

Abstract
Objective
Patients with inflammatory arthritis (IA) associated with immune checkpoint inhibitor (ICI) treatment for cancer are typically seronegative for anti-cyclic citrullinated peptide (CCP) antibodies and rheumatoid factor, but little is known about the presence of other autoantibodies in this patient population. We investigated the prevalence and characteristics of anti-RA33 antibodies in patients with ICI-induced IA.
Methods
Anti-RA33 ELISAs were performed on sera from four groups of patients: 79 with ICI-induced IA, 52 with rheumatoid arthritis (RA), 35 treated with ICIs without IA during follow-up and 50 healthy controls. Anti-RA33 positivity and level, clinical and demographic data were compared across groups.
Results
Anti-RA33 antibodies were found in 9/79 (11.4%) patients with ICI-induced IA but in 0/35 patients treated with ICIs who did not develop IA (0%; p=0.04). Of the patients positive for anti-RA33, two had sera available from before ICI treatment; anti-RA33 antibodies were present in both pre-ICI treatments. In patients with RA, 7.7% were positive for anti-RA33 antibodies as were 2% of healthy controls. In ICI-induced IA, anti-RA33 antibodies were associated with anti-CCP antibodies (p=0.001). We found no statistically significant differences in other clinical characteristics in those with and without anti-RA33 antibodies.
Conclusions
Anti-RA33 antibodies are present in a subset of patients with ICI-induced IA, absent in other ICI-treated patients and may be a biomarker for developing IA. Additional studies evaluating serial samples before and after ICI treatment will further establish the temporal relationship of these antibodies to IA development.
Keywords: Arthritis, Autoimmunity, Rheumatoid Factor
Key messages.
What is already known on this topic
Patients with inflammatory arthritis (IA) due to immune checkpoint inhibitor (ICI) therapy have been primarily seronegative for anti-cyclic citrullinated peptide and rheumatoid factor, but studies have not evaluated the presence of other antibodies associated with early IA, like anti-RA33 antibodies, in this patient population.
What this study adds
The study showed over 11% of patients with ICI-induced IA had anti-RA33 antibodies, while none of the ICI-treated patients without IA had these antibodies; some patients had anti-RA33 antibodies prior to receiving ICI therapy.
How this study might affect research, practice or policy
If validated in future studies, anti-RA33 antibodies could be a biomarker for risk of developing ICI-induced IA.
Introduction
Immune checkpoint inhibitors (ICIs) improve survival across malignancies.1 ICIs target regulatory molecules such as cytotoxic lymphocyte antigen-4, programmed cell death protein 1 and programmed death ligand 1 and can cause excess immune activation leading to immune-related adverse events (irAEs).2 IrAEs may resemble rheumatic diseases such as inflammatory arthritis (IA), polymyalgia rheumatica, sicca syndrome and myositis.3 4
There are key differences, however, between rheumatic irAEs and traditional autoimmune diseases in clinical characteristics, treatment and biomarkers.5 ICI-induced IA is heterogeneous and can persist after cessation of ICI therapy.6–8 Imaging has shown synovitis, tenosynovitis, and erosions typical of rheumatoid arthritis (RA), but also enthesitis, enthesophytes, and axial inflammation more characteristic of spondyloarthritis.9–11 Greater than 90% of patients with ICI-induced IA lack traditional autoantibodies associated with RA and are negative for HLA-B27, a genetic marker for spondyloarthritis.8
Antibodies to heterogeneous nuclear ribonucleoprotein (hnRNP) A2/B1, termed anti-RA33 antibodies, have been described in RA, undifferentiated IA, systemic lupus erythematosus and mixed connective tissue disorder.12 These antibodies target a nuclear protein involved in mRNA splicing. A meta-analysis showed that anti-RA33 has high specificity (0.90) for diagnosing RA but low sensitivity (0.33).13 RA33 antibodies can be detected in patients with RA who are seronegative for rheumatoid factor (RF) and anti-cyclic citrullinated peptide (CCP) antibodies.14 Although the RA33 antigen can be targeted in its native or citrullinated form, antibodies that prefer the native form of hnRNP A2/B1 have been detected in early RA and in patients with low erosion scores on imaging.15 The presence of anti-RA33 antibodies targeting the native antigen in early IA and the paucity of antibodies to citrullinated antigens in patients with ICI-induced IA16 led us to hypothesise that antibodies to the native RA33 antigen could be present in ICI-induced IA.
