

Abstract
Objective:
Mutations in γ-aminobutyric acid (GABA) transporter 1 (GAT-1)-encoding SLC6A1 have been associated with myoclonic atonic epilepsy and other phenotypes. We determined the patho-mechanisms of the mutant GAT-1, in order to identify treatment targets.
Methods:
We conducted whole-exome sequencing of patients with myoclonic atonic epilepsy (MAE) and characterized the seizure phenotypes and EEG patterns. We studied the protein stability and structural changes with homology modeling and machine learning tools. We characterized the function and trafficking of the mutant GAT-1 with 3H radioactive GABA uptake assay and confocal microscopy. We utilized different models including a knockin mouse and human astrocytes derived from induced pluripotent stem cells (iPSCs). We focused on astrocytes because of their direct impact of astrocytic GAT-1 in seizures.
Results:
We identified four novel SLC6A1 variants associated with MAE and 2 to 4 Hz spike-wave discharges as a common EEG feature. Machine learning tools predicted that the variant proteins are destabilized. The variant protein had reduced expression and reduced GABA uptake due to endoplasmic reticular retention. The consistent observation was made in cortical and thalamic astrocytes from variant-knockin mice and human iPSC-derived astrocytes. The Slc6a+/A288V mouse, representative of MAE, had increased 5–7 Hz spike-wave discharges and absence seizures.
Interpretation:
SLC6A1 variants in various locations of the protein peptides can cause MAE with similar seizure phenotypes and EEG features. Reduced GABA uptake is due to decreased functional GAT-1, which, in thalamic astrocytes, could result in increased extracellular GABA accumulation and enhanced tonic inhibition, leading to seizures and abnormal EEGs.
Keywords: SLC6A1, GABA transporter 1 (GAT-1), Protein misfolding, Myoclonic atonic epilepsy (MAE), Endoplasmic reticulum (ER), Astrocytes, Thalamus
1. Introduction
SLC6A1 variants are associated with a wide spectrum of neurodevelopmental phenotypes, with myoclonic atonic epilepsy (MAE) being the most prominent seizure phenotype (Carvill et al., 2015; Mattison et al., 2018; Johannesen et al., 2018). MAE is characterized by abrupt falls, also called drop attacks, due to sudden loss of muscle tone. MAE patients can display multiple seizure types including absence, myoclonic jerks, and generalized tonic clonic seizure with or without fever. In SLC6A1 variant mediated epilepsy, over 70% of patients experience absence seizures (Goodspeed et al., 2020). It is intriguing that the inhibition of SLC6A1-encoding GABA transporter 1 (GAT-1) increases tonic inhibition and gives rise to absence seizures in animal models (Cope et al., 2009; Crunelli et al., 2020; Walker and Kullmann, 2012), while pharmacological inhibition of GAT-1 with tiagabine has been used to treat focal epilepsy (Bresnahan et al., 2019). Thus, it is of the utmost importance to understand if SLC6A1 variants cause a loss-of-function or a gain-of-function and how each contributes to the disease phenotype.
We have previously identified that altered protein stability due to protein misfolding is a major pathology for GAT-1 variants, which would result in impaired protein trafficking (Cai et al., 2019; Wang et al., 2020). Consequently, trafficking-deficient transporters result in reduced protein presence at the cell surface and synapse and compromised GABA uptake, likely leading to increased extracellular GABA level and tonic inhibition in the brain. Enhanced tonic inhibition in thalamocortical neurons is sufficient to elicit both electrographic and behavioral seizures in animals, including the Genetic Absence Epilepsy Rat from Strasbourg (GAERS), Stargazer mice, and lethargic mice (Cope et al., 2009).
It has been established that GAT-1 is expressed in both neurons and astrocytes (Minelli et al., 1995; Mermer et al., 2021) with regional or cell type variations. In the thalamus, GAT-1 is mainly expressed in astrocytes in both humans and rodents (De Biasi et al., 1998). Recent work reappraised the localization of GAT-1 in the neocortex and indicated that GAT-1 is also expressed in microglial cells and oligodendrocytes (Fattorini et al., 2020). One study using immunohistochemistry and electron microscopy for ultrastructural investigations demonstrated that GAT-1 and GABA transporter 3 (GAT-3) immunoreactivity is present throughout the thalamus in small punctate structures scattered in the neuropil among unlabeled neuronal perikaryal. In contrast to other brain regions, such as the cortex (Minelli et al., 1995; Minelli et al., 1996) or hippocampus (Ribak et al., 1996), GAT-1 and GAT-3 staining is absent from GABAergic synaptic terminals (Minelli et al., 1995). Ultrastructural investigations confirm that GAT-1 and GAT-3 labelling is restricted to astrocytes (Minelli et al., 1995; De Biasi et al., 1998). Labeled astrocytes are adjacent to terminals making either symmetric or asymmetric synaptic contacts, and are close to neuronal profiles that do not form synaptic contacts in the plane of the section. These findings demonstrate that in the rodent thalamus, the GABA uptake mediated by GAT-1 and GAT-3 is localized exclusively to astrocytes near the synapses and in the neuropil and absent from GABAergic terminals. This implicates the critical role of thalamic and astrocytic GAT-1 in seizure genesis. Considering the contribution of cortico-thalamic-cortical circuitry in absence seizure generation, it is important to determine the functional consequence of the mutant GAT-1 in astrocytes. Thus, characterizing the impact of SLC6A1 variants in astrocytes could provide direct evidence explaining seizure genesis in patients bearing the SLC6A1 variants.
In this study, we report novel SLC6A1 variants associated with MAE and characterize the clinical history, EEG patterns, and molecular defects of these variants. We used machine learning tools to predict protein stability and determined the hydrophobicity score of the protein surface. We also determined the function and trafficking pattern of variants in the physiologically-relevant cell models: mouse cortical and thalamic astrocytes and human astrocytes derived from human induced pluripotent stem cells (iPSCs). We compared and validated the molecular findings from these novel variants with a previously characterized recurrent variant A288V associated with MAE and other phenotypes (Mermer et al., 2021). Our study provides novel findings and suggests a unifying disease mechanism for MAE and absence epilepsy with 2–4 Hz spike wave discharges (SWDs) associated with SLC6A1 variants in accordance with previous studies in animal models of absence seizures.
2. Materials and methods
2.1. Patient history
The patients and unaffected family members were evaluated at the Pediatric Epilepsy Program at Children’s Hospital of Shanghai Fudan University and Xiangya Hospital of the Central-Southern University in Changsha, China. The collected clinical data included age of seizure onset, a detailed developmental history, seizure types and frequency, response to antiseizure drugs, family history, and general and neurological examination results. Video electroencephalography (EEG) was conducted, and the results were reviewed by certified electroencepha-lographers. Written informed consent for the sharing of clinical and genetic information was obtained from the parents.
2.2. Whole-exome sequencing (WES), variant interpretation and confirmation
The exome was captured from peripheral blood DNA using Agilent SureSelectV6 (Agilent Technologies, Santa Clara, California) or IDT xGen Exome Research Panel (Integrated DNA Technologies, Coralville, Iowa). Subsequent paired-end sequencing was carried out with Illumina HiSeq4000 or NovaSeq 6000 (Illumina, Santa Clara, California). Data processing, alignment (using a Burrows-Wheeler algorithm, BWA-mem), and variant calling were performed using Genome Analysis Tool Kit (GATK v4) best practices (https://software.broadinstitute.org/gatk/best-practices/) from the Broad Institute according to the reference genome GRCh38. Variants were annotated using ANNOVAR (http://www.openbioinformatics.org/annovar/). Variants were absent from the population database gnomAD (https://gnomad.broadinstitute.org/). Multiple bioinformatic analysis results indicated that the variants were deleterious. Variants were classified in accordance with the guidelines of the American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015). Sanger sequencing was performed for variant confirmation and segregation analysis for family members.
2.3. The cDNAs for coding GABA transporter 1
The plasmid cDNA encoding enhanced yellow fluorescent protein (EYFP)-tagged rat GAT-1 was sub-cloned into the expression vector pCMV (Cai et al., 2019; Scholze et al., 2002). Replications of patient GAT-1 mutations were cloned via a standard molecular cloning process. A QuikChange Site-Directed Mutagenesis Kit was utilized to introduce the GAT-1 mutation into the wildtype GAT-1 coding sequence. The product from the polymerase chain reaction was transformed using DHα competent cells and finally plated. A clone was chosen from the kanamycin-containing agar plate and grown overnight, replicating the cDNA. All GAT-1 mutations were confirmed by DNA sequencing. Both the wildtype and the mutant cDNAs were prepared with Qiagen Maxiprep kit.
2.4. Slc6a1+/A288V knockin mouse and EEG recordings
We generated the novel Slc6a1+/A288V global knockin mouse model in collaboration with University of Connecticut Health Center. The CRISPR-CAS9 strategy was utilized to mediate 863C to T knockin in exon 9 of the coding sequence. The heterozygous mouse was bred with the C57BL/6 J wildtype mouse (stock 000664). The mice used for biochemistry were between 2 and 4 months old. The pups used for astrocytes culture were at postnatal day 0–3. Mice used for EEG recordings were 1–2 months old. Both male and female mice were included except for EEG recordings, for which only males were included.
Video-monitoring synchronized EEG recordings were conducted based on our standard lab protocol (Warner et al., 2016; Kang et al., 2015; Warner et al., 2017). Mice were surgically implanted with prefabricated skull head mounts (Pinnacle Technology) that have two bipolar EEG channels and one subcutaneous nuchal EMG channel. Four holes were drilled through the skull to dura, with two placed 1 mm anterior to the bregma and two placed 7 mm posterior to the bregma and each being 1.5 mm lateral to the central sulcus. These holes accommodated a prefabricated mouse head mount, which was fastened to the skull with stainless steel screws (Small Parts, Miami Lakes, FL). Loose skin was sutured around the implant, and at least seven days were allowed for recovery before EEG recording. Following the recovery period, mice were placed individually in cylindrical chambers and allowed ad libitum access to food and water. EEG recordings lasted for at least 48 h for each mouse.
2.5. EEG analysis with Seizure Pro software
The EEG recording sampling rate is set at 400 Hz with a pre-amplifier gain of 100 Hz. EEG and EMG channels have a filter set at 25 Hz. EEG recordings are scored blindly by a skilled scorer using the Sirenia Seizure Pro software. A power analysis is performed using the theta frequency band of 5–7 Hz. We chose to measure 5–7 Hz SWDs because they correlate with the typical 2–4 Hz SWDs observed in humans (Seki et al., 2002; Savojardo et al., 2016). An average power is calculated using baseline recordings and applied to seizure analysis for treatment recordings. Parameters for seizures including both morphology and duration identified by the software were confirmed with video recordings. The Racine Scale (Stage 1: mouth/facial movements; Stage 2: head nodding; Stage 3: forelimb clonus; Stage 4: rearing; Stage 5: rearing and falling) was used for seizure identification. The identified SWDs were then confirmed with video monitoring and compared across treated and non-treated recordings.
2.6. Cultures of HEK293T cells, Chinese hamster ovary cells, and mouse astrocytes
The human embryonic kidney (HEK) 293 T cell line and Chinese hamster ovary (CHO) cell line were purchased from American Type Culture Collection (ATCC), and the cells were used under passage 20 for experiments. Cells were seeded at a density of 0.4 × 106 per 60 mm dish and passaged every 3–4 days. HEK293T cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS and 1% penicillin/streptomycin (Gibco). CHO cells were used as a control because we identified that their endogenous GAT-1 expression is lower than in HEK293T cells. CHO cells were maintained in a F12(1×) nutrient mixture supplemented with 10% FBS and 1% penicillin/streptomycin (Gibco). For the culture of mouse cortical and thalamic astrocytes, we used the same protocol as in our previous study (Mermer et al., 2021). Briefly, the cortices or thalami of postnatal day 0–3 pups were dissected. The tissues were minced after removing the meninges and then digested with 0.25% trypsin for 10 min at 37 °C. The debris and large tissue chunks were removed, and the remaining cell suspension was plated in uncoated 100 mm dishes at a density of 3–6 million cells. The plated cells were taken as passage 0 and the astrocytes used for experiments were prepared from passage 2. The culture media was refreshed the next day and then every 4–5 days. At least 95% of the cell population was astrocytes, as confirmed by anti-glial fibrillary acidic protein antibody. The culture medium used for astrocytes is the same as is used in the HEK293T cells, which is Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS and 1% penicillin/streptomycin. As in HEK293T cells, the astrocytes were transfected with PEI.
2.7. Human astrocytes derived from induced pluripotent stem cells (iPSCs)
The human induced pluripotent stem cell (iPSC) line was purchased from Thermo Fisher (A18945). We followed our previous protocol used for the astrocyte differentiation from the SLC6A1(S295L) patient cell line (Mermer et al., 2021). We first prepared the neural progenitor cells (NPCs) and then differentiated the NPCs into astrocytes. The differentiation of NPCs was induced with the STEMdiff SMADi Neural induction kit from STEMCELL. Differentiation of human astrocytes started from NPCs at day 5 of passage 1 (P1). The differentiation of astrocytes was initiated by using the Astrocyte medium (ScienCell) for 25–30 days (doi: https://doi.org/10.1101/2020.04.21.054361) (Romero-Morales et al., 2021) and the culture medium was refreshed every other day. Astrocytes were passaged at ~70% confluence and were validated based on the protocol described in our previous study by staining with S100β and Glial Fibrillary Acidic Protein (GFAP) (Mermer et al., 2021). >95% of cells adopted astrocytic morphology and about 80% of the total cell population was positively stained with GFAP and S100B, consistent with previous studies on astrocyte differentiation (Mermer et al., 2021).
