

Abstract
After the global spread of the SARS-CoV-2 Omicron BA.2, some BA.2 subvariants, including BA.2.9.1, BA.2.11, BA.2.12.1, BA.4, and BA.5, emerged in multiple countries. Our statistical analysis showed that the effective reproduction numbers of these BA.2 subvariants are greater than that of the original BA.2. Neutralization experiments revealed that the immunity induced by BA.1/2 infections is less effective against BA.4/5. Cell culture experiments showed that BA.2.12.1 and BA.4/5 replicate more efficiently in human alveolar epithelial cells than BA.2, and particularly, BA.4/5 is more fusogenic than BA.2. We further provided the structure of the BA.4/5 spike receptor-binding domain that binds to human ACE2 and considered how the substitutions in the BA.4/5 spike play roles in ACE2 binding and immune evasion. Moreover, experiments using hamsters suggested that BA.4/5 is more pathogenic than BA.2. Our multiscale investigations suggest that the risk of BA.2 subvariants, particularly BA.4/5, to global health is greater than that of original BA.2.
Keywords: SARS-CoV-2, COVID-19, Omicron, BA.4, BA.5, BA.2.12.1, BA.2, transmissibility, immune resistance, pathogenicity
Graphical abstract
The SARS-CoV-2 Omicron variants BA.4 and BA.5 are currently causing infections and COVID-19 morbidities worldwide. Compared with the earlier variant BA.2, BA.4/5 shows more efficient replication and is more fusogenic. Structural views as well as in vivo studies in hamsters explain the antibody evasion and increased pathogenicity of BA.4/5 over BA.2.
Introduction
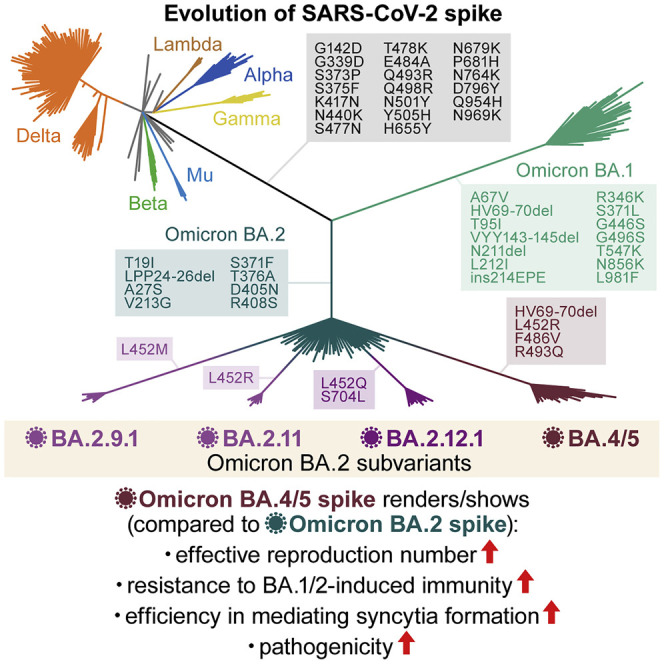
Since the end of November 2021, the SARS-CoV-2 Omicron variant (B.1.1.529 and BA lineages) has spread worldwide and has outcompeted prior SARS-CoV-2 variants of concern (VOCs), such as Delta. After the surge of the Omicron BA.1 variant, another Omicron variant, BA.2, outcompeted BA.1 and has become the most dominant variant in the world (Ito et al., 2022; UKHSA, 2022; Yamasoba et al., 2022b). Thereafter, as of May 2022, the BA.2 subvariants that harbor the substitution at the L452 residue of the spike (S) protein, such as BA.4 and BA.5, were frequently detected (Tegally et al., 2022; WHO, 2022). These observations suggest that novel BA.2 subvariants bearing substitutions at the L452 residue of the S protein are more transmissible than the original BA.2. These recent developments led the World Health Organization (WHO) to consider these BA.2 subvariants bearing substitutions at the L452 residue of the S protein, BA.4, BA.5, BA.2.12.1, BA.2.9.1, and BA.2.11, as VOC lineages under monitoring (VOC-LUM) on May 18, 2022 (WHO, 2022).
