

Abstract
Background
Fibroblast growth factor receptor (FGFR) signaling influenced tumour occurrence and development. Overexpression of FGFR had been observed in many types of cancers, including colon cancer. FGFR inhibitor is considered to be effective in treating colon cancer patients.
Methods
First, the kinase inhibition rate was determined. MTT, western blotting, colony formation, EdU and comet assays were performed to evaluate the anti-tumour effects of F1-7 in vitro. RNA-seq and bioinformatics analysis were used for further verification. Additionally, a xenograft model was generated to investigate the anti-tumour effect of F1-7.
Results
F1-7 can inhibit the proliferation of colon cancer cells in vitro. It could significantly inhibit FGFR phosphorylation and its downstream signaling pathway. Whole-genome RNA-seq analysis found that the changed genes were not only functionally focused on MAPK signaling pathway but also related to cell apoptosis and ferroptosis. Experimental evidence demonstrated that F1-7 can directly increase the level of cellular DNA damage. The occurrence of DNA damage led to cell cycle arrest and inhibition of cell metastasis and cell apoptosis. Mouse model experiments also confirmed that F1-7 could inhibit tumour growth by inhibiting the FGFR pathway.
Conclusions
F1-7 exhibits anti-tumour activity by inhibiting the FGFR pathway. It could be a novel therapeutic agent for targeting colon cancer cells.
Subject terms: Cell proliferation, Apoptosis, Cancer
Background
Colon cancer is a prevalent malignancy in people with irregular eating habits and is harmful to the digestive tract. In 2012, more than 1.4 million patients were diagnosed with colon cancer, and more than 700,000 patients died as a result. Some people have predicted that by 2030, the overall situation will worsen: its incidence is expected to increase by 60%, with 2.2 million cases of colon cancer per year worldwide [1]. Today, surgical resection or chemotherapy drugs are used to treat colon cancer. However, surgical resection may cause a series of complications and is only suitable for early-stage tumours [2]; chemotherapy drugs are unique in treating cancers that have spread and prevented tumour recurrence, but their usage is limited due to their toxicity to normal cells. Therefore, it is vital to develop a new targeted drug to specifically target colon cancer cells [3].
The fibroblast growth factor/fibroblast growth factor receptor (FGF/FGFR) signaling pathway is widely involved in cell metabolism regulation, including proliferation, differentiation, migration and survival [4]. Mutations in the FGFR genome have been reported in a variety of tumour types, especially in colon cancer. Some colon cancer patients harbour FGFR genetic alterations, such as copy number gain, mutation and mRNA overexpression. These mutations usually lead to tumour formation and resistance to some tyrosine kinase inhibitors [5]. As a result, colon cancers characterised by FGF/FGFR mutations are often challenging to treat and have a poor prognosis. FGFR is a potential new target for treating colon cancers [6].
Previously, researchers have tried tyrosine kinase inhibitors (TKIs), such as ponatinib [7], dovtinib [8] and lucitanib [9] to treat FGFR-deregulated tumours. Although these inhibitors have good kinase inhibitory activity against FGFR, the toxicity profiles of those inhibitors relative to their on-target activity against other kinases have restricted their further development for the treatment of cancer [10]. In the past few years, several selective FGFR inhibitors have been reported, and some of them have been used in clinical studies to evaluate their therapeutic effect on patients. Most of them, such as AZD4547 [11], NVP-BGJ398 [12], LY2874455 [13], competitively bind the ATP-binding pockets of FGFR in a noncovalent form, thereby preventing receptor phosphorylation and blocking signal transmission. However, these inhibitors are only effective for FGFR1-3 and do not show much selectivity for FGFR4. In addition, these drugs are mainly used to treat cholangiocarcinoma and breast cancer but are rarely used for colon cancer. Consistent with this, it is urgent to develop more selective FGFR inhibitors.
In this article, we report a novel inhibitor of FGFR, F1-7, which is subtly selective for FGFR. It inhibits the FGFR pathway in colon cancer cell lines in a dose-dependent manner, thereby causing DNA damage in cells, inhibiting cell growth and metastasis and eventually leading to cell apoptosis.
Methods
Antibodies and reagents
F1-7 was synthesised in the laboratory, and AZD4547 was purchased from Sigma, which was used as a positive control. The cell culture DMEM was purchased from Gibco. The antibody of GAPDH (5174), p-FGFR (3476), FGFR1 (9740), FGFR4 (8562S), p-AKT (4060), AKT (9272), p-MAPK (4370), MAPK (4695), cleaved-PARP (5625), Bcl-2 (15071), BAX (5023), γ-H2AX (80312), CyclinB1 (4135) and α-Tubulin (2144) were purchased from Cell Signaling Technology (Danvers, MA, USA). The antibody of c-Myc (AF6489) and CDK1 (AF0111) was purchased from Beyotime Biotechnology (Shanghai, China). The antibody of MDM2 was purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Anti-rabbit IgG (H + L), Biotinylated Antibody (14708) and Anti-mouse IgG (H + L), Biotinylated Antibody (14709) were purchased from Cell Signaling Technology (Danvers, MA, USA). FITC Annexin V Apoptosis Detection Kit I and Propidium Iodide (PI) were purchased from BD Pharmingen (Franklin Lakes, NJ). BeyoClick™ EdU Cell Proliferation Kit with Alexa Fluor 488 was purchased from Beyotime Biotechnology (Shanghai, China).
Cell lines and culture
The human colon cancer cell lines HCT-116, RKO and SW620 were purchased from the Cell Resources Center of the Shanghai Institutes for Biological Sciences (Chinese Academy of Sciences, Shanghai, China). All cell lines were identified by STR profiling and tested for mycoplasma contamination. The three cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS. All of the cells were incubated at 37 °C in an atmosphere of 5% CO2.
