

ABSTRACT
The mechanism by which avian reovirus (ARV)-modulated suppression of mTORC1 triggers autophagy remains largely unknown. In this work, we determined that p17 functions as a negative regulator of mTORC1. This study suggest novel mechanisms whereby p17-modulated inhibition of mTORC1 occurs via upregulation of p53, inactivation of Akt, and enhancement of binding of the endogenous mTORC1 inhibitors (PRAS40, FKBP38, and FKPP12) to mTORC1 to disrupt its assembly and accumulation on lysosomes. p17-modulated inhibition of Akt leads to activation of the downstream targets PRAS40 and TSC2, which results in mTORC1 inhibition, thereby triggering autophagy and translation shutoff, which is favorable for virus replication. p17 impairs the interaction of mTORC1 with its activator Rheb, which promotes FKBP38 interaction with mTORC1. It is worth noting that p17 activates ULK1 and Beclin1 and increases the formation of the Beclin 1/class III PI3K complex. These effects could be reversed in the presence of insulin or depletion of p53. Furthermore, we found that p17 induces autophagy in cancer cell lines by upregulating the p53/PTEN pathway, which inactivates Akt and mTORC1. This study highlights p17-modulated inhibition of Akt and mTORC1, which triggers autophagy and translation shutoff by positively modulating the tumor suppressors p53 and TSC2 and endogenous mTORC1 inhibitors.
IMPORTANCE The mechanisms by which p17-modulated inhibition of mTORC1 induces autophagy and translation shutoff is elucidated. In this work, we determined that p17 serves as a negative regulator of mTORC1. This study provides several lines of conclusive evidence demonstrating that p17-modulated inhibition of mTORC1 occurs via upregulation of the p53/PTEN pathway, downregulation of the Akt/Rheb/mTORC1 pathway, enhancement of binding of the endogenous mTORC1 inhibitors to mTORC1 to disrupt its assembly, and suppression of mTORC1 accumulation on lysosomes. This work provides valuable information for better insights into p17-modulated inhibition of mTORC1, which induces autophagy and translation shutoff to benefit virus replication.
KEYWORDS: avian reovirus, p17, p53, Akt, PRAS40, TSC2, Rheb, mTORC1, autophagy, translation shutoff
INTRODUCTION
The mammalian target of rapamycin (mTOR) forms two functionally different complexes termed mTORC1 and mTORC2 (1) and modulates many signaling pathways, thereby promoting tumorigenesis via the coordinated phosphorylation of its target proteins that directly regulate translation, cell cycle, cell growth, and proliferation (2–4). mTORC1 consists of a complex that includes mTOR and raptor, whereas mTORC2 is composed of a complex that includes mTOR and rictor (rapamycin-insensitive companion) (3, 4). Both mTORC1 and mTORC2 are activated by growth factors, including insulin, insulin-like growth factor 1, and others (5–7). Growth factors activate mTORC1 through class I phosphatidylinositol 3-kinase (PI3K), PDK1, Akt, TSC1/TSC2 complex, and Rheb (Ras homolog enriched in brain) (8). Akt inhibits several host proteins by phosphorylation/inactivation of these downstream targets such as PRAS40, TSC2, glycogen synthase kinase-3α/β (GSK3α/β), and forkhead transcription factor class O1/3a (FoxO1/3a). Akt activates mTORC1 through multisite phosphorylation of TSC2 within the TSC1/TSC2 complex (9–11), and this inhibits the ability of TSC2 to serve as a GTPase-activating protein (GAP) for Rheb, a Ras-like small GTPase (8), thereby allowing Rheb-GTP to accumulate. Many reports have suggested that Rheb promotes cell growth in an mTOR- and p70S6K-dependent manner (12–14). Akt-mediated phosphorylation can downregulate tuberin’s GTPase-activating potential toward Rheb, which regulates mTOR through FKBP38. FKBP38 binds mTOR and inhibits its activity in a manner similar to that of the FKBP12-rapamycin complex (15). The interaction of FKBP38 with mTOR is regulated by Rheb, which binds directly to FKBP38 and prevents it from association with mTOR (15). Several reports have shown that mTORC1 substrates, such as S6K, 4E-BP1, and PRAS40 possess the TOS motif (16–19). Since PRAS40 has a TOS motif for binding raptor, it inhibits mTORC1-directed phosphorylation of p70S6K and 4E-BP1 by competing with these proteins for raptor binding (19).
Avian reoviruses (ARVs) are nonenveloped and belong to the family Reoviridae. ARV is an oncolytic virus that has been the focus of studies on anticancer treatments (20–22). ARVs contain 10 double-stranded RNA genome segments and replicate in the cytoplasm of infected cells. Genome segment S1 contains three open reading frames that are translated into p10, p17, and σC proteins. p17 has been demonstrated to modulate cellular signal pathways which regulate autophagy, cell cycle, viral protein synthesis, and virus replication (23–28). Although p17 is a nucleocytoplasmic shuttling protein (26) which induces autophagy and cell cycle retardation by activation of p53 and inactivation of mTORC1 (23–25, 27), the mechanisms by which p17 modulates suppression of mTORC1 remain largely unknown. The aim of this work was to perform a comprehensive study to investigate the mechanisms underlying p17-modulated inhibition of mTORC1. This work reveals for the first time that p17 inactivates mTORC1 inducing autophagy in cancer cell lines. The current study provides insight into p17-modulated suppression of mTORC1 through upregulation of the p53/phosphatase and tensin homology deleted on chromosome 10 (PTEN) pathway, downregulation of the Akt/Rheb/mTORC1 pathway, and enhancement of the endogenous mTORC1 inhibitors binding to mTORC1 to dysregulate its assembly and accumulation on lysosomes.
RESULTS
p17 activates PRAS40 and increases the formation of class III PI3K/Beclin 1 complex through the p53-dependent pathway.
Previously, we found that ARV infection and p17 transfection can trigger autophagy in immortalized chicken embryo fibroblast (DF-1) cells and African green monkey kidney (Vero) cells by suppression of mTORC1 through regulation of the p53/PTEN/Akt pathway (23). This finding inspired us to further investigate the mechanisms underlying p17-modulated effects on downstream targets of Akt, PRAS40, TSC2, Rheb, and mTOR. The activity of mTORC1 is negatively regulated by the TSC1/TSC2 complex and PRAS40 (10, 19). Previous reports suggested distinct roles for the TSC complex/Rheb axis and PRAS40 in regulating mTORC1 (29, 30). Genetic ablation of TSC2 is lethal during development in mice or flies, while depletion of PRAS40 is tolerated. This reveals that Rheb activation and PRAS40 inhibition are not redundant mechanisms for mTORC1 activation, revealing that Rheb activity is required for PRAS40 dissociation. Both TSC2 and PRAS40 are substrates of Akt kinase (9–11). PRAS40 is not only a substrate of Akt but also a component of mTORC1; thus, it links the Akt and the mTOR pathway. Phosphorylation of p70S6K, 4E-BP1, and ULK1 by mTORC1 is commonly used as an indicator of mTORC1 activity (16–19). Therefore, we next chose to examine the phosphorylation levels of PRAS40, TSC2, and downstream targets of mTOR (p70S6K and ULK1). As shown in Fig. 1A, increased levels of p-p53, p-PTEN, Beclin1, and LC3-II were observed in p17-transfected cells with concomitant Akt and mTOR dephosphorylation/inactivation, which results in a significant decrease in phosphorylation levels of the downstream targets PRAS40, 70S6K, and ULK1 (Fig. 1A). To further confirm that p17-modulated activation of p53 inactivates mTORC1, depletion of p53 with a short hairpin RNA (shRNA) was carried out. In this study, Vero and DF-1 cells were transfected with the pcDNA3.1-p17 plasmid or cotransfected with p17-pcDNA3.1 and p53 shRNA plasmids. The negative effects of p17 on the Akt/mTORC1 pathway could be reversed in p53 knockdown cells, further confirming that p17-modulated mTOR dephosphorylation/inactivation and PRAS40 dephosphorylation/activation occur via the p53-dependent pathway. Beclin 1, which is involved in the initial step of the formation of autophagosomes, is directly targeted by signaling pathways and is found in a complex with Vps34, a class III PI3K (31). Since Beclin 1 is at the heart of a regulatory complex for class III PI3K/Vps34, whose activity is required for preautophagosome formation (31), the levels of Beclin 1 and the amounts of Beclin 1 and class III PI3K association were assayed. Importantly, the decrease in the level of p-ULK, increase in the level of Beclin 1, and upregulation of an autophagy marker (LC3-II) by p17 were reversed in p53 knockdown cells. Collectively, our findings suggest that p17-modulated upregulation of the upstream activator of autophagy (Beclin 1 and ULK1) occurs in a p53-dependent manner.
FIG 1.

