

SUMMARY
N6-methyladenosine (m6A) is deposited co-transcriptionally on thousands of cellular mRNAs and plays important roles in mRNA processing and cellular function. m6A is particularly abundant within the brain and is critical for neurodevelopment. However, the mechanisms through which m6A contributes to brain development are incompletely understood. RBM45 acts as an m6A-binding protein that is highly expressed during neurodevelopment. We find that RBM45 binds to thousands of cellular RNAs, predominantly within intronic regions. Rbm45 depletion disrupts the constitutive splicing of a subset of target pre-mRNAs, leading to altered mRNA and protein levels through both m6A-dependent and m6A-independent mechanisms. Finally, we find that RBM45 is necessary for neuroblastoma cell differentiation and that its depletion impacts the expression of genes involved in several neurodevelopmental signaling pathways. Altogether, our findings show a role for RBM45 in controlling mRNA processing and neuronal differentiation, mediated in part by the recognition of methylated RNA.
Graphical Abstract
In brief
Choi et al. identify RBM45 as an m6A-binding protein enriched in the developing brain. RBM45 binds to thousands of cellular RNAs, primarily within introns, and regulates constitutive splicing of target transcripts. Loss of RBM45 causes altered expression of neurodevelopmental genes and defects in the proliferation and differentiation of neuroblastoma cells.
INTRODUCTION
Post-transcriptional RNA regulation is an important mechanism for controlling the gene expression programs that underlie brain development. In particular, m6A has emerged in recent years as an abundant RNA modification that is a critical regulator of brain development (Livneh et al., 2020; Flamand and Meyer 2019). For instance, knockout of the N6-methyladenosine (m6A) methyltransferase components METTL3 and METTL14 in the mouse cortex results in slowed neural progenitor proliferation rates, delays in progenitor cell differentiation, and disrupted cortical patterning (Yoon et al., 2017; Engel et al., 2018; Y. Wang et al., 2018). In the cerebellum, loss of METTL3 causes hypoplasia and apoptosis of granule cells (Wang et al., 2018). Despite the importance of m6A for brain development, our understanding of the mechanisms through which m6A controls neurodevelopmental gene expression programs remains limited.
m6A impacts many aspects of RNA processing, primarily through the recruitment of m6A “reader” proteins that preferentially bind to methylated RNAs and carry out diverse functions. (Meyer and Jaffrey 2017; Yue et al. 2015). Among the first m6A readers to be identified were the YTH domain-containing proteins, which directly bind to m6A through a conserved YTH domain (Dominissini et al., 2012). However, subsequent studies have identified other RNA-binding proteins (RBPs) as both direct and indirect m6A readers, indicating that m6Acan influence RNA binding of RBPs beyond just those that contain a YTH domain (Edupuganti et al., 2017; N. Liu et al., 2015; Huang et al., 2018; Edens et al., 2019). Despite these advances in our understanding of m6A:protein interactions, knowledge of tissue-specific m6A readers and their functions remains poor. This is especially true in the brain, where m6A is particularly abundant (Meyer et al., 2012).
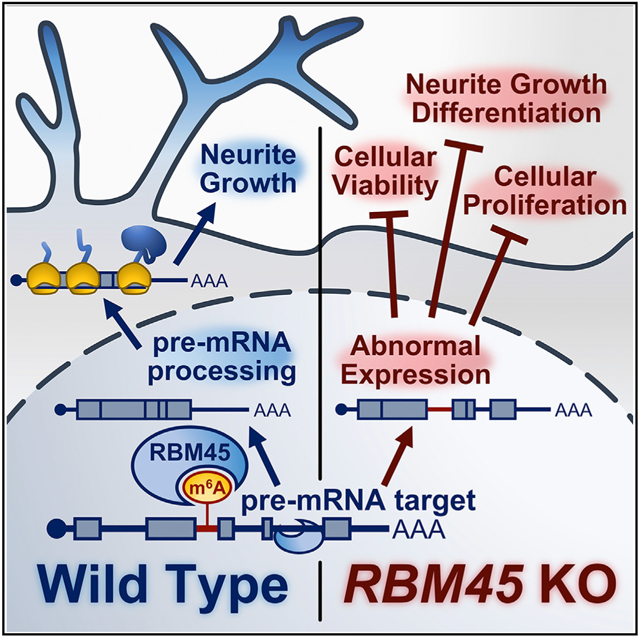
Here, we used an unbiased approach to identify m6A reader proteins in the brain and identified RBM45 as an m6A-binding protein. RBM45 is highly expressed in the developing brain and has been linked to age-related neurodegenerative disease, but its RNA targets and roles in RNA processing are unknown (Collins et al., 2012; Tamada et al., 2002; Mashik et al., 2016; Li et al., 2015). We find that RBM45 is a direct m6A-binding protein that recognizes m6A via two C-terminal RNA-binding domains (RBDs). Further, we identify the cellular RNA targets of RBM45 and show that it binds to introns and impacts constitutive splicing, likely through both m6A-dependent and -independent mechanisms. Finally, we demonstrate that RBM45 depletion compromises the proliferation and differentiation of human SH-SY5Y cells. This is accompanied by dampening of gene expression changes that normally occur during differentiation, an effect that is rescued by RBM45 re-expression. Altogether, our studies identify RBM45 as an m6A-binding protein that controls constitutive splicing to regulate the expression of target RNAs important for neurodevelopment.
RESULTS
RBM45 is an m6A-binding protein
To identify proteins in the brain that preferentially bind to m6A, we performed RNA pulldowns using unmodified and m6A-modified bait RNAs and protein lysates from the mouse brain as well as from mHippoE-2 cells, an immortalized mouse embryonic hippocampal neuron cell line (Gingerich et al., 2010) (Figures S1A and S1B; Table S1). Proteins with preferential binding to methylated RNA were then identified using liquid chromatography-tandem mass spectrometry (LC-MS/MS). This revealed several YTH domain-containing proteins, indicating that our approach reliably detects m6A-binding proteins (Figures 1A and S1C-S1E; Table S1). Interestingly, one of the most highly enriched m6A-binding proteins that we identified was RNA binding motif-containing protein 45 (RBM45), a protein that is abundant in the developing brain and that has been implicated in neurodegenerative disease (Tamada et al., 2002; Collins et al., 2012; Li et al. 2015, 2016; Mashik et al., 2016) (Figure 1A; Table S1). We further confirmed this finding using RNA pulldowns with lysates from HEK293T cells expressing hemagglutinin (HA)-tagged RBM45 (Figure 1B). Preferential m6A binding was retained for both mouse and human RBM45, indicating that the m6A-binding behavior of RBM45 is conserved between mice and humans (Figure 1B).
Figure 1. RBM45 binds to m6A-modified RNA.

(A) Scatterplot with proteins recovered from RNA pulldown LC-MS/MS experiments using A or m6A RNA baits and lysates from mHiPPoE-2 cells, with biological replicates plotted on each axis. RBM45 and known m6A reader proteins YTHDF1, YTHDF2, YTHDF3, and YTHDC2 are labeled. Red, proteins enriched in m6A RNA pulldowns; blue, proteins enriched in A RNA pulldown.
(B) Western blot following RNA pulldowns using lysates from HEK293T cells transfected with human or mouse HA-RBM45 shows preferential binding to m6A-modified RNA. YTHDF2 and GAPDH probed as positive and negative controls, respectively. Ctrl (no RNA), A, and m6A RNA pulldowns are indicated. Results represent 3 biological replicates.
(C) Quantification of RBM45 enrichment by Coomassie stain from RNA pulldown assays using A or m6A bait RNA and purified GST-RBM45. RBM45 enrichment is normalized to A RNA pulldown levels. Mean ± SEM plotted from 4 biological replicates. Significance calculated using a Welch’s t test; **p = 3.1 × 10−3.
(D) Fluorescence polarization assays using GST-RBM45 and A or m6A FAM-labeled RNA shows preferential binding to m6A RNA (Kd m6A = 462 ± 73 nM; Kd A = 1,371 ± 212 nM). Mean ± SD plotted for 3 biological replicates.
(E) Schematic of RBM45 variants used for RNA pulldown assays. Size indicated in amino acids.
(F) RNA pulldown results using A and m6A RNA with lysates from HEK293T cells transfected with the indicated FLAG-tagged RBM45 constructs. YTHDF2 and GAPDH are shown as positive and negative controls, respectively. Data represent 5 biological replicates.
See also Figure S1.
To determine whether RBM45 binds m6A directly, we incorporated a UV crosslinking step and stringent wash conditions to remove proteins that are indirectly recovered in the RNA pulldown assay. The preferential interaction between m6A and RBM45 remained, indicating that RBM45 binds directly to m6A (Figures S1F and S1G). This direct interaction was further confirmed with RNA pulldowns using purified, GST-tagged RBM45, which revealed a ~2.5-fold preferential binding to m6A-modified RNA compared with unmodified RNA (Figures 1C and S1H). We then determined the binding affinity of RBM45 for m6A with fluorescence anisotropy, which showed a ~3-fold preference of RBM45 for m6A-containing RNA (Kd m6A = 462 ± 73 nM; Kd A = 1,371 ± 212 nM) (Figure 1D). Altogether, our results identify RBM45 as an m6A-binding protein that directly interacts with methylated RNA.
C-terminal RBDs of RBM45 work cooperatively to recognize m6A
RBM45 contains three RNA-recognition motifs (RRMs; RRM1, RRM2, and RRM3) and a homo-oligomer assembly (HOA) domain originally predicted to form an RBD (Henderson 2018; Wang et al., 2020; Tamada et al., 2002) (Figure 1E). To determine the region(s) within RBM45 that are responsible for m6A binding, we generated various FLAG-tagged RBM45 deletion mutants and expressed them in HEK293T cells. Cell lysates were then used in RNA pulldown assays to determine the ability of each RBM45 variant to recognize methylated and unmethylated RNA (Figures 1F and S1I-S1K). Deletion of any single RBD alone or any two RRMs together does not prevent m6A binding (Figure S1K). However, deletion of all three RRM domains reduces RNA binding but does not completely abolish m6A recognition, suggesting that the HOA domain may contribute to m6A binding (Figure S1K). Indeed, removal of both the HOA and RRM3 domains compromises RNA recognition and preferential binding of RBM45 to m6A (Figure 1F). This suggests that the C-terminal RBDs of RBM45, comprised of the HOA and RRM3 domains, work cooperatively to recognize m6A. This is consistent with a recent study showing that the N-terminal region of RBM45 does not preferentially bind m6A (Chen et al., 2021).
RBM45 binds to intronic regions of target RNAs
Previous studies have identified RBM45 as a predominantly nuclear RBP enriched in the brain (Tamada et al., 2002; Wang et al., 2020; Chen et al., 2021), but the RNAs that it binds in cells are unknown. To identify RBM45 target RNAs, we performed cross-linking and immunoprecipitation sequencing (CLIP-seq) in both mHippoE-2 and HEK293T cells expressing mouse or human HA-RBM45, respectively (Figure S2A). We identified 16,399 peaks in 6,495 RNAs in mHippoE-2 cells and 13,758 peaks in 5,230 RNAs in HEK293T cells that are present in two biological replicates (Figures 2A-2C and S2B; Table S2). A total of 1,868 RNAs are bound by RBM45 in both cell types, indicating a high degree of target RNA overlap between mouse and human (Figure 2D; Table S2). Strikingly, nearly 90% of peaks in both cell types map to intronic regions (Figures 2A and 2B). These peaks are enriched internally and near the 3′ end of introns and are depleted at splice junctions (Figures S2C and S2D), consistent with a recent report of RBM45 binding to internal intronic regions in the parvovirus (B19) pre-mRNA (Wang et al., 2020).
Figure 2. RBM45 binds to introns in target pre-mRNAs.

(A) Distribution of RBM45 CLIP peaks in HEK293T cells and mHippoE-2 cells.
(B) IGV browser tracks showing input and CLIP reads for the RBM45 targets CDK5RAP3 and Ide. CLIP peaks indicated with shading and bars below each CLIP track.
(C) RNA immunoprecipitation (RIP)-qPCR validation of RBM45 targets identified by CLIP in HEK293T and mHippoE-2 cells. 18S rRNA shown as a control. Mean ± SEM from 3 biological replicates plotted. Statistical significance calculated using a Welch’s t test; **p ≤ 0.01, ***p ≤ 1.0 × 10−3, ****p ≤ 1.0 × 10−4.
(D) Overlapping RBM45 CLIP targets between mHippoE-2 and HEK293T cells.
(E) Most enriched motifs in RBM45 CLIP peaks for HEK293T and mHippoE-2 cells by significance (padj).
(F) Absolute distance density plots for m6A DRACH motif enrichment within 100 nt of HEK293T and mHiPPoE-2 RBM45 CLIP peak summits. Randomly shuffled peaks shown as control.
(G) Absolute distance density plots show enrichment of chromatin-associated RNA (CA-RNA) m6A sites near RBM45 CLIP peak summits compared with nuclear (Nuc) and cytoplasmic (Cyto) RNA m6A sites. Randomly shuffled sites shown as controls.
(H) Pie charts showing RBM45 CLIP peaks in HEK293T cells and mHippoE-2 cells that overlap directly with m6A sites identified by DART-seq (CLIP-DART) or TNT-seq (CLIP-TNT).
(I) Histograms of RBM45 CLIP peak enrichment levels (log2 fold change [FC] CLIP/input) in wild-type (WT; red) and Mettl3 KO (blue) cells.
See also Figures S2 and S3.
We identified GAC or bipartite GAC sequences as the top enriched motifs in RBM45 peak regions in both mHippoE-2 and HEK293T cells (Figures 2E and S2E), which is consistent with in vitro selection-based studies of RBM45 binding sequences (Dominguez et al., 2018; Cook et al., 2017). GAC is also the predominant minimal consensus sequence for m6A (Meyer et al., 2012; Desrosiers et al. 1974), suggesting that RBM45 binding to cellular RNAs may be driven at least in part by m6A. Indeed, we observed enrichment of the m6A consensus near RBM45 CLIP peak summits in both mouse and human cells (Figure 2F).
To investigate whether RBM45 target sites are enriched for m6A, we used DART-seq (Meyer 2019; Zhu et al., 2022) to profile m6A on chromatin-associated RNA (CA-RNA), nuclear RNA, and cytoplasmic RNA from mHippoE-2 and HEK293T cells (Figures S3A-S3C). As expected, the CA-RNA fraction contains a substantially higher proportion of intronic m6A sites compared with the nuclear and cytoplasmic RNA fractions (Figures S3D and S3E), and the m6A sites identified in all three RNA fractions are enriched for the m6A consensus sequence (Figure S3F; Table S3). We found that m6A sites are enriched near RBM45 CLIP peaks, an effect that is highest for CA-RNA (Figure 2G). Additionally, RBM45 binding sites and m6A sites share a similar distribution within introns of target pre-mRNAs (Figure S3G).
To determine whether RBM45 binding sites are methylated, we analyzed the overlap between RBM45 CLIP peaks and m6A sites. We found 1,440 CLIP peaks in mHippoE-2 cells and 2,023 CLIP peaks in HEK293T cells that overlap with m6A sites, many of which are located within introns (Figure 2H; Table S2). A similar result was obtained when comparing RBM45 CLIP peaks with intronic m6A sites previously identified by antibody-based m6A profiling of nascent RNA in HEK293T cells (TNT-seq) (Louloupi et al., 2018) (Figure 2H). Of the 1,868 conserved RBM45 target RNAs in mHippoE-2 and HEK293T cells, 683 RNAs contain m6A sites within CLIP peak regions (Table S2). Gene Ontology (GO) analysis of these RNAs shows enrichment of transcripts involved in synaptic function, cellular homeostasis, and neurite projection morphogenesis, suggesting potential roles for m6A-modified RBM45 target RNAs in brain development and function (Table S2).
To determine the influence of m6A on RBM45 binding to cellular RNAs, we performed RBM45 CLIP-seq in METTL3-depleted mHippoE-2 and HEK293T stable cells (Figures S2F-S2H). We observed significantly reduced RBM45 binding at CLIP peak regions in METTL3-depleted cells (Figure 2I; Table S2). Specifically, we found that 5% of peaks in mHippoE-2 cells and 15% of peaks in HEK293T cells exhibit reduced RBM45 binding after METTL3 depletion, indicating that RBM45 binding to a subset of target RNAs is m6A dependent (Table S2).