Resistance to antiviral humoral immunity can be mainly determined by substitutions in the S protein. For instance, Omicron BA.1 (Cao et al., 2021; Cele et al., 2021; Dejnirattisai et al., 2022; Garcia-Beltran et al., 2021; Liu et al., 2021; Meng et al., 2022; Planas et al., 2021; Takashita et al., 2022a; VanBlargan et al., 2022) and BA.2 (Bruel et al., 2022; Takashita et al., 2022b; Yamasoba et al., 2022b; Yamasoba et al., 2022c) exhibit profound resistance to neutralizing antibodies induced by vaccination, natural SARS-CoV-2 infection, and therapeutic monoclonal antibodies. In addition to immune evasion, substitutions in the S protein potentially modulate viral pathogenicity. In particular, the fusogenicity of the S protein in in vitro cell cultures is closely associated with viral pathogenicity in an experimental hamster model. For example, the Delta S protein is highly fusogenic in cell cultures and highly pathogenic in hamsters when compared with ancestral D614G-bearing B.1.1 S proteins (Saito et al., 2022). In contrast, the Omicron BA.1 S protein is less fusogenic and pathogenic than the B.1.1 S protein (Meng et al., 2022; Suzuki et al., 2022). Furthermore, we have recently demonstrated that the Omicron BA.2 S protein is more fusogenic and potentially confers the virus with higher pathogenicity than the Omicron BA.1 S protein (Yamasoba et al., 2022b).
Newly emerging SARS-CoV-2 variants need to be carefully and rapidly assessed for a potential increase in their growth efficacy in the human population, their pathogenicity, and/or their evasion from antiviral immunity. The substitution at the L452 residue of the SARS-CoV-2 S protein was detected in the Delta (L452R) and Lambda (L452Q) variants, which were previously classified as a VOC and a variant of interest (VOI), respectively (WHO, 2022). Importantly, we previously demonstrated that the L452R (Motozono et al., 2021) and L452Q (Kimura et al., 2022a) increase viral infectivity by promoting the binding of the S receptor-binding domain (RBD) to human angiotensin converting enzyme 2 (ACE2). We have recently characterized the virological features of SARS-CoV-2 Omicron BA.1 (Meng et al., 2022; Suzuki et al., 2022) and BA.2 (Yamasoba et al., 2022b). However, the impact of the substitution of the L452 residue of the S protein on the virological characteristics of Omicron BA.2 remains unclear. Altogether, these observations suggest that novel BA.2 subvariants bearing substitutions at the L452 residue of the S protein, particularly those classified as VOC-LUM, could pose a potential risk for global health, and we herein elucidate the virological characteristics of these novel BA.2 subvariants.
Results
Emergence of BA.2 subvariants bearing the L452R/Q/M substitution
Omicron substantially diversified during the epidemic. In South Africa, where Omicron was first reported at the end of November 2021 (NICD, 2021a, NICD, 2021b), a variety of Omicron sublineages (BA.1–BA.5) have emerged (Figures 1 A and S1 A) (Tegally et al., 2022). The Omicron BA.4 and BA.5 variants are closely related to each other and bear identical S proteins (Figure 1A). Because BA.4 and BA.5 form a monophyletic clade with BA.2 (Figure 1A), we herein refer to BA.4 and BA.5 as BA.2 subvariants. Compared with the BA.2 S protein, BA.4 and BA.5 S proteins harbor the L452R, HV69-70del, and F486V, as well as a revertant R493Q. Notably, in addition to the BA.4 and BA.5 lineages, several BA.2 subvariants that bear the substitutions at the L452 residue of the S protein have also emerged (Figures 1B and S1B; Table S1). In-depth tracing of the emergence of BA.2 subvariants bearing substitutions at the L452 residue of the S protein detected seven common ancestry groups of the BA.2 variants bearing L452R, L452Q, or L452M in the S protein (Figures 1B, S1C, and S1D; Table S2). As of May 15, 2022, the Phylogenetic Assignment of Named Global Outbreak (PANGO) lineage (https://cov-lineages.org) annotates four out of the seven BA.2 subvariants bearing substitutions at the L452 residue of the S protein: BA.2.9.1 (BA.2 S:L452M) in Denmark, BA.2.11 (BA.2 S:L452R) in France, BA.2.12.1 (BA.2 S:L452Q/S704L) in the USA, and BA.2.13 (BA.2 S:L452M) in Belgium (Figures 1B, 1C, and S1C). However, the other three lineages were not annotated as of May 15, 2022 (Figure 1B). On May 18, 2022, the WHO classified these six L452R/M/Q-bearing BA.2 subvariants, BA.4, BA.5, BA.2.9.1, BA.2.11, BA.2.12.1, and BA.2.13, as VOC-LUM (WHO, 2022). Most importantly, these BA.2 subvariants had higher effective reproduction numbers (Re) than the original BA.2 subvariant (Figure 1D; Table S3). In particular, the Re values of BA.4 and BA.5 (in South Africa) and BA.2.12.1 (in the USA) are 1.19-, 1.21-, and 1.13-fold higher than that of BA.2, respectively (Figure 1D), and these three subvariants have begun outcompeting the original BA.2 in several countries (Figures 1E and S1E). As of May 15, 2022, BA.4, BA.5, and BA.2.12.1 have been detected in 20, 19, and 36 countries, respectively. Altogether, our data indicate that multiple subvariants bearing substitutions at the L452 residue of the S protein have independently emerged in several countries. From these data, at the time of writing the initial manuscript of this study in May 2022, we predicted that these BA.2 subvariants, particularly BA.4 and BA.5, would spread worldwide and become the next predominant variants in the near future. As of July 7, 2022, since BA.4, BA.5, and BA.2.12.1 began circulating in the same countries (e.g., the USA), we directly compared the Re values in certain countries among these variants. The estimated Re values of BA.4, BA.5, and BA.2.12.1 in the USA were 1.28-, 1.36-, and 1.10-fold higher than that of BA.2, respectively, suggesting that BA.5 shows the highest Re value among the variants tested (Figure S1F; Table S3). As of July 2022, BA.5 has begun outcompeting the original BA.2 in multiple countries and will be the next predominant variant in the world in the near future.