The ARV p17 protein induces autophagy via activation of p53. (A) The levels of the respective proteins and their phosphorylated forms were examined by Western blotting assays with the indicated antibodies. Western blots were quantitated by densitometric analysis using ImageJ and normalized to actin. Numbers below each lane are percentages of the control level of a specific protein in mock-treated cells. Similar results were obtained in three independent experiments. (B) Vero cells were transfected with the indicated vectors for 24 h. The levels of p-p70 S6k, p-ULK1, and LC3-I/II were examined by Western blotting assays with the indicated antibodies. β-Actin was used as an internal control. (C) (Top) Vero cells were transfected or cotransfected with the indicated vectors for 24 h. Rabbit IgG was used as a negative control. (Bottom) Western blots from the upper panel were quantitated by densitometric analysis using ImageJ and normalized to actin. Data are means and SE from three independent experiments. (D) Coimmunoprecipitation experiments with class III PI3k and Beclin 1. Vero cells were transfected or cotransfected with the indicated vectors for 24 h. In p53 knockdown cells, Vero cells were transfected with p53 shRNA for 6 h followed by infection with ARV at an MOI of 10 for 24 h. Densitometry analysis results for Western blotting are expressed as the amount (fold) of class III PI3k and Beclin 1 association as shown on the left. The levels of indicated proteins in cells alone or with mock transfection was considered 1-fold. Similar results were obtained in three independent experiments. Significance between the treatments was determined by DMRT using SPSS software. Values followed by the same letter are not significantly different (P < 0.05). Each value is the mean (with SE) from three independent experiments.
As shown in Fig. 1A, phosphorylation of Akt at T308 and S473 was dramatically reduced in p17-transfected cells with concomitant decreased levels of phosphorylated PRAS40 at T246 compared to the mock-treated control. PRAS40 contains a TOS motif binding to the raptor site (16–19) and suppresses mTORC1-directed phosphorylation of p70S6K and 4E-BP1 by competing with these proteins for raptor binding (19). It was reported that PRAS40 phosphorylation at T246 by Akt forms a docking site for 14-3-3, which binds to raptor and sequesters raptor from mTORC1 (32, 33). To examine whether p17 modulates inhibition of mTORC1 by PRAS40 binding to raptor, overexpression of PRAS40 and PRAS40T24A mutant proteins was carried out to examine the levels of downstream targets of mTOR1 (p70S6K and ULK) and the autophagy marker LC3-II. Data shown in Fig. 1B reveal a decrease in p-p70S6K and p-ULK levels in p17-transfected cells. Importantly, the levels of LC3-II were elevated in p17-transfected Vero cells (Fig. 1B) and could be enhanced with coexpression of the PRAS40T24A mutant protein (Fig. 1B). Furthermore, coimmunoprecipitation assays were carried out to investigate whether p17 promotes the binding of PRAS40 to raptor. The results reveal that increased amounts of PRAS40 and raptor association were observed in the p17-transfected and mock-transfected control groups, respectively (Fig. 1C). p17-modulated elevation of PRAS40 binding to raptor could be enhanced along with coexpression of PRAS40 or PRAS40T24A mutant proteins accompanied by reduction of the association of p70S6K with the raptor (Fig. 1C), suggesting that there is less interactive effect between raptor and p70S6K in p17-transfected cells. Taken together, our findings suggest that p17 can promote the interaction of PRAS40 and raptor. Furthermore, since the expression level of Beclin1 is upregulated by p17 (Fig. 1A), we next aimed to study whether p17 promotes the interaction of Beclin 1 and class III PI3K. The amounts of Beclin 1 and class III PI3K association in cellular extracts from untreated, ARV-infected, and p17- and p53 shRNA-transfected cells were analyzed by Western blotting assays. We found that increased amounts of Beclin 1 and class III PI3K association were observed in ARV-infected and p17-transfected cells (Fig. 1D). p17-modulated elevated interaction of Beclin1 and class III PI3K could be reversed in p53 knockdown cells (Fig. 1D), suggesting that p17 enhances the formation of the Beclin 1/class III PI3K complex in a p53-dependent manner.
Reversion of insulin-modulated activation of the Akt/mTORC1 pathway by the ARV p17 protein.
An earlier study suggested that the phosphorylation of p70S6K and 4E-BP1 stimulated by insulin depends on activation of Akt and that Akt directly phosphorylates mTOR (5). In this work, increased levels of p-Akt, p-RAS40, p-TSC2, p-mTOR, p-p70S6K, p-eIF4E, and p-ULK1 were observed in insulin-treated cells while the expression levels of Beclin 1 were decreased. The LC3-II levels were unchanged compared to the mock-treated group (Fig. 2). We next wanted to investigate whether p17 is capable of antagonizing insulin-stimulated PI3K signaling that upregulates the Akt/mTORC1 pathway. Importantly, our results reveal that the positive effect of insulin on the Akt/mTORC1 pathway could be reversed by p17 (Fig. 2). The increased LC3-II levels were seen in p17-transfected cells but reversed in the presence of insulin (Fig. 2). Since this work focuses on p17-modulated regulation of PRAS40 and TSC2, it is worth noting that insulin-modulated phosphorylation/inactivation of PRAS40 and TSC2 could be reversed in p17-transfected cells. Because p70S6K is an important factor downstream of mTOR and eIF4E phosphorylation is regulated by mTORC1, as expected, a decrease in p-p70S6K and p-eIF4E levels was seen in p17-transfected cells (Fig. 2). This work has yielded promising results, which show for the first time that insulin-stimulated activation of mTORC1 could be reversed by p17 through a p53/PTEN-dependent mechanism.
FIG 2.

Reversion of p17-modulated mTORC1 inhibition by insulin. The effect of insulin on the phosphorylation levels of Akt, PRAS40, mTOR, and downstream molecules in p17-transfected Vero and DF-1 cells was analyzed. Vero and DF-1 cells were pretreated with insulin (0.2 μM) for 2 h, followed by transfection with either pcDNA3.1-Flag or pcDNA3.1-Flag-p17 for 24 h. Densitometry analysis results for Western blotting are expressed as percentages representing levels of Akt, PRAS40, mTOR, and downstream molecules, normalized to β-actin. Western blots were quantitated by densitometric analysis using ImageJ and normalized to actin. Numbers below each lane are percentages of the control level of a specific protein in cells alone. Similar results were obtained in three independent experiments.
p17 activates TSC2 and PRAS40 through inactivation of Akt.
As mentioned above, insulin-modulated PRAS40 and TSC2 phosphorylation/inactivation could be restored by p17 (Fig. 2). Because of our interest in p17 modulating mTORC1 regulators TSC2, PRAS40, Rheb, FKBP12, and FKBP38, we next examined whether ARV infection and pcDNA3.1-p17 and pcDNA3.1-p171-118 transfection influence the levels of these host proteins. To rule out the possibility of overexpression artifacts, all assays were carried out in both ARV-infected and p17-transfected cells. Since the p17(1–118) mutant protein cannot reach the nucleus to exert its effect on activation of p53 (24), it was used as a negative control. Importantly, a marked decrease in p-TSC2 and p-PRAS40 levels was seen in p17-transfected cells in a time-dependent manner, while the levels of p-TSC2 and p-PRAS40 in the mock-treated controls (untreated and p17 mutant-transfected cells) were not changed (Fig. 3A and B). Interestingly, a decrease in in the level of p-TSC2 was observed in ARV-infected cells at 12 and 24 h postinfection (Fig. 3A and B). In addition, the levels of Rheb, FKBP38, and FKBP12 were not altered (Fig. 3A and B). We also found that decreased levels of p-raptor were observed in p17-transfected cells in a time-dependent manner (Fig. 3C).
FIG 3.

Analysis of expression levels of TSC2, PRKS40, Rheb, FKBP12, FKBP38, and raptor. (A and B) Levels of the respective proteins and their phosphorylated forms in the mock-treated control and different treatments. (C) Levels of p-raptor and raptor in pcDNA3.1 (vector only)- and pcDNA3.1-p17-transfected cells. Whole-cell lysates were collected at the indicated time points for Western blotting. Western blots were quantitated by densitometric analysis using ImageJ and normalized to actin. Numbers below each lane are percentages of the control level of a specific protein in mock-treated cells. Similar results were obtained in three independent experiments.
p17-modulated mTORC1 inhibition through its negative effect on Rheb enhances FKBP38 interaction with mTORC1.