RBM45 regulates gene expression by influencing RNA splicing
To investigate the consequences of RBM45 depletion on gene expression, we generated Rbm45 knockout (KO) stable mHippoE-2 cell lines using CRISPR-Cas9 (Figures 3A, S4A, and S4B). We also generated control cell lines expressing Cas9 but no single guide RNA (sgRNA; Cas9 control), as well as Rbm45 KO-rescue cell lines in which we restored RBM45 expression in Rbm45 KO cells (Figure 3A). We identified 668 RNAs with significant gene-expression changes between Rbm45 KO and Cas9 control cells, including 427 RNAs with increased expression and 241 RNAs with decreased expression in Rbm45 KO cells (Figures 3B and S4C; Table S4). We further identified a subset of these RNAs that are differentially expressed between Rbm45 KO and KO-rescue cells and that contain RBM45 CLIP peaks (Figures 3C and S4C; Table S4). This resulted in a set of 46 high-confidence RNAs that are bound by RBM45 and exhibit RBM45-dependent gene expression, an effect we further validated using qRT-PCR (Figures 3D and S4D; Table S4).
Figure 3. RBM45 regulates target RNA expression in mHippoE-2 cells.

(A) Rbm45 levels in Cas9 control, Rbm45 KO, and KO-rescue mHippoE-2 cells. Mean ± SEM from 3 biological replicates plotted. Statistical significance calculated using a Welch’s t test. ***p = 3.0 × 10−4, ****p < 1.0 × 10−6.
(B) Venn diagram shows a subset of differentially expressed RNAs (adjusted p value [padj[ ≤ 0.05, ∣log2FC∣≥log2(1.2)) bound by RBM45 in mHippoE-2 cells.
(C) Scatterplot of significant (padj ≤ 0.05) differential gene-expression changes (∣log2FC∣≥log21.2) between Cas9 control and Rbm45 KO mHiPPoE-2 cells (x axis) or Rbm45 KO and KO-rescue mHiPPoE-2 cells (y axis). Blue, up-regulated CLIP targets, green; up-regulated non-target (NT) RNAs; red, down-regulated CLIP targets; orange, down-regulated NT RNAs. Targets (Cpne8, Ide, and Neat1) chosen for validation in (D) are labeled alongside Rbm45.
(D) Relative expression of Cpne8, Ide, and Neat1 mRNAs in Cas9 control, Rbm45 KO, and KO-rescue mHippoE-2 cells. Mean ± SEM from 3 biological replicates plotted. Significance calculated using a Welch’s t test; *p ≤ 0.05, **p ≤ 0.01, ****p ≤ 1.0 × 10−4.
(E) RNA-seq alignments for Ide in Cas9 control, Rbm45 KO, and KO-rescue mHippoE-2 cells. RBM45 CLIP peaks and m6A sites are indicated.
(F) Western blot shows an RBM45-dependent effect on IDE expression in mHippoE-2 cells. Results represent 5 biological replicates.
(G) Densitometry quantification of differential IDE expression in (F). Mean ± SEM plotted; significance determined using a Welch’s t test; *p < 0.05, **p < 0.01.
(H) qRT-PCR quantification along the Ide pre-mRNA using primer pairs targeting 5′ or 3′ splice junctions (SJs). The exon 11 3′ SJ and exon 12 5′ SJ flank intron 11, where RBM45 binds. Mean ± SEM from 3 biological replicates plotted. Significance calculated using an adjusted Welch’s t test (Holm-Sidak); *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 1.0 × 10−3, ****p ≤ 1.0 × 10−4.
(I) Ide expression in CA, Nuc, and Cyto RNA from mHiPPoE-2 cell lines. Mean ± SEM from 3 biological replicates is plotted; expression normalized to Cas9 control cells. Significance calculated by adjusted two-way ANOVA (Tukey’s); *p ≤ 0.05, **p ≤ 0.01.
See also Figure S4.
We next sought to determine the mechanism through which RBM45 regulates target RNAs to control gene expression. Our finding that RBM45 binds preferentially to intronic regions, as well as previous reports that RBM45 is localized primarily to the nucleus and interacts with splicing factors (Collins et al., 2012; Li et al., 2015, 2016), prompted us to explore potential roles for RBM45 in the regulation of RNA splicing. We first investigated whether RBM45 regulates alternative splicing by analyzing RNA-seq data from Cas9 control and Rbm45 KO cells using both DEXSeq and rMATS (Anders et al. 2012; Shen et al., 2014). This revealed very few transcripts exhibiting RBM45-dependent alternative splicing (Figure S4E; Table S4). Similar trends were observed when we examined retained introns (Figure S4E; Table S4), suggesting that RBM45 does not directly impact alternative splicing or intron retention for most of its target RNAs.
The finding that RBM45 depletion alters the abundance of target transcripts without influencing alternative splicing prompted us to explore whether RBM45 directs constitutive splicing of its target RNAs instead. We focused on the high-confidence set of RNAs that are bound by RBM45 and that show RBM45-dependent changes in expression (Figure 3C; Table S4). Among these RNAs is Ide, which is one of the most highly enriched transcripts in our RBM45 CLIP-seq data. Ide encodes for insulin-degrading enzyme (IDE), a protein with potential therapeutic value in the treatment of neurodegenerative disease due to its ability to degrade amyloid beta (Aβ) peptides (Farris et al., 2003; Kurochkin et al. 2018). We observed a ~10-fold reduction in Ide mRNA levels and decreased IDE protein levels in Rbm45 KO cells, both of which are rescued by RBM45 overexpression (Figures 3D-3G). When we examined RNA-seq reads throughout the Ide gene, we found elevated read density within intron 12 in Rbm45 KO cells, an effect that is largely restored in KO-rescue cells (Figures 3E; Table S4). Interestingly, introns 11 and 12 are where RBM45 CLIP-seq peaks are located (Figure 3E).
We saw no evidence of RBM45 depletion impacting alternative Ide splicing (Figure S4E), so we investigated the possibility that RBM45 regulates constitutive splicing of the Ide pre-mRNA. Targeted qRT-PCR across several intron-exon boundaries throughout Ide revealed increased abundance of amplicons containing intron 11 as well as other intron-exon boundaries, consistent with a general defect in pre-mRNA processing (Figure 3H). We also observed a substantial drop off in the splicing efficiency of exons downstream of exon 10 in Rbm45 KO cells (Figure S4F). This effect was not caused by intron retention, as we failed to identify any Ide variants that include partially or fully retained intron 11 or 12 (Figures S4G and S4H). Moreover, the reduced Ide mRNA levels in Rbm45 KO cells are not due to introduction of a premature termination codon and nonsense-mediated decay (NMD) since inhibition of NMD with cycloheximide does not alter Ide abundance (Figure S4I). Instead, we observed reduced nuclear Ide levels in Rbm45 KO cells, suggesting that Rbm45 depletion leads to nuclear degradation of aberrantly processed Ide pre-mRNA (Figure 3I). This effect is also mimicked in Mettl3-depleted cells, further suggesting that both m6A and RBM45 contribute to proper Ide splicing (Figure 3I).
RBM45 regulates proliferation and differentiation of SH-SY5Y cells
RBM45 targets in mHippoE-2 and HEK293T cells are enriched for RNAs involved in developmental processes (Table S2), and Rbm45 expression in the brain peaks around embryonic day 13.5 (E13.5), a time when neurogenesis and neuronal differentiation are highly active (Semple et al., 2013; Bond et al. 2015 (Figures S5A and S5B). This prompted us to investigate whether RBM45 might regulate neurodevelopmental processe such as cellular proliferation or neuronal differentiation. For these studies, we used SH-SY5Y cells, a human neuroblas toma cell ferentiates into mature neuronal-like cell in the presence of retinoic acid (RA) (Krishna et al., 2014; Pi et al., 2017).
First, we generated Cas9 control, RBM45 KO, and RBM41 KO-rescue SH-SY5Y stable cell lines, which we validated using sequencing, quantitative MS, and qRT-PCR (Figures S5C and S5E). We observed a 1.8-fold increase in cell death in RBM45 KO cells relative to Cas9 control cells, which is rescued wit RBM45 re-expression (Figure 4A). RBM45 KO cells also have reduced growth rates and lower expression of proliferatio markers compared with Cas9 control cells, effects that ar restored in KO-rescue cells (Figures 4B-4D, S5F, and S5G) Thus, RBM45 contributes to both the viability and proliferate of SH-SY5Y cells.
Figure 4. RBM45 is required for SH-SY5Y proliferation and differentiation.

(A) Viability of Cas9 control, RBM45 KO, and KO-rescue SH-SY5Y cells. Mean ± SEM for percentage of dead cells is indicated (n = 3 biological replicates). Significance calculated using a two-tailed Kolmogorov-Smirnov (KS) test; ****p < 2.2 × 10−16.
(B) Cell counts over 72 h for SH-SY5Y cell lines. Mean ± SEM shown for 3 biological replicates. Significance calculated using two-way ANOVA; ****p < 1.0 × 10−6.
(C) Ki67 immunofluorescence in SH-SY5Y cell lines. n = biological replicates. Scale bar: 50 μm.
(D) Quantification of Ki67+ cells in Cas9 control, RBM45 KO, and KO-rescue SH-SY5Y cells. Boxplot with medians from 3 biological replicates shown. Significance calculated using a Welch’s t test; *p = 2.5 × 10−2, **p = 6.3 × 10−3.
(E) Cas9 control, RBM45 KO, and KO-rescue SH-SY5Y cells treated with DMSO or 10 μM RA. Scale bar: 50 μm. Arrows, extended neurites.
(F) Violin plots of neurite lengths in SH-SY5Y cell lines. Medians and quartiles for 4 biological replicates shown. Significance calculated using a two-tailed KS test; ***p ≤ 1.0 × 10−3, ****p ≤ 1.0 × 10−4.
(G) Quantification of GAP-43 protein expression in response to RA. Mean ± SEM for relative western blot band intensity plotted. Significance calculated using a Welch’s t test; ***p = 3.0 × 10−4, ****p < 1.0 × 10−4.
(H) Quantification of TrkB protein expression in response to RA. Mean ± SEM for relative western blot band intensity plotted. Significance calculated using a Welch’s t test; **p = 1.8 × 10−3, ****p = 1.0 × 10−4.
See also Figure S5.
We next investigated whether RBM45 influences neuronal differentiation. Treatment of SH-SY5Y cells with RA induces the extension of long neurites and expression of mature neuronal markers, such as TrkB and GAP-43 (Korecka et al., 2013; Shipley et al. 2016) (Figures S5H-S5K). We found that RA-treated RBM45 KO cells grow shorter and fewer neurites and exhibit reduced viability compared with Cas9 control cells and KO-rescue cells (Figures 4E, 4F, and S5K-S5N). Additionally RBM45 KO cells fail to exhibit proper induction of the neuronal markers GAP-43 and TrkB in response to RA, whereas inductio of these proteins in RBM45 KO-rescue cells is similar to that of Cas9 control cells (Figures 4G and 4H). Altogether, these data indicate that RBM45 is necessary for the differentiation of SH-SY5Y cells into mature neurons.
RBM45 regulates the expression of RNAs involved in neurodevelopment
We next sought to determine the gene expression changes that underlie the effects of RBM45 depletion on SH-SY5Y cell differentiation. We first performed RBM45 CLIP-seq in SH-SY5Y cells to identify the target RNAs bound by RBM45. We identified 12,406 RBM45 CLIP peaks in 5,086 RNAs and observed an enrichment of RBM45 binding sites within introns, similar to what we found in mHippoE-2 cells and HEK293T cells (Figure 5A; Table S5). The preference for intronic binding in SH-SY5Y cells was also seen following RA treatment (Figure 5A). Examination of RBM45 target mRNAs revealed several transcripts encoding important neurodevelopmental proteins, consistent with the effects of RBM45 depletion on SH-SY5Y cell proliferation and differentiation (Figure 5B; Table S5).
Figure 5. RBM45 regulates gene expression during neuronal differentiation.

(A) Distribution of high-confidence RBM45 CLIP peaks identified in DMSO- or RA-treated SH-SY5Y cells.
(B) RIP-qPCR validation of RBM45-bound mRNAs identified by CLIP in DMSO- and RA-treated SH-SY5Y cells. 18S rRNA shown as a control. CPNE8 and NTRK2 are examples of RA-induced transcripts bound by RBM45. Mean ± SEM from 3 biological replicates. Significance calculated using a Welch’s t test; *p ≤ 0.05, ***p ≤ 1.0 × 10−3, ****p ≤ 1.0 × 10−4.
(C) Volcano plots show gene expression in SH-SY5Y cells after RA treatment (log2FC RA:DMSO). Statistically significant changes (padj ≤ 0.05, ∣log2FC∣ ≥log2(1.2)) are colored in blue.
(D) Scatterplot of differentially expressed genes in DMSO- versus RA-treated Cas9 control and KO-rescue SH-SY5Y cells. Blue, up-regulated CLIP targets; green, up-regulated NT RNAs; red, down-regulated CLIP targets; orange, down-regulated NT RNAs. RBM45-regulated target RNAs chosen for validation in are labeled.
(E) Venn diagram shows a subset of differentially expressed RNAs in Cas9 control and KO-rescue stable cells that are bound by RBM45.
(F) Clustered heatmap of differentially expressed genes after differentiation in Cas9 control, RBM45 KO, and KO-rescue SH-SY5Y cells.
(G) qRT-PCR validation of gene expression for select target RNAs in SH-SY5Y cell lines. Mean ± SEM from 6 biological replicates. Significance calculated using an adjusted Welch’s t test (Holm-Sidak); **p ≤ 0.01, ***p ≤ 1.0 × 10−3, ****p ≤ 1.0 × 10−4.
See also Figure S6.
To determine how RBM45 impacts gene expression during differentiation, we performed RNA-seq on Cas9 control, RBM45 KO, and KO-rescue SH-SY5Y cells cultured with or without RA for 7 days. In Cas9 control cells, RA treatment causes widespread changes in gene expression (3,532 differentially expressed RNAs in RA-treated versus DMSO-treated cells) (Figure 5C; Table S5). However, we identified only 28 differentially expressed RNAs in RBM45 KO cells in response to RA (Figure 5C; Table S5). The RA-induced gene-expression changes in Cas9 control cells and KO-rescue cells are highly similar, indicating that RBM45 re-expression largely compensates for the effects of RBM45 depletion at the RNA level (Figure 5C). These effects are not limited to RBM45 target RNAs, indicating that both direct and indirect changes are responsible for RBM45-dependent alterations in gene expression during differentiation (Table S5).
To determine which RNAs are likely to be directly impacted by the loss of RBM45, we identified transcripts whose expression levels are altered by RA treatment in RBM45 KO cells compared with Cas9 control cells and subsequently restored in KO-rescue cells. This includes 1,110 total RNAs, 608 of which are down-regulated after RA treatment in Cas9 control and KO-rescue cells and 492 of which are up-regulated (Figures 5D-5F; Table S5). We further refined this list by focusing on a set of 405 high-confidence RBM45-dependent RNAs identified as RBM45 binding targets in our human CLIP-seq dataset (Figures 5E and 5F), a subset of which we validated using qRT-PCR (Figures 5G and S6A). Many of these RNAs encode proteins important for neurodevelopment, such as those involved in Wnt signaling, neurotrophic signaling, and transcriptional regulation (Table S5).
We next sought to understand how RBM45 regulates the expression of neurodevelopment-associated RNAs to influence SH-SY5Y cell differentiation. Global splicing analysis in Cas9 control, RBM45 KO, and KO-rescue SH-SY5Y cells revealed few RBM45-dependent changes in exon or intron usage at baseline or after RA treatment (Figures S6B and S6C; Table S5). These results are similar to what we observed in immortalized hippocampal neurons and suggest that loss of RBM45 does not lead to widespread changes in alternative splicing of its target RNAs. We therefore explored the possibility that RBM45 regulates constitutive splicing of differentially expressed transcripts in SH-SY5Y cells. We focused on two mRNAs, WNT3 and NTRK2, which show RBM45-dependent gene-expression changes and which contain intronic RBM45 CLIP peaks (Figures 6A and 6B). These targets were chosen both for the important roles they play in neurodevelopment as well as the fact that they exhibit opposite gene expression changes after RBM45 depletion, a pattern that is reflected in the larger pool of high-confidence RBM45-dependent target RNAs (Table S5).
Figure 6. RBM45 regulates splicing of neurodevelopmental target RNAs.

(A) RNA-seq alignments for NTRK2 in SH-SY5Y cell lines.
(B) RNA-seq alignments for WNT3 in SH-SY5Y cell lines.
(C) NTRK2 expression by qRT-PCR in untreated SH-SY5Y cell lines. Significance calculated using an adjusted Welch’s t test (Holm-Sidak); ***padj = 3.6 × 10−4, ****Padj ≤ 1.0 × 10−6.
