

Abstract
Genes involved in the anaerobic metabolism of phenol in the denitrifying bacterium Thauera aromatica have been studied. The first two committed steps in this metabolism appear to be phosphorylation of phenol to phenylphosphate by an unknown phosphoryl donor (“phenylphosphate synthase”) and subsequent carboxylation of phenylphosphate to 4-hydroxybenzoate under release of phosphate (“phenylphosphate carboxylase”). Both enzyme activities are strictly phenol induced. Two-dimensional gel electrophoresis allowed identification of several phenol-induced proteins. Based on N-terminal and internal amino acid sequences of such proteins, degenerate oligonucleotides were designed to identify the corresponding genes. A chromosomal DNA segment of about 14 kbp was sequenced which contained 10 genes transcribed in the same direction. These are organized in two adjacent gene clusters and include the genes coding for five identified phenol-induced proteins. Comparison with sequences in the databases revealed the following similarities: the gene products of two open reading frames (ORFs) are each similar to either the central part and N-terminal part of phosphoenolpyruvate synthases. We propose that these ORFs are components of the phenylphosphate synthase system. Three ORFs showed similarity to the ubiD gene product, 3-octaprenyl-4-hydroxybenzoate carboxy lyase; UbiD catalyzes the decarboxylation of a 4-hydroxybenzoate analogue in ubiquinone biosynthesis. Another ORF was similar to the ubiX gene product, an isoenzyme of UbiD. We propose that (some of) these four proteins are involved in the carboxylation of phenylphosphate. A 700-bp PCR product derived from one of these ORFs cross-hybridized with DNA from different Thauera and Azoarcus strains, even from those which have not been reported to grow with phenol. One ORF showed similarity to the mutT gene product, and three ORFs showed no strong similarities to sequences in the databases. Upstream of the first gene cluster, an ORF which is transcribed in the opposite direction codes for a protein highly similar to the DmpR regulatory protein of Pseudomonas putida. DmpR controls transcription of the genes of aerobic phenol metabolism, suggesting a similar regulation of anaerobic phenol metabolism by the putative regulator.
Phenolic compounds are important plant constituents. For example, lignin represents about 20 to 30% of the biomass of higher plants and consists of phenylpropane units with various hydroxyl or methoxyl groups. Phenol is formed from lignin and several other natural or synthetic substrates during microbial degradation. The best-known phenol-generating enzyme is tyrosine phenol lyase, whose activity is used in bacterial taxonomic studies. Because of its toxic effects on cells, phenol was used as an antiseptic in clinical applications for a long time. In the chemical industry, phenol is a basic substance for the production of phenolic compounds from coal. Because of its broad application in industry, phenolic compounds are among the most prominent man-made groundwater contaminants derived from coal treatment processes, such as gasifying coal.
The aerobic metabolism of phenol has been studied extensively; in all known aerobic pathways, mono-oxygenases initiate the degradation of phenol by hydroxylation to catechol (for a recent review on phenol ortho hydroxylases, see reference 60). Among ortho-hydroxylating phenol hydroxylases, one-component flavoproteins or multicomponent enzyme systems are recognized. The following ring cleavage is a key reaction in microbial degradation of aromatic compounds. Catechol is oxygenolytically cleaved by dioxygenases by either ortho or meta cleavage.
Anaerobic growth on phenol has been observed for various bacteria (4, 24, 37, 55, 59, 62, 63, 64, 68). In all cases studied, phenol appeared to be carboxylated to 4-hydroxybenzoate and growth on phenol was dependent on the presence of CO2 (62). Consortia of fermenting bacteria convert phenol to benzoate (24, 68) and decarboxylate 4-hydroxybenzoate to phenol (24, 68). They also catalyze an isotope exchange between D2O and the proton at C-4 of the aromatic ring of phenol (23).
Phenol carboxylation to 4-hydroxybenzoate is a paradigm for a new type of biological carboxylation reaction. The process has been studied in the denitrifying bacterium Thauera aromatica (3, 32–34, 63) (Fig. 1). The gram-negative, rod-shaped bacterium belongs to the β-group of the proteobacteria and was enriched and isolated under denitrifying conditions with phenol as substrate (1, 62; for similar strains, see references 61 and 64). It has a broad substrate spectrum and is able to grow anaerobically on many aromatic compounds. Cells grown with phenol were simultaneously adapted to growth with 4-hydroxybenzoate, whereas 4-hydroxybenzoate-grown cells did not metabolize phenol. Induction of the capacity to metabolize phenol required several hours.
FIG. 1.

Postulated pathway of anaerobic phenol metabolism in the denitrifying bacterium T. aromatica. E1, phenylphosphate synthase; E2, phenylphosphate carboxylase (the mechanism by which the phenolate anion is bound to the enzyme E2 is unknown so far); E3, 4-hydroxybenzoate-CoA ligase; E4, 4-hydroxybenzoyl-CoA reductase (dehydroxylating, ferredoxin dependent); E5, benzoyl-CoA reductase (dearomatizing, ferredoxin dependent). For further metabolism of benzoyl-CoA to three acetyl-CoA, one CO2, and six H molecules, see reference 25. Fd, ferredoxin. X∼P, phosphoryl donor, unknown so far.
Phenol carboxylation proceeds in two steps and seems to involve formation of phenylphosphate as the first intermediate (equation 1) (32, 33). An enzyme activity catalyzing an isotope exchange between [14C]phenol and the phenyl moiety of phenylphosphate was identified in extracts of phenol-grown cells (equation 3) and was lacking in 4-hydroxybenzoate-grown cells. [32P]Phosphate did not exchange with phenylphosphate. This suggests a phosphorylating enzyme, E1, with a ping-pong mechanism which becomes phosphorylated in an essentially irreversible step (equations 2 and 3). The phosphorylated enzyme E1 is postulated to transform phenol to phenylphosphate in a reversible reaction (equation 3). The whole reaction is understood as the sum of equations 2 and 3. Unfortunately, the phosphoryl donor X∼P is unknown so far. The enzyme E1 was termed “phenylphosphate synthase.”
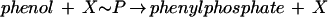 |
1 |
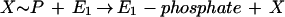 |
2 |
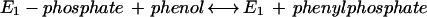 |
3 |
Phenylphosphate is postulated to be the substrate of a second enzyme, E2 (32). It requires Mn2+ and catalyzes the carboxylation of phenylphosphate to 4-hydroxybenzoate (equation 4). An enzyme activity catalyzing an isotope exchange between 14CO2 and the carboxyl group of 4-hydroxybenzoate (equation 6) was present in phenol-grown cells; [14C]phenol did not exchange with 4-hydroxybenzoate. It was assumed that the carboxylation reaction (equation 4) and the CO2 isotope-exchange reaction (equation 6) are catalyzed by the same enzyme, reaction 6 being a partial reaction of reaction 4. The CO2 isotope-exchange activity again suggests a ping-pong mechanism for enzyme E2 (equations 5 and 6), involving an enzyme E2-phenolate intermediate which is formed in a (presumably) exergonic reaction (equation 5) followed by a reversible carboxylation (equation 6).
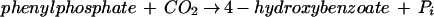 |
4 |
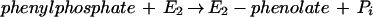 |
5 |
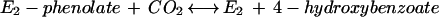 |
6 |
The actual substrate is CO2 rather than bicarbonate (as is the case in biotin-dependent carboxylases), and the carboxylating enzyme was not inhibited by avidin, a potent inhibitor of biotin-dependent carboxylases. Both results suggest that biotin is not involved in carboxylation. The enzyme E2 was termed “phenylphosphate carboxylase.” Synthesis of both the phosphorylating and the carboxylating enzymes is strictly regulated. All activities were only present in cells grown anoxically on phenol and were lacking, e.g., in 4-hydroxybenzoate-grown cells (29, 34). The phenol-carboxylating enzyme in T. aromatica does not belong to any group of the studied carboxylases, as it seems to proceed via a phosphorylated free intermediate, it is extremely oxygen sensitive and sensitive to radical-trapping agents (S. Breinig, M. Hügler, and G. Fuchs, unpublished results), and it is not dependent on biotin or thiamine diphosphate. It differs from most known carboxylases by using carbon dioxide as substrate and by the requirement of a metal as cocatalyst.
Further metabolism of 4-hydroxybenzoate proceeds in two steps to the intermediate benzoyl coenzyme A (CoA) (Fig. 1). A specific CoA ligase forms 4-hydroxybenzoyl-CoA (8), which becomes reductively dehydroxylated to benzoyl-CoA by a molybdo-flavo-iron-sulfur protein, 4-hydroxybenzoyl-CoA reductase (11, 14). A 2 [4Fe-4S] ferredoxin serves as electron donor for this reaction (14). The ligase and reductase are both induced in phenol- and in 4-hydroxybenzoate-grown cells, but not in benzoate-grown cells (29). Benzoyl-CoA is a common intermediate in the metabolism of many aromatic compounds. It is reductively dearomatized (9, 10) and further metabolized to three acetyl-CoA molecules and one CO2 molecule (note that one CO2 was originally introduced into phenol) (reviewed in reference 28). The anaerobic metabolism of phenol is shown in Fig. 1.
Nothing was known so far about the molecular background of anaerobic phenol metabolism. Based on the N-terminal and internal peptide sequence information of five phenol-induced proteins, it was our aim to find the genes coding for phenol-induced proteins. Analysis of the DNA sequences should provide new insights into the function of the phenol-induced proteins and into the regulation of anaerobic phenol metabolism.
MATERIALS AND METHODS
Materials.
Chemicals were reagent grade and purchased from Fluka (Neu-Ulm, Germany), Merck (Darmstadt, Germany), Serva (Heidelberg, Germany), or Sigma-Aldrich (Deisenhofen, Germany). Biochemicals were from Boehringer (Mannheim, Germany). Scintillation cocktail and acrylamide stock solution were purchased from Roth (Karlsruhe, Germany). [14C]Na2CO3 (2 GBq mmol−1) was purchased from American Radiolabeled Chemicals Inc./Biotrend Chemikalien GmbH (Cologne, Germany). Nitrogen, helium, and a N2-H2 gas mixture (95% nitrogen, 5% hydrogen) were purchased from Linde (Höllriegelskreuth, Germany).
Bacterial strains, growth conditions, cell harvesting, and storage.
