

Abstract
Background
A vaccine containing 6 melanoma-associated peptides to stimulate helper T cells (6MHP) is safe, immunogenic, and clinically active. A phase I/II trial was designed to evaluate safety and immunogenicity of 6MHP vaccines plus programmed death 1 (PD-1) blockade.
Participants and methods
Participants with advanced melanoma received 6MHP vaccines in an incomplete Freund’s adjuvant (6 vaccines over 12 weeks). Pembrolizumab was administered intravenously every 3 weeks. Tumor biopsies at baseline and day 22 were analyzed by multiplex immunohistochemistry. Primary end points were safety (Common Terminology Criteria for Adverse Events V.4.03) and immunogenicity (ex vivo interferon-γ ELISpot assay). Additional end points included changes in the tumor microenvironment (TME) and clinical outcomes.
Results
Twenty-two eligible participants were treated: 6 naïve to PD-1 antibody (Ab) and 16 PD-1 Ab-experienced. Median follow-up was 24.4 months. Most common treatment-related adverse events (any grade) included injection site reactions, fatigue, anemia, lymphopenia, fever, elevated aspartate aminotransferase, pruritus, and rash. Treatment-related dose-limiting toxicities were observed in 3 (14%) participants, which did not cross the study safety bound. A high durable T cell response (Rsp) to 6MHP was detected in only one participant, but twofold T cell Rsps to 6MHP were detected in 7/22 (32%; 90% CI (16% to 52%)) by week 13. Objective clinical responses were observed in 23% (1 complete response, 4 partial responses), including 4/6 PD-1 Ab-naïve (67%) and 1/16 PD-1 Ab-experienced (6%). Overall survival (OS) was longer for PD-1 Ab-naïve than Ab-experienced participants (HR 6.3 (90% CI (2.1 to 28.7)). In landmark analyses at 13 weeks, OS was also longer for those with T cell Rsps (HR 6.5 (90% CI (2.1 to 29.2)) and for those with objective clinical responses. TME evaluation revealed increased densities of CD8+ T cells, CD20+ B cells, and Tbet+ cells by day 22.
Conclusions
Treatment with the 6MHP vaccine plus pembrolizumab was safe, increased intratumoral lymphocytes, and induced T cell Rsps associated with prolonged OS. The low T cell Rsp rate in PD-1 Ab-experienced participants corroborates prior murine studies that caution against delaying cancer vaccines until after PD-1 blockade. The promising objective response rate and OS in PD-1 Ab-naïve participants support consideration of a larger study in that setting.
Keywords: immunogenicity, vaccine; programmed cell death 1 receptor; combined modality therapy; immunotherapy; CD4-positive T-lymphocytes
What is already known on this topic
Primary resistance to checkpoint blockade is commonly attributed to a lack of pre-existing T cell responses (Rsps) to cancer antigens.
Cancer vaccines designed to induce CD4+ T-helper cell responses are emerging as promising cancer immunotherapies.
A key question in patient management is whether vaccines given after prior PD-1 antibody (Ab) failure may induce clinical responses when combined with renewed PD-1 Ab, or whether, as suggested in murine studies, vaccines are preferably administered concurrently with PD-1 blockade.
What this study adds
Among PD-1 Ab-naïve and PD-1 Ab-experienced participants, treatment with the six melanoma helper peptides vaccine plus pembrolizumab was safe, increased intratumoral lymphocytes, and induced T cell Rsps associated with prolonged overall survival.
The low T cell Rsp rate seen in the PD-1 Ab-experienced participants (3/16; 19%) compared with that seen in the PD-1 Ab-naïve participants (4/6; 67%) corroborates prior murine studies and thus sheds important light that supports initiating vaccines concurrent with, rather than after, administration of PD-1 blockade therapy.
How this study might affect research, practice or policy
This may be relevant for investigators to consider in analysis and design of ongoing clinical trials of neoantigen vaccines, where PD-1 Ab is given prior to creation of the neoantigen vaccine.
The promising objective response rate and overall survival in PD-1 Ab-naïve participants support consideration of a larger study to assess definitive benefit in that setting.
Introduction
Programmed death 1 (PD-1) blockade induces durable clinical responses in patients with advanced melanoma1 2; however, monotherapy PD-1 blockade is only effective in a minority of patients.3 Primary resistance to checkpoint blockade is commonly attributed to a lack of pre-existing T cell responses (Rsps) to cancer antigens.4 5 Cancer vaccines targeting mutated neo-antigens or shared tumor antigens to induce CD4+ T-helper cell responses6–8 are emerging as promising immunotherapies.9–11 A melanoma vaccine using six melanoma helper peptides (6MHP), restricted by class II major histocompatibility complex (MHC), emulsified in incomplete Freund’s adjuvant (IFA) can induce Th1-dominant CD4+ T cell Rsps in 40%–80% of patients, induce peptide-specific antibody (Ab) responses in 77% of patients,12–14 induce epitope spreading to CD8 epitopes,14 and induce objective clinical responses.13 15 Response Evaluation Criteria in Solid Tumors (RECIST)-defined objective response rates (ORR) and disease control rates (DCRs) with 6MHP vaccines alone have been observed in 8% and 30%, respectively, with stable disease (SD) and clinical responses lasting 1–7 years.13 15 Overall survival (OS) is also significantly associated with CD4+ T-helper cell response to 6MHP.15 It is critical to support T cell infiltration and function in the tumor microenvironment (TME), both to improve the clinical benefit of this and other vaccines, and to enhance the benefit of PD-1 blockade.
PD-1 expression is increased on exhausted CD4+ T cells16; thus, blocking PD-1/programmed death-ligand 1 (PD-L1) may restore and support CD4+ T cell function in the TME, and combining it with 6MHP vaccines may improve tumor targeting and infiltration by enhancing the CD4+ T cell Rsps to melanoma antigens. Two clinical trials combined nivolumab plus a peptide vaccine restricted by class I MHC and demonstrated their co-administration to be safe and immunogenic17 18; however, among ipilimumab-refractory patients, the ORRs were not increased by adding that vaccine—designed to stimulate CD8+ T cells—(27% vs 26%).17 Another recent clinical trial by Kjeldsen et al combined nivolumab plus a vaccine containing two peptides from indoleamine 2,3-dioxygenase (IDO) and PD-L1 that induced both CD4 + and CD8 + T cell Rsps.19 This trial only enrolled PD-1 Ab-naïve participants with metastatic cutaneous melanoma and reported an 80% ORR (90% CI (63% to 91%)),19 providing evidence that a vaccine inducing CD4+ T cell is associated with high ORR in PD-1 Ab-naïve patients. It is unclear if that promising outcome was due to the selection of peptide antigens from checkpoint molecules, or whether peptide vaccines that induce T-helper cells may also induce favorable T cell Rsps in combination with checkpoint blockade. Furthermore, a key question in patient management is whether vaccines given after prior PD-1 Ab failure may induce clinical responses when combined with renewed PD-1 Ab, or whether, as suggested in prior murine studies,20 21 vaccines are preferably administered concurrently with PD-1 blockade. To our knowledge, we are reporting the first trial using a vaccine with class II MHC-restricted peptides co-administered with PD-1 blockade for participants with advanced melanoma who are PD-1 Ab naïve as well as participants who have previously been treated with PD-1 Ab (Ab experienced).