(D) Expression of RBM45 targets TrkB, TLE3, and WNT3 in DMSO- and RA-treated SH-SY5Y cell lines. Data represent 4 biological replicates.
(E) Relative expression along the length of the NTRK2 pre-mRNA using qRT-PCR with primer pairs targeting 5′ or 3′ SJs. Exon-intron junctions flanking RBM45 target introns are bold red. Mean ± SEM from 3 biological replicates. Significance calculated using an adjusted Welch’s t test (Holm-Sidak); ***p ≤ 1.0 × 10−3, ****p ≤ 1.0 × 10−4.
(F) Expression of NTRK2 RNA by qRT-PCR in subcellular fractions from DMSO- and RA-treated SH-SY5Y cells. Expression normalized to CA-RNA within genotypes. Mean ± SEM from 2 biological replicates. Significance calculated by two-way ANOVA; ****p ≤ 1.0 × 10−4.
(G) WNT3 expression by qRT-PCR in SH-SY5Y cell lines. Significance calculated using an adjusted Welch’s t test (Holm-Sidak); ****padj = 7.0 × 10−6.
(H) Relative expression of unspliced WNT3 pre-mRNA measured by qRT-PCR with primer pairs targeting 5′ or 3′ SJs along the length of the pre-mRNA. Exon-intron junctions flanking target introns are bold red. Mean ± SEM from 3 biological replicates. Significance calculated using an adjusted Welch’s t test (Holm-Sidak); *p ≤ 0.05, ***p ≤ 1.0 × 10−3.
(I) Expression of WNT3 RNA by qRT-PCR in subcellular fractions from DMSO- and RA-treated SH-SY5Y cells. Expression normalized to CA-RNA within genotypes. Mean ± SEM from 2 biological replicates plotted. Significance calculated by two-way ANOVA; ****p ≤ 1.0 × 10−4.
See also Figure S6.
NTRK2 encodes the TrkB protein, which mediates neurotrophin signaling and contributes to synaptic function and brain development (Minichiello 2009; Gupta et al., 2013). NTRK2 mRNA levels are substantially reduced in RBM45 KO cells, an effect that is reversed in KO-rescue cells (Figure 6C). This is accompanied by a reduction in RA-induced TrkB protein levels in RBM45 KO cells, which is rescued after RBM45 re-introduction (Figure 6D). To determine whether the effects of RBM45 depletion on NTRK2 expression reflect defects in constitutive splicing, we used targeted qRT-PCR across intron-exon junctions of NTRK2 to investigate splicing efficiency in the vicinity of RBM45 binding sites. RBM45 KO cells have elevated levels of unspliced NTRK2 pre-mRNA products including and adjacent to introns 14 and 16, which are the regions of RBM45 CLIP peaks (Figure 6E; Table S2). These effects are rescued after re-introduction of RBM45 (Figure 6E). This is not the result of alternative splicing, as our global analysis did not identify alternative NTRK2 exon usage in RBM45 KO cells, and we did not observe alternative splicing in the mature NTRK2 mRNA sequence (Figure S6D; Table S5). In addition, we found that RBM45 KO causes a buildup of NTRK2 transcripts in the nucleus without affecting the amount of chromatin-associated NTRK2 (Figure 6F). This suggests that RBM45 depletion does not impact nascent NTRK2 transcription but instead leads to deficient processing that results in nuclear accumulation of NTRK2 pre-mRNA.
We used a similar approach to examine the RBM45 target WNT3, which is a member of the Wnt family of signaling proteins important for many aspects of neurodevelopment, including anterior-posterior patterning and neural stem cell proliferation and differentiation (Biechele et al., 2013). WNT3 mRNA and protein levels are significantly up-regulated in RBM45 KO cells and restored in KO-rescue cells (Figures 6D and 6G). Targeted qRT-PCR at splice junctions surrounding intron 2, the site of RBM45 CLIP peaks, revealed decreased levels of intron 1- and 2-containing pre-mRNA in RBM45 KO cells, which are largely restored in KO-rescue cells (Figure 6H). Similar to NTRK2, we saw no evidence of alternative WNT3 splicing in RBM45 KO cells (Figure S6D; Table S5), and we found that RBM45 depletion causes nuclear accumulation of WNT3 mRNA without impacting CA-RNA levels (Figure 6I). Together, these data suggest that RBM45 KO leads to accelerated splicing of the WNT3 pre-mRNA and more efficient production of mature WNT3 mRNA, which subsequently contributes to elevated levels of WNT3 protein.
The effects we observed for the NTRK2 and WNT3 mRNAs were recapitulated in other RBM45-bound transcripts (Figures S6E-S6I) and support the finding that RBM45 depletion can lead to both accelerated and delayed processing of its target pre-mRNAs. Collectively, these data indicate that RBM45 controls the expression of target mRNAs both in undifferentiated and differentiated SH-SY5Y cells. RBM45 KO causes defects in constitutive splicing efficiency that are most prominent near the intronic binding sites of RBM45, leading to altered mature mRNA levels and deficiencies in cell proliferation and differentiation.
A subset of RBM45 target RNAs is co-regulated by m6A
Our biochemical studies of RBM45 suggest that its recognition of target RNAs is enhanced by the presence of m6A. Examination of our DART-seq datasets revealed that 56% of differentially expressed target transcripts (184 of 327 RNAs) in RBM45 KO SH-SY5Y cells contain at least one RBM45 CLIP peak that overlaps with an m6A site (Figures 7A and S7A; Tables S2 and S4). We therefore wondered whether m6A contributes to RBM45-dependent gene-expression changes and differentiation in SH-SY5Y cells. To investigate this, we generated METTL3 KO SH-SY5Y cells (Figures S7C and S7D). Interestingly, METTL3 KO cells largely phenocopy RBM45 KO cells following RA treatment, extending fewer and shorter neurites relative to Cas9 control cells (Figures 7B, S7E, and S7F).
Figure 7. Effects of METTL3 depletion in SH-SY5Y cells.

(A) Gene-expression changes in Cas9 control and KO-rescue SH-SY5Y cells after RA treatment. Differentially expressed genes are colored as follows: red, methylated CLIP target; green, CLIP target; blue, NT. A subset of neurodevelopment-associated target RNAs is labeled.
(B) Violin plots of neurite lengths for DMSO- and RA-treated Cas9 control and METTL3 KO SH-SY5Y cells. Medians and quartiles from 4 biological replicates shown. Significance calculated using a two-tailed KS test; **p ≤ 0.01, ****p ≤ 1.0 × 10−4.
(C) Venn diagram shows a subset of differentially expressed genes after RA treatment shared by Cas9 control and KO-rescue SH-SY5Y cells (purple, 1,100 RNAs) that are also bound by RBM45 in human cells (green, 5,421 RNAs) and co-regulated by both RBM45 and METTL3 (cyan, 1,586 RNAs, ∣log2FC∣≥log21.2).
(D) METTL3 KO dampens gene expression for many of the 405 RBM45 CLIP targets that exhibit sufficient coverage (padj ≤ 1) and are differentially expressed following RA treatment in Cas9 control SH-SY5Y cells. Histogram generated using log2FC (RA:DMSO) values for each RNA.
(E) Violin plots show log2FC (RA:DMSO) gene-expression changes for RBM45 targets in SH-SY5Y cell lines. 239 RNAs are up-regulated and 166 RNAs are down-regulated in differentiated Cas9 control cells (padj ≤ 0.05), and these changes are muted in METTL3 KO cells. Medians and quartiles of log2FC values indicated. Significance calculated using a two-tailed KS test; ****p ≤ 1.0 × 10−4.
(F) Cumulative distribution of log2FC (RA:DMSO) gene-expression changes for RBM45 CLIP targets (top) and NT RNAs (bottom) that are m6A modified and responsive to differentiation (dark blue) or unmodified and responsive to differentiation (cyan). Significance calculated using a two-tailed KS test between gene-expression profiles for Cas9 controlRA:DMSO and METTL3 KORA:DMSO conditions.
(G) qRT-PCR validation of gene-expression changes for select target RNAs in DMSO- and RA-treated SH-SY5Y cell lines. Mean ± SEM from 6 biological replicates plotted. Significance calculated using an adjusted Welch’s t test (Holm-Sidak); *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 1.0 × 10−3.
See also Figure S7.
To determine the effects of METTL3 depletion on gene expression, we performed RNA-seq on DMSO- and RA-treated METTL3 KO SH-SY5Y cells. We first examined the set of RNAs that exhibit significant gene expression changes following RA treatment in Cas9 control cells and divided them into RBM45 CLIP targets and non-targets. Non-target RNAs have a similar gene expression response to RA treatment in METTL3-depleted cells and Cas9 control cells (Figure S7G; Table S6). However, a subset of RBM45 targets (101 of 405 RNAs, 24.9%) have a significantly dampened response to RA treatment in METTL3 KO cells (Figures 7C and 7D; Table S6). Moreover, the subset of RBM45 target RNAs whose CLIP peaks overlap with m6A sites are more likely to be down-regulated in response to RA in METTL3 KO cells than unmethylated CLIP targets (Figure 7F). This effect is not seen for m6A-modified non-target RNAs, indicating a specific effect of loss of METTL3 on the response of methylated RBM45 target transcripts to RA (Figure 7F).
To identify the subset of target RNAs co-regulated by both RBM45 and METTL3, we looked at the difference in RA-induced gene-expression changes of RBM45 CLIP targets in Cas9 control cells compared with RBM45 KO cells or METTL3 KO cells. This revealed 514 CLIP targets that are co-regulated by the depletion of RBM45 or METTL3 following RA treatment, several of which we validated by qRT-PCR (Figures 7F, 7G, and S7H; Table S6). Interestingly, many of these RNAs encode proteins involved in neurodevelopmental processes, such as neuron projection development, cellular differentiation, and Wnt signaling (Table S6). Examination of splicing in one such neurodevelopmental gene, WNT3, revealed similar defects in the splicing efficiency of exon 3 in METTL3 KO cells and RBM45 KO cells, suggesting that RBM45-dependent splicing of Wnt3 is co-regulated by m6A (Figure S7I).
Although the effects of METTL3 depletion on global gene expression following RA treatment are not as widespread as those observed in RBM45 KO cells, these data indicate that a subset of RBM45 target RNAs is co-regulated by both METTL3 and RBM45 during SH-SY5Y cell differentiation. Thus, the impact of RBM45 on SH-SY5Y cell differentiation is likely due to both m6A-dependent and -independent RNA targeting. Both methylated and unmethylated RBM45 targets that are abnormally expressed following RBM45 depletion include mRNAs involved in neurodevelopmental signaling pathways; thus, the effects of RBM45 on SH-SY5Y cell proliferation and differentiation are likely mediated at least in part by the regulation of such transcripts, including WNT3 and NTRK2, which in turn impact downstream gene expression programs important for neurodevelopment.
DISCUSSION
RNA:protein interactions are important for controlling the gene expression programs that contribute to brain development. One mechanism through which such interactions are regulated is by the presence of m6A. However, our understanding of how m6A influences RNA:protein interactions in the developing brain remains limited. Here, we identified RBM45 as a direct m6A-binding protein that is highly expressed in the developing brain. RBM45 exhibits a ~3-fold greater affinity for methylated RNA compared with unmethylated RNA and recognizes m6A through two RBDs at its C-terminal end: the HOA domain and the RRM3 domain. The HOA domain is an intrinsically disordered region that contributes to RBM45 oligomerization and interactions with neurodegenerative disease-associated RBPs (Li et al., 2016), but there is also evidence that it facilitates RBM45 binding to RNA (Wang et al., 2020). RRMs, on the other hand, are a well-characterized class of RBD found in numerous RBPs, but they have not been widely associated with m6A recognition. A previous study found that the HNRNPA2B1 protein binds m6A, presumably through its RRM domains (Alarcón et al., 2015), but subsequent work suggests that the RRMs in this protein do not bind m6A directly (Wu et al., 2017). A recent structural study showed that the N-terminal RRM1 and RRM2 domains of RBM45 do not bind m6A (Chen et al., 2021), which is consistent with our data. However, potential roles of the HOA domain or RRM3 in m6A recognition were not examined. Thus, it will be interesting to determine the structure of full-length RBM45 to better understand how RRM3 and the upstream HOA domain coordinate to mediate m6A binding. Such studies would also provide insights into how m6A interacts with the RRM domain in RBM45 and may provide hints as to how prevalent RRM-mediated m6A binding is among other RBPs that contain this domain.
Our studies reveal that RBM45 binds to thousands of cellular RNAs, primarily within introns, and we uncover a role for RBM45 in regulating the constitutive splicing of its targets. Interestingly, our data suggest that RBM45 can have both positive and negative impacts on the splicing efficiency of target RNAs, leading to both increased and decreased mature mRNA expression. The precise mechanism through which RBM45 accelerates or impedes splicing of target RNAs is unclear. Previous studies have identified components of the splicing machinery as interacting partners of RBM45, so it is likely that one or more of these interactors mediates these effects (Li et al., 2016). Further exploration of the interaction between RBM45 and splicing factors will undoubtedly deepen our understanding of the mechanisms through which RBM45 controls the splicing process. In addition, although our data suggest that RBM45 depletion effects constitutive splicing of RBM45-bound RNAs, we did identify many genes with differentially expressed exons in RBM45-depleted cells, most of which were non-target RNAs. Thus, it is possible that RBM45 may influence alternative splicing as well as constitutive splicing, perhaps through both direct and indirect mechanisms.
RBM45 was initially discovered as an RBP expressed in the developing brain, but its roles in regulating neurodevelopment have not been explored. We find that RBM45 impacts proliferation and differentiation of human neuroblastoma cells and that loss of RBM45 leads to widespread dampening of the gene expression changes that normally occur during differentiation. A subset of the RNAs that are differentially expressed in RBM45 KO cells during differentiation are RBM45 CLIP targets, and several of these encode proteins involved in neurodevelopmental pathways that influence the expression of downstream genes. Thus, we expect that the widespread gene expression changes observed in RBM45 KO cells reflect both direct effects on RBM45 target RNAs as well as indirect effects caused by altered transcription regulatory networks. Among the RBM45 direct targets that undergo RBM45-dependent changes in expression during differentiation, we find that a subset of these contain m6A and exhibit similar expression changes in METTL3 KO cells. This co-regulation by both m6A and RBM45 suggests that the binding of RBM45 to a subset of target RNAs is mediated at least in part by m6A. These data are also consistent with our biochemical studies, which demonstrate that RBM45 can bind to both methylated and unmethylated transcripts but that the presence of m6A enhances RNA recognition.
Finally, previous studies have shown that RBM45 accumulates in cytoplasmic inclusions in the brains of patients with amyotrophic lateral sclerosis (ALS) or frontotemporal lobar degeneration (FTLD) and that it interacts with other RBPs that form aggregates in diseased brains, including FUS and TDP-43 (Collins et al., 2012; Li et al., 2015, 2016). Interestingly, both of these proteins also play roles in neurodevelopment, as do other RBPs that have been implicated in neurodegenerative disease (Wolozin and Apicco 2015). One model for how aggregation of RBPs may cause neurotoxicity is by sequestering these proteins out of the nucleus or other subcellular locations to prevent their normal function (Hanson et al. 2012; Conlon and Manley 2017). Thus, it will be interesting to investigate whether the effects of RBM45 on mRNA splicing that we observe here are also maintained in the mature brain, as well as whether RBM45 aggregation compromises its ability to modulate splicing and contributes to disease pathogenesis.
Limitations of the study
Our studies used immortalized cell lines to identify RBM45 target RNAs and to examine the effects of RBM45 depletion and rescued expression. Although this has revealed important new insights into RBM45 function, immortalized cell lines may not fully recapitulate what occurs in vivo. Our studies also uncovered a role for RBM45 in the differentiation of SH-SY5Y cells into neuronal-like cells. However, neural stem cells may respond differently to RBM45 depletion. Thus, future studies using mouse models will be important for further understanding how RBM45 controls brain development and for elucidating the RNA targets of RBM45 in living organisms. Additionally, we used CLIP-seq targeting an epitope-tagged version of RBM45, which may not fully represent the targets of endogenous RBM45. This can be further investigated with transcriptome-wide identification of native RBM45 target transcripts. Finally, our studies have discovered a role for RBM45 in mediating pre-mRNA splicing, and we have found that the effects of RBM45 on gene expression are m6A dependent for some mRNAs. However, the mechanism through which RBM45 controls pre-mRNA splicing and the factors that contribute to m6A-dependent versus -independent effects remain unknown. Thus, future work exploring how RBM45 interacts with the splicing machinery and how this may be impacted by m6A will be informative.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kate Meyer (kate.meyer@duke.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact without restriction.
