

ABSTRACT
Following acute infection, herpes simplex virus 1 (HSV-1) establishes lifelong latency in neurons. The latency associated transcript (LAT) is the only viral gene abundantly expressed during latency. Wild-type (WT) HSV-1 reactivates more efficiently than LAT mutants because LAT promotes establishment and maintenance of latency. While sensory neurons in trigeminal ganglia (TG) are important sites for latency, brainstem is also a site for latency and reactivation from latency. The principal sensory nucleus of the spinal trigeminal tract (Pr5) likely harbors latent HSV-1 because it receives afferent inputs from TG. The locus coeruleus (LC), an adjacent brainstem region, sends axonal projections to cortical structures and is indirectly linked to Pr5. Senescent cells accumulate in the nervous system during aging and accelerate neurodegenerative processes. Generally senescent cells undergo irreversible cell cycle arrest and produce inflammatory cytokines and chemokines. Based on these observations, we hypothesized HSV-1 influences senescence and inflammation in Pr5 and LC of latently infected mice. This hypothesis was tested using a mouse model of infection. Strikingly, female but not age-matched male mice latently infected with a LAT null mutant (dLAT2903) exhibited significantly higher levels of senescence markers and inflammation in LC, including cell cycle inhibitor p16, NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3), IL-1α, and IL-β. Conversely, Pr5 in female but not male mice latently infected with WT HSV-1 or dLAT2903 exhibited enhanced expression of important inflammatory markers. The predilection of HSV-1 to induce senescence and inflammation in key brainstem regions of female mice infers that enhanced neurodegeneration occurs.
IMPORTANCE HSV-1 (herpes simplex virus 1), an important human pathogen, establishes lifelong latency in neurons in trigeminal ganglia and the central nervous system. In contrast to productive infection, the only viral transcript abundantly expressed in latently infected neurons is the latency associated transcript (LAT). The brainstem, including principal sensory nucleus of the spinal trigeminal tract (Pr5) and locus coeruleus (LC), may expedite HSV-1 spread from trigeminal ganglia to the brain. Enhanced senescence and expression of key inflammatory markers were detected in LC of female mice latently infected with a LAT null mutant (dLAT2903) relative to age-matched male or female mice latently infected with wild-type HSV-1. Conversely, wild-type HSV-1 and dLAT2903 induced higher levels of senescence and inflammatory markers in Pr5 of latently infected female mice. In summary, enhanced inflammation and senescence in LC and Pr5 of female mice latently infected with HSV-1 are predicted to accelerate neurodegeneration.
KEYWORDS: brainstem, HSV-1, LAT, immune senescence, inflammation, latency
INTRODUCTION
Following herpes simplex virus 1 (HSV-1) acute infection of craniofacial mucosal surfaces, latency is established in neurons. For example, sensory neurons within trigeminal ganglia (TG) are a major site for latency (reviewed in 1, 2). Furthermore, neurons within the central nervous system also harbor viral DNA during latency (3–6). In contrast to productive infection of cultured cells, abundant viral protein expression and infectious virus are not readily detected during latency. The only abundantly expressed viral transcript expressed during latency is the latency-associated transcript (LAT) (1, 2). LAT is considered to be a noncoding RNA (2) because it expresses an unstable 8.3 kb transcript, a stable intron, six micro-RNAs, two small noncoding RNAs, and 3 transcripts antisense to the major LAT. LAT products promote lifelong latency by inhibiting apoptosis (7, 8), promoting neuronal differentiation (9, 10), and impairing productive infection (11, 12). Genes that inhibit apoptosis can substitute for LAT and restore wild-type reactivation levels (13–15), confirming that the anti-apoptosis functions of LAT are crucial for the latency-reactivation cycle. The latency-reactivation cycle is operationally divided into three distinct steps: establishment, maintenance, and reactivation (1, 2). In general, LAT promotes establishment and maintenance of latency (1, 2, 16): consequently, a pool of latently infected neurons can support reactivation from latency in small animal models of infection.
HSV-1 encephalitis (HSE) has a predilection for temporal and frontal lobes but can also affect the brainstem (17–20). Approximately 2/3 of HSE cases are due to reactivation from latency (21), which increases morbidity and reduces the effectiveness of antiviral treatment (22–24). Some patients with a predisposition of encephalitis in the brainstem have mutations in the debranching RNA lariats 1 gene (DBR1), the only RNA lariat debranching enzyme that has been identified (25). This study also revealed that cells expressing the DBR1 mutation are more susceptible to HSV-1 infection.
In mouse models of HSV-1 infection, brainstem is a significant site for viral replication and reactivation from latency (3, 26, 27). Previous studies used the entire brainstem for their studies and did not focus on brainstem regions that have synaptic connections with TG. For example, the principal sensory nucleus of the spinal trigeminal tract (Pr5) receives afferent projections from trigeminal ganglia (TG) (see Fig. 1A and B, for schematic), suggesting HSV-1 readily spreads to this brainstem region. Synaptic connections exist between the locus coeruleus (LC) and the spinal trigeminal tract, which contains Pr5 (28), suggesting synaptic projections of LC are indirectly linked to TG. Notably, LC projections reach all parts of the brain except basal ganglia (29). Furthermore, LC is the principal site for norepinephrine synthesis in the brain, and primarily contains medium-sized neurons with melanin granules (30). Conversely, Pr5 contains many motor neurons (31). Threatening or stressful stimuli trigger norepinephrine release from LC and adrenal production of glucocorticoids, including cortisol (32, 33). These observations are relevant because HSV-1 productive infection and reactivation from latency are enhanced by stress, as mimicked by the synthetic corticosteroid dexamethasone (34, 35).
FIG 1.

Schematic of mouse brain, TG, and brainstem. (A) Sagittal section showing mouse brain, TG, LC, and Pr5. (B) Coronal section of mouse brain and location of LC and Pr5 in brainstem.
Based on the observations discussed above, the objectives of this study were focused on addressing two questions: (i) Does HSV-1 influence senescence and neuroinflammation in Pr5 and LC brainstem regions; and (ii) Does LAT influence virus–host interactions in Pr5 and LC? The logic for comparing the effects of a LAT null mutant and that of wild-type (WT) HSV-1 is that LAT mediates the latency-reactivation cycle by enhancing survival of infected neurons.
RESULTS
HSV-1 enhances senescence in the locus coeruleus of female mice.
Initial studies examined senescence in Pr5 and LC in 8-week-old C57BL/6 mice latently infected with a neurovirulent strain of HSV-1 (McKrae strain) or a LAT null mutant (dLAT2903). Cellular senescence is a multistep process by which proliferating cells, but generally not neurons, undergo irreversible growth arrest and resistance to apoptosis (reviewed in 36, 37). Senescence is induced by cellular damage or stress: for example, telomere shortening, epigenetically induced senescence, oxidative stress, oncogene activation, unresolved DNA damage, or mitochondrial dysfunction. Interestingly, many senescent cells secrete proinflammatory proteins that produce chronic inflammation (reviewed in 38). A recent study demonstrated glial cells are more likely to senesce during the aging process (39). A senescence-associated (SA) β-gal staining procedure was used for these initial studies because it is a reliable method to identify senescent cells in tissue (40). When compared to LC from brainstems of uninfected female (Fig. 2A) and male mice (Fig. 2D), SA β-gal staining was clearly detected in LC of male and female mice latently infected with WT HSV-1 (Fig. 2B and E). Approximately 40 times more β-gal+ cells were detected in LC of female mice latently infected with dLAT2903 (Fig. 2C) when compared to LC from uninfected mice. The number of SA β-gal+ cells in LC of female mice latently infected with dLAT2903 was approximately 2 times higher than LC of female mice latently infected with WT HSV-1. In LC of male mice latently infected with dLAT2903, there were approximately 20-fold more SA β-gal+ cells relative to uninfected mice. However, the difference between dLAT2903 versus WT HSV-1 was not as dramatic in LC of latently infected male mice.