Methyl thiazolyl tetrazolium (MTT) assay
The MTT experiment was performed as described in the previous report [14]. Colon cancer cells were seeded in 96-well plates at 3000–5000 cells per well. After cells adhered to the wall, the tested compound (F1-7 and AZD4547) was dissolved with DMSO and added to the well. After 48 h of reaction, MTT was added to wells for 4 h for reaction crystallisation. After that, DMSO was used to dissolve crystals, the absorbance of the solution at a wavelength of 490 nm was measured with a microplate reader. The half-maximal inhibitory concentrations (IC50) value was calculated by GraphPad 7.0.
Western blot
The western blot (WB) assay experiment was performed as described in the previous report [15]. Cells were seeded in a six-well plate and treated with different concentrations of the compound for 24 h. Then the cells were lysed with a low-temperature lytic solution. Tissue and cellular proteins were extracted by protein lysate buffer (Total Protein Extraction Kit). Proteins were separated with a 10% sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE). The protein was then transferred to a polyvinylidene fluoride (PVDF) membrane and blocked with 5% skim milk for 1.5 h. The blots were incubated with specific primary antibodies. The membranes were incubated with the antibodies, visualisation of bands was recorded by enhanced chemiluminescence.
mRNA library construction and sequencing
Total RNA was isolated and purified using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s procedure to construct mRNA library. The average insert size for the final cDNA library was 300 ± 50 bp. We performed the 2×150 bp paired-end sequencing (PE150) on an Illumina Novaseq™ 6000 (LC-Bio Technology CO., Ltd., Hangzhou, China) following the vendor’s recommended protocol.
Colony formation assay
Colon cancer cells were seeded in six-well plates at 10000 cells per well. F1-7 and AZD4547 were added to cells with different concentrations for 24 h. Then culture medium was replaced with a fresh medium for 7 days. The colonies were washed with PBS, fixed at room temperature with 4% paraformaldehyde, washed and dyed with 0.1% crystal violet for 15 min.
Flow cytometry
Cells were treated with different concentrations of F1-7 for about 48 h. Flow cytometry for detection of apoptotic cells using the apoptosis detection kit I. All samples were analysed in flow cytometry (BD Biosciences) and the data were evaluated using FlowJo software.
Comet assay
The comet assay method has been described in a previously published article [16]. Cells were cultured with a medium and treated with different concentrations of F1-7 for 24 h. 1% normal melting point agarose was spread on the rough surface of a slide, curing it for 8 h. Next, single-cell suspending with 0.7% low melting point agarose spread on the gel and was covered with a coverslip, after curing it at 4 °C for 30 min, removed the coverslip. Slides were placed in the pre-cool lysate for 8 h then moved to a horizontal electrophoresis tank containing electrophoresis liquid and equilibrated for 40 min at room temperature. Electrophoresis was carried out for 15 min at 20 V, following that the slides were washed with neutralisation buffer, water and alcohol for 10 min and then stained with PI and viewed under a fluorescence microscope.
5-Ethynyl-20-deoxyuridine (EdU) incorporation assay
Colon cancer cells were seeded at a density of 1 × 105 cells per well in six-well plates and cultured overnight. The newly synthesised DNA of the cells was assessed by the EdU incorporation assays using a Cell-Light EdU DNA Cell Proliferation Kit (Beyotime Biotechnology, China), following the manufacturer’s instructions. The EdU incorporation rate was expressed as the ratio of EdU-positive cells (red cells) to total Hoechst33342 positive cells (blue cells).
Cell adhesion assay
Cell adhesion assay was performed as described previously. Briefly, cells were pretreated with different concentrations of F1-7, and then trypsinised. Cells (5 × 105 cells/well) were added into 96-well plates which were precoated with fibronectin (Sigma). Plates were incubated for 1 h at 37 °C, then nonadherent cells were removed by a gentle washing three times with PBS, and the remaining cells were stained with 0.1% crystal violet for 15 min. After washing, the precipitates were dissolved with the addition of 30% acetic acid, and the absorption was obtained at 590 nm. The percentage of inhibition was expressed using control wells as 100%.
Immunofluorescence assays
The comet assay method has been described in a previously published article [17]. Cells were seeded in six-well plates with a density of 1 × 106 cells/well, treated with different concentrations of F1-7 for 12 h, then washed three times with PBS and fixed in 4% paraformaldehyde for 20 min, washed three times with PBS, permeabilised with 0.5% Triton-100 for 20 min and blocked in 5%. BSA for 90 min. Primary antibodies (α-Tubulin, diluted at 1:25) were incubated with cells overnight at 4 °C, washed three times with PBS and incubated with fluorescent secondary antibodies at room temperature for 60 min. Then the cells were co-stained with DAPI to visualise the nuclei. All fluorescent samples were captured using a confocal fluorescence microscope.
Transwell assay
Transwell chambers (Corning, Corning, NY, USA) equipped with 8 μm pore insets were used for the invasion assay. The insets were coated with 70 μL of 1:8-diluted Matrigel (BD Biosciences), and 1 × 105 cells were plated in the serum-free medium described above for an incubation period of 48 h. Quantities of 600 μL of culture medium containing 20% FBS (Invitrogen) were added to the lower chamber. No-invaded cells were removed, and the cells that were attached to the bottom of the membrane were fixed with 4% paraformaldehyde, stained with 5% crystal violet (Sigma–Aldrich), crystals dissolved with 150 μL acetic acid and analysed in a Microplate Reader at 560 nm.