Methods
Inclusion and exclusion criteria
Four groups of patients were included. All patient biospecimens were collected after approval by the Johns Hopkins institutional review board.
Patients with ICI-induced IA (n=79) were included if they had no history of IA or other systemic autoimmune disease before starting ICI therapy, had evidence of IA on examination by a board-certified rheumatologist and had at least one serum sample after diagnosis of ICI-induced IA.
Healthy control sera (n=50) was obtained from volunteers who were 18 years of age or older, not pregnant and did not have a history of autoimmune disease, cancer or active HIV, tuberculosis or hepatitis infection.
Patients with RA (n=52) were participants in the Johns Hopkins Arthritis Center’s longitudinal database. Patients had rheumatologist-diagnosed RA and were included if they had sera and clinical data available on the same date.
Sera from ICI-treated patients who did not develop IA (n=35) were collected through the Johns Hopkins upper aerodigestive malignancy group’s tissue collection protocol. Absence of ICI-induced IA was confirmed with medical record review.
Anti-RA33 antibody assay
Anti-RA33 IgG antibodies were measured in serum diluted 1:100 using the IMTEC-RA33-Antibodies ELISA kit for the Quantitative Determination of Anti-RA33-IgG Antibodies (#ITC60015, IMETC) according to the manufacturer’s instructions. The cut-off for positivity was set at 3 SD above the mean of the healthy control group.
Clinical data collection for ICI-induced IA and RA
Patient-reported and physician-reported data were collected in separate longitudinal studies of rheumatic irAEs and RA. Joint counts, patient/physician ratings of disease activity, patient ratings of pain and stiffness, laboratory and imaging findings are recorded at each clinical evaluation.
Statistical analysis
Descriptive statistics were calculated for demographic and clinical variables. χ2 tests were used to compare frequencies of anti-RA33 antibodies. Student’s t-tests or Wilcoxon rank sum tests were used to compare continuous variables in ICI-induced IA patients with and without anti-RA33 antibodies. χ2 tests were used to compare categorical variables in those positive and negative for anti-RA33.
Results
Demographic features and anti-RA33 frequencies and level
Overall, 216 patients were tested for anti-RA33 IgG antibodies; 79 patients had ICI-induced IA, 50 were healthy controls, 52 had RA and 35 were treated with ICIs without developing IA (table 1). The healthy control participants were the youngest group (p<0.01). The ICI-treated patients without IA had the highest prevalence of former/current smokers and were significantly older than those with IA (p<0.01). The ICI-treated patients with and without IA had a different distribution of tumour types (table 1).
Table 1.
Characteristics of four included groups
| ICI-IA (N=79) Reference group |
ICI without IA (N=35) |
RA (N=52) |
Healthy controls (N=50) |
|
| Age (years), mean±SD | 60.4±14.2 | 68.2±9.1*** | 57.9±12.6 | 39.9±10.0*** |
| Female, N (%) | 41 (51.9) | 13 (37.1) | 28 (53.9) | 29 (58) |
| Smoking history, N (%) Never Former Current |
43 (58.8) 28 (37.8) 3 (4.5) |
3 (8.6)*** 29 (82.8) 3 (8.6) |
35 (67.3) 13 (25) 4 (7.7) |
34 (68) 15 (30) 1 (2) |
| Tumour types, N (%) | Melanoma: 27 (34.1) NSCLC: 16 (20.3) GU: 5 (6.3) GI: 11 (13.9) Other: 20 (25.3) |
NSCLC: 27 (77%) Mesothelioma: 2 (5.7%) Poorly differentiated: 3 (8.6%) Other: 3 (8.6%) |
N/A | N/A |
| Other irAEs, N (%) None 1 2 or more |
35 (44.3) 26 (32.9) 18 (22.8) |
21 (60) 12 (34.3) 2 (5.7) |
N/A | N/A |
| Anti-RA33 IgG positive, N (%) | 9 (11.4) | 0 (0)** | 4 (7.7) | 1 (2)* |
| Anti-RA33 level, median (IQR) | 0.27 (0, 4.18) | 0 (0, 1.69)* | 0 (0, 3.89) | 0.61 (0, 2.75) |
T-test for age. χ2 tests for categorical variables. Wilcoxon rank sum test for anti-RA33 level. *p<0.10, **p <0.05 and ***p <0.01.