FIG 2.

Comparison of senescence in LC of mice latently infected with HSV-1 versus uninfected controls. Representative images of SA-β-Gal staining showing positive cells in the LC of the brainstem in females (top panel) versus males (bottom panel). Age-matched uninfected mice were compared to mice latently infected with wild-type HSV-1 (McKRae strain) or dLAT2903 (dLAT).
In contrast to LC, SA β-gal staining was not as pronounced in Pr5 of female or male mice latently infected with WT or dLAT2903 (Fig. 3). However, there appeared to be slightly more SA β-gal+ cells in female or male mice latently infected with dLAT2903. These findings revealed WT HSV-1 and dLAT2903 preferentially induced senescence in the LC brainstem region of female mice and to a lesser extent in LC of male mice.
FIG 3.

Comparison of senescence in Pr5 of mice latently infected with HSV-1 versus uninfected controls. Representative images of SA-β-Gal showing positive cells in the Pr5 of the brainstem in females (top panel) versus males (bottom panel). Age-matched uninfected mice were compared to mice latently infected with wild-type HSV-1 (McKRae strain) or dLAT2903 (dLAT).
HSV-1 increases expression of key senescence markers in the LC of female mice.
Additional studies measured RNA levels of cellular genes (p16, p21, and p53) that mediate senescence. The p16 and p53 genes encode proteins that inhibit cell cycle progression, and these proteins are tumor suppressors. Hence, these genes are reliable markers for the senescence phenotype (41, 42). Consequently, we tested whether HSV-1 (WT or dLAT2903) induced expression of p16 or p21/p53. In females latently infected with WT HSV-1, there was an approximately 20-fold increase in p16 mRNA expression in the Pr5 region, but only a 3-fold increase in Pr5 of female mice latently infected with dLAT2903 (Fig. 4). Significant increases in p21 or p53 RNA levels were not detected in Pr5 of male or female mice latently infected with WT or dLAT2903.
FIG 4.

Effects of HSV-1 latent infection on senescence in Pr5 or LC. Eight-week-old mice (males and females) were infected with ~2 × 105 infectious virus particles as described in Materials and Methods. We compared gene expression between uninfected mice (U) and those infected with wild type (WT; McKRae strain) or dLAT2903 (dLAT) using primers described in Table 1. Gene expression changes in senescence markers p16, p21, or p53 were examined (n = 3-4 mice/group). * denotes significant difference (P < 0.05) between uninfected aged mice; a denotes significant difference (P < 0.05) between WT.
In LC of female but not male mice latently infected with dLAT2903, there was a striking 87-fold increase in p16 mRNA expression compared to a 10-fold increase with WT virus. Significant increases of p21 mRNA expression in LC were not identified in male or female mice latently infected with WT or dLAT2903. p53 RNA levels were significantly upregulated in LC of female, but not male, mice latently infected with dLAT2903. It was surprising to find that p53, but not p21, was stimulated in LC of female mice latently infected with dLAT2903, because the p21 promoter is transactivated by p53 (43). The ability of p53 to transactivate the p21 promoter, versus the Bax or Fas promoter, is dependent on tissue- and cell-specific factors (44). For example, the monocytic leukemia zinc finger (MOZ) transcriptional coactivator must interact with p53 to activate p21 expression (45), confirming p53 expression can increase dramatically but not activate p21 expression. In summary, these findings confirmed HSV-1 selectively induced senescence in LC of female mice latently infected with dLAT2903, as judged by significant increased levels of p16 and p53 mRNA. These studies also revealed that dLAT2903 preferentially induced senescence in LC of latently infected female mice whereas dLAT2903 or WT HSV-1 induced expression of key senescence genes in Pr5.
Preferential increase of NLRP3 expression in LC of female mice latently infected with dLAT2903.
NLRP3 (NOD-, LRR-, and pyrin domain-containing protein 3) RNA levels were examined because this mRNA is translated into an intracellular sensor that detects pathogen associated molecular patterns (PAMPS) and damage associated molecular patterns (DAMPS), which can activate the NLRP3 inflammasome (reviewed in 46) (Fig. 5A). In addition to activation by viral infections (47, 48), NLRP3 activation correlates with functional decline during aging, including Alzheimer’s disease (AD), interferon-alpha induction (IFN-α), and increased levels of reactive oxygen species (ROS) (46). NLRP3 inflammasome activation leads to caspase 1-dependent release of proinflammatory cytokines, interleukin 1 beta (IL-1β), and IL-18, for example, and gasdermin d-mediated pyroptotic cell death, a pro-inflammatory form of cell death. Strikingly, NLRP3 RNA expression was significantly upregulated in the Pr5 region of female mice latently infected with WT or dLAT2903 but not in age-matched males or uninfected mice (Fig. 5B). Female mice latently infected with dLAT2903, but not WT HSV-1, exhibited a significant increase in NLRP3 RNA expression (approximately 30-fold increase) in LC. In male mice latently infected with dLAT2903, a modest but significant increase in NLRP3 expression was detected in LC (Fig. 5B).
FIG 5.
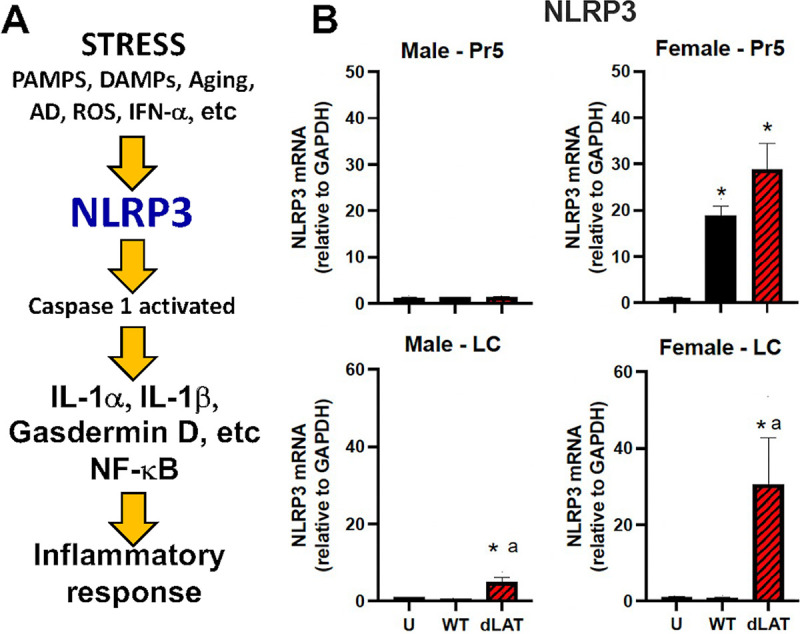
Effects of HSV-1 latent infection on NLRP3 expression in Pr5 or LC. (A) Schematic of key events leading to NLRP3 activation and the resulting inflammation. (B) NLRP3 RNA expression (n = 3–4 mice/group) was examined using primers described in Table 1. Expression was compared between uninfected mice (U) and those infected with WT HSV-1 (McKRae strain) or dLAT2903 (dLAT). * denotes significant difference (P < 0.05) between uninfected aged mice; a denotes significant difference (P < 0.05) between WT.
Preferential increase of senescence-associated secretory phenotype expression in latently infected female mice.
Senescent cells can affect neighboring cells in a paracrine manner through the senescence-associated secretory phenotype (SASP; Fig. 1A) (41). For example, interleukin-1 alpha (IL-1α), IL-1β, and tumor necrosis factor alpha (TNF-α) are key SASP and inflammatory markers. Since NLRP3 RNA levels were preferentially increased in Pr5 and LC of female mice (Fig. 5), we predicted IL-1α and IL-1β would also be increased in latently infected female mice but to a lesser degree in males. Monocyte chemoattractant protein-1 (MCP1), another SASP factor, is an important chemokine that regulates migration and infiltration of monocytes/macrophages (reviewed in 49), and was also examined.