2.8. Polyethylenimine (PEI) transfection
Standard transfection protocols were performed in HEK293T cells (Cai et al., 2019) and astrocytes. Briefly, 24 h before transfection, HEK293T cells were split equally into plates of the desired size. Astrocytes at passage 2 were prepared and grown in 35 mm dishes for GABA uptake and 100 mm dishes for western blot. The experiments were conducted in cells with 80–90% confluence. Transfection for both HEK293T cells and astrocytes was based on our standard PEI protocol with PEI reagent (40 kD, Polysciences) at a DNA: transfection reagent ratio of 1:2.5, and the cells were harvested 48 h after transfection. For radiolabeling GABA uptake, 1 μg of the cDNAs with PEI at a ratio of 1:2.5 μl was transfected in each 35 mm dish. For total protein expression, 3 μg cDNAs were used for transfection in each 60 mm dish, while 10 μg cDNAs were used for transfection in each 100 mm dish. The cDNAs were combined with Dulbecco Modified Eagle Medium (DMEM) and a PEI/DMEM mixture. For iPSC derived human astrocytes, both PEI and lipofectamine 2000 transfection protocols were used. Lipofectamine transfection was conducted per the manufacturer’s protocol. The experiments were conducted 48 h after transfection.
2.9. Western blot analysis of total GAT-1 protein
Live transfected HEK293T cells were washed with phosphate-buffered saline (1 × PBS, pH 7.4) three times and were then lysed in RIPA buffer (20 mM Tris, 20 mM EGTA, 1 mM DTT, 1 mM benzamidine) supplemented with 0.01 mM PMSF, 0.005 μg/ml leupeptin, and 0.005 μg/ml pepstatin for 30 min at 4 °C. The samples were then subject to protein concentration determination followed by SDS-PAGE. Membranes were incubated with primary rabbit polyclonal antibodies against GAT-1 (Alomone Labs, AGT-001 or Synaptic System, 274102 at 1:200 dilution). Primary mouse monoclonal antibodies against ATPase (Developmental Studies Hybridoma Bank) were used as a loading control. Blots were imaged with a digital fluorescence scanner, Odyssey DLx (LI-COR), and analysis of blots was performed in the program used to acquire the images: Image Studio Lite Version 5.3. To avoid variation, we normally measure the protein band twice and take the mean of their integrated density values (IDVs). The raw values of the targeted protein in both the wildtype and the mutant conditions were normalized to the loading control. The mutant condition was then normalized to the wildtype.
2.10. Radioactive 3H-labeled GABA uptake assay
The radioactive 3H-labeled GABA uptake assay in HEK293T, mouse astrocytes and human astrocytes was modified from previous studies (Cai et al., 2019; Mermer et al., 2021). Briefly, cells were cultured in 35 mm dishes three days before the GABA uptake experiment in DMEM with 10% fetal bovine serum and 1% penicillin/streptomycin. The cells were then transfected with equal amounts of the wildtype or the mutant GAT-1 cDNAs (1 μg) with PEI at a ratio of 1 μg cDNA:2.5 μl of PEI for each condition 24 h after cell seeding. GABA uptake assay was carried out 48 h after transfection. The cells were incubated with preincubation solution (140 mM NaCl, 5 mM CaCl2, 1 mM MgSO4, 2 mM glucose and 2.5 mM HEPES, pH 7.4, Osmolarity 320) for 15 min and then incubated with preincubation solution containing 1μci/ml 3H GABA and 10 μM unlabeled GABA for 30 min at room temperature. After washing, the cells were lysed with 0.25 N NaOH for 1 h. Acetic acid glacial was added and lysates were then determined on a liquid scintillator with QuantaSmart. The flux of GABA (pmol/μg/min) was averaged with duplicates or triplets for each condition at each transfection. The average counting was taken as n = 1. The untransfected condition was included as a reference. The pmol/μg/min in the mutant was then normalized to the wildtype from each experiment, which was arbitrarily taken as 100%.
2.11. Live cell confocal microscopy and image acquisition
Live cell confocal microscopy was performed using an inverted Zeiss laser scanning microscope (Model 510) with a 63 × 1.4 NA oil immersion lens, 2–2.5 × zoom, and multi-track excitation. Astrocytes were plated on poly-d-lysine-coated, glass-bottom imaging dishes at the density of 1–2 × 105 cells and cotransfected with 1 μg of the wildtype or the mutant GAT-1 plasmids and 1 μg ERCFP with PEI based on our standard lab protocol. Cells were examined with excitation at 458 nm for ECFP and 514 nm for EYFP. All images were single confocal sections averaged from 8 times to reduce noise, except when otherwise specified. The images were acquired using a LSM 510 invert confocal microscope with 63× objective.
2.12. Protein structural modeling and machine learning tools
We predicted the impact of the variant on the GAT-1 protein with multiple machine learning tools. Tertiary structures of both the wildtype and mutated GAT-1 proteins were modeled with I-TASSER (Zhang, 2008) and analyzed by MAESTRO web (Laimer et al., 2016). Details in structural differences between the wildtype and the mutant GAT-1 were illustrated using the modeled structure with DynaMut (Rodrigues et al., 2018). Analysis of self-aggregation or co-aggregation was conducted using PASTA 2.0 (Walsh et al., 2014).
2.13. Data analysis
Numerical data were expressed as mean ± SEM. A one-way analysis of variance (ANOVA) and Newman-Keuls test was used to determine significance compared to the wildtype condition and between variants if there are more than two conditions. An unpaired t-test was used between the wildtype and the mutant condition for the studies in the Slc6a1+/A288V knockin mice. Proteins were quantified by Image Studio Lite 5.3 software and data were normalized to loading controls and then to wildtype transporter proteins, which were arbitrarily taken as 1 in each experiment. The radioactivity of GABA uptake was measured in a liquid scintillator with QuantaSmart. The flux of GABA (pmol/μg/min) in the wildtype GAT-1 samples was arbitrarily taken as 1 in each experiment. The baseline activity of HEK cells was not subtracted. The fluorescence intensities from confocal microscopy experiments were determined using MetaMorph imaging software and the measurements were carried out in ImageJ, as modified from previous descriptions (Warner et al., 2016; Kang et al., 2015; Kang et al., 2004). All statistical analysis was conducted in GraphPad version 8 (GraphPad Prism, La Jolla, CA). Statistical significance was taken as p < 0.05.
3. Results
3.1. Molecular genetics identified novel variants in SLC6A1 associated with absence epilepsy and MAE
Variants in SLC6A1 have been associated with a spectrum of epilepsy syndromes and neurodevelopmental disorders (Carvill et al., 2015; Mattison et al., 2018; Johannesen et al., 2018; Cai et al., 2019; Wang et al., 2020). We have reported variants in SLC6A1 associated with Lennox-Gastaut syndrome and epilepsy with autism due to partial loss of GAT-1 function. Here we report four de novo variants that are associated with myoclonic atonic epilepsy (Fig. 1A). The variants (46G > T, 1379 T > G, 1084G > A, and 1485G > A) were identified in the patients but not in the unaffected parents (Fig. 1B) using whole-exome sequencing. The variants are located at different functional domains of the GAT-1 protein peptide (Fig. 1C). Two variants result in truncated proteins—GAT-1 (E16X) and GAT-1(W495X)—while the other two missense variants alter amino acids that are conserved across species (Fig. 1D).
Fig. 1.
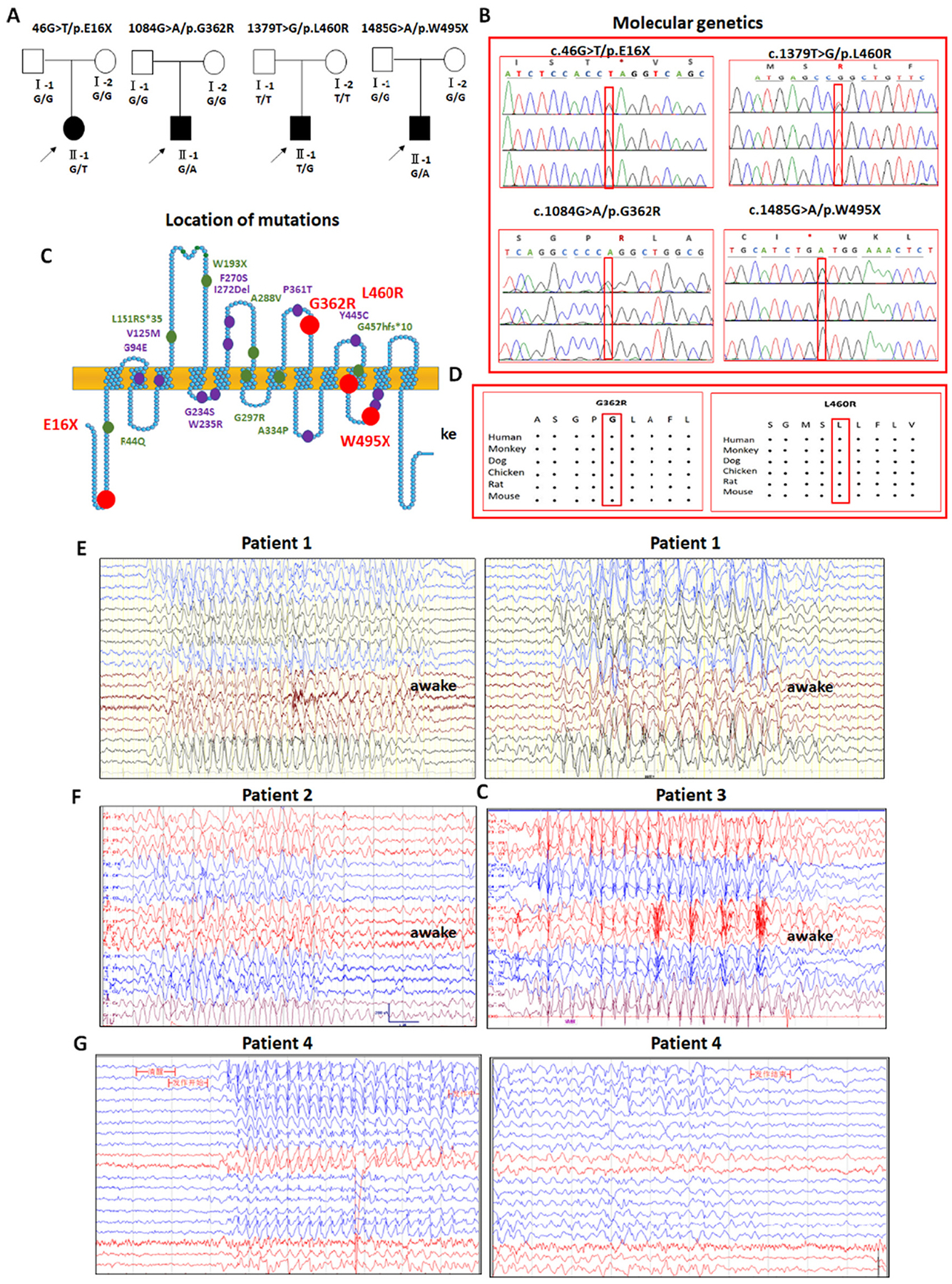
Molecular genetics and electroencephalogram (EEG) patterns of four novel variants in SLC6A1 associated with myoclonic atonic epilepsy.
(A) Four patients from different families carrying novel variants in SLC6A1. (B). Electropherograms showing identified novel variants in SLC6A1 from patients. (C). Schematic representation of GAT-1 protein topology and locations of novel GAT-1 variants (red dots) identified in patients associated with a spectrum of epilepsy syndromes. The purple dots represent the other variants in SLC6A1 that are previously reported for GABA uptake. It is predicted that GAT-1 contains 12 transmembrane domains. The positions of variants are based on the published LeuT crystal structure. (D) Amino acid sequence homology shows that glycine (G) at residue 362 and leucine (L) at residue 460 are highly conserved in SCL6A1 in humans (Accession NO.NP_003033.3) and across species as shown in the boxed region. (E). In the patient carrying the E16× variant (patient 1), video EEG recordings showed long-term discharges of medium-high amplitudes of 2–3 Hz spike-slow waves and 4–7 Hz theta activity mixed with fast activity with a very low amplitude during both wakefulness (left panel) and sleep (right panel). The epileptiform discharges are prominent in the occipital region or generalized. (F) In the patient carrying the G362R variant (patient 2), video EEG recordings showed many medium-high amplitudes of 2.5–3.5 Hz interictal spike-slow waves, clusters or continuous ictal discharges in the bilateral parietal, occipital, and posterior temporal areas during wakefulness. (G) In the patient carrying the L460R variant (Patient 3), there were long-term, generalized 2.5–3.5 Hz spike-slow wave discharges with high amplitude during wakefulness. (H). In the patient carrying the W495X variant (Patient 4), there were long-term, generalized 2.5–3.5 Hz spike-slow wave discharges with medium amplitude, especially in the bilateral frontal regions during wakefulness.