Strains of T. aromatica, K172 (1, 62), S100 (62), SP (54), LG356 (unpublished results), and T1 (61), and Azoarcus evansii KB740 (12) were cultured anaerobically at 30°C in a mineral salt medium with 0.5 mM phenol, 10 mM bicarbonate, and 2 mM nitrate or with 5 mM 4-hydroxybenzoate and 15 mM NaNO3 as described elsewhere (11, 62). Growth was measured by following the optical density at 578 nm (OD578). An OD578 of 1.0 corresponded to a cell concentration of approximately 0.3 g of dry cell mass per liter. The harvested cells were immediately frozen and stored in liquid nitrogen. Growth conditions of Escherichia coli strains (XL1-blue MRF′ [Δ(mcrA)183 Δ(mcr-CB-hsdSMR-mrr)173 endA1 supE44 thi-1 recA1 gyrA96 relA1 lac [F′ proAB lacIq ZΔM15 Tn10 (Tetr)]]; K38 [hfrC phoA4 pit-10 tonA22 ompF627 relA1 spoT λ−]; P2392 [E14− (McrA−) hsdR514 supE44 supF58 lacYl or Δ(lacIZY)6 galK2 galT22 metB1 trpR55 (P2 lysogen)] and XLOLR [Δ(mcrA) 183 Δ(mcrCB-hsdSMR-mrr)173 endA1 thi-1 recA1 gyrA96 relA1 lac [F′ proAB lacIq ZΔM15 Tn10 (Tetr)] Su− (nonsuppressing) λr (lambda resistant)]) were as previously described (50).
Preparation of cell extract.
All steps were performed under strictly anaerobic conditions. Frozen cells (20 g) were suspended in 20 ml of 20 mM imidazole-HCl (pH 6.5), 10% glycerol, 0.5 mM dithionite, and traces of DNase I, disrupted by a passage through a French press (137 MPa), and ultracentrifuged (1.5 h at 100,000 × g and 4°C). The supernatant containing the soluble protein fraction (55 mg of protein/ml) was used immediately or stored at −20°C for later experiments. The soluble protein fraction (cell extract) contained all 4-hydroxybenzoate: 14CO2 exchange and phenylphosphate carboxylase activity.
Enzymatic tests.
All tests were conducted at 30°C under strictly anaerobic conditions. The assays used 1 ml of assay mixture in a stoppered vial with a gas phase of approximately 250 μl. The assays for 14CO2:4-hydroxybenzoate isotope exchange and carboxylation of phenylphosphate were performed as described elsewhere (32, 43). The amount of radioactivity in acid-stable products (4-hydroxybenzoate) in a sample of 250 μl was analyzed by liquid scintillation counting. The amount of labeled 4-hydroxybenzoate formed was calculated from the amount of fixed radioactivity, taking into account the known specific radioactivity of bicarbonate in the assay (80 Bq/nmol).
Partial purification of three dominant phenol-induced proteins, F1, F2, and F3.
All chromatographic steps were performed under anaerobic conditions in a glove box under an N2-H2 atmosphere (95%/5% [vol/vol]) at 4°C. All buffers were filtered and degassed and then supplemented with dithionite to a final concentration of 0.5 mM. Generally used buffers were as follows. Buffer A contained 20 mM imidazole-HCl (pH 6.8), 20 mM KCl, 0.5 mM MnCl2, and 10% glycerol. Buffer B contained buffer A plus 500 mM KCl. The purification was started with extract (approximately 20 ml) from 20 g of frozen cells grown on phenol and nitrate.
Ion-exchange chromatography 1.
Cell extract (20 ml) was applied on a DEAE-Sepharose fast-flow column (35 ml; Pharmacia), which was run at a flow rate of 0.5 ml/min. The column was washed with 50 ml of buffer A, followed by a flat linear gradient from 20 mM KCl (100% buffer A) to 200 mM KCl (60% buffer A, 40% buffer B) in 100 ml and a steeper gradient from 200 mM KCl (60% buffer A, 40% buffer B) to 500 mM KCl (100% buffer B) in another 100 ml. Fractions of 2.5 ml were collected, and 6-μl samples of every second fraction were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Compared with extract of 4-hydroxybenzoate-grown cells, three dominant phenol-induced proteins, F1 (60 kDa), F2 (58 kDa), and F3 (67 kDa), were separated. F1 eluted between 60 and 100 mM KCl, F2 eluted between 140 and 190 mM KCl, and F3 eluted between 250 and 350 mM KCl. Fractions containing F1, F2, and F3 were pooled and tested for phenylphosphate carboxylase and CO2 isotope exchange activities.
Ion-exchange chromatography 2.
Two milliliters of the pooled fractions of F1, F2, and F3 after DEAE-Sepharose fast-flow chromatography was applied to a high-performance liquid chromatography (HPLC) system equipped with a MonoQ HR5/5 column (1 ml; Pharmacia), which was run at a flow rate of 0.5 ml/min. Fractions of 1 ml were collected in each run, and aliquots of 10 μl were analyzed by SDS-PAGE. For F1 purification, the column was equilibrated with buffer A containing 60 mM KCl and eluted with this buffer until the UV absorption baseline at 280 nm was stable. Samples of every fraction were tested for both activities and analyzed by SDS-PAGE. F1 eluted after 5 to 10 column volumes. For F2 purification, the column was equilibrated with buffer A containing 150 mM KCl. The column was washed with this buffer until the UV absorption baseline at 280 nm was stable. F2 eluted after 10 to 14 column volumes. For F3 purification, the column was equilibrated with buffer A containing 260 mM KCl and washed until the baseline at 280 nm was stable. Then, a linear gradient of 260 to 400 mM KCl in buffer A (8 column volumes) was applied. F3 eluted early in the gradient between 260 and 330 mM KCl.
Affinity chromatography on blue Sepharose.
Ten milliliters of the F2 pool after chromatography on DEAE was loaded on a blue Sepharose column (48 ml; Pharmacia), equilibrated with 20 mM Tris-HCl, pH 7.5, at a flow rate of 2 ml/min. Because of the instability of the color matrix in the presence of reducing agents, no dithionite was added. The column was washed with 1 volume of the same buffer, and then F2 was eluted with 10 mM phenylphosphate in 20 mM Tris-HCl, pH 7.5, in 3 volumes. Fractions of 5 ml were collected, and 100-μl samples of each fraction were analyzed by SDS-PAGE (10% acrylamide).
SDS-PAGE.
Polyacrylamide gels (normally 11.5% acrylamide) were prepared as previously described (35). Staining of the proteins was done according to a previously described method (67).
Two-dimensional gel electrophoresis.
The first dimension (isoelectric focusing) was done with cell extract (120 μg of protein) according to Görg et al. (25) by using the Immobiline Dry Strip system (linear pH gradient 3 to 10; Pharmacia) according to the manufacturer's protocol. The second dimension (SDS-PAGE) was performed as described above. Comparison of extracts of cells grown on phenol and 4-hydroxybenzoate allowed the identification of phenol-induced proteins.
Protein transfer onto polyvinylidene difluoride membranes.
Proteins of cell extract and of fractions after chromatography were electrophoretically separated on SDS-polyacrylamide gels and were then transferred to an Immobilon-Psq transfer membrane (Millipore, Bedford, Mass.) for N-terminal sequencing of the phenol-induced proteins by using the Nova Blot System (Multiphor II; Pharmacia LKB, Freiburg, Germany) according to the manufacturer's protocol.
Amino acid sequencing and tryptic digest.
Protein spots from cell extract of phenol-grown cells obtained after two-dimensional electrophoresis and fractions containing the phenol-induced proteins F1 to F3 after chromatography on MonoQ were transferred to a polyvinylidene difluoride membrane. Induced proteins were excised and sequenced using an Applied Biosystems 473A sequencer or an automated gas phase sequencer. The sequencing was performed according to the Edman method. The separated amino acid derivatives were analyzed by C8 reversed-phase HPLC. For peptide sequencing, the pooled fractions containing F2 after chromatography on blue Sepharose were digested with modified trypsin (Promega, Mannheim, Germany) as follows: 500 μg of protein in 200 μl of 20 mM Tris-HCl, pH 7.5, was adjusted to pH 8 with 3 μl of triethylamine, and 10 μg of trypsin in 10 μl of H2O (Promega sequencing grade modified) was added. The digest was carried out at 37°C for 4 h. The reaction was stopped by heating for 5 min to 100°C. After centrifugation, volumes of 5, 70, and 100 μl were sequentially applied to the HPLC. The peptides generated were separated on a reversed-phase C18 Superpac-Sephasil HPLC column (Amersham Pharmacia Biotech, Uppsala, Sweden).
Cloning, DNA sequencing, and nucleic acid manipulations.
Plasmids used for transformation of E. coli strains were pBK-CMV phagemid [fl(−)ori lacZ CMV SV40 poly(A) colE1 ori Neor Kanr], pBluescript II KS/SK(±) [fl(±)ori lacZ colE1 ori Ampr], and pGP1-2 [Kanr T7 Gen1]. Oligonucleotides used for amplification of the probes for screening via PCR were as follows (>, forward; <, reverse; all in 5′ to 3′ direction): fubrei p374 (>GCGGCGCTTTCGGCACA), fubrei 375 (<GCGACTTCGCGCATCTTGTT), F2H (>ATGGAYCTSCGSTACTTCATC), F2t43R (<CATSAGGAAYTCSGCCTGCTG), λ15H (>TCGCCGGCGACGACGCCG), and λ15R (<CCGCGCGCTGCGCCGCCG). PCR products used as probes were labeled with [α-32P]dCTP (Amersham). Standard protocols were used for DNA cloning, transformation, amplification, and purification (50). Recombinant plasmids were generated in pBluescript vectors and were maintained in E. coli XL1-blue. T. aromatica DNA has a high GC content (67%). This made sequencing difficult because of the compression phenomenon. Three methods were applied. First, sequencing of plasmid DNA by the Sanger method was performed with 35S-dATP. Second, sequencing of a PCR product with internal Cy5-dATP labeling, 0.5 μg of template/kbp, was performed according to the manufacturer's protocol and analyzed with an ALF express (Pharmacia). Third, cycle sequencing of PCR DNA according to the manufacturer's protocol (Pharmacia) was performed. Sequences were analyzed with an ALF express (Pharmacia). Part of the sequencing was performed with an ALF Automated Sequencer (Pharmacia).
Cloning strategy.
Based on the N-terminal amino acid sequences of F2 and of two internal fragments, degenerate oligonucleotides were designed according to the codon usage of T. aromatica. A 500-bp PCR product (F2 forward and F2T6 reverse primers) of the N-terminal sequence of F2 was labeled with [32P]dCTP and used as a probe for screening the λ EMBL3 gene library. One phage hybridized with the probe. From the positive λ clone, two adjacent BamHI fragments of 2.7 and 4.0 kbp and an EcoRI fragment of 5.25 kbp were subcloned in pBluescript and sequenced (for restriction sites, see Fig. 5). The boundary between the BamHI fragments was verified by amplification and sequencing of an overlapping DNA fragment. A double-stranded contiguous sequence of ca. 9 kbp was obtained from the insert of this λ clone. To obtain sequence information of the 5′ and 3′ flanking regions of the first λ clone, gene libraries of T. aromatica were rescreened. A 500-bp probe of the 5′ boundary of the known sequence was amplified and used to screen the λ EMBL3 library again. Two positive clones were obtained whose inserts were subcloned in pBluescript. Plasmids containing parts of the known sequence were identified by PCR assays and used for DNA sequencing. A total of 6.2 kbp of DNA was sequenced from two overlapping subclones containing either a 3.7-kbp PstI fragment or a 9-kbp BamHI fragment. A 600-bp probe of the 3′ boundary of the known sequence was amplified and used to screen a λ ZAP library. One positive phage was detected and converted to a phagemid as recommended by the supplier (Stratagene). It contained an insert of 8 kbp which overlapped with the known sequence and was used to sequence a further 400 bp of the 3′ flanking region.