An earlier report suggested that activity of mTOR is regulated by Rheb and that Rheb regulates mTOR by antagonizing its endogenous inhibitor, FKBP38 (15). Rheb interacts directly with FKBP38 and prevents its association with mTOR in a GTP-dependent manner. Having shown that p17-modulated Akt and mTOR dephosphorylation/inactivation results in a significant decrease in phosphorylation levels of p70S6K and ULK1 accompanied by increased levels of LC3II (Fig. 1A), we next employed a rescue assay by overexpression of Rheb in p17-transfected Vero cells to study whether the effect of p17 on mTORC1 could be reversed. The negative effect of p17 on mTORC1 could be moderately reversed in cells overexpressing Rheb (Fig. 4A). Furthermore, coimmunoprecipitation assays were carried out to study p17-modulated interaction of Rheb/FKBP38, Rheb/mTOR1, and FKBP38/mTORC1. As shown in Fig. 4B, the binding of Rheb to FKBP38 or mTOR was dramatically reduced in p17-transfected cells compared to the mock-treated control (vector alone) (Fig. 4B). The negative effect of p17 could be reversed in the presence of insulin (Fig. 4B). Consistent with results shown in Fig. 1A, p17 exerts a negative effect on Rheb through activation of the p53/PTEN pathway, which inactivates Akt, thereby reducing Rheb interaction with mTOR. Similar to the negative effect of p17 on the interaction of Rheb/FKBP38 and Rheb/mTOR, the binding of Rheb to FKBP38 or mTOR was dramatically reduced in cells treated with the Akt inhibitor Akt III (Fig. 4C). We found that overexpression of Rheb enhanced the interaction of Rheb/FKBP38 or Rheb/mTOR and that this effect could be reversed in cells coexpressing p17 and Rheb (Fig. 4D). Since p17 reduces the binding of Rheb to FKBP38 or mTOR, we next investigated whether p17 can enhance the binding of FKBP38 to mTOR via activation of TSC2. The amounts of FKBP38, mTOR, and TSC2 in cellular extracts from different treatments were analyzed by Western blotting assays. Coimmunoprecipitation results reveal that p17 increases the association of FKBP38 and mTOR, while this effect could be reversed in cells overexpressing Rheb or TSC2 shRNA (Fig. 4E). Since the levels of TSC2, Rheb, FKBP38, and FKBP12 were unchanged in ARV-infected or p17-transfected cells (Fig. 3A and B), this rules out the possibility that p17 weakens the interaction of Rheb/FKBP38, Rheb/mTOR, and FKBP38/mTOR due to the availability of interaction partners. In this work, p17 was analyzed to determine whether it can interact with Rheb or FKB38. We found that p17 does not associate with either Rheb or FKBP38 (Fig. 4B to E). In addition, we found that p17 does not coprecipitate with PRAS40 and FKBP12 (data not shown).
FIG 4.

The ARV p17 protein exerts a negative effect on Rheb-mTORC1 interaction. (A) To study whether the effect of p17 on mTORC1 could be reversed by overexpression of Rheb in p17-transfected Vero cells, cells were transfected or cotransfected with the indicated vectors for 24 h. The levels of the respective proteins and their phosphorylated forms were examined by Western blotting assays with the indicated antibodies. Western blots were quantitated by densitometric analysis using ImageJ and normalized to actin. Numbers below each lane are percentages of the control level of a specific protein in mock-treated cells. Similar results were obtained in three independent experiments. (B to D) To understand whether p17, insulin, or Akt inhibitor (Akt III) affects the interaction between Rheb and other host cell proteins, coimmunoprecipitation experiments of Rheb, p17, mTOR, FKBP38, and FKBP12 were performed. (B) To study whether insulin reversed p17-modulated inhibition of Rheb/mTORC1 interaction, cells were treated with insulin (0.2 μM) for 2 h followed by transfection of cells with either pcDNA3.1-Flag-p17 plasmid for 24 h at 37°C. The interaction of Rheb and other host cell proteins was examined by coimmunoprecipitation experiments. (C) Vero cells were treated with the Akt inhibitor Akt III (2.5 μM) and transfected with pcDNA3.1-Flag-p17 plasmid DNA for 24 h. (D) Vero cells were transfected or cotransfected with the indicated vectors for 24 h. (E) To investigate whether p17 can enhance the binding of FKBP38 to mTOR via activation of TSC2, a coimmunoprecipitation assay was carried out. Vero cells were transfected or cotransfected with the indicated vectors for 24 h. The amounts of FKBP38, mTOR, and TSC2 in cellular extracts from different treatments were analyzed by Western blotting assays. Densitometry analysis results for Western blotting shown in panels B to E are expressed as the amount of protein and protein association (fold). Values for mock-treated cells were considered 1-fold. Similar results were obtained in three independent experiments.
p17 dysregulates mTORC1 assembly.
Insulin and a well-studied negative regulator of mTORC1, rapamycin, were used to explore how p17 modulates inhibition of mTORC1 assembly. Inhibition of mTORC1 by rapamycin or ARV reduced raptor and mTOR association, as determined by a reduction in the amount of raptor that coimmunoprecipitated with mTOR (Fig. 5A). Furthermore, in the absence of rapamycin, raptor interaction with mTOR was elevated and FKBP12/mTOR binding was blunted (Fig. 5A). Having shown that p17 promotes FKBP38 interaction with mTOR (Fig. 4E), we next examined whether p17 modulates FKBP12 interaction with mTOR. Interestingly, the interaction of FKBP12 with mTOR was elevated in ARV-infected and rapamycin-treated cells (Fig. 5A). The effect of rapamycin on the interaction of FKBP12 and mTORC1 could be enhanced in ARV-infected Vero cells (Fig. 5A). Our results reveal that once FKBP12 binds more to mTORC1 in rapamycin-treated Vero cells, FKBP38 binds less mTORC1. This finding is supported by a previous study suggesting that FKBP38 is structurally related to FKBP12 and is able to bind mTOR to inhibit its activity in a manner similar to that of the FKBP12-rapamycin complex (15). As shown in Fig. 5A, the binding of PRAS40 to mTORC1 was also reduced in ARV-infected Vero cells. These data are consistent with a report revealing that PRAS40 interaction with mTORC1 requires the presence of both mTOR and raptor (19).
FIG 5.

The ARV p17 protein disrupts the mTORC1 assembly. (A) Coimmunoprecipitation of p17, mTOR, raptor, FKBP38, FKBP12, and PRAS40 was carried out. Vero cells were pretreated without or with rapamycin (5 μM) for 2 h, followed by infection with ARV at an MOI of 10 or without ARV infection. The cellular proteins were incubated with mTOR antibody, and the immunoprecipitated proteins were detected using the indicated antibodies by Western blotting assays. (B and C) In order to study the effects of insulin (0.2 μM) or rapamycin (5 μM) on mTORC1 assembly, coimmunoprecipitation of p17, mTOR, raptor, FKBP38, FKBP12, PRAS40, Rheb, p70 S6k, and 4EBP1 was carried out. Vero cells were pretreated with either insulin or rapamycin for 2 h, followed by transfection with either pcDNA3.1-p17 or pcDNA3.1 plasmid for 24 h, respectively. The cellular proteins were incubated with mTOR or raptor antibodies, and the immunoprecipitated proteins were detected using the indicated antibodies by Western blotting assays. Densitometry analysis results for Western blotting in panels A to C are expressed as the amount of protein and protein association (fold). Values for mock-treated cells were considered 1-fold. Signals for all blots were quantified using ImageJ software. Data in panels A, B, and C are means and SE from three independent experiments.
Since ARV infection or p17 transfection increased the binding of FKBP12 or FKBP38 to mTOR, the binding of raptor to mTOR was reduced (Fig. 5B). The increased amounts of FKBP38 and mTOR association are consistent with data shown in Fig. 4B and C revealing p17-modulated reduction of Rheb and mTORC1 interaction. The binding of FKBP12 or FKBP38 to mTOR was increased with concomitant decreased binding of p70S6K and 4E-BP1 to raptor or mTOR in p17-transfected cells (Fig. 5B and C). p17-modulated inhibition of raptor and mTOR interaction was reversed in the presence of insulin, thereby reducing the binding of FKBP12 or FKBP38 to mTOR, which results in increased binding of p70S6K and 4E-BP1 to raptor or mTOR (Fig. 5B and C). Inhibition of mTORC1 by rapamycin diminished raptor interaction with mTOR, as determined by a reduction in the amount of raptor that coimmunoprecipitated with mTOR (Fig. 5A to C). Since PRAS40 disrupts mTORC1, similar to the effect of rapamycin, the association of PRAS40 to raptor or mTORC1 was significantly reduced in rapamycin-treated cells (Fig. 5A and C). Based on our findings, we next investigated whether the positive effect of insulin on mTORC1 could be reversed by p17. In the absence of insulin, increased binding of PRAS40 to raptor was seen in p17-transfected Vero cells (Fig. 5C), thereby reducing the binding of 4E-BP1 and p70S6K to raptor and presentation to mTORC1, which results in a decrease in phosphorylation levels of p70S6K and 4E-BP1 (Fig. 1A and B). As expected, p17-modulated increased interaction of PRAS40 and raptor was moderately reversed in the presence of insulin, thereby increasing 4E-BP1 and p70S6K binding to raptor or mTOR (Fig. 5C). Since mTORC1 is bound by rapamycin/FKBP12 complex in rapamycin-treated Vero cells, PRAS40 interaction with raptor was dramatically reduced in p17-transfected Vero cells (Fig. 5C). Additionally, our results reveal that p17 does not coprecipitate with either mTOR or raptor (Fig. 5B and C).
p17-modulated inhibition of mTORC1 accumulation on lysosomes.
In the presence of amino acids, mTORC1 is recruited to the lysosomal surface and is necessary for its activation by Rheb (34). The above-described results prompted us to explore whether p17 inhibits mTORC1 accumulation on lysosomes through regulation of PRAS40, TSC2, and Rheb. Therefore, Vero cells were transfected with the different vectors or shRNAs for 24 h. Cells were fixed and processed for immunofluorescence staining of mTOR and LAMP1 (lysosome marker) and observed under a confocal fluorescence microscope. The yellow dots in the merged panel show that mTORC1 is located on the lysosomes (Fig. 6A). Our results suggest that p17 inhibits mTORC1 recruitment to lysosomes and that this effect could be moderately reversed in the presence of insulin (Fig. 6A and B) and enhanced by the PRAS40T24A mutant protein. Conversely, mTORC1 accumulation on lysosomes could be enhanced in cells overexpressing Rheb. This effect was moderately reversed with coexpression of p17 (Fig. 6 and B). In this study, we also examined the effect of TSC2, an important inhibitor of mTORC1, on mTORC1 accumulation on lysosomes. As shown in Fig. 6A and B, mTORC1 accumulation on lysosomes was elevated in TSC2 knockdown cells compared to the mock-treated group, and this effect could be reversed in cells cotransfected with the pcDNA3.1-p17 and TSC2 shRNA vectors. Collectively, our results suggest that p17 can inhibit mTORC1 accumulation on lysosomes through modulation of PRAS40, TSC2, and Rheb.
