62] with default parameters, different conformers were generated for the pre-filtered library of compounds. OMEGA implements an algorithm that breaks small molecules into fragments and reassembles them into 3D conformations subjecting each conformer to energy evaluations [62]. Conformers are finally clustered after meeting a particular energy threshold [62, 63].
The conformer-generated library was then docked into the active site of the prepared receptor using HYBRID 3.5.0.4 docking program of the OEDocking tools. The same OEDocking receptor prepared during the docking validation was used for the docking. The Chemgauss4 scoring function (OpenEye Scientific Software, Inc., Santa Fe, NM, USA, www.eyesopen.com) was used to score the best binding conformations. This was the first phase of the structure-based virtual screening in the consensus docking methodology.
Virtual screening with AutoDock Vina
The pre-filtered library of 3942 compounds was also virtually screened against the human furin protease using AutoDock Vina [64]. The structure-data file (SDF) format of the 3942 compounds was loaded into PyRx (version 0.8) [65] and energy minimized using its integrated Open Babel [66]. The energy minimization was done using the United Force Field (UFF) with a limit of 200 iterations for each ligand. The energy minimized ligands were each converted to PDBQT format, an acceptable input for AutoDock Vina. PyRx was again used to prepare the receptor into an acceptable AutoDock format. The same grid box dimensions used for the docking validation were selected for the screening of the hits. These dimensions alongside a specified exhaustiveness of eight were registered in a configuration file. The configuration file was retrieved along with the PDBQT files of the receptor and the ligands. The docking was performed on a Dell EMC high-performance computing (HPC) system which consists of CentOS 7 operating system, 6 nodes, 12 GPUs, 216 CPUs, and 277 TB of storage situated at the West African Centre for Cell Biology of Infectious Pathogens (WACCBIP), University of Ghana, Accra. After docking, nine different poses were generated for each compound. For each compound, the pose with the most negative binding energy and zero RMSD value was selected. Compounds which had binding energies equal to or below − 7 kcal.mol−1 were considered for further analysis [41].
Root mean square deviation comparisons
Docking poses of hit compounds obtained from AutoDock Vina screening were compared to their respective poses obtained from screening with HYBRID docking tool using the LigAlign [67] program interfaced with PyMOL (v 2.0.6). The root mean square deviation (RMSD) calculation was used to score how well poses align with each other. An RMSD cut-off of 2 Å was used [42] since this cut-off value is widely regarded as the most effective threshold value for validating correctly posed molecules [42, 48]. Previous studies have used this threshold to validate docking protocols [42, 68, 69], rank ligands [70], align ligands to reference structures [71, 72], and for comparative studies [73–75]. Herein, compounds with pose alignment scoring below the RMSD cut-off were retained for further analysis.
Selection of potential lead compounds
Compounds were selected as potential leads based on three criteria. Firstly, the compounds must have reasonably good binding energies from both docking protocols and be considered top hit after each screening. Secondly, their ability to align well in the pose comparison analysis, that is, compound alignment RMSD score must meet the cut-off of 2 Å [42]. Lastly, occupation of active site regions based on pose visualization analysis and compound interactions with critical residues (His194, Ser368, and Asp153) of the active site. The selected potential leads were then subjected to stability and binding free energy analysis using molecular dynamics (MD) simulation and MM-PBSA calculations in GROningen MAchine for Chemical Simulations (GROMACS) version 2018 [76].
Molecular dynamics simulation
The potential lead compounds in complex with the receptor were subjected to 100 ns MD simulation. This was done to study the stability of the interactions between the compounds and the receptor. GROMACS (version 2018) was used to carry out the MD simulations for the unbound protein and protein–ligand complexes [77, 78]. The GROMOS96 43a1 force field was employed for each simulation. The ligand topology files were generated using PRODRG2 server [79]. The complexes were solvated within an SPC water model in a dodecahedron box, with box-solute distance of 1.0 nm. Due to the negative charges on the protein, the system was neutralized using 14 sodium ions. The steepest descent algorithm was then used to energy minimize the system over 1000 steps. The system was equilibrated using the constant-temperature constant-pressure (NPT) and constant-temperature, constant-volume (NVT) ensembles. A 100 ns production MD was finally run on each complex system. The simulations were performed on the same computing resource used for the docking procedure. Xmgrace was used to plot the simulation graphs after the production run was completed [80].
MM-PBSA calculations
The Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) binding energy calculations method was used to calculate the binding free energies of each protein–ligand complex of the potential leads with g-mmpbsa [81, 82] after they had been subjected to MD simulations via GROMACS (version 2018) [76] using the GROMOS96 43a1 force field. The total binding free energies of the complexes were calculated from the van der Waals, electrostatic, polar solvation, and non-polar solvation energies. The polar solvation and non-polar solvation energies are estimated from the Poisson–Boltzmann equation and the solvent accessible surface area (SASA) methods. The total binding energy () equation is given [83] as Eq. 1.
| 1 |
Structural similarity search and activity prediction of hits
A search for compounds similar in structure to the predicted hits was done via DrugBank [84] with a threshold of 0.7 in order to find close analogues of the predicted hits. Literature was then searched for the use of these analogues in the treatment of similar disease conditions or in any experimental studies relevant to our findings. Furthermore, the biological activities of the predicted hits were determined using Prediction of Activity Spectra for Substances (PASS) [37, 85]. PASS uses Bayesian models to predict biological activities of compounds and their mechanisms of action [37, 85]. Hence, PASS can estimate the likelihood of a compound belonging to a specific class of active compounds using the probable activity (Pa) and probable inactivity (Pi) measures, which range between 0 and 1. The SMILES of the predicted hits were used as inputs for the PASS predictions.
Pharmacokinetic and toxicity profiling of potential leads
The absorption, distribution, metabolism, excretion, and toxicity (ADME/Tox) properties of the potential leads were profiled using SwissADME [86], pkCSM [87], and ProTox-II [88]. It is crucial to determine the ADME/Tox profiles of compounds since exposure to some compounds and their combinations may be harmful [88]. In silico toxicity models complement existing experimental models by predicting the effects of compounds, which in turn reduce the time, cost, and number of animal models required for testing [88]. SwissADME employs multiple linear regression, BOILED-Egg, and various binary classification models for compound pharmacokinetic predictions [86]. Likewise, the pkCSM server uses distance-based graph modeling approach to predict and optimize the pharmacokinetic properties and toxicity of compounds [87]. Graph modeling is an approach where atoms of compounds are represented as nodes and their bonds as edges [89]. However, ProTox-II is solely for toxicity prediction of chemicals focusing mainly on cytotoxicity, acute toxicity, hepatotoxicity, carcinogenicity, adverse outcomes pathways (Tox21), mutagenicity, immunotoxicity, and toxicity targets. ProTox-II relies on molecular similarity, pharmacophores, fragment propensities, and machine-learning predictive models for the various toxicity predictions [88].
Results and discussion
The availability of a crystallized 3D furin protease structure and a bound ligand makes it convenient to employ structure-based virtual screening (SBVS) methods in this study. SBVS is a computational approach used at the early stage of drug discovery that makes it easier and faster to sample out potential bioactive compounds from a large library of compounds/chemical space. With SBVS, the best interaction mode between compounds and their targets are easily predicted [90]. In view of this, top-ranked hit compounds which bind to a target must achieve a particular conformation, position, and orientation (pose) in order to attain the desired interaction with residues of the protein active site [91]. Improvement of poses has become very important now in virtual screening because the accuracy of most scoring functions of docking algorithms depends on them. Due to this, the consensus docking methodology which is similar to the consensus scoring approach was proposed [48]. This study used the approach to predict inhibitors for the human furin protease, combining OEDocking HYBRID [39] and AutoDock Vina [40]. We compared poses of 45 hit compounds obtained through consensus docking from both docking programs. Twenty compounds had an RMSD below 2 Å, a threshold below which poses are said to align properly [42]. Subsequently, seven potential leads were selected based on their interactions with active site residues.
Docking validation
This study evaluated the docking performance of HYBRID and AutoDock Vina via Screening Explorer [61] in order to ascertain how effectively these docking tools can distinguish between active and inactive molecules for the furin target. Their performances were thus evaluated using metrics including the area under the curve (AUC) of the receiver operating characteristic curve (ROC) and Boltzmann-enhanced discrimination of ROC (BEDROC). The ROC AUC metric, with values ranging between 0 and 1, measures the relationship between sensitivity (fraction of selected true active compounds out of the total actives) and specificity (fraction of false positives out of the total inactive compounds). An AUC value close to 0 and less than 0.5 means poor discrimination [67, 68]. The greater the AUC, the higher the likelihood of the virtual screening protocol to discriminate between active and inactive compounds. AUC values of approximately 0.50 and 0.72 were achieved for HYBRID and AutoDock Vina, respectively.