FIG. 5.

Organization of the clusters of genes possibly involved in anaerobic phenol metabolism in T. aromatica. Phenol-induced proteins F1 to F5 and the corresponding number of amino acids (aa) are shown below the ORFs. The reactions catalyzed by PEP synthase and 3-octaprenyl-4-hydroxybenzoate carboxy lyase (isoenzymes UbiD and UbiX) are shown below; these enzymes show similarity to some of the proteins possibly involved in phenol metabolism. Like colors (black and grey) stand for genes and gene products with similarities. Black arrows indicate the direction of transcription. The coding regions (sizes in base pairs) are indicated, as well as the restriction sites of various enzymes.
Southern hybridization.
DNA fragments resolved by agarose gel electrophoresis were partially hydrolyzed by gently rinsing in 0.25 M HCl for about 20 min, followed by denaturing in 0.5 M NaOH–1.5 M NaCl for another 20 min. Finally, the gel was neutralized by excessively rinsing it in 1 M Tris-HCl–3 M NaCl, pH 7, three times for 10 min each. Then, the DNA was vacuum blotted onto a nylon/NC membrane with the help of a vacuum gel dryer system (Slab Gel Dryer 2003; Pharmacia LKB). After the blot procedure, the slots of the gel were marked on the membrane and the membrane was allowed to dry at room temperature. Fixation of the DNA on the membrane was achieved by incubating for 2 h at 80°C. In addition, chromosomal DNA was also blotted by capillary blotting, which guaranteed a better transfer of the large fragments (50).
Screening of gene banks of T. aromatica.
A λEMBL3 gene bank (Stratagene) and a λZAPExpress gene bank (Stratagene) were prepared and screened according to the manufacturer's protocol.
PCR.
PCR conditions were as follows: 100 ng of target DNA, 200 nM concentrations of each primer, 200 μM (each) dATP, dCTP, dTTP, and dGTP, 50 mM KCl, 1.5 mM MgCl2, 10 mM Tris-HCl (pH 9.0), 1 U of Taq DNA polymerase (Amersham Pharmacia Biotech). The assay was overlaid with 75 μl of paraffin oil. PCR parameters were as follows: 30 s at 95°C, 1 min at 40°C, 2.5 min at 72°C, 30 cycles. For amplification of fragments larger than 3 kbp, Taq DNA polymerase from Qiagen (Hilden, Germany) was used under the following conditions (Taq PCR kit from Qiagen): 100 ng of target DNA, a 200 nM concentration of each primer, 200 μM (each) dATP, dCTP, dTTP, dGTP, (NH4)2SO4, and KCl, 4.5 mM MgCl2, 10 mM Tris-HCl (pH 8.7), 1× Q solution, and 1 U of Taq DNA polymerase (Qiagen). PCR parameters were as follows: 30 s at 95°C, 1 min at 45°C, 2.5 min at 72°C, 30 cycles.
Expression of the T. aromatica genes.
Heterologous expression of the genes was performed as previously described (13) for E. coli K38 containing the pGP1-2 plasmid, which contains the gene for the heat-inducible T7 RNA polymerase. Strain K38 was transformed according to a previously described method (18) with the pBluescript vector containing different parts of the phenol-induced gene cluster of T. aromatica under the control of a T7 promoter.
Computer analysis.
The nucleotide and amino acid sequences were analyzed using the PC/gene software package (Genofit) and the Open Reading Frame Finder (ORF Finder; http://www.ncbi.nlm.nih.gov/gorf/gorf.html). Similar sequences were identified using the BLAST search using the TBLASTN algorithm provided by the National Center for Biotechnology Information.
Nucleotide sequence accession number.
All sequence data have been deposited in the EMBL nucleotide sequence database under accession no. AJ272115.
RESULTS
Detection of phenol-induced proteins.
Growth on phenol and nitrate obviously requires induction of proteins that are required for anaerobic phenol metabolism. This follows from the observation that 4-hydroxybenzoate-grown cells were not able to metabolize phenol immediately, but only after a prolonged adaptation period of several hours. Furthermore, phenol-grown cells contained several protein bands in SDS-PAGE that were missing in 4-hydroxybenzoate-grown cells (not shown). Therefore, the soluble fraction (supernatant obtained after centrifugation at 100,000 × g) and the membrane fraction (pellet obtained after centrifugation at 100,000 × g, not shown) of extracts of cells that were grown anaerobically on phenol and 4-hydroxybenzoate, respectively, were compared in more detail by two-dimensional gel electrophoresis (Fig. 2).
FIG. 2.

Two-dimensional gel electrophoresis of soluble proteins (supernatant obtained after centrifugation at 100,000 × g) of T. aromatica anaerobically grown on 4-hydroxybenzoate (A) and phenol and CO2 (B). Triangles indicate phenol-induced proteins. F1 to F5 were isolated and their N-terminal amino acid sequences were determined.
The soluble fraction contained up to 11 phenol-induced proteins. Three of them, called F1, F2, and F3, showing apparent molecular masses of about 60, 58, and 67 kDa, respectively, were strongly visible and were stained with similar intensity on SDS-polyacrylamide gels. The pI values were estimated to be 6.5, 7.0, and 5.5, respectively. Two further induced proteins, called F4 and F5, with apparent molecular masses of about 20 kDa and pI values of approximately 6.5 and 8.0, respectively, could be identified. Another two protein spots (no. 10 and 11 in Fig. 2) were only weakly stained and, because of their size (apparently about 14 kDa), were hardly visible. Their pI values were estimated to be 5.5 and 9.0. Up to four additional phenol-induced proteins were not clearly resolved from neighbor proteins; their apparent molecular masses were in the range of 30 to 55 kDa, and their pI values were between 5.8 and 7.0. The membrane fraction did not contain proteins that were strongly phenol induced.
Partial purification of phenol-induced proteins.
The high content of phenol-induced proteins encouraged us to purify phenylphosphate carboxylase and CO2 isotope exchange activity, despite its oxygen sensitivity. A chromatographic separation of cell extract on a DEAE-Sepharose fast-flow column is shown in Fig. 3A. Both activities coeluted in two fractions, and both contained phenol-induced proteins F1 and F2, although at different ratios. Both activities seemed to require the presence of some amount of the protein F1. Neither activity was measured in a third fraction containing the protein F3. The subfractions of the first two fractions were pooled and tested for both activities. In the first fraction, 35% of the residual activity of the CO2 isotope exchange reaction and 15% of the phenylphosphate carboxylase activity could be detected, and in the second fraction, 20 and 10% of the activities, respectively, were detected. No combination of the three fractions restored full activity.
FIG. 3.

Partial purification of phenol-induced proteins. (A) DEAE-Sepharose fast-flow chromatography of 15 ml of the soluble fraction (supernatant obtained after centrifugation at 105 × g) of T. aromatica grown on phenol and nitrate. Fractions containing the F1, F2, and F3 proteins (identified by SDS-PAGE) that were pooled are indicated as black bars (1, 2, and 3, respectively) below the x axis. Only 10 to 20% of both the phenylphosphate carboxylating activity and the isotope exchange were detectable in the F1 and F2 pools. In the F3 pool, none of the activities were measurable. Circle, phenylphosphate carboxylase activity (values in parentheses). Rhombus, isotope-exchange activity. (B) SDS–10% PAGE of the fractions obtained by chromatography of the soluble protein fraction on a DEAE-Sepharose fast-flow column. Lane 1, extract of cells grown with phenol and nitrate; lane 2, pooled fractions containing F1; lane 4, pooled fractions containing F2; lane 6, pooled fractions containing F3; lanes 3, 5, and 7, F1 to F3 were further purified by chromatography on MonoQ. The three proteins eluted with 60, 200, and 330 mM KCl, respectively. (C) SDS–10% PAGE of F2 (fractionated by DEAE-Sepharose fast flow) after chromatography on blue Sepharose. Lane 1, DEAE fraction of F2; lane 2, main fraction of F2 eluting with 10 mM phenylphosphate from blue Sepharose.
The two pooled DEAE fractions containing mainly F1 and F2 were chromatographed isocratically on MonoQ HR5/5, and the pooled DEAE fraction containing mainly F3 was chromatographed on MonoQ HR5/5 under refined conditions by using a linear KCl gradient. This resulted in further purification of the three proteins (Fig. 3B), but activity was completely lost. When the three DEAE fractions were applied to blue Sepharose at pH 7.0, F1 to F3 bound to the column and were eluted with 1 M KCl. At pH 7.5, F2 was specifically eluted with 10 mM phenylphosphate (Fig. 3C).
N-terminal sequencing of five phenol-induced proteins (F1 to F5), peptide sequencing of F2, and synthesis of degenerate primers.
Three phenol-induced proteins, F1 to F3, were enriched by chromatographic techniques in the enzymatically inactive form, and two other phenol-induced proteins, F4 and F5, were obtained from two-dimensional gels. The material was sufficient for N-terminal sequencing and peptide sequencing of F2. Up to 40 amino acids were determined, with only a few ambiguous amino acid residues. Comparison of the experimentally determined sequences with the sequences deduced from the genes (see below) showed very good correspondence, with only very few mismatches, and most uncertain amino acids were confirmed to be correct (Fig. 4). The results of the N-terminal sequencing were used to design degenerate primers, taking into account the known codon usage of T. aromatica. Amino acid sequences that were used as templates are underlined in Fig. 4.
FIG. 4.

Determined and deduced N-terminal amino acid sequences of phenol-induced proteins F1 to F5 and internal sequences of protein F2. X, amino acids that could not be sequenced; small letters, amino acids that were uncertain. Underlined amino acids were used as templates for primer design (names of the forward and reverse primers are given in parentheses). F1 corresponds to Orf6, F2, to Orf4, F3 to Orf1, F4 to Orf5, and F5 to Orf8.
Screening of a λ EMBL3 and a λ ZAP express gene library.
Based on the N-terminal amino acid sequences of F2 and of an internal fragment of F2, degenerate oligonucleotides were designed. The primers used for the generation of labeled probes to screen the libraries are listed in Materials and Methods. A labeled 500-bp PCR product corresponding to the N-terminal sequence of F2 (F2H/F2T43R) was used as a probe for screening a λ EMBL3 gene library. One phage hybridized with the probe. The insert DNA was prepared and subcloned in pBluescript vectors. About 9 kbp of sequence information of both DNA strands was obtained.