Animal model
All animal care and experiments were performed according to the guidelines and approval of the Wenzhou Medical University Animal Policy and Welfare Committee. Female BALB/c (nu/nu) mice (4–6 weeks) were purchased from the Vital River Experimental Animal Center (Beijing, China) and maintained at the animal experimental centre at Wenzhou Medical University. All the mice were housed under 12 h light–dark cycles at 25 °C and free of water and diet. HCT-116 cancer cells were planted (5 × 106 cells in 100 μL of PBS) into the back of mice. Once tumour volumes reached ~100 mm3, mice were divided into three different groups (no differences in mean body weights or tumour volumes between the groups, and the investigator was blinded to the group allocation). The treatment groups (six mice per group) were treated with F1-7 20 mg/kg or 40 mg/kg by intraperitoneal injection every day. The tumour volumes were measured in length (L), width (W) and calculating volume (V = 0.5 × L × W2) before every injection. After 2 weeks, all mice were executed. The tumours were removed and prepared for western blot analysis and IHC. Tumour weight was measured.
Immunohistochemistry (IHC)
Tumour tissue sections (4 μm) were deparaffinised, rehydrated and incubated with primarily Ki-67 and γH2AX antibodies. HRP-conjugated secondary antibodies were used for detection. Images were obtained with Leica microscope.
Apoptosis assay (TUNEL staining)
The cell apoptosis in vivo was assessed by the ONE STEP TUNEL Apoptosis Assay Kit (Beyotime, China), following the manufacturer’s instructions. Cell nuclei was stained with DAPI, and fluorescence was evaluated by fluorescence microscopy.
Haematoxylin and eosin staining
The hearts, lungs, kidneys and livers of animals were fixed in 4% paraformaldehyde and embedded in paraffin. The paraffin tumour tissue sections (5 μm) were deparaffinised and rehydrated and then stained with eosin and haematoxylin. The images were captured using a light microscope.
Bioinformatic analysis and data visualisation
All statistical analyses were performed with R software (version 4.0.4). The genes were ordered according to log2 (Foldchange), and the gene list was subjected to gseGO function from clusterProfiler [18] package for KEGG and GO analysis. Data visualisation was performed based on ggplot2 and pheatmap packages.
Statistical analysis
All experiments were assayed as three independent experiments. Unpaired Student’s t-tests were used to compare the means of two groups. One-way analysis of variance (ANOVA) was used for comparison among the different groups by GraphPad Prism 7.0 (GraphPad Software, CA, USA). P-value <0.05 was considered statistically significant. For every figure, statistical tests are justified as appropriate. We did not exclude samples or animals. All data meet the assumptions of the tests. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those generally used in the field.
Results
F1-7 is a potent FGFR inhibitor and exhibits anti-tumour activity in colon cancer
FGFR1-4 are receptor tyrosine kinases (RTKS), which consist of extracellular regions, transmembrane domains and intracellular tyrosine kinase regions [19]. In a previous study, we designed and synthesised F1-7 using computer-aided drug design (CADD) and structure-based design strategies (Fig. 1a and additional file 1). F1-7 shows potent inhibition of the kinase of recombinant FGFR1, 2, 3 and 4 in a dose-dependent manner, with half-maximal inhibitory concentration (IC50) values of 10, 21, 50 and 4.4 nmol/L, respectively (Fig. 1b). Since FGFRs are generally overexpressed in patients with colon cancer, we carried out an MTT assay to test the anti-tumour activity of F1-7 in colon cancer. The data demonstrated that F1-7 inhibited colon cancer cell (HCT-116, RKO and SW620) viability in a concentration-dependent manner, with an IC50 of 1–2 µM. Compared to the positive control AZD4547, a novel and selective inhibitor of the FGFR1, 2 and 3 tyrosine kinases, F1-7 showed greater cytotoxicity to colon cancer cells (Fig. 1c).
Fig. 1. F1-7 inhibiting phosphorylation of FGFR in colon cancer.

a The chemical structure of F1-7. b Inhibition of FGFR kinase activity in biochemical assays. c IC50 values of F1-7 in colon cancer cells. Cell lines were seeded in 96-well plate and treated with various concentrations of F1-7. Proliferation was measured after 48 h of treatment by the MTT assay. Data are expressed as the mean ± SD of three independent experiments. d Cells were incubated with F1-7 at different concentrations for 24 h, cell lysates were collected for western blot analysis to determine protein expression of FGFR signaling pathway, GAPDH was used as loading control.
Next, we examined the effect of F1-7 on FGFR phosphorylation and signaling in colon cancer cells by western blotting (Fig. 1d). Treatment of the HCT-116, RKO and SW620 cell lines with increasing doses of F1-7 for 12 h resulted in dose-dependent inhibition of FGFR phosphorylation, while the total amounts of their target proteins remained unchanged. However, AZD4547 at the same concentration did not significantly change the phosphorylation of FGFR (Supplementary Fig. 1A). The same decreasing trend was also observed in protein kinase B (AKT) and mitogen-activated protein kinase (MAPK) phosphorylation levels. Both of these proteins are downstream of FGFR signaling and have a vital effect on the development of cancer [20].
Together, these data indicated that the possible mechanism by which F1-7 inhibits colon cancer cell proliferation is potentially related to the inhibition of FGFR and/or downstream pathway phosphorylation by F1-7.
F1-7 affects the physiological processes of cancer cells at the transcriptomic level
In the present study, we treated HCT-116 with F1-7 for 12 h and then performed RNA-seq. The results showed that 187 mRNAs were significantly upregulated and 130 mRNAs were significantly downregulated (≥2.0-fold) in the dosing group compared to the vehicle control group (Fig. 2a). The heatmap showed the top 50 most significantly different genes (Fig. 2b). Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) is a database predicting functional associations between proteins [21]; we used the functional protein association network to show how these genes interact (Supplementary Fig. 1B). The specific functions of these genes require further research.