To test these predictions, male and female C57BL/6 mice were infected with WT HSV-1 or dLAT2903 and mRNA levels of the denoted SASP factors examined. IL-1α mRNA levels were significantly increased in Pr5 of female mice latently infected with WT HSV-1 when compared to males or females latently infected with dLAT2903 and uninfected mice (Fig. 6). IL-1β, TNF-α, and MCP1 mRNA levels were also significantly higher in Pr5 of female mice latently infected with WT or dLAT2903.
FIG 6.

Effects of HSV-1 latent infection on inflammation in Pr5 or LC. Gene expression of IL-1α, IL-1β, TNF-α, and MCP1 (n = 3-4 mice/group) was compared in uninfected mice (U), and those infected with WT HSV-1 (McKRae strain) or dLAT2903 (dLAT) using primers described in Table 1. * denotes significant difference (P < 0.05) between uninfected aged mice; a denotes significant difference (P < 0.05) between WT.
In contrast to the results observed in Pr5, IL-1α, IL-1β, and TNF-α RNA levels were significantly increased in LC of female mice latently infected with dLAT2903 but not WT HSV-1. Conversely, MCP-1 RNA levels were not significantly different in LC of uninfected males or females or latently infected males or females. Collectively, these studies revealed that expression of inflammatory cytokines (IL-1α, IL-1β, and TNF-α) were detected in LC of female mice latently infected with dLAT2903. Except for IL-1α, WT HSV-1 and dLAT2903 induced higher levels of inflammatory cytokines in Pr5 of latently infected female mice. Male mice latently infected with WT or dLAT2903 did not induce dramatic differences in the SASP factors that were examined. These studies support the concept that HSV-1 preferentially induces expression of inflammatory cytokines in LC and Pr5 of female mice. These studies also suggest LAT products impair expression of senescence and inflammatory cytokines in LC, but not as consistently in Pr5, of latently infected female mice.
HSV-1 increases expression of a neuronal activity related gene and ROS in brainstem.
To test whether neuronal activity was altered in mice latently infected with HSV-1, we measured Fos-related antigen 1 (Fra-1) RNA levels because it is a well-accepted marker for neuronal activation (50). Significantly higher levels of Fra-1 mRNA expression were detected in Pr5 of female C57BL/6 mice latently infected with WT HSV-1 or dLAT2903. In LC, only females latently infected with dLAT2903 exhibited significantly higher levels of Fra-1 RNA levels (Fig. 7). HSV-1 infection was reported to increase reactive oxygen species ROS (51, 52). To investigate the defensive antioxidant response to viral infection, we measured expression of the master transcriptional regulator of ROS, nuclear factor erythroid 2-related factor 2 (Nrf2), and its target antioxidant gene, NAD(P)H:quinone acceptor oxidoreductase type 1 (NQO1). Both Nrf2 and NQO1 mRNA expression were significantly upregulated in Pr5 of female, but not male, mice latently infected with WT HSV-1 or dLAT2903. In contrast to the Pr5 results, Fra-1, Nrf2, and NQO1 RNA expression was significantly higher in the LC of female mice latently infected with dLAT2903, but not WT HSV-1.
FIG 7.

Effects of HSV-1 latent infection on neural activity and ROS in Pr5 or LC. Gene expression of Fra-1, NRF2, and NQO1 was compared in uninfected mice (U) and those infected with WT HSV-1 (McKRae strain) or dLAT2903 (dLAT) (n = 3–4 mice/group). * denotes significant difference (P < 0.05) between uninfected aged mice; a denotes significant difference (P < 0.05) between WT.
Comparison of HSV-1 acute replication in males versus females.
To examine whether enhanced senescence and inflammation in LC of female mice infected with dLAT2903 was merely due to sex-specific differences in viral replication during acute infection, virus shedding was examined in the ocular cavity during acute infection (Fig. 8). Significant differences in viral replication were not detected in females versus males following infection with WT HSV-1 or dLAT2903. Furthermore, significant differences in WT HSV-1 versus dLAT2903 replication were not observed, which is consistent with previously published studies (53, 54).
FIG 8.
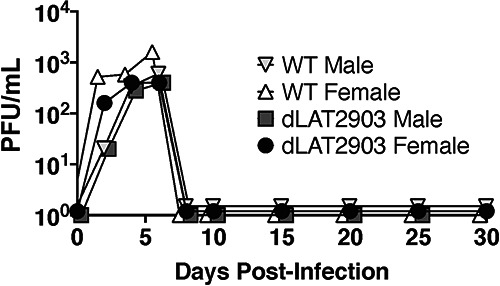
Ocular swabs from HSV-1 infected mice. Eight-week-old C57BL/6 mice were ocularly infected with ~2 × 105 PFU either HSV-1 strain McKrae or dLAT2903 without scarification as previously described (34). Ocular swabs were obtained every other day for 10 days and then every 5 days for a total of 30 days postinfection (n = 5 mice/group). Samples were freeze-thawed three times prior to plaquing on Vero cells to measure titers of infectious virus.
Quantification of viral loads in TG, Pr5, and LC of latently infected mice.
Additional studies compared viral DNA levels in TG, LC, and Pr5 during acute infection, 4 days after infection (4 dpi) and latency. With respect to TG, there were no significant differences in TG at 4 dpi or latency when males or females were infected with WT HSV-1 versus dLAT2903 (Fig. 9A). However, there were higher viral DNA levels at 4 dpi when compared to latency, which is a trend we consistently find. With respect to LC, significantly higher levels of viral DNA were detected in male mice infected with dLAT2903 at 4 dpi when compared to male mice infected with WT HSV-1 (Fig. 9B). Furthermore, significantly more viral DNA was detected in LC of male mice versus females infected with dLAT2903. During latency, significant differences in viral DNA levels were not observed in LC of males or females regardless of whether mice were infected with WT HSV-1 or dLAT2903. We consistently detected similar levels of viral DNA (WT or dLAT2903) in Pr5 of acutely infected male mice (Fig. 9C). Conversely, viral DNA was only detected in 2 out of 5 female mice acutely infected with HSV-1 (WT or dLAT2903). In summary, these studies revealed viral DNA levels were not dramatically different in males and females of LC or TG during latency. Conversely, viral DNA was sporadically detected in Pr5 of male and particularly in female mice during latency.
FIG 9.

Quantification of viral DNA in Pr5 and LC during acute infection and latency. Eight-week-old male (M) or female (F) C57BL/6 mice were ocularly infected with WT HSV-1 (n = 10) or dLAT2903 (n = 10). At 4 or 30 days postinfection, mice were euthanized and TG (Panel A), LC (Panel B), and Pr5 (Panel C) were harvested as described in materials and methods (n = 4 or 5 per group, per time point). DNA was prepared from tissues and qPCR performed using primers against HSV-1 gB and mouse GAPDH. Data are shown as the ratio of gB to GAPDH, max to min. *, P < 0.05 using one-way ANOVA with Tukey's multiple-comparison test.
DISCUSSION
These studies revealed that complex virus- and sex-specific interactions mediate the effect HSV-1 has on senescence and inflammation in Pr5 and LC of latently infected mice. Since Pr5 contains many motor neurons (31) but LC primarily contains medium-sized neurons with melanin granules (30), these compelling differences are likely to impact virus–host interactions. For example, detection of viral DNA in Pr5 during latency was not as consistent as in LC. This result supports the prediction that differences in neurons in Pr5 versus LC are important for mediating these differences. A schematic that summarizes the salient features of female- and LAT-specific effects of these novel findings is shown in Fig. 10. LC in female rats is larger, dendrites are denser and longer, and they establish more synaptic contacts than in males (reviewed in 55, 56). With respect to functional differences, corticotropin-releasing factor (CRF) receptors are expressed at higher levels in LC of female rats and mice (57, 58). This difference is significant because stressful stimuli increase levels of norepinephrine (NE), CRF release, adrenocorticotropic hormone (ACTH), and production of cortisol via the hypothalamic-pituitary-adrenal axis (32, 33). Consequently, CRF is 10–30 times more potent with respect to activating LC neurons in females following stressful stimuli (59). The enhanced effects of CRF in female LC neurons also activate the cyclic adenosine monophosphate (cAMP) and protein kinase A signaling cascade (60). Collectively, the heightened response to stressful stimuli in females is predicted to trigger viral activity in LC of females because of increased stress (35, 61, 62), and cyclic AMP levels (63) can increase viral replication and incidence of reactivation from latency. Although the mice used for this study were not purposely stressed, handling of mice during cage changing or infection and confinement in cages will likely increase stress levels.