3.2. Common seizure and EEG features with 2–4 Hz spike wave discharges in patients and related treatment regimens
Our study is consistent with previous studies (Carvill et al., 2015; Johannesen et al., 2018) indicating that MAE is the major seizure phenotype for SLC6A1 variant mediated disorders, and all four patients were diagnosed with MAE due to the de novo variants. No positive family history of seizures was reported. Compared with mutations in GABAA receptor subunits that are associated with a wide range of epilepsy phenotypes and various epilepsy syndromes, individuals with SLC6A1 variants showed relative phenotypic homogeneity of epilepsy with MAE and childhood absence epilepsy (CAE). MAE is characterized by the onset of myoclonic, myoclonic-atonic, and atonic seizures between 1 and 4 years of age and the presence of generalized spike-wave discharges (SWDs) or polyspike-wave discharges. Besides common drop attacks or atonic seizures, all four patients were reported to have 2–4 Hz generalized SWDs on their EEGs either during sleep, wakefulness, or both (Fig. 1E to G). The common EEG feature of 2–4 Hz in this cohort of patients is consistent with previous studies on SLC6A1 variant associated epilepsies (Goodspeed et al., 2020; Cai et al., 2019; Wang et al., 2020; Poliquin et al., 2021; Johannesen et al., 2016). The patient bearing the GAT-1(E16X) variant also had fast activity (15–25 Hz) with a low amplitude in EEG. Similar fast activity in EEG has been reported in a GAT-1(G234S) patient (Cai et al., 2019). Three out of four patients had neurodevelopmental delay before seizure onset, and all had impaired cognition after seizure onset. The patient with the GAT-1(G362R) variant also had idiopathic short stature syndrome. It is of note that seizures in all patients were controlled with valproic acid alone or in combination with levetiracetam (Lev) (Table 1). However, cognitive dysfunction was not improved with seizure control. This is consistent with our previous study on the patients with GAT-1(V125M) variant (Poliquin et al., 2021) and suggests no direct correlation of seizure control and cognitive improvement for SLC6A1 variants. Brain magnetic resonance imaging (MRI) was unremarkable for all four patients.
Table 1.
Clinical information and patient history of four SLC6A1 variants associated with myoclonic atonic epilepsy.
| Gender/age at inclusion | Family history | Cognition before epilepsy onset | Age at epilepsy onset | Seizure type | EEG | Epilepsy syndrome at diagnosis | Cognition after seizure onset | Behavior problems | Neurological findings | Effective AED | Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 Female/8 yr | None | normal | 2 yr | myoclonic seizure, absence | large amount of slow wave discharges from back of head with small amount of generalize slow wave (2.5–3.5 Hz) | DD/ID | none | normal | VPA | c.46G > T (p.Glu16X) de novo | |
| Patient 2 Male/7 yr | None | DD | 4 yr | myoclonic atonic epilepsy | sharp, spike, and multiple spike and slow wave discharges from occipital, parietal, temporal lobe region (2–4 Hz) | ID | none | normal | VPA | c.1084G > A (Gly362Arg) de novo | |
| Patient 3 Male/3 yr | None | DD | 2 yr | absence seizures, myoclonia, GTCS | asynchronous spike and slow wave from bilateral frontal pole and frontal regions (2–4 Hz) | DD | possible ASD | normal | VPA, LEV | c.1379 T > G (L460R) de novo | |
| Patient 4 Male/4 yr | None | DD | 1 yr | myoclonic-atonic seizures, myoclonia, GTCS | polyspike and wave complexes especially in frontal and occipital regions (2–3.5 Hz) | Doose syndrome | DD | none | hypotonia | VPA, LEV | c.1485G > A (W495X) de novo |
Abbreviations: DD (developmental delay); ID (intellectual disability); GTCS (generalized tonic clonic seizures); ASD (autistic spectrum disorder); VPA (valproic acid); LEV (levetiracetam); AED (antiepileptic drugs).
3.3. Machine learning based protein structural modeling suggests that the two missense variants destabilize the protein conformation
Our previous studies on SLC6A1 variants reported reduced protein stability and enhanced endoplasmic reticulum (ER) associated degradation (ERAD). The GAT-1(E16X) and GAT-1(W495X) variants generate truncated GAT-1 that are usually unstable, consistent with previous findings (Cai et al., 2019; Wang et al., 2020). We then predicted the impact of the two missense variants GAT-1(G362R) and GAT-1(L460R) on the transporter protein stability via several machine learning tools. Homology modeling of the GAT-1(G362R) and GAT-1(L460R) variants in GAT-1 protein (Fig. 2A–B) was conducted using I-TASSER (Zhang, 2008) with homology template PDB ID 4m48. The mutant residue arginine in both locations may trigger several conformational changes on GAT-1 by adding a 3-carbon aliphatic straight chain. Different from previously reported SLC6A1 variants (Cai et al., 2019; Wang et al., 2020), these variants add a positive charge to the side chains, which may disturb the equilibrium of the transmembrane protein conformation and destabilize the protein structure. This destabilization hypothesis is also supported by predicting the ΔΔG of the variant using the machine learning-based protein structure stability prediction methods SDM (Pandurangan et al., 2017), mCSM (Pires et al., 2014a), DUET (Pires et al., 2014b), INPS (Savojardo et al., 2016), DynaMut (Rodrigues et al., 2018) and MAESTROweb (Laimer et al., 2016). As indicated in Fig. 3C–D and Supplementary Table 1, five out of seven tools predicted that the GAT-1(G362R) variant destabilized the GAT-1 protein and six out of seven predicted that GAT-1(L460R) variant destabilized the GAT-1 protein (Supplementary Table 1). Although unpredicted, the variant protein GAT-1(E16X) and GAT-1(W495X) resulting from nonsense variants are also likely destabilized due to the generation of a premature stop codon. Reduced protein stability consequently results in enhanced ERAD.
Fig. 2.
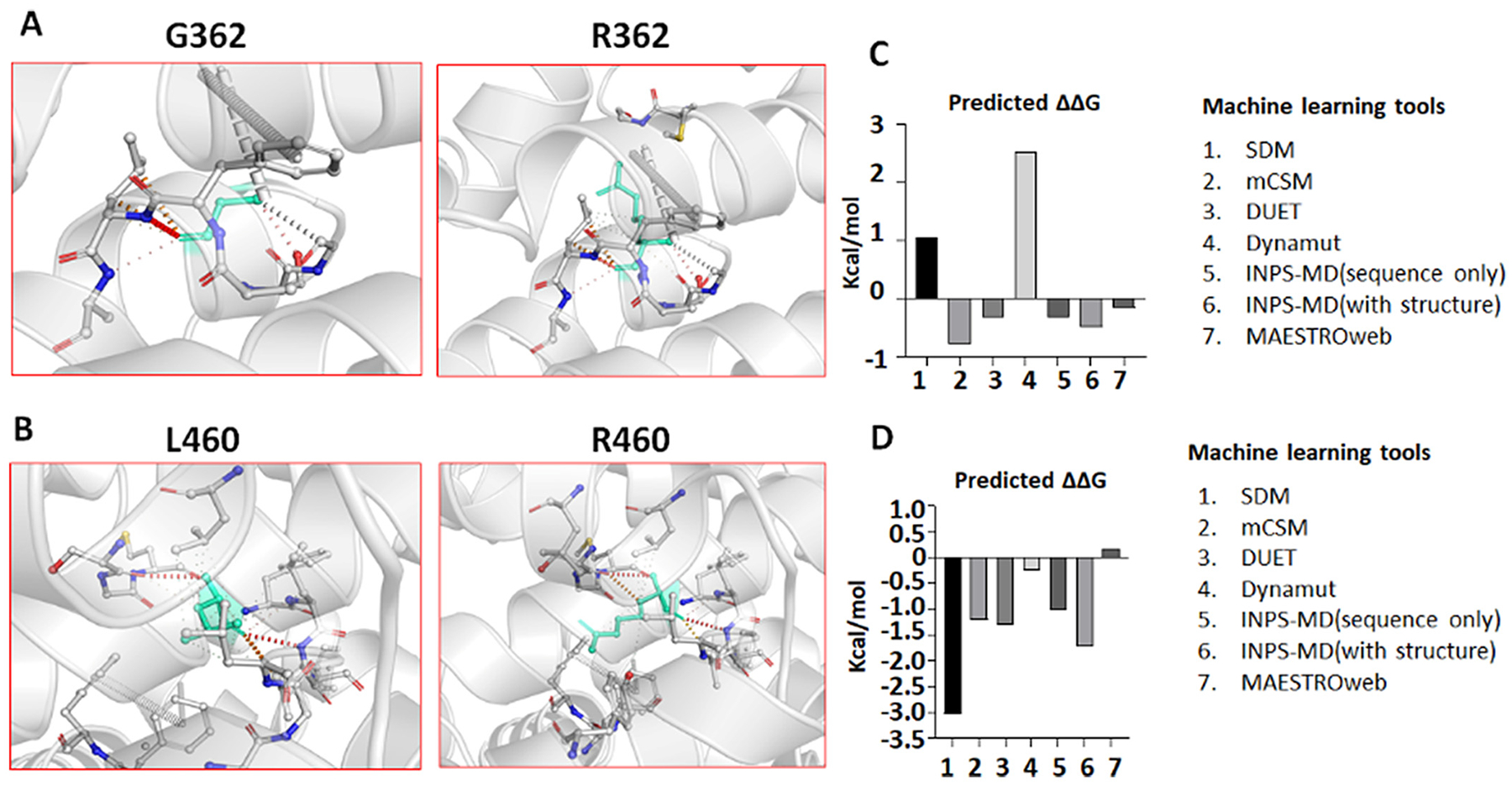
Machine learning tools predict reduced protein stability of the missense variant GAT-1(G362R) and GAT-1(L460R) protein.
(A-B). Tertiary structures of both the wildtype and variant GAT-1 (G362R) (A) or GAT-1(L460R) (B) protein are predicted by I-TASSER and DynaMut. In A, the glycine at residue 362 is mutated to arginine, both highlighted in light green, alongside the surrounding residues. DynaMut predicted the interatomic interactions, where halogen bonds are depicted in blue and hydrogen bonds are in red. In B, leucine at residue 460 is mutated to arginine. (C, D). Machine learning tools predicted ΔΔG (Kcal/mol) of the mutant GAT-1(G362R) or GAT-1(L460R)) protein. Bars in the positive direction are predicted as stabilizing while bars in the negative direction are predicted as destabilizing.
Fig. 3.
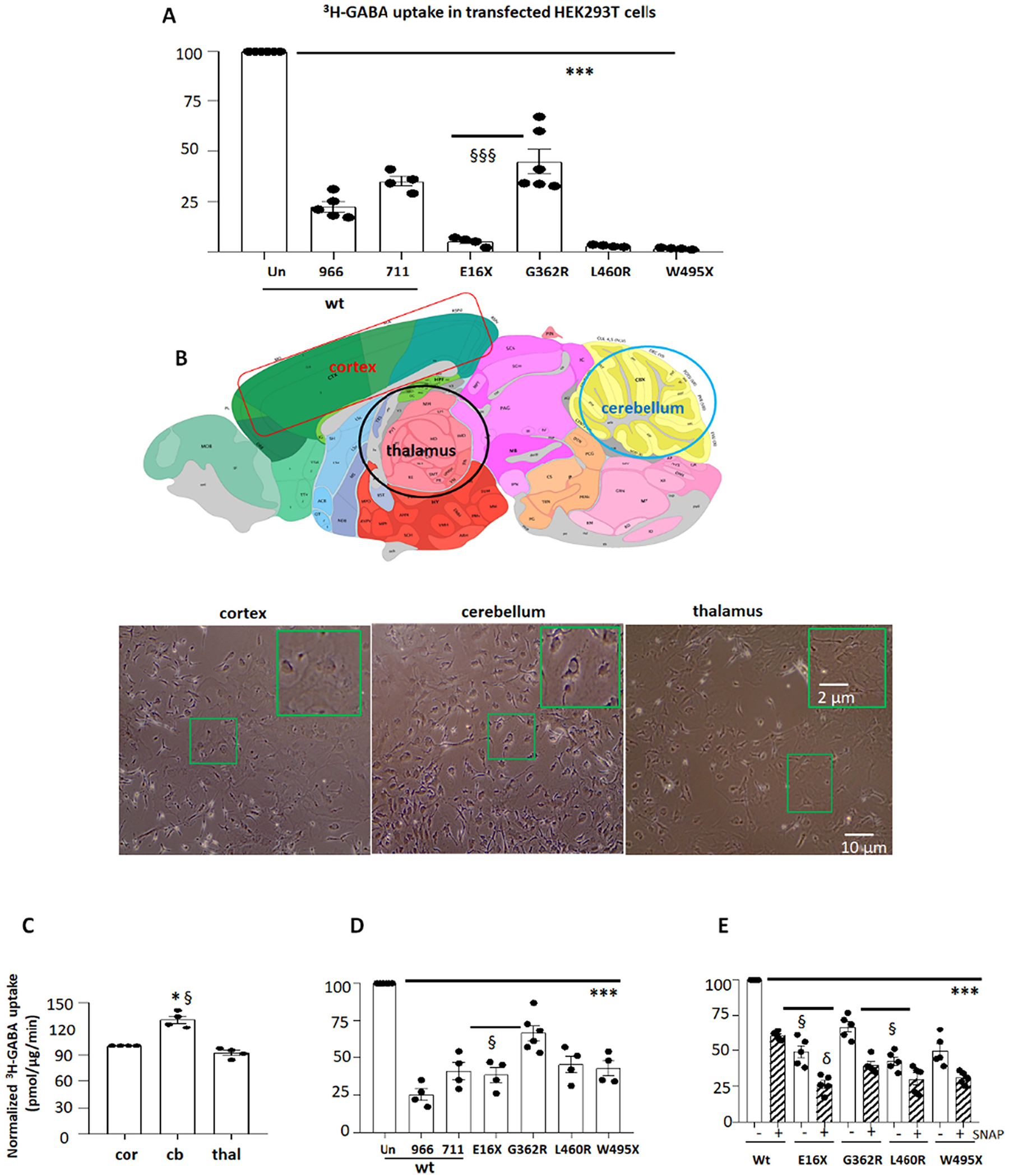
The GABA reuptake function of the variant GAT-1 transporters was reduced in HEK 293 T cells and mouse astrocytes.