To get more sequence information 5′ of the first segment, a new probe was amplified by PCR (fubrei p374 and fubrei 375) and used for gene bank screening. Two positive phages whose DNA was subcloned in pBluescript vectors were obtained. The sequences overlapped with the 5′ sequences of the clone obtained by the first screen.
To obtain more sequence information 3′ of the sequences of the first screen, a λZAPExpress gene library was screened with a 600-bp probe derived from the 3′ end of the sequences (λ15H/λ15R). One positive clone was detected, and the phagemid DNA was prepared. The size of the DNA insert was estimated to be at least 8 kbp by agarose gel electrophoresis. The 5′ sequences of the phagemid insert overlapped with the 3′ sequences of the first screen.
DNA sequencing and identification of the genes coding for putative proteins involved in anaerobic phenol metabolism.
Screening of the gene libraries and sequencing of the subcloned DNA resulted in sequence information for 14,272 bp. Eleven open reading frames (ORFs) were detected (Fig. 5). Table 1 summarizes the location and orientation of ORFs 1 to 11 and, the number of amino acids encoded by each ORF, as well as the percent similarity and percent identity of the amino acid sequences of ORFs 1 to 11 with known sequences in the databases.
TABLE 1.
Properties of the gene products involved in anaerobic phenol metabolism and similarities with other proteins in the databasesa
| Gene product | Similar proteinb | Gene propertiesc
|
Protein propertiesd
|
Protein correspondencee
|
Protein with highest similarityf
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dir | Range | Length (aa) | Mol mass (kDa)
|
pI
|
% Identity | % Similarity | E value | References | Protein | ||||
| Deduced | Exp | Deduced | Exp | ||||||||||
| Orf11 | (7) | ← | 688–2436 | 582 | 63.756 | 55 | 6.14 | 6.5 | 47.2 | 72.3 | 1E−20 | gnl|PID|d1010531 (D63814) | DmpR (transcriptional activator) |
| Orf1 | F3 | → | 2864–4702 | 612 | 70.261 | 67 | 5.06 | 5.5 | 16.7 | 39.3 | 4E−10 | gi|147146 (M69116) | PEP synthase (central part) |
| Orf2 | (6) | → | 4716–5840 | 374 | 40.423 | 40 | 5.38 | 5.5 | 21.8 | 34.5 | 1E−63 | gi|147146 (M69116) | PEP synthase (N-terminal part) |
| Orf3 | (9) | → | 5853–6524 | 223 | 24.244 | 30 | 6.19 | 6.5 | 14.5 | 30.2 | 1E−8 | gi|2621183 (AE000803) | Inosine-5′-monophosphate dehydrogenase |
| Orf4 | F2 | → | 6587–8005 | 472 | 52.605 | 58 | 6.06 | 7.0 | 30.8 | 58.95 | 5E−47 | gi|549586|sp|P26615|UbiD | 3-Octaprenyl-4-hydroxybenzoate carboxy lyase |
| Orf5 | F4 | → | 8070–8579 | 169 | 18.450 | 18 | 5.62 | 6.5 | 38.8 | 63.8 | 2E−25 | gi|2851406|sp|P45396|YrbI | Hypothetical protein |
| Orf6 | F1 | → | 8616–10073 | 485 | 53.940 | 60 | 5.64 | 6.5 | 29.4 | 57.1 | 1E−31 | gi|549586|sp|P26615|UbiD | 3-Octaprenyl-4-hydroxybenzoate carboxy lyase |
| Orf7 | (8) | → | 10731–11804 | 357 | 38.198 | 35 | 6.68 | 7.0 | 24.7 | 47.5 | 7E−25 | gi|549586|sp|P26615|UbiD | 3-Octaprenyl-4-hydroxybenzoate carboxy lyase |
| Orf8 | F5 | → | 11819–12403 | 194 | 21.262 | 20 | 6.97 | 8.0 | 60.3 | 86.8 | 5E−56 | gi|2507150|sp|P09550|UbiX | 3-Octaprenyl-4-hydroxybenzoate carboxy lyase |
| Orf9 | (11) | → | 12414–12845 | 143 | 15.209 | 17 | 7.60 | 9.0 | 40.0 | 64.8 | 8E−13 | gi|2622617 (AE000910) | Hypothetical protein |
| Orf10 | (10) | → | 12884–13432 | 182 | 19.936 | 18 | 5.76 | 5.5 | 36.1 | 62.7 | 2E−9 | gi|2129134|pir|D64443| | Mutator protein MutT |
The deduced properties of the gene products of orf1 to orf11 (as determined by diverse computer programs) are compared with the properties of phenol-induced proteins detected in two-dimensional gels.
Numbers in parentheses refer to the numbers of the spots of corresponding proteins in a two-dimensional gel in Fig. 2.
Dir, direction of transcription. The range is of the nucleotides of the corresponding gene.
Mol mass, molecular mass; pI, isoelectric point; Exp., experimentally determined (two-dimensional gels); aa, amino acids. The length is the number of amino acids of the corresponding translated gene product.
Overall similarity and identity of the amino acid sequence to proteins in the databases are given. % Identity, the percentage of amino acids that are identical between the two proteins; % Similarity, the percentage of amino acids that are identical or conserved between the two proteins; E value, Expect value. The Expect value estimates the statistical significance of the match, specifying the number of matches, with a given score, that are expected in a search of a database of this size absolutely by chance.
Given are references of similar proteins. Database accession numbers are in parentheses.
Ten ORFs are transcribed in the same direction. The first six ORFs (orf1 to orf6) are separated by less than 65 bp and amount to 7,210 bp. This cluster of putative genes is followed by a 658-bp noncoding region whose transcript contains two putative hairpin mRNA secondary structures. Another cluster of four putative genes with fewer than 40 bp of intergenic regions amounts to 2,702 bp. Downstream of orf10, 840 bp which did not code for proteins were sequenced. Upstream of orf1 and transcribed in the opposite direction, another putative gene was found, orf11, which was separated by 428 bp from orf1.
Comparison of deduced amino acid sequences of ORFs 1 to 11 with the experimentally determined N-terminal and internal amino acid sequences of phenol-induced proteins revealed that the following genes code for known proteins: orf1 for F3, orf4 for F2, orf5 for F4, orf6 for F1, and orf8 for F5 (Fig. 4). The predicted molecular masses agree reasonably well with the experimentally determined masses (Table 1). The similarities of the gene products possibly involved in anaerobic phenol metabolism with other proteins in the databases, comparison of deduced and experimentally determined sequences, and some extrapolated parameters of the proteins compared to those of the phenol-induced protein spots are summarized in Table 1.
Protein F3 (encoded by orf1) and the orf2 gene product show similarity to parts of phosphoenolpyruvate synthase (PEP synthase) of E. coli. The reaction catalyzed by PEP synthase is shown in Fig. 5. The C-terminal half of F3 is similar to the central part of PEP synthases and contains a highly conserved histidine (amino acid 569) which is known to become phosphorylated in the PEP synthase reaction (histidine 421 in the central part of PEP synthase of E. coli). The orf2 gene product shows similarity to the N-terminal domain of PEP synthase. In E. coli, this domain is probably involved in ATP binding. We propose that ORFs 1 and 2 code for two proteins belonging to the phenylphosphate synthase system, whereby phenol ultimately becomes phosphorylated. This proposal seems plausible, considering the structural similarity between the enol form of pyruvate and phenol, respectively.
ORFs 4, 6, and 7 code for F2, F1, and a not-yet-identified protein, respectively. Sequence alignment shows that each of these gene products is overall similar to the ubiD gene product of E. coli, 3-octaprenyl-4-hydroxybenzoate carboxy lyase. This enzyme is involved in the biosynthesis of ubiquinone; the reaction catalyzed is shown in Fig. 4. orf8 codes for F5 and shows high similarity to ubiX, a gene which codes for an isoenzyme of 3-octaprenyl-4-hydroxybenzoate carboxy lyase (UbiD). UbiX and F5 were also similar to phenylacrylic acid decarboxylase from Saccharomyces cerevisiae. An alignment of a conserved amino acid sequence motif showed that all three proteins have a common E-X-P motif. We propose that the proteins encoded by orf4 and orf6 to orf8 are involved in the carboxylation of phenylphosphate or similar phenolic compounds; this proposal is based on the similarity of the known reactions of the similar proteins, decarboxylation of 3-octaprenyl-4-hydroxybenzoate or phenylacrylic acid, to the phenylphosphate carboxylase reaction.
The function of four gene products is enigmatic. Orf3 showed similarity to a putative inosine-5′-monophosphate dehydrogenase of Methanobacterium thermautotrophicum. Orf5 (F4) and Orf9 showed similarities to hypothetical and conserved proteins whose functions are unknown. Orf10 has high similarity to the putative Mutator T protein (MutT) of Methanococcus jannaschii.
orf11 codes for a protein that shows strong similarity to the DmpR gene regulator of Pseudomonas putida, which regulates expression of genes involved in aerobic metabolism of phenolic compounds (41, 57, 58). Therefore, we assume that Orf11 is the regulator of anaerobic phenol metabolism.
Expression of F1 to F5 proteins in E. coli.
Five of the 11 ORFs were identified as genes coding for F1 to F5. However, there was no proof that the other putative genes were expressed as well. Therefore, several DNA fragments were ligated into pBluescript KS under the control of the heat-inducible T7 polymerase promoter and transformed into E. coli K38, and expression was induced. The following DNA fragments were used (compare with Fig. 5): 3.7-kbp PstI fragment containing orf1 (which codes for the F3 protein) and orf2; a 2.7-kbp BamHI fragment containing orf3 and orf4 (which codes for F2); a 4.0-kbp BamHI fragment containing orf5 (which codes for F4), orf6 (which codes for F1), and orf7; and a 5.25-kbp EcoRI fragment containing the 3′ end of orf6, the intergenic region between orf6 and orf7, and the complete sequences of orf7, orf8 (coding for F5), orf9, and orf10. The proteins were separated by SDS-PAGE and localized by autoradiography (Fig. 6).
FIG. 6.

Expression of ORFs 1 to 6 in E. coli (T7 polymerase experiment). A volume of 25 μl was loaded onto each lane. Lanes 1, 4, and 7, marker proteins; lane 2, 3.7-kbp PstI fragment containing ORF1 and ORF2; lane 3, 2.7-kbp BamHI fragment containing ORF3 and ORF4; lane 5, 5.25-kbp EcoRI fragment containing ORF7, ORF8, ORF9, and ORF10; lane 6, 4.0-kbp BamHI fragment containing ORF5, ORF6, and ORF7.
orf1 and orf2 were obviously transcribed by the T7 polymerase. On the SDS-polyacrylamide gel, the resulting proteins F3 and Orf2 migrated at apparent molecular masses of 67 and 41 kDa, respectively, which agreed reasonably well with the predicted molecular masses. orf3 and orf4 were also both transcribed by the T7 polymerase. F2 (encoded by orf4) was strongly induced and had an apparent molecular mass of approximately 58 kDa. This was slightly larger than expected according to the nucleotide sequence (52 kDa), but agreed with the apparent molecular mass of F2 prepared from T. aromatica. In contrast, expression of orf3 was not strongly induced and ORF3 was hardly visible on the autoradiogram. However, the apparent molecular mass of 24 kDa agreed with the predicted molecular mass. The experiment with the 4.0-kbp BamHI fragment containing orf5 to orf7 showed the expression of only two proteins. Their apparent sizes of approximately 19 and 60 kDa corresponded to those of the phenol-induced proteins F4 and F1, respectively. As in cell extract, F1 ran with a molecular mass larger than that expected from the nucleotide sequence (54 kDa). No expression was observed for orf7, which would have resulted in a 39-kDa protein. The putative secondary mRNA structures between orf6 and orf7 may have prevented translation of orf7.