FIG 6.

The ARV p17 protein inhibits mTORC1 translocation to lysosomes. (A) To study whether p17, PRAS40, TSC2, and insulin regulate mTORC1 translocation to lysosomes, Vero cells were fixed and processed for immunofluorescence staining with DAPI. Vero cells were pretreated with insulin (0.2 μM) for 2 h, followed by transfection with the pcDNA3.1-p17 vector for 24 h. All transfections or cotransfections were performed for 24 h. Stained mTOR (red) and lysosome (green) were observed under a fluorescence microscope. The yellow dots in the merged panel show that the mTOR complex is located on the lysosome. (B) Quantification of colocalization was quantified with ImageJ software. The amount of colocalized fluorescence in the mock-treated control group was considered to be 1-fold. At least 10 fields of 2 cells per field were taken for each sample per experiment. All experiments were performed in three independent experiments.
p17 induces autophagy in cancer cell lines.
Our previous studies showed that p17 induces autophagy through upregulation of the p53/PTEN pathway and inactivation of the Akt/mTORC1 pathway and mTORC2 in Vero and DF-1 cells (23, 35). In this work, we further investigated whether p17 can trigger autophagy in two cancer lines, non-small cell lung carcinoma (A549) and human cervical carcinoma (HeLa). These two cancer cell lines could be infected with the ARV S1133 strain at a multiplicity of infection (MOI) of 10 for 48 h. Virus titers of ARV-infected A549 and HeLa cancer cells reached 106.4 and 106.04, respectively, at 48 h postinfection, while ARVs do not infect normal human lung fibroblast (HFL-1) cells. The results suggest that oncolytic ARVs can infect and replicate in these cancer cell lines. Western blot results reveal that p17 increases the levels of p-p53 (S15) and p-PTEN (S380/T382/383) accompanied by decreased levels of p-Akt (T308 and S473), p-mTOR (S2488), p-70S6K (T308), and p-ULK (S757) (Fig. 7A). Importantly, the decrease in the level of p-ULK, increase in the level of autophagy regulator (Beclin 1), and upregulation of LC3-II (36) by p17 were observed in HeLa and A549 cancer cells (Fig. 7A). The results reveal that the Akt and mTORC1 inhibition is correlated with the conversion of LC3-I to LC3-II, suggesting a role for Akt and mTORC1 as negative mediators of p17-induced autophagy. Formation of autophagosomes can be analyzed by immunofluorescence analysis, as the staining of LC3-I is diffusely cytoplasmic, whereas the staining of LC3-II is punctate (37). Since the green fluorescent protein (GFP)-LC3 construct is unstable under lysosomal acidic and degradative conditions, the relatively stable construct mCherry-LC3 was used in this study (37). Assays performed in both ARV-infected and p17-transfected HeLa and A549 cancer cells showed a significant increase in the numbers of mCherry-LC3 puncta in a time-dependent manner (Fig. 7B). Thapsigargin (TG) can induce LC3-II puncta (23) and was used as a positive control. As shown in Fig. 7B, numbers of mCherry-LC3 puncta significantly increased in a time-dependent manner in TG-treated cancer cells. The numbers of mCherry-LC3 puncta were significantly increased at 18 h posttreatment (Fig. 7B and C). This is consistent with Fig. 7A revealing decreased levels of p-mTORC1 and p-ULK1 accompanied by increased levels of LC3-II in p17-transfected cells. This is the first report to suggest that p17 induces autophagy in HeLa and A549 cancer cell lines.
FIG 7.

ARV p17-induced autophagy in cancer cell lines. (A) HeLa and A549 cancer cell lines were transfected with the pcDNA3.1-p17 and pcDNA3.1 vectors, respectively. The levels of the respective proteins and their phosphorylated forms were examined by Western blotting assays with the indicated antibodies. (B) HeLa and A549 cancer cells were infected with ARV at an MOI of 10 or transfected with the pcDNA3.1-p17 vectors at the indicated time points. Aside from these treatments, cells were also treated with TG (5 μM) at the indicated time points. The mCherry-GFP-LC3 puncta (red) in cells were observed under fluorescence microscopy. Cell nuclei were stained with DAPI. Bars, 25 mm. (C) The numbers of LC3 puncta were calculated from the results in panel B. Each value is the mean (with SE) from three independent experiments.
p17 induces autophagy and translation shutoff to benefit virus replication.
Our findings reveal that p17 inhibits mTORC1 and reduces the level of the phosphorylated form of eIF4E. We next investigated whether p17 regulates cellular protein synthesis. Our results reveal that p17 reduces host protein synthesis in a time-dependent manner (Fig. 8A). The increase in transfection time was inversely proportional to the levels of cellular protein translation (Fig. 8A). The data are consistent with Fig. 2 revealing p17-modulated inhibition of p70S6K and eIF4E, which play important roles in translation initiation control. Cell viability in p17-transfected DF-1 cells was assessed by a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay and was only slightly reduced compared to that of the mock-treated cells (Fig. 8B). Modulation of the activity of various translation initiation factors is a critical strategy by which viruses regulate cellular protein production. Our findings are supported by our earlier studies (23, 38). Previously, we suggested that ARV shuts off cellular protein synthesis by modulating phosphorylation of eIF2α (23), eukaryotic translation initiation factors, and elongation factor (eEF2) but not its own protein production (38). Collectively, this work further suggests that p17 is the major protein that is responsible for ARV-induced translation shutoff. Furthermore, our previous work suggested that depletion of p53 and PTEN reduces virus yield while knockdown of Tpr, CDK2, and CDK4 increases virus production (24, 35). To investigate whether p17-modulated inhibition of mTORC1 affects virus replication, knockdown of p53, TSC2, and LC3, rapamycin treatment, and overexpression of Rheb and PRAS40T24A mutant proteins in Vero cells were carried out. We found that knockdown of p53, TSC2, and LC3 as well as overexpression of Rheb in cells reduced virus yield, while overexpression of the PRAS40T24A mutant protein or rapamycin treatment increased virus production (Fig. 8C). Collectively, our results reveal that p17-modulated mTORC1 inhibition triggers autophagy and translation shutoff, which benefits virus replication.
FIG 8.

ARV p17 induces translation shutoff and autophagy, benefiting virus replication. (A) Relative translation levels of cellular proteins at the indicated time points in different treatments in DF-1 cells were assayed using pulse-chase labeling. Cellular protein band intensity was assessed in relation to actin to quantify p17-regulated cellular translation shutoff. Data are means of triplicate results. (B) Cell viability in pcDN3.1 vector- or pcDN3.1-p17-transfected DF-1 cells was assessed by MTT assay. (C) The influence of the p53/Akt/mTOR pathway on ARV replication was analyzed by measuring the virus titers of different treatment groups. Vero cells were transfected with shRNAs or overexpressed PRAS40, PRAS40T24A, and Rheb for 6 h, followed by infection with ARV at an MOI of 10 for 24 h. Aside from these treatments, cells were also pretreated with rapamycin (5 μM) for 2 h, followed by infection with ARV at an MOI of 10 for 24 h. The treated- and untreated-cell lysates were collected to determine virus titers 24 h postinfection.
DISCUSSION
Although p17 has been shown to modulate mTORC2, autophagy, cell cycle, viral protein synthesis, and virus replication (21, 23–28, 35), the precise mechanisms by which p17 modulates host factors or signal pathways to suppress mTORC1 remains largely unknown. The present study provides novel insights into p17-modulated inactivation of mTORC1 by upregulation of p53 and TSC2, two negative regulators of the pathway, enhancement of endogenous mTORC1 inhibitor (FKBP38, FKBP12, and PRAS40) interaction with mTORC1, and suppression of mTORC1 accumulation on lysosomes. Our findings reveal that p17-modulated inhibition of mTORC1 relies on several different mechanisms. First, p17 disrupts mTORC1 assembly by promoting PRAS40 interaction with raptor and sequestering raptor from mTORC1, thereby disrupting the mTORC1 complex. Through this mechanism, PRAS40 disrupts the mTORC1 complex, similar to the effect of rapamycin. Raptor is involved in recruiting substrates for phosphorylation by the kinase domain of mTOR. PRAS40 interaction with raptor competes with the binding of raptor to p70S6K and 4E-BP1, which inhibits mTORC1-directed phosphorylation of p70S6K and 4E-BP1, thereby causing translation shutoff. Akt can activate mTORC1 by phosphorylating PRAS40 at T246, thereby relieving the PRAS40-mediated inhibition of mTORC1 (32, 33). Therefore, p17 elevates PRAS40 interaction with raptor due to its effect on inactivation of Akt. This notion is further supported by our finding that p17 antagonizes insulin-stimulated p70S6K and 4EBP1 phosphorylation by preventing these proteins from raptor binding (19). In addition, our results reveal that p17 triggers dephosphorylation of raptor in the tested cell lines, very likely reducing affinity of the binding of raptor to mTOR.