Early recognition of active molecules by both docking protocols was assessed using BEDROC. BEDROC is an enrichment metric that uses an exponential function to assign more weights to early ranked molecules than late ones. BEDROC values range between 0 and 1, with 0.5 being the value expected for random selection. The BEDROC parameter α was set to 20.0 which means that 80% of the maximum contribution to the metric comes from 8% of the data list [67]. BEDROC values of 0.024 and 0.649 were observed for OEDocking and AutoDock Vina, respectively. BEDROC values greater than 0.5 are considered best performance for early enrichment. HYBRID had relatively low AUC and BEDROC values because the tool was unable to generate poses for 32 of the active compounds and 81 decoys using the bound MI-1148 as reference. The AUC value for AutoDock Vina which was above 0.7 (Fig. 2), implies that AutoDock Vina has a highly acceptable discrimination rate for our data compared to HYBRID.
Fig. 2.
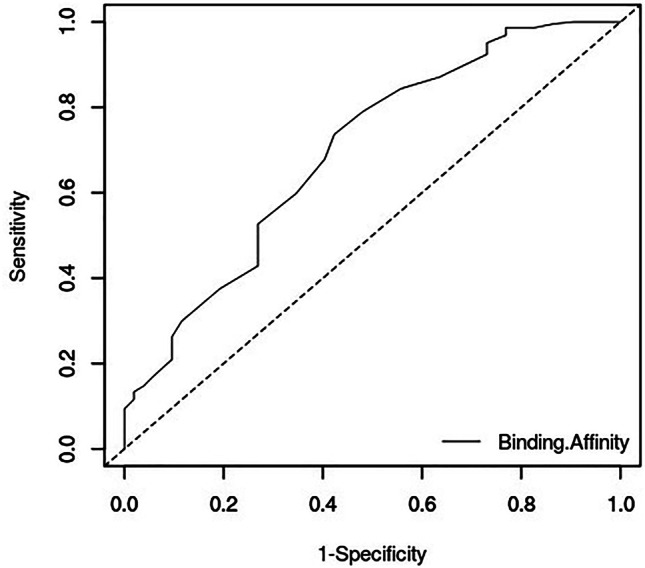
ROC AUC assessment of AutoDock Vina after screening 52 active compounds and 224 decoys against the furin protease. A reasonable AUC value of 0.72 was achieved
HYBRID has the advantage of using the reference ligand to enhance the docking performance by selecting compounds with similar poses while AutoDock Vina has been predicted to have a high active-decoy discrimination rate. Thus using these two docking protocols complement the other’s strengths. Values obtained for the assessment analysis are suggestive that AutoDock Vina is a good docking tool for selecting actives against the protein target.
Molecular docking studies
Virtual screening with HYBRID
A library of 3942 pre-filtered natural product compounds was docked into the active site of the crystal structure of human furin protease. The spatial disposition of the known ligand (MI-1148) was used to define the active site by using Spruce4Docking (OpenEye Scientific Software, Inc., Santa Fe, NM, USA, www.eyesopen.com). A bound ligand is important when using HYBRID because its docking and scoring algorithm work based on how well docked molecules mimic the shape and 3D positioning of chemical features of the co-crystallized ligand in the protein active site. Docked poses of compounds are usually biased towards the pose of the crystallographic ligand. This is one reason why this docking application was employed for our initial docking protocol to obtain compounds which might dock with similar binding poses as MI-1148 (Fig. 3).
Fig. 3.

Chemical structure of the bound ligand, para-guanidinomethyl-Phac-R-Tle-R-Amba (MI-1148)
HYBRID 3.5.0.4 was used to run the initial molecular docking taking as inputs the receptor-ligand complex and the pre-filtered library. This formed the first phase of the consensus docking process. After this phase of virtual screening with HYBRID, compounds with binding energies below − 7 kcal.mol−1 were considered [41]. A total of 89 compounds were found in all, with the most negative binding energy being − 12.04 kcal.mol−1, implying the highest binding affinity. Upon visual inspections with VIDA (OpenEye Scientific Software, Inc., Santa Fe, NM, USA, www.eyesopen.com), all 89 compounds were found to dock in the active site. Compound ZINC000001530775 had the most negative binding energy with the furin protease with a value of − 12.035076 kcal.mol−1, followed by smirnovine and tubastrine with binding energies of − 10.141556 and − 9.564701 kcal.mol−1, respectively. Naringenin 7-p-coumaroylglucoside and ( +)-lariciresinol-9-O-beta-D-glucopyranoside had binding energies of − 9.401727 and − 9.345255 kcal.mol−1, respectively. Caulindole A, 6-bromo-N-methylaplysinopsin and pinobanksin_3-(E)-caffeate also had strong binding affinity to the furin protease with binding energies of − 9.247932, − 9.177244, and − 8.666552 kcal.mol−1, respectively.
Virtual screening with AutoDock Vina
The pre-filtered library was also docked against the furin protease using AutoDock Vina. A total of 3911 out of the 3942 compounds were successfully screened against the furin protease. In all, 1754 compounds had binding energies of − 7.0 kcal.mol−1 or lower. ZINC000095486208 demonstrated the most negative binding energy of − 10.0 kcal.mol−1 against the human furin protease. The known inhibitor, MI-1148, 10′-hydroxyusambarensine and 3-O-(beta-D-glucopyranosyl)_etioline had a binding energy of − 9.7 kcal.mol−1 when docked against the protease. Comparatively, a previous study docked 163 ligands against the furin protease and the TMPRSS2 using AutoDock Vina [92]. The top 23 compounds found from the study were reported to bind with the catalytic residues of furin at the active site with binding energies ranging from − 8.7 to − 7.0 kcal.mol−1 [92]. Also, in the quest to find SARS-CoV-2 inhibitors, 29 phytocompounds from Acacia pennata (L.) Willd were docked against the SARS-CoV-2 Mpro and the furin protease [93]. The 29 compounds had binding energies ranging from − 9.0 to − 2.0 kcal.mol−1 [93]. Another study docked mozenavir with the furin protease using AutoDock and reported a binding energy of − 12.05 kcal.mol−1 as against the reference furin inhibitor decanoyl-RVKR-chloromethyl ketone which had a binding energy of − 6.89 kcal.mol−1 [94].
Selecting hits from the docking protocol
Compounds which had met the − 7.0 threshold when docked using both OEDocking HYBRID and AutoDock Vina were shortlisted for further studies. From the top 89 and 1754 compounds from HYBRID and AutoDock Vina, respectively, 45 compounds demonstrated binding energies of − 7.0 kcal.mol−1 or lower with both docking protocols and were thus selected as potential hits. The binding energies of the top 45 compounds ranged from − 9.7 to − 7.0 kcal.mol−1. The compound, 10′-hydroxyusambarensine had the least binding with the furin protease with a binding energy of − 9.7 kcal.mol−1, followed by ZINC000095486212 with − 9.4 kcal.mol−1. ZINC000085967772 and apigenin-7-O-6″-E-p-coumaroyl-beta-D-glucopyranoside_beta_2 both had a binding energy of -9.3 kcal.mol−1. The compounds identified herein have relatively high binding affinity to the furin protease than those predicted using AutoDock Vina in previous studies [92, 93] and are worthy of further exploration. Furthermore, visualization of the selected hit compounds in PyMOL (v 2.0.6) showed that all the selected compounds docked well in the active site.
RMSD comparison
The poses of the top 45 compounds from AutoDock Vina were compared to their respective HYBRID poses. Out of the 45 selected compounds from the docking with AutoDock Vina, 20 of them had RMSD values ≤ 2 Å when compared to their corresponding HYBRID pose. The compound with the best aligned pose (malvinidin-3-arabinoside) had a RMSD score of 0.616, whereas pinobanksin_3-(E)-caffeate had the highest RMSD value of 1.993 Å among the top 20 compounds (Table 1). Compound 10′-hydroxyusambarensine which had the highest affinity to the furin protease (as predicted via AutoDock Vina) also had a RMSD of 0.772 Å (Table 1).
Table 1.
