

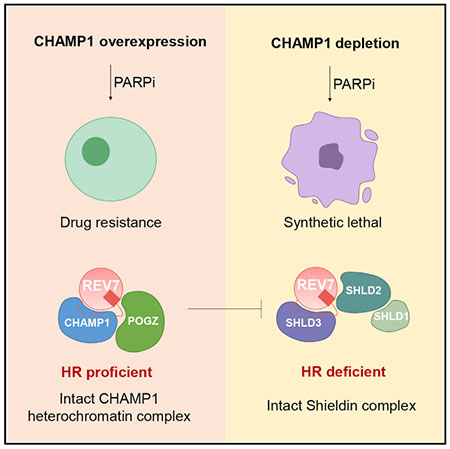
SUMMARY
A critical determinant of DNA repair pathway choice is REV7, an adaptor that binds to various DNA repair proteins through its C-terminal seatbelt domain. The REV7 seatbelt binds to either REV3, activating translesion synthesis, or to SHLD3, activating non-homologous end joining (NHEJ) repair. Recent studies have identified another REV7 seatbelt-binding protein, CHAMP1 (chromosome alignment-maintaining phosphoprotein 1), though its possible role in DNA repair is unknown. Here, we show that binding of CHAMP1 to REV7 activates homologous recombination (HR) repair. Mechanistically, CHAMP1 binds directly to REV7 and reduces the level of the Shieldin complex, causing an increase in double-strand break end resection. CHAMP1 also interacts with POGZ in a heterochromatin complex further promoting HR repair. Importantly, in human tumors, CHAMP1 overexpression promotes HR, confers poly (ADP-ribose) polymerase inhibitor resistance, and correlates with poor prognosis. Thus, by binding to either SHLD3 or CHAMP1 through its seatbelt, the REV7 protein can promote either NHEJ or HR repair, respectively.
In brief
Feng et al. demonstrate that CHAMP1 promotes homologous recombination by binding to REV7 and reducing the level of the Shieldin complex, causing an increase in double-strand break end resection. CHAMP1 and POGZ form a complex to further promote HR. Upregulation of CHAMP1 expression is a mechanism of acquired PARP inhibitor resistance.
Graphical Abstract
INTRODUCTION
REV7 (also known as MAD2L2, MAD2B, or FANCV) is a highly conserved member of the HORMA family of proteins, named for its three founding members: HOp1, a meiotic chromosome axis factor, REV7, and MAD2, a spindle assembly checkpoint protein (Clairmont and D’Andrea, 2021; de Krijger et al., 2021a). REV7 is an abundant cellular protein and is unique among HORMA proteins, both in its large number of binding partners and in its involvement in multiple distinct pathways. Germline biallelic mutations in the REV7 gene can cause the inherited chromosome instability syndrome Fanconi anemia (Bluteau et al., 2016). REV7 adopts the two classic closed and open seatbelt conformations of HORMA proteins, and SHLD3 and REV3 are among its seatbelt-dependent binding partners (Clairmont et al., 2020).
REV7 is an important determinant of DNA repair pathway choice (Clairmont and D’Andrea, 2021). When closed REV7 (c-REV7) binds to SHLD3, this interaction promotes the assembly of the Shieldin complex (Findlay et al., 2018; Ghezraoui et al., 2018; Gupta et al., 2018; Tomida et al., 2018). The Shieldin complex in turns blocks double-strand break (DSB) end resection, promotes the fill-in of resected DSBs, and promotes non-homologous end joining (NHEJ) (Dev et al., 2018; Gao et al., 2018; Mirman et al., 2018; Noordermeer et al., 2018). When c-REV7 binds to REV3 in the POLζ complex, the interaction promotes error-prone translesion synthesis (TLS) repair.
The AAA + ATPase, TRIP13, along with its substrate adaptor p31comet, can open REV7 and release SHLD3 or REV3 (Clairmont et al., 2020; Sarangi et al., 2020). Similarly, TRIP13 and p31comet are known to open other HORMA proteins, such as MAD2 (Brulotte et al., 2017; Miniowitz-Shemtov et al., 2015; Ye et al., 2015). The mechanism by which REV7 is converted from the inactive open conformation back to the active closed form is less well understood, and it may be DNA damage dependent. The mechanism may also involve the binding of another seatbelt-binding motif (SBM)-containing protein or a new post-translational modification.
REV7 has at least one additional seatbelt-binding partner, CHAMP1 (also known as C13orf80, CAMP, or ZNF828). CHAMP1 is a little-known but highly conserved zinc finger protein first identified as a REV7 interactor (Itoh et al., 2011). CHAMP1 localizes to chromosomes, recruits REV7 to spindles, and plays a role in kinetochore-microtubule interactions. Disruption of CHAMP1 leads to characteristic defects in chromosome alignment in mitosis. Germline heterozygous mutations in CHAMP1 are associated with a rare syndromic form of intellectual disability in humans (Isidor et al., 2016). Crystallographic analysis of the REV7/CHAMP1 complex (Hara et al., 2017) revealed a strong similarity to the REV7/REV3 and REV7/SHLD3 interaction surface (Hara et al., 2010); however, some structural features are distinct, suggesting dissimilar binding affinities of the interacting partners. Despite the clear role of REV7 in DNA repair pathway choice, little is known about the role of its interactor CHAMP1 in DNA repair.
Here, we demonstrate that the binding of CHAMP1 to REV7 promotes DSB end resection and error-free HR repair. CHAMP1 binds directly to the seatbelt domain of REV7, thereby increasing HR repair and competing with the binding of SHLD3 to REV7 in the Shieldin complex. High cellular levels of CHAMP1 protein favor HR repair over NHEJ and are often observed in human tumors with acquired HR proficiency. Moreover, CHAMP1 is a component of a large, multisubunit heterochromatin complex, containing HP1α, LEDGF, HDGFRP2, and POGZ, previously shown to promote HR activity (Baude et al., 2016; Clairmont et al., 2020; Daugaard et al., 2012; Nozawa et al., 2010; Vermeulen et al., 2010). One function of this complex may be to sequester REV7 away from other error-prone repair pathways, perhaps under specific cellular conditions, after specific DNA damaging events, and at specific regions of the genome.
RESULTS
CHAMP1 promotes homologous recombination repair
In order to determine the possible involvement of CHAMP1 in DNA repair, we knocked down CHAMP1 expression with siRNA in U2OS cells (Figure 1). Interestingly, CHAMP1 knockdown resulted in a reduction in HR activity, based on the decrease in GFP fluorescence generated by the DR-GFP template versus the EJ5-GFP template (Pierce et al., 1999; Stark et al., 2004) (Figures 1A, 1B, and S1A). Since an early step in HR repair is DSB end resection (Symington, 2014), we used the single-molecule analysis of resection tracks (SMART) assay (Huertas and Cruz-Garcia, 2018) to quantify resection. Indeed, two siRNAs to CHAMP1 decreased DSB end resection (Figure 1C). Cells with an HR deficiency have a defect in RAD51 foci assembly and exhibit sensitivity to poly (ADP-ribose) polymerase (PARP) inhibitors (PARPi) (Bryant et al., 2005; Farmer et al., 2005). Accordingly, RPE-1 cells or U2OS cells with a CRISPR-Cas9-mediated knockout of CHAMP1 exhibited reduced RAD51 foci (Figure 1D) and were sensitive to the PARPi Olaparib (Figures 1E, 1F, and S1B–S1D).
Figure 1. CHAMP1 promotes homologous recombination.
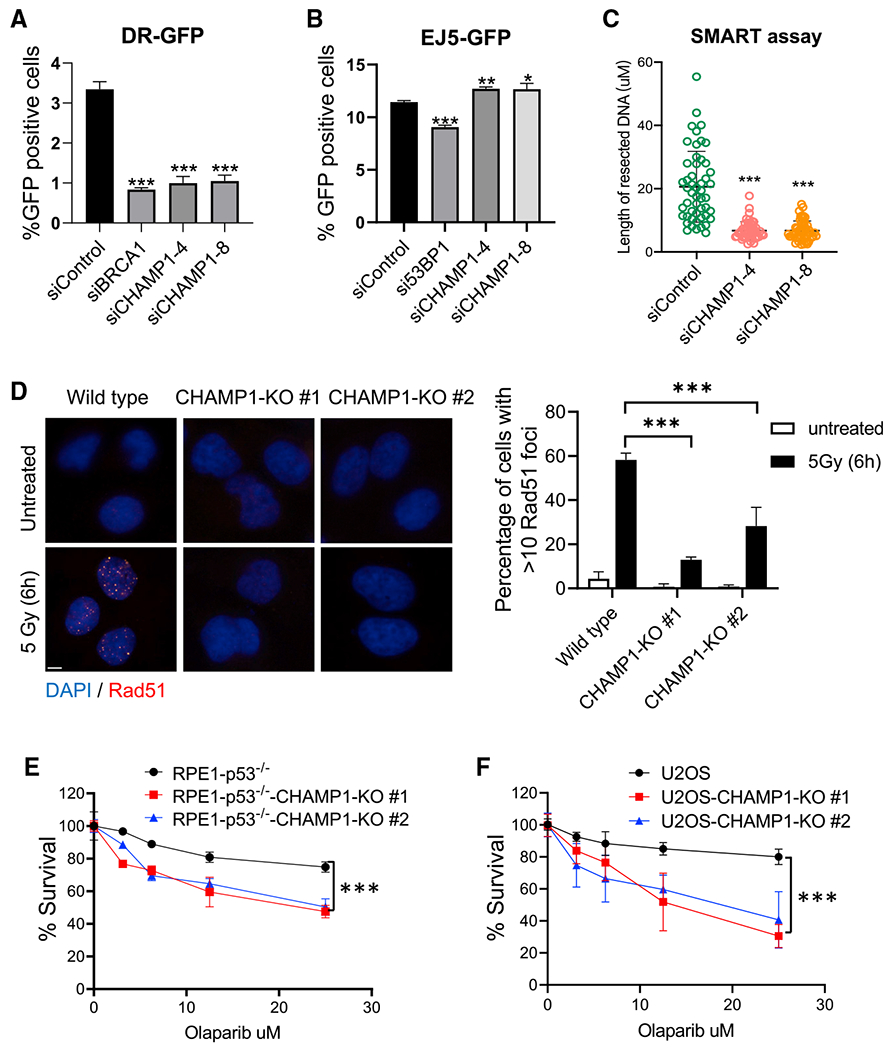
(A) Graph showing the percentage of GFP-positive cells after DR-GFP analysis. U2OS cells were infected with I-SceI adenovirus and knocked down for BRCA1 or CHAMP1 using siRNA. N = 3 biologically independent experiments. Error bars indicate SD, and p values were calculated using two-tailed Student’s t test, ***p < 0.001.
(B) Graph showing the percentage of GFP-positive cells after EJ5-GFP analysis. U2OS cells were infected with I-SceI adenovirus and knocked down for 53BP1 or CHAMP1 using siRNA. N = 3 biologically independent experiments. Error bars indicate SD, and p values were calculated using two-tailed Student’s t test, ***p < 0.001, **p < 0.01, *p < 0.05.