Second, in addition to the Akt/PRAS40 signaling axis, our results suggest a mechanism whereby p17 exerts a negative effect on Rheb through inactivation of Akt, which weakens Rheb and mTORC1 interaction and promotes the inhibitory interaction of the endogenous mTORC1 inhibitor FKBP38 and mTORC1, thereby inhibiting mTORC1. It has been reported that Akt phosphorylates the downstream target TSC2, which inhibits its GTPase activating protein activity toward the GTP-binding protein Rheb (10). This study suggests that Akt-mediated phosphorylation of TSC2 is an important strategy for p17 to inactivate mTORC1. Furthermore, a compelling finding in this work is that p17 enhances FKBP12 interaction with mTORC1 in rapamycin-untreated or -treated Vero cells. Rapamycin binds to FKBP12, creating a drug-receptor complex that binds and inhibits mTORC1 (39). Since p17 does not directly interact with FKBP12, p17 seems to act indirectly in promoting FKBP12 binding to mTORC1, which disrupts the mTORC1 complex by preventing mTOR interaction with raptor. Based on our findings, this work suggests a mechanism whereby p17 functions as a negative regulator of mTORC1 in a manner different from that of rapamycin. Clinical data obtained so far suggest that rapamycin treatment shows promise against some tumors (40, 41). A recent study by our group suggested that p17 is able to retard cell growth and cell cycle of several cancer cell lines and reduces tumor size in nude mice (21). It is of highest importance to learn more about the mechanism with the aim of predicting the types of tumors that will respond to p17. We believe that our description of p17 modulating cellular factors and signaling pathways to inactivate mTORC1 provides new insight into viral nonstructural proteins regulating autophagy, cellular translation, cell cycle, and tumorigenesis. mTORC1 must be recruited to lysosome membranes and activated by Rheb, a direct mTORC1 activator (34). In addition to the above-mentioned strategies, we found that p17 regulates the TSC2/Rheb pathway and PRAS40 to suppress mTORC1 accumulation on lysosomes. Importantly, we found that the negative effect of p17 on mTORC1 accumulation on lysosomes could be reversed in the presence of insulin and enhanced by the PRAS40T24A mutant protein (Fig. 6A and B). Furthermore, mTORC1 accumulation on lysosomes could be further elevated in Rheb-overexpressing or TSC2 knockdown cells, and this effect could be moderately reversed with coexpression of p17 and Rheb. Collectively, these findings suggest that p17 modulates accumulation of mTORC1 on lysosomes and inhibits the mTORC1 complex.
Our findings suggest that ARV-induced host translation shutoff is due to the effect of ARV p17. The shutoff of host protein synthesis in virus-infected cells is the important mechanism for supporting viral replication (42, 43). During an early step in the initiation of protein synthesis, eIF4E recognizes and binds the 7-methylguanosine-containing mRNA cap (44). In this work, we demonstrate that p17 inactivates mTORC1, which in turn reduces the phosphorylated levels of p70S6K and eIF4E, resulting in translation shutoff. This is supported by our previous reports suggesting that ARV inactivates several translation initiation and elongation factors (23, 38). The results presented here suggest that two changes occur in the host translation machinery regulated by ARV. First, several translation initiation and elongation factors are inactivated, and second, the eIF4F complex may be disrupted by the ARV p17 protein. The alteration in the eIF4F complex involves dephosphorylation of the cap-binding protein IF4E and dissociation of eIF4E from the eIF4F complex. Many viruses were found to impede cap-dependent translation that potentially facilitates translation of viral transcripts or slows production of cellular proteins (42, 43). Several reports suggested that rapamycin specifically inhibits cap-dependent translation by inhibiting phosphorylation of 4E-BP1 and accelerates the shutoff of host protein synthesis (45, 46). In this work, enhanced virus yield and elevated dephosphorylation of eIF4E, 4E-BP1, eIF4B, and eIF4G (38) after rapamycin treatment suggest that ARV replication was not hindered by inhibition of cap-dependent translation, suggesting that ARV can withstand cap-dependent translation inhibition. ARV may exploit these mechanisms to govern the expression of viral or cellular genes that are important for the completion of the virus life cycle. This work provides a novel insight into the mechanism by which p17 triggers autophagy and host translation shutoff through activation of p53 signaling and inactivation of mTORC1, which is beneficial for virus replication.
In the current and previous studies, the interplay of p17, the p53/PTEN and Akt/TSC2/Rheb/mTORC1 pathways, and mTORC2 have been studied (21, 23, 24, 35). Several reports suggest that rapamycin blocks G1-S progression by decreasing the level of cyclin D1, showing that mTORC1 controls this stage of the cell cycle (47). It was demonstrated that p53-independent inhibition of mTORC1 suppresses senescence (48). Conversely, p17 inactivates mTORC1 and causes cell cycle retardation in a p53-dependent manner (21, 24, 25), implying that p17 may suppress cellular senescence by its ability to inhibit the mTORC1 pathway. The RTK/PI3K/Akt pathway is one of the most potent driving forces promoting tumor progression. Since p17 upregulates the p53/PTEN pathway (23, 24), inactivates the Akt/mTORC1 pathway, and induces cell cycle retardation (21) and autophagy in several cancer cell lines, targeting p53, PTEN, and mTORC1 using p17 may be an effective strategy for the virotherapy of human cancers. More basic and mechanistic studies will further elucidate the role of p17 in the complexity of the cancer-signaling pathway networks.
As alluded to earlier, the process of autophagosome formation is tightly controlled by mTOR and Beclin 1, which are central in the regulation of autophagy (31, 49). mTOR is one of the key regulators of autophagy and shuts off autophagy (49). Downstream of mTORC1, Beclin 1 is at the heart of a regulatory complex for class III PI3K/Vps34 whose activity is required for preautophagosome formation (27). In this work, we found that p17 induces autophagy in HeLa and A549 cancer cells by the p53/Akt/mTORC1 pathway. Our recent report suggested that p17 weakens the interaction between Beclin 1 and 14-3-3 through inactivation of Akt, thereby inducing autophagy (35). This work further suggests a mechanism whereby p17 triggers autophagy through upregulation of the expression level of Beclin1 and promoting the formation of Beclin1/class III PI3K complex in a p53-dependent manner. These data reveal that p17 functions as an activator of Beclin 1 by at least two independent mechanisms. Collectively, our results provide novel insight into p17-triggered autophagy by modulating the upstream autophagy activators through activation of p53 and inactivation of Akt and mTORC1. A clearer understanding of the molecular basis for virus-induced changes can shed light on normal cellular events and on the specific ways that viruses manipulate their hosts. A model of the interplay between p17 and the mTORC1 activators and inhibitors is depicted in Fig. 9.
FIG 9.
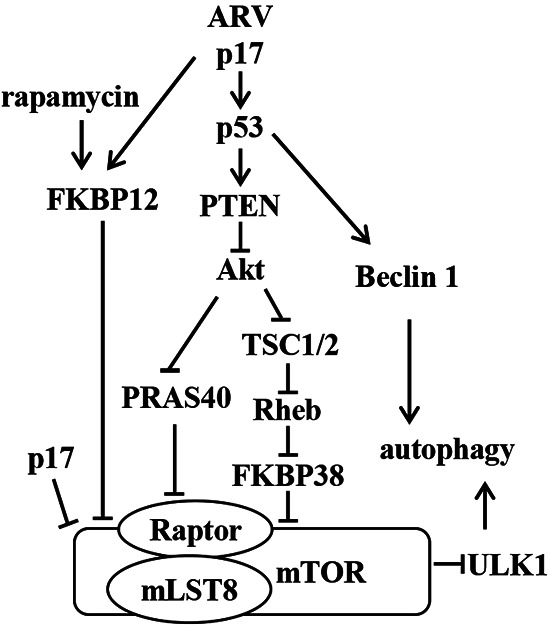
Model depicting ARV p17-modulated inhibition of mTORC1, which induces autophagy and translation shutoff. p17 upregulates the p53/PTEN pathway, which in turn downregulates the Akt/Rheb/mTORC1 pathway. p17 activates PRAS40 and FKBP38 to bind mTORC1 and disrupts mTORC1 assembly through inactivation of Akt. p17 also promotes FKBP12 interaction with mTORC1 and inhibits mTORC1. The p17 protein activates the upstream regulators of autophagy, ULK1 and Beclin1, to induce autophagy. Arrows indicate activation; bars indicate repression.
MATERIALS AND METHODS
Virus and cells.
In this work, DF-1 and Vero cells as well as two human cancer cell lines (A549 and HeLa) were cultured in minimum essential medium (MEM) or Ham's F-12K (Kaighn's) medium supplemented with 10% fetal bovine serum (FBS) and 10 mM HEPES (pH 7.2). One day before each experiment, all tested cells were seeded in 6-cm cell culture dishes at 1 × 106 cells in a 37°C incubator with 5% CO2. Except for insulin treatment or coimmunoprecipitation assays, all cells used in the study were maintained in serum-free medium for 2 h and then refreshed with medium containing 5 to 10% FBS overnight once cell confluence reached about 75%.