The purpose of this study was to evaluate the safety and immunogenicity of a class II MHC-restricted 6MHP vaccine plus pembrolizumab in patients with advanced melanoma with or without prior PD-1 blockade therapy and to obtain preliminary data on clinical outcomes and on changes in the TME. We hypothesized that co-administration of a 6MHP vaccine with pembrolizumab would be safe, would increase the magnitude, duration, and number of participants with circulating CD4+ T cell Rsps compared with a prior trial incorporating 6MHP vaccines alone,22 and would improve ORRs and OS, notably among participants with circulating T cell Rsps to the vaccine. We also hypothesized that 6MHP plus pembrolizumab would modify the TME, specifically by increasing densities of tumor infiltrating lymphocytes (TILs).
Materials and methods
Participant eligibility
In this open-label, phase I/II study, participants 18 years of age and older with biopsy-proven advanced melanoma (unresectable stage IIIB/C or IV based on American Joint Committee on Cancer V.7,23 at initial presentation or at recurrence), which was measurable by RECIST V.1.1 criteria,24 25 were eligible for inclusion. Participants may have had cutaneous, uveal, mucosal primary melanoma, or an unknown primary melanoma. Staging was confirmed by cytological or histological examination. For those with at least one additional site of metastasis accessible for biopsy pretreatment and on day 22, consent included tumor biopsies (needle biopsy, incisional or excisional biopsy, with or without image guidance): this was required for at least 12 participants. The lesions to be biopsied were specified at study enrollment and not included as target lesions for RECIST calculations, except that clear progression in those lesions would not be consistent with objective response. The full protocol is provided as online supplemental file 1.
jitc-2022-005424supp001.pdf (1.7MB, pdf)
Participants had to be eligible for treatment with pembrolizumab based on treating clinician judgment. Eligible patients included those naïve to PD-1 blockade (PD-1 Ab-naïve cohort) and those previously treated with PD-1 blockade but experienced disease progression during or after treatment or did not experience an objective clinical response (PD-1 Ab-experienced cohort). Participants who previously received other immunotherapy agents (eg, interferon (IFN)-α, ipilimumab, interleukin-2, or a prior cancer vaccine other than 6MHP) were eligible. Additional inclusion criteria included: Eastern Cooperative Oncology Group (ECOG) performance status 0–1, at least two intact regional lymph node basins, normal organ function, and absence of major autoimmune disorders. Participants were excluded for pregnancy, other concurrent cancer therapy, uncontrolled diabetes, or autoimmune disorders requiring therapy. This study was designed to accrue a total of 22 evaluable participants. All participants were assigned a Virginia Malignant Melanoma (VMM) number, which is a patient identifier that is compliant with the Health Insurance Portability and Accountability Act of 1996 (HIPAA).
Vaccine components and treatment regimen
All participants were treated at one academic institution and were vaccinated with a mixture of 200 μg each of 6MHP restricted by class II MHC26 27 emulsified in IFA (figure 1). The peptides in 6MHP range in length from 14-mers to 23-mers (online supplemental text). The IFA used was Montanide ISA-51VG (Seppic, Fairfield, New Jersey, USA). For each vaccine, peptides in a 1 mL aqueous solution were emulsified 1:1 with 1 mL of IFA (total 2 mL). Vaccines were administered half-subcutaneously and half-intradermally in one skin location (ie, upper arm or thigh) for vaccines 1–3 (days 1, 8, 15). Vaccines 4–6 were given in a different extremity (days 43, 64, 85) to mitigate the risk of skin toxicity. Pembrolizumab 200 mg was administered intravenously every 3 weeks for up to 2 years, beginning day 1 (figure 1).
Figure 1.
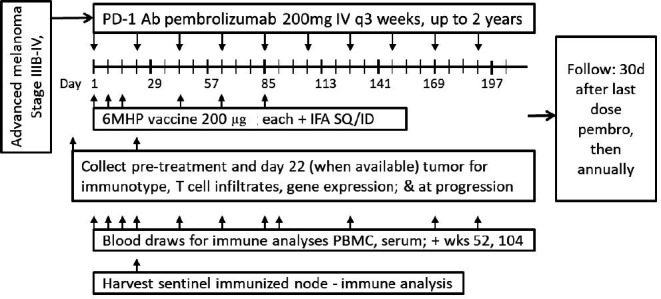
Study schema. 6MHP, six melanoma helper peptides; Ab, antibody; ID, intradermally; IFA, incomplete Freund’s adjuvant; IV, intravenously; PBMC, peripheral blood mononuclear cells; PD-1, programmed cell death protein 1; SQ, subcutaneously.
jitc-2022-005424supp002.pdf (9.5MB, pdf)
Blood was collected from each participant at multiple time points (figure 1), and an excisional biopsy of a vaccine-site draining lymph node (sentinel immunized node (SIN)28) was performed on day 22 for the first 10 participants (figure 1) to assess T cell Rsps. Peripheral blood mononuclear cells (PBMCs) and SIN cells were viably cryopreserved in 90% fetal bovine serum/10% dimethyl sulfoxide, and serum was also frozen for subsequent immunological assays in batch. Tumor biopsies were collected pretreatment and day 22 (n=12), with portions preserved in formalin and frozen in liquid nitrogen.
Objective clinical response was assessed at screening and on days 57, 127, and as indicated per standard care, using cross-sectional imaging. Participants were followed 30 days after last dose of pembrolizumab, then annually for disease status and survival.
Objectives
The primary objectives of this study were (1) to determine whether administration of 6MHP plus pembrolizumab is safe, and (2) to estimate the immunogenicity of 6MHP in the blood, and SIN when available, when co-administered with pembrolizumab. An optimal treatment combination was defined as one with an acceptable toxicity profile as measured by dose-limiting toxicities (DLTs) and a high rate of high durable immune response (hdRsp) as measured by CD4+ T cell Rsp to 6MHP during the time period of vaccine administration.
Secondary objectives included determining whether 6MHP plus pembrolizumab modifies the TME by increasing infiltration of CD4+ and CD8+ T cells into tumor metastases, and increasing Th1-dominant immune signatures in the TME.
Exploratory objectives included obtaining preliminary data on whether 6MHP plus pembrolizumab: (1) induces objective clinical responses in patients with or without prior PD-1 blockade therapy, (2) improves OS relative to published data, and (3) improves OS in patients with versus without T cell Rsps to melanoma antigens.
Summary of protocol revisions
The original protocol (V.5/18/2015) was only for patients offered pembrolizumab second-line therapy after ipilimumab, but approval of pembrolizumab as first-line therapy led to revised inclusion criteria and objectives (V.3/21/2016) prior to the first patient enrolled. These were maintained through the study. The only other significant change was to reduce target enrollment from 42 to 22 eligible participants.