GI, gastrointestinal; GU, genitourinary; IA, inflammatory arthritis; ICI, immune checkpoint inhibitor; IgG, immunoglobulin; irAEs, immune-related adverse events; NSCLC, non small cell lung cancer; RA, rheumatoid arthritis.
Of the 79 patients with ICI-induced IA, 9 had anti-RA33 antibodies (11.4%, table 1). This was a larger percentage of anti-RA33 positivity than for healthy controls (2%, p=0.05) and ICI-treated patients without IA (0%, p=0.04). There was no statistically significant difference in anti-RA33 positivity between those with ICI-induced IA and RA (7.7%, p=0.48).
Patients with ICI-induced IA had most of the higher level measurements as visualised in figure 1. For those above the cut-off for anti-RA33 positivity, patients with ICI-induced IA had a mean level of 61.8 units and a median level of 29.3 units, while patients with RA had a mean level of 49.3 units and a median level of 23.4 units.
Figure 1.
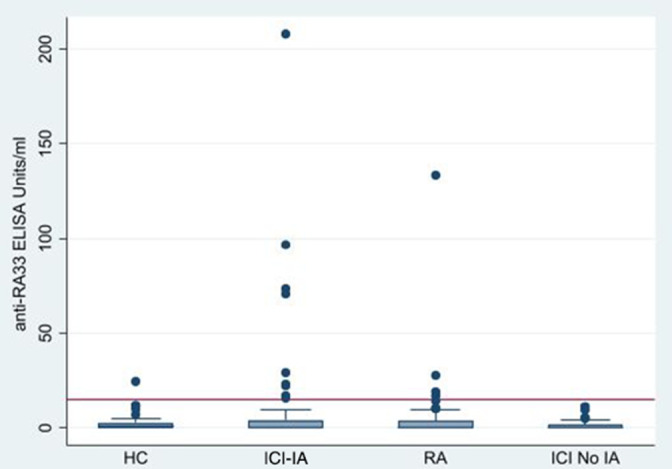
Anti-RA33 levels in immune checkpoint inhibitor-induced Inflammatory arthritis (ICI-IA), healthy controls (HC), rheumatoid arthritis (RA) and ICI-exposed patients without IA (ICI No IA).
RA patients and anti-RA33
The mean duration of RA at the time of sample was 9.9 years (SD 10.5). Of the 52 patients, 31 (59.6%) had erosions on imaging, 34 had anti-CCP (65.4%), 30 were positive for RF (57.7%) and 32 had at least one shared epitope allele (61.5%). There were no statistically significant differences in clinical or demographic features between the four patients with anti-RA33 antibodies from the overall population of RA patients tested.
Clinical features of anti-RA33-positive and anti-RA33-negative patients with ICI-induced IA
ICI-induced IA patients with and without anti-RA33 antibodies did not have significant differences in tumour type or immunotherapy regimen (table 2). People with anti-RA33 antibodies were more likely to have anti-CCP antibodies than anti-RA33-negative individuals (22% vs 1.4%, p=0.001), but had a similar prevalence of RF and antinuclear antibodies (ANA) (p=0.54 and 0.82). The time to develop IA from ICI initiation was similar in both groups, 189 days on average in the anti-RA33-positive group and 209 days in the anti-RA33-negative group (p=0.79). Patients with and without anti-RA33 had similar clinical features at their first rheumatology visit (table 2). Of patients seen in follow-up at least 6 months after ICI cessation, 70.5% still had symptoms of IA or were on medications to control IA symptoms, and there were no differences in persistence of IA by anti-RA33 status (table 2).