Using the 5.25-kbp EcoRI fragment, none of the ORFs from orf7 to orf10 were expressed. This may be due to the fact that the secondary structures downstream of orf6 may have prevented transcription of orf7 to orf10. Despite several attempts, we did not succeed in subcloning the DNA sequence of orf7 to orf10 without the putative secondary structures and thus could not express these genes. However, expression of orf8 is clearly induced in T. aromatica in the presence of phenol, as F5 is one of the phenol-induced proteins. This suggests that the second gene cluster consisting of orf7 to orf10 indeed codes for structural phenol-induced proteins.
Genomic Southern hybridization of chromosomal DNA of T. aromatica and other bacterial strains.
Genetic work with T. aromatica is troublesome because of poor growth on agar plates. To obtain data on a possible correlation of the phenol-induced genes and their function in anaerobic phenol metabolism, a 32P-labeled 750-bp PCR product (F2H/F2t43R) that corresponded to the 5′ part of the gene coding for F2 (orf4) was used as a probe for Southern hybridization. The probe should only hybridize with the DNA of those strains that were closely related to T. aromatica and able to metabolize phenol. The 16S rRNA of Thauera sp. strain S100 is 100% identical and that of strain SP is more than 98.9% identical to that of T. aromatica type strain K172 (E. Stackebrandt and G. Fuchs, unpublished results). The phenol-induced genes should not be present in strains that could not have been shown to grow on phenol, like Thauera sp. strain LG356 (which has 100% 16S rRNA sequence identity with strain SP) and Azoarcus evansii type strain KB740.
The result of the genomic Southern experiment was surprising (Fig. 7). DNA of all organisms hybridized with the labeled probe. Moreover, the sizes of the hybridizing fragments seemed to be very similar in most of the organisms. In all organisms, the probe hybridized with the undigested DNA and/or fragments larger than 15 kbp. The experimentally determined sizes of hybridizing fragments of the digested chromosomal DNA of T. aromatica were consistent with the expected sizes. Only after EcoRI digestion did the probe hybridize with two fragments instead of one, as expected. The strongly hybridizing fragment of about 5 kbp corresponded to the expected one, and the 7-kbp fragment hybridized only weakly, which may be due to unspecific binding of the probe. T. aromatica type strain K172 and strain S100 showed the same sizes and patterns of hybridizing fragments; strain SP differed slightly in its hybridization pattern. Hybridization of strain SP DNA was expected, because this organism is also able to grow on phenol. Slight differences in the hybridization patterns may be explained with slight differences in the genome sequences of the three organisms.
FIG. 7.

Southern blot of genomic DNA of T. aromatica strains (type strain K172, strains S100, SP, and LG356) and A. evansii type strain KB740. The DNA was restricted with BamHI and EcoRI. After separation with agarose gel electrophoresis, DNA fragments were blotted onto a nylon membrane and hybridized with a 32P-labeled probe for the 5′-terminal part of the gene of F2. Arrows indicate the positions of the marker DNA, whose sizes are given in kilobase pairs (kbp).
Unexpectedly, chromosomal DNA of strain LG356 also hybridized. Moreover, the hybridization pattern of strains LG356 and SP were quite different, although these organisms have 100% identical 16S rRNA sequences. Even A. evansii DNA hybridized; however, the hybridization pattern was totally different from those of the Thauera strains, which may be due to major differences in the genome sequences of Thauera and Azoarcus species. The results of the genomic Southern experiment showed that there is no simple correlation between the presence of the F2 gene and reported growth on phenol.
DISCUSSION
Genes and gene organization.
Eleven ORFs were cloned and sequenced, and up to 11 phenol-induced proteins were identified by two-dimensional gel electrophoresis. Table 1 summarizes the deduced sizes and isoelectric points of these gene products. Five of the genes coded for the identified phenol-induced proteins F1 to F5. Comparison of the experimentally determined and deduced values showed that the molecular masses agreed reasonably well with the prediction.
However, the deduced pI values (as determined by diverse computer programs) did not fit exactly to those estimated experimentally. In general, the deduced pI values were 0.5 to 1.0 U lower than the experimentally determined values. This discrepancy may be due to inaccurate determination of the pI values, which were extrapolated assuming an immobilized linear pH gradient of 3 to 10. Six phenol-induced protein spots could not be directly attributed to the corresponding genes by purification or extraction mainly due to the poor separation of the proteins. Although the estimated molecular masses and pI values fit reasonably well with those predicted from ORFs 2, 3, 7, and 9 to 11, the identification is very preliminary and may be incorrect in some cases. For example, protein spot no. 7 may essentially fit to the gene product of orf11, which encodes the putative regulator protein of phenol metabolism, but it is unlikely that a regulator protein is induced in such amounts.
Ten of the 11 genes are transcribed in the same direction and are organized in two gene clusters. To underline the correlation between the genes and the induced proteins, the genes were cloned under the control of the T7 polymerase promoter and were expressed in E. coli. All genes of the first gene cluster (orf1 to orf6) were transcribed and expressed. Probably due to the possible RNA hairpin secondary structures between the two clusters, we did not observe expression of the second cluster. However, the fact that the induced protein F5 (which is encoded by orf8 of the second gene cluster) is expressed in the presence of phenol in T. aromatica leaves no doubt that the second gene cluster is expressed as well.
Possible functional assignment of the genes.
The two essential steps in phenol metabolism are the phosphorylation of phenol to phenylphosphate and the subsequent carboxylation of phenylphosphate to 4-hydroxybenzoate. Two gene products showed similarity to PEP synthase and four gene products showed similarity to 3-octaprenyl-4-hydroxybenzoate carboxy lyase, respectively, both of E. coli. Because of the similar chemistry involved in the respective reactions, we suggest that these genes are coding for phenol-phosphorylating and phenylphosphate-carboxylating enzymes, respectively.
Orf1 and Orf2, homologues of PEP synthase.
Two ORFs, orf1 (coding for F3) and orf2, code for proteins of 70.2 and 40.4 kDa with similarity to the central and the N-terminal parts, respectively, of PEP synthase. Thus, the gene products appear to belong to the family of the structurally and functionally related PEP-utilizing enzymes, which comprises three groups of proteins (19, 20): (i) PEP synthase (or pyruvate:water dikinase [EC 2.7.9.2]), (ii) pyruvate:orthophosphate dikinase (EC 2.7.9.1), and (iii) PEP-protein phosphotransferase (EC 2.7.3.9), which is enzyme I of the PEP-dependent phosphotransferase system. Two sequence signatures are shared by all these proteins and by no other proteins in the current protein databases. The first sequence signature (PROSITE, accession no. PS00370) contains an active-site histidine residue that mediates phosphoryl transfer. The second sequence signature (PROSITE, accession no. PS00742) is a conserved region, the exact role of which is unknown. The functional similarity of the enzymes lies in the transfer of a phosphoryl group via a phospho-histidine intermediate and in the utilization of PEP or pyruvate as a substrate; the corresponding active sites are located on independently folded domains. The E. coli PEP synthase (175 kDa) is a dissociable dimer of identical subunits (87.4 kDa), and histidine residue 442 has been identified as the phosphorylation site (6, 42). In the thermophilic archaebacterium Staphylothermus marinus, PEP synthase is a homomultimeric enzyme complex whose subunits have a similar structure as the subunits of the homodimeric E. coli enzyme (20).
T. aromatica Orf1 (protein F3) shows 54% similarity to the central part of PEP synthase of E. coli and carries the conserved sequence motif of PEP-utilizing enzymes (PROSITE, accession no. PS00370) that contains the active-site histidine residue. The PEP synthase of B. subtilis contains the (putative) active-site histidine at the C-terminal part of the protein, as does F3. The N-terminal part of F3 shows no similarity to any region of the known PEP synthase sequences or to other sequences in the databases. Orf2 showed 60% similarity to the N-terminal part of PEP synthase of E. coli. We believe that the gene products of orf1 and orf2 together form an active enzyme complex responsible for phosphorylation of phenol. The phosphoryl donor for this reaction remains unknown, although a number of potential cosubstrates have been tested (33).
Orf4, -6, -7, and -8, homologues of 3-octaprenyl-4-hydroxybenzoate carboxy lyase.
The gene products of orf4 (F2), orf6 (F1), and orf7 showed 47 to 63.8% similarity to UbiD (55.2 kDa) (Table 1 and Fig. 8), and the product of orf8 (F5) showed 86.8% similarity to UbiX (20.7 kDa). UbiD catalyzes the third reaction in ubiquinone biosynthesis, namely, the decarboxylation of 3-octaprenyl-4-hydroxybenzoate to 3-octaprenylphenol (Fig. 5). The gene ubiX encodes an isoenzyme of UbiD that catalyzes the same reaction. So far, only a few biochemical data are available on both enzymes. The holoenzyme UbiD of E. coli has a molecular mass of 340 kDa, and its activity is dependent on the presence of Mn2+ and an unidentified, heat-stable factor with a mass of <10 kDa (36). Moreover, the activity was increased by adding membrane preparations or phospholipids, indicating that the enzyme normally functions in association with the membrane.
FIG. 8.

Alignment of the conserved regions of F1, F2, and Orf7 possibly involved in phenol metabolism showing similarity to 3-octaprenyl-4-hydroxybenzoate carboxy lyase of E. coli and to 4-hydroxybenzoate decarboxylase of C. hydroxybenzoicum (A) and regions of conserved amino acid sequences in several decarboxylases (B). (A) Boxes with like colors (black and grey) represent similar domains. UbiD, 3-octaprenyl-4-hydroxybenzoate carboxy lyase; 4-OHBDC, 4-hydroxybenzoate decarboxylase; F1, F2, and Orf7, phenol-induced proteins of T. aromatica. (B) PDC, p-coumaric acid decarboxylase; PAD, phenylacrylic acid decarboxylase; FDC, ferulate decarboxylase. Bold letters represent amino acids that are identical in more than 50% of the sequences. The numbers refer to the first and last positions of the motif in the corresponding sequence. E.c., E. coli; T.a., T. aromatica; C.h., C. hydroxybenzoicum; S.c., Saccharomyces cerevisiae; L.p., Lactobacillus plantarum; B.s., Bacillus subtilis; B.p., Bacillus pumilus.