(A). HEK293T cells under passage 20 were transfected with wildtype (wt) or the variant GAT-1YFP cDNAs (1 μg per a 35 mm dish) for 48 h before 3H radioactive GABA uptake assay. GAT-1-specific inhibitors Cl-966 (50 μM) and NNC-711 (30 μM) were added during flux time for each experiment. (B). Diagram adapted from the Allen Brain Atlas, demonstrating the areas that were dissected for astrocyte culture. The astrocytes from the cortex, cerebellum, and thalamus of wildtype C57BL/6 J mouse pups at postnatal day 0–3 were prepared and the live cells at passage 2 were imaged before experiment of GABA uptake assay. (C). The graph showing relative GABA uptake assay in astrocytes from cortex, cerebellum and thalamus. The GABA uptake from cerebellum (cb) and thalamus (thal) was normalized to the GABA uptake in the cortex (cor). Mouse cortical (D) or thalamic (E) astrocytes at passage 2 were transfected with wildtype (wt) or the variant GAT-1YFP cDNAs (1 μg per a 35 mm dish) for 48 h before 3H radioactive GABA uptake assay. In C, GABA uptake in astrocytes from the cerebellum and thalamus was normalized to that from the cortex (=100%) each time. In D, GABA uptake in astrocytes expressing the variant or wildtype cDNAs treated with GAT-1 inhibitors Cl-966 or NNC-711 was normalized to the astrocytes expressing the wildtype cDNAs without GAT-1 inhibition. “Un” stands for the astrocytes transfected with the wildtype GAT-1YFP without treatment of GAT-1 inhibitors. (966 stands for Cl-966 while 711 for NNC-711). In E, GABA uptake in astrocytes expressing the variant or wildtype cDNAs treated with GAT-3 inhibitor (SNAP5114 (30 μM)) was normalized to that in astrocytes expressing the wildtype cDNAs without GAT-3 inhibition. GAT-3 inhibitor was applied 1 h before and during uptake assay. In C, D and E, N = 4–5 transfections from 4 batches of cultured astrocytes, ***P < 0.001 vs astrocytes expressing the wildtype GAT-1YFP. In A, §§§P < 0.001 vs G362R. In C, *P < 0.05 vs cortex; §P < 0.05 vs thalamus. In D, §P < 0.05 vs G362R. In E, §P < 0.05 vs G362R without treatment; δ P < 0.05 vs G362R with SNAP5114 treatment. In C, D and E, N = 4–5 experiments, cells were prepared from 3 different litters of mouse pups. One-way analysis of variance (ANOVA) and Newman-Keuls test was used to determine significance compared to the wt condition and between mutations. Values were expressed as mean ± S.E.M.
3.4. All four variants had impaired GABA uptake in HEK293T and mouse astrocytes
We then determined the function of the wildtype and the mutant GAT-1 in HEK cells and cultured mouse cortical astrocytes by radioactive 3H-GABA uptake assay. The measurements in the variant transporter were then normalized to that of the cells expressing the wildtype GAT-1YFP, which was taken as 1. Compared with the wildtype, all four GAT-1 variants had reduced 3H-GABA uptake in HEK293T (wt = 100% vs 5 ± 1.1% for E16X, 44.7 ± 6.1% for G362R; 2.8 ± 0.25% for L460R and 1.5 ± 0.2.% for W495X) (Fig. 3A). We then determined the GABA uptake function of the variant GAT-1 in cultured mouse astrocytes from cortex, cerebellum, and thalamus. We included the cerebellum because some patients with SLC6A1 variants display ataxia (Johannesen et al., 2018). We compared astrocytes cultured from different brain regions such as the cerebral cortex, cerebellum and thalamus (Fig. 3B). GABA uptake in astrocytes cultured from the cortex and thalamus was similar, while the GABA uptake activity in astrocytes culture from the cerebellum was slightly higher (Fig. 3C) (130.7 ± 4% vs 1 for cortex vs 92 ±2.8% for thalamus). We chose to study astrocytes from the cortex and thalamus because of the cortico-thalamic pathway’s involvement in absence seizures. All variants had reduced GABA uptake in cortical astrocytes (wt = 100% vs 38.25 ± 4.86% for E16X, 66.5 ± 5.1% for G362R; 45.25 ± 5.6% for L460R and 42.75 ± 4.9% for W495X) (Fig. 3D). GAT-1 inhibitors Cl-966 (50 μM) and NNC-711 (35 μM) reduced GABA uptake, and, similarly, all four variant GAT-1 showed reduced GABA uptake compared with the astrocytes transfected with wildtype GAT-1YFP in the absence of a GAT-1 inhibitor. However, the extent of reduction was smaller than that in HEK293T cells. This is likely due to the endogenous GAT-1 expression in astrocytes, as some GABA uptake activity in the mutant condition is contributed by the endogenous expression of GAT-1.
Consistent with the findings in cortical astrocytes, the variant GAT-1 had reduced GABA uptake activity in thalamic astrocytes. Because GABA uptake activity is impacted by both GAT-1 and GAT-3, we applied GAT-3 inhibitor SNAP5114 (30 μM) to the cells so the GABA uptake activity would be solely contributed by GAT-1. We tested GABA uptake in thalamic astrocytes cultured from wildtype pups treated with different concentrations of SNAP 5114 including 15 μM, 30 μM, 60 μM, 120 μM. We did not observe any difference of GABA uptake in cells treated with 30 μM, and 120 μM of SNAP5114, s we thus applied 30 μM of SNAP5114 in subsequent experiments. The GAT-3 inhibitor reduced the GABA uptake to ~60% of the untreated in the wildtype. Additionally, when we compared the cells transfected with GAT-1(E16X) YFP and enhanced YFP alone, the GABA uptake activity was comparable in the two conditions (Supplementary Fig. 1). In thalamic astrocytes, regardless of SNAP5114 treatment, the variant conditions showed reduced GABA uptake activity (for thalamic astrocytes without GAT-3 inhibition, wt = 100% vs 49 ± 4.3% for E16X, 67.0 ± 3.7% for G362R; 42.6 ± 2.9% for L460R and 49.8 ± 4.7% for W495X; for thalamic astrocytes with GAT-3 inhibition, wt = 100% vs 26.4 ± 2.7% for E16X, 39.6 ± 2.4% for G362R; 30.00 ± 4.1% for L460R and 30.8 ± 1.9% for W495X) (Fig. 3E). Similar to cortical astrocytes, thalamic astrocytes expressing GAT-1 (G362R) had greater GABA uptake activity than other mutations. SNAP5114 treatment reduced GABA uptake by ~40% in the variant condition, consistent with the observation in the wildtype.
3.5. Variant GAT-1 transporters had reduced GAT-1 total protein expression in both HEK 293 T cells and mouse astrocytes
Machine learning tools suggested that the variant proteins were misfolded and had reduced stability. Reduced protein stability reduces total protein expression and causes reduced GABA uptake. We then determined the total level of variant protein expression in HEK293T cells or mouse cortical astrocytes. In HEK293T cells, the expression of variant GAT-1 proteins is heterogeneous. The wildtype GAT-1YFP protein runs as three bands with a main band of ~108 KDa (band 1) and two small bands of 96 KDa and 90 KDa (band 2 and 3), as previously reported (Cai et al., 2019; Wang et al., 2020; Cai et al., 2005). In the untreated GAT-1(W495X), two small bands with lower molecular mass were observed. The upper band was likely the ER glycosylated or unglyco-sylated immature species. Since only fully glycosylated protein can reach the cell surface and exert GABA uptake functions, we then treated the protein lysates with Endo-H to determine if the protein maturation is altered. With Endo-H digestion that removes the ER attached glycans, bands 1 and 2 were not shifted because they are mature, in which the glycans were not added in ER. Band 3 in both the wildtype and the variant conditions was shifted to a lower molecular mass because the protein isoform is immature, in which the glycans were added inside ER (Fig. 4A). We included all mature and immature isoforms and quantified the total protein amount. Compared with the wildtype, the GAT-1 (E16X) expression was almost undetectable (0.002 ± 0.0002), and GAT-1(W495X) also had minimal expression (0.096 ± 0.029). GAT-1 (G362R) had reduced expression (0.50 ± 0.07), while GAT-1(L460R) protein expression was unaltered (0.96 ± 0.06). The wildtype was arbitrarily taken as 1 (Fig. 4B U). However, with Endo-H digestion, all the variant proteins including the GAT-1(L460R) had reduced expression (0.39 ± 0.04 for G362R, 0.73 ± 0.06 for L460R and 0.32 ± 0.015 for W495X) (Fig. 5B Endo-H). The variant GAT-1 differentially affected the expression of the wildtype allele (Supplementary Fig. 3).
Fig. 4.
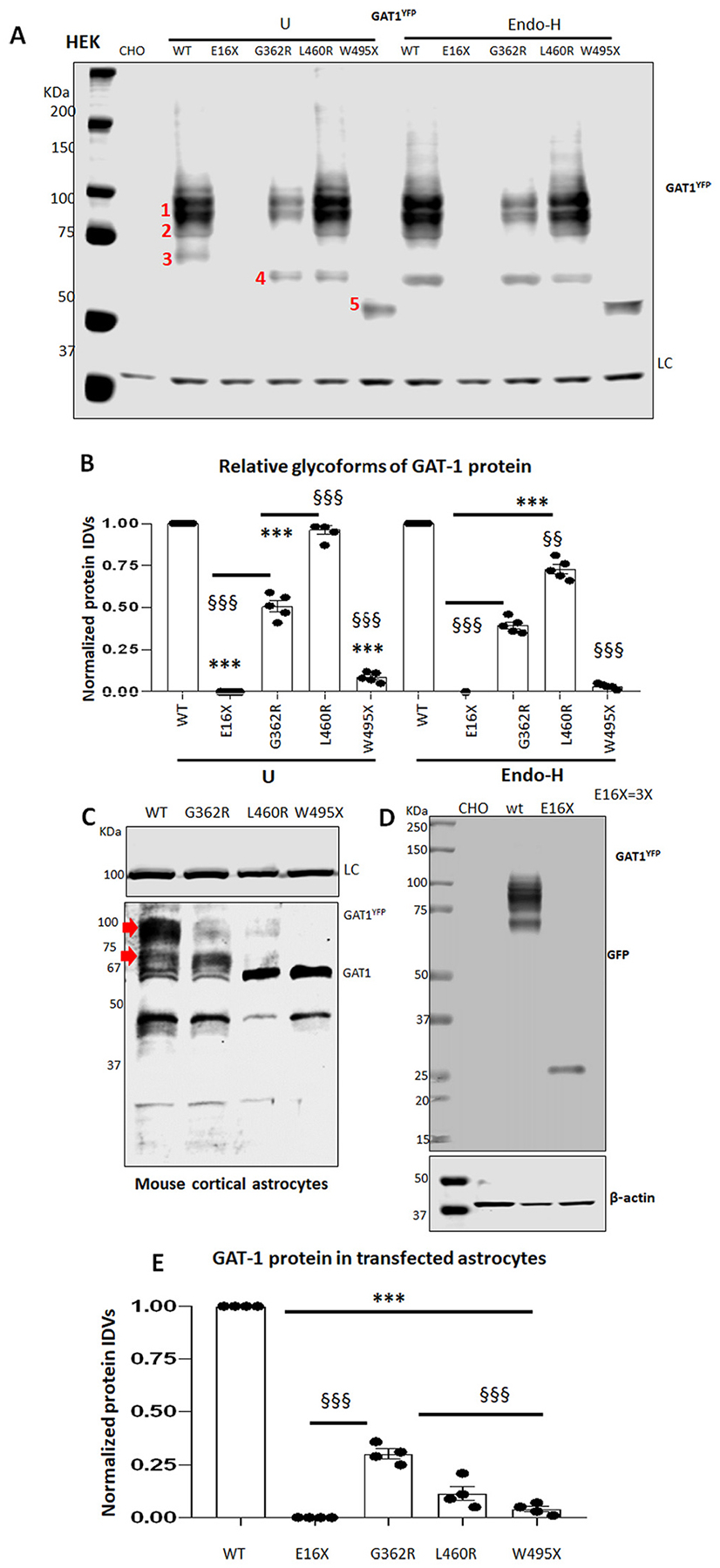
The mature and functional GAT-1 protein was reduced in the variant GAT-1.