(C) Quantification of resected ssDNA measured by SMART assay in U2OS cells treated by siControl or siRNAs targeting CHAMP1 for 48 h. Approximately 50 fibers were counted per experiment. Error bars indicate SD, and p values were calculated using Student’s t test, ***p < 0.001.
(D) (Left) Representative images of RAD51 foci formation in wild-type and two CHAMP1 knockout U2OS cell lines 6 h after 5Gy IR treatment. Scale bar, 5 μm. (Right) Quantification of >10 RAD51 foci. n = 3 biologically independent experiments. Error bars indicate SD, ***p < 0.001. Statistical analysis was performed using two-tailed Student’s t test.
(E) 3-day cytotoxicity analysis of wild-type and two CHAMP1 knockout RPE1(p53−/−) cell lines treated with various doses of olaparib; n = 3 independent experiments. Wild-type versus CHAMP1-KO#1, ***p < 0.001; wild-type versus CHAMP1-KO#2; error bars indicate SD, ***p < 0.001; statistical analysis was performed using two-way ANOVA.
(F) 3-day cytotoxicity analysis of wild-type and two CHAMP1 knockout U2OS cell lines treated with various doses of olaparib. Cell viability was detected by CellTiter-Glo (Promega), n = 3 independent experiments. Wild-type versus CHAMP1-KO#1, ***p < 0.001; wild-type versus CHAMP1-KO#2; error bars indicate SD, ***p < 0.001; statistical analysis was performed using two-way ANOVA.
DNA damage activates the binding of REV7 to CHAMP1
Previous studies have demonstrated that CHAMP1 interacts directly with REV7 (Hara et al., 2017), a known regulator of DNA HR repair (Boersma et al., 2015; Xu et al., 2015). To confirm and extend these findings, we showed that DNA damage with ionizing radiation (IR) activated the binding of CHAMP1 and REV7 and stimulated the colocalization of REV7 and CHAMP1 in nuclear foci (Figures 2A–2C). In contrast, there was relatively little change in the level of this complex during cell cycle progression (Figure S2A). IR exposure increased the REV7/CHAMP1 interaction, reaching maximum levels after 1 hour, followed by a rapid decline (Figures 2D and 2E), and CHAMP1-deficient cells were sensitive to IR (Figure S2B). Taken together, we reasoned that the DNA damage-inducible interaction of CHAMP1 and REV7 may be required for HR repair.
Figure 2. DNA damage activates CHAMP1 binding to REV7.
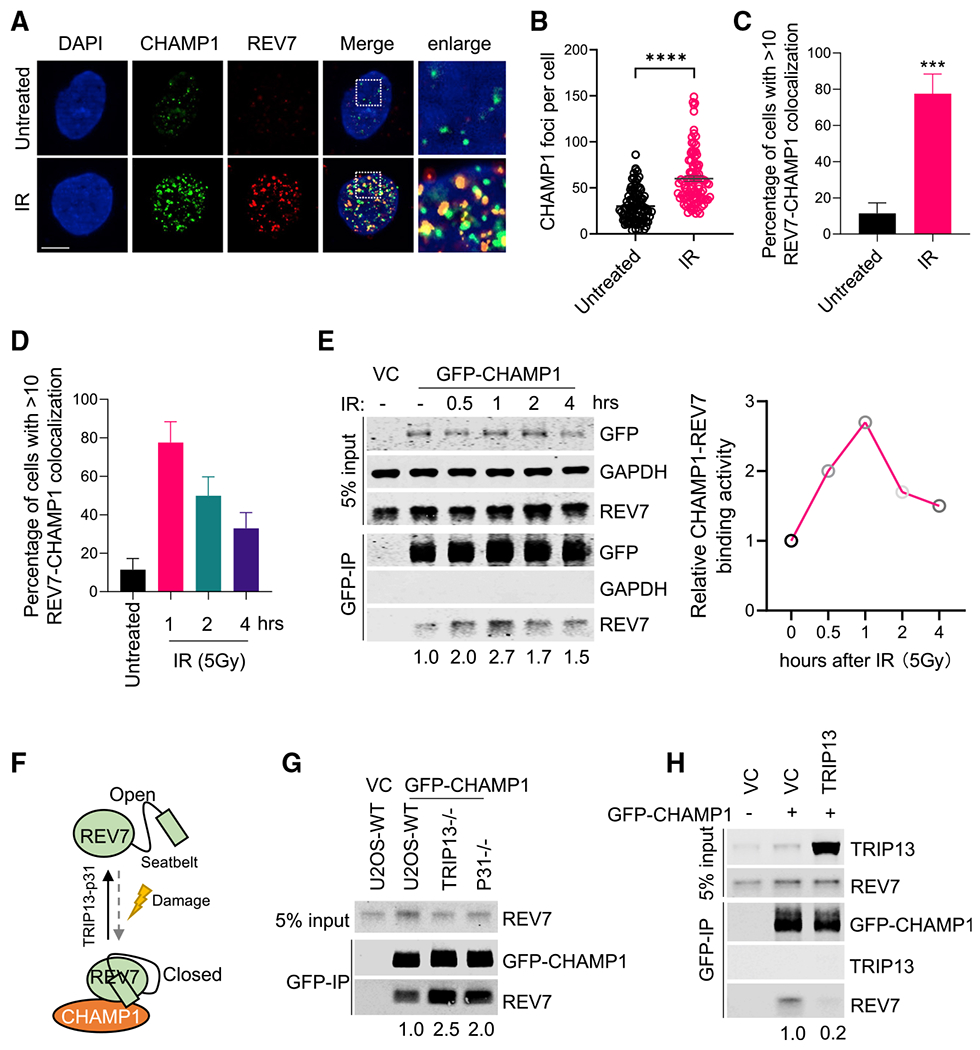
(A) Representative images of CHAMP1 and REV7 foci 1 h after IR (5Gy) treatment in RPE1 cells. Colocalizations of CHAMP1 and REV7 are shown as indicated. DAPI was used to stain the nuclei. Scale bar, 5 μm.
(B) Quantification of CHAMP1 foci shown in (A). Error bars indicate SEM, and p values were calculated using two-tailed Student’s t test. ****p < 0.0001.
(C) Quantification of CHAMP1-REV7 colocalized foci shown in (A). Error bars indicate SD (n = 3), and p values were calculated using two-tailed Student’s t test. ***p < 0.001.
(D) Quantification of CHAMP1-REV7 colocalized foci with or without IR (5Gy) treatment as indicated. Error bars indicate SD.
(E) (Left) Western blot showing GFP-immunoprecipitation of GFP-tagged CHAMP1 in 293T cells after IR (5Gy) treatment as indicated and the co-immunoprecipitation of endogenous REV7. VC, vector control. (Right) Quantification of the relative CHAMP1-REV7 binding activity.
(F) A cartoon showing that CHAMP1 interacts with the closed REV7, which is regulated by TRIP1-p31 complex and DNA damage.
(G) Western blot showing GFP-immunoprecipitation of GFP-CHAMP1 in wild-type (WT), TRIP13−/−, and p31−/− U2OS cells, and the co-immunoprecipitation of endogenous REV7.
(H) Western blot showing GFP-immunoprecipitation of GFP-CHAMP1 in U2OS-vector control (VC) and TRIP13 overexpressed U2OS cells, and the co-immunoprecipitation of endogenous TRIP13 and REV7. All of the immunoblots are representative of at least two independent experiments.
Previous studies have shown that REV7 exists in two distinct conformations, open and closed. The closed conformation of REV7 binds to SHLD3 via the REV7-SBM. The TRIP13/p31 complex can open REV7 and release seatbelt-binding proteins (Clairmont et al., 2020; Sarangi et al., 2020) (Figure 2F). To further confirm that REV7 binds to CHAMP1 via the SBM, we analyzed the co-immunoprecipitation of REV7 and CHAMP1 in the presence or absence of p31 or TRIP13. As predicted, overexpression of TRIP13 reduced the co-immunoprecipitation of REV7 and CHAMP1, while TRIP13 or p31 knockdown increased the co-immunoprecipitation (Figures 2G and 2H). Moreover, TRIP13 knockout, like CHAMP1 knockout, results in a decrease in HR activity (Clairmont et al., 2020; Sarangi et al., 2020). Interestingly, knockdown of TRIP13 in CHAMP1 knockout cells resulted in a greater decrease in HR activity, as determined by enhanced PARPi sensitivity (Figures S2C and S2D), indicating that the two proteins have non-epistatic activity.
CHAMP1 is a suppressor of the REV7/Shieldin complex
Previous studies have shown that REV7 and the subunits of the Shieldin complex promote NHEJ and are antagonists of HR repair. Knockout of any of these proteins rescues DSB end resection, RAD51 loading, and HR repair, resulting in PARPi resistance of BRCA1-deficient cells. Since CHAMP1 promotes HR repair, we reasoned that CHAMP1 may function as an antagonist of the REV7 and the Shieldin complex. To test this hypothesis, we reduced CHAMP1 expression in wild-type U2OS cells or in cells knocked out for REV7 (Figure S3A). HR activity was scored by measuring the level of RAD51 (Figures 3A and 3B) foci or pRPA foci (Figures S3B and S3C), both known to be elevated in the setting of HR. As predicted, siRNA knockdown of CHAMP1 in wild-type RPE1 cells reduced HR activity. Interestingly, siRNA knockdown of CHAMP1 in cells knocked out for REV7 failed to reduce HR activity, based on the assessment of PARPi sensitivity (Figures 3C and 3D), suggesting that CHAMP1 can promote HR only in the presence of REV7, through its direct interaction.
Figure 3. CHAMP1 regulates HR through REV7.
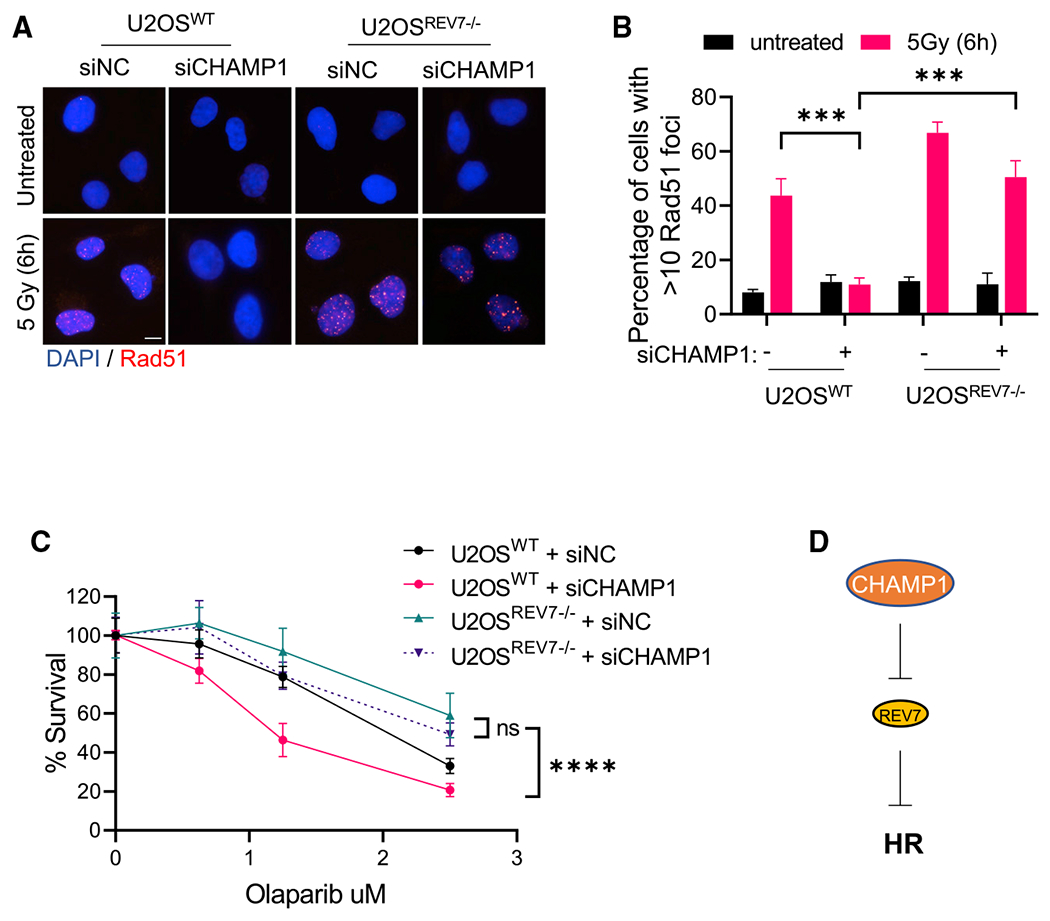
(A) Representative images of RAD51 foci formation in wild-type and REV7−/− U2OS cells treated with siRNA negative control (siNC) and siCHAMP1, and 6 h after 5Gy IR treatment. Scale bar, 5 μm.