Dose-limiting toxicities
A DLT was defined as any unexpected adverse event (AE) that was possibly, probably, or definitely related to treatment with 6MHP and/or pembrolizumab. A DLT of 6MHP was defined as grade ≥3 hematological or non-hematological toxicity, except ulceration at a vaccine site was considered a DLT only if the ulcer was >2 cm in diameter, required antibiotics, or required surgical debridement. A DLT of pembrolizumab was defined as toxicity requiring permanent discontinuation per specified dose modification guidelines for drug-related AEs.29 Selected grade ≥2 ocular AEs were also considered DLTs, regardless of the agent to which the event may be attributed. AEs that led to treatment discontinuation were also considered DLTs even if they did not meet prespecified criteria for DLT. The DLT tolerance level was chosen to be 25%. Participants were monitored for toxicities during and for 30 days after last study treatment using National Cancer Institute Common Terminology Criteria for Adverse Events V.4.03 criteria. Stopping rules for observed toxicities were based on serious AEs rate of 7.9% (95% CI (3.2% to 15.5%)) reported among patients treated with pembrolizumab alone.30 The rate of DLTs for early stopping included assessment up to day 43. DLTs were monitored for 30 days after last study treatment, and late observed rates that cross the bound would be reason to suspend accrual for safety review by the sponsor-investigator and the Data Safety Monitoring Committee (DSMC). A sequential probability ratio test (SPRT) based on a binomial test of proportions for DLTs was used. Only the upper boundary was used for monitoring to protect against excessive failure rates. The stopping boundary was for a SPRT contrasting a 15% vs 30% DLT rate, with nominal type I and II errors of 10% and 10%, respectively.
Immune response
Circulating CD4+ T cell Rsps to 6MHP peptides were assessed at each specified time point (figure 1) using ELISpot assays. IFN-γ ELISpot assays were performed directly ex vivo, after cryopreservation (direct ELISpot) as previously reported,22 for PBMC and, when available, SIN lymphocytes. Briefly, 200,000 PBMC were plated per well, and pulsed with synthetic peptide (10 µg/mL of each peptide), in quadruplicate. Negative controls included irrelevant peptide from HIV gag protein, and no peptide. Positive controls included a mixture of viral peptides (CEF peptide pool),31 phorbol myristate acetate (PMA)-ionomycin and phytohaemagglutinin (PHA). Evaluation of T cell Rsps was based on the following definitions at each assay time point: Nvax=number of T cells responding to vaccine peptide; Nneg=number of T cells responding to maximum negative control; Rvax=Nvax/Nneg. For evaluations of PBMC, a participant was considered to have a T-cell Rsp to vaccination (binary yes/no) at each time point after baseline, by direct ELISpot assay only if all the following previously used criteria22 were met: (1) Nvax exceeded Nneg by at least 20/100,000 CD4+ cells (0.02%), (2) (Nvax–1 SD)≥(Nneg+1 SD), (3) Rvax ≥2×, and (4) Rvax postvaccination ≥2× Rvax prevaccination. The criteria for a CD4+ Rsp in a SIN was the same as for PBMCs, but without criterion (4) above. The proportion of responding cells per 100 000 CD4+ T cells was calculated based on the proportion of CD4+ T cells (CD3+CD4+ by flow cytometry) among PBMC or SIN. Fold-increases <1 were set to 1 to indicate no response and to prevent overinflating adjusted fold-increases due to prevaccine ratios <1, or division by 0, while not affecting the determination of response. An hdRsp required a 5× increase above baseline in any two or more time points (through day 85), and a durable immune response (dRsp) required only a 2× increase over background. For comparison with our prior Mel44 trial,22 we further report on single detections of immune response at 2× (Rsp) and 5× (hRsp) increases above background. Continuous measures of immune response denoted as fold-increase must satisfy conditions (1)–(4) and were defined as the amount of Rvax.
The proportion of participants with an hdRsp, dRsp, Rsp, and hRsp in blood and, when available, Rsp in SIN was calculated (±90% CIs).32 Participants who discontinued protocol therapy early were considered immune response failures if no response was observed in evaluable samples. Hypothesis testing was based on the number of participants that satisfied the criteria of an hdRsp in the blood, as specified in the initial protocol. The target of 22 participants was chosen based on having sufficient information to test for a null hdRsp rate of 20% vs an alternative hdRsp rate of 45% with a one-side 10% level binomial test. At study conclusion, the null hypothesis would be rejected and the treatment regimen considered worthy of further study if at least 8/22 (36%) eligible participants experience a hdRsp.
Assay consistency is represented by interassay coefficients of variation (CVs) calculated for the response of two normal donors to the CEF peptide pool. For the high responder normal donor, mean number of spots was 165/100,000 cells plated, and the CV was 27%. A low responder normal donor was included in 4 of the 16 assays, for which the mean was 18 and the CV was 24%.
TME—multiplex immunohistochemistry and analysis
Changes in the TME were assessed by multiplex immunohistochemistry (mIHC) and image analysis for quantification of melanoma tertiary lymphoid structures (TLSs) and TILs; 5 µm thick sections were cut from formalin-fixed, paraffin-embedded tumor specimens, with human LN samples as positive controls, and mIHC was performed. Details for the mIHC are mentioned in online supplemental text.
Stained slides were scanned using the PerkinElmer Vectra V.3.0 system. Regions of interest were selected in Phenochart software analyzing the entire tumor biopsy, and these regions were matched between the TLS and TIL panels; ×20 magnification images were acquired. These images were spectrally unmixed using single stain positive control images in the InForm software (Akoya Biosciences), and immune cells were quantified using HALO software (Indica Labs, Albuquerque, New Mexico, USA). Lymphocyte densities were quantified (cells/mm2).
The proportion of participants with increased infiltration of CD4+ and CD8+ T cells into tumor metastases and increased Th1-dominant immune signatures in the TME were estimated. If increased infiltration of immune cells on day 22 compared with pretreatment is observed in the tumor in 9/12 eligible participants then the lower limit of a one-sided 90% CI exceeds 50%, then this would be considered a promising result and supportive of further study of the treatment regimen.
Clinical efficacy
Participants were assessed for objective clinical responses (RECIST V.1.1) and were classified by best overall response. ORR was estimated. Progression-free survival (PFS) and OS distributions were estimated using Kaplan-Meier curves (±95% CI); HR (±90% CIs) were used to compare PD-1 Ab-experienced to PD-1 Ab-naïve participants.
Results
Clinical characteristics
The study opened to accrual November 19, 2015, and closed to accrual June 26, 2019 after meeting the enrollment target. Among 27 participants assessed, 22 eligible participants were enrolled and treated (online supplemental figure 1): 6 naïve to PD-1 Ab and 16 PD-1 Ab experienced. The melanomas had arisen from a non-acral skin primary (12, 54%), acral skin primary (5, 23%), uveal primary (3, 14%), or anal mucosal primary (2, 9%). Most participants had ECOG performance status of 0 (59%) and stage IV disease at registration (95%). Participant demographics are provided in online supplemental table 1. Clinical and immunological details for each participant are shown in table 1. Follow-up data for calculating OS and PFS were collected up to December 1, 2021.
Table 1.