HEK293T (A, B) or mouse cortical astrocytes at passage 2 (C, D, E) were transfected with the wildtype or the variant GAT-1YFP cDNAs (3 μg per 60 mm dish or 9 μg per 100 mm dish) for 48 h before harvest. Equal protein amounts were loaded for SDS-PAGE. The membrane was immunoblotted with anti-rabbit GAT-1. In A, protein lysates from HEK293T cells expressing the wildtype and the variant GAT-1 cDNAs were either undigested (U) or digested with Endo-H (H) followed by SDS-PAGE fractionation. Bands 1, 2, 3, 4 and 5 represent the main bands detected in lysates without Endo-H treatment, while band 3 was shifted down to a lower molecular mass than band 4 after Endo-H digestion. To better show both mature and immature bands of GAT-1YFP, a slightly overexposed blot is presented, but note that this has no effect on the quantification data in Image Studio. (B). Graph showing normalized total protein IDVs in HEK293T cells. The variant GAT-1 protein integrated density values (IDVs) were normalized to the wildtype untreated (U) or Endo-H treated (H). In C, Total lysates from mouse cortical astrocytes transfected with wildtype or the variant GAT-1YFP cDNAs (9 μg per a 100 mm dish) for 48 h before harvest were subject to SDS-PAGE. The red arrow pointed bands were quantified. The band run at 67 KDa is likely the endogenous GAT-1 from astrocytes. In D, the protein samples of the wildtype and the GAT-1(E16×) were run separately due to the small protein mass of GAT-1(E16X). The loaded protein amount of the GAT-1E(16X) was tripled for better visualization. The membrane was immunoblotted by a rabbit anti-GAT-1 in A and C and by a mouse anti-GFP in D because GAT-1(E16X) is undetectable by anti-GAT-1 antibody. (E). The total protein IDVs in the mouse cortical astrocytes were plotted. In A and D, CHO stands for Chinese hamster ovary cells. CHO cells were used for loading control because of the low level of endogenous GAT-1 expression (N = 4–5 transfections from 4 to 5 batches of cultured astrocytes, ***P < 0.001 vs astrocytes expressing the wildtype GAT-1YFP; §§§ < 0.001 vs G362R. One-way analysis of variance (ANOVA) and Newman-Keuls test was used to determine significance compared to the wt condition and between variants. Values were expressed as mean ± S.E.M).
Fig. 5.
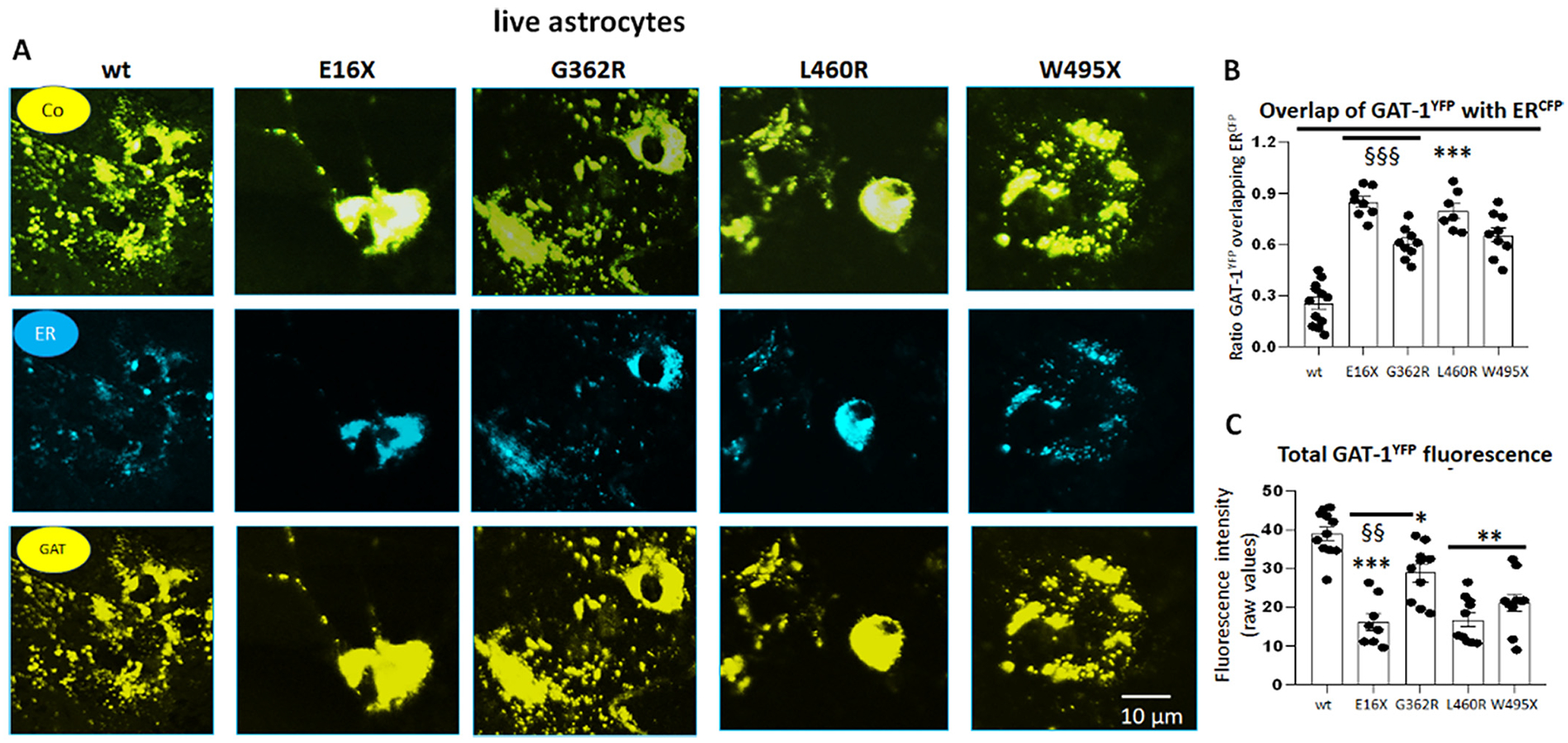
The mutant GAT-1 had reduced total protein expression but increased localization inside the endoplasmic reticulum in astrocytes.
A. Mouse cortical astrocytes under passage 2 were transfected with wildtype GAT-1YFP or the variant GAT-1 (E16X)YFP, GAT-1 (G362R)YFP, GAT-1 (L460R)YFP and GAT-1 (W495X)YFP with the pECFP-ER marker (ERCFP) at a 1:1 ratio (1 μg:1 μg cDNAs) for 48 h. Live cells were examined under a confocal microscopy with excitation at 458 nm for CFP and 514 nm for YFP. All images were single confocal sections averaged from 8 times to reduce noise, except when otherwise specified. “Co” stands for overlay, “ER” stands for endoplasmic reticulum marker (ERCFP) marker, and “GAT” stands for GAT-1YFP. (B). The GAT-1YFP fluorescence overlapping with ERCFP fluorescence was quantified by Metamorph with colocalization percentage. (C). Total fluorescence intensity (raw values) over the area was measured for each whole field and the raw mean values were plotted. In B and C, ***p < 0.001 mutant vs. wt, §§ P < 0.01, §§§ < 0.001 vs G362R. N = 7–8 representative fields from 4 different transfections. One-way analysis of variance (ANOVA) and Newman-Keuls test was used to determine significance compared to the wt condition and between mutations. Values were expressed as mean ± S.E.M.
Consistent with the findings from HEK293T cells, the variant GAT-1 transporters displayed reduced total protein expression (Fig. 4C, D, E) in mouse cortical astrocytes. We could discriminate the GAT-1YFP from the endogenous GAT-1 in astrocytes by the protein’s molecular mass. The recombinant GAT-1YFP is ~27 KDa larger than the endogenous GAT-1 because of the addition of the YFP tag. Because the GAT-1(E16X) could not be detected with GAT-1, we used a GFP antibody to target the YFP tag. Additionally, due to the much smaller size of the truncated protein, GAT-1(E16X) was run separately from the other variants. The magnitude of the variant protein reduction was larger in mouse cortical astrocytes than in HEK293T cells. The variant GAT-1(E16X) and GAT-1 (W495X) proteins were almost undetectable. The total amount of all variant transporter protein was reduced (0.31 ± 0.05 for G362R, and 0.12 ± 0.07 for L460R, and 0.04 ± 0.03 for the wildtype), while the GAT-1(E16X) protein amount was reduced to be undetectable. We then tripled the amount of GAT-1(E16X) protein compared with the wildtype, and a faint band was detected (Fig. 4D). The large reduction of the variant protein in astrocytes is likely due to the relatively low expression of the mutant protein, which can be more efficiently degraded than in HEK293T cells. A similar phenomenon has been reported in a GABRA1 variant associated with childhood absence epilepsy (Kang et al., 2009a). The reduction of the variant GAT-1 was consistent when the variant cDNAs were expressed alone or with the wildtype allele (Supplementary Fig. 3). The variant GAT-1 could cause dominant negative suppression on the wildtype allele as demonstrated in our previous study (Nwosu et al., 2022).
3.6. Variant GAT-1 transporters had increased localization in endoplasmic reticulum in mouse astrocytes
To our knowledge, there is no study on how a variant protein is processed in the ER of astrocytes. We have previously identified that increased ER retention of variant protein due to misfolding and glycosylation arrest (Kang et al., 2009a; Kang et al., 2009b) is a common phenomenon for variants across genes affecting the GABAergic pathway. Those ER retention-prone variant proteins can have either a higher or lower proportion of the total protein level compared with its wildtype counterpart (Kang et al., 2009a; Kang et al., 2009b). To evaluate the subcellular localization of the variant GAT-1, we coexpressed the wildtype GAT-1YFP or the variant GAT-1YFP with an ER marker, ERCFP. When compared to wildtype, each of the four variant GAT-1 transporters had a stronger presence intracellularly, colocalizing with the ER marker (Fig. 5A). The percent fluorescence signal of GAT-1 overlapping with ER marker ERCFP was higher in the variant GAT-1 compared to the wildtype (25.5 ± 3.6% for wt vs 84.88 ± 3.07 for E16X; 60.56 ± 3.03 for G362R; 79.57 ± 4.33 for L460R, and 65.56 ± 4.31 for W495X) (Fig. 5B). The total GAT-1YFP fluorescence signal was lower in cells expressing all mutant GAT-1 transporters (16.24 ± 2.18 for E16X; 29.98 ± 2.27 for G362R; 16.84 ± 1.85 for L460R and 21.22 ± 2.26 for W495X) than in the cells expressing the wildtype (39.02 ± 1.75 for wt) (Fig. 5C). It is worth noting that we compared the subcellular localization of the GAT-1 (E16X)YFP and YFP alone, the protein distribution pattern was indistinguishable between astrocytes expressing the variant GAT-1(E16X)YFP and YFP alone. Interestingly, the variants, especially the GAT-1(E16X) and GAT-1(W495X), had reduced cell numbers with detectable YFP signal (65.4 ± 3.3% for wt, 11 ± 1.5% for E16X, 55.5 ± 6.0% for G362R,52.2 ± 3.7% for L460R and 16.7 ± 2.0% for W495X, Supplementary 2). Together, this indicates that all GAT-1 variants were retained inside the ER, which subsequently caused ER-associated degradation and reduced the total GAT-1 protein level.
3.7. Variant GAT-1 transporters had impaired GABA reuptake in human astrocytes derived from iPSCs
We then determined the function of the wildtype and the variant GAT-1 in human astrocytes using a radioactive 3H GABA uptake assay based on our previous study in human and mouse astrocytes (Mermer et al., 2021). Human astrocytes derived from iPSCs were validated with glial fibrillary acid protein (GFAP) 27 to 30 days after differentiation and were transfected with the wildtype or the variant GAT-1 cDNAs (Fig. 6A). The GABA uptake was measured 48 h later. Compared with the wildtype, all four GAT-1 variants had reduced 3H GABA uptake in human astrocytes (wt = 100% vs 39.67 ± 1.99% for E16X; 58.83 ±4.36% for G362R; 49.33 ± 4.26% for L460R and 49.67 ± 3.9% for W495X) (Fig. 6B). There was robust GABA uptake activity in differentiated astrocytes as demonstrated by the steep decrease in uptake counts when GAT-1 inhibitor Cl-966 (50 μM) and NNC-711 (35 μM) were added. The GABA uptake activity in astrocytes transfected with the wildtype GAT-1YFP was ~1.5–2 fold higher than mock transfected with the empty vector pcDNA. Both the protein expression level of the transfected GAT-1YFP and GABA uptake activity were much lower in astrocytes compared with the HEK293T cells transfected with the same amount of cDNAs.
Fig. 6.