(B) Quantification of RAD51 in (A). More than 10 RAD51 foci were counted. Error bars indicate SD, n = 3 biologically independent experiments, ***p < 0.001. Statistical analysis was performed using two-tailed Student’s t test.
(C) A 14-day clonogenic survival of wild-type and REV7−/− U2OS cells treated with various doses of olaparib after siControl or siCHAMP1 treatment. Error bars indicate SD, n = 3, ***p < 0.001. Statistical analysis was performed using two-way ANOVA.
(D) A cartoon shows that CHAMP1 inhibits REV7 to promote HR.
To further assess the effect of CHAMP1 on HR in the context of the Shieldin complex, we next knocked down CHAMP1 expression in SHLD2-deficient or SHLD3-deficient RPE cells. RPE cells with a CRISPR knockout of BRCA1 are defective in HR repair, as shown by their reduced RAD51 foci (Figures 4A and 4B), reduced pRPA foci (Figures S4A and S4B), and olaparib sensitivity (Figure 4C). CRISPR knockout of SHLD2 in these BRCA1-deficient cells resulted in increased RAD51 foci, pRPA foci, and olaparib resistance. While CHAMP1 knockdown in the BRCA1-deficient cells further reduced HR proficiency, CHAMP1 knockdown failed to reduce the HR activity in the RPE-BRCA1(−/−) SHLD2(−/−) cells. Consistent with these results, knockdown of CHAMP1 in a PARPi-resistent SHLD2-deficient breast cancer cell line, HCC1937 (Scully et al., 1999; Tomida et al., 2018), also failed to result in olaparib sensitivity (Figures S4C and S4D). Similarly, CHAMP1 knockdown failed to reduce the HR activity in RPE-BRCA1(−/−) SHLD3(−/−) cells, as determined by the olaparib resistance of these cells (Figure 4D). Taken together, CHAMP1 is a suppressor of the REV7/Shieldin complex, which in turn is a suppressor of HR repair (Figure 4E).
Figure 4. CHAMP1 regulates HR through Shieldin complex.
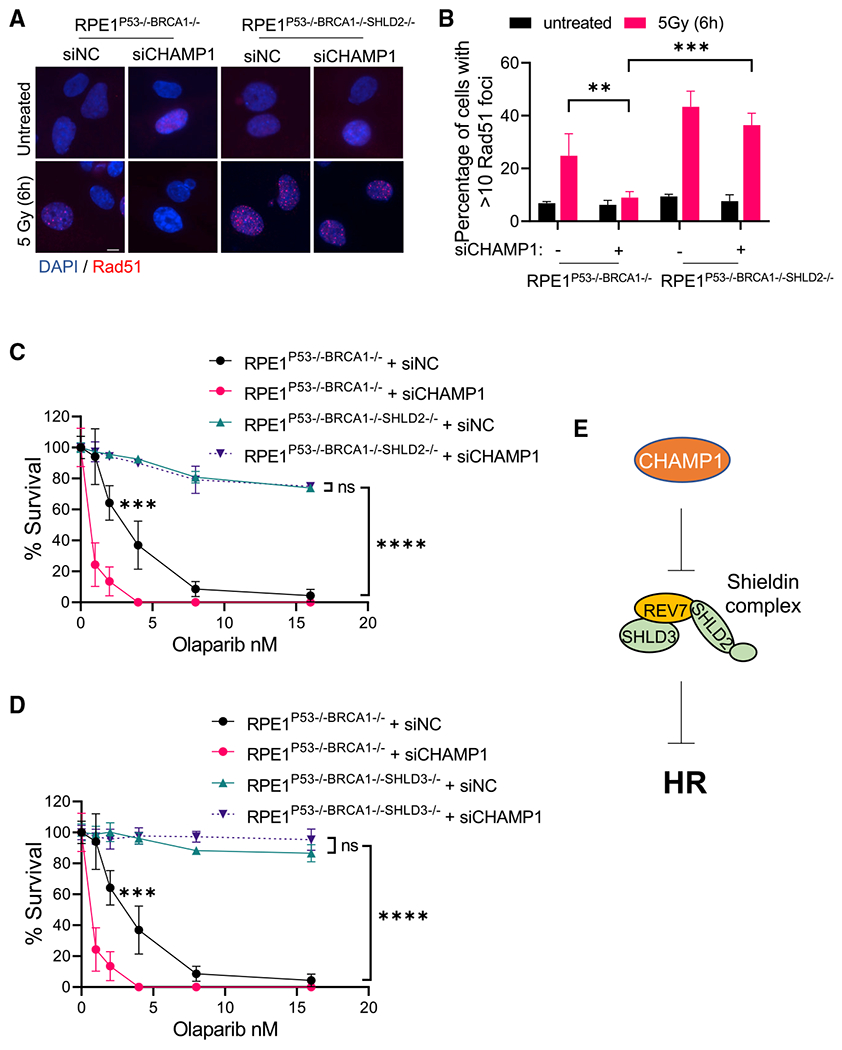
(A) Representative images of RAD51 foci formation in RPE1P53−/−BRCA1−/− and RPE1P53−/−BRCA1−/−SHLD2−/− cells treated with siRNA negative control (siNC) or siCHAMP1, and 6 h after 5Gy IR treatment. Scale bar, 5 μm.
(B) Quantification of RAD51 foci in (A). More than 10 RAD51 foci were counted. Error bars indicate SD, n = 3, **p < 0.01, ***p < 0.001. Statistical analysis was performed using two-tailed Student’s t test.
(C) A 14-day clonogenic survival of RPE1P53−/−BRCA1−/− and RPE1P53−/−BRCA1−/−SHLD2−/− cells treated with various doses of olaparib after siControl (siNC) or siCHAMP1 treatment. Error bars indicate SD, n = 3 independent experiments, *p < 0.05, ****p < 0.0001. Statistical analysis was performed using two-way ANOVA.
(D) A 14-day clonogenic survival of RPE1P53−/−BRCA1−/− and RPE1P53−/−BRCA1−/−SHLD3−/− cells treated with various doses of olaparib after siControl (siNC) or siCHAMP1 treatment. Error bars indicate SD, n = 3 independent experiments, ***p < 0.001, ****p < 0.0001. Statistical analysis was performed using two-way ANOVA.
(E) A cartoon shows that CHAMP1 inhibits Shieldin complex to promote HR.
CHAMP1 increases HR activity by binding to REV7 and reducing the level of the REV7/SHLD3 complex
In its primary sequence, CHAMP1 has non-overlapping N-ZNF (C2H2-Zn finger domains), SPE (PxxSPExxK motifs), WK (SPxxWKxxP motifs), FPE (FPExxK motifs), and C-ZNF regions (Itoh et al., 2011) (Figures 5A and 5B). Based on the crystal structure, the CHAMP1-WK region binds directly to the REV7 seatbelt domain (Hara et al., 2017). Indeed, the WKPAKPAPS domain of CHAMP1 corresponds to the known consensus of a REV7 SBM. We therefore generated a mutant form of CHAMP1 that disrupts this REV7 binding interaction (i.e., the W334A/K335A double mutation, referred to as the CHAMP1-2A mutation). As predicted, the CHAMP1-2A mutant failed to bind to REV7, unlike wild-type CHAMP1 (Figure 5C). Interestingly, the CHAMP1-2A mutant failed to correct the PARPi sensitivity of CHAMP1 −/− cells, further confirming that CHAMP1 binding to REV7 is required for enhancement of HR activity (Figure 5D). The failure of CHAMP1-2A to restore PARPi resistance was confirmed in transfected U2OS-CHAMP1 (−/−) cells (Figures S5A and S5B).
Figure 5. CHAMP1 increases HR activity by binding to REV7 and reducing the level of the REV7/SHLD3 complex.
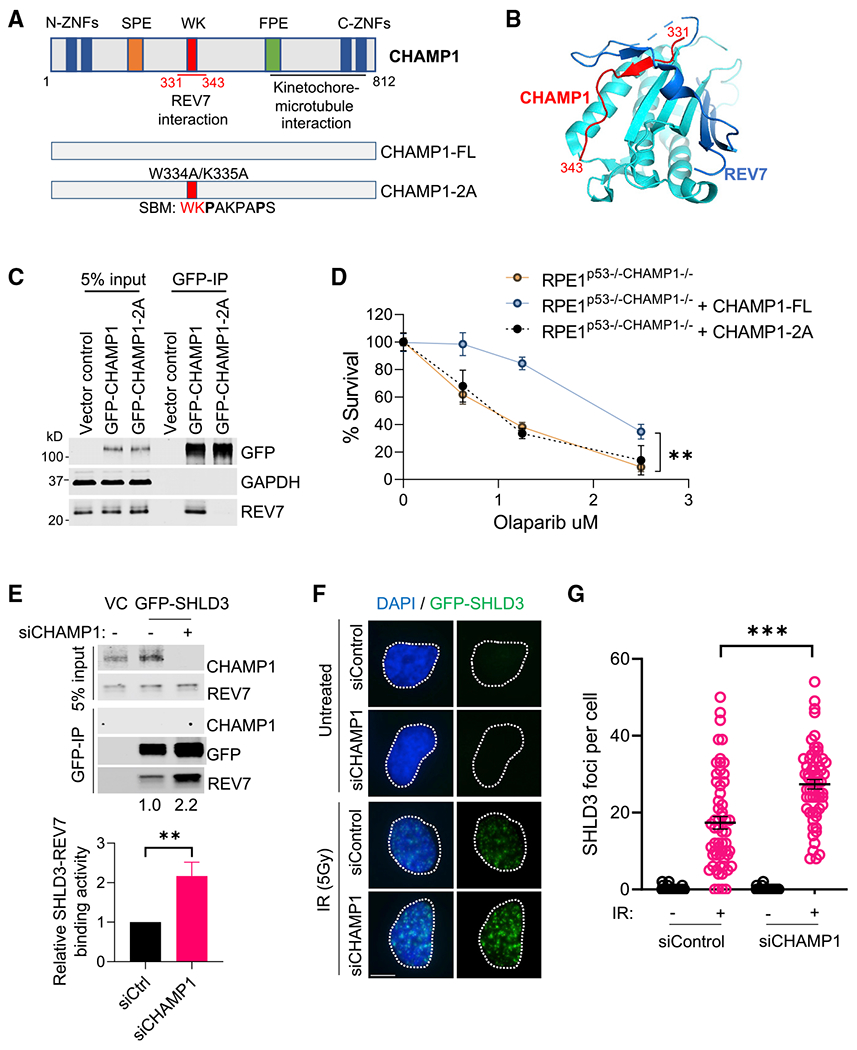
(A) (Top) Schematic of CHAMP1 protein showing its various domains and REV7 binding region. (Bottom) Schematic of CHAMP1-full-length (FL) and W334A/K335A (2A) mutants. SBM, REV7 seatbelt-binding motif.