Fig. 2. Colon cancer cells transcriptome level was influenced by F1-7.

a Log22-fold differentially expressed genes (p < 0.05) in HCT-116 cells treated with F1-7 2 µM for 12 h. b Heatmap of top 50 differentially expressed genes. c KEGG enrichment analyses on the differentially genes.
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed on the genes in the last transcriptomic analysis to predict metabolism and signal transduction pathways. In the KEGG report (Fig. 2c), the top 20 enriched pathways of the genes were presented. Coincidentally but not surprisingly, the most significantly enriched pathway was MAPK, which is located downstream of FGFR. This finding further validated the pharmacological effect of F1-7. These genes were also enriched in pathways associated with apoptosis. Interestingly, ferroptosis-related gene expression was also altered, as shown in Fig. 2c and Supplementary Table 1. These results suggest that F1-7 can induce apoptosis and ferroptosis by inhibiting FGFR.
F1-7 induces apoptosis and ferroptosis in colon cancer cells
To verify the previous hypothesis, we performed a series of fundamental experiments. Compared to AZD4547 (positive control) at 4 µM, F1-7 more efficiently reduced the colony formation of three colon cancer cell lines in a dose-dependent manner (Fig. 3a). Next, we treated the three cell lines with the positive control (4 µM) and F1-7 at different concentrations (0, 1, 2 and 4 µM) for 48 h and then analysed apoptosis by flow cytometry (FCM). Treatment with F1-7 increased the proportion of apoptotic cells in a dose-dependent manner (Fig. 3b, c). Since the protein expression levels of cleaved-PARP, Bcl-2 and Bax are associated with apoptosis activity in tumour cells, a western blot assay was performed to assess the effects of F1-7 on these proteins. As shown in Fig. 3d, the expression of cleaved-PARP [22] and Bax [23] (apoptotic markers) clearly increased, while the expression of Bcl-2 [24] (a proto-oncogene that protects against Bax-induced apoptosis) decreased.
Fig. 3. F1-7 induced colon cancer cells apoptosis and ferroptosis.
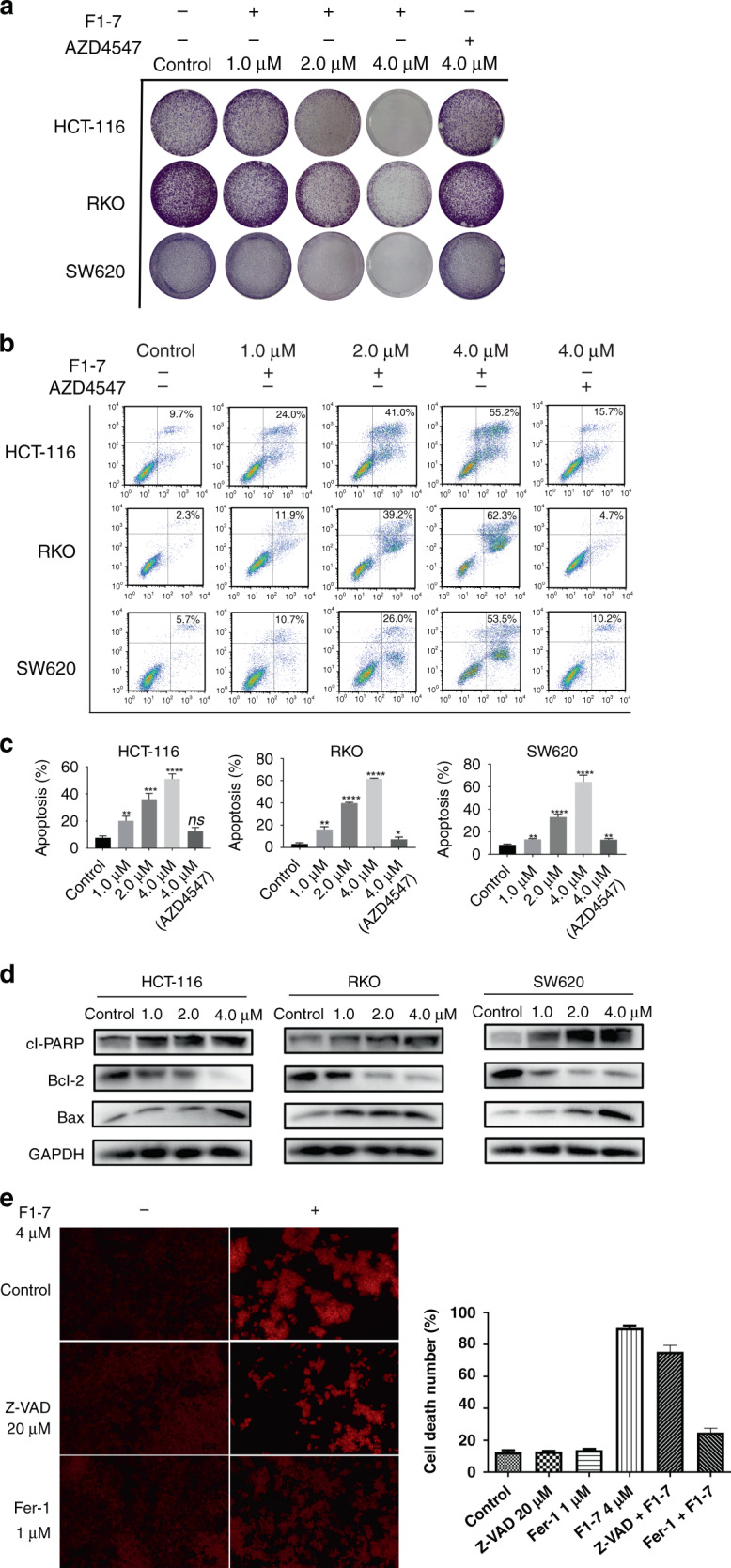
a Colony forming assay of colon cancer cell lines. Cells were incubated with different concentrations of F1-7 for 24 h. On day 7, colonies were fixed and photographed. b, c Colon cancer cells were treated with F1-7 and AZD4547 for 48 h. Cells were stained with Annexin V and propidium iodide (PI), and then analysed by flow cytometry. Samples were measured in triplicate and experiments were independently repeated three times d Cells were incubated with F1-7 at different concentrations as indicated for 24 h, the cell lysates were prepared for western blot analysis to determine protein expression of cl-PARP, Bcl-2, Bax and GAPDH. e PI staining of HCT-116 cells with and without f1-7 with inhibitors of apoptosis and ferroptosis. The positive of cells were calculated. Data are expressed as mean ± SD (n = 3).