Clinical and immunological data for each participant
| VMM | Age at enrollment | Sex | ECOG PS | Primary melanoma site |
Stage at enrollment | LDH level at enrollment (U/L)* | Biopsiable tumor† | PD-1 Ab naïve | PD-L1 status‡ | BRAF V600E/K mutation status | High durable T cell response | Durable T cell Rsp | T cell Rsp | On treatment month | Best RECIST response | Survival status | Survival duration (months)§ |
| 1014 | 72 | M | 1 | Skin, acral | IV | 202 | Yes | No | N/A | WT | No | No | Yes¶ | May 2018 | PD | Deceased | 31.51 |
| 1088 | 63 | F | 0 | Skin, non-acral | IV | 146 | Yes | No | 20% | WT | No | No | No | February 2018 | PD | Alive | 45.77 |
| 1265 | 72 | M | 0 | Skin, non-acral | IV | 231 | Yes | No | N/A | WT | No | No | No | October 2016 | PD | Deceased | 10.45 |
| 1274 | 36 | F | 0 | Skin, non-acral | IV | 177 | No | No | N/A | V600E | No | No | No | March 2017 | SD | Deceased | 47.38 |
| 1275 | 62 | F | 1 | Skin, non-acral | IV | 515 | No | No | N/A | V600E | No | No | No | March 2017 | PD | Deceased | 4.34 |
| 1284 | 48 | F | 1 | Anal, mucosal | IV | 334 | No | No | N/A | WT | No | No | No | November 2017 | PD | Deceased | 10.22 |
| 1287 | 75 | F | 0 | Skin, acral | IV | 221 | No | No | N/A | V600E | No | No | No | November 2017 | SD | Deceased | 20.86 |
| 1290 | 54 | F | 0 | Skin, non-acral | IV | 156 | No | No | 0% | V600E | No | No | Yese, f | March 2018 | PR | Alive | 45.01 |
| 1291 | 48 | M | 1 | Skin, non-acral | IV | 3177 | Yes | No | <1% | V600E | No | No | No | April 2018 | PD | Deceased | 2.50 |
| 1293 | 57 | M | 0 | Uveal | IV | 400 | No | No | 0% | WT | No | No | No | May 2018 | PD | Deceased | 6.77 |
| 1296 | 64 | M | 0 | Anal, mucosal | IV | 198 | No | No | 50%–60% | WT | No | No | No | August 2018 | PD | Deceased | 10.55 |
| 1297 | 50 | F | 1 | Skin, acral | IV | 404 | Yes | No | N/A | WT | No | No | No | August 2018 | PD | Deceased | 3.48 |
| 1301 | 56 | M | 0 | Skin, acral | IV | 280 | Yes | No | >95% | WT | No | No | No | January 2019 | SD | Deceased | 26.38 |
| 1302 | 70 | M | 1 | Skin, non-acral | IV | 195 | Yes | No | 75% | WT | No | No | No | March 2019 | PD | Deceased | 3.61 |
| 1305 | 67 | F | 1 | Skin, acral | IV | 274 | Yes | No | 2%–3% | WT | No | No | No | Jun 2019 | PD | Deceased | 24.41 |
| 1306 | 63 | M | 0 | Uveal | IV | 186 | Yes | No | 5% | WT | No | Yes | Yes¶ | Jun 2019 | SD | Deceased | 19.48 |
| 1272 | 74 | F | 0 | Skin, non-acral | IV | 155 | Yes | Yes | <1% | V600E | No | No | No | December 2016 | PD | Deceased | 43.33 |
| 1276 | 81 | M | 0 | Skin, non-acral | IV | 250 | Yes | Yes | N/A | N/A | No | No | Yes | April 2017 | CR | Alive | 54.44 |
| 1278 | 44 | M | 0 | Skin, non-acral | IIIC | 184 | No | Yes | 20%–25% | V600E | Yes | Yes | Yes¶ | June 2017 | PR | Alive | 52.70 |
| 1285 | 69 | M | 0 | Skin, non-acral | IV | 177 | No | Yes | 5%–10% | WT | No | No | Yes¶ | November 2017 | PR | Alive | 47.54 |
| 1295 | 79 | M | 1 | Skin, non-acral | IV | 223 | No | Yes | 10% | WT | No | No | No | July 2018 | PR | Deceased | 37.49 |
| 1299 | 60 | M | 1 | Uveal | IV | 992 | Yes | Yes | <1% | WT | No | No | Yes¶ | October 2018 | PD | Alive | 37.32 |
Bolded text highlights participants who were PD-1 Ab naïve, had a T cell Rsp, or had a PR or CR.
*The normal range for blood LDH level is between 125 and 250 U/L.
†Participants who had one or more additional sites of metastasis available for biopsy pretreatment and on day 22.
‡Per cent of tumor cells expressing PD-L1.
§Survival duration was calculated from the start of study treatment through the date of last follow-up or date of death.
¶Participants who had a fivefold T cell Rsp (hRsp).
**VMM 1290 had no T cell Rsp in peripheral blood mononuclear cells, but did have a T cell Rsp (Rsp and hRsp) in the sentinel immunized node.
Ab, antibody; CR, complete response; ECOG, Eastern Cooperative Oncology Group; F, female; hRsp, high T cell response; LDH, lactic acid dehydrogenase; M, male; N/A, not applicable or unknown; PD-1, programmed cell death protein 1; PD, progressive disease; PD-L1, programmed death-ligand 1; PR, partial response; PS, performance status; RECIST, Response Evaluation Criteria in Solid Tumors; response, Rsp; SD, stable disease; VMM, Virginia Malignant Melanoma; WT, wild type.
Toxicities and adverse events
The most common treatment-related adverse events (trAEs) of any grade were injection site reaction (91%), fatigue (82%), skin induration at vaccine sites (73%), anemia (41%), lymphopenia (27%), fever (27%), elevated aspartate aminotransferase (AST) (27%), pruritus (27%), and maculopapular rash (27%) (online supplemental table 2A). Maximum grades for the systemic trAEs were primarily grade 1 and were transient. Vaccine site reactions, induration, and ulceration were grade 2 in 5%, 23%, and 0% of participants, respectively, and were grade 3 in 5%, 0%, and 5%, respectively.
Four treatment-related DLTs were observed in three (14%) participants, which did not cross the study safety bound (online supplemental table 2B). All three participants were PD-1 Ab experienced. Two DLTs were attributed to pembrolizumab alone (VMM 1088, grade 3 elevated AST; VMM 1301, grade 3 uveitis), one was attributed to vaccine alone (VMM 1290, grade 3 skin ulceration at vaccine site), and one was attributed to pembrolizumab and vaccine (VMM 1290, grade 3 uveitis). Grade 3 elevated AST of VMM 1088 returned to pretrial baseline after a prednisone taper. VMM 1301 developed uveitis in the right eye after cataract surgery while on pembrolizumab; the last vaccine was 1-year prior to the DLT occurrence. That uveitis resolved with steroids; however, right eye vision remained poor due to pseudophakic cystoid macular edema secondary to cataract surgery. VMM 1290 had been treated with four cycles of ipilimumab/nivolumab and 27 cycles of pembrolizumab prior to study enrollment (PR as best RECIST response prior to subsequent disease progression) and is the one PD-1 Ab-experienced participant who had a PR on this study and remains alive still after 45 months. The uveitis of VMM 1290 resolved with steroids, and vision returned to pretrial baseline; the grade 3 skin ulceration at the vaccine site healed without antibiotics or surgery.
All 22 participants received one or more vaccinations. Nine (41%) completed all protocol treatment. Two (9%) stopped early for DLTs, while the third with a DLT (VMM 1301) completed all vaccinations over a year before DLT occurrence. Eleven (50%) discontinued early due to melanoma disease progression during active treatment (online supplemental figure 1).