(B) Structure of the REV7-CHAMP1 complex. REV7 is shown in cyan and blue (seatbelt domain), and the CHAMP1 fragment (residues 331–343) is shown in red.
(C) Western blot showing GFP-immunoprecipitation of GFP-empty vector, GFP-CHAMP1 wild-type, and GFP-CHAMP1-2A mutant, and the co-immunoprecipitation of endogenous REV7 in 293T cells.
(D) RPE1P53−/−CHAMP1−/− cells were transfected with GFP-empty vector, GFP-CHAMP1 wild-type, or GFP-CHAMP1-2A mutant. The GFP-positive cells were sorted for clonogenic assay. A 14-day clonogenic assay of indicated cell lines treated with various doses of olaparib; error bars indicate SD, n = 3 independent experiments, **p < 0.01. Statistical analysis was performed using two-way ANOVA.
(E) (Top) Western blot showing GFP-immunoprecipitation of GFP-SHLD3 in 293T cells, treated with or without siCHAMP1, and the co-immunoprecipitation of endogenous CHAMP1 and REV7. (Bottom) Quantification of the relative SHLD3-REV7 binding activity from three independent immunoprecipitation western blots. Error bars indicate SD, n = 3. **p < 0.01.
(F) Representative images of GFP-SHLD3 foci formation in GFP-SHLD3 stable expressed RPE1 cells treated with siRNA negative control or siCHAMP1, and 1 h after 5Gy IR treatment. Scale bar, 5 μm.
(G) Quantification of RAD51 foci in (F). More than 60 cells were counted per treatment. ***p < 0.001. Statistical analysis was performed using two-tailed Student’s t test. Error bar means SEM.
Since the ability of CHAMP1 to regulate HR repair requires the presence of the REV7/Shieldin complex, we reasoned that CHAMP1 expression might directly affect the interaction of proteins in the Shieldin complex. Compared with the parental U2OS cells, U2OS cells with a knockout of CHAMP1 exhibited increased binding of REV7 and SHLD3 (Figure 5E). Complementation of these CHAMP1(−/−) cells with wild-type CHAMP1 reduced the level the REV7/SHLD3 complex, providing a mechanism for the CHAMP1-induced increase in HR in cells with a functional Shieldin complex. This reduction in the Shieldin complex correlated with the increased DSB end resection and the increased HR. We also examined the effect of siCHAMP1 on the IR-mediated induction of GFP-SHLD3 foci. The assembly of these foci is a marker for the activity of a functional Shieldin complex (Zhao et al., 2021). Indeed, CHAMP1 depletion increased GFP-SHLD3 foci formation after IR treatment (Figures 5F and 5G). Taken together, the binding of CHAMP1 to REV7 accounts for the decreased level of the Shieldin complex and in the upregulation of DSB end resection and HR repair.
CHAMP1 overexpression is common in tumors with underlying HR deficiency and correlates with poor cancer patient prognosis
To further validate this model, we next sought clinical evidence that CHAMP1 expression might affect cancer patient survival (Figure 6). For patients with HR-deficient tumors, such as BRCA1-deficient tumors, a high level of CHAMP1 expression correlates with a worse prognosis (Figures 6A and 6B). This result further suggests that an acquired upregulation of CHAMP1 expression can partially correct the HR deficiency of these tumors, leading to a more aggressive tumor phenotype. Another recent study indicated that the CHAMP1 gene is amplified in a large fraction of human cancers, including breast and ovarian cancers, and that CHAMP1 upregulation enhances tumor cell proliferation (Sack et al., 2018). Also consistent with these observations, the level of CHAMP1 mRNA expression in cancer cell lines strongly correlates with the level of CCNE1 mRNA expression (Figures 6C and 6D). Cells with a high degree of replication stress resulting from CCNE1 amplification may therefore rely on CHAMP1-mediated HR for their survival. Indeed, breast cancer cell lines with high expression of CCNE1 mRNA are more dependent on CHAMP1 for their proliferation and survival.
Figure 6. CHAMP1 overexpression is common in tumors with underlying HR deficiency and correlates with poor cancer patient prognosis.
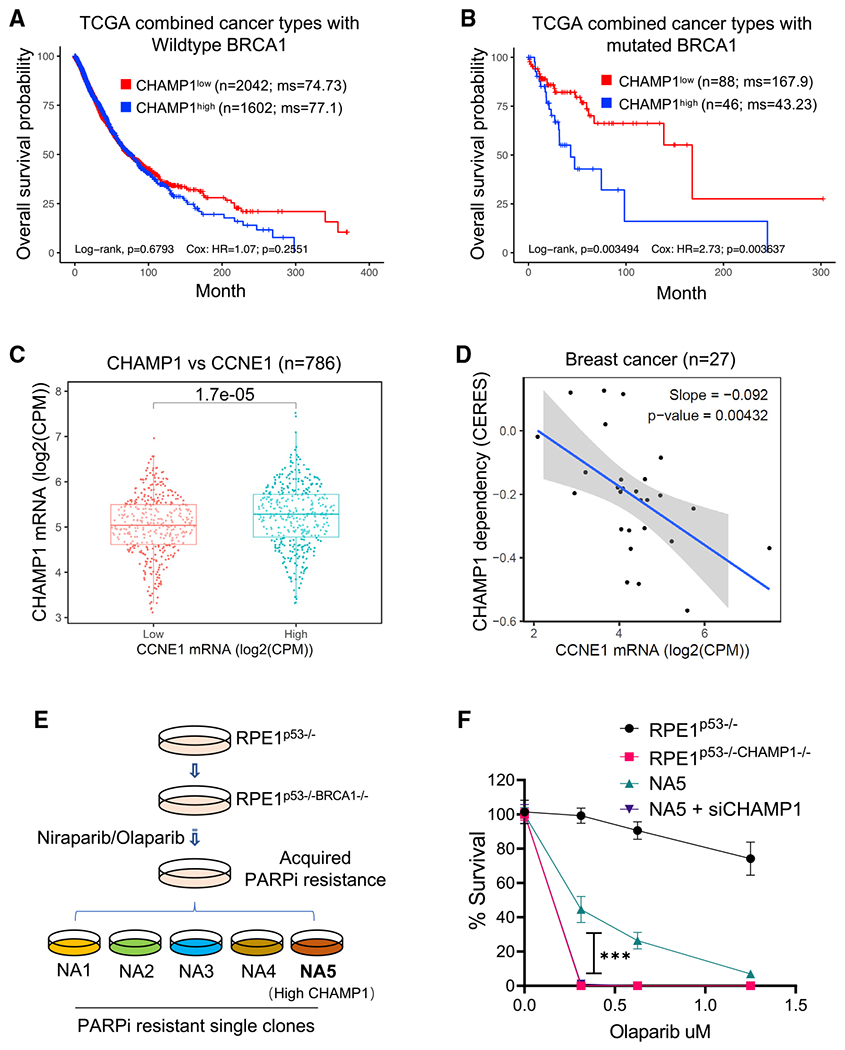
(A and B) Kaplan-Meier curves depicting overall survival of patients from TCGA with CHAMP1 expression and wild-type BRCA1 (A), and mutated BRCA1 (B). This analysis combines tumors from these TCGA studies: BLCA (bladder), BRCA (breast), COAD/READ (colorectal), LUAD (lung), LUSC (lung squamous), OV (ovarian), and SKCM (skin). N represents number of patients.
(C) CHAMP1 expression positively correlates with cyclin E expression. N represents number of cancer cell lines.
(D) Breast cancer cells with high expression of CCNE1 are more dependent on CHAMP1 for survival. N represents number of cancer cell lines.
(E) Schematic of PARPi-resistant RPE1p53−/−BRCA1−/− cells generation. RPE1p53−/−BRCA1−/− cells were treated with increasing concentrations of the PARPis niraparib/olaparib over 3 months, and then isolated by single-cell clones from the niraparib- and olaparib-resistant pools.
(F) A 14-day clonogenic survival of RPE1p53−/−, RPE1p53−/−BRCA1−/−, and niraparib/olaparib-resistant RPE1p53−/−BRCA1−/− NA5 cell clone treated with various doses of olaparib after siControl or siCHAMP1 treatment. Error bars indicate SD, n = 3 independent experiments. ***p < 0.001. Statistical analysis was performed using two-way ANOVA.
We next reasoned that human tumors with an underlying defect in an HR pathway might upregulate CHAMP1 as a compensatory mechanism to tolerate their low HR and their replication stress. To test this hypothesis, we correlated the level of CHAMP1 expression in ovarian tumors with patient survival. For patients with tumors with low REV7 expression, the level of expression of CHAMP1 did not affect survival (Figures S6A and S6B). Thus, consistent with the cellular data, the elevated HR activity in cells with low or absent REV7 expression was unaffected by CHAMP1 expression levels. Interestingly, for patients with tumors with high REV7 expression, the level of CHAMP1 expression significantly affected patient survival. The high CHAMP1 expression correlated with a more aggressive tumor and poor patient prognosis, perhaps resulting from the improved HR activity of these tumors. Thus, consistent with the epistasis data in Figure 3, the ability of CHAMP1 to enhance HR is directly dependent on the presence of the REV7 protein in the tumor cell.
We next sought additional evidence that CHAMP1 upregulation correlates with PARPi resistance. We used a panel of BRCA1-deficient cell lines with acquired PARPi resistance, collected through serial selection in increasing concentrations of PARPi (Figure 6E) (Farkkila et al., 2021). These cells exhibited multiple independent mechanisms of PARPi resistance, including downregulation of the Shieldin complex or upregulation of ATR/CHK1 pathway activity (Farkkila et al., 2021). Interestingly, one of these PARPi-resistant clones (NA5) exhibited high CHAMP1 protein expression compared with the parental PARPi-sensitive cell line (Figure S6C). Knockdown of CHAMP1 in these cells restored PARPi sensitivity (Figure 6F). Taken together, BRCA1-deficient cells can acquire PARPi resistance, at least in part, by upregulating CHAMP1 expression.