Reagents and antibodies.
Insulin was purchased from Life Technologies (Carlsbad, USA). Rapamycin was purchased from Merck Co. (Darmstadt, Germany). The Akt III inhibitor, specific for Akt, was purchased from Enzo Life Science (New York, USA). Polyclonal antibodies against the p17 protein of ARV were from our laboratory stock. The catalog numbers and dilution factors of the primary and secondary antibodies used in this study are shown in Table 1.
TABLE 1.
Catalog numbers and dilution factors of the antibodies used in this study
| Antibody | Catalog no. | Clone name | Dilution factor | Manufacturer |
|---|---|---|---|---|
| Mouse anti-p17a | 2,000 | Our laboratory | ||
| Mouse anti-p53 | 2527 | 7F5 | 3,000 | Cell Signaling |
| Rabbit anti-p-p53 (S15)a | 9284 | 2,000 | Cell Signaling | |
| Rabbit anti-PTEN | 9559 | 138G6 | 3,000 | Cell Signaling |
| Rabbit anti-p-PTEN (S380/T382/383) | 9554 | 2,000 | Cell Signaling | |
| Rabbit anti-Akt | 4691 | C67E7 | 3,000 | Cell Signaling |
| Rabbit anti-p-Akt (T308) | 2965 | C31E5 | 2,000 | Cell Signaling |
| Rabbit anti-p-Akt (S473) | 3787 | 736E11 | 2,000 | Cell Signaling |
| Rabbit anti-TSC2 | 3990 | D57A9 | 2,000 | Cell Signaling |
| Rabbit anti-p-TSC2 (S939) | 3615 | 1,000 | Cell Signaling | |
| Rabbit anti-p-TSC2 (T1462) | 3617 | 5B12 | 1,000 | Cell Signaling |
| Rabbit anti-PRAS40 | 2691 | D23C7 | 3,000 | Cell Signaling |
| Rabbit anti-p-PRAS40 (T246) | 13175 | D4D2 | 3,000 | Cell Signaling |
| Mouse anti-Rheb | sc-271509 | B-12 | 1,000 | Santa Cruz |
| Rabbit anti-FKBP38 | sc-66894 | H-220 | 1,000 | Santa Cruz |
| Rabbit anti-raptor | 2280 | 24C12 | 3,000 | Cell Signaling |
| Rabbit anti-p-raptor (S863) | sc-130214 | 1,000 | Santa Cruz | |
| Mouse anti-FKBP12 | sc-100282 | L-17 | 1,000 | Santa Cruz |
| Rabbit anti-mTOR | 2983 | 7C10 | 3,000 | Cell Signaling |
| Mouse anti-p-mTOR (S2448)a | 2971 | 2,000 | Cell Signaling | |
| Rabbit anti-LAMP1 | 9091 | D2D11 | 3,000 | Cell Signaling |
| Rabbit anti-eIF4Ea | 3591 | 2,000 | Cell Signaling | |
| Rabbit anti-p-eIF4E (S209) | 9741 | 2,000 | Cell Signaling | |
| Rabbit anti-p70 S6ka | 9202 | 3,000 | Cell Signaling | |
| Mouse anti-p-p70 S6k (T389) | 9234 | 108D2 | 1,000 | Cell Signaling |
| Rabbit anti-ULK1a | 4773 | R600 | 3,000 | Cell Signaling |
| Rabbit anti-p-ULK1 (S757)a | 6888 | 2,000 | Cell Signaling | |
| Mouse anti-Beclin 1 | 3495 | D40C5 | 3,000 | Cell Signaling |
| Rabbit anti-PI3k class III | 4263 | D9A5 | 2,000 | Cell Signaling |
| Rabbit anti-4EBP1 | 9644 | 53H11 | 2,000 | Cell Signaling |
| Rabbit anti-LC3Ba | 2775 | 2,000 | Cell Signaling | |
| Mouse anti-β-actin | MAB1501 | C4 | 10,000 | Millipore |
| Goat anti-mouse IgG (H + L) HRP | 5220-0341 | 5,000 | SeraCare | |
| Goat anti-rabbit IgG (H + L) HRP | 5220-0336 | 5,000 | SeraCare |
Polyclonal antibody.
Plasmid construction and transfection.
pcDNA3.1-p17 and its mutant pcDNA3.1-p171-118 of ARV were described previously (24). In order to understand how PRAS40 and Rheb are regulated by ARV p17 to cause mTORC1 inhibition, pCI-neo-PRAS40, pCI-neo-PRAS40T246A, and pCI-neo-Rheb vectors were constructed. To prepare cDNA for PRAS40 and Rheb genes, total RNA was extracted from Vero cells using TRIzol solution (Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the manufacturer's protocol. The PRAS40, PRAS40T246A, and Rheb gene fragments were amplified by PCR using the primers shown in Table 2. Reverse transcription (RT) was carried out at 42°C for 15 min and 72°C for 15 min. PCR was performed with 1 μL of cDNA, 1 μL of each primer, 2 μL of PCR mix, and 15 μL of double-distilled water (ddH2O), in a total volume of 20 μL. The PCR conditions for amplification were 95°C for 5 min, 35 cycles of 95°C for 30 s, 55°C for 30 s (Rheb) or 57°C for 30 s (PRAS40), and extension at 72°C for 30 s (Rheb) or 1.5 min (PRAS40), followed by 72°C for 10 min for a final extension. For transfection, cells were seeded into 6-cm cell culture dishes. At about 75% confluence, cells were transfected with respective constructs by using the Lipofectamine reagent based on the manufacturer’s protocol (Invitrogen, Carlsbad, CA, USA). In this work, the pLKO-AS1-puro plasmids encoding shRNA were from the National RNAi Core Facility of Academia Sinica, Taiwan. Sequences for p53, TSC2, and LC3 are as follows: p53, CTCAGACTGACATTCTCCACTTCTTGTTC (catalog no. TG320558); TSC2, GAGGGTAAACAGACGGAGTTT (catalog no. TRCN000028 8686); and LC3, TGGACAAGACCAAGTTCCTGGTGCCTGCAC (catalog no. TG503902). In this work, cells were transfected with their respective shRNAs for 6 h, followed by infection with ARV at MOI of 10 for 24 h. The whole-cell lysates were collected for Western blot analysis.
TABLE 2.
Primers used for amplification of targeted genesa
| Gene | Accession no. | Directionb | Sequence (5′–3′)c | Location | Expected size (bp) |
|---|---|---|---|---|---|
| p17 | AF330703 | F | CGGAATTCATGCAATGGCTCCGCCATACGA (EcoRI) | 293–314 | 441 |
| R | GCTCTAGATCATAGATCGGCGTCAAATCGC (XbaI) | 733–712 | |||
| p171-118 | AF330703 | F | CGGAATTCACAATGCAATGGCTCCGCCATACG (EcoRI) | 293–313 | 354 |
| R | AAACTCGAGTCAGGATTGAGACCCGCCATCCCAATG (XhoI) | 646–623 | |||
| PRAS40 | NM_032375.5 | F | TAGAATTCAAGATGGCGTCGGGGCGC (EcoRI) | 793–807 | 782 |
| R | CGGTCGACCTCCCTGGACTTCAATATTTCC (SalI) | 1574–1553 | |||
| PRAS40T246A | NM_032375.5 | F | TAGAATTCAAGATGGCGTCGGGGCGC (EcoRI) | 793–807 | 782 |
| R | TAGTCGACTCAATATTTCCGCTTCAGCTTCTGGAAGTCGCTGGCGTTAAGCCG (SalI) | 1563–1519 | |||
| Rheb | NM_005614.4 | F | CAGAATTCAAGATGCCGCAGTCCAAGTC (EcoRI) | 382–401 | 558 |
| R | TTGTCGACTCACATCACCGAGCATGAAG (SalI) | 939–920 |
Primers used for creating pcDNA3.1-p17, pcDNA3.1-p-171-118, pCI-neo-PRAS40, pCI-neo-PRAS40 T246A, and pCI-neo-Rheb vectors for transfection.
F, forward; R, reverse.
Underlines indicate restriction enzyme digestion sites in the primers.
Coimmunoprecipitation assays.
To understand whether ARV p17 protein affects the interaction between mTOR complex proteins as well as Beclin 1 and class III PI3K, immunoprecipitation was performed using Millipore's Catch & Release V2.0 (Upstate Biotechnology, Lake Placid, NY, USA) according to the manufacturer's protocol. Vero cells were seeded in a 6-well plate containing 10% FBS in MEM until the cells formed a monolayer. The cells were transfected with the p17 gene for 24 h. The transfected cells were washed twice with phosphate-buffered saline (PBS) to remove the medium. Trypsin was added at a concentration of 0.5%, cultures were placed in a 37°C incubator to digest the cell adhesion proteins, and then medium containing 10% fetal bovine serum was added to terminate the trypsin activity. After the cells were evenly dispersed, samples were centrifuged at 500 × g for 8 min at 4°C, the supernatant was removed, and the culture medium was removed by washing with PBS. The protein was quantified and stored in a refrigerator at 4°C. Five hundred micrograms of total proteins collected from each sample was incubated with 4 μg of p17, Rheb, mTOR, and raptor antibodies or rabbit or mouse IgG (negative control) at 4°C for 24 h. The immunoprecipitated proteins were separated by 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) followed by Western blotting analysis with the indicated antibodies.