We assume that some or all of those four genes code for enzymes involved in carboxylation of phenylphosphate to 4-hydroxybenzoate because decarboxylation of 3-octaprenyl-4-hydroxybenzoate to 3-octaprenyl-phenol can be compared to the reverse reaction of phenylphosphate carboxylase. Interestingly, the four genes of T. aromatica are distributed on two different clusters of genes. F1 and F2 are encoded in one cluster together with the genes possibly encoding the phenol-phosphorylating enzymes (F3 and Orf2), whereas Orf7 and F5 are encoded in a second gene cluster where no phosphorylating enzymes seem to be encoded (Fig. 5). Both clusters are expressed in the presence of the substrate phenol, as gene products of both were identified as strongly phenol-induced proteins.
Like the E. coli enzyme UbiD, the phenylphosphate carboxylase and isotope exchange activities were both dependent on Mn2+. Phenylphosphate carboxylase of T. aromatica could be purified by gel filtration chromatography with little loss of activity. This indicated that, unlike 3-octaprenyl-4-hydroxybenzoate decarboxylase, no low-molecular-weight factor is required for enzyme activity.
There are recent reports that enzymes involved in anaerobic degradation of vanillic acid (4-hydroxy-3-methoxybenzoic acid) (17), 3,4-dihydroxybenzoic acid (28a) and 4-hydroxybenzoic acid (30) show similarity to the UbiD/UbiX system of E. coli, and this similarity is extended to the phenol system in T. aromatica (Fig. 8). The vdcBCD gene cluster in Streptomyces sp. strain D7 codes for proteins specifically involved in vanillic acid decarboxylation to 2-methoxyphenol (guaiacol). VdcC is similar to UbiD in E. coli and to Orf4, -6, and -7 in T. aromatica. VdcB is similar to UbiX and phenylacrylic acid decarboxylase of S. cerevisiae and to Orf8 of T. aromatica. VdcD shows similarity to an unknown protein of Sphingomonas and YclD (function not known) of Bacillus subtilis; in T. aromatica, no gene product similar to VdcD was found. Wiegel and coworkers identified the gene coding for the (reversible) 4-hydroxybenzoate decarboxylase (54.4 kDa) in Clostridium hydroxybenzoicum (30). This enzyme was similar to the three UbiD-like gene products of T. aromatica and to UbiD from E. coli (53.4 kDa) (17). Two other ORFs, orf2 and orf3, downstream of the gene for the 4-hydroxybenzoate decarboxylase were similar to the vdcDB genes of Streptomyces sp.; however, orf3 seems not to be complete. A sequence motif with highly conserved amino acids was also found in three decarboxylases from Lactobacillus plantarum (15), B. subtilis (16), and Bacillus pumilus (66). In all listed protein sequences, an E-X-P motif is conserved and may therefore play a role in substrate binding or catalysis of (hydroxy-) arylic acid decarboxylation or carboxylation of the corresponding phenolic compounds.
Obviously, in the Clostridium, Streptomyces, and Thauera species, the genes coding for UbiD-like decarboxylases are arranged in one or two clusters together with ubiX homologues. In E. coli, however, ubiD and ubiX are widely separated on the chromosome. There remain open questions about T. aromatica: why there are four genes with similarity to (reversible) decarboxylases organized in two clusters and why both clusters are transcribed in the presence of phenol. Perhaps phenol acts as a gratuitous inducer of the second gene cluster that may play a role in the metabolism of other phenolic compounds (for a classical example of gratuitous induction, see reference 5).
Orf10, homologue of a mutator T protein.
The gene product of orf10 showed approximately 60% similarity to the mutator protein MutT of M. jannaschii (Table 1 and Fig. 9). Mutator proteins are enzymes involved in DNA metabolism which are widespread in prokaryotes and eukaryotes. The 15 members of the MutT family of proteins are believed to act in nucleoside phosphate metabolic pathways. 8-Oxo-7,8-dGTP (8-oxo-dGTP) is formed in the nucleotide pool of a cell during normal cellular metabolism and causes AT-to-GC transversion mutations when it is incorporated into DNA (7, 22, 53). The MutT protein of E. coli and related mammalian enzymes specifically hydrolyze 8-oxo-dGTP to 8-oxo-dGMP and PPi, thereby preventing the occurrence of transversion mutation (38, 43). Members of this class of enzymes require two divalent cations per active site for activity, one coordinated by the enzyme and the other by the enzyme-bound nucleoside triphosphate (39). The substrate of proteins of the MutT family varies from 8-oxo-dGTP and diphosphoinositol polyphosphate (49) to adenosine(5′)triphospho(5′)adenosine, ADP-ribose, and NADH (43). One can only speculate which role Orf10 plays in the anaerobic metabolism of phenol in T. aromatica, e.g., in phenol activation.
FIG. 9.

Alignment of highly conserved regions of a protein of T. aromatica encoded by orf10 possibly involved in anaerobic phenol metabolism showing similarity to putative 8-oxo-dGTPase/ADP-ribose pyrophosphatase (MutT) of M. jannaschii. In parentheses are given the amino acid regions of the domains. !, either I or V; %, either F or Y; #, either N, D, Q, or E.
Orf11, a putative regulator protein.
One ORF located 5′ of the two gene clusters and transcribed in the opposite direction codes for a protein with 72% similarity to the regulator protein DmpR of P. putida CF600, which regulates aerobic (dimethyl)phenol metabolism (Fig. 10). DmpR is a member of the NtrC family of transcriptional activators and activates transcription of the dmp operon, which contains all 15 structural genes needed for the degradation of (dimethyl)phenol, in response to aromatic effector compounds (58). These effectors are phenol, o-cresol, m-cresol, p-cresol, and 3,4-dimethylphenol, and also ethylphenol, catechol, and 3- and 4-methylcatechol. MopR, the regulator of phenol degradation in Acinetobacter calcoaceticus NCIB8250, also belongs to the NtrC family (51). Like all regulators of the NtrC family, Orf11 contains several conserved protein domains (40). The highly conserved central (C) domain probably binds and hydrolyzes ATP and promotes open-complex formation with RNA polymerase. The C-terminal (D) domain is expected to bind to the corresponding target DNA sequence (“enhancer”). The highly variable sensory domain is joined to the C domain by short interdomain linker B. NtrC-like regulators sense their respective signals by direct binding of the effector to the A domain (56). Effector binding to the A domain then leads to release of the C domain (21, 56), thus permitting the binding of ATP followed by an ATP-driven cycle of multimerization and demultimerization of the regulator at the enhancer sites (45). This supports open-complex formation of ς54-containing RNA polymerase with the promoter (65). We propose that Orf11 controls the expression of the first or both gene clusters by a similar mechanism and acts as a transcriptional activator, when phenol or a similar compound plus ATP is present.
FIG. 10.

Orf11 of T. aromatica showing similarity to a regulator protein in aerobic (dimethyl)phenol metabolism (DmpR) of P. putida: comparison of the domain structures of DmpR of P. putida CF600 and Orf11 of T. aromatica (A) and alignment of the conserved amino acid regions (B). Structures were adapted from reference 35. The domains A, C, and D and the hinge region B of DmpR are indicated. In parentheses are given the amino acid regions of the conserved regions. !, either I or V; $, either L or M; %, either F or Y; #, either N, D, Q, or E.
In P. putida strains P35X and CF600, the difference in the spectrum of aromatic growth substrates seems to be due to the different specificity of effector binding to the regulator protein rather than due to the different substrate specificity of the catabolic enzymes (58). However, this hypothesis has yet to be proven. All DmpR-like effector proteins (>20 known) mediate their action through a single binding site within the A domain (44). Depending on the effector, this results in differential effects on ATPase activity of the C domain and its productive coupling to the transcriptional apparatus.
Promoter region.
Transcriptional activators of the NtrC-like family usually regulate transcription from ς54-dependent promoters, as was shown in P. putida for the positive regulator DmpR (57). The structural dmp genes of the aerobic phenol degradation pathway are clustered in a single operon that lies downstream of a nif/ntr-like promoter sequence. Upon inspection of the first gene cluster in T. aromatica, a similar nif/ntr-like promoter sequence was indeed found 5′ of orf1 (Fig. 11). Primer extension analysis has to be performed to show whether transcription really starts at the expected site. So far, nothing is known about promoter sequences in T. aromatica, so the −24 and −12 consensus sequences are speculative. Upstream of the second gene cluster, no nif/ntr-like promoter sequences were found. All ς54-dependent promoters analyzed so far are regulated by positive transcriptional activators that usually bind to specific DNA sequences (“enhancer sequences,” mostly inverted repeats) located relatively far upstream of the promoter (100 to 200 bp) (31).
FIG. 11.

(A) Putative nif/ntr-like promoter sequences upstream of the first gene cluster in T. aromatica. The putative −24 and −12 promoter elements (open boxes) and ribosome binding site (RBS; underlined) are indicated. The ATG initiation codon is in bold. The vertical arrow indicates the putative start site of transcription. The numbers at the beginning and at the end give the positions in the whole sequence of 14,272 bp. (B) Nucleotide sequences of nif- and ntr-like promoters. a, The transcriptional start sites of these three promoters has not yet been reported, and the promoter location is assumed on the basis of sequence similarity only. y, C or T; w, A or T.
Regulation of transcription.
We suggest that phenol acts as an effector of a transcriptional activator protein, as there is an ntr-like regulator gene present upstream of the two gene clusters and a nif/ntr-like promoter sequence upstream of the first cluster of genes. In addition to the regulation by phenol, there may be additional catabolite repression, but no experimental data are available. As T. aromatica could not be shown to metabolize phenol in the presence of oxygen, it has to be shown whether O2 represses the induction of transcription of the genes involved in phenol metabolism.
Presence of genes possibly involved in anaerobic phenol metabolism in different strains of T. aromatica and various Azoarcus species.
So far, it has not been possible to unequivocally correlate genes coding for phenol-induced proteins and their function. Southern hybridization of a labeled probe of orf4 (coding for F2) with chromosomal DNA of different strains seemed to indicate that all tested Thauera and Azoarcus strains have a gene similar to orf4, whether or not they have been shown to metabolize phenol. One plausible assumption is that all strains are able to utilize phenol, but so far the appropriate growth conditions have not been applied. Alternatively, rearrangement and deletion may be the reason why some Thauera and Azoarcus strains are able to metabolize phenol and some are not.
Related carboxylases acting on aromatic compounds.