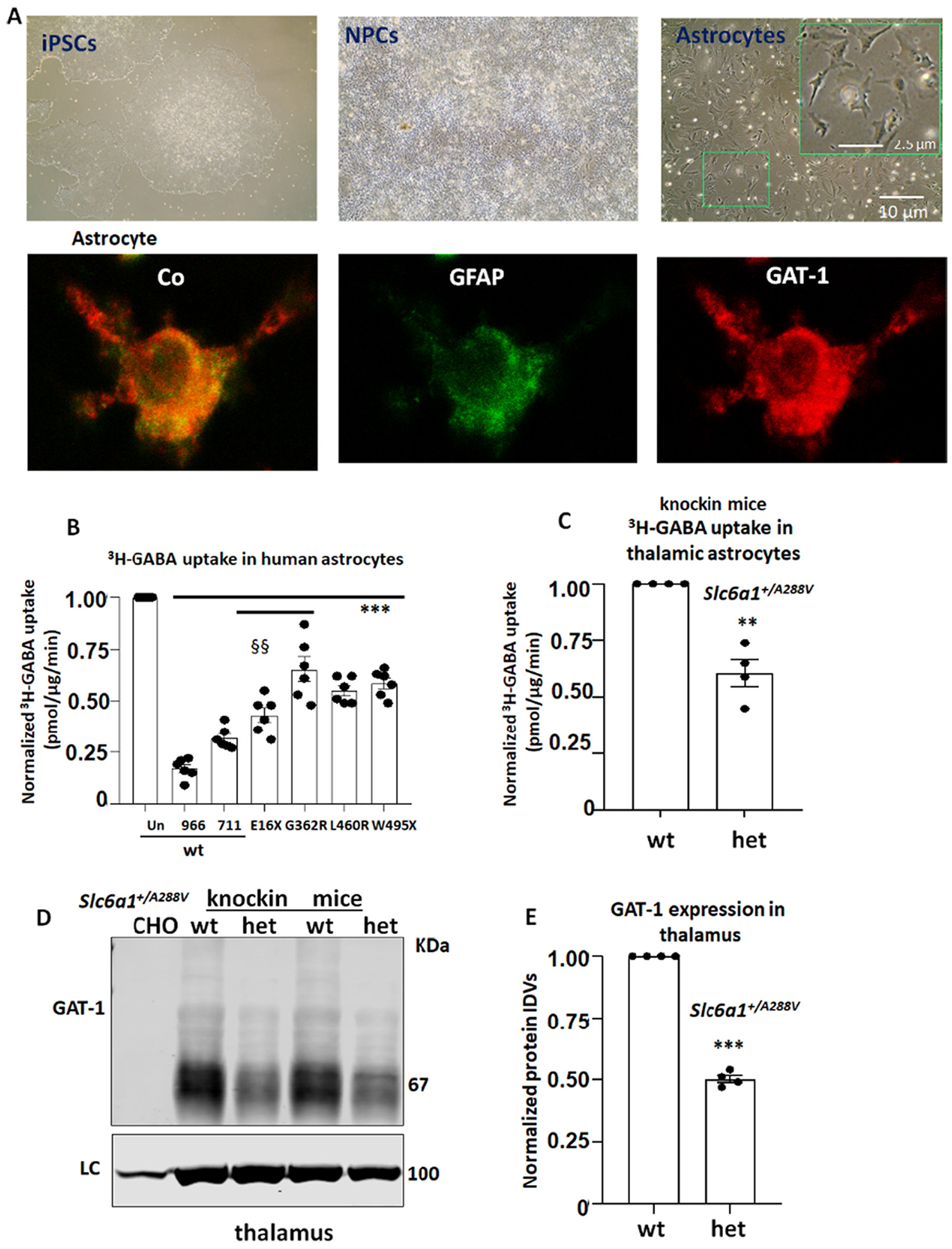
The GABA uptake function of the mutant GAT-1 transporters was reduced in human iPSC derived astrocytes and in the Slc6a1+/A288V mice.
(A). Images showing human induced pluripotent stem cells, neural progenitor cells (NPCs) and the 30th day old astrocytes after differentiation from NPCs. The inserted box shows astrocytes with typical star-like morphology (upper panel). The 30th day old astrocytes were fixed with freshly made 4% Paraformaldehyde (4% PFA) and immuno-stained with mouse monoclonal anti-glial fibrillary acidic protein (GFAP) and rabbit polyclonal anti-GAT-1 antibodies. The mouse IgG was visualized with Alexa 488 and rabbit IgG was visualized with Alexa 555 (Lower panel). (B). Human astrocytes derived from human iPSCs 30 days after differentiation were transfected with the wildtype or the mutant GAT-1YFP cDNAs (1 μg per 35 mm dish) for 48 h before 3H radioactive GABA uptake assay. GABA flux was measured after 30–45 min transport at room temperature. The uptake of GABA, expressed in pmol/μg protein/min, was averaged from duplicates for each condition and for each transfection. The average counting was taken based on normalization to wt = 100%. The pmol/μg protein/min in the variant was then normalized to the wildtype from each experiment, which was arbitrarily set as 100%. ***p < 0.01 vs. wt, §§ P < 0.01 vs G362R, n = 6 different transfections from 4 batches of differentiations. Cl-966 (100 μm) or NNC-77 (70 μm) was applied 30 min before preincubation and removed during preincubation in C, The GABA uptake (pmol/μg protein/min) from thalamic astrocytes cultured from the heterozygous Slc6a1+/A288V pups were normalized to the wildtype (n = 4 pairs of wildtype and heterozygous pups, unpaired t-test). (D, E). Brain tissue from thalamic region was dissected from 2 to 4 months old Slc6a1+/A288V mice. Lysates were analyzed by SDS-PAGE and immunoblotted with a polyclonal anti rabbit GAT-1 (C). The protein IDVs of the heterozygous (het) were normalized to the loading control ATPase and then to the wildtype (wt) (n = 4 pairs of mice) (D). E. In B, C and E, **p < 0.01, ***p < 0.001 variant or het vs. wt, in C, §§ P < 0.01 E16X vs G362R. One-way analysis of variance (ANOVA) and post hoc Newman-Keuls test. Values were expressed as mean ± S.E.M.
3.8. A representative mouse of MAE, Slc6a1+/A288V knockin, had impaired GABA uptake in thalamic astrocytes and increased seizure activity
We then evaluated the total GAT-1 expression in the thalamus of the Slc6a1+/A288V, a novel mouse model of MAE and absence epilepsy. We chose the A288V variant because it has been identified in multiple epilepsy cohorts and thus has been extensively characterized (Carvill et al., 2015; Johannesen et al., 2018; Mermer et al., 2021). Patients heterozygous for the A288V variant display MAE, absence epilepsy, and autism (Seki et al., 2002). We dissected the thalamus from 2 to 4 months old wildtype and Slc6a1+/A288V mice. The total GAT-1 in thalamus was reduced (0.5025 ± 0.015 vs wt = 1, Fig. 6C, D) in the variant heterozygous mice. We cultured the astrocytes from the thalamus and treated the astrocytes with GAT-3 inhibitor (SNAP5114 30 μM) to ensure that only GAT-1 activity was measured. The thalamic astrocytes displayed reduced GABA uptake compared with the wildtype (60.75% ± 6.01% vs wt = 100%, Fig. 6E).
Based on a previous study, impaired GAT-1 in thalamic astrocytes can directly cause absence seizures (Pirttimaki et al., 2013). To confirm this, we conducted video-monitoring synchronized EEG recordings on a novel mouse model Slc6a1+/A288V knockin. We chose the A288V variant because it has been identified in multiple cohorts (Carvill et al., 2015; Johannesen et al., 2018; Mermer et al., 2021). Additionally, we have extensively characterized the variant and identified that the GABA uptake function of GAT-1(A288V) is similar to that of GAT-1(G362R), which is ~30% of the wildtype (Mermer et al., 2021). With Sirenia Seizure Pro software, we measured the occurrence of 5–7 Hz spike-wave discharges (SWDs). We chose to measure 5–7 Hz SWDs because it is the mouse correlate of 2–4 Hz SWDs in humans (Seki et al., 2002). The heterozygous mice had increased 5–7 Hz spike-wave-discharges over 48 h of recordings (69.6 ± 5.30 for het, 2.0 ± 0.71 for wt) and displayed behavioral arrest, suggesting absence seizures (Fig. 7 A, B and C). The detection of a low level of absence-like activity with a short time duration (1.351 ± 0.12 vs 4.50 ± 0.43 s) in the wildtype mice is consistent with a previous study (Arain et al., 2012) and has been commonly observed in studies for other epilepsy mice. The SWDs in the variant mice were much more frequent and with a longer time duration than in the wildtype mice. It is not surprising as we have previously demonstrated that the GABA uptake of the GAT-1(A288V) is only ~30% of the wildtype (Mermer et al., 2021). Together, our data provided evidence that the GAT-1 activity is reduced in thalamus and may contribute to seizures in the Slc6a1+/A288V mice.
Fig. 7.
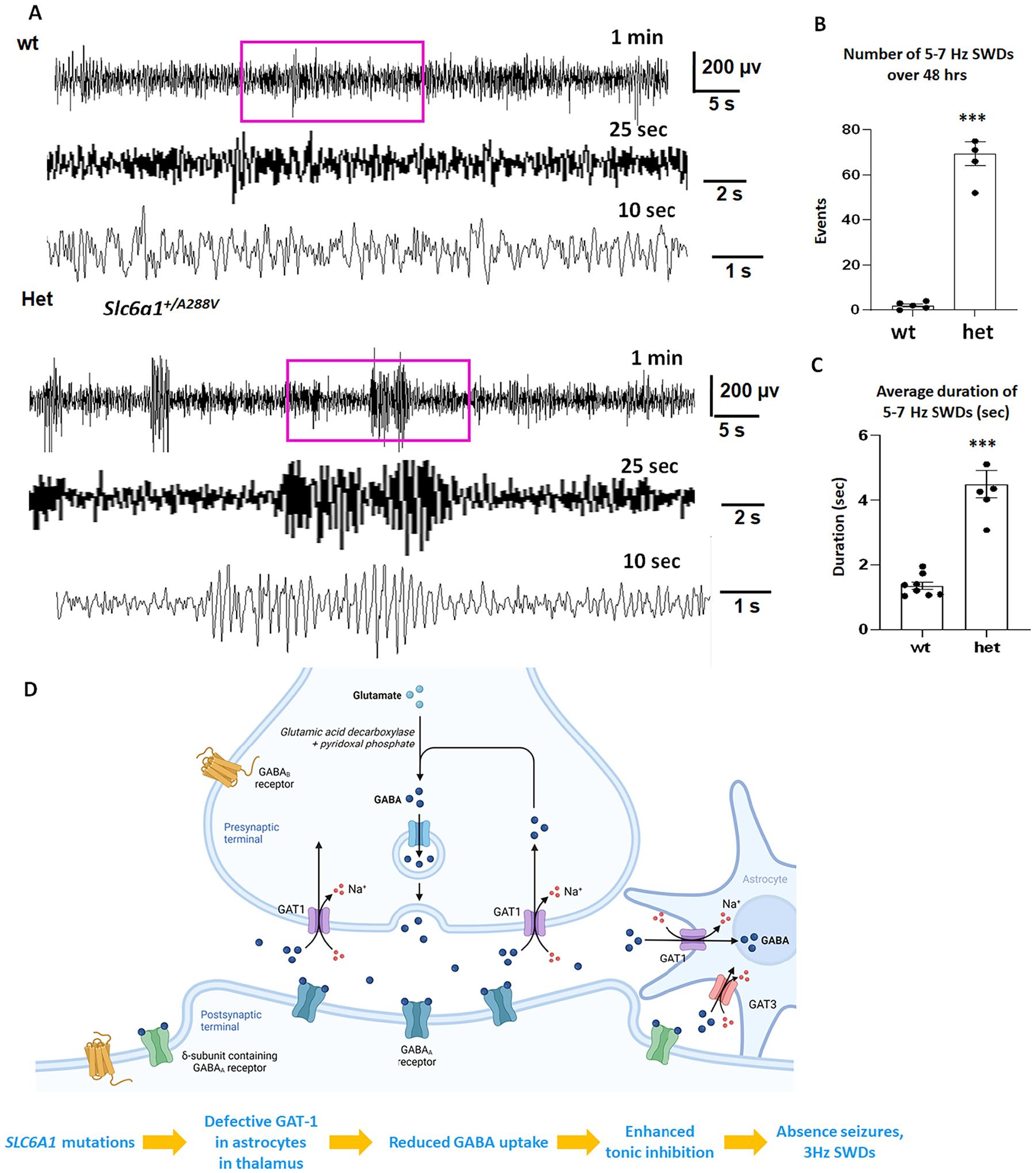
The Slc6a1+/A288V knockin mice had increased 5–7 Hz spike wave discharges and absence seizures.
(A). Representative EEG recordings show the baseline recordings from the wildtype and the heterozygous Slc6a1+/A288V (het) KI mice at 1–2 months old. The EEG traces from Channel 1 were presented. Both the wildtype and the heterozygous mice were awake during that period of recordings, the purple boxed segments were expanded. (B, C). Graph showing the total number of 5–7 Hz SWDs or average seizure duration during 48 h recordings in the wildtype and the het KI mice. ***p <0.001; vs wt, unpaired t-test, N = 8 for wildtype and 6 for heterozygous mice, Values were expressed as mean ± S.E.M. (D). Cartoon illustrates that variant GAT-1 results in reduced GABA uptake function in its host cells including astrocytes. In thalamus, reduced GABA uptake in astrocytes directly leads to increased extracellular GABA, enhanced tonic current and seizures. In summary, defective GAT-1 in the astrocytes could result in increased extracellular GABA and increased tonic inhibition, leading to increased absence seizures and 3 Hz SWDs in patients.