POGZ binds to CHAMP1 and cooperates in HR repair
Recent studies have shown that CHAMP1 is a subunit of a large multisubunit complex of HP1α heterochromatin binding proteins. This complex includes HP1α, POGZ, LEDGF, and HDGFRP2 (Baude et al., 2016; Clairmont et al., 2020; Daugaard et al., 2012; Nozawa et al., 2010; Vermeulen et al., 2010). REV7 co-immunoprecipitates with multiple components of this complex (Noordermeer et al., 2018), further suggesting a functional link with DNA repair regulation. Interestingly, knockdown or knockout of many of the subunits of this complex, such as HP1α (Soria and Almouzni, 2013), LEDGF (Daugaard et al., 2012), or HDGFRP2 (Baude et al., 2016), similar to the knockdown of CHAMP1, reduces DSB end resection and HR and increases PARPi sensitivity (Olivieri et al., 2020). To confirm and extend these studies, we next evaluated the POGZ subunit of this complex. A recent report indicated that POGZ also functions in HR repair (Heath et al., 2021). Consistent with this report, knockdown of POGZ, like knockout of CHAMP1, resulted in decreased DSB end resection, decreased RAD51 foci, and increased PARPi sensitivity, consistent with a decrease in HR activity (Figures 7A–7C, S7A, and S7B). Moreover, IR-induced DNA damage activated the co-immunoprecipitation of POGZ, CHAMP1, and REV7 (Figure 7D). Knockdown of POGZ reduced the interaction of REV7 and CHAMP1 (Figure 7E), and POGZ binding to CHAMP1 did not require a functional REV7 seatbelt interaction (Figure S7C). Importantly, knockdown of POGZ in CHAMP1(−/−) cells or knockdown of CHAMP1 in POGZ(−/−) cells resulted in no additional impairment of HR repair or PARPi sensitivity (Figures 7F and 7G), demonstrating that POGZ and CHAMP1 are epistatic in HR repair. Finally, POGZ expression, like CHAMP1 expression, is increased in many human cancers, consistent with its compensatory role in promoting HR repair (Figure S7D). Taken together, these results demonstrate that a CHAMP1-containing multisubunit complex has a functional role in reducing the level of the REV7/SHLD3 complex and the Shieldin complex and in promoting HR repair (Figure S7E).
Figure 7. POGZ is epistatic with CHAMP1 in the regulation of homologous recombination.
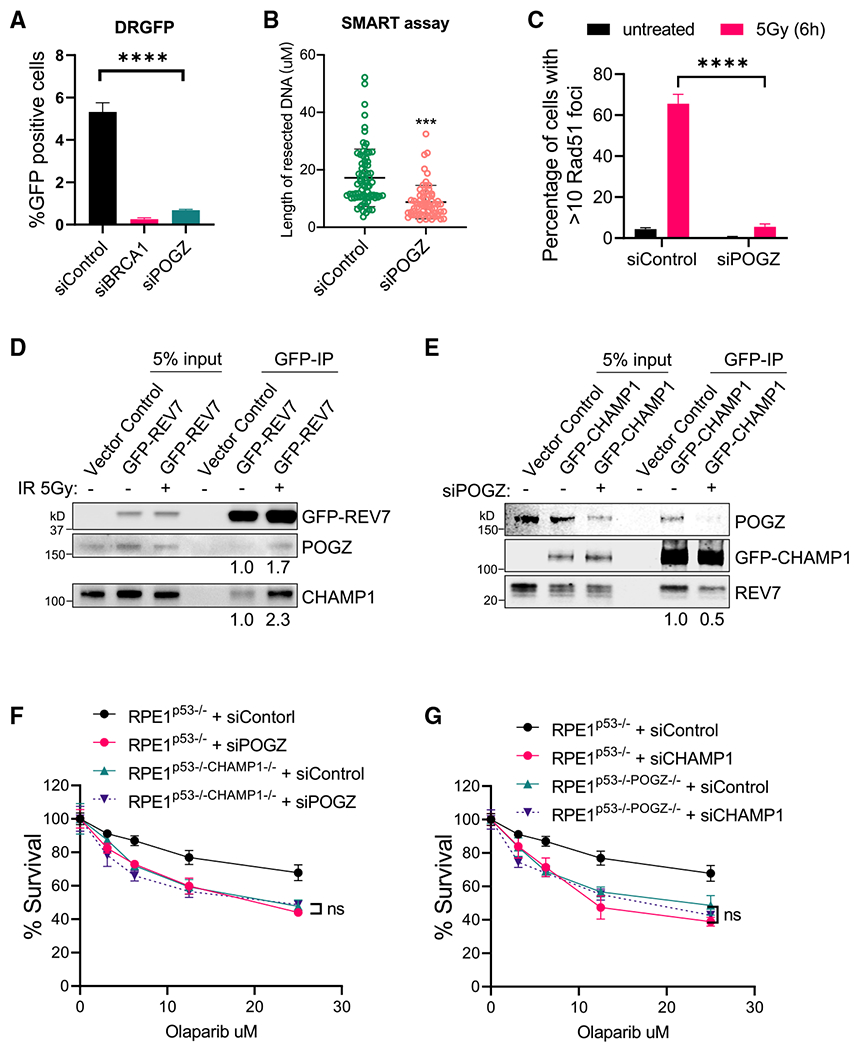
(A) Graph showing the percentage of GFP-positive cells after DR-GFP analysis. U2OS cells were infected with I-SceI adenovirus and knocked down for BRCA1 or POGZ using siRNA. N = 3 biologically independent experiments. Error bars indicate SD, and p values were calculated using two-tailed Student’s t test, ****p < 0.0001.
(B) Quantification of resected ssDNA measured by SMART assay in U2OS cells treated by siControl or siRNAs targeting CHAMP1 for 48 h. Approximately 50 fibers were counted per experiment. Error bars indicate SD, and p values were calculated using Student’s t test, ***p < 0.001.
(C) Quantification of >10 RAD51 foci in wild-type and two CHAMP1 knockout U2OS cell lines 6 h after 5Gy IR treatment. Error bars indicate SD, n = 3 biologically independent experiments. ****p < 0.0001. Statistical analysis was performed using two-tailed Student’s t test.
(D) Western blot showing GFP-immunoprecipitation of GFP-REV7 in HEK293T cells treated with or without irradiation (5Gy), and the co-immunoprecipitation of endogenous CHAMP1 and POGZ.
(E) Western blot showing GFP-immunoprecipitation of GFP-CHAMP1 in HEK293T cells treated with or without siPOGZ, and the co-immunoprecipitation of endogenous CHAMP1 and POGZ.
(F) 3-day cytotoxicity analysis of RPE1p53−/− and RPE1p53−/−CHAMP1−/− cells treated with various doses of olaparib after 48 h siControl or siPOGZ treatment. Cell viability was detected by CellTiter-Glo (Promega). Error bars indicate SD, n = 3 independent experiments. Statistical analysis was performed using two-way ANOVA.
(G) 3-day cytotoxicity analysis of RPE1p53−/− and RPE1p53−/−POGZ−/− cells treated with various doses of olaparib after 48 h siControl or siCHAMP1 treatment. Cell viability was detected by CellTiter-Glo (Promega). Error bars indicate SD, n = 3 independent experiments. Statistical analysis was performed using two-way ANOVA. All of the immunoblots are representative of at least two independent experiments.
DISCUSSION
In the current report, we demonstrate that CHAMP1 is a positive regulator of HR repair. Knockout of CHAMP1 in RPE cells or U2OS cells results in a decrease in double-strand end resection and a decrease in pRPA and RAD51 loading. Moreover, knockout of CHAMP1 results in an increase in sensitivity of BRCA1-deficient cells to the PARPi olaparib.
Mechanistically, CHAMP1 promotes HR repair by its direct binding to REV7. In the current study, we generated a mutation in the REV7 binding site of CHAMP1. The corresponding mutant CHAMP1 protein, referred to as CHAMP-2A, failed to coimmunoprecipitate with REV7 and failed to correct the HR defect of CHAMP1 knockout cells. The HR defect of the CHAMP-2A protein corresponded to its inability to compete with the binding of SHLD3 to REV7. Accordingly, the CHAMP1-2A protein failed to reduce the cellular level of the REV7/SHLD3 complex and the REV7/Shieldin complex, thus resulting in increased NHEJ activity and decreased HR activity.
Although CHAMP1 may have additional, independent roles in the regulation of HR repair, its suppression of the Shieldin complex appears to be its primary mechanism of promoting HR. Indeed, cells with a knockout of REV7, versus cells with a double knockout of REV7 and CHAMP1, have equally high levels of DSB end resection, RAD51 foci, and PARPi resistance. Similarly, a knockout of SHLD2 and a double knockout of SHLD2 and CHAMP1 have comparable elevated levels HR activity. These results argue for an epistatic relationship of CHAMP1 with components of the Shieldin complex (Figure 4E).
Previous studies indicate that TRIP13, like CHAMP1, is a positive regulator of HR repair (Clairmont et al., 2020; de Krijger et al., 2021b). Furthermore, both of these proteins upregulate HR repair, at least in part, through their downregulation of the Shieldin complex. TRIP13/p31comet opens the conformation of REV7 and thereby releases SHLD3 from the Shieldin complex. CHAMP1, in contrast, binds REV7 directly through the REV7 seatbelt and competes with SHLD3 binding, thus reducing the Shieldin complex. TRIP13/p31comet appears to be non-epistatic with CHAMP1 in HR repair, since knockdown of TRIP13/p31comet in CHAMP1 (−/−) cells results in a further decline in HR activity.
DNA damage activates the closure of REV7 seatbelt and the increased interaction of REV7 with either SHLD3, REV3, or CHAMP1. How and when REV7 selectively chooses one binding partner versus another is mostly unknown. IR appears to preferentially activate the REV7/CHAMP1 complex, while other DNA damaging agents, such as UV light, may preferentially activate the REV7/REV3 complex. Also, the choice of a specific REV7 complex may also be influenced by either the local organization of the genome, cell cycle cues, or cell type specificity. The specific interaction of REV7 with SHLD3, REV3, or CHAMP1 may also be determined by the activity of the TRIP13/p31comet enzyme. While all three complexes are disassembled by TRIP13/p31comet, cells may choose to selectively release one complex or another, depending on specific cellular demands for NHEJ, TLS, or HR, respectively.
Additional factors may influence the relative levels of REV7/SHLD3, REV7/REV3, and REV7/CHAMP1 in the cell. First, REV7 may preferentially bind to CHAMP1 in heterochromatic regions of the genome, such as centromeres and telomeres, resulting in higher local levels of HR repair. Second, CHAMP1 may also have a distinct binding affinity for the REV7 seatbelt. Indeed, based on the corresponding crystal structures, the molecular interactions of the REV7 seatbelt with either CHAMP1 or REV3 are distinct (Hara et al., 2010, 2017) and likely to result in distinct binding affinities and off rates. Third, some tumor cells with an underlying genetic deficiency in HR repair or high replication stress exhibit a higher level of expression of CHAMP1. Interestingly, this increase in CHAMP1 may provide these cells with a compensatory increase in HR and a higher capacity for tolerating replication stress. The specific interaction of REV7 with these various binding partners may be highly regulated by post-translational modifications and under distinct cellular conditions. Future studies will be required to further assess the spatial and temporal control of the REV7 interaction with CHAMP1, SHLD3, and REV3.