Ferroptosis is a new form of cell death which has different properties and functions in physical conditions or various diseases including cancers. It points to a new future for developing new generation small molecule compounds. RNA-seq indicated F1-7 may influence ferroptosis by FGFR pathway. To further verify whether F1-7 can induce cell ferroptosis, we added inhibitors of ferroptosis (ferrostatin-1) and apoptosis (Z-VAD) to F1-7-treated cells and then assessed cell death (PI staining, which only stained dead cells). The results (Fig. 3e) showed that cell death was reduced after the addition of ferroptosis and apoptosis inhibitors, indicating that cell death occurred via ferroptosis and apoptosis. However, it is not clear how F1-7 affects ferroptosis through FGFR, further research is needed to determine.
Together, these results confirmed that F1-7 significantly promotes apoptosis and ferroptosis in colon cancer cells.
F1-7 induces DNA damage-mediated cell death in colon cancer cells
To further investigate the molecular mechanism of F1-7 in colon cancer, we used an in silico GO-BP analysis to predict and score the physiological changes that occurred after the addition of the drug. As a result, cells mainly focused on apoptosis and DNA damage (Fig. 4a). We assumed that F1-7 induces DNA damage-mediated apoptosis in colon cancer cells. Consequently, we treated HCT-116 and RKO cells with different concentrations for 24 h and then checked the expression of γ-H2AX (which is a DNA damage biomarker [25]) by immunoblotting. The results showed that with increasing F1-7 concentrations, the expression of γ-H2AX was significantly increased (Fig. 4b). Next, a comet assay was used to quantify DNA damage in vitro. The results showed that the comet tail of F1-7-treated cells was much longer and had higher DNA intensity with increasing doses (Fig. 4c), and quantitative analysis for the comet assay exhibited significantly increased comet tail formation after F1-7 treatment (Fig. 4d), suggesting the substantial accumulation of fragmented DNA due to F1-7. Finally, we used an EdU experiment to evaluate cell proliferation ability by measuring the DNA synthesis function of cells. As shown in Fig. 4e and Supplementary Fig. 2A, with increasing drug concentrations, the number of EdU-positive cells decreased. Together, these data suggested that inhibition of FGFR activation induces the death of colon cancer cells due to DNA damage.
Fig. 4. F1-7 induced colon cancer cells DNA damage.

a GO-BP enrichment analyses on the differentially genes. b Cells were incubated with F1-7 at different concentrations as indicated for 24 h, the cell lysates were prepared for western blot analysis to determine protein expression of γH2AX and GAPDH. c, d The results of comet assay after treatment with F1-7 for 24 h, showing the presence of comet-like tail DNA (400 ×) and the density of DNA tail were calculated. e The results of EdU assay after treatment with F1-7 for 24 h, EdU-positive cell (red) immunofluorescence assay was performed, DAPI-stained nuclei blue. The positive of cells were calculated. Data are expressed as mean ± SD (n = 3).
Effect of F1-7 on cell cycle distribution
The above research proved that F1-7 can induce cell DNA damage and block synthesis, and the expression of some factors that drive the normal cell cycle might be affected. We first used FCM to examine its effect on cell cycle distribution. Treatment with different concentrations of F1-7 increased the number of cells in the G2/M phase (Fig. 5a, b). Next, the expression of G2/M phase cell-cycle-associated proteins was detected by western blotting. When cells were incubated with F1-7 for 24 h, the expression of cyclin B1, MDM2 and CDK1 decreased with increasing treatment concentration (Fig. 5c). Some researchers reported that c-Myc, which was regulated by MAPK, could regulate G2/M cell cycle progression [26]. We thought maybe it is c-Myc that caused cell cycle arrest. To clarify the above conclusion, we conducted an overexpression of c-Myc and the results showed that overexpressed c-Myc did have an antagonistic effect on F1-7 induced G2/M arrest (Supplementary Fig. 2B, C). Therefore, we affirmed that F1-7 regulates the expression of G2/M cell-cycle-related proteins and induces G2/M cell cycle arrest by down-regulating the expression of c-Myc.
Fig. 5. Effects of F1-7 on colon cancer cell cycle.

a, b Effects of 24 h incubation with different concentrations of F1-7 on cell cycle. With the increase of F1-7 concentration, cells were arrested in the G2/M phase. Data are expressed as mean ± SD (n = 3). c Cells were incubated with F1-7 at different concentrations as indicated for 24 h, the cell lysates were prepared for western blot analysis to determine protein expression of CyclinB1, MDM2, CDK1 and GAPDH.