CD4+ T cell response
T cell Rsps were evaluated against the pool of 6MHP using direct ex vivo IFN-γ ELISpot assays. An example direct ELISpot response is shown in online supplemental figure 2. Only 1/22 (4.5%; 90% CI (0% to 20%)) participants met criteria for a hdRsp, a PD-1 Ab-naïve participant (VMM 1278; table 2). At least twofold T cell Rsps were detected in 7/22 participants (32%; 90% CI (16% to 52%)) overall by week 13, including 4/6 PD-1 Ab naïve (67%; 90% CI (27% to 94%)) and 3/16 PD-1 Ab experienced (19%; 90% CI (5% to 42%)) (figure 3A). This included six with Rsp in PBMC and one with Rsp in a SIN. Among these seven participants with a Rsp, sites of primary melanoma were primary cutaneous non-acral (four), uveal (two), and cutaneous acral melanoma (one). Six of these same seven participants also met criteria for an hRsp (five in PBMC and one in a SIN). Two participants had a dRsp. The best immune response for each participant and the detailed ELISpot data are shown in table 1 and online supplemental table 3, respectively.
Table 2.
Participants with CD4+ T cell Rsps to 6 melanoma helper peptides vaccine
| High durable T cell Rsp in PBMC | Durable T cell Rsp in PBMC | Rsp in PBMC | hRsp in PBMC | Rsp in SIN | Rsp in PBMC by week 13 or in SIN | |||||
| #/n (%) | (90% CI)** | #/n (%) | (90% CI)** | #/n (%) | (90% CI)** | #/n (%) | (90% CI)** | #/n (%) | #/n (%) | |
| PD-1 Ab naïve | 1/6 (17%) | (1 to 58) | 1/6 (17%) | (1 to 58) | 4/6 (67%) | (27 to 94) | 3/6 (50%) | (15 to 85) | 0/3 (0%) | 4/6 (67%) |
| PD-1 Ab experienced | 0/16 (0%) | (0 to 17) | 1/16 (6%) | (0 to 26) | 2/16 (12.5%) | (2 to 34) | 2/16 (12.5%) | (2 to 34) | 1/7 (14%)† | 3/16 (19%) |
| All participants | 1/22 (4.5%) | (0 to 20) | 2/22 (9%) | (2 to 26) | 6/22 (27%) | (13 to 47) | 5/22 (23%) | (9 to 42) | 1/10 (10%)† | 7/22 (32%) |
*The Clopper-Pearson exact method was used to generate 90% CIs.
†This participant met criteria for both Rsp and hRsp in the SIN.
Ab, antibody; hRsp, high T cell response; PBMC, peripheral-blood mononuclear cell; PD-1, programmed cell death protein 1; Rsp, any T cell response; SIN, sentinel immunized node.
To explore whether the PBMC for the two cohorts differed in immune responsiveness at baseline, we compared the IFN-γ producing cells per 100,000 PBMC at baseline for the two cohorts, by Mann-Whitney U test, p values were 0.66, 0.08, and 0.75 for reactivity to CEF peptides, PHA, and PMA, respectively.
TME—multiplex immunohistochemistry and analysis
Tumor biopsies obtained pretreatment and day 22 for 12 participants (table 1) were evaluated with mIHC (representative images in online supplemental figures 3 and 4). CD4+ T cells were not reported because of technical challenges with that antibody. Total DAPI+ cells, pretreatment and day 22, for each of these 12 participants are provided in online supplemental table 4. TME evaluation in 12 participants revealed increased densities (cells/mm2) of CD8+ T cells (10/12, 83%), CD20+ B cells (10/12, 83%), and Tbet+ cells (9/12, 75%) by day 22 (table 3), among others. The median fold increases of these cells were 2.58, 2.62, and 4.02, respectively. Tumor biopsies were also evaluated for a classical TLS presence (yes/no), which was defined as a structure containing discrete B and T cell regions and containing PNAd+ vasculature indicative of high endothelial venules. No classical TLS structures were observed in pre-treatment or post-treatment biopsies for the 12 participants (online supplemental table 5). However, TLS-like structures containing several key features of TLS were identified in three participant specimens (VMM 1014, VMM 1265, and VMM 1301) post-treatment only (online supplemental table 5, figure 4B). These TLS-like structures contained partially organized B and T cell clusters and PNAd+ vasculature. Additionally, TLS-like structures consisting of distinct B and T cell regions but lacking PNAd+ vasculature, were observed in one of those same patient specimens post-treatment only (VMM 1301) (online supplemental table 5 and figure 4A). Many of the evaluated participant biopsies contained peritumoral stroma—where TLS are commonly found; however, most were core biopsies, and this may contribute to undersampling for TLS (online supplemental table 5). Additionally, TLS assessments were made in day 22 biopsies; thus, TLS maturation may occur at a later time point that was not sampled.
Table 3.
Infiltration of CD8+ T cells into the tumor microenvironment
| Cell type | Median baseline (cells/mm2) | Median day 22 (cells/mm2) | Median fold increase | Number of participants with fold increases (%) | Lower limit of 90% CI (% increasing) |
| CD8+ cells | 107.8 | 194.7 | 2.58 | 10* (83.3) | 61.4 |
| CD8+GzmB+ cells | 5.0 | 31.6 | 3.63 | 9* (75.0) | 52.5 |
| CD8+Tbet+ cells | 0.48 | 3.71 | 4.83 | 9* (75.0) | 52.5 |
| FoxP3+ cells | 11.63 | 13.75 | 1.15 | 8 (66.7) | 44.1 |
| Granzyme B+ cells | 9.11 | 37.6 | 2.93 | 8 (66.7) | 44.1 |
| Tbet+ cells | 1.34 | 7.45 | 4.02 | 9* (75.0) | 52.5 |
| CD20+ cells | 0.83 | 6.40 | 2.62 | 10* (83.3%) | 61.4 |
| CD8+Ki67+ cells | 59.5 | 104.6 | 2.07 | 10* (83.3%) | 61.4 |
| CD20+Ki67+ cells | 0.35 | 1.08 | 2.11 | 9* (75.0%) | 52.5 |
| CD83+ cells | 0.31 | 0.19 | 0.50† | 4 (33.3%) | 15.4 |
| Ki67+ cells | 980.0 | 1280.7 | 0.89 | 4 (33.3%) | 15.4 |
*Increased infiltration in 9/12 participants was designated as a promising result in the study design.
†Median fold increases calculated on 10 patients. Two were excluded for values of 0 at both baseline and Day 22.
Clinical outcome
Objective clinical responses were observed in 23% (one complete response (CR), four partial responses (PR)) of participants (table 4). All five participants with an objective clinical response had a primary cutaneous (non-acral) melanoma. Time to best clinical response was 9.5 (CR, VMM 1276), 7.8 (PR, VMM 1295), 2.1 (PR, VMM 1290), 1.8 (PR, VMM 1278), and 1.7 (PR, VMM 1285) months. VMM 1276 first achieved a PR at 1.8 months and subsequently a CR at 9.5 months. T cell Rsps to 6MHP were observed in 4 of 5 participants with CR/PR (80%), but in only 3 of 17 participants with SD or progressive disease (PD) as best clinical response (17.6%). However, among those 17 participants, 11 discontinued study treatment prior to administration of the last vaccine and collection of week 13 PBMC; 2 of 3 participants with a T cell Rsp and SD or PD completed all vaccinations with PBMC obtained through at least week 13 (VMM 1299 had PBMC collected through week 12 and thus missed one vaccine/blood draw). Among the six participants naïve to PD-1 blockade, four experienced objective clinical responses, including one CR and three PRs (4/6; ORR 67%) (figure 3A). These six participants included one with uveal melanoma. Thus, objective responses were observed in 4/5 participants with melanoma metastatic from a cutaneous primary (80%). PD-L1 expression by melanoma cells for these six participants was negative (<1%) for two, positive (≥1%) for three, and unknown for one. BRAF status was V600E mutated for two, wild-type for three, and unknown for one (table 1). Among the 16 participants who had previously had PD-1 antibody therapy, there was 1 PR (1/16; ORR 6%) and 4 SD for an overall DCR of 31% (5/16). Duration of SD for these four participants was 2.4 (VMM 1287), 2.9 (VMM 1274), 5.7 (VMM 1306), and 8.0 (VMM 1301) months.