Table 2.
Clinical features of anti-RA33-positive and anti-RA33-negative ICI-IA patients
| Total (N=79) |
Anti-RA33 positive (N=9) |
Anti-RA33 negative (N=70) |
P-value | |
| Age (years), mean±SD | 60.4±14.2 | 62.3±9.5 | 60.1±14.7 | 0.66 |
| Female, N (%) | 41 (51.9) | 7 (77.8) | 34 (48.6) | 0.10 |
| Type of cancer, N (%) Melanoma NSCLC GU GI Other |
27 (34.1) 16 (20.3) 5 (6.3) 11 (13.9) 20 (25.3) |
3 (33.3) 2 (22.2) 0 (0) 2 (22.2) 2 (22.2) |
24 (34.3) 14 (20) 5 (7.1) 9 (12.9) 18 (25.7) |
0.88 |
| ICI regimen, N (%) Anti-PD-1 Anti-PD-L1 Combo PD-1/CTLA-4 |
52 (65.8) 6 (7.6) 21 (26.6) |
8 (88.9) 0 (0) 1 (11.1) |
44 (62.9) 6 (8.6) 20 (28.6) |
0.29 |
| Time to develop ICI-IA from ICI start in days, median (IQR) | 153 (59, 304) | 153 (31, 273) | 167 (61, 304) | 0.79 |
| Prior chemo, N (%) | 37 (46.8) | 6 (66.7) | 31 (44.3) | 0.21 |
| Prior radiation, N (%) | 27 (34.2) | 5 (55.6) | 22 (31.4%) | 0.15 |
| Additional irAEs, N (%) None 1 2 or more |
35 (44.3) 26 (32.9) 18 (22.8) |
2 (22.2) 4 (44.4) 3 (33.3) |
33 (47.1) 22 (31.4) 15 (21.4) |
0.36 |
| CDAI at presentation, median (IQR), N=47 | 18.1 (11.3, 23) | 19.75 (9.25, 33.5) | 18.1 (11.3, 23) | 0.87 |
| Swollen joint count, median (IQR), N=72 | 6 (3, 10) | 4 (2, 9) | 6 (3, 11) | 0.73 |
| Patient global median (IQR), N=63 | 35 (15, 50) | 35 (17, 70) | 35 (12, 50) | 0.53 |
| Stiffness VAS, median (IQR), N=67 | 50 (33, 75) | 70 (45, 75) | 50 (30, 75) | 0.41 |
| Pain VAS, median (IQR), N=68 | 50 (21.5, 75) | 71 (20, 75) | 50 (23, 70) | 0.33 |
| Enthesitis, N (%) | 20 (25.3) | 2 (22.2) | 18 (25.7) | 0.82 |
| Required corticosteroid, N (%) | 65 (82.3) | 7 (77.8) | 58 (82.9) | 0.71 |
| Required csDMARD, N (%) | 26 (32.9) | 4 (44.4) | 22 (31.4) | 0.43 |
| Required biologic, N (%) | 14 (17.7) | 2 (22.2) | 12 (17.1) | 0.71 |
| RF positive, N (%), N=75 | 3 (4) | 0 (0) | 3 (4.5) | 0.54 |
| CCP positive, N (%), N=75 | 3 (4) | 2 (22) | 1 (1.4) | 0.001 |
| ANA positive, N (%), N=69 | 12 (17.4%) | 1 (14.2) | 11 (17.7) | 0.82 |
| ICI persistence (6 months after ICI cessation), N (%), N=51 | 36 (70.5) | 3 (75) | 33 (70) | 0.84 |
χ2 test for categorical variables. T-test for age. Wilcoxon rank sum for other continuous variables.