Electrophoresis and Western blotting assays.
The cells were seeded in a 6-well cell culture dish 1 day before infection with virus or transfection with plasmid as described above. The collected cells were washed twice with 1× PBS and lysed with lysis buffer (Cell Signaling, Danvers, MA, USA). The Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA) was used according to the manufacturer's protocol to determine the concentration of solubilized protein in cell lysates. An equal amount of sample was mixed with 2.5× Laemmli loading buffer and boiled in a water bath for 10 min. The samples were electrophoresed by 10% SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane. The appropriate primary antibody and horseradish peroxidase secondary antibody conjugate were used to check protein expression. After membrane incubation with enhanced chemiluminescence (ECL Plus) reagent (Amersham Biosciences, Little Chalfont, United Kingdom), the results were detected on X-ray film (Kodak, Rochester, NY, USA). ImageJ was used to calculate the amount of target protein.
Immunofluorescence staining.
An active mTOR is located on the surface of lysosomes. To study whether p17 affects the accumulation of mTOR on lysosome, Vero cells were mock transfected or transfected with the pcDNA3.1-p17 vector for 24 h. The cells were washed thoroughly with PBS and fixed in 4% formaldehyde. The cells were incubated with primary antibodies (mTOR and the lysosome marker LAMP1) at 4°C overnight. The cells were washed with PBST (PBS containing Triton 20) and incubated with a fluorescent dye-conjugated secondary antibody (1:500) for 2 h at room temperature. Then, after five washes with PBST, the cells were fixed with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA) and observed under confocal microscopy. For quantifying the colocalization of mTOR and lysosome, at least 10 regions were taken from each sample in each experiment, with 1 or 2 cells in each region. The quantification of colocalization was performed using ImageJ software.
LC3 puncta.
To study whether ARV or p17 can induce autophagy of A549 and HeLa cancer cells, cells were transfected with the mCherry-LC3 vector, and LC3 puncta were observed under fluorescence microscopy. Cancer cell lines were infected with ARV at an MOI of 10 or transfected with the pcDNA3.1-p17 vectors. Aside from these treatments, cells were also treated with TG (5 μM). Cells were fixed with 4% paraformaldehyde in PBS for 1 h at room temperature, followed by soaking in PBS with 0.3% Triton X-100 for 10 min. After washing with PBS, the cells were blocked with SuperBlock T20 (PBS) blocking buffer (Thermo Fisher Scientific, Waltham, MA, USA) at 4°C for 30 min. Cell nuclei were stained with DAPI for 10 min in the dark, followed by observation with a BX51 fluorescence microscope. The coverslips were washed with PBS three times at room temperature and then mounted onto glass slides using ibidi mounting medium (ibidi GmbH, Lochhamer Schlag, Germany).
Isotope labeling.
Relative translation levels of cellular proteins at the indicated time points in mock-, pcDNA3.1-, and pcDNA3.1-p17-transfected DF-1 cells were determined using pulse-chase labeling. Cells that had been mock transfected or transfected with the pcDNA3.1-p17 vector at different time points were labeled with [35S]methionine. After labeling, cells were washed twice with PBS and lysed with 70 μL of 5× Laemmli loading dye. Cells were harvested by scraping and boiled for 10 min. Equal amounts of samples were analyzed by 10% SDS-PAGE. A set of gels were transferred to polyvinylidene fluoride membranes to detect actin content and the expression level of p17. Labeling results were obtained from exposing dried gels to X-ray film (Kodak, Rochester, NY, USA). Cellular protein band intensity was assessed in relation to actin to quantify p17-modulated cellular translation shutoff.
Determination of virus titer.
To further investigate whether ARVs infect the cancer cell lines A549 and HeLa, cells were infected with the ARV S1133 strain at an MOI of 10 for 48 h. Normal HFL-1 cells were included as a negative control. To determine the effect of the mTOR pathway on ARV replication, shRNAs were used to knock down p53, TSC2, and LC3 in ARV-infected Vero cells. Cells were transfected with shRNAs or induced for overexpression of PRAS40, PRAS40T246A, and Rheb for 6 h, followed by infection with ARV S1133 at an MOI of 10 for 24 h. In this study, cells were also pretreated with rapamycin (5 μM) for 2 h, followed by infection with ARV at MOI of 10 for 24 h. After collection of the extracellular supernatant, an equal volume of MEM or F-12K was added to each well, and the cell membrane was destroyed by freezing and thawing three times. The supernatant was centrifuged at 12,000 × g for 10 min at 4°C to collect the intracellular fraction and stored at −80°C for further virus titration. Virus titer was determined by an agar-covered plaque assay performed in triplicate as described previously (27).
Statistical analysis.
Duncan’s multiple range test (DMRT) using SPSS software (version 20.0) was used to evaluate the statistical significance of all data obtained in this study. Each value represents the mean and standard error (SE) from three independent experiments.
ACKNOWLEDGMENTS
This study was supported by grants from the Ministry of Science and Technology in Taiwan (109-2313-B-005-006-MY3), Taichung Veterans General Hospital and National Chung Hsing University (TCVGH-NCHU1117608), and the iEGG and Animal Biotechnology Center from The Feature Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan (111S0023A).
All authors made substantive intellectual contributions to the present study and approved the final manuscript. H.-J.L. conceived of the study and created the original hypothesis, wrote the paper, and supervised the project; J.-Y.L. and W.-R.H. performed most of the experiments. W.-R.H., J.-Y.L., T.-L.L., B.L.N., and H.-J.L. analyzed data; H.-J.L. and B.L.N. revised and edited the manuscript.
We declare that we have no competing interests.
Contributor Information
Hung-Jen Liu, Email: hjliu5257@nchu.edu.tw.
Susana López, Instituto de Biotecnologia/UNAM.
REFERENCES
- 1.Betz C, Hall MH. 2013. Where is mTOR and what is it doing there? J Cell Biol 203:563–574. 10.1083/jcb.201306041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fingar DC, Blenis J. 2004. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 23:3151–3171. 10.1038/sj.onc.1207542. [DOI] [PubMed] [Google Scholar]
- 3.Wullschleger S, Loewith R, Hall MN. 2006. TOR signaling in growth and metabolism. Cell 124:471–484. 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 4.Guertin DA, Sabatini DM. 2007. Defining the role of mTOR in cancer. Cancer Cell 12:9–22. 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 5.Sekulic A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM, Abraham RTA. 2000. direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res 60:3504–3513. [PubMed] [Google Scholar]
- 6.Wang L, Rhodes CJ, Lawrence JC. 2006. Activation of mammalian target of rapamycin (mTOR) by insulin is associated with stimulation of 4EBP1 binding to dimeric mTOR complex 1. J Biol Chem 281:24293–24303. 10.1074/jbc.M603566200. [DOI] [PubMed] [Google Scholar]
- 7.Zinzalla V, Stracka D, Oppliger W, Hall MN. 2011. Activation of mTORC2 by association with the ribosome. Cell 144:757–768. 10.1016/j.cell.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 8.Dibble CC, Cantley LC. 2015. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol 25:545–555. 10.1016/j.tcb.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dan HC, Sun M, Yang L, Feldman RI, Sui XM, Ou CC, Nellist M, Yeung RS, Halley DJ, Nicosia SV, Pledger WJ, Cheng JQ. 2002. Phosphatidylinositol 3-kinase/Akt pathway regulates tuberous sclerosis tumor suppressor complex by phosphorylation of tuberin. J Biol Chem 277:35364–35370. 10.1074/jbc.M205838200. [DOI] [PubMed] [Google Scholar]
- 10.Inoki K, Li Y, Zhu T, Wu J, Guan KL. 2002. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4:648–657. 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 11.Potter CJ, Pedraza LG, Xu T. 2002. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol 4:658–665. 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- 12.Patel PH, Thapar N, Guo L, Martinez M, Maris J, Gau CL, Lengyel JA, Tamanoi F. 2003. Drosophila Rheb GTPase is required for cell cycle progression and cell growth. J Cell Sci 116:3601–3610. 10.1242/jcs.00661. [DOI] [PubMed] [Google Scholar]
- 13.Saucedo LJ, Gao X, Chiarelli DA, Li L, Pan D, Edgar BA. 2003. Rheb promotes cell growth as a component of the insulin/TOR signaling network. Nat Cell Biol 5:566–571. 10.1038/ncb996. [DOI] [PubMed] [Google Scholar]
- 14.Stocker H, Radimerski T, Schindelholz B, Wittwer F, Belawat P, Daram P, Breuer S, Thomas G, Hafen E. 2003. Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat Cell Biol 5:559–566. 10.1038/ncb995. [DOI] [PubMed] [Google Scholar]
- 15.Bai X, Ma D, Liu A, Shen X, Wang QJ, Liu Y, Jiang Y. 2007. Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science 318:977–980. 10.1126/science.1147379. [DOI] [PubMed] [Google Scholar]