Table 4.
Best clinical response on study treatment
| PD-1 Ab naïve (n=6) |
PD-1 Ab experienced (n=16) |
All participants (n=22) |
|
| Complete response | 1 (16.7%) (0.4% to 64.1%) |
0 (0.0% to 20.6%) |
1 (4.5%) (0.1% to 22.8%) |
| Partial response | 3 (50.0%) (11.8% to 88.2%) |
1 (6.2%) (0.2% to 30.2%) |
4 (18.2%) (5.2% to 40.3%) |
| Stable disease | 0 (0.0% to 45.9%) |
4 (25%) (7.3% to 52.4%) |
4 (18.2%) (5.2% to 40.3%) |
| Progressive disease | 2 (33.3%) (4.3% to 77.7%) |
11 (68.8%) (41.3% to 89.0%) |
13 (59.1%) (36.4% to 79.3%) |
| Number progression-free | 2 (33.3%) (4.3% to 77.7%) |
0 (0.0% to 20.6%) |
2 (9.1%) (1.1% to 29.2%) |
| Number alive | 4 (66.7%) (22.3% to 95.7%) |
2 (12.5%) (1.6% to 38.4%) |
6 (27.3%) (10.7% to 50.2%) |
Number of participants, % of total, (95% CI).
OS and PFS outcomes for these two patient subsets are shown in figure 2. The median OS for PD-1 Ab-experienced participants was 10.6 months (95% CI (4.34 to 47.38)), and was not reached for PD-1 Ab-naïve participants (HR 6.3 (90% CI (2.1 to 28.7)). Median PFS was 1.9 months (95% CI (1.64 to 12.03)) and 6.2 months (95% CI (1.31 to 16.79)) for PD-1 Ab-experienced and Ab-naïve participants, respectively (HR 3.7 (90% CI (1.4 to 12.4)). Median follow-up for the two subsets was 24.4 months. After a median follow-up of 45.4 months (range 37–55 months), 4/6 PD-1 Ab-naïve participants remain alive. Additional exploratory analyses were performed to understand if clinical response or T cell Rsp was associated with OS (figure 3). Since both are time-dependent variables, these were assessed in a landmark analysis at 13 weeks, since the vaccines were completed by then. OS based on landmark analysis was prolonged for those with objective clinical responses by week 13 (CR/PR vs SD/PD: 3-year OS was 100% (90% CI (100% to 100%) given no deaths for CR/PR vs 22% (90% CI (8% to 41%) for SD/PD; figure 3B). VMM 1295 had a PR at 7.8 months and thus had not experienced a PR before the landmark at 13 weeks. The four participants who developed an objective response by week 13 continue to be alive at last follow-up, with one of them going on to develop a CR by 9.5 months. The landmark analysis was based on participants with at least a 2× T cell Rsp over background consistent with the definition used in other prior vaccine trials,22 and the more stringent hdRsp was only observed in one participant. Interestingly, OS was significantly longer for those with T cell Rsps by week 13 in this landmark analysis (HR 6.5 (90% CI (2.1 to 29.2)), figure 3C).
Figure 2.

Clinical outcomes overall and as a function of clinical cohort. Kaplan-Meier survival curves show estimated overall survival (A) and progression-free survival (B) for the full dataset (n=22), and overall survival (C) and progression-free survival (D) stratified by clinical cohort (programmed death 1 (PD-1) antibody (Ab) naïve or experienced).
Figure 3.

(A) Immune response and objective clinical response. (B) Landmark analysis of overall survival by complete/partial response (CR/PR) by week 13. (C) Landmark analysis of overall survival by twofold T cell responses by week 13. Ab, antibody; PD-1, programmed death 1.
Discussion
To our knowledge, this is the first trial reporting safety, immunogenicity, and clinical outcomes of a peptide vaccine designed to stimulate CD4+ T cells co-administered with PD-1 blockade for participants with advanced melanoma who were PD-1 Ab naïve or PD-1 Ab experienced. Treatment-related DLTs were observed in three (14%) participants, which did not cross the study safety bound, supporting safety of this combination. An hdRsp to 6MHP was detected in only one participant, but twofold T cell Rsps to 6MHP were detected in 7/22 by week 13. Objective clinical responses were observed in 23%, including 4/6 PD-1 Ab naïve (67%) and 1/16 PD-1 Ab experienced (6%, table 1). OS was longer for PD-1 Ab-naïve than Ab-experienced participants. In landmark analyses at 13 weeks, OS was also longer for those with T cell Rsps and for those with objective responses. TME evaluation revealed increased densities of CD8+ T cells, CD20+ B cells, and Tbet+ cells by day 22.
Our prior work has demonstrated that vaccines using 6MHP, restricted by class II MHC, emulsified in IFA induce Th1-dominant CD4+ T cell Rsps in 40%–80% of patients and induce objective clinical responses.13 15 However, some T cell Rsps with IFA are transient, and not all participants develop high and/or durable responses. Thus, there is still a need to enhance immune responses to vaccines. In the present study, we have found that vaccination with 6MHP in IFA plus pembrolizumab induced T cell Rsps in 7/22 participants (32%) overall, of which 4/6 were PD-1 Ab naïve (67%) and 3/16 were PD-1 Ab experienced (19%). Only 1 of 22 participants met criteria for hypothesis testing of a hdRsp, which was less than the 18% hdRsp rate with 6MHP plus IFA alone in the prior Mel44 trial.22 The lower than anticipated immune response rate reported in the current trial may in part be attributable to 73% of study participants (16/22) being PD-1 Ab experienced. Recent data in mice and humans have shown that the timing of PD-1 blockade relative to the administration of a cancer vaccine may be crucial in dictating the therapeutic responses observed.20 21 33 Among PD-1-resistant mouse models, simultaneous PD-1 blockade with vaccine therapy reversed PD-1 resistance and consequently reduced tumor volume and prolonged OS. However, when PD-1 blockade was given before antigen priming, the therapeutic benefit seen from their simultaneous administration was eliminated.21 In a human trial for metastatic prostate cancer, prostate-specific antigen levels decreased with concurrent, but not sequential, administration of PD-1 blockade plus a cancer vaccine, and measurable tumor volume decreased for 4/5 patients treated concurrently.33 Since most participants develop an immune response to vaccination by week 13,13 22 the prior murine study21 raises the possibility that the most optimal timing for initiation of PD-1 blockade is after an immune response has occurred, rather than before the cancer vaccine. PD-1 blockade prior to vaccine generated dysfunctional PD-1+CD38hiCD8+ T cells, which mediated resistance to anti-PD-1 Ab.21 Administering PD-1 blockade before vaccine may create a negative feedback loop by which more IFN-γ is released, PD-L1 and IDO expression is upregulated, and regulatory T cells are recruited to the TME,34 collectively blunting new T cell proliferation on exposure to a subsequent vaccine antigen. Our results corroborate this hypothesis as 4/6 PD-1 Ab-naïve participants (67%) had a T cell Rsp, which is within range of immune response rates of prior studies,12–14 and the only participant with an hdRsp was also PD-1 Ab naïve. Our data, combined with that from the aforementioned mice and human studies, support initiating vaccines prior to, or concurrent with, administration of PD-1 blockade therapy. This may be relevant to consider in analysis and design of ongoing clinical trials of neoantigen vaccines, where PD-1 Ab is given prior to creation of the neoantigen vaccine. Furthermore, given the induction of dysfunctional PD-1+CD38hiCD8+ T cells reported by Verma et al,21 investigating the presence of PD-1+CD38hi cells in CD8+ and CD4+ cells would be interesting to investigate in future studies and with a larger sample size.