In summary, our data from HEK293T cells, mouse cortical and thalamic astrocytes as well as human astrocytes demonstrated that the variants caused almost a complete loss of function for GAT-1(E16X), GAT-1(L460R) and GAT-1(W495X), and a partial loss of function for GAT-1(G362R). Loss or reduced GAT-1 function in the thalamus may cause increased extracellular GABA levels, thus leading to enhanced tonic inhibition and absence seizures. Absence seizure is a main seizure type and EEG feature of 2–4 Hz is a primary EEG feature in MAE patients associated with SLC6A1 variants. Consistent with the seizure type and EEG feature in patients bearing SLC6A1 variants, the Slc6a1+/A288V mice also had absence seizures as reflected by increased 5–7 Hz SWDs over 48 h of EEG recordings compared with the wildtype littermates (Fig. 7A and B).
As illustrated in the cartoon (Fig. 7C), variants in GAT-1 result in reduced GABA uptake function in astrocytes and neurons. Previous study has demonstrated that GAT-1 is mainly expressed in astrocytes in thalamus (De Biasi et al., 1998) in humans and in rodents. Our data from Slc6a1+/S295L mice indicated that GAT-1 is abundantly expressed in thalamus as in other brain regions such as cortex and hippocampus (Nwosu et al., 2022). Reduced GABA uptake in thalamic astrocytes can directly lead to increased extracellular GABA, enhanced tonic current, seizure activity and increased 3 Hz in humans and 5–7 Hz SWDs in mice.
4. Discussion
4.1. SLC6A1 variants of various genetic abnormalities and in different locations of protein peptide, could result in similar epilepsy phenotypes in patients
SLC6A1 variants are associated with variable epilepsy syndromes and neurodevelopmental disorders. Despite the phenotypic heterogeneity, MAE and intellectual disability (ID) are consistently reported as prominent phenotypes for SLC6A1 variants (Carvill et al., 2015; Johannesen et al., 2018). Here we report four novel variants in unrelated patients of MAE who are heterozygous for SLC6A1 variants, further supporting MAE as a major phenotype for SLC6A1 variants as originally reported (Carvill et al., 2015). The SLC6A1 variants reported here include two nonsense and two missense variants. It is worth noting that the patients with nonsense variants had very similar clinical phenotypes even though the premature stop codons are located at opposite ends of the full-length protein peptide. The truncation at the N terminus removes much of the protein peptide, in contrast to truncation in the C terminus that preserves most of the peptide. It is possible the variant proteins with different lengths of the protein peptide may adopt distinct conformations that differentially affect the remaining wildtype allele and modify the disease phenotype (Johannesen et al., 2018). However, the similar consequence of GABA uptake activity and behavioral phenotypes suggests the variant protein itself from either variant had minimal effect on the remaining wildtype allele function and overall phenotype manifestation. It is likely the MAE phenotype across the variants is caused by GAT-1 haploinsufficiency and GABA uptake deficit.
4.2. Common EEG features of ~3 Hz spike-wave discharges in MAE associated with SLC6A1 variants
All patients were diagnosed with MAE presenting with sudden falls and demonstrated a strong EEG correlate with absence seizure based on previous studies (Goodspeed et al., 2020; Cai et al., 2019; Poliquin et al., 2021). Electrophysiological features are similar to those described in GAT-1 deficient mice (Chiu et al., 2005; Jensen et al., 2003). All patients had 2 to 4 Hz SWDs that were dominant in frontal or centro-occipital regions. It was noticed that the EEG patterns during sleep and wakefulness were different for the patient with the GAT-1(G362R) variant. The seizure activity was responsive to valproic acid treatment alone or in combination with levetiracetam. However, cognition was not improved with seizure control, suggesting that GAT-1 deficiency may directly contribute to impaired cognition, instead of being a consequence of seizure activity. This notion is supported by the fact that some patients with SLC6A1 variants only display neurodevelopmental delay or intellectual disability either without seizures or before seizure onset.
4.3. Machine learning tools identified reduced stability and altered hydrophobicity of the surface of the mutant GAT-1 protein
Based on existing knowledge of thermology effects, we used machine learning based methods to predict the impact of missense variants using the changes of ΔΔG, a metric for measuring how a single point variant alters protein stability. It measures the change in energy between the folded and the unfolded states when a point variant is present. A ΔΔG of less than zero, means the point variant destabilizes the protein. Although the performances of these computational methods still need improvement due to limited training sets and imperfect prediction models, they could give fast and stable predictions without requiring expensive, labor-intensive wet-lab experiments. Our data indicate that the mutant GAT-1 proteins had reduced stability, which is consistent with our biochemical study. Together, this suggests that the GAT-1 variants, regardless of whether nonsense or missense, can destabilize the variant proteins and cause enhanced protein disposal inside the ER, consequently reducing GABA uptake function.
4.4. Reduced GABA reuptake function of SLC6A1 variants in astrocytes
It is believed that GAT-1 is mainly located in the axon and nerve terminals of GABAergic interneurons and in astrocytes (Minelli et al., 1995; Chiu et al., 2005; Conti et al., 1998). At the pre-synaptic terminal, GAT-1 is responsible for the re-uptake of GABA from the synaptic cleft. Although the data of the GABA reuptake from GAT-1 variant knockin mice is not available, it has been reported that in GAT-1-deficient knockout mice (Jensen et al., 2003), GABA reuptake is compromised, resulting in both increased ambient GABA levels and spontaneous spike-wave discharges. Increased ambient GABA would cause enhanced tonic inhibition, which is likely mediated by GABAA receptor δ subunit containing receptors (Cope et al., 2009). We demonstrated that all four variants resulted in reduced GABA reuptake in both human and mouse astrocytes. This is likely due to reduced total amounts of the functional protein in astrocytes (Fig. 6).
4.5. Conserved protein quality control machinery in the ER of the astrocytes and ER-associated degradation of the mutant GAT-1
The loss of GAT-1 function in astrocytes is likely due to the ER retention of GAT-1. ER retention and enhanced degradation is likely a common molecular mechanism for many pathological variants. The findings were similar to the expression of the wildtype GAT-1 treated with tunicamycin, an ER stress inducer, in which GAT-1 was subject to ER retention and reduced presence at the cell surface (Wang et al., 2020). Misfolded GAT-1 will be subject to enhanced degradation without further translocation to other cellular compartments or the cell surface (Kang et al., 2015; Kang et al., 2009b). Our data indicate that the astrocytes have conserved protein quality control and the mutant protein was likely subject to ER retention and associated degradation.
4.6. Defective GAT-1 in astrocytes in the thalamus can cause enhanced tonic inhibition
We demonstrated that the mutant GAT-1 caused reduced GABA uptake in both cortical and thalamic astrocytes. It is known that the thalamus is a rhythmogenic structure, which is responsible for generating and maintaining oscillatory activity. Oscillatory activity underlies key brain functions such as sleep, sensation, perception and consciousness (Gent et al., 2018) while increased oscillation has been well documented in epilepsy including the Gabrg2+/Q390X mouse model of epilepsy (Huang et al., 2017). Astrocytic GAT-1 deficit could directly cause enhanced tonic inhibition because of the excessive extracellular GABA. Enhanced tonic inhibition in the thalamus has been shown to cause absence epilepsy and 5–7 Hz SWDs in multiple animal models. The 5–7 Hz SWDs in rodents are equivalent to the 2–4 Hz SWDs in human epilepsy patients. Previous studies have attributed a dysfunction of the astrocytic GAT-1 as the basis of absence seizures in experimental animals (Pirttimaki et al., 2013). Our finding of GAT-1 deficiency in thalamic astrocytes may explain the EEG activity and seizures in SLC6A1 mutation patients. As mentioned above, in the thalamus of humans and rodents, GAT-1 is mainly expressed in astrocytes (De Biasi et al., 1998). This study thus provides direct evidence of GAT-1 deficiency in thalamic astrocytes, a likely cause for seizure genesis in patients.
Our data is consistent with previous studies from multiple models of absence seizures (Cope et al., 2009) and suggests that GAT-1 functional deficit in astrocytes may result in enhanced tonic inhibition in SLC6A1 variants associated epilepsy. Multiple lines of evidence from protein trafficking, expression, ER retention and transporter channel function suggest that loss-of-function – not gain of function – is likely the major mechanism for epilepsy associated with SLC6A1 variants.
4.7. GAT-1 functional deficit in astrocytes may contribute to seizure phenotypes in SLC6A1 variant-mediated disorders
It is known that GAT-1 is expressed in the nerve endings of GABAergic interneurons and astrocytes. We have previously compared the variant GAT-1 in both neurons and astrocytes (Mermer et al., 2021) and believe the variant protein metabolism is conserved across cell types. This is especially important given GAT-1 is expressed in both neurons and astrocytes. It is established that GAT-1 inhibition causes enhanced tonic inhibition (Cope et al., 2009; Crunelli et al., 2012). In contrast, enhanced astroglial GABA uptake attenuates tonic GABAA inhibition. The changes of GABA uptake, GABA release, and GABAA receptor inhibition could eventually lead to epileptogenesis (Brooks-Kayal et al., 1998; During et al., 1995; Semyanov et al., 2003; Borg et al., 1995). Along the same line, it has been reported that GAT-1 currents are reduced in a rat model of absence seizures (Pirttimaki et al., 2013). We thus speculate that defective GAT-1 in astrocytes contributes to reduced GABA reuptake, making the brain prone to seizure generation.
However, GAT-1 is expressed in both neurons and astrocytes, and the GAT-1 deficit in neurons could also contribute to disease phenotype. Additionally, the seizures we observed in the Slc6a1+/A288V mice and in the clinical EEG in patients may not specifically represent MAE, but rather a wide spectrum of disease phenotypes. Indeed, the GAT-1 (A288V) mutation in patients is not only associated with MAE, but is also associated with CAE, autism, and neurodevelopmental delay. Furthermore, other factors such as sex and GABAB receptors may also contribute to the disease pathophysiology and phenotype. It is well demonstrated that blocking either GAT-1 or GAT-3 can prolong oscillations (Lu et al., 2020) and enhanced GABAB receptor-mediated signaling is critical for generation of thalamocortical seizures. It is likely that GABAB receptor signaling is augmented by GAT-1 inhibition due to mutant GAT-1 during a chronic disease course such as in SLC6A1 variant-mediated disorders, thus potentiating absence-like epileptiform oscillations in the thalamus (Lu et al., 2020; Dodgson and Watford, 1990).
In summary, this study has reported novel SLC6A1 variants associated with MAE and the related molecular defects in the physiologically relevant cell model, thalamic astrocytes. All patients displayed 2 to 4 Hz SWDs. All four variants result in reduced GABA uptake of the mutant GAT-1 in HEK293T, mouse and human astrocytes due to reduced protein stability and expression. This study, in combination with previous studies (Mattison et al., 2018; Cai et al., 2019; Wang et al., 2020), demonstrates that partial or complete loss-of-function is a major mechanism for SLC6A1 variants mediated epilepsy as well as other disease phenotypes. Astrocytic GAT-1 functional deficiency in the thalamus may contribute to seizure generation and 2–4 Hz SWDs in EEG as identified in a rat model of absence seizures (Pirttimaki et al., 2013). The study identified an apparently converging pathology between that previously characterized for absence seizures in experimental animal models and absence epilepsy and MAE mediated by SLC6A1 variants (Lee et al., 2013; Dlugos et al., 2013; Gencpinar et al., 2016).
Supplementary Material
Acknowledgements
We would like to thank the patients and their families who participated in this study for their cooperation. We are very grateful for Dr. Vivian Gama’s lab and Mr. Anuj Rastogi for consultation on human astrocyte culture and differentiation.
Funding
The study on functional assay was conducted at Vanderbilt University Medical Center and supported by research grants from SLC6A1 Connect, NINDS R01 082635 and R01 121718 (to K.J.Q.). The study on structural modeling was supported by the United States of America National Institutes of Health (grant No. R35-GM126985 to DX). Imaging data were performed in part through the VUMC Cell Imaging Shared Resource.
The funders had no role in study design, data collection and analysis, and interpretation of data, decision to publish or preparation of the manuscript.
Footnotes
CRediT authorship contribution statement
Felicia Mermer: Investigation. Sarah Poliquin: Investigation, Writing – review & editing. Shuizhen Zhou: Investigation. Xiaodong Wang: Investigation. Yifeng Ding: Investigation. Fei Yin: Investigation. Wangzhen Shen: Investigation. Juexin Wang: Software. Kathryn Rigsby: Investigation. Dong Xu: Funding acquisition, Software. Taralynn Mack: Investigation. Gerald Nwosu: Investigation. Carson Flamm: Investigation. Matthew Stein: Investigation. Jing-Qiong Kang: Funding acquisition, Investigation, Writing – original draft, Writing – review & editing.
Declaration of Competing Interest
None.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2022.105810.