F1-7 inhibits colon cancer cell invasion
FGFR-dependent signaling pathways involve cell invasion, migration and proliferation [27]. Several experiments were performed to assess the effect of F1-7 on colon cancer cell migration and invasion. α-Tubulin, as a part of microtubules, serves as an essential component of the cell cytoskeleton. It plays a vital role in maintaining cell shape, cell division and invasion [28]. We stained colon cancer cells with α-tubulin antibodies to observe the morphological changes after treating them with F1-7. The results indicated that after F1-7 treatment, the size and state of the cells changed after being stimulated by the drug, the area of the cells decreased and the cells gradually aggregated rather than spread out (Fig. 6a, Supplementary Fig. 2B). The adhesion of metastatic cancer cells to the target organ vascular endothelium is critical in transendothelial migration [29]. In this study, an adhesion assay was performed to test whether F1-7 could reduce cell adhesion. As shown in Fig. 6b, compared to the control, F1-7 significantly reduced the adhesion ability of colon cancer cell lines. Furthermore, Transwell invasion assays were used to explore the effect of F1-7 on the invasion ability of colon cancer cells. Compared to the control, F1-7 at different concentrations (0, 1, 2 and 4 µM) significantly inhibited colon cancer cell invasion (Fig. 6c, d).
Fig. 6. F1-7 showed promising efficacy to suppress colon cancer cells proliferation.

a RKO were incubated with the F1-7 at different concentrations for 24 h. The expression of α-Tubulin (green) in RKO cells was determined by immunofluorescence assay. DAPI-stained nuclei blue. b Cell adhesion assay was performed to evaluate the migration impact of F1-7 on colon cancer cells. The results were representative of three replicate experiments and data are expressed as mean ± SD. c, d Transwell assay was performed to evaluate the migration impact of F1-7 on HCT-116 and RKO. The violet was dissolved by 30% acetic acid and detected absorbance at 590 nm, the percentage of inhibition was expressed using control wells as 100%. Data are expressed as mean ± SD (n = 3).
F1-7 inhibited colon tumour growth in a xenograft model
It was confirmed that F1-7 has an anti-tumour effect in vitro. Then, we established xenograft models to identify the activity of F1-7 in vivo. Eighteen BALB/c (nu/nu) mice (4–6 weeks) were maintained at the animal experimental centre until they reached 18 g in weight. HCT-116 cancer cells were plated (5 × 106 cells) into the backs of mice. Once tumour volumes reached ~100 mm3, the mice were intraperitoneally injected with F1-7 (20 mg/kg and 40 mg/kg) once daily for two weeks. Over time, the volume of tumours treated with F1-7 was markedly reduced compared to that of tumours treated with vehicle control (Fig. 7a, b). Additionally, tumour tissues were weighed 2 weeks after intraperitoneal injection, and we observed that the weight of tumours decreased after treatment with F1-7 (Fig. 7c). Next, tumour phosphorylation of FGFR, MAPK and some apoptosis markers was measured by western blotting. As shown in Fig. 7d, F1-7 exhibited a dose-dependent inhibition of FGFR and MAPK phosphorylation in HCT-116 tumours. Dose-dependent changes in Bcl-2 and Bax expression were also observed. These phenomena indicated that FGFR phosphorylation and downstream signaling were inhibited in xenograft models, thus inducing cancer cell apoptosis.
Fig. 7. The anti-tumour activity of F1-7 in vivo.

a Tumour volume was measured every day. b The graph of tumours in different groups. c The weight of the tumours was measured. d The tumour tissues were extracted in lysis buffer, and western blot analysis was performed. e Representative immunohistochemical staining images of cell proliferation marker (Ki-67) and DNA damage marker (γH2AX) in tumour tissues. f TUNEL staining results of the tumour tissues.
To investigate DNA damage in vivo after F1-7 treatment, we took advantage of immunohistochemical analysis of γ-H2AX and Ki-67 (a well-known marker for the evaluation of cell proliferation); the results showed that tumour cells suffered serious DNA damage with drug treatment (Fig. 7e). TUNEL staining showed increased cell death in vivo with F1-7 treatment (Fig. 7f), and hematoxylin and eosin (H&E) staining (Supplementary Fig. 3) indicated cell deformation and shrinkage. Together, these data showed that F1-7 is a promising drug that exhibits potent anti-tumour activity by inhibiting the FGFR signaling pathway.
Discussion
FGFRs play an irreplaceable role in the occurrence and progression of tumours, particularly colon cancer. The current priority is to develop a novel and highly effective FGFR inhibitor for the treatment of colon cancer. Some FGFR inhibitors are being investigated. PRN1371 showed strong FGFR1-3 inhibitory activity in preclinical trials but has limited activity against common resistant gatekeeper mutants (V561M) of FGFR1 [30]. FINN-2 and FINN-3 irreversibly inhibit FGFR and have also been confirmed to be effective against FGFR1 and FGFR2 mutations but have no similar effects on clinical testing [31]. Other inhibitors under development, such as BLU9931 and BLU554, inhibit FGFR4 by targeting Cys522 in the hinge region but have weak inhibitory activity against FGFR1-3 [32]. More efforts are needed to develop FGFR inhibitors.
In previous experiments, we synthesised a novel FGFR inhibitor (F1-7) by using CADD and structure-based design strategies. In the kinase experiment, F1-7 showed potent inhibition of four FGFR isoforms. Considering that approximately one-third of colon cancer patients carry FGFR gene alterations and may benefit from FGFR inhibitors [33], we assessed the anti-tumour effect of F1-7 in colon cancer.
Since the FGFR signaling pathway mainly regulates the migration and invasion of cancer cells [34], when cells are treated with F1-7, FGFR signaling is inhibited, and the ability of cell migration and invasion were suppressed. F1-7 inhibited cell polarisation and thus affected cell migration. Also, we proved that F1-7 could restrict tumour growth in vivo. When injected with F1-7, the FGFR pathway in the tumour was inhibited, DNA damage occurred in the cells, the expression of some apoptotic-related proteins was changed and the cells lost growing activity. Compared with the control group, the tumour volume increased slowly at the low concentration (20 mg/kg), and the tumour barely grew at a high concentration (40 mg/kg). These consequences indicated that F1-7 has a terrific anti-tumour effect on colon cancer.