ANA, antinuclear antibodies; CCP, anti-cyclic citrullinated peptide; CDAI, Clinical Disease Activity Index; csDMARD, conventional synthetic disease modifying antirheumatic drugs; CTLA-4, cytotoxic lymphocyte antigen-4; GI, gastrointestinal; GU, genitourinary; IA, inflammatory arthritis; ICI, immune checkpoint inhibitor; irAEs, immune-related adverse events; NSCLC, non small cell lung cancer; PD-1, programmed cell death protein 1; PD-L1, programmed death ligand 1; RF, rheumatoid factor; VAS, 100-point Visual Analogue Scale.
Anti-RA33 pre-ICI treatment
Pre-ICI treatment sera within 1 month of ICI start was available for 15 subjects with ICI-induced IA. Of these 15, 2 had positive anti-RA33 antibodies after ICI treatment, while the rest were negative. Both patients with anti-RA33 antibodies after ICI treatment were also positive for anti-RA33 before treatment, while none of the other 13 were positive in pretreatment sera. Neither patient who was positive for anti-RA33 pretreatment and post treatment was positive for anti-CCP.
Discussion
This is the first evaluation of autoantibodies beyond ANA, anti-CCP and RF in patients with ICI-induced IA. Anti-RA33 antibodies were detected in 11.4% of patients with ICI-induced IA, whereas none of the ICI-treated controls without IA were positive. For two patients with ICI-induced IA with sera from before ICI treatment, anti-RA33 antibodies were present before ICI exposure. Anti-RA33 antibodies did not correlate with clinical features of ICI-induced IA, except for anti-CCP antibodies. Notably, anti-RA33 antibodies were more prevalent in this cohort of ICI-induced IA than anti-CCP antibodies.
The presence of anti-RA33 antibodies in a subset of patients with ICI-induced IA suggests two main possibilities for their role in pathogenesis. First, anti-RA33 antibodies newly develop as an event in IA initiation post-ICI treatment. Alternatively, anti-RA33 antibodies are present before ICI therapy and are a marker of risk for developing IA. Since antibodies were present pre-ICI in both of the patients who were positive for anti-RA33 and had pre-ICI treatment sera available, it is possible that, as has been reported in patients with anti-CCP antibodies,17 there are patients with preclinical IA who manifest clinical IA only after ICI treatment.
There is a question of whether this autoantibody represents an immune response to a tumour antigen since several tumour types overexpress RA33 including glioblastoma, hepatocellular carcinoma and non-small cell lung cancer.18–20 Anti-RA33 antibodies were not found in ICI-treated patients without IA, however, suggests that there is some specificity for anti-RA33 antibodies to ICI-induced IA.
While this pilot study revealed anti-RA33 antibodies as potential biomarkers of IA in ICI-treated individuals, there are several limitations to consider, which should be addressed in future studies. One limitation was that ICI-treated controls without IA primarily had lung cancer, while patients with ICI-induced IA had diverse tumours. The predominance of lung cancer in the group of ICI-treated controls likely explains their older age and higher prevalence of smoking. Since RA33 expression has been observed in a range of tumour types, future study of ICI-treated patients with different cancers will be informative. Healthy controls were also significantly younger than other groups which may have affected the rates of autoantibodies detected in these individuals since autoantibodies can accumulate with age. The younger age of healthy controls may have also affected the cut-offs for positivity as this was determined from the healthy control group. Additionally, the study was primarily cross-sectional in nature. It is unclear when anti-RA33 antibodies develop in the course of ICI-induced IA or how long they persist.
Importantly, anti-RA33 antibodies were present in 7.7% of patients with RA in our cohort, which is within the wide range of positivity reported in the literature (6.2%–41%).14 15 21 22 While the reasons for the heterogeneity in prevalence between cohorts are unclear, it may reflect differences in ethnic background, disease duration or assay used, since there is no standardised or clinically available anti-RA33 assay. In addition, the rate of positivity in the ICI-IA group was higher than that of the RA group in our study. Other studies interrogating the specificity of anti-RA33 in non-RA disease and healthy controls has found a prevalence of about 3% these other populations, similar to our healthy control prevalence.14 Patients with ICI-IA had the highest prevalence of antibodies as compared with all other groups using our assay conditions, which suggests its potential utility as a biomarker in this patient population.