Figure 1.

Epidemic of BA.2 subvariants bearing the L452R/Q/M substitutions in S
(A) A maximum likelihood (ML) tree of the Omicron lineages sampled from South Africa. The mutations acquired in the S proteins of BA.4 and BA.5 lineages are indicated in the panel. Note that R493Q is a reversion (i.e., back mutation from the BA.1–BA.3 lineages [R493] to the B.1.1 lineage [Q493]). Bootstrap values, ∗ ≥0.85; ∗∗ ≥0.9.
(B) An ML tree of BA.2. The BA.2 subvariants bearing substitutions at the L452 residue of the S protein are indicated as colored dots, and the estimated common ancestry groups of the variants are indicated as vertical bars. The PANGO lineages are indicated in the panel. The substitutions in the S proteins of each group are shown in parentheses.
(C) Heatmap summarizing the frequency of amino acid substitutions. Substitutions detected in >50% of sequences of any lineage are shown.
(D) Estimated relative Re of each viral lineage, assuming a fixed generation time of 2.1 days. The Re value of BA.2 is set at 1. The posterior (violin), posterior mean (dot), and 95% Bayesian confidence interval (CI) (line) are shown.
(E) Epidemic dynamics of SARS-CoV-2 lineages. The results for up to five predominant lineages in South Africa (top) and the USA (bottom) are shown. The observed daily sequence frequency (dot) and the dynamics (posterior mean, line; 95% CI, ribbon) are shown. The dot size is proportional to the number of sequences. The BA.2 subvariants without substitutions at the L452 residue of the S protein are summarized as “BA.2.”
In (D) and (E), the SARS-CoV-2 genome surveillance data downloaded from GISAID (https://www.gisaid.org/) on May 15, 2022, was used.
See also Figure S1 and Table S1. Summary of the mutations among Wuhan-Hu-1, original BA.2 and BA.2 subvariants, related to Figure 1, Table S2. The estimated common ancestry groups of BA.2 variants bearing the S L452 substitutions, related to Figure 1, Table S3. The estimated relative Re values of viral lineages in each country, related to Figure 1.
Figure S1.

Phylogenetic analysis of BA.2 subvariants, related to Figure 1
(A) The mutation profile of the Omicron lineages in South Africa, related to Figure 1A. Mutations detected in ≥5 sequences in the ML tree are summarized.
(B) Comparison of mutations in S protein among BA.2 subvariants. Mutations detected in ≥50% sequences of at least one lineage are summarized.
(C) The country and PANGO lineage of the BA.2 sequences in the ML tree, related to Figure 1B.
(D) Estimation of each common ancestry group of the S protein L452 substitution-bearing BA.2 variants. The amino acid at position 452 in the S protein in each ancestral node was estimated by a Markov model, and the branches where the L452 substitution was acquired (red branches with asterisks) were estimated.
(E) Epidemic dynamics of SARS-CoV-2 lineages. The results for up to five predominant lineages in Denmark (left), France (middle), and Belgium (right) where the BA.2-related Omicron variants bearing the S protein L452R/Q/M substitution circulating are shown. The observed daily sequence frequency (dot) and the dynamics (posterior mean, line; 95% CI, ribbon) are shown. The dot size is proportional to the number of sequences. The BA.2 sublineages without substitution at the L452 residue of the S protein are summarized as “BA.2.”
(F) Estimated relative Re of each viral lineage, assuming a fixed generation time of 2.1 days. The Re value of BA.2 is set at 1. The posterior (violin), posterior mean (dot), and 95% Bayesian confidence interval (CI) (line) are shown. Unlike Figure 1D, the SARS-CoV-2 genome surveillance data downloaded on July 7, 2022, was used.