However, F1-7 still has great prospects for development. Its exploitation strategy is to be able to competitively bind to the ATP-binding pocket, thereby inhibiting FGFR phosphorylation and blocking the activation of the downstream signaling pathway. It is a second-generation TKI, so there will inevitably be off-target effects that lead to decreased inhibition effects and toxicity to the body. FGFR structural mutations may also lead to decreased efficacy. Therefore, its application still needs to be further optimised.
In conclusion, F1-7, a novel small molecule pan-FGFR inhibitor, inhibits FGFR and its downstream pathways and inhibits colon cancer cell migration and apoptosis by causing DNA damage (Fig. 8). Our results clearly show that F1-7 could be developed as a novel anticancer drug to treat colon cancer. These studies can clarify the anti-tumour mechanism of FGFR inhibitors and provide a new strategy for colon cancer treatment.
Fig. 8.

Schematic representation of the potential anti-tumour mechanism of F1-7 via inhibiting FGFR and activating DNA damage in colon cancer cells.
Supplementary information
Author contributions
YC and XZ contributed to the conception of the study; YL, LZ and XC performed the experiment; FH and XS revised and complemented the figure; DC and JS contributed significantly to analysis and manuscript preparation; YL performed the data analyses and wrote the manuscript; JW, QX and YX helped perform the analysis with constructive discussions.
Funding
This study was supported by the National Natural Science Funding of China (81473242, 21877085 and 21602159).
Data availability
The data used to support the findings of this study are available from the corresponding author upon request.
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Our study was approved by ‘the Laboratory Animal Centre, Wenzhou Medical University’ (wydw2020-0886).
Consent for publication
No personal data or identifying information are being submitted.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Yanan Liu, Liting Zhang.
Contributor Information
Youqun Xiang, Email: tom502@sina.com.
Xiaohui Zheng, Email: zhengxh@wmu.edu.cn.
Yuepiao Cai, Email: ypcai@wmu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-022-01878-4.
References
- 1.Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66:683–91. doi: 10.1136/gutjnl-2015-310912. [DOI] [PubMed] [Google Scholar]
- 2.Murakawa T. Past, present, and future perspectives of pulmonary metastasectomy for patients with advanced colorectal cancer. Surg Today. 2021;51:204–11. doi: 10.1007/s00595-020-02119-y. [DOI] [PubMed] [Google Scholar]
- 3.Cheng YD, Yang H, Chen GQ, Zhang ZC. Molecularly targeted drugs for metastatic colorectal cancer. Drug Des Devel Ther. 2013;7:1315–22. doi: 10.2147/DDDT.S52485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Touat M, Ileana E, Postel-Vinay S, Andre F, Soria JC. Targeting FGFR signaling in cancer. Clin Cancer Res. 2015;21:2684–94. doi: 10.1158/1078-0432.CCR-14-2329. [DOI] [PubMed] [Google Scholar]
- 5.Formisano L, Lu Y, Servetto A, Hanker AB, Jansen VM, Bauer JA, et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat Commun. 2019;10:1373. doi: 10.1038/s41467-019-09068-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Porta R, Borea R, Coelho A, Khan S, Araujo A, Reclusa P, et al. FGFR a promising druggable target in cancer: Molecular biology and new drugs. Crit Rev Oncol Hematol. 2017;113:256–67. doi: 10.1016/j.critrevonc.2017.02.018. [DOI] [PubMed] [Google Scholar]
- 7.Gozgit JM, Wong MJ, Moran L, Wardwell S, Mohemmad QK, Narasimhan NI, et al. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol Cancer Ther. 2012;11:690–9. doi: 10.1158/1535-7163.MCT-11-0450. [DOI] [PubMed] [Google Scholar]
- 8.Ma WW, Xie H, Fetterly G, Pitzonka L, Whitworth A, LeVea C, et al. A phase Ib study of the FGFR/VEGFR inhibitor dovitinib with gemcitabine and capecitabine in advanced solid tumor and pancreatic cancer patients. Am J Clin Oncol. 2019;42:184–9. doi: 10.1097/COC.0000000000000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guffanti F, Chila R, Bello E, Zucchetti M, Zangarini M, Ceriani L, et al. In vitro and in vivo activity of lucitanib in FGFR1/2 amplified or mutated cancer models. Neoplasia. 2017;19:35–42. doi: 10.1016/j.neo.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steegmann JL, Baccarani M, Breccia M, Casado LF, Garcia-Gutierrez V, Hochhaus A, et al. European LeukemiaNet recommendations for the management and avoidance of adverse events of treatment in chronic myeloid leukaemia. Leukemia. 2016;30:1648–71. doi: 10.1038/leu.2016.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gavine PR, Mooney L, Kilgour E, Thomas AP, Al-Kadhimi K, Beck S, et al. AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. 2012;72:2045–56. doi: 10.1158/0008-5472.CAN-11-3034. [DOI] [PubMed] [Google Scholar]
- 12.Komla-Ebri D, Dambroise E, Kramer I, Benoist-Lasselin C, Kaci N, Le Gall C, et al. Tyrosine kinase inhibitor NVP-BGJ398 functionally improves FGFR3-related dwarfism in mouse model. J Clin Invest. 2016;126:1871–84. doi: 10.1172/JCI83926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu D, Guo M, Min X, Dai S, Li M, Tan S, et al. LY2874455 potently inhibits FGFR gatekeeper mutants and overcomes mutation-based resistance. Chem Commun (Camb) 2018;54:12089–92. doi: 10.1039/C8CC07546H. [DOI] [PubMed] [Google Scholar]
- 14.Liu Q, Yu S, Zhao W, Qin S, Chu Q, Wu K. EGFR-TKIs resistance via EGFR-independent signaling pathways. Mol Cancer. 2018;17:53. doi: 10.1186/s12943-018-0793-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin S, Yang L, Yao Y, Xu L, Xiang Y, Zhao H, et al. Flubendazole demonstrates valid antitumor effects by inhibiting STAT3 and activating autophagy. J Exp Clin Cancer Res. 2019;38:293. doi: 10.1186/s13046-019-1303-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qiu Y, Xiao Z, Wang Y, Zhang D, Zhang W, Wang G, et al. Optimization and anti-inflammatory evaluation of methyl gallate derivatives as a myeloid differentiation protein 2 inhibitor. Bioorg Med Chem. 2019;27:115049. doi: 10.1016/j.bmc.2019.115049. [DOI] [PubMed] [Google Scholar]
- 17.Lu HR, Meng LH, Huang M, Zhu H, Miao ZH, Ding J. DNA damage, c-myc suppression and apoptosis induced by the novel topoisomerase II inhibitor, salvicine, in human breast cancer MCF-7 cells. Cancer Chemother Pharmacol. 2005;55:286–94. doi: 10.1007/s00280-004-0877-z. [DOI] [PubMed] [Google Scholar]
- 18.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wenger T, Miller D, Evans K. FGFR Craniosynostosis Syndromes Overview. In: MP Adam et al. (eds). GeneReviews®. October 20, 1998.