Evaluating immunological factors in ICI-induced IA that are associated with other forms of early IA, such as the presence of anti-RA33 antibodies, may help determine aetiology and define treatment targets. Future studies elucidating the temporal relationship of these antibodies to ICI treatment and validating the presence of anti-RA33 in ICI-induced IA in separate and larger cohorts will help determine the utility of these antibodies in clinical care and their role in pathogenesis.
rmdopen-2022-002511supp001.xlsx (69KB, xlsx)
Footnotes
Contributors: LCC, ED, COB and AAS planned the study. LCC and ED obtained the data. LCC performed the statistical analysis. All authors interpreted the data and drafted and revised the manuscript.
Funding: This work was supported by National Institute for Musculoskeletal and Skin Disorders (K23AR075872, P30AR070254, R01AR073208) and Bristol Myers Squibb.
Competing interests: LCC and COB have received research funding from Bristol Myers Squibb. LCC has consulted for Bristol Myers Squibb. JB has served on advisory boards for Bristol Myers Squibb, Merck, Astra Zeneca and Regeneron and has received research funding from Bristol Myers Squibb. VA has received research funding from Bristol Myers Squibb and Astra Zeneca. PMF has received honoraria or consulting fees from Amgen, AstraZeneca, Bristol Myers Squibb, Delfi, Novartis, Janssen, Iteos, Sanofi, Genentech, F-Star, G1 Therapeutics and Surface Oncology; research funding from AstraZeneca, BMS, Kyowa, Corvus and Novartis; and is a DSMB member for Flame and Polaris. EJL has received institutional grant/research support from Bristol Myers Squibb, Merck and Sanofi/Regeneron and has been a consultant for Array BioPharma, Bristol Myers Squibb, Eisai, EMD Serono, Genentech, Macrogenics, Merck, Novartis, Odonate Therapeutics, OncoSec and Sanofi-Regeneron. MS, ED, AAS and JM have no competing interests to disclose.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Datasets will be made available upon reasonable request.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
This study involves human participants and was approved by Johns Hopkins institutional review board (IRB #s 00123172, 0040493, 00066509, 00100653) Participants gave informed consent to participate in the study before taking part.
References
- 1.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 2015;27:450–61. 10.1016/j.ccell.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Postow MA, Sidlow R, Hellmann MD. Immune-Related adverse events associated with immune checkpoint blockade. N Engl J Med 2018;378:158–68. 10.1056/NEJMra1703481 [DOI] [PubMed] [Google Scholar]
- 3.Cappelli LC, Gutierrez AK, Bingham CO, et al. Rheumatic and musculoskeletal immune-related adverse events due to immune checkpoint inhibitors: a systematic review of the literature. Arthritis Care Res 2017;69:1751–63. 10.1002/acr.23177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdel-Wahab N, Suarez-Almazor ME. Frequency and distribution of various rheumatic disorders associated with checkpoint inhibitor therapy. Rheumatology 2019;58:vii40–8. 10.1093/rheumatology/kez297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calabrese LH, Calabrese C, Cappelli LC. Rheumatic immune-related adverse events from cancer immunotherapy. Nat Rev Rheumatol 2018;14:569–79. 10.1038/s41584-018-0074-9 [DOI] [PubMed] [Google Scholar]