Immune resistance of BA.2 subvariants
We have recently demonstrated that BA.4/5 is more resistant to a therapeutic monoclonal antibody, cilgavimab, which is a component of Evusheld, than BA.2 (Yamasoba et al., 2022c). Additionally, recent studies have demonstrated that BA.4 and BA.5 are relatively resistant to the antiviral humoral immunity induced by BA.1 infection (Khan et al., 2022; Tuekprakhon et al., 2022). To investigate the sensitivity of BA.2 subvariants to antiviral humoral immunity, we prepared pseudoviruses bearing the S proteins of these BA.2 subvariants, including BA.2.9.1/BA.2.13 (BA.2 S:L452M; we herein refer to it as BA.2.9.1), BA.2.11 (BA.2 S:L452R), BA.2.12.1 (BA.2 S:L452Q/S704L), and BA.4/5 (BA.2 S:L452R/HV69-70del/F486V/R493Q), as well as their derivatives and the original BA.2. Consistent with recent studies, including ours (Evans et al., 2022; Khan et al., 2022; Rössler et al., 2022a; Rössler et al., 2022b; Turelli et al., 2022; Wang et al., 2022b; Yamasoba et al., 2022b), the convalescent sera from individuals who were infected with BA.1 (8–21 days after testing; Figure 2 A) and BA.2 (8–27 days after testing; Figure 2B) were poorly antiviral against all pseudoviruses tested. The sera from 16 individuals infected with BA.1 who were 2-dose vaccinated convalescents (i.e., BA.1 breakthrough infection; 10–27 days after testing) were obtained (Table S4), and the sensitivity of BA.2.9.1 and BA.2.11 to these antisera was comparable to that of BA.2 (Figure 2C). On the other hand, consistent with recent studies (Khan et al., 2022; Tuekprakhon et al., 2022), BA.4/5 was significantly (2.3-fold) more resistant to BA.1 breakthrough infection sera than BA.2 (p < 0.0001 by the Wilcoxon signed-rank test), and the F486V conferred resistance (Figure 2C). In the case of the sera from 14 individuals infected with BA.2 who were 2-dose (9 individuals) or 3-dose (5 individuals) vaccinated convalescents (i.e., BA.2 breakthrough infection; 11–61 days after testing), BA.4/5 exhibited a significant (1.8-fold) resistance compared with BA.2 (Figure 2D; p = 0.0059 by the Wilcoxon signed-rank test). In contrast to BA.1 breakthrough infection sera (Figure 2C), the S704L but not the F486V contributed to the resistance to BA.2 breakthrough infection sera (Figure 2D). Interestingly, the insertion of HV69-70del and R493Q made the pseudovirus more sensitive to both BA.1 (Figure 2C) and BA.2 (Figure 2D) breakthrough infection sera.
Figure 2.

Immune resistance of BA.2 subvariants
Neutralization assays were performed with pseudoviruses harboring the S proteins of B.1.1 (the D614G-bearing ancestral virus), BA.1, BA.2 subvariants and BA.2-based derivatives, and the following sera.
(A) Convalescent sera from not fully vaccinated individuals who had been infected with BA.1 (14 non-vaccinated donors).
(B) Convalescent sera from not fully vaccinated individuals who had been infected with BA.2 (9 non-vaccinated and 1 1-dose vaccinated. 10 donors in total).
(C) Convalescent sera from fully vaccinated individuals who had been infected with BA.1 after full vaccination (16 2-dose vaccinated donors).
(D) Convalescent sera from fully vaccinated individuals who had been infected with BA.2 after full vaccination (9 2-dose vaccinated and 5 3-dose vaccinated. 14 donors in total).
(E–G) BNT162b2 vaccine sera (15 donors) collected at 1 month after 2nd-dose vaccination (E), 1 month after 3rd-dose vaccination (F), and 4 months after 3rd-dose vaccination (G).
(H) Sera from mice immunized with BA.2 S RBD (11 mice).
(I–K) Sera from hamsters infected with BA.2 (18 hamsters) (I), BA.2.12.1 (6 hamsters) (J), and BA.4/5 (6 hamsters) (K).
Assays for each serum sample were performed in triplicate to determine the 50% neutralization titer (NT50). Each dot represents one NT50 value, and the geometric mean and 95% CI are shown. The numbers in red indicate the fold change resistance of BA.4/5 versus BA.2 (C, D, F–I, and K) or BA.2.12.1 (J). The horizontal dashed line indicates the detection limit (120-fold). Statistically significant differences were determined by two-sided Wilcoxon signed-rank tests. The p values between BA.4/5 and BA.2 (C, D, F–I, and K) or BA.2.12.1 (J) are indicated in the panels. Asterisks in the panels indicate statistically significant differences (p < 0.05) between BA.2 and BA.2-based derivatives. Red and blue asterisks, respectively, indicate decreased and increased NT50s. Information on the vaccinated/convalescent donors is summarized in Table S4.
See also Table S4.