Top 20 potential hit compounds seeded for analysis after consensus docking of human furin protease. Shown in the table are their binding energy scores obtained from HYBRID and AutoDock Vina docking. RMSD values obtained from pose comparison are also presented
| Compound name | Binding energy (HYBRID docking (kcal.mol−1)) | Binding energy (AutoDock Vina docking (kcal.mol−1)) | RMSD value (Å) |
|---|---|---|---|
| Malvinidin-3-arabinoside | − 7.47553 | − 8.0 | 0.616 |
| 10′-hydroxyusambarensine | − 7.65450 | − 9.7 | 0.772 |
| Z-hymenialdisine | − 7.43515 | − 8.3 | 0.899 |
| Quercetin 3-O-arabinopyranoside | − 7.13269 | − 7.9 | 1.245 |
| ZINC000085967772 | − 7.13617 | − 9.3 | 1.252 |
| Z-debromohymenialdisine | − 7.07985 | − 7.4 | 1.405 |
| Abyssinone_IV | − 7.78757 | − 8.8 | 1.432 |
| Teucrol | − 7.23971 | − 8.2 | 1.499 |
| 6-bromo-N-methylaplysinopsin | − 9.17724 | − 7.4 | 1.554 |
| ( ±)-enterofuran | − 7.06213 | − 7.2 | 1.558 |
| ZINC000095485902 | − 7.07927 | − 7.3 | 1.589 |
| ZINC000014780903 | − 7.62793 | − 7.6 | 1.671 |
| Caulindole B | − 7.32471 | − 8.0 | 1.796 |
| ZINC000095486083 | − 7.91902 | − 8.4 | 1.800 |
| Rosmarinic acid | − 7.58047 | − 8.8 | 1.841 |
| N-E-caffeoyl tyramine | − 7.75178 | − 8.2 | 1.850 |
| Bidwillon_A | − 7.62793 | − 8.1 | 1.895 |
| Quercitrin | − 7.03509 | − 8.1 | 1.930 |
| Caulindole A | − 9.24793 | − 9.1 | 1.937 |
| Pinobanksin_3-(E)-caffeate | − 8.66655 | − 9.1 | 1.993 |
Furin-ligand interactions
Inhibitors which bind to the human furin protease were observed to bind at certain interaction sites on the catalytic domain of the protein (Fig. 4) [95]. These interaction sites within the active site contain the catalytic triad Ser368, His194, and Asp153 which are important for ligand interactions. Aside these catalytic residues, other ligand binding residues include Trp254, Pro256, Val231, Asn295, Glu257, Gly255, Ala267, Asp233, Asp236, Tyr308, and Asp264 [93, 94]. Mozenavir was previously shown in silico to interact with Tyr308, Gly265, Gly255, Asp154, and Val231 via hydrogen bonds and formed hydrophobic contacts with Glu236, Pro256, Trp254, Leu227, His194, Gly229, Asp264, Asp191, Glu230, and Gly229 [94]. Imatinib forms two hydrogen bonds with Glu236 and Gly255, and interacts with the furin protease via weak hydrophobic contacts with Val231, Pro256, Trp254, and Gly294 [29].
Fig. 4.

Furin protease binding pocket. (A) Active site region of furin protease. Labels S1, S2, S3, S4, and S5 are regions within the active site where active ligands are supposed to occupy. Regions S1 and S2 are areas where catalytic triad Ser368 (green), His194 (yellow), and Asp153 (purple) are found, and these were areas most occupied by potential inhibitors. (B) The active site catalytic triad are rendered in sticks representation
After visual inspection, most of the compounds occupied sites S1 to S4 (Fig. 4). None of the compounds was found to occupy position S5. Also, none of the compounds was found to occupy all the five sites. This may be due to the smaller sizes of the compounds when compared to the co-crystallized ligands.
A total of seven compounds were shortlisted based on the following criteria: (i) visual inspection of compound occupation of the active site mainly in the areas S1, S2, S3, S4, and S5; and (ii) compound interaction with important active site residues, with emphasis being placed on the catalytic triad. The seven shortlisted compounds include quercitrin, teucrol, malvinidin-3-arabinoside, N-E-caffeoyl tyramine, ZINC000085967772, pinobanksin 3-(E)-caffeate, and abyssinone IV. It is worthy to note that all seven selected potential leads (Fig. 5) occupied mostly S1 and S2 as these are the areas where majority of the interacting residues are found including the catalytic triad. Two compounds namely quercitrin and abyssinone IV were also found to form hydrogen bond interactions with critical residue Ser368 and hydrophobic bond interactions with His194 (Table 2 and Fig. 6A and B). The other five compounds did not share hydrogen bond interactions with any of the catalytic triad; however, they shared hydrophobic bond interactions with Ser368 and His194 (Figure S1). They also shared strong hydrogen bond contacts with other important surrounding residues (Table 2).
Fig. 5.

Chemical structures of the potential lead compounds predicted against furin protease. (A) Quercitrin. (B) Teucrol. (C) Malvinidin-3-arabinoside. (D) N-E-caffeoyl tyramine. (E) Pinobanksin 3-(E)-caffeate. (F) Abyssinone IV. (G) ZINC000085967772
Table 2.
Potential lead compounds and interacting residues via hydrogen bond (H-bond) interactions. Also shown is their binding energies obtained via AutoDock Vina comparable to that of MI-1148 which was used as control
| Compound name | Binding energy (kcal.mol−1) | Number of H-bond contacts | Residues involved in H-bond interactions | Residues involved in hydrophobic interactions |
|---|---|---|---|---|
| Quercitrin | − 8.1 | 9 | Ser368, Leu227, Asp258, Asn295, Thr365, Asp191 | His194, Ser253, Trp254, Gly294, Gly366, Thr367 |
| Teucrol | − 8.2 | 7 | Pro256, Asp154, Asn192, Asp191, Ala292, Leu227, Asp306 | Asp228, Trp254, Gly255, Asp258, Gly294, Ser293, Thr309, Thr367 |
| Malvinidin-3-arabinoside | − 8.0 | 6 | Pro256, Gly255, Asp258, Asn295 | Asp154, His194, Leu227, Val231, Glu236, Ser253, Trp254, Glu257, Gly294, Thr367 |
| N-E-caffeoyl tyramine | − 8.2 | 5 | Asp154, Asn192, Ala292, Asp191, Asp306, Leu227 | His194, Asp228, Ser253, Trp254, Gly255, Pro256, Asp258, Ser293, Gly294, Thr367, Ser368 |
| ZINC000085967772 | − 9.3 | 5 | Pro256, Asp306, Asp258, Ala292, Asn295 | His194, Leu227, Val231, Trp254, Gly255, Trp291, Ser293, Gly294, Thr309, Thr367 |
| Pinobanksin 3-(E)-caffeate | − 9.1 | 5 | Pro256, Asp154, Asn192, Ala292, Asp191 | His194, Leu227, Asp228, Ser253, Trp254, Gly255, Asp258, Ser293, Gly294 |
| Abyssinone IV | − 8.8 | 2 | Ser368, Leu227 | Asp153, Asp154, Asp191, His194, Val231, Ser253, Asp258, Asn295, Trp254, gly255, Gly294, Thr367 |
| MI-1148 (control) | − 9.7 | 13 | Asp191, Asn192, His194, Ala292, Ser368, Asp258, Pro256, Asp306, Tyr308, Asp264, Glu236, Leu227 | Asp154, Thr232, Asp233, Ser253, Trp254, Gly255, Glu257, Ala267, Asp288, Asp264, Ser293, Gly294, Thr309, Thr367 |
Fig. 6.

Poses of selected potential lead compounds. Almost all the compounds were found to bind in S1 and S2 regions where catalytic residues are located. (A) Abyssinone IV and (B) Quercitrin had hydrogen or hydrophobic interactions with catalytic residues His194 and Ser368. Residues involved in hydrogen bond interactions with ligands are labelled in black while residues involved in hydrophobic interactions are labelled red. Images were generated with PyMOL (v 2.0.6)
Molecular dynamics simulation
Understanding the dynamic behavior of the proposed potential leads within the target’s binding pocket involves studying factors such as stability, structure compactness, and residue fluctuations. These factors were assessed via the root mean square deviation (RMSD) and radius of gyration (Rg) plots for 100-ns simulation time.
Root mean square deviation (RMSD)
The RMSD measures the flexibility/stability of the protein. This was monitored both for the unbound protein and the protein–ligand complexes. At the initial stages of the simulation, both the unbound protein and the complexes had their RMSD values peak to about 0.25 nm. The RMSDs of all the complexes rose until about 80 ns where they were observed to remain stable till the end of the 100-ns simulation time (Fig. 7). These results were consistent with the RMSD plots for furin-mozenavir, furin-folic acid, and furin-folinic acid complexes [94, 96]. However, same observation cannot be said about the unbound protein as the RMSD continued to rise even after 80 ns to levels around 0.3 nm. Similar observations have been reported in a 10-ns MD study, where the RMSD of the furin protease rose towards the end of the simulation time to about 0.4 nm [94]. This is indicative that the presence of the ligands may have brought stability to the protein. Nonetheless, complexes with ligands pinobanksin_3-(E)-caffeate (yellow colored) and teucrol (cream colored) achieved stability at RMSDs of 0.3 nm and higher.