Our results further demonstrate that a multisubunit heterochromatin protein complex may function to upregulate HR repair and downregulate NHEJ repair. This complex contains the proteins HP1α (Soria and Almouzni, 2013), LEDGF (Daugaard et al., 2012), HDGFRP2, POGz, and CHAMP1. Our results demonstrate that DNA damage with IR can activate the assembly of these proteins. IR activated the binding of POGz, CHAMP1, and REV7. At some DSBs, this complex may result in a reduction of REV7/SHLD3 complexes and may thereby result in the choice between HR repair and NHEJ repair.
Our results suggest that the level of CHAMP1 protein may be a determinant of the level of HR repair in human cancer cells. Tumor cells with germline or acquired BRCA1/2 mutations resulting in an HR deficiency, or tumors with increased replication stress, undergo a selective pressure to increase their HR activity. Amplification of the CHAMP1 gene, or upregulation of CHAMP1 expression, can provide an enhancement of HR activity. Consistent with this notion, human tumor databases reveal an increase in CHAMP1 expression, which may account for the relatively poor prognosis of cancer patients bearing these tumors.
A recent study published during the review of our manuscript independently confirmed our finding that CHAMP1 and REV7 interact and promote HR (Fujita et al., 2022). According to this work, CHAMP1 is recruited to laser stripes following DNA damage, and CHAMP1 promotes DNA DSB end resection and pRPA loading. Moreover, CHAMP1 interacts with POGZ in the promotion of HR repair. Interestingly, the C-terminal region of CHAMP1 was also responsible for the increase in HR repair. Since C-terminally truncated versions of the CHAMP1 protein still retain the REV7 binding motif, the C terminus may contain additional features required for functional HR repair.
Limitations of the study
In current work, we have shown that CHAMP1 can bind to REV7, through the seatbelt domain of REV7, and it can promote HR repair. The CHAMP1/REV7 interaction reduces the level of the SHLD3/REV7 interaction, thereby reducing the Shieldin complex and promoting DSB end resection. While CHAMP1 expression promotes the overall reduction of Shieldin and competes with NHEJ repair, the precise details of the CHAMP1/REV7 interaction are less clear. For instance, we still do not know whether the CHAMP1/REV7 interaction is confined to specific regions of the genome, such as heterochromatic regions. Also, whether the interaction is regulated by local or cell cycle timed events, perhaps through post-translational modifications of CHAMP1 or REV7, is unknown. In the future, it will be important to assess the regulation of the CHAMP1/REV7 interaction at the single-cell level. Finally, we have shown that CHAMP1 upregulation correlates in a cell culture with overall PARPi resistance. Still, PARPi resistance is governed by several cellular processes, and the relevance or importance of CHAMP1 upregulation to drug resistance remains to be determined.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alan D. D’Andrea (Alan_Dandrea@dfci.harvard.edu).
Materials availability
The knockout cell lines generated in this study are available from the lead contact without restrictions.
Data and code availability
This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table.
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| C13orf8/CHAMP1 | Abnova | Cat# H00283489-B01P; RRID:AB_1673079 |
| Mad2L2/REV7 | Abcam | Cat# ab180579; RRID:AB_2890174 |
| POGZ | Bethyl Laboratories | Cat# A302-509A; RRID:AB_1966082 |
| TRIP13 | Abcam | Cat# ab128171; RRID:AB_11145670 |
| H3 | Cell Signaling Technology | Cat# 3638; RRID:AB_1642229 |
| GFP | Cell Signaling Technology | Cat# 2956; RRID:AB_1196615 |
| β-Actin | Cell Signaling Technology | Cat# 3700; RRID:AB_2242334 |
| RPA2-P-Ser33 | Fisher Scientific | Cat# NB100544; RRID:AB_526631 |
| RAD51 | Santa Cruz | Cat# sc-8349; RRID:AB_2253533 |
| GAPDH | Cell Signaling Technology | Cat# 5174; RRID:AB_10622025 |
| Goat anti-Rat IgG (Alexa Fluor 488) | Invitrogen | Cat# A-11006; RRID:AB_2534074 |
| Goat anti-Mouse IgG (Alexa Fluor 488) | Invitrogen | Cat# A32723; RRID:AB_2633275 |
| Chicken anti-mouse IgG (Alexa Fluor 594) Anti-rabbit IgG, HRP-linked | Invitrogen | Cat# A-21201; RRID:AB_2535787 |
| Flag | Millipore-Sigma | F3165; RRID:AB_259529 |
| Chemicals, peptides, and recombinant proteins | ||
| Olaparib | Selleckchem | S1060 |
| BrdU | Millipore-Sigma | B5002 |
| Anti-FLAG® M2 Magnetic Beads | Millipore-Sigma | M8823 |
| GFP-Trap_A | Chromotek | Gta-20 |
| Critical commercial assays | ||
| CellTiter-Glo Luminescent Viability Assay | Promega | G7570 |
| Subcellular protein fractionation kit | ThermoFisher | 78840 |
| Deposited data | ||
| Raw imaging data | This paper; Mendeley data | https://doi.org/10.17632/fkh6v4dw6y.1 |
| TCGA Pan-Cancer Atlas Studies’ RNASeq expression data | TCGA (PMID: 24071849) | https://www.cbioportal.org/ |
| Cancer cell lines’ RNASeq expression data | Cancer Cell Line Encyclopedia (Ghandi et al., 2019) | https://depmap.org/portal/ |
| Cancer cell lines’ dependency data (CRISPR DepMap Public 19Q4 dataset) | Broad Institute Cancer Dependency Map (Meyers et al., 2017) | https://depmap.org/portal/ |
| Experimental models: Cell lines | ||
| 293T | ATCC | N/A |
| U2OS | ATCC | N/A |
| RPE-1 | ATCC | N/A |
| HCC1937 | D’Andrea lab | N/A |
| U2OS-DR-GFP | D’Andrea lab | N/A |
| U2OS-EJ5 | D’Andrea lab | N/A |
| Oligonucleotides | ||
| siCHAMP1-4: CAGGAATCAAGCAGTGATCAA | Qiagen | SI00318038 |
| siCHAMP1-8: CAGGAACTTCGTAAACCATCA | Qiagen | SI04282159 |
| SiPOGZ: CCGGGTGTAAGGTCCCTTTAA | Qiagen | SI00688583 |
| siTRIP13: TTGGGACAGCTTGGTATACGA | Qiagen | (Clairmont et al., 2020) |
| AllStars Neg. Control siRNA | Qiagen | 0001027281 |
| Recombinant DNA | ||
| CHAMP1 CRISPR guide RNA 1_pSpCas9 BB-2A-GFP (PX458) | Genscript | N/A |
| CHAMP1 CRISPR guide RNA 3_pSpCas9 BB-2A-GFP (PX458) | Genscript | N/A |
| POGZ CRISPR guide RNA 1_pSpCas9 BB-2A-GFP (PX458) | Genscript | N/A |
| POGZ CRISPR guide RNA 2_pSpCas9 BB-2A-GFP (PX458) | Genscript | N/A |
| Software and algorithms | ||
| ImageJ | U. S. National Institutes of Health | https://imagej.nih.gov/ij/ |
| Graphpad Prism 9 | Graphpad | https://www.graphpad.com/ |
| R (version 4.0.3) | R Foundation for Statistical Computing | https://www.R-project.org/ |
| R package survival (version 3.2–10) | R Foundation for Statistical Computing | https://CRAN.R-project.org/package=survival |
| R package survminer (version 0.4.9) | R Foundation for Statistical Computing | https://CRAN.R-project.org/package=survminer |
| R package edgeR (version 3.32.1) | Bioconductor | https://www.bioconductor.org/packages/release/bioc/html/edgeR.html |
| R package ggplot2 (version 3.3.5) | R Foundation for Statistical Computing | https://ggplot2.tidyverse.org |
| R package ggpubr (version 0.4.0) | R Foundation for Statistical Computing | https://CRAN.R-project.org/package=ggpubr |
| R package ggpmisc (version 0.4.3) | R Foundation for Statistical Computing | https://CRAN.R-project.org/package=ggpmisc |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human U2OS, RPE1-hTERT, HCC1937 and HEK293T cells were cultured in DMEM/F12 + Glutamax (Invitrogen) supplemented with 10% FBS (Sigma) and 1% penicillin-streptomycin (Invitrogen) at 37°C with 5% CO2. Each of these cell lines were obtained from the American Type Culture Collection (ATCC) and validated by short tandem repeat (STR) testing. U2OS-DR-GFP and U2OS-EJ5 cell line has been genetically modified to harbor a DR-GFP or EJ5-GFP reporter genes in U2OS cells, and can be used to monitor homologous recombination and NHEJ efficiency using the GFP reporter (Clairmont et al., 2020).
METHOD DETAILS
RNA interference and over-expression
DNA transfections and siRNA knockdowns were carried out using Lipofectamine LTX (Invitrogen) and RNAiMax (Invitrogen) respectively according to the manufacturer’s protocols. The individual siRNAs used are: AllStar negative siControl (1027281); siCHAMP1 #4 (SI00973084); siCHAMP1 #8 (SI04282159); siBRCA1 (SI00930510); si53BP1 (SI01456539) were purchased from Qiagen. ON-target Human siPOGZ (L-006953-01-0005) were purchased from Horizon Discovery.
Generation of knockout cell lines with CRISPR-Cas9
CHAMP1 and POGZ guide RNA sequences were cloned into the pSpCas9 BB-2A-GFP (PX458) vector (GenScript). U2OS and RPE1p53−/− cells were transfected with Cas9-gRNA plasmids. After 48 h GFP positive cells were selected using a BD FACSAria II cell sorter. Single cells from GFP positive pool were cultured for three to four weeks and colonies were screened for knockouts by western blotting using the anti-p31comet antibody (Millipore-Sigma). The guide RNA sequences targeting CHAMP1 in this study were: #1 TCGTAAACCATCAGCACGTT and #2 CCAGAGATCCGTAGTCCAGC. The guide RNA sequences targeting POGZ in this study were: #1 CAGTTTGTTAAGCCGACAGT and #2 TCTGCTGATCGAGTTCTACG.
GFP-based DNA repair assays
For DR- and EJ5-GFP reporter assays, U2OS cells carrying the respective GFP expression cassette were transfected with the indicated siRNAs. 24 h after transfection, cells were infected with or without I-SceI lentivirus. After 48 h, cells were harvested and detected by flow cytometry. The data was analyzed using the FlowJo software.
Cellular fractionation and immunoblot analysis
Cells were lysed with NP40 buffer (1% NP40, 300 mM NaCl, 0.1 mM EDTA, 50 mM Tris (pH 7.5)) supplemented with phosphatase and protease inhibitor cocktail (Roche). Cell lysates were resolved by NuPAGE 4–12% Bis-Tris gels (Invitrogen), and transferred onto nitrocellulose membranes. Membranes were blocked with 5% BSA in TBST and were sequentially incubated with primary and secondary antibodies and detected using chemiluminescence or fluorescence (LI-COR Biosciences). For chromatin extraction, chromatin-bound extracts were got using subcellular protein fractionation kit (Thermo). The band intensities were measured by ImageJ.