- 6.Braaten TJ, Brahmer JR, Forde PM, et al. Immune checkpoint inhibitor-induced inflammatory arthritis persists after immunotherapy cessation. Ann Rheum Dis 2020;79:332–8. 10.1136/annrheumdis-2019-216109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cappelli LC, Brahmer JR, Forde PM, et al. Clinical presentation of immune checkpoint inhibitor-induced inflammatory arthritis differs by immunotherapy regimen. Semin Arthritis Rheum 2018;48:553–7. 10.1016/j.semarthrit.2018.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghosh N, Tiongson MD, Stewart C. Checkpoint inhibitor-associated arthritis: a systematic review of case reports and case series. J Clin Rheumatol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Albayda J, Dein E, Shah AA, et al. Sonographic findings in inflammatory arthritis secondary to immune checkpoint inhibition: a case series. ACR Open Rheumatol 2019;1:303–7. 10.1002/acr2.1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bronstein Y, Ng CS, Hwu P, et al. Radiologic manifestations of immune-related adverse events in patients with metastatic melanoma undergoing anti-CTLA-4 antibody therapy. AJR Am J Roentgenol 2011;197:W992–1000. 10.2214/AJR.10.6198 [DOI] [PubMed] [Google Scholar]
- 11.Feist J, Murray A, Skapenko A, et al. A rare side effect of checkpoint inhibitor Therapy-Nivolumab-Induced axial polyarthritis of the facet and Costovertebral joints. Arthritis Rheumatol 2019;71:1823. 10.1002/art.41036 [DOI] [PubMed] [Google Scholar]
- 12.Steiner G, Smolen J. Autoantibodies in rheumatoid arthritis and their clinical significance. Arthritis Res 2002;4 Suppl 2:S1–5. 10.1186/ar551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang X, Wang M, Zhang X, et al. Diagnostic accuracy of anti-RA33 antibody for rheumatoid arthritis: systematic review and meta-analysis. Clin Exp Rheumatol 2016;34:539–47. [PubMed] [Google Scholar]
- 14.Sieghart D, Platzer A, Studenic P, et al. Determination of autoantibody isotypes increases the sensitivity of Serodiagnostics in rheumatoid arthritis. Front Immunol 2018;9:876. 10.3389/fimmu.2018.00876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konig MF, Giles JT, Nigrovic PA, et al. Antibodies to native and citrullinated RA33 (hnRNP A2/B1) challenge citrullination as the inciting principle underlying loss of tolerance in rheumatoid arthritis. Ann Rheum Dis 2016;75:2022–8. 10.1136/annrheumdis-2015-208529 [DOI] [PubMed] [Google Scholar]
- 16.Cappelli LC, Darrah E, Shah AA. Patients with checkpoint inhibitor-induced inflammatory arthritis do not become seropositive for anti-cyclic citrullinated peptide when followed over time. ACR Open Rheumatol 2021. 10.1002/acr2.11363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belkhir R, Burel SL, Dunogeant L, et al. Rheumatoid arthritis and polymyalgia rheumatica occurring after immune checkpoint inhibitor treatment. Ann Rheum Dis 2017;76:1747–50. 10.1136/annrheumdis-2017-211216 [DOI] [PubMed] [Google Scholar]
- 18.Golan-Gerstl R, Cohen M, Shilo A, et al. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res 2011;71:4464–72. 10.1158/0008-5472.CAN-10-4410 [DOI] [PubMed] [Google Scholar]
- 19.Shilo A, Ben Hur V, Denichenko P, et al. Splicing factor hnRNP A2 activates the Ras-MAPK-ERK pathway by controlling A-Raf splicing in hepatocellular carcinoma development. RNA 2014;20:505–15. 10.1261/rna.042259.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou J, Nong L, Wloch M, et al. Expression of early lung cancer detection marker: hnRNP-A2/B1 and its relation to microsatellite alteration in non-small cell lung cancer. Lung Cancer 2001;34:341–50. 10.1016/S0169-5002(01)00254-9 [DOI] [PubMed] [Google Scholar]
- 21.Syed Mohamed Suhail SM, Nur Atiqah I, Nur Salimi Zakirah ZA, et al. Diagnostic performance of anti-RA33 antibody as a serological marker for rheumatoid arthritis. Malays J Pathol 2019;41:259–65. [PubMed] [Google Scholar]
- 22.Al-Mughales JA. Immunodiagnostic significance of anti-RA33 autoantibodies in Saudi patients with rheumatoid arthritis. J Immunol Res 2015;2015:604305. 10.1155/2015/604305 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
rmdopen-2022-002511supp001.xlsx (69KB, xlsx)
Data Availability Statement
Datasets will be made available upon reasonable request.