Fig. 7.

Root mean square deviation (RMSD) plot of furin-ligand complexes over 100-ns simulation. The backbone after least-squares (lsq) fit to the backbone
Radius of gyration (Rg)
Rg is used to evaluate the compactness of the protein structures throughout the simulation period. A steady Rg for a particular structure indicates that the protein is stably folded [97, 98]. Most of the complexes including the unbound protein had relatively stable Rg values revolving between 2.15 and 2.2 nm (Fig. 8), consistent with previous Rg studies on furin-folic acid and furin-folinic acid complexes [96]. Only quercitrin and teucrol complexes had their Rg values below this range; however, they maintained a steady value of about 2.14 and 2.125 nm, respectively, beyond 60 ns (Fig. 8).
Fig. 8.

Radius of gyration (Rg) plot of furin-ligand complexes over 100-ns simulation
Binding free energy
MM-PBSA calculations have been deemed to be more accurate in estimating binding free energies of compounds than scoring functions of most docking programs [99]. Estimating binding free energies for protein–ligand interactions has been made robust using g-mmpbsa [81]. The g-mmpbsa integrates both GROMACS and APBS for its energy calculations. Herein, binding energies for each of the eight complexes were estimated separately after each undergoing molecular dynamics simulation. After the calculations, pinobanksin 3-(E)-caffeate had the strongest binding with a binding free energy score of − 189.219 ± 25.602 kJ/mol (Table 3 and Fig. 9). Comparatively, the same compound had a better binding affinity score from the molecular docking results with a value of − 9.1 kcal.mol−1 (Table 2). ZINC000085967772 which had the most negative binding energy (− 9.3 kcal.mol−1) in the docking results among the potential leads also showed strong binding from the MM-PBSA calculations with a binding free energy score of − 172.892 ± 24.913 kJ/mol (Table 3 and Fig. 9). All the other potential leads had reasonable binding free energies (Table 3) which correlated well with their binding affinities obtained after molecular docking. A previous study investigated the molecular interactions between the furin protein and two ligands comprising folic and folinic acids using the MM-PBSA method. Folic and folinic acids had binding free energies of − 27.90 and − 12.84 kcal.mol−1, respectively [96]. Although, the interacting residues of the furin protease with folic and folinic acids are different from those observed herein, the predicted compounds in this study have higher affinities to the furin protease than the two folate analogues [96].
Table 3.
Contributing energies of the potential leads estimated from molecular Mechanics/Poisson-Boltzmann Surface Area (MM-PBSA) calculations. The energy values are stated in average with their standard deviations
| Compound name | van der Waals energy (kJ/mol) | Electrostatic energy (kJ/mol) | Polar solvation energy (kJ/mol) | Nonpolar solvation energy (kJ/mol) | Binding free energy |
|---|---|---|---|---|---|
| Quercitrin | − 197.532 ± 20.459 | − 107.316 ± 28.560 | 229.403 ± 40.315 | − 19.562 ± 1.354 | − 95.007 ± 22.816 |
| Teucrol | − 182.988 ± 21.019 | − 50.614 ± 22.080 | 128.654 ± 27.686 | − 16.986 ± 1.193 | − 121.935 ± 25.297 |
| Malvinidin-3-arabinoside | − 158.093 ± 33.267 | − 70.333 ± 36.322 | 113.350 ± 45.573 | − 15.654 ± 1.857 | − 130.731 ± 35.454 |
| N-E-caffeoyl tyramine | − 188.480 ± 12.010 | − 46.255 ± 15.189 | 92.678 ± 23.135 | − 14.939 ± 1.065 | − 156.996 ± 19.169 |
| ZINC000085967772 | − 244.543 ± 24.925 | − 66.744 ± 16.519 | 158.873 ± 34.790 | − 20.478 ± 1.494 | − 172.892 ± 24.913 |
| Pinobanksin_3-(E)-caffeate | − 220.695 ± 17.948 | − 72.870 ± 25.096 | 122.875 ± 34.294 | − 18.529 ± 1.343 | − 189.219 ± 25.602 |
| Abyssinone IV | − 172.210 ± 31.278 | − 39.115 ± 16.107 | 74.818 ± 33.845 | − 16.395 ± 2.274 | − 152.902 ± 27.063 |
Fig. 9.

Binding free energies of protein–ligand complexes of potential lead compounds from MM-PSA calculations
Herein, MM-PBSA calculations on the control (MI-1148) showed very strong binding with the target detailing a binding energy of − 2178.767 kJ/mol ± 67.643 kJ/mol. This is particularly not surprising as the compound was synthetically designed to specifically target the protein [19]. Nonetheless, the binding free energies exhibited by the predicted compounds are reasonable. Therefore, structural modifications or utilization as scaffolds for the design of furin-targeted compounds could elicit stronger binding with the furin protease.
Biological activity prediction and structural similarity search
Quercitrin, one of the potential leads is a glycoside formed from the flavonoid quercetin and deoxy sugar rhamnose. Quercetin has also been found to show therapeutic effects against Enterovirus 71 protease, therefore blocking the virus replication in hosts [100]. Enterovirus 71 is also a single-stranded RNA virus known to cause hand, foot, and mouth disease in under-aged children (< 5 years) [100]. Quercitrin is found in some medicinal plants used by folk medicine in the treatment of inflammation [101]. Additionally, it was predicted to be anti-inflammatory with Pa of 0.754 and Pi of 0.010. Inflammation is a key feature in COVID-19 disease, accounting for some unforeseen circumstances or even death in patients [102]. A study identified anti-inflammatory compounds which targeted p38 MAPK receptor in the quest to salvage high concentrations of pro-inflammatory cytokines in COVID-19 mechanisms [103]. Quercitrin was also predicted to be a membrane permeability inhibitor (Pa = 0.986 and Pi = 0.001), RNA synthesis inhibitor (Pa = 0.558 and Pi = 0.004), as anti-influenza virus (Pa = 0.683 and Pi = 0.007), and anti-herpes (Pa = 0.518 and Pi = 0.008).
Teucrol, abyssinone IV, and ZINC000085967772 were all predicted to be membrane permeability inhibitors with Pa values of 0.759, 0.644, and 0.714 and Pi values of 0.018, 0.065, and 0.033, respectively. Teucrol was also predicted to be a membrane integrity agonist (Pa = 0.948 and Pi = 0.004) with anti-inflammatory properties (Pa = 0.569 and Pi = 0.039). Abyssinone IV and ZINC000085967772 were predicted to be cytoprotectant (Pa = 0.694 and Pi = 0.005) and hepatoprotectant (Pa = 0.589 and Pi = 0.013). Abyssinone IV has antiviral properties against the rhinovirus (Pa = 0.600 and Pi = 0.006). Pinobanksin 3-(E)-caffeate which is a propolis component was predicted to be anti-inflammatory (Pa = 0.821, Pi = 0.005) and hepatoprotectant (Pa = 0.829, Pi = 0.004). Interestingly, propolis components have recently been studied as possible therapeutics for COVID-19, and it showed inhibitory effects on ACE2, TMPRSS2, and PAK1 signaling pathways [104, 105]. Propolis is also used in traditional medicine worldwide due to its reported biological activities which include antibacterial, antiviral, anti-inflammatory, and anticancer [106–108].
In addition, rosmarinic acid, which was part of the 20 selected hits, has also been shown to have anti-influenza effects when its activity was tested on mice infected with A/FM/1/47 H1N1 virus. Significant improvements were observed in the infected mice treatment [109]. Rosmarinic acid is a compound isolated from a plant known as Sarcandra glabra (Thunb.) Nakai. Pursuing these compounds for antiviral, anti-inflammatory, and cell protective activities hold promising future especially against single-stranded viruses like SARS-CoV-2. Therefore, the potential leads and some of the hit compounds could be explored via in vitro and in vivo studies for antiviral purposes.