Data and code availability
Proteomics data generated in this study have been deposited to the ProteomeXchange consortium via the Proteomics Identifications Database (PRIDE) partner repository. Sequencing data generated in this study have been deposited to the National Center for Biotechnology Information Gene Expression Omnibus (GEO) database repository. All data generated in this study are publicly available as of the date of publication. This paper also reanalyzes existing, publicly available sequencing data deposited to the Encyclopedia of DNA Elements (ENCODE) project repository (Luo et al., 2020). Accession numbers for all deposited datasets are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-FLAG® M2 mouse monoclonal antibody (WB 1:2000) | Millipore Sigma | Cat# F1804; RRID: AB_262044 |
| Anti-HA rabbit monoclonal antibody (WB 1:2000) | Cell Signaling | Cat# 3724; RRID: AB_1549585 |
| Anti-HA mouse monoclonal antibody (WB 1:2000, IP 1:100) | Millipore Sigma | Cat# H3663; RRID: AB_262051 |
| Anti-YTHDF2 rabbit polyclonal antibody (WB 1:2000) | Aviva | Cat# ARP67917; RRID: AB_2861185 |
| Anti-GAPDH rabbit polyclonal antibody (WB 1:5000) | ProteinTech | Cat# 10494-1-AP; RRID: AB_2263076 |
| Anti-IDE rabbit polyclonal antibody (WB 1:1000) | Abcam | Cat# ab32216; RRID: AB_775686 |
| Anti-KI67 rabbit polyclonal antibody (WB 1:1000, IF 1:1000) | Abcam | Cat# ab16667; RRID: AB_302459 |
| Anti-Phospho-Histone H3 (Ser10) rabbit polyclonal antibody (WB 1:1000) | Cell Signaling | Cat# 9701; RRID: AB_331535 |
| Anti-TrkB (NTRK2) rabbit monoclonal antibody (WB 1:1000) | Abcam | Cat# ab134155; RRID: AB_2857962 |
| Anti-SFRP1 rabbit monoclonal antibody (WB 1:500) | Abcam | Cat# ab126613; RRID: AB_11128257 |
| Anti-TLE3/ESG rabbit polyclonal antibody (WB 1:1000) | Abcam | Cat# ab94972; RRID: AB_10860535 |
| Anti-RBM45 rabbit polyclonal antibody (discontinued) (WB 1:1000) | Abcam | Cat# ab123912; RRID: AB_10973537 |
| Anti-Histone H3 rabbit polyclonal antibody (WB 1:5000) | Cell Signaling | Cat# 9715; RRID: AB_331563 |
| Anti-m6A rabbit polyclonal antibody (WB 1:1000) | Abcam | Cat# ab151230; RRID: AB_2753144 |
| Anti-WNT3/WNT3A rabbit monoclonal antibody (WB 1:1000) | Abcam | Cat# ab172612; RRID: AB_2725748 |
| Anti-Rabbit HRP goat polyclonal antibody (WB 1:2500) | Abcam | Cat# ab6721; RRID: AB_955447 |
| Anti-Mouse HRP goat polyclonal antibody (WB 1:5000) | Millipore Sigma | Cat# GENXA-931 |
| Anti-Mouse Alexa 647 goat polyclonal antibody (IF 1:3000) | ThermoFisher | Cat# A-21235; RRID: AB_2535804 |
| Anti-Mouse Alexa 488 goat polyclonal antibody (IF 1:3000) | ThermoFIsher | Cat# A-11029; RRID: AB_138404 |
| Anti-Rabbit Alexa 568 goal polyclonal antibody (IF 1:3000) | ThermoFisher | Cat# A-11036; RRID: AB_10563566 |
| Anti-Rabbit Alexa 488 goal polyclonal antibody (IF 1:3000) | ThermoFisher | Cat# A-11008; RRID: AB_143165 |
| Bacterial and virus strains | ||
| MAX Efficiency™ DH5α Competent Cells | ThermoFisher | Cat# 18258012 |
| Rosetta™ 2(DE3) Competent Cells | Millipore Sigma | Cat# 71397-3 |
| Chemicals, peptides, and recombinant proteins | ||
| Glutathione Sepharose™ 4B Affinity Chromatography Media (GE Healthcare) | GE Healthcare | Cat# 17-0756-01 |
| L(−)-Glutathione (reduced form), ≥98% | VWR | Cat# 97061-574 |
| DAPI (4′,6-Diamidine-2′-phenylindole dihydrochloride) | Roche | Cat# 10236276001 |
| 6xHis-GST-RBM45 | This paper | See STAR Methods |
| Critical commercial assays | ||
| NEBNext® rRNA Depletion Kit (Human/Mouse/Rat) with Sample Purification Beads | NEB | E6350 |
| High Sensitivity DNA Kit, for Bioanalyzer 2100 platform | Agilent | 5067-4626 |
| RNA 6000 Pico Kit, for Bioanalyzer 2100 platform | Agilent | 5067-1513 |
| Invitrogen™ Dynabeads™ Oligo(dT)25 mRNA isolation beads | ThermoFisher | 61005 |
| Invitrogen™ RiboMinus™ Transcriptome Isolation Kit, human/mouse | ThermoFisher | K155002 |
| NEBNext® Ultra™ II RNA Library Prep Kit for Illumina® | NEB | E7770 |
| SMARTer® Stranded Total RNA-Seq Kit v3 | Takara Bio | 634485 |
| LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit | ThermoFisher | L34975 |
| RNeasy Micro Kit | QIAGEN | 74004 |
| Qproteome Bacterial Protein Prep Kit | QIAGEN | 37900 |
| QIAEX II Gel Extraction Kit | QIAGEN | 20021 |
| QIAGEN Plasmid Plus Midiprep Kit | QIAGEN | 12943 |
| QIAprep Spin Miniprep Kit | QIAGEN | 27106 |
| QIAquick Gel Extraction Kit | QIAGEN | 28704 |
| QuickLyse Miniprep Kit | QIAGEN | 27406 |
| Deposited data | ||
| RNA-pulldown LC-MS/MS in mHippoE-2 cell lysate | This paper, PRIDE | PXD034161 |
| RNA-pulldown LC-MS/MS in adult mouse (C57BLK6/J) brain lysate | This paper, PRIDE | PXD034159 |
| HA-tagged RBM45 CLIP-seq in mHippoE-2 cells | This paper, GEO | GSE190292 |
| HA-tagged RBM45 CLIP-seq in HEK293T cells | This paper, GEO | GSE190287 |
| HA-tagged RBM45 CLIP-seq in DMSO-/RA-treated SH-SY5Y cells | This paper, GEO | GSE205625 |
| in vitro subcellular fractionation DART-seq in mHippoE-2 cells | This paper, GEO | GSE190311 |
| in vitro subcellular fractionation DART-seq in HEK293T cells | This paper, GEO | GSE190303 |
| Ribo-depleted RNA-seq data in mHippoE-2 cells | This paper, GEO | GSE190322 |
| Ribo-depleted RNA-seq data in DMSO-/RA-treated SH-SY5Y cells | This paper, GEO | GSE190316 |
| SRSF7 eCLIP in HepG2 cells | Gene Yeo, UCSD ENCODE | ENCSR513NDD |
| SRSF7 eCLIP in K562 cells | Gene Yeo, UCSD ENCODE | ENCSR468FSW |
| poly(A) RNA-seq, embryonic mouse (B6NCrl) forebrain timecourse | Barbara Wold, CalTech ENCODE | ENCSR443OEA |
| poly(A) RNA-seq, embryonic mouse (B6NCrl) midbrain timecourse | Barbara Wold, CalTech ENCODE | ENCSR505AHT |
| poly(A) RNA-seq, embryonic mouse (B6NCrl) hindbrain timecourse | Barbara Wold, CalTech ENCODE | ENCSR328UYN |
| Experimental models: Cell lines | ||
| Immortalized mouse hippocampal cell line (mHippoE-2) | Cedarlane Labs | CVCL_D377 |
| Human embryonic kidney (HEK) cell line (HEK293T) | ATCC | CVCL_0063 |
| Human neuroblastoma cell line (SH-SY5Y) | ATCC | CVCL_0019 |
| Experimental models: Organisms/strains | ||
| C57BL/6J, mus musculus | Jackson Labs | IMSR_JAX:000664 |
| Oligonucleotides | ||
| Biotin-labeled non-m6A Dlg4 RNA oligo: 5′-Biotin-GGGCCCUUGGUCUGGACU GAAUAGCCCAAGCCC-3′ | This paper | N/A |
| Biotin-labeled m6A Dlg4 RNA oligo: 5′-Biotin-GGGCCCUUGGUCUGGm6A CUGAAUAGCCCAAGCCC-3′ | This paper | N/A |
| Biotin-labeled non-m6A ssRNA oligo: 5′-Biotin-UUCUUCUGUGGACUGUG-3′ | This paper | N/A |
| Biotin-labeled m6A ssRNA oligo: 5′-Biotin-UUCUUCUGUGGm6ACUGUG-3′ | This paper | N/A |
| Biotin-labeled non-m6A randomer RNA oligo: 5′-Biotin-GGGGAGUUCUACAGUCCGA CNNNNNNNNNNNRACNNNNNNNNN NNGGAAUUCUCGGGUGUCAAGG-3′ | This paper | N/A |
| Biotin-labeled m6A randomer RNA oligo: 5′-Biotin-GGGGAGUUCUACAGUCCG ACNNNNNNNNNNNRm6ACNNNNNN NNNNNGGAAUUCUCGGGUGUCAAGG-3′ | This paper | N/A |
| Fluorescein-labeled (FAM) non-m6A ssRNA oligo: 5′-FAM-UUCUUCUGUGGACUGUG-3′ | This paper | N/A |
| Fluorescein-labeled (FAM) m6A ssRNA oligo: 5′-FAM-UUCUUCUGUGGm6ACUGUG-3′ | This paper | N/A |
| For DNA oligonucleotides used in this study, see Table S1 | This paper | Table S1 |
| Recombinant DNA | ||
| pcDNA™3.1 (+) Mammalian Expression Vector | Invitrogen | Cat# V79020 |
| pcDNA™4/TO Mammalian Expression Vector | Invitrogen | Cat# V102020 |
| pET-GST | Addgene | Plasmid #42049 |
| pCMV-VSV-G | Addgene | Plasmid #8454 |
| psPAX2 | Addgene | Plasmid #12260 |
| lentiCRISPR v2 | Addgene | Plasmid #52961 |
| pLenti-puro | Addgene | Plasmid #39481 |
| Software and algorithms | ||
| Prism 8 (v8.2.1) | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ (v1.53a) | NIH | https://imagej.nih.gov/ij/ |
| NeuronJ (v1.4.3) | E. Meijering et al., 2004 | https://imagescience.org/meijering/software/neuronj/ |
| IGV (v2.8.9, v2.9.4) | J.T. Robinson et al., 2011 | https://igv.org |
| SeqAn (v2.3.2), FLEXBAR (v3.0.3) | M. Dodt et al., 2012 | https://github.com/seqan/flexbar |
| Novoalign (v3.09.00) | Novocraft | http://www.novocraft.com/products/novoalign/ |
| Python (v2.7.11) | Python | https://www.python.org |
| homer (v4.11.1) | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| MEME (v5.0.5) | Bailey and Elkan, 1994 | https://meme-suite.org/meme/index.html |
| R (v3.6.0) | R | https://www.r-project.org |
| DESeq2 (v1.32.0) | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Picard (v2.18.2) | “Picard Toolkit.” 2019. Broad Institute | https://broadinstitute.github.io/picard/ |
| rMATS (v4.0.2) | Shen et al., 2014 | http://rnaseq-mats.sourceforge.net |
| DEXSeq (v1.38.0) | Anders et al., 2012 | https://bioconductor.org/packages/release/bioc/html/DEXSeq.html |
| Subread (v1.6.3) | Liao et al., 2014 | http://subread.sourceforge.net |
| deeptools (v3.2.1) | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| HiSAT2 (v2.1.0) | Kim et al., 2019 | http://daehwankimlab.github.io/hisat2/ |
| Bedtools (v2.25.0) | Quinlan Lab, Univ. of Utah | https://bedtools.readthedocs.io/en/latest/ |
| Trimmomatic (v0.38) | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| MACS2 (v2.1.1.20160309) | Y. Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| Bullseye (unreleased) | Tegowski et al., 2022 | https://github.com/mflamand/Bullseye |
This paper does not report original code. This paper used existing, publicly available code to analyze sequencing data. Relevant identifying information for software and algorithms used in this study listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL METHOD AND SUBJECT DETAILS
Cell culture
The female human embryonic kidney cell line HEK293T (Cat#CRL-3216; RRID: CVCL_0063) was obtained from the American Type Culture Collection (ATCC) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 4.5 g/L glucose, L-glutamine, and sodium pyruvate (Corning) supplemented with 10% (v/v) fetal bovine serum (Avantor Seradigm) and Penicillin-Streptomycin (Gibco). Immortalized mouse hippocampal cell line mHippoE-2 (Cat# CVCL_D377; RRID: CVCL_D377) was obtained from Cedarlane Laboratories and cultured in DMEM with 4.5 g/L glucose, L-glutamine, and sodium pyruvate (Corning) supplemented with 10% (v/v) fetal bovine serum (Avantor Seradigm) and Penicillin-Streptomycin (Gibco). The sex of mHippoE-2 cells was not disclosed by the manufacturer and may be a mixed population of male and female. The female human neuroblastoma cell line SH-SY5Y (Cat#CRL-2266; RRID: CVCL_0019) was obtained from the ATCC and cultured in equal parts Minimum Essential Medium Eagle (EMEM) with Earle′s salts, L-glutamine and sodium bicarbonate (Sigma-Aldrich) and Ham’s F12 Nutrient Mix (ThermoFisher), supplemented with 10% (v/v) gamma-irradiated and heat-treated fetal bovine serum (Avantor Seradigm) and Penicillin-Streptomycin (Gibco). Cell lines used in this study have not been authenticated. Cell lines used in this study were cultured in a humidified 37°C incubator with 5% CO2.
Mouse primary tissue
Mice used in these studies were housed in the Duke University mouse facility overseen by the Duke Division of Laboratory Animal Resources (DLAR). Mice were group housed after weaning and fed ad libitum. RNA pulldown-MS was performed using primary mouse brain tissue extracts acquired from two adult female C57BL/6J mice (B6; The Jackson Laboratory, Cat#000664; RRID: IM-SR_JAX:000664). Mice were euthanized with CO2 asphyxiation and decapitated as a secondary method. Mouse brains were dissected and placed in Lysis Buffer (10 mM NaCl, 2 mM EDTA, 0.5% Triton X-100, 0.5 mM DTT, 10 mM Tris, pH 7.4) for tissue lysis by dounce homogenization. Lysates were cleared of tissue debris by centrifugation (10,000 x g, 15 min, 4°C) and used for RNA pulldown. For RT-qPCR experiments, total RNA was extracted from mouse brain tissues acquired from prenatal, post-natal, and adult C57BL/6J mice. Pregnant dams were euthanized with CO2 asphyxiation and decapitated. Embryonic mice of both sexes aged E13.5, E15.5, and E18.5 were isolated, and primary brain tissue was collected and placed in TRIzol reagent for RNA extraction, isolation, and purification (Invitrogen). Post-natal and adult (≥6 weeks) primary brain tissue was collected from euthanized mice of both sexes in similar fashion.
METHOD DETAILS
RNA oligonucleotides sample preparation
RNA oligonucleotides were synthesized according to previously established methods (B. Liu et al., 2018). In brief, modified (N6-methyladenosine (m6A), 5′-biotin, and 5′-fluorescein (5′-FAM)-labeled) and unmodified RNA oligonucleotides were synthesized with the MerMade 6 Oligo Synthesizer system, using 2′-tBDSilyl protected phosphoramidites (ChemGenes Corporation) on 1 μmol standard synthesis columns (BioAutomation). m6A phosphoramidites were obtained from ChemGenes. 5′-biotin phosphoramidites, and 5′-fluorescein (5′-FAM) phosphoramidites were obtained from Glen Research. Post-synthesis, oligonucleotides were cleaved and subjected to base deprotection, then quenched with Glen-Pak RNA quenching buffer and loaded onto Glen-Pak RNA cartridges (Glen Research) for purification according to the manufacturer’s instructions. Samples were then ethanol precipitated and buffer exchanged into nuclease-free water, then stored at −80°C until usage. DMT-off 2′O deprotected samples were directly ethanol precipitated and purified using large-scale 20% (w/v) denaturing TBE-PAGE, followed by electroelution into 20 mM Tris buffer, pH 8.0 and buffer exchange into nuclease-free water, then stored at −80°C until usage.