FIG 10.

Schematic of working model for virus–host interactions in Pr5 and LC. Following a stressful stimulus, LC in female (♀) mice exhibit higher responses because they express higher levels of corticotropin-releasing factor (CRF) receptors. Hence, they release more norepinephrine (NE), CRF, and adrenocorticotropic hormone (ACTH). This culminates in higher levels of glucocorticoids, including cortisol via the hypothalamic-pituitary-adrenal (HPA) axis. Enhanced expression of “stress-mediated” signaling pathways are predicted to increase lytic cycle viral gene expression and inflammation. Whether reactivation from latency occurs is not known. LAT expression in LC neurons, but not Pr5 neurons, is important for restricting programmed cell death (PCD), including apoptosis, inflammation, and viral gene expression. These differences in LC neurons are predicted to enhance expression of key inflammatory and SASP transcripts relative to LC or Pr5 in males (♂). Paracrine effects are predicted to enhance inflammation in Pr5 of latently infected female mice.
While increased glucocorticoid levels are typically associated with an anti-inflammatory response (64), increased glucocorticoid levels transiently activate NLRP3 leading to production of IL-β and TNF-α (65). The proinflammatory properties of glucocorticoids are believed to sensitize innate immune responses following cellular stressors, including viral infections. Increased glucocorticoid levels can also lead to apoptosis (66), including neurons (67). The proximity of LC to Pr5 and other adjacent brainstem regions is expected to increase stress in the brainstem of females due to paracrine effects. Additional unknown sex-specific cellular factors in Pr5 and LC are also expected to mediate the effects observed in our studies.
In addition to cellular factors mediating the sex-specific events described in this study, expression of LAT products, directly or indirectly, restrict programmed cell death, inflammation, and viral gene expression in LC of female mice (Fig. 10). Support for this prediction comes from higher expression of IL-1-β, TNF-α, Fra-1, NLRP3, NRF2, and NQO1 in LC of female mice latently infected with the LAT null mutant (dLAT2903) versus WT HSV-1. Conversely, these dramatic differences were not observed in LC of age-matched males latently infected with dLAT2903 or WT HSV-1. Furthermore, a LAT-specific effect was not generally observed in Pr5 of latently infected mice, which correlates with sporadic establishment and/or maintenance of latency in Pr5. Support for this prediction comes from studies demonstrating WT HSV-1 and dLAT2903 induced expression of p16, p53, IL-α, IL-1-β, TNF-α, Fra-1, NLRP3, NRF2, and NQO1 to similar levels in Pr5 of latently infected female, but not male, mice. Certain LAT functions are predicted to restrict inflammation and cell death in LC of latently infected female mice. For example, LAT influences the interferon response in mouse TG and a human neuronal cell line (SK-N-SH), but not in human embryonic lung cells (68). This finding was confirmed by an independent study that concluded LAT downregulates certain components of the type I interferon pathway (69), which correlates with the ability of LAT to impair apoptosis (7, 8, 11). Further support for the concept that LAT impairs innate immune responses comes from the finding that LAT small noncoding RNAs interact with retinoic acid-inducible gene I (RIG-I) (70), a cytosolic pattern recognition receptor that triggers type-1 interferon responses (71). Finally, the ability of LAT to interfere with lytic cycle viral gene expression (11, 12, 72) may reduce inflammation and senescence via an indirect mechanism. Since inflammatory and senescence markers were consistently expressed in Pr5 of latently infected mice regardless of LAT expression, we believe differences in neuronal composition in LC versus Pr5 were important for the LAT-specific effects observed. Additional studies are necessary to fully understand the complex virus host interactions in Pr5 versus LC regions of brainstem during latency.
Emerging evidence suggests pathological changes in key brainstem regions occur during early stages of AD development (73–76). For example, neurofibrillary tangles are detected in LC of nondemented people decades prior to clinical onset of AD. Furthermore, hyperphosphorylated tau, the major component of neurofibrillary tangles and β-amyloid deposition in AD, is detected in LC prior to clinical onset of AD. Disruption of LC functions would reduce neuronal survival because LC secretes brain-derived neurotrophic factor (BDNF) and nerve growth factor (77–79). Notably, women have a 1 in 5 chance of developing AD, whereas 1 in 11 men develop AD (80, 81). Infectious agents, including HSV-1, have been suggested to contribute to neurodegeneration in a subset of AD cases (82, 83). This topic is controversial, and it is unlikely HSV-1 is the primary causative agent of AD. However, HSV-1 may serve as a cofactor in a small subset of people, in particular women with predisposing factors. While LAT null mutants have not been identified in nature, we speculate the lifelong HSV-1 latency-reactivation cycle in humans enhances senescence and neuroinflammation in LC and Pr5 during the aging process.
In conclusion, these studies revealed a sexual predilection in LC and Pr5 regions of brainstem in female C57BL/6 mice latently infected with HSV-1. Comparing virus–host interactions in brainstem of mice infected with a LAT null mutant versus the parental WT viral strain may provide new insight into LAT functions and how these viral products regulate the latency-reactivation cycle. Finally, these studies may identify additional cellular factors in LC and Pr5 that mediate HSE and neurodegeneration.
MATERIALS AND METHODS
Virus and cell lines.
HSV-1 strain McKrae dLAT2903R (WT; LAT+/+) and dLAT2903 (LAT−/−) were obtained from the late Dr. Steven Wechsler and grown in Vero cell (ATCC CCL-81) monolayers in minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 100 IU/mL penicillin, 100 μg/mL Streptomycin at 37°C, and 5% CO2 until >80% CPE (cytopathic effect) was observed. The dLAT2903R virus was rescued from dLAT2903 by restoring LAT sequences by homologous recombination: this virus is identical to the parental WT McKrae strain of HSV-1 with respect to replication and reactivation from mice and rabbits (54, 84). Viral aliquots were obtained through serial freeze-thawing of cells and subsequent centrifugation to remove cellular debris. Virus was titered on Vero cell monolayers to determine PFU/mL for each stock prior to infection of mice.
Mouse studies.
Eight-week old (male or female) C57BL/6J mice were purchased from Jackson Laboratories. These mice were housed and handled in agreement with the Oklahoma State University Institutional Animal Care and Use Committee, approved animal use protocol VM15-26. Mice were acclimated to standard laboratory conditions (12 h light/dark cycles, 5 animals/cage) for 1 week prior to infection. Mice were anesthetized via standard isoflurane/oxygen vaporization and infected with ~2 × 105 PFU WT HSV-1 (dLAT2903R) or the LAT−/− mutant (dLAT2903, n = 50) in 2 μL MEM per eye without scarification. As described previously (34), virus was instilled into the ocular cavity and then the eyelid was closed and rubbed gently to enhance infection. After infection, mice were monitored for herpes simplex virus induced encephalitis (HSE)-related symptoms such tremors, swollen forehead, or weight loss. Animals exhibiting these symptoms were promptly and humanely euthanized via isoflurane overdose and subsequent cervical dislocation. Infected mice were not allowed to reach HSE-induced death as an endpoint. Since C57BL/6J mice are highly resistant to the effects of HSV-1 (85, 86), surviving animals frequently displayed symptoms of acute ocular herpes infections such as redness, discharge, and fur loss at the site of inoculation. As needed, mice were treated twice daily with Neomycin/polymyxin B/Bacitracin zinc triple antibiotic ophthalmic ointment (Bausch and Lomb, Tampa, FL) to prevent secondary bacterial infection and minimize pain and distress. Mice infected for 30 days were operationally defined as being latently infected.