To assess the sensitivity of BA.2-related subvariants to vaccine-induced humoral immunity, we used vaccine sera collected from 15 individuals 1 month after the 2nd-dose vaccination (Figure 2E), 1 month after the 3rd-dose vaccination (Figure 2F), and 4 months after the 3rd-dose vaccination (Figure 2G). As recently reported (Gruell et al., 2022; Qu et al., 2022), 2nd-dose vaccine sera did not work against all BA.2-related subvariants tested (Figure 2E). Although the sera after the 3rd-dose vaccinations were effective against BA.2-related subvariants, BA.4/5 exhibited 1.8- and 1.6-fold more resistance to vaccine sera 1 month after the 3rd-dose vaccination (Figure 2F; p = 0.0039 by the Wilcoxon signed-rank test) and 4 months after the 3rd-dose vaccination (Figure 2G; p = 0.0039 by the Wilcoxon signed-rank test), respectively. Similar to BA.2 breakthrough infection sera (Figure 2D), the S704L contributed to the resistance to 3rd-dose vaccine sera (Figures 2F and 2G). Moreover, similar to both BA.1 (Figure 2C) and BA.2 (Figure 2D) breakthrough infection sera, the HV69-70del and R493Q increased the sensitivity to 3rd-dose vaccine-induced antiviral sera (Figures 2F and 2G).
To further address the possibility of evasion of BA.2-related subvariants from the immunity induced by the infection of the original BA.2, we used the sera obtained from both BALB/c mice immunized with recombinant BA.2 S RBD (Figure 2H) and hamsters infected with BA.2 (Figure 2I) (Yamasoba et al., 2022b). BA.4/5 was 2.3-fold more resistant to BA.2 S RBD-immunized murine sera (Figure 2H; p < 0.0001 by the Wilcoxon signed-rank test) and 3.8-fold more resistant to the BA.2-infected hamster sera collected at 16 days post infection (d.p.i.) (Figure 2I; p = 0.0009 by the Wilcoxon signed-rank test). In contrast to the results using human sera, the L452Q and R493Q contributed to the resistance to BA.2-induced rodent antisera (Figures 2H and 2I). Moreover, we used the sera collected from BA.2.12.1-infected (Figure 2J) and BA.4/5-infected (Figure 2K) hamsters at 16 d.p.i. Although the antiviral activity of BA.2.12.1-infected hamster sera against BA.2 was comparable to that against 2.12.1, BA.4/5 exhibited 3.6-fold higher resistance than BA.2.12.1 (Figure 2J; p = 0.031 by the Wilcoxon signed-rank test). Moreover, BA.2 was 10.9-fold more resistant to the BA.4/5-infected hamster sera than BA.4/5 (Figure 2K; p = 0.031 by the Wilcoxon signed-rank test). Altogether, these results suggest that BA.4/5 is resistant to the immunity induced by BA.1 and BA.2, and that the antigenicity of BA.4/5 is different from that of BA.2 and BA.2.12.1.
Virological features of BA.2 subvariants
To investigate the virological characteristics of the L452R/Q/M-bearing BA.2 subvariants, we measured pseudovirus infectivity using HOS cells expressing ACE2 and transmembrane protease, serine 2 (TMPRSS2) (Kimura et al., 2022a; Motozono et al., 2021; Saito et al., 2022; Suzuki et al., 2022; Uriu et al., 2021; Yamasoba et al., 2022b). As shown in Figure 3 A, all BA.2 subvariants tested exhibited significantly higher infectivity than BA.2. The pseudovirus infectivity of BA.2.9.1, BA.2.11, and BA.2.12.1 was comparable to that of ancestral D614G-bearing B.1.1 and, notably, the infectivity of the BA.4/5 pseudovirus was 18.3-fold higher than that of the BA.2 pseudovirus (Figure 3A). The BA.2 derivatives bearing L452Q, HV69-70del, and F486V exhibited increased infectivity (Figure 3A). These results suggest that multiple mutations in the BA.4/5 S proteins, including HV69-70del, L452R, and F486V, increase pseudovirus infectivity. However, when we used both HEK293-ACE2/TMPRSS2 cells and HEK293-ACE2 cells, on which endogenous surface TMPRSS2 is undetectable (Yamasoba et al., 2022b), as target cells, the fold increase in pseudovirus infectivity of BA.2 subvariants caused by TMPRSS2 expression on the target cells was not observed (Figure S2 A). These results suggest that TMPRSS2 is not associated with an increase in pseudovirus infectivity. A yeast surface display assay using the SARS-CoV-2 S RBD and soluble human ACE2 (Dejnirattisai et al., 2022; Kimura et al., 2022a, 2022b; Motozono et al., 2021; Yamasoba et al., 2022b; Zahradník et al., 2021a) showed that the KD value of the BA.4/5 S RBD is comparable to that of BA.2 S RBD (Figure 3B), suggesting that the binding affinity of the S RBDs of BA.2 and BA.4/5 is similar. The L452R and L452M substitutions decreased KD values, while the F486V substitution increased KD value (Figure 3B). Because the KD value of the L452R/F486V BA.2 was significantly lower than that of the F486V BA.2 (Figure 3B), our results suggest that the L452R compensates for the decreased binding affinity to human ACE2 caused by the F486V and, therefore, the binding affinity of BA.2 and BA.4/5 to human ACE2 are similar.