Western blotting
Protein concentrations for all samples were quantified by Bradford assay using a Nanodrop 1000 UV-visible spectrophotometry system and BSA standards within an order of magnitude of the desired concentration (1-2 mg/mL). Samples were prepared to the same volume and concentration (typically 0.5–1 μg/μL protein) using 4x NuPAGE™ LDS Loading Dye and separated using gel electrophoresis on NuPAGE™ 4-12% Bis-Tris SDS-PAGE gels (Invitrogen), then transferred onto PVDF membranes (Amersham) using a wet electrophoretic transfer system (BioRad). Membranes were trimmed and washed three times in 0.1% PBST (0.1% Tween-20 in PBS, pH 7.4). Membranes were initially blocked with 5% non-fat dry milk or 1% bovine serum albumin (BSA) in 0.1% PBST for 1 hr at room temperature (RT) under constant agitation on a rotator, or overnight at 4°C on a rotator if necessary. Membranes were then incubated in primary antibody solution (1:500-1:5,000 primary antibody [See key resources table, Primary Antibodies], 1% BSA in 0.1% PBST) for 1 hr at RT or overnight at 4°C on a rotator. Membranes were then washed three times with 0.1% PBST on a rotator (5 min, RT for each wash), and incubated with secondary antibody solution (1:2,500-1:5,000 secondary antibody [See key resources table, Secondary Antibodies] in 0.1% PBST) for 1 hr at RT on a rotator. After three more washes with 0.1% PBST, membranes were incubated with 1:1 chemiluminescent reagent solution (Amersham) for 2-3 min at RT before imaging with the ChemiDoc MP imager system (BioRad).
Dot blot assays
RNA oligonucleotide concentrations were quantified using a Nanodrop 1000 UV-visible spectrophotometry system. Methylated and unmodified RNA samples of equal volume (3 μL) and concentration (50 ng/μL RNA oligonucleotide) were spotted onto a Hybond-N+ membrane (Amersham) and then fixed using direct UV-irradiation (150-300 mJ/cm2 UV254 nm). Post-crosslinking, membranes were washed twice in 0.1% PBST and subject to immunodetection for 5′-biotinylation and/or m6A, as described above for Western blotting.
In cellulo RNA Pulldown-WB
Cultured cells were mechanically or enzymatically (ThermoFisher Gibco™ TrypLE™) dissociated at ~75 to 85% confluency, washed and concentrated with cold phosphate buffered saline (1x PBS, 2,000 xg for 1 min at 4°C), then resuspended in an appropriate amount of Lysis Buffer (10 mM NaCl, 2 mM EDTA, 0.5% Triton X-100, 0.5 mM DTT, 10 mM Tris, pH 7.4) containing freshly added mammalian protease/phosphatase inhibitor, typically 1 mL per 2 × 106 cells. Cells were transferred to a dounce homogenizer and lysed using 20 to 30 strokes on ice. Whole cell extracts were then brought to final concentrations of 150 mM KCl and 5% Glycerol (v/v), and centrifuged to pellet insoluble cellular debris (10,000 x g for 15 min at 4°C). Whole cell lysate supernatant concentrations were measured using the Bradford assay (ThermoFisher) against known BSA standards. An appropriate volume of Streptavidin-Agarose binding matrix slurry, 30-45 μL per pulldown sample and 100 μL/1 mg lysate for pre-clearing, was aliquoted and batch-washed (4,000 xg for 1 min at 4°C) three times in 900 μL of Wash Buffer (10 mM Tris, pH 7.5, 1.5 mM MgCl2, 150 mM KCl, 0.5 mM DTT, 0.05% (v/v) NP-40 substitute). Pre-washed slurry was then further divided based on sample or usage (Lysate pre-clearing: 100 μL/1 mg of lysate, Control: no RNA oligo, A: 5′-biotin unmodified RNA oligo, m6A: 5′-biotin m6A-modified RNA oligo, scr: 5′-biotin m6A-modified RNA oligo with a scrambled m6A consensus sequence). Pulldown samples were batch-incubated with 1–2 μg/sample of RNA oligo in Binding Buffer +100 U/mL RNase inhibitor for 1 hr at 4°C on a rotator. Lysates were concurrently pre-cleared for 1 hr at 4°C on a rotator. After incubation, pulldown samples were washed twice (4,000 xg for 1 min at 4°C) with Wash Buffer to remove any unbound RNA oligo from solution (control/mock samples were treated and washed identically). Pre-cleared lysate slurry mixtures were centrifuged gently (2,000 xg for 1 min at 4°C), with pre-cleared lysate supernatant being carefully collected for use in RNA pulldown reactions. RNA pulldown reactions (1 mL total volume, 2 μg RNA oligo-bound volume of slurry, 1-2 mg of pre-cleared whole cell lysate, and Lysis/Wash Buffer) were prepared and incubated on a rotator, either for 2 hr at room temperature (RT) or overnight at 4°C. RNA pulldown reactions were then washed five times with Wash Buffer (4,000 xg for 1 min at 4°C), and resuspended in 30–50 μL of mild elution buffer (50 mM Tris, pH 7.5, 200mM NaCl, 2% SDS (w/v), and 1 mM biotin) for 30 min at 60°C, 1100 rpm on a thermal shaker.
RNA pulldown eluents were diluted 1:10 using NuPAGE™ LDS Loading Dye (ThermoFisher, 4x) and denatured at 95°C for 5 min. 10 μL of eluent and 10 μg of whole cell lysate were loaded on NuPAGE™ 4–12% Bis-Tris PAGE gels (ThermoFisher) and then transferred onto PVDF membranes (GE Healthcare, Amersham) using a wet electrophoretic transfer system (BioRad). The membrane was then blocked with 5% nonfat dry milk or 1% purified bovine serum albumin (BSA) in 0.1% PBST for 1 hr at RT on a rotator. Membranes were incubated in solution containing primary antibody (1:1,000-1:3,000 antibody, 1% BSA in 0.1% PBST) for 1 hr at RT on a rotator or overnight at 4°C on a rotator. Membranes were then washed three times with 0.1% PBST and incubated with secondary antibody solution (1:2,500-1:5,000 appropriate secondary antibody, 0.1% PBST) for 1 hr at RT on a rotator. After three more washes with 0.1% PBST, membranes were incubated with 1:1 chemiluminescent reagent solution (Amersham) for 2-3 min at RT before imaging with the ChemiDoc MP imager system (BioRad).
In cellulo UV-crosslinked RNA pulldown-WB
For ultraviolet radiation-induced RNA:protein crosslinking pulldowns, RNA pulldown reactions were subjected to 300 mJ/cm2 UV254nm irradiation post-incubation of lysates and RNA oligonucleotides, immediately prior to washing. The RNA pulldown Wash Buffer (10 mM Tris, pH 7.5, 1.5 mM MgCl2, 150 mM KCl, 0.5 mM DTT, 0.05% (v/v) NP-40 substitute) was supplemented with 1 M NaCl. After washes, the RNA pulldown samples were boiled at 95°C with 1x NuPAGE™ LDS Loading Dye for 5 min and used for western blotting.
Densitometry analysis
To quantify expression levels, WB lanes intensities were isolated and specific bands quantified by ImageJ (NIH) (Schneider et al. 2012). Band intensities were then normalized to those of housekeeping or loading control proteins, and then plotted as a mean expression value ± standard error measurement (SEM) relative to a designated control sample or condition.
In cellulo RNA pulldown-LS-MS/MS and data analysis
RNA pulldown samples were eluted into 60 μL of mild elution buffer (30 min at 60°C, 1100 rpm). 10 μL of each RNA pulldown sample and 10 μg of input lysate were then loaded onto a NuPAGE™ 4–12% Bis-Tris PAGE gel (Invitrogen) and transferred using electrophoresis to a PVDF membrane (GE Healthcare, Amersham) for Western blot. After validation, the remaining volume of eluent was stored at −80°C. LC-MS/MS was performed by the Duke Proteomics and Metabolomics Shared Resource. Samples were thawed and subject to acute PAGE separation for in-gel tryptic digestion. After in-gel digestion and lyophilization, peptides were reconstituted in 12 μL of 1% (v/v) trifluoroaceteic acid (TFA)/2% (v/v) acetonitrile (MeCN) containing 12.5 fmol/μL yeast alcohol dehydrogenase surrogate standard. A QC pool was prepared by mixing 2 μL of each samples, ~40% of each sample was analyzed by qualitative LC-MS/MS. Briefly, the sample was first trapped on a Symmetry C18 180 μm × 20 mm trapping column (5 μL/min at 99.9/0.1 v/v H2O/MeCN) followed by an analytical separation using a 1.7 μm Acquity HSS T3 C18 75 μm × 250 mm column (Waters) with a 90 min gradient of 5 to 30% MeCN with 0.1% formic acid at a flow rate of 400 nL/min and column temperature of 55°C. Data collection on the QExactive HF MS was performed in data-dependent acquisition (DDA) mode with a 120,000 resolution (at m/z 200) full MS scan from m/z 375 to 1600 with a target AGC value of 3e6 ions and 50 ms IT, followed by 12 MS/MS scans at 30,000 resolution (@ m/z 200) at a target AGC value of 5e4 ions, 45 ms IT and minimum AGC threshold of 5e3. A 20 s dynamic exclusion was employed, and an exclusion list for high abundance streptavidin peptides was employed. Unique peptides, identified at 1% peptide false discovery rate, were then indexed against the mouse Uniprot database and loaded into the Scaffold software suite. Endogenously-biotinylated carboxylases were identified in all of the samples, which suggested that the elution conditions were sufficient to disrupt the streptavidin-biotin interaction.
Data from two biological replicates were analyzed, with unique spectral counts of peptides reported for each experimental replicate in the MS data. We adjusted the A RNA and m6A RNA spectral counts through a pseudocount (+1) to avoid mathematical constraints associated with logarithmic compression of 0/zero (ie., log2 = undefined, log2 = 0) and reported the m6A-dependent enrichment of RBPs bound as function of the pseudocount: log2 . Statistical significance for RBP enrichment via unique spectral peptide counts between A and m6A pulldown samples for both replicates were calculated using a conservative variant of the student’s t-test (two-tail distribution, homoscedastic variance) between A-RNA and m6A-RNA spectral counts.
In vitro purification of GST-tagged human RBM45
The full-length human RBM45 protein coding sequence was cloned into the pET-GST plasmid (Addgene, plasmid #42049), which encodes for an N-terminal GST-tagged recombinant protein. The plasmid was then transformed into Rosetta™ 2(DE3) Competent Cells (Sigma). Clonal isolates bearing the correct recombinant protein sequence were then tested for 1 mM IPTG-induced GST-hRBM45 expression, and chosen based on the amount of recombinant GST-hRBM45 expression observed by Coomassie stain post-induction at 4 and 24 hr in 37°C.
To obtain bacterial cultures expressing GST-hRBM45, 250 mL small-scale cultures of SuperBroth (3.5% tryptone, 2.0% yeast extract, 0.5% NaCl, 1 N NaOH) were first innoculated with transformed Rosetta™ 2(DE3) cells and then allowed to expand for distribution into large-scale bacterial cultures (1.5–2 L SuperBroth). Large scale cultures were then allowed to grow to a pre-determined optical density (O.D.) of 0.600. At O.D. 0.600, large-scale protein expression was induced with the addition of 1 mM IPTG (v/v), followed by incubation at 25°C, 225 rpm overnight (16-24 hr). After induction, bacterial cultures were centrifuged (6,000 xg, 15 min at 4°C), isolated, and used immediately for purification of GST-hRBM45 or stored at −80°C as bacterial pellets until purification.
Bacterial pellets were lysed using lysozyme-mediated lysis for 30 min at 4°C or on ice with the Qproteome Lysis Kit (QIAGEN), according to the manufacturer’s instructions. Lysates were centrifuged (15,000 xg, 30 min at 4°C) to isolate the lysate supernatant fraction and pellet insoluble debris. Supernatants were allowed to bind to 0.5 mL pre-washed GST-Sepharose matrix (GE Healthcare) per 50 mL lysate supernatant for 1 hr or overnight at 4°C with constant, but gentle rotation. Post-binding, GST-Sepharose matrix was collected in PolyPrep® filtration columns (BioRad) and allowed to flow by gravity filtration. GST beads were then washed twice using 10 mL cold 1x PBS through gravity filtration. Washed beads were then eluted in small volumes using five volumes of 5 mL elution buffer (50 mM Tris, 10 mM reduced glutathione, pH 8.0). Small volumes of sample collected at each step were analyzed by gel electrophoresis and Coomassie staining to verify successful purification of GST-RBM45, defined as >80% by gel imaging analysis (ImageJ). Elution volumes were then spin-concentrated and buffer exchanged using three washes of Final Buffer (20 mM Tris, 300 mM KCl, 1 mM DTT, 5 mM EDTA, 10% glycerol) to a final volume of 500 to 1000 μL. Protein concentration was determined using the Bradford assay on the Nanodrop 1000 system and confirmed using appropriately concentrated BSA standards on a Coomassie-stained SDS-PAGE gel.
In vitro RNA pulldown-SDS-PAGE-coomassie
For in vitro RNA pulldowns utilizing purified recombinant GST-RBM45, the RNA pulldown protocol was followed as described above, but native Final Buffer solution (20 mM Tris, 300 mM KCl, 1 mM DTT, 5 mM EDTA, 10% glycerol) was substituted for both reaction/ lysis and wash buffers. RNA pulldown samples were loaded on NuPAGE™ 4–12% Bis-Tris PAGE gels (ThermoFisher), subjected to gel electrophoresis (180 V, 50-60 min at RT) and then stained with 0.1% (w/v) Brilliant Blue G (Coomassie G-250) dye in 40% (v/v) methanol, 10% (v/v) acetic acid for 1 hr or overnight at RT on a rotator. Stained gels were then destained using mild and harsh de-staining buffers (Destain I (harsh): 40% methanol, 10% acetic acid (v/v); Destain II (mild): 10% methanol, 5% acetic acid (v/v)) over 16-48 hours before imaging and quantification using the ChemiDoc MP imager system (BioRad).
Fluorescence polarization
5′-FAM labeled RNA oligonucleotides were diluted to a 1 nM concentration in a final volume of 190 μL Final Buffer in Fisherbrand™ round bottom disposable borosilicate glass tubes (ThermoFisher). A Lambda 25 UV/vis spectrometer (PerkinElmer) was blanked against Final Buffer, and initial polarization values were measured at room temperature (25°C). Concentrated GST-RBM45 was titrated into sample tubes at pre-determined concentrations ranging from 100 nM to 6 μM to obtain fluorescence polarization values for reactions containing no RNA oligonucleotide, 5′-FAM unmodified RNA, or 5′-FAM m6A-modified RNA. The range of collected polarization values (mP) was then normalized to the range of experimentally determined values for all replicates across each sample and fit to a one-site total binding model to determine binding affinity (dissociation constant, Kd) and other binding parameters.
DNA plasmid transfection
Transformed DH5α cells were grown in 25 to 35 mL Lysogeny Broth (LB) containing ampicillin at 37°C, 225 rpm in a thermal shaker overnight. Cultures were centrifuged (6,000 xg, 15 min at 4°C) and decanted, then subject to DNA plasmid isolation using the Plasmid Plus midiprep kit (QIAGEN), according to the manufacturer’s instructions. For mammalian cell culture transfection, the appropriate amount of DNA plasmid was prepared in a volume of pre-warmed OptiMEM™ reduced-serum medium (ThermoFisher). An equal volume of pre-warmed reduced-serum medium containing PEI MAX (PolySciences) at a 2:1 PEI:DNA ratio was identically prepared. The two solutions were mixed and allowed to incubate at RT for 20 min, after which time the transfection reaction mixture was pipetted drop-wise onto the culture. Culture media was replaced ~4 hr post-transfection with fresh pre-warmed media.