Structural similarity search was performed for each of the selected compounds via DrugBank using a similarity threshold of 0.7. This procedure was performed to decipher if any of the potential leads have similar structures or analogues studied in vitro, in vivo, or in clinical tests. Quercitrin (quercetin 3-O-α-l-rhamnopyranoside) had a DrugBank similarity index (SI) of 1.0 with isoquercetin, a quercetin derivative which is monoglycosylated. Malvinidin-3-arabinoside which was also identified as a potential lead compound had a similarity index of 0.822 with isoquercetin. Isoquercetin is a drug that is currently being tested clinically as medication for moderate to severe COVID-19 patients [110]. This supports the possible evaluation of quercitrin with a broad antiviral activity as a potential COVID-19 therapeutic agent. Likewise, teucrol was structurally similar to amiloxate with similarity index of 0.722. Amiloxate is a derivative of cinnamic acid which is known to have anti-inflammatory properties [111]. Abyssinone IV and pinobanksin 3-(E)-caffeate were also found to be structurally similar to taxifolin, a compound known to be an anti-inflammatory agent [112, 113] with similarity indexes of 0.749 and 0.767, respectively. Abyssinone IV also has similarities to hesperetin (SI: 0.805). Hesperetin is known to have anti-cancer and anti-inflammatory effects [114, 115]. Pinobanksin 3-(E)-caffeate is also structurally similar to silibinin with SI of 0.767. Silibinin is a flavonolignan which is said to have hepatoprotective effects and is used in managing hepatitis [116, 117]. Hepatitis is caused by the hepatitis virus which is known to exploit furin family of proteins (proprotein convertases) in its disease mechanism [21].
Pharmacokinetic and toxicity profiling of potential leads
SwissADME and pkCSM predictions showed that all the potential lead compounds have reasonable solubility and intestinal absorption with an absorption rate greater than 45% (Table 4 and Fig. 10). Also, none of the compounds were predicted to cross the blood–brain barrier (BBB). This is specifically significant because earlier studies have established that drugs that target sections of the human body other than the nervous system should not cross the BBB in order to avoid psychotropic side effects [118]. The compounds were also favorable towards cytochrome P450 enzymes, having shown none or few possible inhibitions to CYP2D6, CYP2C19, CYP2C9, and CYP1A2. However, four of the ligands namely Teucrol, N-E-caffeoyl tyramine, Pinobanksin 3-(E)-caffeate, and Abyssinone IV were predicted as possible substrates of CYP3A4 enzyme. Hepatic and renal clearance were low for all the compounds. For the toxicity predictions, the compounds passed the Ames test for toxicity and none but N-E-caffeoyl tyramine was found to be hepatotoxic (Table 4). Protox-II toxicity predictions revealed that the compounds were immunotoxic with probability greater than 0.6. Since furin is induced in T cells and is crucial for maintaining peripheral immune tolerance [119], this may be expected. All the compounds, except for N-E-caffeoyl tyramine had plausible lethal doses (LD50) making them favorably less toxic (Table 5). The higher the lethal dose per kilogram of a compound, the lesser its toxicity. Three of the compounds namely Quercitrin, ZINC000085967772, and Pinobanksin 3-(E)-caffeate were predicted to have about 50% chance of being carcinogenic. Teucrol was predicted to have three other active targets with each having a probability above 0.7 (Table 5). However, the fact that most of these compounds were predicted to be within the toxicity classes of 4 and 5 gives strong indications that these compounds may nonetheless be non-toxic.
Table 4.
ADMET property predictions of potential leads using pkCSM server
| Parameters | Quercitrin | Teucrol | Malvinidin-3-arabinoside | N-E-caffeoyl tyramine | ZINC000085967772 | Pinobanksin 3-(E)-caffeate | Abyssinone IV | |
|---|---|---|---|---|---|---|---|---|
| Absorption | Water solubility (log mol/L) | − 3.087 | − 3.183 | − 3.139 | − 3.18 | − 4.273 | − 4.534 | − 4.457 |
| Caco2 permeability (log Papp 10–6 cm/s) | 0.121 | − 0.183 | − 0.831 | 0.868 | 1.362 | − 0.025 | 1.07 | |
| Intestinal absorption (% absorbed) | 62.005 | 65.224 | 48.562 | 90.997 | 93.397 | 73.62 | 91.499 | |
| Skin permeability (log Kp) | − 2.735 | − 2.735 | − 2.735 | − 2.735 | − 2.992 | − 2.735 | − 2.774 | |
| P-glycoprotein I inhibitor | No | No | No | No | Yes | Yes | Yes | |
| P-glycoprotein II inhibitor | No | No | No | No | No | Yes | Yes | |
| Distribution | VDss (human, log L/Kg) | 0.451 | 0.292 | 0.45 | 0.62 | 0.36 | − 0.692 | 0.136 |
| BBB permeability (log BB) | − 1.69 | − 1.228 | − 1.986 | − 0.962 | − 0.991 | − 1.196 | − 0.183 | |
| CNS permeability (log PS) | − 4.196 | − 3.195 | − 4.176 | − 2.634 | − 2.664 | − 3.327 | − 1.737 | |
| Metabolism | CYP2D6 substrate | No | No | No | No | No | No | No |
| CYP3A4 substrate | No | Yes | No | Yes | No | Yes | Yes | |
| CYP2C19 inhibitor | No | No | No | No | No | No | Yes | |
| CYP2C9 inhibitor | No | No | No | No | No | Yes | Yes | |
| CYP1A2 inhibitor | No | No | No | Yes | No | No | No | |
| Excretion | Total clearance (log ml/min/kg) | 0.479 | 0.253 | 0.728 | 0.195 | 0.004 | − 0.024 | 0.801 |
| Renal OCT2 substrate clearance | No | No | No | No | No | No | No | |
| Toxicity | Ames toxicity | No | No | No | No | No | No | No |
| Hepatotoxicity | No | No | No | Yes | No | No | No | |
| Oral rat acute toxicity (LD50, mol/kg) | 2.889 | 2.358 | 2.648 | 2.499 | 2.564 | 2.372 | 2.217 | |
| Skin sensitization | No | No | No | No | No | No | No |
Fig. 10.

Predicted ADME properties of potential lead compounds via SwissADME. (A) Quercitrin. (B) Teucrol. (C) Malvinidin-3-arabinoside. (D) N-E-caffeoyl tyramine. (E) ZINC000085967772. (F) Pinobanksin 3-(E)-caffeate. (G) Abyssinone IV
Table 5.
Toxicity prediction of potential lead compounds using Protox-II
| Compound name | LD50 mg/Kg | Toxicity class | Predicted active targets | Probability |
|---|---|---|---|---|
| Quercitrin | 5000 | 5 |
Carcinogenicity Immunotoxicity Aryl hydrocarbon receptor |
0.50 0.97 0.55 |
| Teucrol | 5000 | 5 |
Immunotoxicity Estrogen receptor ligand-binding domain Phosphoprotein (tumor supressor) p53 ATPase family AAA domain-containing protein 5 |
0.68 0.84 0.85 0.77 |
| Malvinidin-3-arabinoside | 5000 | 5 |
Immunotoxicity Cytotoxicity |
0.99 0.50 |
| N-E-caffeoyl tyramine | 500 | 4 | Immunotoxicity | 0.64 |
| ZINC000085967772 | 5000 | 5 |
Carcinogenicity Immunotoxicity |
0.51 0.97 |
| Pinobanksin 3-(E)-caffeate | 2000 | 4 |
Carcinogenicity Immunotoxicity |
0.55 0.97 |
| Abyssinone IV | 2000 | 4 |
Immunotoxicity Mitochondrial membrane potential |
0.88 0.69 |
Conclusion
The study used the consensus docking approach via the structure-based virtual screening of human furin protease to effectively identify seven potential novel anti-COVID-19 compounds consisting of quercitrin, teucrol, malvinidin-3-arabinoside, N-E-caffeoyl tyramine, ZINC000085967772, pinobanksin_3-(E)-caffeate, and abyssinone IV. This is in a bid to support ongoing efforts to identify effective therapeutics against the SARS-CoV-2 by targeting the furin protease [29, 30]. Furin protease is a plausible COVID-19 target due to its cleavage site on the spike protein and its role in facilitating the entry of SARS-CoV-2 into host cells. The molecules showed strong active site interactions with the catalytic residues of the furin with plausibly high binding affinity than previously identified compounds from earlier studies [92, 93]. They also had favorable results when subjected to MD simulations including MM-PBSA calculations, biological activity prediction, and structural similarity search [78, 94–96]. The compounds were predicted as antiviral, anti-inflammatory, anti-cancer, hepatoprotective, cytoprotective, RNA synthesis inhibitors, and membrane permeability inhibitors with reasonable ADMET properties. Isoquercetin, a structurally similar compound to quercitrin is currently undergoing clinical trials as COVID-19 drug. Once the biological activities of these compounds are reinforced experimentally, they could nonetheless serve as scaffolds for the design of new and improved furin protease inhibitors with potent antiviral properties.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors are grateful to the West African Centre for Cell Biology of Infectious Pathogens (WACCBIP), University of Ghana for allowing us to use Zuputo, a DELL high-performance computing systems for the computational studies.