Figure 3.

Virological features of BA.2 subvariants in vitro
(A) Pseudovirus assay. The percent infectivity compared with that of the virus pseudotyped with the BA.2 S protein are shown.
(B) Binding affinity of the RBD of SARS-CoV-2 S protein to ACE2 by yeast surface display. The KD value indicating the binding affinity of the RBD of the SARS-CoV-2 S protein to soluble ACE2 when expressed on yeast is shown.
(C) Crystal structure of the BA.4/5 S RBD-human ACE2 complex. Characteristic substitutions in the BA.4/5 S RBD are shown in brown sticks. In the close-up view, the ACE2 residues surrounding these substitutions are shown in dark gray sticks. Corresponding residues in the BA.1 S RBD-human ACE2 complex structure are also shown in green (RBD) and light gray (ACE2) sticks. Dashed lines represent hydrogen bonds.
(D and E) S-based fusion assay. (D) S protein expression on the cell surface. Representative histograms stained with an anti-S1/S2 polyclonal antibody are shown in Figure S2B, and the summarized data are shown. In the left panel, the number in the histogram indicates the mean fluorescence intensity (MFI). Gray histograms indicate isotype controls. (E) S protein-based fusion assay in Calu-3 cells. The recorded fusion activity (arbitrary units) is shown. The dashed green line indicates the results of BA.2.
Assays were performed in quadruplicate (A and E) or triplicate (B and D), and the presented data are expressed as the average ± SD. Each dot indicates the result of an individual replicate. In (A), (B), and (D), statistically significant differences between BA.2 and other variants (∗ p < 0.05 in D) were determined by two-sided Student’s t tests. In (E), statistically significant differences between BA.2 and other variants across time points were determined by multiple regression. The familywise error rates (FWERs) calculated using the Holm method are indicated in the figures.
Figure S2.

Virological features of the S proteins of BA.2 subvariants in vitro, related to Figure 3
(A) Fold increase in pseudovirus infectivity based on TMPRSS2 expression.
(B) Electron density map of the three substituted amino acid residues, compared with those of BA.2, in the RBD of BA.4/5. Polder maps (Liebschner et al., 2017) omitting the R452, V486 or Q493 in the RBD and surrounding residues contoured at the level of 3.5σ, 2.5σ, or 3.1σ, are respectively shown.
(C) S protein expression on the cell surface. Representative histograms stained with an anti-S1/S2 polyclonal antibody are shown. The number in the histogram indicates MFI. Gray histograms indicate isotype controls. The summarized data are shown in Figure 3D.
(D) S-based fusion assay in VeroE6/TMPRSS2 cells. The recorded fusion activity (arbitrary units) is shown. The dashed green line indicates the results of BA.2.
(E) Coculture of S-expressing cells with HEK293-ACE2/TMPRSS2 cells. Left, representative images of S-expressing cells cocultured with HEK293 cells (top) or HEK293-ACE2/TMPRSS2 cells (bottom). Nuclei were stained with Hoechst 33342 (blue). Right, size distribution of syncytia (green). The numbers in parentheses indicate the numbers of GFP-positive syncytia counted. Scale bars, 200 μm.
In (A) and (D), assays were performed in quadruplicate, and the presented data are expressed as the average ± SD. In (A) and (E), each dot indicates the result of an individual replicate.
In (D), statistically significant differences between BA.2 and the other variants across time points were determined by multiple regression. The FWERs calculated using the Holm method are indicated in the figures.
In (E), statistically significant differences between BA.2 and the other variants (∗ p < 0.05) were determined by two-sided Mann-Whitney U tests.