Lentiviral generation and purification
Low-passage HEK293T cells (<P15) were plated onto 15 cm tissue culture plates and allowed to grow to ~60% confluence before being transfected with lentiviral packaging (Δ8.9+VSVg, PMD2.g+PsPax2) and transfer vectors (lentiCRISPR_v2 or pLenti) using PEI (4.28:3:1 transfer:packaging:envelope plasmid ratio, 50 μg total DNA plasmid per 15 cm culture). After 72 h, virus-containing media from transfected cultures were collected, centrifuged to pellet cellular debris (3,000 xg, 5 min at 4°C), and layered on top of a 3 mL 20% sucrose/PBS cushion in an ultracentrifuge tube (Beckman Coulter®) and centrifuged in an L8-60M ultracentrifuge (Beckman Coulter®) using the SW-28 rotor (Beckman Coulter®) for 2 hr at 19,700 rpm, 17°C under vacuum. After centrifugation, tubes were decanted and allowed to dry at RT for 5-10 min. Lentivirus pellets were resuspended in 100 μL of cold PBS and allowed to resuspend in solution overnight at 4°C on a rotator. Purified lentivirus was aliquoted separately into small volumes (5-15 μL) and stored at −80°C until used for lentiviral transduction.
Stable cell line generation
Cultured cells were infected with lentivirus expressing the desired transgene and a cistronic selection marker. 48 hr post-transduction, cultures were passaged into fresh media containing an appropriate concentration of selection antibiotic, based on previously conducted antibiotic kill curves, and allowed to incubate to select for infected cells. The selected infected cellular pools were then plated using the limiting dilution assay (LDA) on a 96-well plate. After two weeks in culture, plates were inspected for single-cell clonal isolates, which were expanded into larger culture vessels. After clonal isolate expansion, cells were tested to ensure stable expression of the transgene using WB or, as necessary, targeting of a particular genetic locus (performed using Sanger sequencing, RT-qPCR, and RNA-seq).
m6A quantification by UPLC-MS/MS
Total RNA was first isolated and extracted from wild-type (WT), Cas9 Control, or METTL3-depleted (METTL3 KO) HEK293T, mHippoE-2, or SH-SY5Y stable cells using Trizol (Invitrogen) according to the manufacturer’s instructions. Total RNA was then treated with DNase I (NEB) to eliminate DNA contamination, followed by two rounds of poly-A+ mRNA isolation using the Dynabeads mRNA Purification Kit (ThermoFisher) and 1-2 rounds of RiboMinus treatment to remove rRNA (ThermoFisher). mRNA purification was then confirmed by Bioanalyzer analysis (Agilent). For each sample replicate, 200 ng of mRNA was first digested by 2 U of Nuclease P1 (Sigma) in 50 μL nuclease free water with 2.5 mM ZnCl and 25 mM NaCl for 2 h at 37°C, followed by treatment with 5 U of Antarctic phosphatase (NEB) for 2 h at 37°C. Samples were analyzed using the Xevo TQ-S mass spectrometry system.
Protein expression quantification by nanoLC-MS/MS
~40 μg of protein lysate from each clonal stable SH-SY5Y cell line was reduced, alkylated, and then digested using an S-trap micro device (Protifi). A proteomic strategy to measure RBM45 and GAPDH protein expression was developed using frequently observed peptides from ProteomicsDB and MS/MS spectra prediction with Prosit in Skyline. 1 ug of each sample was analyzed by nanoLC-MS/MS using scheduled parallel reaction monitoring on Exploris480 (Thermo). Using a dotp cutoff of 0.9, 3 peptides from RBM45 and 4 peptides from GAPDH were used to measure protein expression levels in Cas9 Control, RBM45 KO, and RBM45 KO-Rescue SH-SY5Y stable cells.
CLIP-seq
We performed a variation of the iCLIP protocol (I. Huppertz et al., Methods, 2014), with the addition of a paired size-matched (SM) input RNA control library for each CLIP sample, as detailed in the eCLIP protocol (E.L. Van Nostrand et al., Nature Methods, 2016). Briefly, stable cell lines expressing HA-tagged RBM45 were crosslinked with 300 mJ/cm2 UV254 nm light. Cultures were then washed, lysed, and partially digested (RNase I, Ambion) to create protein-RNA fragments of suitable size (50-75 bp) for sequencing. Lysates were used for immunoprecipitation of HA-RBM45 using a monoclonal anti-HA tag antibody (Cell Signaling, #3724) and magnetic Protein A/G beads (ThermoFisher). 10 μg of antibody was used in 100 μL bead slurry per 1 mg of input lysate. Input (1%) and HA-RBM45 CLIP fractions were then separated by SDS-PAGE and transferred to a nitrocellulose membrane. Regions of the membrane containing the crosslinked protein of interest (MW + ~75 kDa) were then excised from both input and CLIP lanes using a sterile razor blade and diced into fine squares. Protein-RNA complexes were dissociated from the membrane through Proteinase K (Invitrogen) treatment, leaving purified input RNA and CLIP RNA fractions that were subsequently used to generate sequencing libraries using ligation of RNA adapters bearing unique molecular identifiers (UMIs) to assist in identification of duplicated reads, followed by circularization, enzymatic digestion, and cDNA library amplification, as referenced in the iCLIP protocol (I. Huppertz et al., Methods 2014). Purified cDNA libraries diluted to 20 nM were submitted to the Duke Center for Genomic and Computational Biology (GCB) Sequencing shared resource facility. mHiPPoE-2 CLIP libraries were processed on a HiSeq 4000 instrument (Illumina) using 50 bp single-end sequencing and HEK293T CLIP libraries were processed on a NovaSeq 6000 platform (Illumina) using 50 bp paired-end sequencing.
CLIP-seq data analysis
Reads were trimmed (FLEXBAR 3.0.3), de-multiplexed (Python 2.7.11), and aligned (Novoalign 3.09.000) to the desired genome (mouse, mm10; human, hg19) (Dodt et al., 2012). Alignments were then subject to de-duplication based on UMI headers (CTK 1.1.3). De-duplicated alignments were then used to call statistically significant peaks (MACS2 2.1.1, p < 5 × 10−3) based on comparative enrichment between paired input and CLIP RNA alignments (Zhang et al., 2008). Replicate peak sets were compared to identify a high-confidence subset of RBM45 CLIP peaks common to both biological replicates and then assigned strand based on local transcript direction. Peaks were then annotated (HOMER 4.11.1) to provide target gene, binding site information and used for de novo motif discovery (HOMER 4.11.1, MEME 5.0.5) to identify the most enriched sequences in RBM45 CLIP peaks (Heinz et al., 2010). Relative and absolute distance plots were generated using custom scripts.
Subcellular fractionated in vitro DART-seq
mHiPPoE-2 and HEK293T Cas9 Control stable cells were grown to confluence, dissociated, washed and collected in cold PBS. Cells were then subject to subcellular fractionation as previously described (Ke, et al., Genes and Dev., 2017). Fractionation efficiency was verified using RT-qPCR on RNA from each subcellular fraction, testing for changes in the relative expression levels of intronic RNA (Ccbe1 intron), a nuclear long ncRNA (Neat1), and the 3′UTR of a housekeeping gene (Actb) between subcellular fractions. 50 ng of total RNA from each fraction were prepared in a 50 μL reaction volume containing 1x DART Buffer (100 mM Tris-HCl, pH 7.4, 500 mM KCl, 1 μM ZnCl2), 1 μL of RNaseOUT (ThermoFisher), and 250ng of purified APOBEC-YTH or APOBEC-YTHmut protein. Samples were incubated for 4 hr at 37°C in a thermal shaker. DART-treated RNA was then purified using the RNeasy Micro Kit (QIAGEN). Half of each sample was used for construction of sequencing libraries with the SMARTer-Seq pico v3 low input RNA library kit (Takara). Purified cDNA libraries diluted to 20 nM were then sequenced on a NovaSeq 6000 instrument (Illumina) using 150 bp paired-end sequencing.
DART-seq data analysis
DART-seq data were analyzed using Bullseye, a custom editing analysis pipeline. First, reads were trimmed (Trimmomatic 0.38) and aligned (HiSAT2 2.1.0) to the appropriate genome (mouse, mm10; human, hg19) (Bolger et al. 2014; Kim et al., 2019). Alignments were then subject to deduplication (Perl 5.30.1) and positional coordinate matrices were generated for each, with a minimum of >10 reads required at each position. To examine subcellular fractionation efficiency, alignments from biological replicates were first subject to analysis using the CollectRnaSeqMetrics function (Picard 2.18.2), in order to verify enrichment of intronic reads in CA-fractions alongside comparative reductions of intronic RNA in nuclear and cytoplasmic fractions. Each APOBEC-YTH replicate matrix was then compared to two biological replicates of the appropriate APOBEC-YTHmut control matrices to identify C2U mutations with mutation rates ranging from 5% to 90%, and 1.25-fold more edited in the APOBEC-YTH condition. C2U mutation sites were then filtered for “RAC” sequences to identify C2U mutations directly adjacent m6A sites. For each set of biological replicates, C2U sites were merged to reveal a “low-stringency” dataset of m6A sites and overlapped to reveal a “high-stringency” dataset of m6A sites. Sites were then annotated (HOMER 4.11.1) to identify which genes were methylated and where, as well as used for de novo motif discovery (HOMER 4.11.1, MEME 5.0.5) to identify the most enriched sequences. Relative and absolute distance plotting functions were provided by M.F. and validated against existing metagene profiling algorithms (HOMER 4.11.1), mRNA metagene profiling algorithms were also taken from publicly available resources (metaPlotR). Downstream analyses were performed in R (R 3.6.0).
Ribo-depleted RNA-seq
mHiPPoE-2 stable cell lines or cultured, undifferentiated, and differentiated SH-SY5Y stable cell lines were grown to confluence, dissociated, washed, and resuspended in 500 μL TRIzol Reagent (ThermoFisher). Total RNA was extracted, isolated, and purified according to the manufacturer’s recommendations, treated with DNase I (NEB), and then purified by phenol:chloroform:isoamyl alcohol (PCI) extraction) and ethanol (EtOH) precipitation. Integrity of total RNA samples was checked using the 2100 Bioanalyzer (Agilent™) with RNA samples of RIN > 8.0 used for subsequent RNA library prep and sequencing. To generate sequencing libraries, 1 μg of total RNA was subject to ribosomal RNA depletion using the NEBNext rRNA depletion kit (NEB) and used for library preparation with the NEBNext Ultra II RNA Library prep kit (NEB), both according to the manufacturer’s instructions. cDNA libraries were amplified, purified, and analyzed by Bioanalyzer (Agilent™) prior to submission. Purified cDNA libraries diluted to 20 nM were then submitted to the Duke Center for Genomic and Computational Biology (GCB) Sequencing shared resource facility and processed on a NovaSeq 6000 instrument (Illumina) using 150 bp paired-end sequencing.
Ribo-depleted RNA-seq data analysis
Reads were trimmed (FLEXBAR 3.0.3) and aligned (HiSAT2 2.1.0) to the appropriate genome (mouse, mm10; human, hg19). Alignments were converted to bigwig coverage tracks (deeptools 3.2.1) for viewing in IGV and used to generate read counts (feature-Counts 1.6.5, SubRead 1.6.3) of exons, introns, and transcripts from a provided reference file (ENSEMBL human GENCODE 38 or mouse GENCODE M27; and NCBI RefSeq human GRCh37/hg19 or GRCm38/mm10) for differential exon usage, intron usage, and gene expression analyses (Liao et al. 2014; Ramírez et al., 2016; Robinson et al., 2011). Differential gene expression (DESeq2) (Love et al. 2014) analyses were performed in R (R 3.6.0). Differential gene expression analyses utilized default parameters and default significance cut-off values (padj ≤ 0.05). Alternative splicing analyses were performed using two platforms, rMATS (Shen et al., 2014) and DEXSeq (Anders et al. 2012; Reyes et al., 2013) to identify transcripts with significant changes in alternative splicing, as well as those containing differential exon usage or intron retention events. Alternative splicing events were identified using default parameters (rMATS) or significance cut-off values of praw ≤ 0.01 (DEXSeq).
Splicing efficiency analysis
To calculate splicing efficiency (SE), bipartite junctional reads between exons for a given RNA were isolated and normalized to read depth of bipartite reads between exons 1 and 2 for each biological sequencing replicate, and then averaged across replicates for each genotype.
SH-SY5Y differentiation
SH-SY5Y stable cell lines were plated in 6-well or 10-cm plates and cultured to ~50% confluence in growth media (1:1 Eagle’s Minimal Essential Medium, Ham’s F12 Nutrient Mix, 10% heat-inactivated FBS, Penicillin/Streptomycin). To initiate the differentiation time courses, control cultures were grown in differentiation media (Neurobasal A Medium, 1x B27 nutrient supplement, 1x GlutaMax, Penicillin/Streptomycin) containing 0.1% (v/v) DMSO and experimental cultures were grown in differentiation media containing 10 μM all-trans retinoic acid (RA, Enzo) in 0.1% (v/v) DMSO. Treated cultures were allowed to incubate (37°C, 5.0% CO2) for seven days, with half of the culture media supplemented with fresh differentiation media containing DMSO only or 10 μM RA at days 3 and 5. On day 7, cultures were used to generate whole cell lysates for Western blot analysis or resuspended in TRIzol Reagent for RNA extraction, isolation, and purification.
Reverse-transcription PCR (RT-PCR)
1 μg of DNA-free total RNA was subject to reverse transcription using iScript RT Supermix (BioRad) to generate cDNA, which was then further diluted (1:20) in nuclease-free water. cDNA synthesis was performed using SuperScript III RT to specifically target and reverse transcribe poly(A)+ RNA with oligo(dT) primers. cDNAs were then used for PCR (CloneAmp HiFi PCR master mix, Takara), using gene-specific primers at terminal ends of the transcript to amplify mature mRNAs, inclusive of any potential isoforms. PCR reactions were then analyzed using agarose gel electrophoresis.
Real-time quantitative PCR (RT-qPCR)
Relative expression levels were measured using RT-qPCR reactions prepared with iTaq SYBR Green master mix (BioRad) on a CFX-Connect Real-Time system (BioRad), according to the manufacturer’s recommendations. 10 μL technical replicate reactions for each biological replicate sample were plate across two wells in a MicroAmp® 96-well plate (ThermoFisher), with three or more biological replicate samples for any given experimental or control condition. For a gene of interest, qPCR primers were generated using Primer3Plus and checked for specificity using PrimerBLAST. Relative expression levels calculated using the ΔΔCq method, where reported experimental gene Cq values were normalized to ribosomal RNA (18s rRNA) Cq values for each sample. Plots and statistical analyses generated using the GraphPad Prism (8.2.1) software suite. Primer efficiencies were experimentally determined using relative expression measurements over a five-fold range of magnitudes in cDNA concentration (1:1 to 1:10,000), with primer amplification efficiencies ranging between 1.86 and 2.04.
Splice junction RT-qPCR
RT-qPCR was performed as described above. For any given splice junction, primers flanking the exon-intron junction were generated using Primer3Plus and checked for specificity using PrimerBLAST. Cq values of splice junctions were normalized to Cq values of total expression for a given gene using the ΔΔCq method. Plots and statistical analyses were generated using the GraphPad Prism (8.2.1) software suite.
RNA immunoprecipitation (RIP)
RIP was performed as previously described (A.M. Khalil et al., 2009). Briefly, 1×107 cells were lysed in RIP Buffer (150 mM KCl, 25 mM Tris pH 7.4, 5 mM EDTA, 0.5 mM DTT, 0.5% NP-40 substitute) with freshly added RNase inhibitor (100 U/mL) and protease inhibitor by dounce homogenization and centrifuged to pellet insoluble cellular debris. Magnetic Protein A/G beads (ThermoFisher) were pre-washed using magnetic separation with RIP Buffer and incubated with either 10 μg of Rabbit IgG isotype control (ThermoFisher) or 10 μg of anti-HA antibody (Cell Signaling) for 1 hr at 4°C. Antibody-conjugated beads were then washed twice with RIP Buffer to remove any unbound antibody and incubated with lysate for 2 h at RT or overnight at 4°C on a rotator. RIP reactions were then washed five times with RIP Buffer and resuspended in 100 μL of PBS. 10 μL (10%) of the RIP reactions were used for WB analysis against 0.1% input lysate (v/v) to assess RIP efficiency and the remaining 90 μL (90%) was resuspended in TRIzol LS Reagent (ThermoFisher) for RNA extraction and isolation.
RNA immunoprecipitation real-time quantitative PCR (RIP-RT-qPCR)
RNA was extracted from RIP reactions and from 0.9% input RNA. Input and RIP RNAs were used for cDNA synthesis with half of the RNA used for mock reverse transcription (no RT enzyme) and the other half used for reverse transcription with the iScript RT Supermix (BioRad). cDNA samples which were then further diluted (1:5 to 1:20). RT-qPCR was performed as described above, using iTaq SYBR Green master mix (BioRad) on the CFX-Connect Real-Time system (BioRad). Relative RIP enrichments were calculated as previously described (Sigma, Imprint® RIP kit): HA RIP Cq values were first compared to IgG control Cq values using the ΔCq method and then normalized across biological replicates by normalizing for input expression levels (1% input, ΔΔCq method). Primer pairs targeting 18s rRNA were used as a non-target control (NT) as well as to assess RIP efficiency by quantification of 18s rRNA de-enrichment between paired input to IP samples. Plots and statistical analyses were generated using the GraphPad Prism (8.2.1) software suite.