It is not unexpected for PD-1 blockade alone to increase intratumoral T and B cells, as well as Th1 (Tbet+) cells in the TME35; however, peptide vaccines may further enhanced tumor targeting and infiltration.19 36 The impact of the vaccines in this trial may be direct, by enhancing CD4+ T cells reactive to tumor antigens and possibly by inducing epitope spreading to CD8+ T cells, and indirect by secretion of IFN-γ by those T cells, which in turn enhances expression of critical homing receptor ligands by tumor-associated endothelium.37 The impact of vaccines is supported by murine models of colon and breast carcinoma which demonstrated the addition of a cancer vaccine to PD-L1 inhibition significantly enhanced CD4+ and CD8+ T cell infiltration into the TME and promoted the Th1 phenotype of TILs.36 In humans, Kjeldsen et al demonstrated increases in vaccine-specific CD3+ and CD8+ T cells in tumors of 4/5 patients, 3 of whom had an objective clinical response.19 We have not yet evaluated for epitope spreading in this study and have not infiltrating T cells or B cells for reactivity to melanoma antigens generally or to peptides in our vaccines more specifically. In ongoing studies, we are working to understand whether CD4+ T cells induced by vaccination infiltrate melanoma metastases, whether they remain functional in situ, and whether they may explain in part the infiltration by other CD4+ or CD8+ T cells.
In the present study, objective clinical responses were seen in 4/6 PD-1 Ab-naïve participants (67%; 1 CR, 3 PR), which compares favorably to that of 33% published for pembrolizumab alone.3 Since uveal melanoma tends to respond poorly to PD-1 blockade,38 39 it may be more relevant that the objective clinical response rate among PD-1 Ab-naïve participants with non-uveal melanomas was 4/5 (80%; 90% CI (34% to 99%)). Although a limitation of our study is that the sample size is small among PD-1 Ab-naïve participants, our results are nonetheless provocative. Interestingly, the 80% ORR among PD-1 Ab-naïve participants with cutaneous melanomas is concordant with data from a recent clinical trial using a peptide vaccine of similar length plus nivolumab among PD-1 Ab-naïve participants with metastatic cutaneous (non-uveal) melanoma, which reported an 80% ORR (90% CI (63% to 91%)).19 While the power in the current study is too small to draw a conclusion, it nevertheless provides insight into a possible synergistic effect between 6MHP plus pembrolizumab. Since the CI of the 4/5 participants with non-uveal melanomas who had an objective clinical response was 34%–99%, a larger sample size is needed to provide a more precise estimate of the response rate in those naïve to PD-1 blockade.
In our study, OS was prolonged for PD-1 Ab-naïve versus Ab-experienced participants as well as for those with T cell Rsps and for those with objective responses in landmark analyses. This prolonged OS may in part be attributable to the higher immune response rates seen among the PD-1 Ab-naïve patients, more optimal antigen priming prior to PD-1 blockade as discussed in the aforementioned murine21 and human33 studies, or due to the nature of disease among PD-1 Ab-experienced patients that, by definition, have progressed on prior PD-1 and Cytotoxic T Lymphocyte Associated Protein 4 (CTLA-4) therapies.
Conclusion
In participants with advanced melanoma, combined treatment with the 6MHP vaccine plus pembrolizumab was safe, increased intratumoral T and B cells, as well as Th1 (Tbet+) cells, and induced T cell Rsps that were associated with prolonged OS. The low T cell Rsp rate in PD-1 Ab-experienced participants corroborates prior murine studies that caution against delaying cancer vaccines until after PD-1 blockade. The promising ORR and OS in PD-1 Ab-naïve participants support consideration of a larger study to assess definitive benefit.
Acknowledgments
We thank Merck for providing pembrolizumab at no charge for participants on this trial.
Footnotes
Twitter: @vkmd2
Correction notice: This article has been corrected since it was first published online. The author Rick Daniel Vavolizza has been updated to Rick D Vavolizza.
Contributors: All authors contributed to the manuscript writing. CLS, GRP, RDV, and KAC-B contributed to the study design. WCO, KTS, and ISM performed the laboratory studies. CLS, KH, EMG, WWG, and VK enrolled study participants. CLS, KH, EMG, WWG, and VK assessed and reported clinical toxicities and other clinical data and outcomes. CLS, KH, and LTD performed skin and lymph node biopsies. RDV, CLS, GRP, and NV provided data analysis and final data reporting. CLS is the guarantor, who accepts full responsibility for the finished work and/or the conduct of the study, had access to the data, and controlled the decision to publish.
Funding: NCI R01 CA178846; T32 CA163177 (RDV); Merck; P30 CA044579 (Molecular, Immunologic, and Translational Sciences Core, Biorepository and Tissue Research Facility, Office of Clinical Research, and Biostatistics Shared Resource); gifts from Alice and Bill Goodwin and the Commonwealth Foundation for Cancer Research.
Competing interests: CLS has the following disclosures: research support to the University of Virginia from Celldex (funding, drug), GSK (funding), Merck (funding, drug), 3M (drug), Theraclion (device staff support); funding to the University of Virginia from Polynoma for PI role on the MAVIS Clinical Trial; funding to the University of Virginia for roles on Scientific Advisory Boards for Immatics and CureVac. CLS also receives licensing fee payments through the UVA Licensing and Ventures Group for patents for peptides used in cancer vaccines.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
The clinical trial MEL64 was performed with IRB-HSR (#18174) and FDA approval (IND #10825) and is registered with ClinicalTrials.gov (NCT02515227). Participants gave informed consent to participate in the study before taking part.