Author contribution
B.D., S.K.K., and M.D.W. conceptualized and designed the project. S.K.K. and D.B. performed the computational analysis with contributions from E.B., K.S.E., and M.D.W. B.D. wrote the first draft with contributions from S.K., E.B., K,S.E., and M.D.W. All authors have read and agreed to the submitted version of the manuscript.
Data availability
Not applicable.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hasöksüz M, Kiliç S, SARAÇ F, Coronaviruses and SARS-COV-2. Turkish J Med Sci. 2020;50:549–556. doi: 10.3906/sag-2004-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartenian E, Nandakumar D, Lari A, et al. The molecular virology of coronaviruses. J Biol Chem. 2020;295:12910–12934. doi: 10.1074/jbc.REV120.013930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Y, Peng F, Wang R, et al. The deadly coronaviruses: the 2003 SARS pandemic and the 2020 novel coronavirus epidemic in China. J Autoimmun. 2020;109:102434. doi: 10.1016/J.JAUT.2020.102434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seyed Hosseini E, Riahi Kashani N, Nikzad H, et al. The novel coronavirus Disease-2019 (COVID-19): mechanism of action, detection and recent therapeutic strategies. Virology. 2020;551:1–9. doi: 10.1016/J.VIROL.2020.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valencia DN. Brief review on COVID-19: the 2020 pandemic caused by SARS-CoV-2. Cureus. 2020 doi: 10.7759/cureus.7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kadkhoda K (2020) COVID-19: an immunopathological view. mSphere 5. 10.1128/mSphere.00344-20 [DOI] [PMC free article] [PubMed]
- 7.Cascella M, Rajnik M, Aleem A et al (2022) Features, evaluation, and treatment of coronavirus (COVID-19) [PubMed]
- 8.Sadeghifar J, Jalilian H, Momeni K, et al. Outcome evaluation of COVID-19 infected patients by disease symptoms: a cross-sectional study in Ilam Province. Iran BMC Infect Dis. 2021;21:903. doi: 10.1186/s12879-021-06613-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geier MR, Geier DA. Respiratory conditions in coronavirus disease 2019 (COVID-19): important considerations regarding novel treatment strategies to reduce mortality. Med Hypotheses. 2020;140:109760. doi: 10.1016/j.mehy.2020.109760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y-C, Lu M-C, Yang S-F, et al. Respiratory care for the critical patients with 2019 novel coronavirus. Respir Med. 2021;186:106516. doi: 10.1016/j.rmed.2021.106516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Omokhua-Uyi AG, Van Staden J. Natural product remedies for COVID-19: a focus on safety. South African J Bot. 2021;139:386–398. doi: 10.1016/J.SAJB.2021.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shokeen K, Pandey S, Shah M, Kumar S (2020) Insight towards the effect of the multibasic cleavage site of SARS-CoV-2 spike protein on cellular proteases. 10.1101/2020.04.25.061507 [DOI] [PMC free article] [PubMed]
- 13.Belen-Apak FB, Sarialioglu F. The old but new: can unfractioned heparin and low molecular weight heparins inhibit proteolytic activation and cellular internalization of SARS-CoV2 by inhibition of host cell proteases? Med Hypotheses. 2020;142:109743. doi: 10.1016/j.mehy.2020.109743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang T, Bidon M, Jaimes JA, et al. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Res. 2020;178:104792. doi: 10.1016/j.antiviral.2020.104792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nesci S. SARS-CoV-2 first contact: Spike-ACE2 interactions in COVID-19. Chem Biol Drug Des. 2021;98:207–211. doi: 10.1111/CBDD.13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoffmann M, Kleine-Weber H, Schroeder S et al (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 10.1016/j.cell.2020.02.052 [DOI] [PMC free article] [PubMed]
- 17.Hoffmann M, Kleine-Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol Cell. 2020 doi: 10.1016/j.molcel.2020.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Couture F, DAnjou F, Day R (2011) On the cutting edge of proprotein convertase pharmacology: from molecular concepts to clinical applications. Biomol Concepts [DOI] [PMC free article] [PubMed]
- 19.Hardes K, Becker GL, Lu Y, et al. Novel furin inhibitors with potent anti-infectious activity. ChemMedChem. 2015;10:1218–1231. doi: 10.1002/cmdc.201500103. [DOI] [PubMed] [Google Scholar]
- 20.Braun E, Sauter D. Furin-mediated protein processing in infectious diseases and cancer. Clin Transl Immunol. 2019;8:e1073. doi: 10.1002/CTI2.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Izaguirre G. The proteolytic regulation of virus cell entry by furin and other proprotein convertases. 2019;11:837. doi: 10.3390/V11090837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng Y-WW, Chao T-LL, Li C-LL, et al. Furin inhibitors block SARS-CoV-2 spike protein cleavage to suppress virus production and cytopathic effects. Cell Rep. 2020;33:108254. doi: 10.1016/j.celrep.2020.108254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papa G, Mallery DL, Albecka A, et al. Furin cleavage of SARS-CoV-2 spike promotes but is not essential for infection and cell-cell fusion. PLOS Pathog. 2021;17:e1009246. doi: 10.1371/journal.ppat.1009246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peacock TP, Goldhill DH, Zhou J, et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat Microbiol. 2021;6:899–909. doi: 10.1038/s41564-021-00908-w. [DOI] [PubMed] [Google Scholar]
- 25.Anand P, Puranik A, Aravamudan M et al (2020) SARS-CoV-2 strategically mimics proteolytic activation of human ENaC. Elife 9.10.7554/eLife.58603 [DOI] [PMC free article] [PubMed]
- 26.Zhang T, Wu Q, Zhang Z. Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr Biol. 2020;30:1346–1351.e2. doi: 10.1016/j.cub.2020.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vankadari N. Structure of furin protease binding to SARS-CoV-2 spike glycoprotein and implications for potential targets and virulence. J Phys Chem Lett. 2020;11:6655–6663. doi: 10.1021/acs.jpclett.0c01698. [DOI] [PubMed] [Google Scholar]
- 28.Peinado JR, Kacprzak MM, Leppla SH, Lindberg I. Cross-inhibition between furin and lethal factor inhibitors. Biochem Biophys Res Commun. 2004;321:601–605. doi: 10.1016/j.bbrc.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 29.Wu C, Zheng M, Yang Y et al (2020) Furin: a potential therapeutic target for COVID-19. iScience 23:101642. 10.1016/j.isci.2020.101642 [DOI] [PMC free article] [PubMed]
- 30.Xu Y-M, Inacio MC, Liu MX, Gunatilaka AAL. Discovery of diminazene as a dual inhibitor of SARS-CoV-2 human host proteases TMPRSS2 and furin using cell-based assays. Curr Res Chem Biol. 2022;2:100023. doi: 10.1016/j.crchbi.2022.100023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elmi A, Sayem SA-J, Ahmed M, Abdoul-Latif F (2020) Natural compounds from Djiboutian medicinal plants as inhibitors of COVID-19 by in silico investigations. Int J Curr Pharm Res 52–57. 10.22159/ijcpr.2020v12i4.39051
- 32.Siddiqui AA, Iram F, Siddiqui S, Sahu K. Role of natural products in drug discovery process. Int J Drug Dev Res. 2014;6:172–204. [Google Scholar]
- 33.El Sayed KA (2000) Natural products as antiviral agents. In: Studies in natural products chemistry. Elsevier, pp 473–572
- 34.Ntie-Kang F, Zofou D, Babiaka SB, et al. AfroDb: a select highly potent and diverse natural product library from African medicinal plants. PLoS ONE. 2013;8:e78085. doi: 10.1371/journal.pone.0078085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Müller-Kuhrt L. (2003) Putting nature back into drug discovery. Nat Biotechnol. 2003;216(21):602–602. doi: 10.1038/nbt0603-602. [DOI] [PubMed] [Google Scholar]
- 36.Poroikov VV, Filimonov DA, Ihlenfeldt W-D, et al. PASS biological activity spectrum predictions in the enhanced open NCI database browser. J Chem Inf Comput Sci. 2003;43:228–236. doi: 10.1021/ci020048r. [DOI] [PubMed] [Google Scholar]
- 37.Parasuraman S. Prediction of activity spectra for substances. J Pharmacol Pharmacother. 2011;2:52–53. doi: 10.4103/0976-500X.77119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jamkhande PG, Pathan SK, Wadher SJ. In silico PASS analysis and determination of antimycobacterial, antifungal, and antioxidant efficacies of maslinic acid in an extract rich in pentacyclic triterpenoids. Int J Mycobacteriology. 2016;5:417–425. doi: 10.1016/j.ijmyco.2016.06.020. [DOI] [PubMed] [Google Scholar]