Immunofluorescence and imaging analysis
Stable cell lines were plated at equal density (1.0 × 105 cells/well, 24-well plate) in wells containing a 22 × 22 mm glass coverslip (VWR) or #1 18 mm glass coverslip (Electron Microscopy Sciences) and cultured in growth media for 48 h. Cultures were then fixed (4% paraformaldehyde (PFA) in PBS for 20 min), permeabilized (100 mM sodium citrate, 20% EtOH, 0.1% Triton X-100 for 2 min on ice), and incubated in blocking solution (1% BSA in PBS) for 1 h at RT. Cultures were incubated with a primary antibody solution containing anti-Ki67 antibody (1% BSA/PBS, Abcam ab16667, 1:1000 working concentration) for 2 h at RT or overnight at 4°C with gentle agitation. Cells were then washed three times with PBS, and incubated with a secondary antibody solution (Alexa 568-conjugated anti-Rabbit IgG, 1:2500 in PBS) for 2 h at RT. Cells were stained with DAPI (Roche, 5 min at RT, manufacturer’s recommendations) prior to mounting on 25 × 75 mm micro slides (VWR) with ProLong Gold antifade reagent (Invitrogen). Slides were allowed to cure for 24 h overnight in the dark and then imaged or stored at −20°C until imaging. Fluorescence imaging of immunofluorescence samples was performed using a DMi8 inverted light microscopy system (Leica) and a 20x or 40x objective lens, supported by the LAS imaging software suite (Leica). An ImageJ (Schneider et al. 2012) macro pipeline was designed to quantify Ki67 immunofluorescence signal in DAPI+ cellular nuclei outlined with the internal cell segmentation algorithm. Ki67 expression indices were calculated for each cell line, across three biological replicates with four fields of view each. Values were then exported and analyzed in R. Plots and statistical analyses were generated using R (3.6.0) in the RStudio (1.4.1717) software suite.
SH-SY5Y fluorescent imaging and neurite tracing
SH-SY5Y stable cell lines were first transduced with lentivirus expressing either a green (mNeonGreen, mNG) or red (dTomato) fluorescent reporter to create fluorescent stable cell lines to facilitate neurite imaging. Green and red stable line pools were then mixed at a 1:1 ratio and plated on a 12-well plate. Cultures were imaged (20x objective, 3 fields of view per well/biological replicate) at day 0 (immediately after treatment with either 0.1% DMSO v/v, or 10 μM RA in 0.1% DMSO) and on day 7. Images were taken using the red fluorescence channel to aid in focus and image acquisition, with images and tracings performed independently for both fluorescence channels in a given field of view to control for selective bias during image acquisition. Neurite tracings generated using an ImageJ software plugin (NeuronJ) (Meijering et al., 2004). All neurites for which the originating cell could confidently be identified were traced in a given field of view by M.W. using NeuronJ, which reports characteristics of traces, including neurite length, number of vertices, and intensity values. Values were then exported, analyzed, and plotted using R (3.6.0) in the RStudio (1.4.1717) software suite.
Cellular proliferation
1.0 × 105 cells per well for each SH-SY5Y stable cell line were plated in 24-well tissue culture plates. At each time point (0 h, 24 h, 48 h, 72 h), cells in one well from each isogenic cell line were dissociated with 250 μL TrypLE, incubated at 37°C, 5.0% CO2 for 5 min, and resuspended with the addition of 250 μL of cold 1x PBS. For each biological replicate (n = 3), multiple fields of view (n = 4–6,1 mm2 squares) were used to count the number of cells at each timepoint in 10 μL of resuspended mixture with a hemacytometer. Growth curves are plotted as a function of the mean ± SEM of the number of cells at each timepoint normalized to initial plated counts (0 h). Statistical significance was determined using the student’s t-test (two-tail distribution, homoscedastic variance) to compare RBM45 KO cell counts against Cas9 Control and RBM45 KO-Rescue cell counts at each time point (24 h, 48 h, 72 h).
Flow cytometry
Cell lines were plated and cultured in 6-well plates for 48 h prior to collection or differentiation treatment. Cultures (1.0-2.0 × 106 cells per well) were dissociated and washed in PBS. Cultures were then resuspended and treated with LIVE/DEAD Near-IR dye (ThermoFisher) according to manufacturer’s recommendations. After treatment, cells were washed in PBS, filtered through a 40 μm nylon cell strainer (Corning), and resuspended in 1 mL sorting media (DMEM, 1% FBS, Penicillin/Streptomycin). Treated cultures were then sorted with a BD FACSAria II flow cytometer using 639 nm excitation per biological replicate using the services of the Duke Flow Cytometry shared resource facility at the Duke Human Vaccine Institute (DHVI). Flow cytometry sorting data was imported, parsed, and analyzed using the flowCore package (R 3.6.0, RStudio 1.4.1717). Cell death was defined as any single cell event with IR emission intensity greater than 40x the IR intensity median per sorting experiment, with the percentage of dead cells reported for each biological replicate. To calculate cell death across biological replicates in cultured, undifferentiated, and differentiated SH-SY5Y cell lines, we subsampled 100,000 sorting events from each set of isogenic conditions. Plots were generated and statistical significance analyses were performed using R (3.6.0) in the RStudio (1.4.1717) software suite.
QUANTIFICATION AND STATISTICAL ANALYSIS
When mentioned, n represents the number of independently performed biological replicates for the given experiment. Data are represented as mean ± SEM, unless otherwise noted. When noted, p values were calculated using Welch’s t-test (two-tail distribution, unequal variance) when comparing means across comparisons of a single condition (ie. genotype). When noted, p values were calculated using Kolmogorov-Smirnov test (KS test) when comparison of sample distributions were considered appropriate, with random subsampling used for comparisons between disparately-sized vectors. In all figures, * p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. For sequencing analyses that utilized publicly available software and/or algorithms, quantification of means and statistical significance values were calculated and reported using default settings, unless otherwise stated. Peak calling (MACS2) cut-offs for replicate peaksets were set at padj ≤ 0.005. Differential gene expression (DESeq2) cut-offs were set at padj ≤ 0.05 (default). Alternative splicing analysis by rMATS was performed using default parameters. Differential exon and intron usage (DEXSeq) cut-offs were both set at p ≤ 0.01.
Supplementary Material
Highlights.
RBM45 preferentially binds to m6A-containing RNA
RBM45 binds thousands of RNAs in mouse and human cells
RBM45 regulates splicing through both m6A-dependent and m6A-independent mechanisms
RBM45 is necessary for proliferation and differentiation of SH-SY5Y cells
ACKNOWLEDGMENTS
We thank members of the Meyer laboratory for scientific discussion and helpful commentary. This research was supported by the National Institutes of Health (R01MH118366 and DP1DA046584 to K.D.M.). K.D.M. is also supported by the Rita Allen Foundation, the Kinship Foundation Searle Scholars Program, and the Klingenstein-Simons Fellowship in Neuroscience.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111293.
DECLARATION OF INTERESTS
K.D.M. has filed a patent application for the DART-seq technology through Duke University.
REFERENCES
- Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, and Tavazoie SF (2015). HNRNPA2B1 is a mediator of M6A-dependent nuclear RNA processing events. Cell 162, 1299–1308. 10.1016/j.cell.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Reyes A, and Huber W (2012). Detecting differential usage of exons from RNA-seq data. Genome Res. 22, 2008–2017. 10.1101/gr.133744.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, and Elkan C (1994). Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proceedings of the International Conference on Intelligent Systems for Molecular Biology 2, 28–36. [PubMed] [Google Scholar]
- Biechele S, Cockburn K, Lanner F, Cox BJ, and Rossant J (2013). Porcn-dependent Wnt signaling is not required prior to mouse gastrulation. Development (Camb.) 140, 2961–2971. 10.1242/dev.094458. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond AM, Ming GL, and Song H (2015). Adult mammalian neural stem cells and neurogenesis: five decades later. Cell Stem Cell 17, 385–395. 10.1016/j.stem.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Yang Z, Wang W, Qian K, Liu M, Wang J, and Wang M (2021). Structural basis for RNA recognition by the N-terminal tandem RRM domains of human RBM45. Nucleic Acids Res. 49, 2946–2958. 10.1093/nar/gkab075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins M, Riascos D, Kovalik T, An J, Krupa K, Krupa K, Hood BL, Conrads TP, Renton AE, Traynor BJ, and Bowser R (2012). The RNA-binding motif 45 (RBM45) protein accumulates in inclusion bodies in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) patients. Acta Neuropathol. 124, 717–732. 10.1007/s00401-012-1045-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon EG, and Manley JL (2017). RNA-binding proteins in neurodegeneration: mechanisms in aggregate. Genes Dev. 31, 1509–1528. 10.1101/gad.304055.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook KB, Vembu S, Ha KCH, Zheng H, Laverty KU, Hughes TR, Ray D, and Morris QD (2017). RNAcompete-S: combined RNA sequence/structure preferences for RNA binding proteins derived from a single-step in vitro selection. Methods 126, 18–28. 10.1016/j.ymeth.2017.06.024. [DOI] [PubMed] [Google Scholar]
- Desrosiers R, Friderici K, and Rottman F (1974). Identification of methylated nucleosides in messenger RNA from novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 71, 3971–3975. 10.1073/pnas.71.10.3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodt M, Roehr JT, Ahmed R, and Dieterich C (2012). FLEXBAR-flexible barcode and adapter processing for next-generation sequencing platforms. Biology 1, 895–905. 10.3390/biology1030895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez D, Freese P, Alexis MS, Su A, Hochman M, Palden T, Bazile C, Lambert NJ, Van Nostrand EL, Pratt GA, et al. (2018). Sequence, structure, and context preferences of human RNA binding proteins. Mol. Cell 70, 854–867.e9. 10.1016/j.molcel.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. (2012). Topology of the human and mouse M6A RNA methylomes revealed by M6A-seq. Nature 485, 201–206. 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- Edens BM, Vissers C, Su J, Arumugam S, Xu Z, Shi H, Miller N, Rojas Ringeling F, Ming GL, He C, et al. (2019). FMRP modulates neural differentiation through M6A-dependent MRNA nuclear export. Cell Rep. 28, 845–854.e5. 10.1016/j.celrep.2019.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edupuganti RR, Geiger S, Lindeboom RGH, Shi H, Hsu PJ, Lu Z, Wang SY, Baltissen MPA, Jansen PWTC, Rossa M, et al. (2017). N6-Methyladenosine (M6A) recruits and repels proteins to regulate MRNA homeostasis. Nat. Struct. Mol. Biol 24, 870–878. 10.1038/nsmb.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel M, Eggert C, Kaplick PM, Eder M, Röh S, Tietze L, Namendorf C, Arloth J, Weber P, Rex-Haffner M, et al. (2018). The role of m 6 A/m-RNA methylation in stress response regulation. Neuron 99, 389–403.e9. 10.1016/j.neuron.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, and Guénette S (2003). Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 100, 4162–4167. 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamand MN, and Meyer KD (2019). The epitranscriptome and synaptic plasticity. Curr. Opin. Neurobiol 59, 41–48. 10.1016/j.conb.2019.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingerich S, Kim GL, Chalmers JA, Koletar MM, Wang X, Wang Y, and Belsham DD (2010). Estrogen receptor alpha and G-protein coupled receptor 30 mediate the neuroprotective effects of 17b-estradiol in novel murine hippocampal cell models. Neuroscience 170, 54–66. 10.1016/j.neuroscience.2010.06.076. [DOI] [PubMed] [Google Scholar]
- Gupta VK, You Y, Gupta VB, Klistorner A, and Graham SL (2013). TrkB receptor signalling: implications in neurodegenerative, psychiatric and proliferative disorders. Int. J. Mol. Sci 14, 10122–10142. 10.3390/ijms140510122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson KA, Kim SH, and Tibbetts RS (2012). RNA-binding proteins in neurodegenerative disease: TDP-43 and beyond. Wiley Interdiscip. Rev. RNA 3, 265–285. 10.1002/wrna.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory Elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson J (2018). Molecular genetic analysis of Rbm45/Drbp1: genomic structure, expression, and evolution. Preprint at bioRxiv. 10.1101/274647. [DOI] [Google Scholar]
- Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, Yuan CL, et al. (2018). Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances MRNA stability and translation. Nat. Cell Biol 20, 285–295. 10.1038/s41556-018-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Morales DR, Thomas K, Presser A, Berstein BE, van Oudenaarden A, et al. (2009). Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proceedings of the National Academy of Sciences of the United States of America 106, 11667–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol 37, 907–915. 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korecka JA, van Kesteren RE, Blaas E, Spitzer SO, Kamstra JH, Smit AB, Swaab DF, Verhaagen J, and Bossers K (2013). Phenotypic characterization of retinoic acid differentiated SH-SY5Y cells by transcriptional profiling. PLoS One 8, e63862. 10.1371/journal.pone.0063862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna A, Biryukov M, Trefois C, Antony PMA, Hussong R, Lin J, Heinäniemi M, Glusman G, Köglsberger S, Boyd O, et al. (2014). Systems genomics evaluation of the SH-SY5Y neuroblastoma cell line as a model for Parkinson’s disease. BMC Genom. 15, 1154. 10.1186/1471-2164-15-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurochkin IV, Guarnera E, and Berezovsky IN (2018). Insulin-degrading enzyme in the fight against alzheimer’s disease. Trends Pharmacol. Sci 39, 49–58. 10.1016/j.tips.2017.10.008. [DOI] [PubMed] [Google Scholar]
- Li Y, Collins M, An J, Geiser R, Tegeler T, Tsantilas K, Garcia K, Pirrotte P, and Bowser R (2016). Immunoprecipitation and mass spectrometry defines an extensive RBM45 protein–protein interaction network. Brain Res., 79–93. 10.1016/j.brainres.2016.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Collins M, Geiser R, Bakkar N, Riascos D, and Bowser R (2015). RBM45 homo-oligomerization mediates association with ALS-linked proteins and stress granules. Sci. Rep 5, 14262. 10.1038/srep14262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Liu B, Merriman DK, Choi SH, Schumacher MA, Plangger R, Kreutz C, Horner SM, Meyer KD, and Al-Hashimi HM (2018). A potentially abundant junctional RNA motif stabilized by M6A and Mg2+. Nat. Commun 9, 2761. 10.1038/s41467-018-05243-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Dai Q, Zheng G, He C, Parisien M, and Pan T (2015). N6 -Methyladenosine-Dependent RNA structural switches regulate RNA-protein interactions. Nature 518, 560–564. 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livneh I, Moshitch-Moshkovitz S, Amariglio N, Rechavi G, and Dominissini D (2020). The M6A epitranscriptome: transcriptome plasticity in brain development and function. Nat. Rev. Neurosci 21, 36–51. 10.1038/s41583-019-0244-z. [DOI] [PubMed] [Google Scholar]