Carboxylation of the aromatic ring is widespread among anaerobic microorganisms, but the respective enzymes have not been studied yet. The initial reaction in anaerobic o-cresol (2-methylphenol) metabolism in denitrifying Azoarcus sp. strain U 120 (formerly a Paracoccus strain) (48) is carboxylation in para-position to 3-methyl-4-hydroxybenzoate. Sulfate-reducing and methanogenic bacteria have been reported to carboxylate m-cresol (3-methylphenol) in para-position to the phenolic hydroxy group to 2-methyl-4-hydroxybenzoate (46, 47). A Desulfococcus sp. carboxylated hydroquinone (1,4-dihydroxybenzene) to gentisate (2,5-dihydroxybenzoate) (27). Some sulfate-reducing bacteria carboxylate catechol (1,2-dihydroxybenzene) to protocatechuate (3,4-dihydroxybenzoate), whereas phenol is not metabolized (26). Aniline is carboxylated to 4-aminobenzoate in Desulfobacterium anilini (52). An aerobic ortho-carboxylation of aniline to 2-aminobenzoic acid was reported for Rhodococcus erythropolis (2). In sulfate-reducing consortia, the initial reaction in the anaerobic metabolism of naphthalene is carboxylation specifically at C2 to form 2-naphthalene carboxylic acid; phenanthrene carboxylation leads to phenanthrene carboxylic acid, and the carboxylation site is unknown (69). Growth on all of these substrates is likely to be CO2 dependent.
ACKNOWLEDGMENTS
This work was supported by the Deutsche Forschungsgemeinschaft, the Fonds der Chemischen Industrie, and cooperation with E. I. DuPont de Nemours.
We thank Hans Heider for helpful suggestions and critical reading of the manuscript. Thanks are also due to Hermann Schägger, Frankfurt, Germany, for amino acid sequencing of some of the proteins, to Juliane Alt-Mörbe, DNA-Analytik Freiburg, for DNA sequencing, to Christa Ebenau-Jehle and Victor Gadon for help in growing cells, and to Sima Sariaslani, Heinz Hefter, and coworkers from DuPont for their support.
REFERENCES
- 1.Anders H-J, Kaetzke A, Kämpfer P, Ludwig W, Fuchs G. Taxonomic position of aromatic-degrading denitrifying pseudomonad strains K 172 and KB 740 and their description as new members of the genera Thauera, as Thauera aromatica sp. nov., and Azoarcus, as Azoarcus evansii sp. nov., respectively, members of the beta subclass of the Proteobacteria. Int J Syst Bacteriol. 1995;45:327–333. doi: 10.1099/00207713-45-2-327. [DOI] [PubMed] [Google Scholar]
- 2.Aoki K, Uemori T, Shinke R, Nishira H. Further characterization of bacterial production of anthranilic acid from aniline. Agric Biol Chem. 1985;49:1151–1158. [Google Scholar]
- 3.Aresta M, Quaranta E, Liberio R, Dileo C, Tommasi I. Enzymatic synthesis of 4-hydroxybenzoic acid from phenol and CO2: the first example of a biotechnological application of a carboxylase enzyme. Tetrahedron. 1998;54:8841–8846. [Google Scholar]
- 4.Bak F, Widdel F. Anaerobic degradation of phenol and phenol derivatives by Desulfobacterium phenolicum sp. nov. Arch Microbiol. 1986;146:177–180. [Google Scholar]
- 5.Barnsley E A. Role and regulation of the ortho and meta pathways of catechol metabolism in pseudomonads metabolizing naphthalene and salicylate. J Bacteriol. 1976;125:404–408. doi: 10.1128/jb.125.2.404-408.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berman K M, Cohn M. Phosphoenolpyruvate synthetase of Escherichia coli. Purification, some properties, and the role of divalent metal ions. J Biol Chem. 1970;245:5309–5318. [PubMed] [Google Scholar]
- 7.Bhatnagar S K, Bullions L C, Bessman M J. Characterization of the MutT nucleoside triphosphatase of Escherichia coli. J Biol Chem. 1991;266:9050–9054. [PubMed] [Google Scholar]
- 8.Biegert T, Altenschmidt U, Eckerskorn C, Fuchs G. Enzymes of anaerobic metabolism of phenolic compounds. 4-Hydroxybenzoate-CoA ligase from a denitrifying Pseudomonas species. Eur J Biochem. 1993;213:555–561. doi: 10.1111/j.1432-1033.1993.tb17794.x. [DOI] [PubMed] [Google Scholar]
- 9.Boll M, Albracht S S, Fuchs G. Benzoyl-CoA reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism. A study of adenosinetriphosphatase activity, ATP stoichiometry of the reaction and EPR properties of the enzyme. Eur J Biochem. 1997;244:840–851. doi: 10.1111/j.1432-1033.1997.00840.x. [DOI] [PubMed] [Google Scholar]
- 10.Boll M, Fuchs G. Benzoyl-coenzyme A reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism. ATP dependence of the reaction, purification and some properties of the enzyme from Thauera aromatica strain K172. Eur J Biochem. 1995;234:921–933. doi: 10.1111/j.1432-1033.1995.921_a.x. [DOI] [PubMed] [Google Scholar]
- 11.Brackmann R, Fuchs G. Enzymes of anaerobic metabolism of phenolic compounds. 4-Hydroxybenzoyl-CoA reductase (dehydroxylating) from a denitrifying Pseudomonas species. Eur J Biochem. 1993;213:563–571. doi: 10.1111/j.1432-1033.1993.tb17795.x. [DOI] [PubMed] [Google Scholar]
- 12.Braun K, Gibson D T. Anaerobic degradation of 2-aminobenzoate (anthranilic acid) by denitrifying bacteria. Appl Environ Microbiol. 1984;48:102–107. doi: 10.1128/aem.48.1.102-107.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Breese K, Boll M, Alt-Mörbe J, Schägger H, Fuchs G. Genes coding for the benzoyl-CoA pathway of anaerobic aromatic metabolism in the bacterium Thauera aromatica. Eur J Biochem. 1998;256:148–154. doi: 10.1046/j.1432-1327.1998.2560148.x. [DOI] [PubMed] [Google Scholar]
- 14.Breese K, Fuchs G. 4-Hydroxybenzoyl-CoA reductase (dehydroxylating) from the denitrifying bacterium Thauera aromatica—prosthetic groups, electron donor, and genes of a member of the molybdenum-flavin-iron-sulfur proteins. Eur J Biochem. 1998;251:916–923. doi: 10.1046/j.1432-1327.1998.2510916.x. [DOI] [PubMed] [Google Scholar]
- 15.Cavin J F, Barthelmebs L, Divies C. Molecular characterization of an inducible p-coumaric acid decarboxylase from Lactobacillus plantarum: gene cloning, transcriptional analysis, overexpression in Escherichia coli, purification, and characterization. Appl Environ Microbiol. 1997;63:1939–1944. doi: 10.1128/aem.63.5.1939-1944.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cavin J F, Dartois V, Divies C. Gene cloning, transcriptional analysis, purification, and characterization of phenolic acid decarboxylase from Bacillus subtilis. Appl Environ Microbiol. 1998;64:1466–1471. doi: 10.1128/aem.64.4.1466-1471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chow K T, Pope M K, Davies J. Characterization of a vanillic acid non-oxidative decarboxylation gene cluster from Streptomyces sp. D7. Microbiology. 1999;145:2393–2403. doi: 10.1099/00221287-145-9-2393. [DOI] [PubMed] [Google Scholar]
- 18.Chung C T, Niemela S L, Miller R H. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc Natl Acad Sci USA. 1989;86:2172–2175. doi: 10.1073/pnas.86.7.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cicicopol C, Peters J, Kellermann J, Baumeister W. Primary structure of a multimeric protein, homologous to the PEP-utilizing enzyme family and isolated from a hyperthermophilic archaebacterium. FEBS Lett. 1994;356:345–350. doi: 10.1016/0014-5793(94)01304-7. [DOI] [PubMed] [Google Scholar]
- 20.Cicicopol C, Peters J, Lupas A, Cejka Z, Muller S A, Golbik R, Pfeifer K, Lilie H, Engel A, Baumeister W. Novel molecular architecture of the multimeric archaeal PEP-synthase homologue (MAPS) from Staphylothermus marinus. J Mol Biol. 1999;290:347–361. doi: 10.1006/jmbi.1999.2878. [DOI] [PubMed] [Google Scholar]
- 21.Fernàndez S, de Lorenzo V, Peréz-Martín J. Activation of the transcriptional activator XylR of Pseudomonas putida by release of repression between functional domains. Mol Microbiol. 1995;16:205–213. doi: 10.1111/j.1365-2958.1995.tb02293.x. [DOI] [PubMed] [Google Scholar]
- 22.Fowler R G, Schaaper R M. The role of the mutT gene of Escherichia coli in maintaining replication fidelity. FEMS Microbiol Rev. 1997;21:43–54. doi: 10.1111/j.1574-6976.1997.tb00344.x. [DOI] [PubMed] [Google Scholar]
- 23.Gallert C, Knoll G, Winter J. Anaerobic carboxylation of phenol to benzoate: use of deuterated phenols revealed carboxylation exclusively in the C4 position. Appl Microbiol Biotechnol. 1991;36:124–129. [Google Scholar]
- 24.Gallert V, Winter J. Comparison of 4-hydroxybenzoate decarboxylase and phenol carboxylase activities in crude extracts in a defined, 4-hydroxybenzoate and phenol-degrading anaerobic consortium. Appl Microbiol Biotechnol. 1992;37:119–124. [Google Scholar]
- 25.Görg A, Postel W, Günther S. The current state of two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis. 1988;9:531–546. doi: 10.1002/elps.1150090913. [DOI] [PubMed] [Google Scholar]
- 26.Gorny N, Schink B. Anaerobic degradation of catechol by Desulfobacterium sp. strain Cat2 proceeds via carboxylation to protocatechuate. Appl Environ Microbiol. 1994;60:3396–3400. doi: 10.1128/aem.60.9.3396-3400.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gorny N, Schink B. Complete anaerobic oxidation of hydroquinone by Desulfococcus sp. strain Hy5: indications of hydroquinone carboxylation to gentisate. Arch Microbiol. 1994;162:131–135. [Google Scholar]
- 28.Harwood C S, Burchhardt G, Herrmann H, Fuchs G. Anaerobic metabolism of aromatic compounds via the benzoyl-CoA pathway. FEMS Microbiol Rev. 1999;22:439–458. [Google Scholar]
- 28a.He Z, Wiegel J. Purification and characterization of an oxygen-sensitive, reversible 3,4-dihydroxybenzoate decarboxylase from Clostridium hydroxybenzoicum. J Bacteriol. 1996;778:3539–3543. doi: 10.1128/jb.178.12.3539-3543.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heider J, Boll M, Breese K, Breinig S, Ebenau-Jehle C, Feil U, Gad'on N, Laempe D, Leuthner B, Mohamed M E, Schneider S, Burchhardt G, Fuchs G. Differential induction of enzymes involved in anaerobic metabolism of aromatic compounds in the denitrifying bacterium Thauera aromatica. Arch Microbiol. 1998;170:120–131. doi: 10.1007/s002030050623. [DOI] [PubMed] [Google Scholar]