To gain structural insight into the interaction of the BA.4/5 S RBD with human ACE2 protein, we performed X-ray crystallographic analysis of the BA.4/5 S RBD-human ACE2 complex and determined its structure at a resolution of 3.36 Å (Figures 3C and S2B; Table S5). Three amino acid substitutions (L452R, F486V, and R493Q) in the BA.4/5 S RBD focused on the interaction with human ACE2 were compared with the BA.2 S RBD (Figure 3C). First, the residue R452 of the BA.4/5 S RBD did not form interactions with any residues of ACE2 (Figure 3C). This observation is reminiscent of the findings on the complex structure of the Delta S RBD, which bears the L452R, and human ACE2 (Han et al., 2022). Second, residue V486 of the BA.4/5 S RBD is located in a hydrophobic patch that is formed by residues F28, L79, M82, and Y83 of human ACE2 (Figure 3C). Compared with phenylalanine (F), valine (V) reduces bulkiness. Therefore, the reduced bulkiness induced by the F486V in the BA.4/5 S RBD may decrease the hydrophobic interaction with human ACE2, as observed in in vitro binding experiments (Figure 3B). Third, residue Q493 of the BA.4/5 S RBD formed a hydrogen bond with residue H34 of human ACE2 (Figure 3C). Related to this interaction, a previous study showed that residue R493 of the Omicron BA.1 S RBD forms a hydrogen bond with residue E35 of human ACE2 (Han et al., 2022). Altogether, the overall binding mode of the BA.4/5 S RBD to human ACE2 is similar to those of the original Wuhan-Hu-1 strain (Lan et al., 2020) and the BA.1 S RBD (Han et al., 2022).
We next analyzed the fusogenicity of the BA.2-related Omicron variants by a cell-based fusion assay (Kimura et al., 2022b; Motozono et al., 2021; Saito et al., 2022; Suzuki et al., 2022; Yamasoba et al., 2022b). As shown in Figures 3D and S2C, the cell surface expression of BA.2.11 S was significantly higher than that of BA.2 S. However, the surface expressions of BA.2.12.1 S and BA.4/5 S were significantly lower than those of the BA.2 S (Figure 3D). The decreased surface expression of the BA.2.12.1 and BA.4/5 S proteins was attributed to the S704L (BA.2.12.1) and the HV69-70del, F486V, and R493Q (BA.4/5), respectively (Figure 3D). The cell-based fusion assay using Calu-3 cells as target cells showed that the fusogenicity of the BA.2.11 S and the BA.4/5 S was significantly greater than that of BA.2 S, while the other substitutions did not critically affect S protein-mediated fusogenicity (Figure 3E). When we used VeroE6/TMPRSS2 cells as target cells, all BA.2 derivatives bearing substitutions at the L452 residue that were tested (i.e., L452R/M/Q; BA.2.9.1, BA.2.11, and BA.2 L452Q), as well as BA.4/5, significantly increased fusogenicity compared with that of the original BA.2, while the fusogenicity of the other mutants, including BA.2.12.1, was comparable to that of BA.2 (Figure S2D). Moreover, a coculture experiment using HEK293-ACE2/TMPRSS2 cells as the target cells (Suzuki et al., 2022; Yamasoba et al., 2022b) showed that the S proteins of BA.2.9.1, BA.2.11, and BA.4/5, but not BA.2.12.1, showed significantly increased fusogenicity compared with that of the original BA.2 (Figure S2E). Altogether, these findings suggest that the S proteins of BA.2 subvariants that bear the L452R, including BA.2.11 and BA.4/5, exhibited higher fusogenicity than the BA.2 S in three independent experimental models (Figures 3E, S2D, and S2E).
Growth capacity of BA.2 subvariants in vitro
We next prepared the chimeric recombinant BA.2 subvariants, rBA.2, rBA.2.9.1, rBA.2.11, rBA.2.12.1, and rBA.4/5, based on a clinical isolate of BA.2 (strain TY40-385, GISAID ID: EPI_ISL_9595859) by reverse genetics (Kimura et al., 2022b; Motozono et al., 2021; Saito et al., 2022; Torii et al., 2021; Yamasoba et al., 2022b) (Figure 4 A). The S genes of recombinant viruses were swapped with those of BA.2 subvariants: BA.2.11 (BA.2 S:L452R), BA.2.9.1 (BA.2 S:L452M), BA.2.12.1 (BA.2 S:L452Q/S704L), or BA.4/5 (BA.2 S:HV69-70del/L452R/F486V/R493Q) (Figure 4A). The plaques formed by the infections of rBA.2.11 and rBA.4/5, which bear the L452R, were larger than those formed by rBA.2 infection, while rBA.2.9.1 infection showed significantly smaller plaques than rBA.2 infection (Figure 4B). Combined with the results of the experiments using S expression plasmids (Figures 3E, S2D, and S2E), these data suggest that the S proteins of BA.2 subvariants bearing the L452R substitution (BA.2.11 and BA.4/5) are more fusogenic than the BA.2 S.
Figure 4.

Growth capacity of BA.2 subvariants in vitro
(A) Scheme for the chimeric recombinant SARS-CoV-2 used in this study. The SARS-CoV-2 genome and its genes are shown. The template was SARS-CoV-2 strain TY40-385 (PANGO lineage BA.2, GISAID ID: EPI_ISL_9595859), and the S genes were swapped with those of the BA.2-related Omicron variants. The substitutions based on the BA.2 S protein are summarized in parentheses.