- 39.McGann M. FRED and HYBRID docking performance on standardized datasets. J Comput Aided Mol Des. 2012;26:897–906. doi: 10.1007/s10822-012-9584-8. [DOI] [PubMed] [Google Scholar]
- 40.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficientoptimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang MW, Lindstrom W, Olson AJ, Belew RK. Analysis of HIV wild-type and mutant structures via in silico docking against diverse ligand libraries. J Chem Inf Model. 2007 doi: 10.1021/ci700044s. [DOI] [PubMed] [Google Scholar]
- 42.Alves M, Froufe H, Costa A, et al. Docking studies in target proteins involved in antibacterial action mechanisms: extending the knowledge on standard antibiotics to antimicrobial mushroom compounds. Molecules. 2014;19:1672–1684. doi: 10.3390/molecules19021672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laskowski RA, Swindells MB. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 44.Ntie-Kang F, Telukunta KK, Döring K, et al. NANPDB: a resource for natural products from northern African sources. J Nat Prod. 2017;80:2067–2076. doi: 10.1021/acs.jnatprod.7b00283. [DOI] [PubMed] [Google Scholar]
- 45.Simoben CV, Qaseem A, Moumbock AFA, et al. Pharmacoinformatic Investigation of medicinal plants from East Africa. Mol Inform. 2020;39:2000163. doi: 10.1002/minf.202000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sterling T, Irwin JJ. ZINC 15 – ligand discovery for everyone. J Chem Inf Model. 2015;55:2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Osman EEA, Rehemtulla A, Neamati N. Why all the fury over furin? J Med Chem. 2022;65:2747–2784. doi: 10.1021/acs.jmedchem.1c00518. [DOI] [PubMed] [Google Scholar]
- 48.Houston DR, Walkinshaw MD. Consensus docking: improving the reliability of docking in a virtual screening context. J Chem Inf Model. 2013;53:384–390. doi: 10.1021/ci300399w. [DOI] [PubMed] [Google Scholar]
- 49.Seeliger D, De Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des. 2010;24:417–422. doi: 10.1007/s10822-010-9352-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lang PT, Brozell SR, Mukherjee S, et al. DOCK 6: combining techniques to model RNA-small molecule complexes. RNA. 2009;15:1219–1230. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chaput L, Mouawad L (2017) Efficient conformational sampling and weak scoring in docking programs? Strategy of the wisdom of crowds. J Cheminform. 10.1186/s13321-017-0227-x [DOI] [PMC free article] [PubMed]
- 52.Gimeno A, Mestres-Truyol J, Ojeda-Montes MJ, et al. Prediction of novel inhibitors of the main protease (M-pro) of SARS-CoV-2 through consensus docking and drug reposition. Int J Mol Sci. 2020 doi: 10.3390/ijms21113793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li J, Fu A, Zhang L (2019) An overview of scoring functions used for protein–ligand interactions in molecular docking. Interdiscip. Sci Comput Life Sci [DOI] [PubMed]
- 54.Pantsar T, Poso A. Binding affinity via docking: fact and fiction. Molecules. 2018;23:1899. doi: 10.3390/molecules23081899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Y, Han L, Liu Z, Wang R. Comparative assessment of scoring functions on an updated benchmark: 2. Evaluation methods and general results. J Chem Inf Model. 2014;54:1717–1736. doi: 10.1021/ci500081m. [DOI] [PubMed] [Google Scholar]
- 56.Ortiz CLD, Completo GC, Nacario RC, Nellas RB. Potential inhibitors of galactofuranosyltransferase 2 (GlfT2): molecular docking, 3D-QSAR, and in silico ADMETox studies. Sci Rep. 2019;9:17096. doi: 10.1038/s41598-019-52764-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ahmad S, Waheed Y, Abro A, et al. Molecular screening of glycyrrhizin-based inhibitors against ACE2 host receptor of SARS-CoV-2. J Mol Model. 2021;27:206. doi: 10.1007/s00894-021-04816-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trujillo-Correa AI, Quintero-Gil DC, Diaz-Castillo F, et al. In vitro and in silico anti-dengue activity of compounds obtained from Psidium guajava through bioprospecting. BMC Complement Altern Med. 2019;19:298. doi: 10.1186/s12906-019-2695-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mysinger MM, Carchia M, Irwin JJ, Shoichet BK. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem. 2012;55:6582–6594. doi: 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McGann M. FRED pose prediction and virtual screening accuracy. J Chem Inf Model. 2011;51:578–596. doi: 10.1021/ci100436p. [DOI] [PubMed] [Google Scholar]
- 61.Empereur-Mot C, Zagury J-F, Montes M. Screening explorer–an interactive tool for the analysis of screening results. J Chem Inf Model. 2016;56:2281–2286. doi: 10.1021/acs.jcim.6b00283. [DOI] [PubMed] [Google Scholar]
- 62.Hawkins PCD, Skillman AG, Warren GL, et al. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model. 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wiggers HJ, Rocha JR, Fernandes WB, et al. Non-peptidic Cruzain inhibitors with trypanocidal activity discovered by virtual screening and in vitro assay. PLoS Negl Trop Dis. 2013 doi: 10.1371/journal.pntd.0002370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Trott O, Olson AJ. Software news and update AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010 doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dallakyan S, Olson AJ (2015) Small-molecule library screening by docking with PyRx. In: Methods Mole Biol pp 243–250 [DOI] [PubMed]
- 66.O’Boyle NM, Banck M, James CA, et al. Open Babel: an open chemical toolbox. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heifets A, Lilien RH. LigAlign: flexible ligand-based active site alignment and analysis. J Mol Graph Model. 2010;29:93–101. doi: 10.1016/j.jmgm.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 68.Kwofie S, Dankwa B, Odame E, et al. In silico screening of isocitrate lyase for novel anti-buruli ulcer natural products originating from Africa. Molecules. 2018;23:1550. doi: 10.3390/molecules23071550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jaundoo R, Bohmann J, Gutierrez G, et al. Using a consensus docking approach to predict adverse drug reactions in combination drug therapies for Gulf War illness. Int J Mol Sci. 2018;19:3355. doi: 10.3390/ijms19113355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sasmal S, El Khoury L, Mobley DL. D3R grand challenge 4: ligand similarity and MM-GBSA-based pose prediction and affinity ranking for BACE-1 inhibitors. J Comput Aided Mol Des. 2020;34:163–177. doi: 10.1007/s10822-019-00249-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ramírez D, Caballero J. Is it reliable to take the molecular docking top scoring position as the best solution without considering available structural data? Molecules. 2018;23:1038. doi: 10.3390/molecules23051038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dalton JAR, Jackson RM. Homology-modelling protein–ligand interactions: allowing for ligand-induced conformational change. J Mol Biol. 2010;399:645–661. doi: 10.1016/j.jmb.2010.04.047. [DOI] [PubMed] [Google Scholar]
- 73.Ren X, Shi Y-S, Zhang Y, et al. Novel consensus docking strategy to improve ligand pose prediction. J Chem Inf Model. 2018;58:1662–1668. doi: 10.1021/acs.jcim.8b00329. [DOI] [PubMed] [Google Scholar]
- 74.Thomas BN, Parrill AL, Baker DL. Self-docking and cross-docking simulations of G protein-coupled receptor-ligand complexes: impact of ligand type and receptor activation state. J Mol Graph Model. 2022;112:108119. doi: 10.1016/j.jmgm.2021.108119. [DOI] [PubMed] [Google Scholar]
- 75.Cross JB, Thompson DC, Rai BK, et al. Comparison of several molecular docking programs: pose prediction and virtual screening accuracy. J Chem Inf Model. 2009;49:1455–1474. doi: 10.1021/ci900056c. [DOI] [PubMed] [Google Scholar]
- 76.Abraham MJ, Murtola T, Schulz R, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 77.Kwofie SK, Enninful KS, Yussif JA, et al. Molecular informatics studies of the iron-dependent regulator (ideR) reveal potential novel anti-Mycobacterium ulcerans natural product-derived compounds. Molecules. 2019;24:2299. doi: 10.3390/molecules24122299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kwofie S, Dankwa B, Enninful K, et al. Molecular docking and dynamics simulation studies predict Munc18b as a target of mycolactone: a plausible mechanism for granule exocytosis impairment in buruli ulcer pathogenesis. Toxins (Basel) 2019;11:181. doi: 10.3390/toxins11030181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schüttelkopf AW, Van Aalten DMFF. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr Sect D Biol Crystallogr. 2004 doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 80.Turner P (2005) XMGRACE, Version 5.1. 19. Cent Coast Land-Margin Res Oregon Grad Inst Sci Technol Beavert