- 30.Huang J, He Z, Wiegel J. Cloning, characterization, and expression of a novel gene encoding a reversible 4-hydroxybenzoate decarboxylase from Clostridium hydroxybenzoicum. J Bacteriol. 1999;181:5119–5122. doi: 10.1128/jb.181.16.5119-5122.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kustu S, North A K, Weiss D S. Prokaryotic transcriptional enhancers and enhancer-binding proteins. Trends Biochem Sci. 1991;16:397–402. doi: 10.1016/0968-0004(91)90163-p. [DOI] [PubMed] [Google Scholar]
- 32.Lack A, Fuchs G. Carboxylation of phenylphosphate by phenol carboxylase, an enzyme system of anaerobic phenol metabolism. J Bacteriol. 1992;174:3629–3636. doi: 10.1128/jb.174.11.3629-3636.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lack A, Fuchs G. Evidence that phenol phosphorylation to phenylphosphate is the first step in anaerobic phenol metabolism in a denitrifying Pseudomonas sp. Arch Microbiol. 1994;161:132–139. doi: 10.1007/BF00276473. [DOI] [PubMed] [Google Scholar]
- 34.Lack A, Tommasi I, Aresta M, Fuchs G. Catalytic properties of phenol carboxylase. In vitro study of CO2: 4-hydroxybenzoate isotope exchange reaction. Eur J Biochem. 1991;197:473–479. doi: 10.1111/j.1432-1033.1991.tb15934.x. [DOI] [PubMed] [Google Scholar]
- 35.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 36.Leppik R A, Young I G, Gibson F. Membrane-associated reactions in ubiquinone biosynthesis in Escherichia coli. 3-Octaprenyl-4-hydroxybenzoate carboxy-lyase. Biochim Biophys Acta. 1976;436:800–810. doi: 10.1016/0005-2736(76)90407-7. [DOI] [PubMed] [Google Scholar]
- 37.Li T, Bisaillon J G, Villemur R, Letourneau L, Bernard K, Lepine F, Beaudet R. Isolation and characterization of a new bacterium carboxylating phenol to benzoic acid under anaerobic conditions. J Bacteriol. 1996;178:2551–2558. doi: 10.1128/jb.178.9.2551-2558.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin J, Abeygunawardana C, Frick D N, Bessman M J, Mildvan A S. Solution structure of the quaternary MutT-M2+-AMPCPP-M2+ complex and mechanism of its pyrophosphohydrolase action. Biochemistry. 1997;36:1199–1211. doi: 10.1021/bi962619c. [DOI] [PubMed] [Google Scholar]
- 39.Mildvan A S, Weber D J, Abeygunawardana C. Solution structure and mechanism of the MutT pyrophosphohydrolase. Adv Enzymol Relat Areas Mol Biol. 1999;73:183–207. doi: 10.1002/9780470123195.ch6. [DOI] [PubMed] [Google Scholar]
- 40.Morett E, Segovia L. The ς54 bacterial enhancer-binding protein family: mechanism of action and phylogenetic relationship of their functional domains. J Bacteriol. 1993;175:6067–6074. doi: 10.1128/jb.175.19.6067-6074.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muller C, Petruschka L, Cuypers H, Burchhardt G, Herrmann H. Carbon catabolite repression of phenol degradation in Pseudomonas putida is mediated by the inhibition of the activator protein PhlR. J Bacteriol. 1996;178:2030–2036. doi: 10.1128/jb.178.7.2030-2036.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Narindrasorasak S, Bridger W A. Phosphoenolypyruvate synthetase of Escherichia coli: molecular weight, subunit composition, and identification of phosphohistidine in phosphoenzyme intermediate. J Biol Chem. 1977;252:3121–3127. [PubMed] [Google Scholar]
- 43.O'Handley S F, Frick D N, Dunn C A, Bessman M J. Orf186 represents a new member of the Nudix hydrolases, active on adenosine(5′)triphospho(5′)adenosine, ADP-ribose, and NADH. J Biol Chem. 1998;273:3192–3197. doi: 10.1074/jbc.273.6.3192. [DOI] [PubMed] [Google Scholar]
- 44.O'Neill E, Sze C C, Shingler V. Novel effector control through modulation of a preexisting binding site of the aromatic-responsive ς(54)-dependent regulator DmpR. J Biol Chem. 1999;274:32425–32432. doi: 10.1074/jbc.274.45.32425. [DOI] [PubMed] [Google Scholar]
- 45.Peréz-Martín J, de Lorenzo V. ATP binding to the ς54-dependent activator XylR triggers a protein multimerization cycle catalyzed by UAS DNA. Cell. 1996;86:331–339. doi: 10.1016/s0092-8674(00)80104-x. [DOI] [PubMed] [Google Scholar]
- 46.Ramanand K, Suflita J M. Anaerobic degradation of m-cresol in anoxic aquifer slurries: carboxylation reactions in a sulfate-reducing bacterial enrichment. Appl Environ Microbiol. 1991;57:1689–1695. doi: 10.1128/aem.57.6.1689-1695.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roberts J, Fedorak P M, Hrudey S E. CO2 incorporation and 4-hydroxy-2-methylbenzoic acid formation during anaerobic metabolism of m-cresol by a methanogenic consortium. Appl Environ Microbiol. 1990;56:472–478. doi: 10.1128/aem.56.2.472-478.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rudolphi A, Tschech A, Fuchs G. Anaerobic degradation of cresols by denitrifying bacteria. Arch Microbiol. 1991;155:238–248. doi: 10.1007/BF00252207. [DOI] [PubMed] [Google Scholar]
- 49.Safrany S T, Caffrey J J, Yang X, Bembenek M E, Moyer M B, Burkhart W A, Shears S B. A novel context for the 'MutT' module, a guardian of cell integrity, in a diphosphoinositol polyphosphate phosphohydrolase. EMBO J. 1998;17:6599–6607. doi: 10.1093/emboj/17.22.6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 51.Schirmer F, Ehrt S, Hillen W. Expression, inducer spectrum, domain structure, and function of MopR, the regulator of phenol degradation in Acinetobacter calcoaceticus NCIB8250. J Bacteriol. 1997;179:1329–1336. doi: 10.1128/jb.179.4.1329-1336.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schnell S, Schink B. Anaerobic aniline degradation via reductive deamination of 4-aminobenzoyl CoA in Desulfobacterium anilini. Arch Microbiol. 1991;155:183–190. [Google Scholar]
- 53.Sekiguchi M. MutT-related error avoidance mechanism for DNA synthesis. Genes Cells. 1996;1:139–145. doi: 10.1046/j.1365-2443.1996.d01-232.x. [DOI] [PubMed] [Google Scholar]
- 54.Seyfried B, Tschech A, Fuchs G. Anaerobic degradation of phenylacetate and 4-hydroxyphenylacetate by denitrifying bacteria. Arch Microbiol. 1991;155:249–255. [Google Scholar]
- 55.Sharak-Genthner B R, Townsend G T, Chapman P J. para-Hydroxybenzoate as an intermediate in the anaerobic transformation of phenol to benzoate. FEMS Microbiol Lett. 1991;78:265–270. doi: 10.1016/0378-1097(91)90168-a. [DOI] [PubMed] [Google Scholar]
- 56.Shingler V. Signal sensing by ς54-dependent regulators: derepression as a control mechanism. Mol Microbiol. 1996;19:409–416. doi: 10.1046/j.1365-2958.1996.388920.x. [DOI] [PubMed] [Google Scholar]
- 57.Shingler V, Bartilson M, Moore T. Cloning and nucleotide sequence of the gene encoding the positive regulator (DmpR) of the phenol catabolic pathway encoded by pVI150 and identification of DmpR as a member of the NtrC family of transcriptional activators. J Bacteriol. 1993;175:1596–1604. doi: 10.1128/jb.175.6.1596-1604.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shingler V, Moore T. Sensing of aromatic compounds by the DmpR transcriptional activator of phenol-catabolizing Pseudomonas sp. strain CF600. J Bacteriol. 1994;176:1555–1560. doi: 10.1128/jb.176.6.1555-1560.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shinoda Y, Sakai Y, Ue M, Hiraishi A, Kato N. Isolation and characterization of a new denitrifying spirillum capable of anaerobic degradation of phenol. Appl Environ Microbiol. 2000;66:1286–1291. doi: 10.1128/aem.66.4.1286-1291.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Solyanikova I P, Golovleva L A. Phenol hydroxylases: an update. Biochemistry (Moscow) 1999;64:365–372. [PubMed] [Google Scholar]
- 61.Song B, Young L Y, Palleroni N J. Identification of denitrifier strain T1 as Thauera aromatica and proposal for emendation of the genus Thauera definition. Int J Syst Bacteriol. 1998;48:889–894. doi: 10.1099/00207713-48-3-889. [DOI] [PubMed] [Google Scholar]
- 62.Tschech A, Fuchs G. Anaerobic degradation of phenol by pure cultures of newly isolated denitrifying pseudomonads. Arch Microbiol. 1987;148:213–217. doi: 10.1007/BF00414814. [DOI] [PubMed] [Google Scholar]
- 63.Tschech A, Fuchs G. Anaerobic degradation of phenol via carboxylation to 4-hydroxybenzoate: in vitro study of isotope exchange between 14CO2 and 4-hydroxybenzoate. Arch Microbiol. 1989;152:594–599. [Google Scholar]
- 64.van Schie P M, Young L Y. Isolation and characterization of phenol-degrading denitrifying bacteria. Appl Environ Microbiol. 1998;64:2432–2438. doi: 10.1128/aem.64.7.2432-2438.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wedel A, Kustu S. The bacterial enhancer-binding protein NTRC is a molecular machine: ATP hydrolysis is coupled to transcriptional activation. Genes Dev. 1995;9:2042–2052. doi: 10.1101/gad.9.16.2042. [DOI] [PubMed] [Google Scholar]
- 66.Zago A, Degrassi G, Bruschi C V. Cloning, sequencing, and expression in Escherichia coli of the Bacillus pumilus gene for ferulic acid decarboxylase. Appl Environ Microbiol. 1995;61:4484–4486. doi: 10.1128/aem.61.12.4484-4486.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zehr B D, Savin T J, Hall R E. A one-step, low background Coomassie staining procedure for polyacrylamide gels. Anal Biochem. 1989;182:157–159. doi: 10.1016/0003-2697(89)90734-3. [DOI] [PubMed] [Google Scholar]
- 68.Zhang X, Wiegel J. The anaerobic degradation of 3-chloro-4-hydroxybenzoate in freshwater sediment proceeds via either chlorophenol or hydroxybenzoate to phenol and subsequently to benzoate. Appl Environ Microbiol. 1992;58:3580–3585. doi: 10.1128/aem.58.11.3580-3585.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang X, Young L Y. Carboxylation as an initial reaction in the anaerobic metabolism of naphthalene and phenanthrene by sulfidogenic consortia. Appl Environ Microbiol. 1997;63:4759–4764. doi: 10.1128/aem.63.12.4759-4764.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]