Immunoprecipitation
After transfection for 48h, 293T or U2OS cells were then harvested and lysed in NETN lysis buffer with proteinase & phosphatase inhibitor cocktail (Thermo, 1:100) for 30 min on ice. They were then incubated with antibody-bead conjugate (Anti-FLAG® M2 Magnetic Beads, Millipore & Sigma or GFP-Trap_A, Chromotek) overnight at 4°C. Beads were washed four times with NETN buffer and immunoprecipitates were eluted by boiling. Western blots were performed to detect the immunoprecipitates. The band intensities were measured by ImageJ.
Drug sensitivity assays
Cells were transfected with plasmid or siRNA 24h before being plated for colony formation or CellTiter-Glo assays (Li et al., 2020). To assay clonogenic survival, cells were seeded at 500–1000 cells/well in 6-well plates in triplicates. Drugs at the shown doses were added after 12 h and cells were permitted to grow for 14 days. Colony formation was scored by fixing and staining with 0.5% (w/v) crystal violet in 20% methanol. For short term CellTiter-Glo survival assays, cells were plated in 96-well plates at 800–1000 cells/well, and treated with drugs at the indicated concentrations after 12 h. Three days later, cellular viability was measured using CellTiter-Glo (Promega). Survival at each drug concentration was calculated as a percentage normalized to the corresponding untreated control, for both assays.
Immunofluorescence assays
Cells were plated on glass coverslips in 12-well plates. They were then either left untreated or treated at 20J/m2 UV or 5Gy IR. After 1 or 6 h, they were harvested by pre-extraction with 0.5% Triton X-100 for 5 min, followed 4% paraformaldehyde fixation for 10 min at 4°C. After three PBS washes, blocking was performed with 3% BSA in PBS for 1 h at room temperature, followed by sequential primary and secondary antibody incubations overnight at 4°C and 1 h at room temperature respectively. The coverslips were mounted with DAPI (Vector Laboratories) and captured using a Zeiss AX10 fluorescence microscope and Zen software, and foci were scored. At least 100 cells were counted for each sample.
SMART assay
The SMART DNA fiber assay procedure was performed largely as described previously (Clairmont et al., 2020). In brief, cells were treated with BrdU (sigma) for 24h, and then exposed to X-ray irradiation to induce DSB formation. Cells were collected 6h after irradiation, and embedded in low melting point agarose plugs before lysis with proteinase K overnight at 50°C. The plugs were then washed with TE buffer and digested with agarase (NEB). The sample solution was spread onto silanized coverslips using the FiberComb machine (Genomic Vision). Combed coverslips were blocking with 3% BSA for 30min, and then incubated with anti-BrdU antibody (rat, abcam) overnight at 4°C. After incubation with secondary Alexa 555-labelled goat anti-rat antibodies, the coverslips were washed and mounted with Vectashield mounting medium (Vector laboratories). Images were captured by Zeiss AX10 flurorescence microscope. At least 50 fibers were counted per condition. The fiber lengths were measured using ImageJ and graphed.
TCGA data acquisition and analysis
The survival analyses of the Cancer Genome Atlas (TCGA) patients were performed using the clinical and RNASeq expression and genomic alteration data of TCGA Pan-Cancer study for 32 cancer types downloaded from the cBioPortal for Cancer Genomics (https://www.cbioportal.org; retrieved March 2020). For the survival analysis with mRNA expression of CHAMP1 (CHAMP1) and REV7 (MAD2L2), for each cancer type, samples were grouped into the low- and high-mRNA expressing groups for CHAMP1 and REV7 based on the expression z-scores of less than −0.5 and greater than 0.5. These expression z-scores were computed relative to the diploid samples. Survival analysis was then performed in R for each cancer type to determine whether there was a difference in the overall survival between the two groups, separately for CHAMP1 and REV7, and for REV7 in each of the two CHAMP1 groups. Kaplan-Meier curves were created, and the log rank test was used to test for a difference in overall survival using the survival package in R. The p values were calculated from the chi-square distribution. The survminer R package was used to estimate median survivals, and to plot the Kaplan-Meier curves. Additionally, Cox proportional hazards regression was performed to estimate the hazard ratio between the low- and the high-mRNA groups for each cancer type.
The survival analyses of TCGA patients with CHAMP1 mRNA expression and BRCA1 mutation status were performed as follows. A tumor was considered BRCA1 muted if it had damaging or other non-conserving variants in BRCA1. Tumors were grouped into the low- and high-mRNA expressing groups for CHAMP1 based on the expression Z score of zero. The combined survival analyses with CHAMP1 expression and BRCA1 mutation status were performed using data from these select TCGA projects: BLCA, BRCA, COAD/READ, LUAD, LUSC, OV and SKCM. Also, the Cox proportional hazards regression was performed with accounting for the differences between cancer types.
Cancer cell lines’ data acquisition and analysis
The association analyses between cyclin E (CCNE1) expression and CHAMP1 dependency and CHAMP1 expression in cancer cell lines were performed using the RNASeq expression data from the Cancer Cell Line Encyclopedia (CCLE) project (Ghandi et al., 2019) and the dependency data from the Broad Institute Cancer Dependency Map (DepMap; CRISPR DepMap Public 19Q4 dataset) (Meyers et al., 2017). Both datasets were downloaded from the DepMap Portal (https://depmap.org/portal/). The RNASeq expression counts were normalized by the TMM (weighted trimmed mean of M-values) method using the edgeR package (Robinson et al., 2010) and transformed into log2-counts per million (log2-CPM) values. The significance of the difference in the CHAMP1 mRNA expression between the low- and high-cyclin E mRNA expressing groups was assessed by the Wilcoxon rank-sum test using the ggpubr R package. In this analysis, the low- and high-cyclin E mRNA expressing groups were determined using the median log2-CPM, and cell lines of the skin lineage were excluded. The correlation between CHAMP1 dependency and cyclin E mRNA expression was performed with simple linear regression using the ggpmisc R package. The plots were generated using the ggplot2 package in R.
QUANTIFICATION AND STATISTICAL ANALYSIS
All values are expressed as mean ± standard deviation (SD) or standard error of the mean (SEM) as indicated in figure legends. The statistical significance of differences was assessed by Student’s t-test for comparison of two groups and one-way analyses of variance (ANOVA) with Tukey’s test for comparison of multiple groups using Graphpad Prism 9 (Key resources table).
Supplementary Material
Highlights.
CHAMP1 promotes homologous recombination repair
CHAMP1 binds directly to REV7 and reduces the level of the Shieldin complex
CHAMP1 interacts with POGZ in a heterochromatin complex further promoting HR repair
CHAMP1 overexpression confers PARPi resistance and correlates with poor prognosis
ACKNOWLEDGMENTS
We thank all members of the D’Andrea laboratory for their helpful suggestions and comments. This work was supported by grants from the US National Institutes of Health (R01HL052725), the Gray Foundation, the Breast Cancer Research Foundation, the Fanconi Anemia Research Fund, the Ludwig Center at Harvard, and the Smith Family Foundation (to A.D.D.) and the Claudia Adams Barr Program in Innovative Basic Cancer Research (to F.L. and P.S).
DECLARATION OF INTERESTS
A.D.D. is a consultant/advisory board member for Merck-EMD Serono, Cyteir Therapeutics, Third Rock Ventures, AstraZeneca, and Cedilla Therapeutics Inc. He is also a stockholder in Cedilla Therapeutics Inc., Oncolinea Pharmaceuticals Inc, Impact Therapeutics Inc, and Cyteir Therapeutics Inc, and reports receiving commercial research grants from Merck-EMD Serono, Bristol Meyers Squibb, Moderna Inc, and Tango Therapeutics Inc.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111297.
REFERENCES
- Baude A, Aaes TL, Zhai B, Al-Nakouzi N, Oo HZ, Daugaard M, Rohde M, and Jäättelä M (2016). Hepatoma-derived growth factor-related protein 2 promotes DNA repair by homologous recombination. Nucleic Acids Res. 44, 2214–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluteau D, Masliah-Planchon J, Clairmont C, Rousseau A, Ceccaldi R, Dubois d’Enghien C, Bluteau O, Cuccuini W, Gachet S, Peffault de Latour R, et al. (2016). Biallelic inactivation of REV7 is associated with Fanconi anemia. J. Clin. Invest 126, 3580–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma V, Moatti N, Segura-Bayona S, Peuscher MH, van der Torre J, Wevers BA, Orthwein A, Durocher D, and Jacobs JJL (2015). MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5’ end resection. Nature 521, 537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brulotte ML, Jeong BC, Li F, Li B, Yu EB, Wu Q, Brautigam CA, Yu H, and Luo X (2017). Mechanistic insight into TRIP13-catalyzed Mad2 structural transition and spindle checkpoint silencing. Nat. Commun 8, 1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, and Helleday T (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917. [DOI] [PubMed] [Google Scholar]
- Clairmont CS, and D’Andrea AD (2021). REV7 directs DNA repair pathway choice. Trends Cell Biol. 31, 965–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clairmont CS, Sarangi P, Ponnienselvan K, Galli LD, Csete I, Moreau L, Adelmant G, Chowdhury D, Marto JA, and D’Andrea AD (2020). TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nat. Cell Biol 22, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugaard M, Baude A, Fugger K, Povlsen LK, Beck H, Sørensen CS, Petersen NHT, Sorensen PHB, Lukas C, Bartek J, et al. (2012). LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat. Struct. Mol. Biol 19, 803–810. [DOI] [PubMed] [Google Scholar]
- de Krijger I, Boersma V, and Jacobs JJL (2021a). REV7: jack of many trades. Trends Cell Biol. 31, 686–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Krijger I, Föhr B, Pérez SH, Vincendeau E, Serrat J, Thouin AM, Susvirkar V, Lescale C, Paniagua I, Hoekman L, et al. (2021b). MAD2L2 dimerization and TRIP13 control shieldin activity in DNA repair. Nat. Commun 12, 5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dev H, Chiang TWW, Lescale C, de Krijger I, Martin AG, Pilger D, Coates J, Sczaniecka-Clift M, Wei W, Ostermaier M, et al. (2018). Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol 20, 954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Färkkilä A, Rodríguez A, Oikkonen J, Gulhan DC, Nguyen H, Dominguez J, Ramos S, Mills CE, Pérez-Villatoro F, Lazaro JB, et al. (2021). Heterogeneity and clonal evolution of acquired PARP inhibitor resistance in TP53- and BRCA1-deficient cells. Cancer Res. 81, 2774–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Findlay S, Heath J, Luo VM, Malina A, Morin T, Coulombe Y, Djerir B, Li Z, Samiei A, Simo-Cheyou E, et al. (2018). SHLD2/FAM35A co-operates with REV7 to coordinate DNA double-strand break repair pathway choice. EMBO J. 37, e100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita H, Ikeda M, Ui A, Ouchi Y, Mikami Y, Kanno SI, Yasui A, and Tanaka K (2022). CHAMP1-POGZ counteracts the inhibitory effect of 53BP1 on homologous recombination and affects PARP inhibitor resistance. Oncogene 41, 2706–2718. [DOI] [PubMed] [Google Scholar]