- Louloupi A, Ntini E, Conrad T, and Ørom UAV (2018). Transient N-6-Methyladenosine transcriptome sequencing reveals a regulatory role of M6A in splicing efficiency. Cell Rep. 23, 3429–3437. 10.1016/j.celrep.2018.05.077. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Hitz BC, Gabdank I, Hilton JA, Kagda MS, Lam B, Myers Z, Sud P, Jou J, Lin K, et al. (2020). New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 48, D882–D889. 10.1093/nar/gkz1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashik T, Sakashita E, Kasashima K, Tominaga K, Kuroiwa K, Nozaki Y, Matsuura T, Hamamoto T, and Endo H (2016). Developmentally regulated RNA-binding protein 1 (Drb1)/RNA-binding motif protein 45 (RBM45), a nuclear-cytoplasmic trafficking protein, forms TAR DNA-binding protein 43 (TDP-43)-Mediated cytoplasmic aggregates. J. Biol. Chem 291, 14996–15007. 10.1074/jbc.M115.712232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijering E, Jacob M, Sarria J-CF, Steiner P, Hirling H, and Unser M (2004). Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry A. 58, 167–176. 10.1002/cyto.a.20022. [DOI] [PubMed] [Google Scholar]
- Meyer KD (2019). DART-seq: an antibody-free method for global M6A detection. Nat. Methods 16, 1275–1280. 10.1038/s41592-019-0570-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, and Jaffrey SR (2017). Rethinking m 6 A readers, writers, and erasers. Annu. Rev. Cell Dev. Biol 33, 319–342. 10.1146/annurev-cellbio-100616-060758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, and Jaffrey SR (2012). Comprehensive analysis of MRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149, 1635–1646. 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L (2009). TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci 10, 850–860. 10.1038/nrn2738. [DOI] [PubMed] [Google Scholar]
- Pu J, Mao Y, Xu L, Zheng T, and Zhang B (2017). Stable cell lines of human SH-SY5Y uniformly expressing wild-type or mutant-type FERM domain containing 7 gene. Kues WA, ed. 14, 2277–2283. 10.3892/etm.2017.4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T, Björn g., et al. (2016). DeepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. 10.1093/NAR/GKW257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A, Anders S, Weatheritt RJ, Gibson TJ, Steinmetz LM, and Huber W (2013). Drift and conservation of differential exon usage across tissues in primate species. Proc. Natl. Acad. Sci. USA 110, 15377–15382. 10.1073/pnas.1307202110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH image to ImageJ: 25 Years of image analysis. Nat. Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple BD, Blomgren K, Gimlin K, Ferriero DM, and Noble-Haeusslein LJ (2013). Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol 106–107, 1–16. 10.1016/j.pneurobio.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu ZX, Lan L, Henry MD, Wu YN, Zhou Q, and Xing Y (2014). RMATS: robust and flexible detection of differential alternative splicing from replicate RNA-seq data. Proc. Natl. Acad. Sci. USA 111, E5593–E5601. 10.1073/pnas.1419161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipley MM, Mangold CA, and Szpara ML (2016). Differentiation of the SH-SY5Y human neuroblastoma cell line. J. Vis. Exp 2016, 53193. 10.3791/53193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamada H, Sakashita E, Shimazaki K, Ueno E, Hamamoto T, Kagawa Y, and Endo H (2002). CDNA cloning and characterization of Drb1, a new member of RRM-type neural RNA-binding protein. Biochem. Biophys. Res. Commun 297, 96–104. 10.1016/S0006-291X(02)02132-0. [DOI] [PubMed] [Google Scholar]
- Tegowski M, Flamand MN, and Meyer KD (2022). ScDART-seq reveals distinct M6A signatures and MRNA methylation heterogeneity in single cells. Mol Cell 82, 868–878.e10. 10.1016/j.molcel.2021.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ganaie SS, Cheng F, Xu P, Ning K, Wang X, Kleiboeker S, Cheng S, and Qiu J (2020). RNA binding motif protein RBM45 regulates expression of the 11-Kilodalton protein of Parvovirus B19 through binding to novel intron splicing enhancers. mBio 11, e00192–20. 10.1128/mBio.00192-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Yue M, Wang J, Wechsler-Reya RJ, Zhang Z, Ogawa Y, Kellis M, Duester G, Zhao JC, et al. (2018). N6-Methyladenosine RNA Modification Regulates Embryonic Neural Stem Cell Self-Renewal through Histone Modifications. Nat. Neurosci 21, 195–206. 10.1038/s41593-017-0057-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolozin B, and Apicco D (2015). RNA binding proteins and the genesis of neurodegenerative diseases. Adv. Exp. Med. Biol 822, 11–15. 10.1007/978-3-319-08927-0_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Su S, Patil DP, Liu H, Gan J, Samie RJ, and Ma J (2017). Molecular Basis for the Specific and Multivariate Recognitions of RNA Substrates by Human HnRNPA2/B1. Nat. Commun 9, 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KJ, Ringeling FR, Vissers C, Jacob F, Pokrass M, Jimenez-Cyrus D, Su Y, Kim NS, Zhu Y, Zheng L, et al. (2017). Temporal control of mammalian cortical neurogenesis by M6A methylation. Cell 171, 877–889.e17. 10.1016/j.cell.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Liu J, and He C (2015). RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 29, 1343–1355. 10.1101/gad.262766.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Yin X, Holley CL, and Meyer KD (2022). Improved methods for deamination-based M6A detection. Front. Cell Dev. Biol 10, 888279. 10.3389/fcell.2022.888279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Proteomics data generated in this study have been deposited to the ProteomeXchange consortium via the Proteomics Identifications Database (PRIDE) partner repository. Sequencing data generated in this study have been deposited to the National Center for Biotechnology Information Gene Expression Omnibus (GEO) database repository. All data generated in this study are publicly available as of the date of publication. This paper also reanalyzes existing, publicly available sequencing data deposited to the Encyclopedia of DNA Elements (ENCODE) project repository (Luo et al., 2020). Accession numbers for all deposited datasets are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-FLAG® M2 mouse monoclonal antibody (WB 1:2000) | Millipore Sigma | Cat# F1804; RRID: AB_262044 |
| Anti-HA rabbit monoclonal antibody (WB 1:2000) | Cell Signaling | Cat# 3724; RRID: AB_1549585 |
| Anti-HA mouse monoclonal antibody (WB 1:2000, IP 1:100) | Millipore Sigma | Cat# H3663; RRID: AB_262051 |
| Anti-YTHDF2 rabbit polyclonal antibody (WB 1:2000) | Aviva | Cat# ARP67917; RRID: AB_2861185 |
| Anti-GAPDH rabbit polyclonal antibody (WB 1:5000) | ProteinTech | Cat# 10494-1-AP; RRID: AB_2263076 |
| Anti-IDE rabbit polyclonal antibody (WB 1:1000) | Abcam | Cat# ab32216; RRID: AB_775686 |
| Anti-KI67 rabbit polyclonal antibody (WB 1:1000, IF 1:1000) | Abcam | Cat# ab16667; RRID: AB_302459 |
| Anti-Phospho-Histone H3 (Ser10) rabbit polyclonal antibody (WB 1:1000) | Cell Signaling | Cat# 9701; RRID: AB_331535 |
| Anti-TrkB (NTRK2) rabbit monoclonal antibody (WB 1:1000) | Abcam | Cat# ab134155; RRID: AB_2857962 |
| Anti-SFRP1 rabbit monoclonal antibody (WB 1:500) | Abcam | Cat# ab126613; RRID: AB_11128257 |
| Anti-TLE3/ESG rabbit polyclonal antibody (WB 1:1000) | Abcam | Cat# ab94972; RRID: AB_10860535 |
| Anti-RBM45 rabbit polyclonal antibody (discontinued) (WB 1:1000) | Abcam | Cat# ab123912; RRID: AB_10973537 |
| Anti-Histone H3 rabbit polyclonal antibody (WB 1:5000) | Cell Signaling | Cat# 9715; RRID: AB_331563 |
| Anti-m6A rabbit polyclonal antibody (WB 1:1000) | Abcam | Cat# ab151230; RRID: AB_2753144 |
| Anti-WNT3/WNT3A rabbit monoclonal antibody (WB 1:1000) | Abcam | Cat# ab172612; RRID: AB_2725748 |
| Anti-Rabbit HRP goat polyclonal antibody (WB 1:2500) | Abcam | Cat# ab6721; RRID: AB_955447 |
| Anti-Mouse HRP goat polyclonal antibody (WB 1:5000) | Millipore Sigma | Cat# GENXA-931 |
| Anti-Mouse Alexa 647 goat polyclonal antibody (IF 1:3000) | ThermoFisher | Cat# A-21235; RRID: AB_2535804 |
| Anti-Mouse Alexa 488 goat polyclonal antibody (IF 1:3000) | ThermoFIsher | Cat# A-11029; RRID: AB_138404 |
| Anti-Rabbit Alexa 568 goal polyclonal antibody (IF 1:3000) | ThermoFisher | Cat# A-11036; RRID: AB_10563566 |
| Anti-Rabbit Alexa 488 goal polyclonal antibody (IF 1:3000) | ThermoFisher | Cat# A-11008; RRID: AB_143165 |
| Bacterial and virus strains | ||
| MAX Efficiency™ DH5α Competent Cells | ThermoFisher | Cat# 18258012 |
| Rosetta™ 2(DE3) Competent Cells | Millipore Sigma | Cat# 71397-3 |
| Chemicals, peptides, and recombinant proteins | ||
| Glutathione Sepharose™ 4B Affinity Chromatography Media (GE Healthcare) | GE Healthcare | Cat# 17-0756-01 |
| L(−)-Glutathione (reduced form), ≥98% | VWR | Cat# 97061-574 |
| DAPI (4′,6-Diamidine-2′-phenylindole dihydrochloride) | Roche | Cat# 10236276001 |
| 6xHis-GST-RBM45 | This paper | See STAR Methods |
| Critical commercial assays | ||
| NEBNext® rRNA Depletion Kit (Human/Mouse/Rat) with Sample Purification Beads | NEB | E6350 |
| High Sensitivity DNA Kit, for Bioanalyzer 2100 platform | Agilent | 5067-4626 |
| RNA 6000 Pico Kit, for Bioanalyzer 2100 platform | Agilent | 5067-1513 |
| Invitrogen™ Dynabeads™ Oligo(dT)25 mRNA isolation beads | ThermoFisher | 61005 |
| Invitrogen™ RiboMinus™ Transcriptome Isolation Kit, human/mouse | ThermoFisher | K155002 |
| NEBNext® Ultra™ II RNA Library Prep Kit for Illumina® | NEB | E7770 |
| SMARTer® Stranded Total RNA-Seq Kit v3 | Takara Bio | 634485 |
| LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit | ThermoFisher | L34975 |
| RNeasy Micro Kit | QIAGEN | 74004 |
| Qproteome Bacterial Protein Prep Kit | QIAGEN | 37900 |
| QIAEX II Gel Extraction Kit | QIAGEN | 20021 |
| QIAGEN Plasmid Plus Midiprep Kit | QIAGEN | 12943 |
| QIAprep Spin Miniprep Kit | QIAGEN | 27106 |
| QIAquick Gel Extraction Kit | QIAGEN | 28704 |
| QuickLyse Miniprep Kit | QIAGEN | 27406 |
| Deposited data | ||
| RNA-pulldown LC-MS/MS in mHippoE-2 cell lysate | This paper, PRIDE | PXD034161 |
| RNA-pulldown LC-MS/MS in adult mouse (C57BLK6/J) brain lysate | This paper, PRIDE | PXD034159 |
| HA-tagged RBM45 CLIP-seq in mHippoE-2 cells | This paper, GEO | GSE190292 |
| HA-tagged RBM45 CLIP-seq in HEK293T cells | This paper, GEO | GSE190287 |
| HA-tagged RBM45 CLIP-seq in DMSO-/RA-treated SH-SY5Y cells | This paper, GEO | GSE205625 |
| in vitro subcellular fractionation DART-seq in mHippoE-2 cells | This paper, GEO | GSE190311 |
| in vitro subcellular fractionation DART-seq in HEK293T cells | This paper, GEO | GSE190303 |
| Ribo-depleted RNA-seq data in mHippoE-2 cells | This paper, GEO | GSE190322 |
| Ribo-depleted RNA-seq data in DMSO-/RA-treated SH-SY5Y cells | This paper, GEO | GSE190316 |
| SRSF7 eCLIP in HepG2 cells | Gene Yeo, UCSD ENCODE | ENCSR513NDD |
| SRSF7 eCLIP in K562 cells | Gene Yeo, UCSD ENCODE | ENCSR468FSW |
| poly(A) RNA-seq, embryonic mouse (B6NCrl) forebrain timecourse | Barbara Wold, CalTech ENCODE | ENCSR443OEA |
| poly(A) RNA-seq, embryonic mouse (B6NCrl) midbrain timecourse | Barbara Wold, CalTech ENCODE | ENCSR505AHT |
| poly(A) RNA-seq, embryonic mouse (B6NCrl) hindbrain timecourse | Barbara Wold, CalTech ENCODE | ENCSR328UYN |
| Experimental models: Cell lines | ||
| Immortalized mouse hippocampal cell line (mHippoE-2) | Cedarlane Labs | CVCL_D377 |
| Human embryonic kidney (HEK) cell line (HEK293T) | ATCC | CVCL_0063 |
| Human neuroblastoma cell line (SH-SY5Y) | ATCC | CVCL_0019 |
| Experimental models: Organisms/strains | ||
| C57BL/6J, mus musculus | Jackson Labs | IMSR_JAX:000664 |
| Oligonucleotides | ||
| Biotin-labeled non-m6A Dlg4 RNA oligo: 5′-Biotin-GGGCCCUUGGUCUGGACU GAAUAGCCCAAGCCC-3′ | This paper | N/A |
| Biotin-labeled m6A Dlg4 RNA oligo: 5′-Biotin-GGGCCCUUGGUCUGGm6A CUGAAUAGCCCAAGCCC-3′ | This paper | N/A |
| Biotin-labeled non-m6A ssRNA oligo: 5′-Biotin-UUCUUCUGUGGACUGUG-3′ | This paper | N/A |
| Biotin-labeled m6A ssRNA oligo: 5′-Biotin-UUCUUCUGUGGm6ACUGUG-3′ | This paper | N/A |
| Biotin-labeled non-m6A randomer RNA oligo: 5′-Biotin-GGGGAGUUCUACAGUCCGA CNNNNNNNNNNNRACNNNNNNNNN NNGGAAUUCUCGGGUGUCAAGG-3′ | This paper | N/A |
| Biotin-labeled m6A randomer RNA oligo: 5′-Biotin-GGGGAGUUCUACAGUCCG ACNNNNNNNNNNNRm6ACNNNNNN NNNNNGGAAUUCUCGGGUGUCAAGG-3′ | This paper | N/A |
| Fluorescein-labeled (FAM) non-m6A ssRNA oligo: 5′-FAM-UUCUUCUGUGGACUGUG-3′ | This paper | N/A |
| Fluorescein-labeled (FAM) m6A ssRNA oligo: 5′-FAM-UUCUUCUGUGGm6ACUGUG-3′ | This paper | N/A |
| For DNA oligonucleotides used in this study, see Table S1 | This paper | Table S1 |
| Recombinant DNA | ||
| pcDNA™3.1 (+) Mammalian Expression Vector | Invitrogen | Cat# V79020 |
| pcDNA™4/TO Mammalian Expression Vector | Invitrogen | Cat# V102020 |
| pET-GST | Addgene | Plasmid #42049 |
| pCMV-VSV-G | Addgene | Plasmid #8454 |
| psPAX2 | Addgene | Plasmid #12260 |
| lentiCRISPR v2 | Addgene | Plasmid #52961 |
| pLenti-puro | Addgene | Plasmid #39481 |
| Software and algorithms | ||
| Prism 8 (v8.2.1) | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ (v1.53a) | NIH | https://imagej.nih.gov/ij/ |
| NeuronJ (v1.4.3) | E. Meijering et al., 2004 | https://imagescience.org/meijering/software/neuronj/ |
| IGV (v2.8.9, v2.9.4) | J.T. Robinson et al., 2011 | https://igv.org |
| SeqAn (v2.3.2), FLEXBAR (v3.0.3) | M. Dodt et al., 2012 | https://github.com/seqan/flexbar |
| Novoalign (v3.09.00) | Novocraft | http://www.novocraft.com/products/novoalign/ |
| Python (v2.7.11) | Python | https://www.python.org |
| homer (v4.11.1) | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| MEME (v5.0.5) | Bailey and Elkan, 1994 | https://meme-suite.org/meme/index.html |
| R (v3.6.0) | R | https://www.r-project.org |
| DESeq2 (v1.32.0) | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Picard (v2.18.2) | “Picard Toolkit.” 2019. Broad Institute | https://broadinstitute.github.io/picard/ |
| rMATS (v4.0.2) | Shen et al., 2014 | http://rnaseq-mats.sourceforge.net |
| DEXSeq (v1.38.0) | Anders et al., 2012 | https://bioconductor.org/packages/release/bioc/html/DEXSeq.html |
| Subread (v1.6.3) | Liao et al., 2014 | http://subread.sourceforge.net |
| deeptools (v3.2.1) | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| HiSAT2 (v2.1.0) | Kim et al., 2019 | http://daehwankimlab.github.io/hisat2/ |
| Bedtools (v2.25.0) | Quinlan Lab, Univ. of Utah | https://bedtools.readthedocs.io/en/latest/ |
| Trimmomatic (v0.38) | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| MACS2 (v2.1.1.20160309) | Y. Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| Bullseye (unreleased) | Tegowski et al., 2022 | https://github.com/mflamand/Bullseye |
This paper does not report original code. This paper used existing, publicly available code to analyze sequencing data. Relevant identifying information for software and algorithms used in this study listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.