- 16.Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S, Yoshino K, Hara K, Tanaka N, Avruch J, Yonezawa K. 2003. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem 278:15461–15464. 10.1074/jbc.C200665200. [DOI] [PubMed] [Google Scholar]
- 17.Schalm SS, Fingar DC, Sabatini DM, Blenis J. 2003. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr Biol 13:797–806. 10.1016/s0960-9822(03)00329-4. [DOI] [PubMed] [Google Scholar]
- 18.Oshiro N, Takahashi R, Yoshino K, Tanimura K, Nakashima A, Eguchi S, Miyamoto T, Hara K, Takehana K, Avruch J, Kikkawa U, Yonezawa K. 2007. The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J Biol Chem 282:20329–20339. 10.1074/jbc.M702636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L, Harris TE, Roth RA, Lawrence JC. 2007. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J Biol Chem 282:20036–20044. 10.1074/jbc.M702376200. [DOI] [PubMed] [Google Scholar]
- 20.Kozak RA, Hattin L, Biondi MJ, Corredor JC, Walsh S, Xue-Zhong M, Manuel J, McGilvray ID, Morgenstern J, Lusty E, Cherepanov V, McBey BA, Leishman D, Feld JJ, Bridle B, Nagy É. 2017. Replication and oncolytic activity of an avian orthoreovirus in human hepatocellular carcinoma cells. Viruses 9:90. 10.3390/v9040090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiu HC, Huang WR, Liao TL, Chi PI, Nielsen BL, Liu JH, Liu HJ. 2018. Mechanistic insights into avian reovirus p17-modulated suppression of cell cycle CDK-cyclin complexes and enhancement of p53 and cyclin H interaction. J Biol Chem 293:12542–12562. 10.1074/jbc.RA118.002341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cai R, Meng G, Li Y, Wang W, Diao Y, Zhao S, Feng Q, Tang Y. 2019. The oncolytic efficacy and safety of avian reovirus and its dynamic distribution in infected mice. Exp Biol Med (Maywood) 244:983–991. 10.1177/1535370219861928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chi PI, Huang WR, Lai IH, Cheng CY, Liu HJ. 2013. The p17 nonstructural protein of avian reovirus triggers autophagy enhancing virus replication via activation of phosphatase and tensin deleted on chromosome 10 (PTEN) and AMP-activated protein kinase (AMPK), as well as dsRNA-dependent protein kinase (PKR)/eIF2α signaling pathways. J Biol Chem 288:3571–3584. 10.1074/jbc.M112.390245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang WR, Chiu HC, Liao TL, Chuang KP, Shih WL, Liu HJ. 2015. Avian reovirus protein p17 functions as a nucleoporin Tpr suppressor leading to activation of p53, p21 and PTEN and inactivation of PI3K/AKT/mTOR and ERK signaling pathways. PLoS One 10:e0133699. 10.1371/journal.pone.0133699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiu HC, Huang WR, Liao TL, Wu HY, Munir M, Shih WL, Liu HJ. 2016. Suppression of vimentin phosphorylation by the avian reovirus p17 through inhibition of CDK1 and Plk1 impacting the G2/M phase of the cell cycle. PLoS One 11:e0162356. 10.1371/journal.pone.0162356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiu HC, Huang WR, Wang YY, Li JY, Liao TL, Nielsen BL, Liu HJ. 2019. Heterogeneous nuclear ribonucleoprotein A1 and lamin A/C modulate nucleocytoplasmic shuttling of avian reovirus p17. J Virol 93:e00851-19. 10.1128/JVI.00851-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang WR, Li JY, Wu YY, Liao TL, Nielsen BL, Liu HJ. 2022. p17-modulated Hsp90/Cdc37 complex governs avian reovirus replication by chaperoning p17, which promotes viral protein synthesis and accumulation of proteins σA and σC in viral factories. J Virol 96:e00074-22. 10.1128/jvi.00074-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang WR, Li JY, Liao TL, Yeh CM, Wang CY, Wen HW, Hu NJ, Wu YY, Hsu CY, Chang YK, Chang CD, Nielsen BL, Liu HJ. 2022. Molecular chaperone TRiC governs avian reovirus replication by preventing outer-capsid protein σC and core protein σA and non-structural protein σNS from ubiquitin-proteasome degradation. Vet Microbiol 264:109277. 10.1016/j.vetmic.2021.109277. [DOI] [PubMed] [Google Scholar]
- 29.Pallares-Cartes C, Cakan-Akdogan G, Teleman AA. 2012. Tissue-specific coupling between insulin/IGF and TORC1 signaling via PRAS40 in Drosophila. Dev Cell 22:172–182. 10.1016/j.devcel.2011.10.029. [DOI] [PubMed] [Google Scholar]
- 30.Xiong X, Xie R, Zhang H, Gu L, Xie W, Cheng M, Jian Z, Kovacina K, Zhao H. 2014. PRAS40 plays a pivotal role in protecting against stroke by linking the Akt and mTOR pathways. Neurobiol Dis 66:43–52. 10.1016/j.nbd.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. 2001. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep 2:330–335. 10.1093/embo-reports/kve061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kovacina KS, Park GY, Bae SS, Guzzetta AW, Schaefer E, Birnbaum MJ, Roth RA. 2003. Identification of a proline-rich Akt substrate as a 14–3-3 binding partner. J Biol Chem 278:10189–10194. 10.1074/jbc.M210837200. [DOI] [PubMed] [Google Scholar]
- 33.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. 2007. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 25:903–915. 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 34.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. 2010. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141:290–303. 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang WR, Chi PI, Chiu HC, Hsu JL, Nielsen BL, Liao TL, Liu HJ. 2017. Avian reovirus p17 and sigma A act cooperatively to downregulate Akt by suppressing mTORC2 and CDK2/cyclin A2 and upregulating proteasome PSMB6. Sci Rep 7:5226. 10.1038/s41598-017-05510-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19:5720–5728. 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tseng HH, Huang WR, Cheng CY, Chiu HC, Liao TL, Nielsen BL, Liu HJ. 2020. Aspirin and 5-aminoimidazole-4-carboxamide riboside attenuate bovine ephemeral fever virus replication by inhibiting BEFV-induced autophagy. Front Immunol 11:556838. 10.3389/fimmu.2020.556838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ji WT, Wang L, Lin RC, Huang WR, Liu HJ. 2009. Avian reovirus influences phosphorylation of several factors involved in host protein translation including eukaryotic translation elongation factor 2 (eEF2) in Vero cells. Biochem Biophys Res Commun 384:301–305. 10.1016/j.bbrc.2009.04.116. [DOI] [PubMed] [Google Scholar]
- 39.Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. 1994. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369:756–758. 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y, Feng M, Chen H, Yang G, Qiu J, Zhao F, Cao Z, Luo W, Xiao J, You L, Zheng L, Zhang T. 2020. Mechanistic target of rapamycin in the tumor microenvironment and its potential as a therapeutic target for pancreatic cancer. Cancer Lett 485:1–13. 10.1016/j.canlet.2020.05.003. [DOI] [PubMed] [Google Scholar]
- 41.Maekawa H, Kawai S, Nishio M, Nagata S, Jin Y, Yoshitomi H, Matsuda S, Toguchida J. 2020. Prophylactic treatment of rapamycin ameliorates naturally enveloping and episode-induced heterotopic ossification in mice expressing human mutant ACVR1. Orphanet J Rare Dis 15:122. 10.1186/s13023-020-01406-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhan Y, Yu S, Yang S, Qiu X, Meng C, Tan L, Song C, Liao Y, Liu W, Sun Y, Ding C. 2020. Newcastle disease virus infection activates PI3K/Akt/mTOR and p38 MAPK/Mnk1 pathways to benefit viral mRNA translation via interaction of the viral NP protein and host eIF4E. PLoS Pathog 16:e1008610. 10.1371/journal.ppat.1008610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edgil D, Polacek C, Harris E. 2006. Dengue virus utilizes a novel strategy for translation initiation when cap-dependent translation is inhibited. J Virol 80:2976–2986. 10.1128/JVI.80.6.2976-2986.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, Sonenberg N. 1999. Regulation of 4EBP1 phosphorylation: a novel two-step mechanism. Genes Dev 13:1422–1437. 10.1101/gad.13.11.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beretta L, Gingras AC, Svitkin YV, Hall MH, Sonenberg N. 1996. Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO J 15:658–664. 10.1002/j.1460-2075.1996.tb00398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tahara SM, Dietlin TA, Bergmann CC, Nelson GW, Kyuwa S, Anthony RP, Stohlman SA. 1994. Coronavirus translational regulation: leader affects mRNA efficiency. Virology 202:621–630. 10.1006/viro.1994.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hashemolhosseini S, Nagamine Y, Morley SJ, Desrivières S, Mercep L, Ferrari S. 1998. Rapamycin inhibition of the G1 to S transition is mediated by effects on cyclin D1 mRNA and protein stability. J Biol Chem 273:14424–14429. 10.1074/jbc.273.23.14424. [DOI] [PubMed] [Google Scholar]
- 48.Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. 2010. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci USA 107:9660–9664. 10.1073/pnas.1002298107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Codogno P, Meijer AJ. 2005. Autophagy and signaling. Their role in cell survival and cell death. Cell Death Differ 12:1509–1518. 10.1038/sj.cdd.4401751. [DOI] [PubMed] [Google Scholar]