- Gao S, Feng S, Ning S, Liu J, Zhao H, Xu Y, Shang J, Li K, Li Q, Guo R, and Xu D (2018). An OB-fold complex controls the repair pathways for DNA double-strand breaks. Nat. Commun 9, 3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, Barretina J, Gelfand ET, Bielski CM, Li H, et al. (2019). Next-generation characterization of the cancer cell line Encyclopedia. Nature 569, 503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezraoui H, Oliveira C, Becker JR, Bilham K, Moralli D, Anzilotti C, Fischer R, Deobagkar-Lele M, Sanchiz-Calvo M, Fueyo-Marcos E, et al. (2018). 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 560, 122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Somyajit K, Narita T, Maskey E, Stanlie A, Kremer M, Typas D, Lammers M, Mailand N, Nussenzweig A, et al. (2018). DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell 173, 972–988.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Hashimoto H, Murakumo Y, Kobayashi S, Kogame T, Unzai S, Akashi S, Takeda S, Shimizu T, and Sato M (2010). Crystal structure of human REV7 in complex with a human REV3 fragment and structural implication of the interaction between DNA polymerase zeta and REV1. J. Biol. Chem 285, 12299–12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Taharazako S, Ikeda M, Fujita H, Mikami Y, Kikuchi S, Hishiki A, Yokoyama H, Ishikawa Y, Kanno SI, et al. (2017). Dynamic feature of mitotic arrest deficient 2-like protein 2 (MAD2L2) and structural basis for its interaction with chromosome alignment-maintaining phosphoprotein (CAMP). J. Biol. Chem 292, 17658–17667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath J, Cheyou ES, Findlay S, Luo VM, Carpio EP, Lee J, Djerir B, Chen X, Morin T, Lebeau B, et al. (2021). POGZ promotes homology-directed DNA repair in an HP1-dependent manner. EMBO Rep. 23, e51041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P, and Cruz-García A (2018). Single molecule analysis of resection tracks. Methods Mol. Biol 1672, 147–154. [DOI] [PubMed] [Google Scholar]
- Isidor B, Küry S, Rosenfeld JA, Besnard T, Schmitt S, Joss S, Davies SJ, Lebel RR, Henderson A, Schaaf CP, et al. (2016). De novo truncating mutations in the kinetochore-microtubules attachment gene CHAMP1 cause syndromic intellectual disability. Hum. Mutat 37, 354–358. [DOI] [PubMed] [Google Scholar]
- Itoh G, Kanno S.i., Uchida KSK, Chiba S, Sugino S, Watanabe K, Mizuno K, Yasui A, Hirota T, and Tanaka K (2011). CAMP (C13orf8, ZNF828) is a novel regulator of kinetochore-microtubule attachment. EMBO J. 30, 130–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Kozono D, Deraska P, Branigan T, Dunn C, Zheng XF, Parmar K, Nguyen H, DeCaprio J, Shapiro GI, et al. (2020). CHK1 inhibitor blocks phosphorylation of FAM122A and promotes replication stress. Mol. Cell 80, 410–422.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miniowitz-Shemtov S, Eytan E, Kaisari S, Sitry-Shevah D, and Hershko A (2015). Mode of interaction of TRIP13 AAA-ATPase with the Mad2-binding protein p31comet and with mitotic checkpoint complexes. Proc. Natl. Acad. Sci. USA 112, 11536–11540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirman Z, Lottersberger F, Takai H, Kibe T, Gong Y, Takai K, Bianchi A, Zimmermann M, Durocher D, and de Lange T (2018). 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature 560, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Álvarez-Quilón A, Moatti N, Zimmermann M, et al. (2018). The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560, 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa RS, Nagao K, Masuda HT, Iwasaki O, Hirota T, Nozaki N, Kimura H, and Obuse C (2010). Human POGZ modulates dissociation of HP1alpha from mitotic chromosome arms through Aurora B activation. Nat. Cell Biol 12, 719–727. [DOI] [PubMed] [Google Scholar]
- Olivieri M, Cho T, Álvarez-Quilón A, Li K, Schellenberg MJ, Zimmermann M, Hustedt N, Rossi SE, Adam S, Melo H, et al. (2020). A genetic Map of the response to DNA damage in human cells. Cell 182, 481–496.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AJ, Johnson RD, Thompson LH, and Jasin M (1999). XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 13, 2633–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack LM, Davoli T, Li MZ, Li Y, Xu Q, Naxerova K, Wooten EC, Bernardi RJ, Martin TD, Chen T, et al. (2018). Profound tissue specificity in proliferation control underlies cancer drivers and aneuploidy patterns. Cell 173, 499–514.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarangi P, Clairmont CS, Galli LD, Moreau LA, and D’Andrea AD (2020). p31(comet) promotes homologous recombination by inactivating REV7 through the TRIP13 ATPase. Proc. Natl. Acad. Sci. USA 117, 26795–26803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Ganesan S, Vlasakova K, Chen J, Socolovsky M, and Livingston DM (1999). Genetic analysis of BRCA1 function in a defined tumor cell line. Mol. Cell 4, 1093–1099. [DOI] [PubMed] [Google Scholar]
- Soria G, and Almouzni G (2013). Differential contribution of HP1 proteins to DNA end resection and homology-directed repair. Cell Cycle 12, 422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark JM, Pierce AJ, Oh J, Pastink A, and Jasin M (2004). Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol. Cell Biol 24, 9305–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS (2014). End resection at double-strand breaks: mechanism and regulation. Cold Spring Harb. Perspect. Biol 6, a016436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomida J, Takata KI, Bhetawal S, Person MD, Chao HP, Tang DG, and Wood RD (2018). FAM35A associates with REV7 and modulates DNA damage responses of normal and BRCA1-defective cells. EMBO J. 37, e99543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, and Mann M (2010). Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 142, 967–980. [DOI] [PubMed] [Google Scholar]
- Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M, et al. (2015). REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 521, 541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Rosenberg SC, Moeller A, Speir JA, Su TY, and Corbett KD (2015). TRIP13 is a protein-remodeling AAA+ ATPase that catalyzes MAD2 conformation switching. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F, Kim W, Gao H, Liu C, Zhang Y, Chen Y, Deng M, Zhou Q, Huang J, Hu Q, et al. (2021). ASTE1 promotes shieldin-complex-mediated DNA repair by attenuating end resection. Nat. Cell Biol 23, 894–904. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table.
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| C13orf8/CHAMP1 | Abnova | Cat# H00283489-B01P; RRID:AB_1673079 |
| Mad2L2/REV7 | Abcam | Cat# ab180579; RRID:AB_2890174 |
| POGZ | Bethyl Laboratories | Cat# A302-509A; RRID:AB_1966082 |
| TRIP13 | Abcam | Cat# ab128171; RRID:AB_11145670 |
| H3 | Cell Signaling Technology | Cat# 3638; RRID:AB_1642229 |
| GFP | Cell Signaling Technology | Cat# 2956; RRID:AB_1196615 |
| β-Actin | Cell Signaling Technology | Cat# 3700; RRID:AB_2242334 |
| RPA2-P-Ser33 | Fisher Scientific | Cat# NB100544; RRID:AB_526631 |
| RAD51 | Santa Cruz | Cat# sc-8349; RRID:AB_2253533 |
| GAPDH | Cell Signaling Technology | Cat# 5174; RRID:AB_10622025 |
| Goat anti-Rat IgG (Alexa Fluor 488) | Invitrogen | Cat# A-11006; RRID:AB_2534074 |
| Goat anti-Mouse IgG (Alexa Fluor 488) | Invitrogen | Cat# A32723; RRID:AB_2633275 |
| Chicken anti-mouse IgG (Alexa Fluor 594) Anti-rabbit IgG, HRP-linked | Invitrogen | Cat# A-21201; RRID:AB_2535787 |
| Flag | Millipore-Sigma | F3165; RRID:AB_259529 |
| Chemicals, peptides, and recombinant proteins | ||
| Olaparib | Selleckchem | S1060 |
| BrdU | Millipore-Sigma | B5002 |
| Anti-FLAG® M2 Magnetic Beads | Millipore-Sigma | M8823 |
| GFP-Trap_A | Chromotek | Gta-20 |
| Critical commercial assays | ||
| CellTiter-Glo Luminescent Viability Assay | Promega | G7570 |
| Subcellular protein fractionation kit | ThermoFisher | 78840 |
| Deposited data | ||
| Raw imaging data | This paper; Mendeley data | https://doi.org/10.17632/fkh6v4dw6y.1 |
| TCGA Pan-Cancer Atlas Studies’ RNASeq expression data | TCGA (PMID: 24071849) | https://www.cbioportal.org/ |
| Cancer cell lines’ RNASeq expression data | Cancer Cell Line Encyclopedia (Ghandi et al., 2019) | https://depmap.org/portal/ |
| Cancer cell lines’ dependency data (CRISPR DepMap Public 19Q4 dataset) | Broad Institute Cancer Dependency Map (Meyers et al., 2017) | https://depmap.org/portal/ |
| Experimental models: Cell lines | ||
| 293T | ATCC | N/A |
| U2OS | ATCC | N/A |
| RPE-1 | ATCC | N/A |
| HCC1937 | D’Andrea lab | N/A |
| U2OS-DR-GFP | D’Andrea lab | N/A |
| U2OS-EJ5 | D’Andrea lab | N/A |
| Oligonucleotides | ||
| siCHAMP1-4: CAGGAATCAAGCAGTGATCAA | Qiagen | SI00318038 |
| siCHAMP1-8: CAGGAACTTCGTAAACCATCA | Qiagen | SI04282159 |
| SiPOGZ: CCGGGTGTAAGGTCCCTTTAA | Qiagen | SI00688583 |
| siTRIP13: TTGGGACAGCTTGGTATACGA | Qiagen | (Clairmont et al., 2020) |
| AllStars Neg. Control siRNA | Qiagen | 0001027281 |
| Recombinant DNA | ||
| CHAMP1 CRISPR guide RNA 1_pSpCas9 BB-2A-GFP (PX458) | Genscript | N/A |
| CHAMP1 CRISPR guide RNA 3_pSpCas9 BB-2A-GFP (PX458) | Genscript | N/A |
| POGZ CRISPR guide RNA 1_pSpCas9 BB-2A-GFP (PX458) | Genscript | N/A |
| POGZ CRISPR guide RNA 2_pSpCas9 BB-2A-GFP (PX458) | Genscript | N/A |
| Software and algorithms | ||
| ImageJ | U. S. National Institutes of Health | https://imagej.nih.gov/ij/ |
| Graphpad Prism 9 | Graphpad | https://www.graphpad.com/ |
| R (version 4.0.3) | R Foundation for Statistical Computing | https://www.R-project.org/ |
| R package survival (version 3.2–10) | R Foundation for Statistical Computing | https://CRAN.R-project.org/package=survival |
| R package survminer (version 0.4.9) | R Foundation for Statistical Computing | https://CRAN.R-project.org/package=survminer |
| R package edgeR (version 3.32.1) | Bioconductor | https://www.bioconductor.org/packages/release/bioc/html/edgeR.html |
| R package ggplot2 (version 3.3.5) | R Foundation for Statistical Computing | https://ggplot2.tidyverse.org |
| R package ggpubr (version 0.4.0) | R Foundation for Statistical Computing | https://CRAN.R-project.org/package=ggpubr |
| R package ggpmisc (version 0.4.3) | R Foundation for Statistical Computing | https://CRAN.R-project.org/package=ggpmisc |