- 20.Liu Y, Cao M, Cai Y, Li X, Zhao C, Cui R. Dissecting the role of the FGF19-FGFR4 signaling pathway in cancer development and progression. Front Cell Dev Biol. 2020;8:95. doi: 10.3389/fcell.2020.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.von Mering C, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 2003;31:258–61. doi: 10.1093/nar/gkg034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou L, Wang S, Cao L, Ren X, Li Y, Shao J, et al. Lead acetate induces apoptosis in Leydig cells by activating PPARgamma/caspase-3/PARP pathway. Int J Environ Health Res. 2021;31:34–44. doi: 10.1080/09603123.2019.1625034. [DOI] [PubMed] [Google Scholar]
- 23.Maes ME, Grosser JA, Fehrman RL, Schlamp CL, Nickells RW. Completion of BAX recruitment correlates with mitochondrial fission during apoptosis. Sci Rep. 2019;9:16565. doi: 10.1038/s41598-019-53049-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Willis S, Day CL, Hinds MG, Huang DC. The Bcl-2-regulated apoptotic pathway. J Cell Sci. 2003;116:4053–6. doi: 10.1242/jcs.00754. [DOI] [PubMed] [Google Scholar]
- 25.Kuo LJ, Yang LX. Gamma-H2AX—a novel biomarker for DNA double-strand breaks. Vivo. 2008;22:305–9. [PubMed] [Google Scholar]
- 26.Yang Y, Xue K, Li Z, Zheng W, Dong W, Song J, et al. [Corrigendum] cMyc regulates the CDK1/cyclin B1 dependentG2/M cell cycle progression by histone H4 acetylation in Raji cells. Int J Mol Med. 2019;44:1988. doi: 10.3892/ijmm.2019.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katoh M, Nakagama H. FGF receptors: cancer biology and therapeutics. Med Res Rev. 2014;34:280–300. doi: 10.1002/med.21288. [DOI] [PubMed] [Google Scholar]
- 28.Hall A. The cytoskeleton and cancer. Cancer Metastasis Rev. 2009;28:5–14. doi: 10.1007/s10555-008-9166-3. [DOI] [PubMed] [Google Scholar]
- 29.Fuhrmann A, Banisadr A, Beri P, Tlsty TD, Engler AJ. Metastatic state of cancer cells may be indicated by adhesion strength. Biophys J. 2017;112:736–45. doi: 10.1016/j.bpj.2016.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Venetsanakos E, Brameld KA, Phan VT, Verner E, Owens TD, Xing Y, et al. The irreversible covalent fibroblast growth factor receptor inhibitor PRN1371 exhibits sustained inhibition of FGFR after drug clearance. Mol Cancer Ther. 2017;16:2668–76. doi: 10.1158/1535-7163.MCT-17-0309. [DOI] [PubMed] [Google Scholar]
- 31.Tan L, Wang J, Tanizaki J, Huang Z, Aref AR, Rusan M, et al. Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors. Proc Natl Acad Sci USA. 2014;111:E4869–4877. doi: 10.1073/pnas.1403438111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu X, Chen H, Patterson AV, Smaill JB, Ding K. Fibroblast growth factor receptor 4 (FGFR4) selective inhibitors as hepatocellular carcinoma therapy: advances and prospects. J Med Chem. 2019;62:2905–15. doi: 10.1021/acs.jmedchem.8b01531. [DOI] [PubMed] [Google Scholar]
- 33.Sootome H, Fujita H, Ito K, Ochiiwa H, Fujioka Y, Ito K, et al. Futibatinib is a novel irreversible FGFR 1-4 inhibitor that shows selective antitumor activity against FGFR-deregulated tumors. Cancer Res. 2020;80:4986–97. doi: 10.1158/0008-5472.CAN-19-2568. [DOI] [PubMed] [Google Scholar]
- 34.Kang X, Lin Z, Xu M, Pan J, Wang ZW. Deciphering role of FGFR signalling pathway in pancreatic cancer. Cell Prolif. 2019;52:e12605. doi: 10.1111/cpr.12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.