Ocular swabs were obtained from mice at the designated times postinfection. Briefly, cotton-tipped applicators were rotated superficially against the ocular cavity and immersed in 1 mL MEM containing l-glutamine (2 mM), and antibiotics (penicillin [10 U/mL], streptomycin [100 μg/mL]), and fungizone (5 μg/mL). Samples were freeze-thawed, vortexed, and plaqued on Vero cells as previously described (34). Data are shown as mean for n = 5 mice at each time point.
Senescence associated beta-galactosidase (SA-β-Gal) staining.
Senescence β-galactosidase staining kit (cat. no: 9860S) purchased from Cell Signaling Technology was used according to the manufacturer’s instruction to perform SA-β-Gal staining on coronal brainstem sections containing the LC and Pr5 region. Serial coronal sections (20 μm thick) of LC and Pr5 brainstem regions were obtained from fixed brains on super frost plus slides using a cryostat. These sections were allowed to air dry after removing them from a −80° C freezer. After rinsing each slide with 1× phosphate-buffered saline (PBS), they were fixed with 1× formaldehyde for 5 min at room temperature. X-Gal was dissolved in dimethylformamide (DMF) to prepare a 20 mg/mL stock solution. The β-Galactosidase staining solution was prepared using X-Gal stock solution, and the final pH was adjusted to 6.0 as per the manufacturer’s instruction. The slides were then incubated in the staining solution at 37 °C overnight in a carbon dioxide (CO2) free incubator. The endpoint for the reaction was the development of blue color, which was then imaged using a light microscope.
Microdissection of key brainstem regions.
Serial coronal sections (60 μm thick) of brainstem (Fig. 1B) were obtained on super frost plus slides using a cryostat. The principal sensory nucleus (Pr5) of the spinal trigeminal tract and locus coeruleus (LC) were microdissected by Palkovit’s technique. A mouse brain stereotaxic atlas was used as a reference (87).
Real-time RT-PCR.
Micropunches from brain regions were collected from either side of the 60-μm thick sections based on stereotaxic coordinates. Punches collected from Pr5 and LC were used for RNA extraction and real-time PCR analysis. The tissue micropunches were lysed in TRIzol reagent and RNA extraction performed using Direct-zol RNA microprep kit (Zymo Research, Irvine, CA, USA). A high-capacity cDNA reverse transcription kit (Applied Biosystems) was used in cDNA conversion. Real time PCRs were carried out using iTaq Universal SYBR Green Mix (Bio-Rad) with 10 ng cDNA per reaction. The mouse primers for p16, p21, p53, IL-1α, IL-1β, TNF-α, MCP-1, Fra-1, Nrf2, NQO1, NLRP3, and GAPDH (the housekeeping gene) were designed using Primer-Blast software and synthesized by Integrated DNA technologies (primer sequences are listed in Table 1). Data was analyzed by the 2-ΔΔCT method.
TABLE 1.
Primers used to compare RNA levels in Pr5 and LC in female versus male mice
| Gene | Forward primer | Reverse primer | Gene accession no. |
|---|---|---|---|
| GAPDH | AAGGGCTCATGACCACAGTC | GGATGACCTTGCCCACAG | NM_001289726.1 |
| Fra1 | ACCTTGTGCCAAGCATCGAC | CTGTACTGGGGATAGGCCAG | NM_010235.2 |
| IL-1α | CAAGATGGCCAAAGTTCGTGAC | GTCTCATGAAGTGAGCCATAGC | NM_010554.4 |
| IL-1β | CACAGCAGCACATCAACAAG | GTGCTCATGTCCTCATCCTG | NM_008361.4 |
| MCP1 | GCAGTTAACGCCCCACTCA | TCCAGCCTACTCATTGGGATCA | NM_011333.3 |
| NLRP3 | GCCCGAGAAAGGCTGTATCC | GAGGTCTCGCCTGTTGATCG | NM_145827.4 |
| NQO1 | TATCCTTCCGAGTCATCTCTAGCA | TCTGCAGCTTCCAGCTTCTTG | NM_008706.5 |
| Nrf2 | CGAGATATACGCAGGAGAGGTAAGA | GCTCGACAATGTTCTCCAGCTT | NM_010902.4 |
| p16 | CGTACCCCGATTCAGGTGAT | TTGAGCAGAAGAGCTGCTACGT | NM_001040654.1 |
| p21 | GACAAGAGGCCCAGTACTTC | GCTTGGAGTGATAGAAATCTGTC | NM_007669.5 |
| p53 | CACAGCGTGGTGGTACCTTA | TCTTCTGTACGGCGGTCTCT | NM_001127233.1 |
| TNF-α | GGAACTGGCAGAAGAGGCACTC | GCAGGAATGAGAAGAGGCTGAGAC | NM_001278601.1 |
Quantification of viral DNA in TG, LC, and Pr5 of latently infected mice.
TG, LC, Pr5, and kidney were isolated as described and DNA isolated according to the Jackson Laboratory DNA isolation protocols. Briefly, tissues were digested in 50 mM Tris-HCl pH 8.0, 100 mM EDTA pH 8.0, 100 mM NaCl, and 1% SDS with 0.5 mg/mL proteinase K overnight at 65 °C. DNA was isolated using phenol-chloroform/isoamyl alcohol three times prior to ethanol precipitation. qPCR was performed on a Bio-Rad CFX Opus using PowerTRACK Sybr Green Master Mix, 100 ng template DNA, 500 nM primers for HSV-1 gB Fwd: 5′-AACGCGACGCACATCAAG-3′, Rev 5′-CTGGTACGCGATCAGAAAGC-3′ or GAPDH Fwd 5′-CATCACTGCCACCCAGAAGACTG-3′, Rev 5′-ATGCCAGTGAGCTTCCCGTTCAG-3′. 2-ΔΔCT was calculated for brain sections compared to kidney and gB compared to GAPDH (K. Harrison and C. Jones, data not shown). Data are shown as box and whisker plots for each individual animal per group, Max to Min with center lines delineating the mean for duplicate wells.
Statistical analysis.
All statistical analysis was performed using GraphPad Prism Version 9.3.1. All data were analyzed using one-way ANOVA comparing the means of three groups (uninfected control, WT HSV-1, and dLAT). The gene expression values were expressed as fold change relative to uninfected control mice. A P value of <0.05 is considered statistically significant.
ACKNOWLEDGMENTS
This research was supported by grants from the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under award number R01NS111167 (C.J.) and the Oklahoma Center for Respiratory and Infectious Diseases (National Institutes of Health Centers for Biomedical Research Excellence Grant # P20GM103648), R15HL148844 (M.S.), and funds from the Sitlington Endowment (C.J.).
Contributor Information
Madhan Subramanian, Email: madhan.subramanian@okstate.edu.
Clinton Jones, Email: clint.jones10@okstate.edu.