References
- Arain FM, Boyd KL, Gallagher MJ, 2012. Decreased viability and absence-like epilepsy in mice lacking or deficient in the GABAA receptor α1 subunit. Epilepsia. 53 (8), e161–e165. 10.1111/j.1528-1167.2012.03596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg WP, Sherwin RS, During MJ, Borg MA, Shulman GI, 1995. Local ventromedial hypothalamus glucopenia triggers counterregulatory hormone release. Diabetes. 44 (2), 180–184. 10.2337/diab.44.2.180. [DOI] [PubMed] [Google Scholar]
- Bresnahan R, Martin-McGill KJ, Hutton JL, Marson AG, 2019. Tiagabine add-on therapy for drug-resistant focal epilepsy. Cochrane Database Syst. Rev (10), CD001908. 10.1002/14651858.CD001908.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA, 1998. Selective changes in single cell GABA(A) receptor subunit expression and function in temporal lobe epilepsy. Nat. Med 4 (10), 1166–1172. 10.1038/2661. [DOI] [PubMed] [Google Scholar]
- Cai G, Salonikidis PS, Fei J, et al. , 2005. The role of N-glycosylation in the stability, trafficking and GABA-uptake of GABA-transporter 1. Terminal N-glycans facilitate efficient GABA-uptake activity of the GABA transporter. FEBS J. 272 (7), 1625–1638. 10.1111/j.1742-4658.2005.04595.x. [DOI] [PubMed] [Google Scholar]
- Cai K, Wang J, Eissman J, et al. , 2019. A missense mutation in SLC6A1 associated with Lennox-Gastaut syndrome impairs GABA transporter 1 protein trafficking and function. Exp. Neurol 320, 112973 10.1016/j.expneurol.2019.112973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvill GL, McMahon JM, Schneider A, et al. , 2015. Mutations in the GABA transporter SLC6A1 cause epilepsy with myoclonic-atonic seizures. Am. J. Hum. Genet 96 (5), 808–815. 10.1016/j.ajhg.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CS, Brickley S, Jensen K, et al. , 2005. GABA transporter deficiency causes tremor, ataxia, nervousness, and increased GABA-induced tonic conductance in cerebellum. J. Neurosci 25 (12), 3234–3245. 10.1523/JNEUROSCI.3364-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti F, Melone M, De Biasi S, Minelli A, Brecha NC, Ducati A, 1998. Neuronal and glial localization of GAT-1, a high-affinity gamma-aminobutyric acid plasma membrane transporter, in human cerebral cortex: with a note on its distribution in monkey cortex. J. Comp. Neurol 396 (1), 51–63. . [DOI] [PubMed] [Google Scholar]
- Cope DW, Di Giovanni G, Fyson SJ, et al. , 2009. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat. Med 15 (12), 1392–1398. 10.1038/nm.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Leresche N, Cope DW, 2012. GABA-A receptor function in typical absence seizures. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV (Eds.), Jasper’s Basic Mechanisms of the Epilepsies, 4th ed. National Center for Biotechnology Information (US). Accessed November 18, 2021. http://www.ncbi.nlm.nih.gov/books/NBK98213/. [PubMed] [Google Scholar]
- Crunelli V, Lőrincz ML, McCafferty C, et al. , 2020. Clinical and experimental insight into pathophysiology, comorbidity and therapy of absence seizures. Brain. 143 (8), 2341–2368. 10.1093/brain/awaa072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Biasi S, Vitellaro-Zuccarello L, Brecha NC, 1998. Immunoreactivity for the GABA transporter-1 and GABA transporter-3 is restricted to astrocytes in the rat thalamus. A light and electron-microscopic immunolocalization. Neuroscience. 83 (3), 815–828. 10.1016/s0306-4522(97)00414-4. [DOI] [PubMed] [Google Scholar]
- Dlugos D, Shinnar S, Cnaan A, et al. , 2013. Pretreatment EEG in childhood absence epilepsy: associations with attention and treatment outcome. Neurology. 81 (2), 150–156. 10.1212/WNL.0b013e31829a3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodgson SJ, Watford M, 1990. Mar. Differential regulation of hepatic carbonic anhydrase isozymes in the streptozotocin-diabetic rat. Arch. Biochem. Biophys 277 (2), 410–414. 10.1016/0003-9861(90)90597-r. [DOI] [PubMed] [Google Scholar]
- During MJ, Ryder KM, Spencer DD, 1995. Hippocampal GABA transporter function in temporal-lobe epilepsy. Nature. 376 (6536), 174–177. 10.1038/376174a0. [DOI] [PubMed] [Google Scholar]
- Fattorini G, Melone M, Conti F, 2020. A reappraisal of GAT-1 localization in neocortex. Front. Cell. Neurosci 14, 9. 10.3389/fncel.2020.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gencpinar P, Kalay Z, Turgut S, et al. , 2016. Evaluation of executive functions in patients with childhood absence epilepsy. J. Child Neurol 31 (7), 824–830. 10.1177/0883073815623632. [DOI] [PubMed] [Google Scholar]
- Gent TC, Bandarabadi M, Herrera CG, Adamantidis AR, 2018. Thalamic dual control of sleep and wakefulness. Nat. Neurosci 21 (7), 974–984. 10.1038/s41593-018-0164-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodspeed K, Pérez-Palma E, Iqbal S, et al. , 2020. Current knowledge of SLC6A1-related neurodevelopmental disorders. Brain Commun 2 (2), fcaa170 10.1093/braincomms/fcaa170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Zhou C, Tian M, et al. , 2017. Overexpressing wild-type γ2 subunits rescued the seizure phenotype in Gabrg2(+/Q390X) Dravet syndrome mice. Epilepsia. 58 (8), 1451–1461. 10.1111/epi.13810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K, Chiu CS, Sokolova I, Lester HA, Mody I, 2003. GABA transporter-1 (GAT1)-deficient mice: differential tonic activation of GABAA versus GABAB receptors in the hippocampus. J. Neurophysiol 90 (4), 2690–2701. 10.1152/jn.00240.2003. [DOI] [PubMed] [Google Scholar]
- Johannesen K, Marini C, Pfeffer S, et al. , 2016. Phenotypic spectrum of GABRA1: from generalized epilepsies to severe epileptic encephalopathies. Neurology. 87 (11), 1140–1151. 10.1212/WNL.0000000000003087. [DOI] [PubMed] [Google Scholar]
- Johannesen KM, Gardella E, Linnankivi T, et al. , 2018. Defining the phenotypic spectrum of SLC6A1 mutations. Epilepsia. 59 (2), 389–402. 10.1111/epi.13986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Kang J, Macdonald RL, 2004. The GABAA receptor gamma2 subunit R43Q mutation linked to childhood absence epilepsy and febrile seizures causes retention of alpha1beta2gamma2S receptors in the endoplasmic reticulum. J. Neurosci 24 (40), 8672–8677. 10.1523/JNEUROSCI.2717-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Macdonald RL, 2009a. Two molecular pathways (NMD and ERAD) contribute to a genetic epilepsy associated with the GABAA receptor GABRA1 PTC mutation, 975delC, S326fs328X. J. Neurosci 29 (9), 2833–2844. 10.1523/JNEUROSCI.4512-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Macdonald RL, 2009b. The GABRG2 mutation, Q351X, associated with generalized epilepsy with febrile seizures plus, has both loss of function and dominant-negative suppression. J. Neurosci 29 (9), 2845–2856. 10.1523/JNEUROSCI.4772-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Zhou C, Xu D, Macdonald RL, 2015. The human epilepsy mutation GABRG2(Q390X) causes chronic subunit accumulation and neurodegeneration. Nat. Neurosci 18 (7), 988–996. 10.1038/nn.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laimer J, Hiebl-Flach J, Lengauer D, Lackner P, 2016. MAESTROweb: a web server for structure-based protein stability prediction. Bioinformatics. 32 (9), 1414–1416. 10.1093/bioinformatics/btv769. [DOI] [PubMed] [Google Scholar]
- Lee S, Kruglikov I, Huang ZJ, Fishell G, Rudy B, 2013. A disinhibitory circuit mediates motor integration in the somatosensory cortex. Nat. Neurosci 16 (11), 1662–1670. 10.1038/nn.3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu AC, Lee CK, Kleiman-Weiner M, Truong B, Wang M, Huguenard JR, Beenhakker MP, 2020. Sep 9. Nonlinearities between inhibition and T-type calcium channel activity bidirectionally regulate thalamic oscillations. Elife. 9, e59548 10.7554/eLife.59548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattison KA, Butler KM, Inglis GAS, et al. , 2018. SLC6A1 variants identified in epilepsy patients reduce γ-aminobutyric acid transport. Epilepsia. 59 (9), e135–e141. 10.1111/epi.14531. [DOI] [PubMed] [Google Scholar]
- Mermer F, Poliquin S, Rigsby K, et al. , 2021. Common molecular mechanisms of SLC6A1 variant-mediated neurodevelopmental disorders in astrocytes and neurons. Brain. 144 (8), 2499–2512. 10.1093/brain/awab207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minelli A, Brecha NC, Karschin C, DeBiasi S, Conti F, 1995. GAT-1, a high-affinity GABA plasma membrane transporter, is localized to neurons and astroglia in the cerebral cortex. J. Neurosci 15 (11), 7734–7746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minelli A, DeBiasi S, Brecha NC, Zuccarello LV, Conti F, 1996. GAT-3, a high-affinity GABA plasma membrane transporter, is localized to astrocytic processes, and it is not confined to the vicinity of GABAergic synapses in the cerebral cortex. J. Neurosci 16 (19), 6255–6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwosu G, Mermer F, Flamm C, et al. , 2022. 4-Phenylbutyrate restored γ-aminobutyric acid uptake and reduced seizures in SLC6A1 patient variant-bearing cell and mouse models. Brain Commun. 4 (3), fcac144 doi.org/ 10.1093/braincomms/fcac144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandurangan AP, Ochoa-Montaño B, Ascher DB, Blundell TL, 2017. SDM: a server for predicting effects of mutations on protein stability. Nucleic Acids Res. 45 (W1), W229–W235. 10.1093/nar/gkx439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires DEV, Ascher DB, Blundell TL, 2014a. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics. 30 (3), 335–342. 10.1093/bioinformatics/btt691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires DEV, Ascher DB, Blundell TL, 2014b. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 42 (Web Server issue), W314–W319. 10.1093/nar/gku411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirttimaki T, Parri HR, Crunelli V, 2013. Astrocytic GABA transporter GAT-1 dysfunction in experimental absence seizures. J. Physiol 591 (4), 823–833. 10.1113/jphysiol.2012.242016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliquin S, Hughes I, Shen W, et al. , 2021. Genetic mosaicism, intrafamilial phenotypic heterogeneity, and molecular defects of a novel missense SLC6A1 mutation associated with epilepsy and ADHD. Exp. Neurol 342, 113723 10.1016/j.expneurol.2021.113723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribak CE, Tong WM, Brecha NC, 1996. GABA plasma membrane transporters, GAT-1 and GAT-3, display different distributions in the rat hippocampus. J. Comp. Neurol 367 (4), 595–606. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, et al. , 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 17 (5), 405–424. 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues CH, Pires DE, Ascher DB, 2018. DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 46 (W1), W350–W355. 10.1093/nar/gky300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Morales AI, Robertson GL, Rastogi A, et al. , 2021. Human IPSC-Derived Cerebral Organoids Model Features of Leigh Syndrome and Reveal Abnormal Corticogenesis. 10.1101/2020.04.21.054361, 2020.04.21.054361. [DOI] [PMC free article] [PubMed]
- Savojardo C, Fariselli P, Martelli PL, Casadio R, 2016. INPS-MD: a web server to predict stability of protein variants from sequence and structure. Bioinformatics. 32 (16), 2542–2544. 10.1093/bioinformatics/btw192. [DOI] [PubMed] [Google Scholar]
- Scholze P, Freissmuth M, Sitte HH, 2002. Mutations within an intramembrane leucine heptad repeat disrupt oligomer formation of the rat GABA transporter 1. J. Biol. Chem 277 (46), 43682–43690. 10.1074/jbc.M205602200. [DOI] [PubMed] [Google Scholar]
- Seki T, Matsubayashi H, Amano T, et al. , 2002. Adenoviral gene transfer of aspartoacylase into the tremor rat, a genetic model of epilepsy, as a trial of gene therapy for inherited epileptic disorder. Neurosci. Lett 328 (3), 249–252. 10.1016/s0304-3940(02)00522-0. [DOI] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM, 2003. GABA uptake regulates cortical excitability via cell type-specific tonic inhibition. Nat. Neurosci 6 (5), 484–490. 10.1038/nn1043. [DOI] [PubMed] [Google Scholar]
- Walker MC, Kullmann DM, 2012. Tonic GABAA Receptor-mediated signaling in epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV (Eds.), Jasper’s Basic Mechanisms of the Epilepsies, 4th ed. National Center for Biotechnology Information (US). Accessed November 18, 2021. http://www.ncbi.nlm.nih.gov/books/NBK98181/. [PubMed] [Google Scholar]
- Walsh I, Seno F, Tosatto SCE, Trovato A, 2014. PASTA 2.0: an improved server for protein aggregation prediction. Nucleic Acids Res. 42 (Web Server issue), W301–W307. 10.1093/nar/gku399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Poliquin S, Mermer F, et al. , 2020. Endoplasmic reticulum retention and degradation of a mutation in SLC6A1 associated with epilepsy and autism. Mol. Brain 13 (1), 76. 10.1186/s13041-020-00612-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner TA, Shen W, Huang X, Liu Z, Macdonald RL, Kang JQ, 2016. Differential molecular and behavioural alterations in mouse models of GABRG2 haploinsufficiency versus dominant negative mutations associated with human epilepsy. Hum. Mol. Genet 25 (15), 3192–3207. 10.1093/hmg/ddw168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner TA, Liu Z, Macdonald RL, Kang JQ, 2017. Heat induced temperature dysregulation and seizures in Dravet syndrome/GEFS+ Gabrg2(+/Q390X) mice. Epilepsy Res. 134, 1–8. 10.1016/j.eplepsyres.2017.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 9, 40. 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.