References
- 1. Eggermont AMM, Blank CU, Mandala M, et al. Longer follow-up confirms recurrence-free survival benefit of adjuvant pembrolizumab in high-risk stage III melanoma: updated results from the EORTC 1325-MG/KEYNOTE-054 trial. J Clin Oncol 2020;38:3925–36. 10.1200/JCO.20.02110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hamid O, Robert C, Daud A, et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann Oncol 2019;30:582–8. 10.1093/annonc/mdz011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521–32. 10.1056/NEJMoa1503093 [DOI] [PubMed] [Google Scholar]
- 4. Nowicki TS, Hu-Lieskovan S, Ribas A. Mechanisms of resistance to PD-1 and PD-L1 blockade. Cancer J 2018;24:47–53. 10.1097/PPO.0000000000000303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71. 10.1038/nature13954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schoenberger SP, Toes RE, van der Voort EI, et al. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 1998;393:480–3. 10.1038/31002 [DOI] [PubMed] [Google Scholar]
- 7. Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature 1998;393:474–8. 10.1038/30989 [DOI] [PubMed] [Google Scholar]
- 8. Bennett SR, Carbone FR, Karamalis F, et al. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 1998;393:478–80. 10.1038/30996 [DOI] [PubMed] [Google Scholar]
- 9. Melssen M, Slingluff CL. Vaccines targeting helper T cells for cancer immunotherapy. Curr Opin Immunol 2017;47:85–92. 10.1016/j.coi.2017.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sahin U, Derhovanessian E, Miller M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017;547:222–6. 10.1038/nature23003 [DOI] [PubMed] [Google Scholar]
- 11. Lowenfeld L, Mick R, Datta J, et al. Dendritic Cell Vaccination Enhances Immune Responses and Induces Regression of HER2pos DCIS Independent of Route: Results of Randomized Selection Design Trial. Clin Cancer Res 2017;23:2961–71. 10.1158/1078-0432.CCR-16-1924 [DOI] [PubMed] [Google Scholar]
- 12. Reed CM, Cresce ND, Mauldin IS, et al. Vaccination with melanoma helper peptides induces antibody responses associated with improved overall survival. Clin Cancer Res 2015;21:3879–87. 10.1158/1078-0432.CCR-15-0233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Slingluff CL, Petroni GR, Olson W, et al. Helper T-cell responses and clinical activity of a melanoma vaccine with multiple peptides from MAGE and melanocytic differentiation antigens. J Clin Oncol 2008;26:4973–80. 10.1200/JCO.2008.17.3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu Y, Petroni GR, Olson WC, et al. Immunologic hierarchy, class II MHC promiscuity, and epitope spreading of a melanoma helper peptide vaccine. Cancer Immunol Immunother 2014;63:779–86. 10.1007/s00262-014-1551-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Slingluff CL, Lee S, Zhao F, et al. A randomized phase II trial of multiepitope vaccination with melanoma peptides for cytotoxic T cells and helper T cells for patients with metastatic melanoma (E1602). Clin Cancer Res 2013;19:4228–38. 10.1158/1078-0432.CCR-13-0002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Crawford A, Angelosanto JM, Kao C, et al. Molecular and transcriptional basis of CD4⁺ T cell dysfunction during chronic infection. Immunity 2014;40:289–302. 10.1016/j.immuni.2014.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weber JS, Kudchadkar RR, Yu B, et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J Clin Oncol 2013;31:4311–8. 10.1200/JCO.2013.51.4802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gibney GT, Kudchadkar RR, DeConti RC, et al. Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin Cancer Res 2015;21:712–20. 10.1158/1078-0432.CCR-14-2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kjeldsen JW, Lorentzen CL, Martinenaite E, et al. A phase 1/2 trial of an immune-modulatory vaccine against IDO/PD-L1 in combination with nivolumab in metastatic melanoma. Nat Med 2021;27:2212–23. 10.1038/s41591-021-01544-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Messenheimer DJ, Jensen SM, Afentoulis ME, et al. Timing of PD-1 blockade is critical to effective combination immunotherapy with Anti-OX40. Clin Cancer Res 2017;23:6165–77. 10.1158/1078-0432.CCR-16-2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Verma V, Shrimali RK, Ahmad S, et al. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1+CD38hi cells and anti-PD-1 resistance. Nat Immunol 2019;20:1231–43. 10.1038/s41590-019-0441-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Slingluff CL, Petroni GR, Chianese-Bullock KA, et al. Randomized multicenter trial of the effects of melanoma-associated helper peptides and cyclophosphamide on the immunogenicity of a multipeptide melanoma vaccine. J Clin Oncol 2011;29:2924–32. 10.1200/JCO.2010.33.8053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Balch CM, Gershenwald JE, Soong S-J, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 2009;27:6199–206. 10.1200/JCO.2009.23.4799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–47. 10.1016/j.ejca.2008.10.026 [DOI] [PubMed] [Google Scholar]
- 25. Schwartz LH, Litière S, de Vries E, et al. RECIST 1.1-Update and clarification: from the RECIST Committee. Eur J Cancer 2016;62:132–7. 10.1016/j.ejca.2016.03.081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dillon PM, Olson WC, Czarkowski A, et al. A melanoma helper peptide vaccine increases Th1 cytokine production by leukocytes in peripheral blood and immunized lymph nodes. J Immunother Cancer 2014;2:23. 10.1186/2051-1426-2-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Slingluff CL, Petroni GR, Chianese-Bullock KA, et al. Immunologic and clinical outcomes of a randomized phase II trial of two multipeptide vaccines for melanoma in the adjuvant setting. Clin Cancer Res 2007;13:6386–95. 10.1158/1078-0432.CCR-07-0486 [DOI] [PubMed] [Google Scholar]
- 28. Slingluff CL, Yamshchikov GV, Hogan KT, et al. Evaluation of the sentinel immunized node for immune monitoring of cancer vaccines. Ann Surg Oncol 2008;15:3538–49. 10.1245/s10434-008-0046-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. O'Kane GM, Labbé C, Doherty MK, et al. Monitoring and management of immune-related adverse events associated with programmed cell death protein-1 axis inhibitors in lung cancer. Oncologist 2017;22:70–80. 10.1634/theoncologist.2016-0164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Robert C, Ribas A, Wolchok JD, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 2014;384:1109–17. 10.1016/S0140-6736(14)60958-2 [DOI] [PubMed] [Google Scholar]
- 31. Currier JR, Kuta EG, Turk E, et al. A panel of MHC class I restricted viral peptides for use as a quality control for vaccine trial ELISPOT assays. J Immunol Methods 2002;260:157–72. 10.1016/S0022-1759(01)00535-X [DOI] [PubMed] [Google Scholar]
- 32. Sergeant E. Epitools Epidemiological Calculators. Ausvet. Available: http://epitools.ausvet.com.au
- 33. McNeel DG, Eickhoff JC, Wargowski E, et al. Concurrent, but not sequential, PD-1 blockade with a DNA vaccine elicits anti-tumor responses in patients with metastatic, castration-resistant prostate cancer. Oncotarget 2018;9:25586–96. 10.18632/oncotarget.25387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Spranger S, Spaapen RM, Zha Y, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 2013;5:200ra116. 10.1126/scitranslmed.3006504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Riaz N, Havel JJ, Makarov V, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 2017;171:934–49. 10.1016/j.cell.2017.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Horn LA, Fousek K, Hamilton DH, et al. Vaccine increases the diversity and activation of intratumoral T cells in the context of combination immunotherapy. Cancers 2021;13. 10.3390/cancers13050968. [Epub ahead of print: 25 02 2021]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peske JD, Woods AB, Engelhard VH. Control of CD8 T-cell infiltration into tumors by vasculature and microenvironment. Adv Cancer Res 2015;128:263–307. 10.1016/bs.acr.2015.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rossi E, Pagliara MM, Orteschi D, et al. Pembrolizumab as first-line treatment for metastatic uveal melanoma. Cancer Immunol Immunother 2019;68:1179–85. 10.1007/s00262-019-02352-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jansen YJL, Seremet T, Neyns B. Pembrolizumab for the treatment of uveal melanoma: a case series. Rare Tumors 2020;12:2036361320971983. 10.1177/2036361320971983 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2022-005424supp001.pdf (1.7MB, pdf)
jitc-2022-005424supp002.pdf (9.5MB, pdf)
Data Availability Statement
Data are available on reasonable request.