Jae U. Jung, Lerner Research Institute, Cleveland Clinic
REFERENCES
- 1.Perng G-C, Jones C. 2010. Towards an understanding of the herpes simplex virus type 1 latency-reactivation cycle. Interdiscip Perspect Infect Dis 2010:262415–262418. 10.1155/2010/262415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Phelan D, Barrozo ER, Bloom DC. 2017. HSV1 latent transcription and non-coding RNA: a critical retrospective. J Neuroimmunol 308:65–101. 10.1016/j.jneuroim.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Lewandowski G, Zimmerman MN, Denk LL, Porter DD, Prince GA. 2002. Herpes simplex type 1 infects and establishes latency in the brain and trigeminal ganglia during primary infection of the lip in cotton rats and mice. Arch Virol 147:167–179. 10.1007/s705-002-8309-9. [DOI] [PubMed] [Google Scholar]
- 4.Fraser N, Lawrence WC, Wroblewska Z, Gilden DH, Koprowski H. 1981. Herpes simplex virus type 1 DNA in human brain tissue. Proc Natl Acad Sci USA 78:6461–6465. 10.1073/pnas.78.10.6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cabrera C, Wohlenberg C, Openshaw H, Rey-Mendez M, Puga A, Notkins AL. 1980. Herpes simplex virus DNA sequences in the CNS of latently infected mice. Nature 288:288–290. 10.1038/288288a0. [DOI] [PubMed] [Google Scholar]
- 6.Rock D, Fraser NW. 1983. Detection of HSV-1 genome in central nervous system of latently infected mice. Nature 302:523–525. 10.1038/302523a0. [DOI] [PubMed] [Google Scholar]
- 7.Inman M, Perng G-C, Henderson G, Ghiasi H, Nesburn AB, Wechsler SL, Jones C. 2001. Region of herpes simplex virus type 1 latency-associated transcript sufficient for wild-type spontaneous reactivation promotes cell survival in tissue culture. J Virol 75:3636–3646. 10.1128/JVI.75.8.3636-3646.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perng GC, Jones C, Ciacci-Zanella J, Stone M, Henderson G, Yukht A, Slanina SM, Hofman FM, Ghiasi H, Nesburn AB, Wechsler SL. 2000. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript (LAT). Science 287:1500–1503. 10.1126/science.287.5457.1500. [DOI] [PubMed] [Google Scholar]
- 9.Li S, Carpenter D, Hsiang C, Wechsler SL, Jones C. 2010. The herpes simplex virus type 1 latency-associated transcript (LAT) locus inhibits apoptosis and promotes neurite sprouting in neuroblastoma cells following serum starvation by maintaining active AKT (protein kinase B). J Gen Virol 91:858–866. 10.1099/vir.0.015719-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamza MA, Higgins DM, Feldman LT, Ruyechan WT. 2007. The latency-associated transcript of herpes simplex virus type 1 promotes survival and stimulates axonal regeneration in sympathetic and trigeminal ganglia. J Neurovirol 13:56–66. 10.1080/13550280601156297. [DOI] [PubMed] [Google Scholar]
- 11.Shen W, Silva MSe, Jaber T, Vitvitskaia O, Li S, Henderson G, Jones C. 2009. Two small RNAs encoded within the first 1.5 kb of the herpes simplex virus type 1 (HSV-1) latency-associated transcript (LAT) can inhibit productive infection, and cooperate to inhibit apoptosis. J Virol 90:9131–9139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. 2008. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 454:780–785. 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perng G-C, Maguen B, Jin L, Mott KR, Osorio N, Slanina SM, Yukht A, Ghiasi H, Nesburn AB, Inman M, Henderson G, Jones C, Wechsler SL. 2002. A gene capable of blocking apoptosis can substitute for the herpes simplex virus type 1 latency-associated transcript gene and restore wild-type reactivation levels. J Virol 76:1224–1235. 10.1128/JVI.76.3.1224-1235.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin L, Perng G-C, Mott KR, Osorio N, Naito J, Brick DJ, Carpenter D, Jones C, Wechsler SL. 2005. The baculovirus inhibitor of apoptosis gene (cpIAP) can restore reactivation of latency to a herpes simplex virus type 1 that does not express the latency associated transcript (LAT). J Virol 79:12286–12295. 10.1128/JVI.79.19.12286-12295.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin L, Carpenter D, Moerdyk-Schauwecker M, Vanarsdall AL, Osorio N, Hsiang C, Jones C, Wechsler SL. 2008. Cellular FLIP can substitute for the herpes simplex virus type 1 LAT gene to support a wild type virus reactivation phenotype in mice. J Neurovirol 14:389–400. 10.1080/13550280802216510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thompson RL, Sawtell NM. 2011. The herpes simplex virus type 1 latency associated transcript is required for the maintenance of reactivation competent latent infection. J Neurovirol 17:552–558. 10.1007/s13365-011-0071-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tyler K, Tedder DG, Yamamoto LJ, Klapper JA, Ashley R, Lichtenstein KA, Levin MJ. 1995. Recurrent brainstem encephalitis associated with herpes simplex virus type 1 DNA in cerebrospinal fluid. Neurology 45:2246–2250. 10.1212/wnl.45.12.2246. [DOI] [PubMed] [Google Scholar]
- 18.Benbir G, Goskan B, Kocer N. 2010. Brainstem lesions in herpes encephalitis. Turk Norol Derg 16:211–214. [Google Scholar]
- 19.Livorsi D, Anderson E, Qureshi S, Howard M, Wang YF, Franco-Paredes C. 2010. Brainstem encephalitis: an unusual presentation of herpes simplex virus infection. J Neurol 257:1432–1437. 10.1007/s00415-010-5600-x. [DOI] [PubMed] [Google Scholar]
- 20.Gorospe W, Rawls RA, Kochs KA, Laos LF, Lambiase L, Silliman SL. 2004. Herpes brainstem encephalitis: a cause of intractable emesis. Hospital Physician 4:39–42. [Google Scholar]
- 21.Yamada S, Kameyama T, Nagaya S, Hashizume Y, Yoshida M. 2003. Relapsing herpes simplex encephalitis: pathological confirmation of viral reactivation. J Neurol Neurosurg Psychiatry 74:262–264. 10.1136/jnnp.74.2.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lahat E, Barr J, Barkai G, Paret G, Brand N, Barzilai A. 1999. Long term neurological outcome of herpes encephalitis. Arch Dis Child 80:69–71. 10.1136/adc.80.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGrath N, Anderson NE, Croxson MC, Powell KF. 1997. Herpes simplex encephalitis treated with acyclovir: diagnosis and long term outcome. J Neurol Neurosurg Psychiatry 63:321–326. 10.1136/jnnp.63.3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Skoldenberg B. 1991. Herpes simplex encephalitis. Scand J Infect Dis Suppl 80:40–46. [PubMed] [Google Scholar]
- 25.Zhang S-Y, Clark NE, Freije CA, Pauwels E, Taggart AJ, Okada S, Mandel H, Garcia P, Ciancanelli MJ, Biran A, Lafaille FG, Tsumura M, Cobat A, Luo J, Volpi S, Zimmer B, Sakata S, Dinis A, Ohara O, Garcia Reino EJ, Dobbs K, Hasek M, Holloway SP, McCammon K, Hussong SA, DeRosa N, Van Skike CE, Katolik A, Lorenzo L, Hyodo M, Faria E, Halwani R, Fukuhara R, Smith GA, Galvan V, Damha MJ, Al-Muhsen S, Itan Y, Boeke JD, Notarangelo LD, Studer L, Kobayashi M, Diogo L, Fairbrother WG, Abel L, Rosenberg BR, Hart PJ, Etzioni A, Casanova J-L. 2018. Inborn errors of RNA lariat metabolism in humans with brainstem viral infection. Cell 172:952–965. 10.1016/j.cell.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen S-H, Yao H-W, Huang W-Y, Hsu K-S, Lei H-Y, Shiau A-L, Chen S-H. 2006. Efficient reactivation of latent herpes simplex virus from mouse central nervous system. J Virol 80:12387–12393. 10.1128/JVI.01232-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao H-W, Ling P, Tung Y-Y, Hsu S-M, Chen S-H. 2014. In vivo reactivation of latent herpes simplex virus 1 in mice can occur in the brain before occurring in the trigeminal ganglia. J Virol 88:11264–11270. 10.1128/JVI.01616-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Domínguez-Salazar E, Naser HF, Velázquez-Moctezuma J. 2014. D1-like antagonist blocks conditioned place preference induced by ejaculation in male rats. Behav Brain Res 269:15–19. 10.1016/j.bbr.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 29.Robertson S, Plummer NW, de Marchena J, Jensen P. 2013. Developmental origins of central norepinephrine neuron diversity. Nat Neurosci 16:1016–1023. 10.1038/nn.3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Breton-Provencher V, Drummond GT, Sur M. 2021. Locus coeruleus norepinephrine in learned behavior: anatomical modularity and spatiotemporal integration in targets. Front Neural Circuits 15:638007. 10.3389/fncir.2021.638007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watson C. 2021. Motor nuclei of the cranial nerve, p 490–498. In Watson C, Puelles L, Paxinos G (ed), The mouse nervous system. Elsevier, New York, NY. [Google Scholar]