- 81.Kumari R, Kumar R, Lynn A. g_mmpbsa —A GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model. 2014;54:1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 82.Baker NA, Sept D, Joseph S, et al. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci. 2002 doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Quimque MTJ, Notarte KIR, Fernandez RAT, et al. Virtual screening-driven drug discovery of SARS-CoV2 enzyme inhibitors targeting viral attachment, replication, post-translational modification and host immunity evasion infection mechanisms. J Biomol Struct Dyn. 2021;39:4316–4333. doi: 10.1080/07391102.2020.1776639/SUPPL_FILE/TBSD_A_1776639_SM5101.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lagunin A, Stepanchikova A, Filimonov D, Poroikov V. PASS: prediction of activity spectra for biologically active substances. Bioinformatics. 2000;16:747–748. doi: 10.1093/bioinformatics/16.8.747. [DOI] [PubMed] [Google Scholar]
- 86.Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pires DEV, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58:4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46:W257–W263. doi: 10.1093/nar/gky318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mahmood O, Mansimov E, Bonneau R, Cho K. Masked graph modeling for molecule generation. Nat Commun. 2021;12:3156. doi: 10.1038/s41467-021-23415-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Maia EHB, Assis LC, de Oliveira TA, et al. Structure-based virtual screening: from classical to artificial intelligence. Front Chem. 2020;8:343. doi: 10.3389/fchem.2020.00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Meng X-Y, Zhang H-X, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7:146. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vardhan S, Sahoo SK. Virtual screening by targeting proteolytic sites of furin and TMPRSS2 to propose potential compounds obstructing the entry of SARS-CoV-2 virus into human host cells. J Tradit Complement Med. 2022;12:6–15. doi: 10.1016/j.jtcme.2021.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zothantluanga JH, Gogoi N, Shakya A et al (2021) Computational guided identification of potential leads from Acacia pennata (L.) Willd. as inhibitors for cellular entry and viral replication of SARS-CoV-2. Futur J Pharm Sci 7:201. 10.1186/s43094-021-00348-7 [DOI] [PMC free article] [PubMed]
- 94.Mamidala E, Davella R, Praveen Kumar M, et al. In silico prediction of mozenavir as a potential drug for SARS-CoV-2 infection via binding multiple drug targets. Saudi J Biol Sci. 2022;29:840–847. doi: 10.1016/j.sjbs.2021.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dahms SO, Hardes K, Becker GL, et al. X-ray structures of human furin in complex with competitive inhibitors. ACS Chem Biol. 2014;9:1113–1118. doi: 10.1021/cb500087x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sheybani Z, Heydari Dokoohaki M, Negahdaripour M, et al. The interactions of folate with the enzyme furin: a computational study. RSC Adv. 2021;11:23815–23824. doi: 10.1039/D1RA03299B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Arnittali M, Rissanou AN, Harmandaris V. Structure of biomolecules through molecular dynamics simulations. Procedia Comput Sci. 2019;156:69–78. doi: 10.1016/j.procs.2019.08.181. [DOI] [Google Scholar]
- 98.Lobanov MY, Bogatyreva NS, Galzitskaya OV. Radius of gyration as an indicator of protein structure compactness. Mol Biol. 2008;42:623–628. doi: 10.1134/S0026893308040195. [DOI] [PubMed] [Google Scholar]
- 99.Wang E, Sun H, Wang J, et al. End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem Rev. 2019;119:9478–9508. doi: 10.1021/acs.chemrev.9b00055. [DOI] [PubMed] [Google Scholar]
- 100.Yao C, Xi C, Hu K, et al. Inhibition of enterovirus 71 replication and viral 3C protease by quercetin. Virol J. 2018 doi: 10.1186/s12985-018-1023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zidane A, Tits M, Angenot L, et al. Phytochemical analysis of Tetraclinis articula in relation to its vasorelaxant property. J Mater Environ Sci. 2014;5:1368–1375. [Google Scholar]
- 102.Wong RSY. Inflammation in COVID-19: from pathogenesis to treatment. Int J Clin Exp Pathol. 2021;14:831. [PMC free article] [PubMed] [Google Scholar]
- 103.Asiedu SO, Kwofie SK, Broni E, Wilson MD. Computational identification of potential anti-inflammatory natural compounds targeting the p38 mitogen-activated protein kinase (MAPK): implications for COVID-19-induced cytokine storm. Biomolecules. 2021;11:653. doi: 10.3390/biom11050653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Berretta AA, Silveira MAD, Cóndor Capcha JM, De Jong D. Propolis and its potential against SARS-CoV-2 infection mechanisms and COVID-19 disease. Biomed Pharmacother. 2020;131:110622. doi: 10.1016/j.biopha.2020.110622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Silveira MAD, De Jong D, Berretta AA, et al. Efficacy of Brazilian green propolis (EPP-AF®) as an adjunct treatment for hospitalized COVID-19 patients: a randomized, controlled clinical trial. Biomed Pharmacother. 2021;138:111526. doi: 10.1016/j.biopha.2021.111526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zulhendri F, Chandrasekaran K, Kowacz M, et al. Antiviral, antibacterial, antifungal, and antiparasitic properties of propolis: a review. Foods. 2021;10:1360. doi: 10.3390/foods10061360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Almuhayawi MS. Propolis as a novel antibacterial agent. Saudi J Biol Sci. 2020;27:3079–3086. doi: 10.1016/j.sjbs.2020.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Przybyłek I, Karpiński TM. Antibacterial properties of propolis. Molecules. 2019;24:2047. doi: 10.3390/molecules24112047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu JX, Zhang Y Hu QP et al (2017) Anti-inflammatory effects of rosmarinic acid-4-O-β-D-glucoside in reducing acute lung injury in mice infected with influenza virus. Antiviral Res 144:34–43. 10.1016/J.ANTIVIRAL.2017.04.010 [DOI] [PubMed]
- 110.NCT04733651 (2021) Study to investigate the treatment effect of Isoquercetin in patients with COVID-19. https://clinicaltrials.gov/show/NCT04733651
- 111.Ye H, Zou T, Jiang X et al (2021) Cinnamic acid reduces inflammation and apoptosis in necrotizing Enterocolitis. Curr Top Nutraceutical Res 20:70–75. 10.37290/ctnr2641-452X.20:70-75
- 112.Asmi KS, Lakshmi T, Balusamy SR, Parameswari R (2017) Therapeutic aspects of taxifolin - an update. J Adv Pharm Educ Res
- 113.Kim J, Lee Y, An H, et al. Anti-inflammatory activities of taxifolin from Opuntia humifusa in lipopolysaccharide stimulated RAW 264.7 murine macrophages. J Appl Biol Chem. 2015;58:241–246. doi: 10.3839/jabc.2015.038. [DOI] [Google Scholar]
- 114.Alshatwi AA, Ramesh E, Periasamy VS, Subash-Babu P. The apoptotic effect of hesperetin on human cervical cancer cells is mediated through cell cycle arrest, death receptor, and mitochondrial pathways. Fundam Clin Pharmacol. 2013;27:581–592. doi: 10.1111/j.1472-8206.2012.01061.x. [DOI] [PubMed] [Google Scholar]
- 115.Jo SH, Kim ME, Cho JH, et al. Hesperetin inhibits neuroinflammation on microglia by suppressing inflammatory cytokines and MAPK pathways. Arch Pharm Res. 2019;42:695–703. doi: 10.1007/s12272-019-01174-5. [DOI] [PubMed] [Google Scholar]
- 116.Umetsu T, Inoue J, Kogure T, et al. Inhibitory effect of silibinin on hepatitis B virus entry. Biochem Biophys Reports. 2018 doi: 10.1016/j.bbrep.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Blaising J, Lévy PL, Gondeau C, et al. Silibinin inhibits hepatitis C virus entry into hepatocytes by hindering clathrin-dependent trafficking. Cell Microbiol n/a-n/a. 2013 doi: 10.1111/cmi.12155. [DOI] [PubMed] [Google Scholar]
- 118.Carpenter TS, Kirshner DA, Lau EY, et al. A method to predict blood-brain barrier permeability of drug-like compounds using molecular dynamics simulations. Biophys J. 2014;107:630–641. doi: 10.1016/j.bpj.2014.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pesu M, Watford WT, Wei L, et al. T-cell-expressed proprotein convertase furin is essential for maintenance of peripheral immune tolerance. Nature. 2008;455:246–250. doi: 10.1038/nature07210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.