- 32.Carrasco GA, Van de Kar LD. 2003. Neuroendocrine pharmacology of stress. Eur J Pharmacol 463:235–272. 10.1016/S0014-2999(03)01285-8. [DOI] [PubMed] [Google Scholar]
- 33.Makino S, Smith MA, Gold PW. 2002. Regulatory role of glucocorticoids and glucocorticoid receptor mRNA levels on tyrosine hydroxylase gene expression in the locus coeruleus during repeated immobilization stress. Brain Res 943:216–233. 10.1016/S0006-8993(02)02647-1. [DOI] [PubMed] [Google Scholar]
- 34.Harrison K, Zhu L, Thunuguntla P, Jones C. 2019. Antagonizing the glucocorticoid receptor impairs explant-induced reactivation in mice latently infected with herpes simplex virus 1. J Virol 93:e00418-19. 10.1128/JVI.00418-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ostler J, Harrison KS, Schroeder K, Thunuguntla P, Jones C. 2019. The glucocorticoid receptor (GR) stimulates herpes simplex virus 1 productive infection, in part because the infected cell protein 0 (ICP0) promoter is cooperatively transactivated by the GR and Krüppel-like transcription factor 15. J Virol 93:e02063-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campisi J. 2013. Aging, cellular senescence, and cancer. Annu Rev Physiol 75:685–705. 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumari R, Jat P. 2021. Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front Cell and Developmental Biology 9:645593. 10.3389/fcell.2021.645593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Freund A, Orjalo AV, Desprez P-Y, Campis J. 2010. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med 16:238–246. 10.1016/j.molmed.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Balasubramanian P, Branen L, Sivasubramanian MK, Monteiro R, Subramanian M. 2021. Aging is associated with glial senescence in the brainstem—implications for age-related sympathetic overactivity. Aging (Albany NY) 13:13460–13471. 10.18632/aging.203111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, Kleijer WJ, DiMaio D, Hwang E. 2006. Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell 5:187–195. 10.1111/j.1474-9726.2006.00199.x. [DOI] [PubMed] [Google Scholar]
- 41.Hernandez-Segura A, Nehme J, Demaria M. 2018. Hallmarks of cellular senescence. Trends Cell Biol 28:436–453. 10.1016/j.tcb.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 42.Rayess H, Wang MB, Srivatsan ES. 2012. Cellular senescence and tumor suppressor gene p16. Int J Cancer 130:1715–1725. 10.1002/ijc.27316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yin Y, Tainsky MA, Bischoff FZ, Strong LC, Wahl GM. 1992. Wild-type p53 restores cell cycle control and inhibits gene amplification in cells with mutant p53 alleles. Cell 70:937–948. 10.1016/0092-8674(92)90244-7. [DOI] [PubMed] [Google Scholar]
- 44.Bouvard V, Zaitchouk T, Vacher M, Duthu A, Canivet M, Choisy-Rossi C, Nieruchalski M, May E. 2000. Tissue and cell-specific expression of the p53-target genes: bax, fas, mdm2 and waf1/p21, before and following ionising irradiation in mice. Oncogene 19:649–660. 10.1038/sj.onc.1203366. [DOI] [PubMed] [Google Scholar]
- 45.Rokudai S, Aikawa Y, Tagata Y, Tsuchida N, Taya Y, Kitabayashi I. 2009. Monocytic leukemia zinc finger (MOZ) interacts with p53 to induce p21 expression and cell-cycle arrest. J Biol Chem 284:237–244. 10.1074/jbc.M805101200. [DOI] [PubMed] [Google Scholar]
- 46.Youm Y-H, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A, Pistell P, Newman S, Carter R, Laque A, Münzberg H, Rosen CJ, Ingram DK, Salbaum JM, Dixit VD. 2013. Canonical Nlrp3 inflammasome links systemic low grade inflammation to functional decline in aging. Cell Metab 18:519–532. 10.1016/j.cmet.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pothlichet J, Meunier I, Davis BK, Ting JP-Y, Skamene E, von Messling V, Vidal SM. 2013. Type I IFN triggers RIG-I/TLR3/NLRP3-dependedent inflammasome activation in influenza A virus infected cells. PLoS Pathog 9:e1003256. 10.1371/journal.ppat.1003256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rajan J, Warren SE, Miao EA, Aderem A. 2010. Activation of the NLRP3 inflammasome by intracellular poly I:C. FEBS Lett 584:4627–4632. 10.1016/j.febslet.2010.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deshmane SL, Kremlev S, Amini S, Sawaya BE. 2009. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 29:313–326. 10.1089/jir.2008.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoffman GE, Le WW, Abbud R, Lee W-S, Smith MS. 1994. Use of Fos-related antigens (FRAs) as markers of neuronal activity: FRA changes in dopamine neurons during proestrus, pregnancy and lactation. Brain Res 654:207–2015. 10.1016/0006-8993(94)90481-2. [DOI] [PubMed] [Google Scholar]
- 51.Zygraich N, Lobmann M, Vascoboinic E, Berge E, Huygelen C. 1974. In vivo and in vitro properties of a temperature sensitive mutant of infectious bovine Rhinotracheitis virus. Res Vet Sci 16:328–335. 10.1016/S0034-5288(18)33731-7. [DOI] [PubMed] [Google Scholar]
- 52.Gonzalez-Dosal R, Horan KA, Rahbek SH, Ichijo H, Chen ZJ, Mieyal JJ, Hartmann R, Paludan SR. 2011. HSV infection induces production of ROS, which potentiate signaling from pattern recognition receptors: role for S-glutathionylation of TRAF3 and 6. PLoS Pathog 7:e1002250. 10.1371/journal.ppat.1002250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perng G-C, Dunkel EC, Geary PA, Slanina SM, Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1994. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J Virol 68:8045–8055. 10.1128/jvi.68.12.8045-8055.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perng G-C, Ghiasi H, Slanina SM, Nesburn AB, Wechsler SL. 1996. The spontaneous reactivation function of the herpes simplex virus type 1 LAT gene resides completely within the first 1.5 kilobases of the 8.3- kilobase primary transcript. J Virol 70:976–984. 10.1128/JVI.70.2.976-984.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Valentino RJ, Reyes B, Van Bockstaele E, Bangasser D. 2012. Molecular and cellular sex differences at the intersection of stress and arousal. Neuropharmacology 62:13–20. 10.1016/j.neuropharm.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morris LS, McCall JG, Charney DS, Murrough JW. 2020. The role of the locus coeruleus in the generation of pathological anxiety. Neuroscience Advances 4:2398212820930321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McCall JG, Al-Hasani R, Siuda ER, Hong DY, Norris AJ, Ford CP, Bruchas MR. 2015. CRH engagement of the locus coeruleus noradrenergic system mediates stress-induced anxiety. Neuron 87:605–620. 10.1016/j.neuron.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCall JG, Siuda ER, Bhatti DL, Lawson LA, McElligott ZA, Stuber GD, Bruchas MR. 2017. Locus coeruleus to basolateral amygdala noradrenergic projections promote anxiety-like behavior. Elife 6:2837. 10.7554/eLife.18247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Curtis AL, Bethea T, Valentino RJ. 2006. Sexually dimorphic responses of the brain norepinephrine system to stress and corticotropin-releasing factor. Neuropsychopharmacology 31:544–554. 10.1038/sj.npp.1300875. [DOI] [PubMed] [Google Scholar]
- 60.Bangasser DA, Curtis A, Reyes BAS, Bethea TT, Parastatidis I, Ischiropoulos H, Van Bockstaele EJ, Valentino RJ. 2010. Sex differences in corticotropin-releasing factor receptor signaling and trafficking: potential role in female vulnerability to stress-related psychopathology. Mol Psychiatry 15:877–904. 10.1038/mp.2010.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ostler J, Sawant L, Harrison K, Jones C. 2021. Regulation of neurotropic herpesvirus productive infection and latency-reactivation cycle by glucocorticoid receptor and stress-induced transcription factors. In Litwack G (ed), Hormones, regulators, and viruses. Elsiever, New York, NY. 10.1016/bs.vh.2021.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Erlandsson AC, Bladh L-G, Stierna P, Yucel-Lindberg T, Hammarsten O, Modéer T, Harmenberg J, Wikström A-C. 2002. Herpes simplex virus type 1 infection and glucocorticoid treatment regulate viral yield, glucocorticoid receptor and NF-kB levels. J Endocrinol 175:165–176. 10.1677/joe.0.1750165. [DOI] [PubMed] [Google Scholar]
- 63.Bloom DC, Stevens JG, Hill JM, Tran RK. 1997. Mutagenesis of a cAMP response element within the latency-associated transcript promoter of HSV-1 reduces adrenergic reactivation. Virology 236:202–207. 10.1006/viro.1997.8723. [DOI] [PubMed] [Google Scholar]
- 64.Rhen T, Cidlowski JA. 2005. Antiinflammatory action of glucocorticoids—new mechanisms of old drugs. N Engl J Med 353:1711–1723. 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 65.Busillo JM, Azzam KM, Cidlowski JA. 2011. Glucocorticoids sensitize the innate immune system through regulation of the NLRP3 inflammasome. J Biol Chem 286:38703–38713. 10.1074/jbc.M111.275370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oakley RH, Cidlowski JA. 2013. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol 132:1033–1044. 10.1016/j.jaci.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mitchell IJ, Cooper AJ, Griffiths MR, Barber DJ. 1998. Phencyclidine and corticosteroids induce apoptosis of a subpopulation of striatal neurons: a neural substrate for psychosis? Neuroscience 84:489–501. 10.1016/S0306-4522(97)00534-4. [DOI] [PubMed] [Google Scholar]
- 68.Peng W, Inman M, Henderson G, Wechsler SL, BenMohamed L, Perng G-C, Jones C. 2005. The locus encompassing the latency-associated transcript (LAT) of herpes simplex virus type 1 interferes with and delays interferon expression in productively infected neuroblastoma cells and trigeminal ganglia of acutely infected mice. J Virol 79:6162–6171. 10.1128/JVI.79.10.6162-6171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Allen SJ, Hamrah P, Gate D, Mott KR, Mantopoulos D, Zheng L, Town T, Jones C, von Andrian UH, Freeman GJ, Sharpe AH, Benmohamed L, Ahmed R, Wechsler SL, Ghiasi H. 2011. The role of LAT in increased CD8+ T cell exhaustion in trigeminal ganglia of mice latently infected with herpes simplex virus 1. J Virol 85:4184–4197. 10.1128/JVI.02290-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Frizzo da Silva L, Jones C. 2013. Small non-coding RNAs encoded within the herpes simplex virus type 1 latency associated transcript (LAT) cooperate with the retinoic acid inducible gene I (RIG-I) to induce beta-interferon promoter activity and promote cell survival. Virus Res 175:101–109. 10.1016/j.virusres.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5:730–737. 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 72.Cliffe AR, Garber DA, Knipe DM. 2009. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol 83:8182–8190. 10.1128/JVI.00712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Braun DJ, Van Eldik LJ. 2018. In vivo brainstem imaging in Alzheimer’s disease: potential for biomarker development. Front Aging Neurosci 10:266. 10.3389/fnagi.2018.00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee JH, Ryan J, Andreescu C, Aizenstein H, Lim HK. 2015. Brainstem morphological changes in Alzheimer’s disease. Neuroreport 26:411–415. 10.1097/WNR.0000000000000362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Serra L, D'Amelio M, Di Domenico C, Dipasquale O, Marra C, Mercuri NB, Caltagirone C, Cercignani M, Bozzali M. 2018. In vivo mapping of brainstem nuclei functional connectivity disruption in Alzheimer’s disease. Neurobiol Aging 72:72–82. 10.1016/j.neurobiolaging.2018.08.012. [DOI] [PubMed] [Google Scholar]
- 76.Theofilas P, Ehrenberg AJ, Dunlop S, Di Lorenzo Alho AT, Nguy A, Leite REP, Rodriguez RD, Mejia MB, Suemoto CK, Ferretti-Rebustini REDL, Polichiso L, Nascimento CF, Seeley WW, Nitrini R, Pasqualucci CA, Jacob Filho W, Rueb U, Neuhaus J, Heinsen H, Grinberg LT. 2017. Locus coeruleus volume and cell population changes during Alzheimer's disease progression: a stereological study in human postmortem brains with potential implication for early-stage biomarker discovery. Alzheimers Dement 13:236–246. 10.1016/j.jalz.2016.06.2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Debeir T, Marien M, Ferrario J, Rizk P, Prigent A, Colpaert F, Raisman-Vozari R. 2004. In vivo upregulation of endogenous NGF in the rat brain by the alpha2-adrenoreceptor antagonist dexefaroxan: potential role in the protection of the basalocortical cholinergic system during neurodegeneration. Exp Neurol 190:384–395. 10.1016/j.expneurol.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 78.Choudhary P, Pacholko AG, Palaschuk J, Bekar LK. 2018. The locus coeruleus neurotoxin, DSP4, and/or a high sugar diet induce behavioral and biochemical alterations in wild-type mice consistent with Alzheimers related pathology. Metab Brain Dis 33:1563–1571. 10.1007/s11011-018-0263-x. [DOI] [PubMed] [Google Scholar]
- 79.Counts SE, Mufson EJ. 2010. Noradrenaline activation of neurotrophic pathways protects against neuronal amyloid toxicity. J Neurochem 113:649–660. 10.1111/j.1471-4159.2010.06622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Abendorth A, Arvin AM. 2000. Host responses to primary infection, p 142–156. In Arvin, Gershin AA (ed), Varicella-Zoster virus. Cambridge University Press, Cambridge, England. [Google Scholar]
- 81.Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, Gyorgy B, Breakefield XO, Tanzi RE, Moir RD. 2010. Why women have more Alzheimer’s disease than men: gender and mitochondrial toxicity of amyloid-beta peptide. J Alzheimer’s Dis 20:S527–S533. [DOI] [PubMed] [Google Scholar]
- 82.Seaks CE, Wilcock DM. 2020. Infectious hypothesis of Alzheimer disease. PLoS Pathog 16:e1008596. 10.1371/journal.ppat.1008596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lin WR, Graham J, MacGowan SM, Wilcock GK, Itzhaki RF. 1998. Herpes simplex virus type 1 in brain and risk of Alzheimer's disease. Alzheimer's Report 1:173–178. [Google Scholar]
- 84.Perng G-C, Esmail D, Slanina S, Yukht A, Ghiasi H, Osorio N, Mott KR, Maguen B, Jin L, Nesburn AB, Wechsler SL. 2001. Three herpes simplex virus type 1 latency-associated transcript mutants with distinct and assymetric effects on virulence in mice compared with rabbits. J Virol 75:9018–9028. 10.1128/JVI.75.19.9018-9028.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lopez CB. 1975. Genetics of natural resistance to herpesvirus infections in mice. Nature 258:152–153. 10.1038/258152a0. [DOI] [PubMed] [Google Scholar]
- 86.Lopez CB. 1980. Resistance to herpes simplex virus—type 1 (HSV-1). Curr Top Micropbiol Immunol 92:15–24. [DOI] [PubMed] [Google Scholar]
- 87.Paxinos G, Franklin K. 2019. The mouse brain in stereotaxic coordinates, 5th ed. Elsevier, San Diego, CA. [Google Scholar]