(B) Plaque assay. VeroE6/TMPRSS2 cells were used for the target cells. Representative panels and a summary of the recorded plaque diameters (20 plaques per virus) (lower right) are shown.
(C–F) Growth kinetics of the chimeric recombinant SARS-CoV-2. VeroE6/TMPRSS2 cells (C), Vero cells (D), human iPSC-derived airway epithelial cells (E), and alveolar epithelial cells (F) were infected with the BA.2-based chimeric recombinant SARS-CoV-2, and the copy numbers of viral RNA in the culture supernatant were routinely quantified by RT-qPCR. The dashed green line indicates the results of rBA.2.
In (B) (lower right panel), each dot indicates the result of an individual plaque, and the presented data are expressed as the average ± SD. Statistically significant differences versus rBA.2 (∗ p < 0.05) were determined by two-sided Mann-Whitney U tests.
In (C)–(F), assays were performed in quadruplicate and the presented data are expressed as the average ± SD. Statistically significant differences between rBA.2 and the other variants across time points were determined by multiple regression. The FWERs calculated using the Holm method are indicated in the figures.
To measure the growth kinetics of BA.2 subvariants, recombinant viruses were inoculated into cells. The replication kinetics of the recombinant BA.2 subvariants tested were comparable to those of rBA.2 in VeroE6/TMPRSS2 cells (Figure 4C). In Vero cells, the growth of rBA.4/5 was similar to that of rBA.2, but that of the other BA.2 subvariants was significantly lower than that of rBA.2 (Figure 4D). In the culture of human airway epithelial cells derived from human induced pluripotent stem cells (iPSCs), the growth of recombinant BA.2 subvariants tested was relatively comparable to that of rBA.2 (Figure 4E). Notably, rBA.2.9.1, rBA.2.12.1, and rBA.4/5 were significantly more efficiently replicated than rBA.2 in human iPSC-derived alveolar epithelial cells (Figure 4F). In particular, at 24 h post infection (h.p.i.), the levels of viral RNA in the supernatant of rBA.2.12.1- and rBA.4/5-infected cultures were 61- and 34-fold higher, respectively, than those of the rBA.2-infected culture (Figure 4F). These results suggest that rBA.2.12.1 and rBA.4/5 replicate more efficiently in human alveolar epithelial cells than BA.2.
Virological features of rBA.2.12.1 and rBA.4/5 in vivo
To investigate the dynamics of viral replication of BA.2 subvariants in vivo, we conducted hamster infection experiments using rBA.2 and the two recombinant BA.2 subvariants, rBA.2.12.1 and rBA.4/5, that exhibited robust growth capacity in human lung cell culture (Figure 4F). Consistent with a recent report (Uraki et al., 2022), the rBA.2-infected hamsters did not exhibit apparent alterations in body weight or two surrogate markers of bronchoconstriction and airway obstruction (enhanced pause [Penh] and the ratio of time to peak expiratory follow-up relative to the total expiratory time [Rpef]). Furthermore, there was no decrease in subcutaneous oxygen saturation (SpO2) observed (Figure 5 A). Notably, the body weights of the rBA.2.12.1-infected and rBA.4/5-infected hamsters were significantly lower than those of the rBA.2-infected hamsters (Figure 5A). Additionally, the Rpef value of the rBA.4/5-infected hamsters was significantly lower than that of the rBA.2-infected hamsters (Figure 5A). These data suggest that rBA.2.12.1 and rBA.4/5 exhibit higher pathogenicity than rBA.2.
Figure 5.

Virological features of BA.2.12.1 and BA.4/5 in vivo
Syrian hamsters were intranasally inoculated with rBA.2, rBA.2.12.1, and rBA.4/5 (summarized in Figure 4A).
(A) Body weight and Penh, Rpef, and SpO2 values were routinely measured. Hamsters of the same age were intranasally inoculated with saline (uninfected).
(B) Viral RNA loads in the oral swab (top), lung hilum (middle), and lung periphery (bottom).
(C) Viral titers in the lung periphery.
(D) IHC of the viral N protein in the lungs at 1, 3, and 5 d.p.i. of all infected hamsters (n = 4 per viral strain).
(E) Percentage of N-positive cells in whole lung lobes at 3 d.p.i.. The raw data are shown in Figure S3B.
(F and G) (F) Histopathological scoring of lung lesions. Representative pathological features are reported in our previous studies (Saito et al., 2022; Suzuki et al., 2022; Yamasoba et al., 2022b). (G) H&E staining of the lungs of infected hamsters. Uninfected lung alveolar space and bronchioles are also shown.
(H) Type II pneumocytes in the lungs of infected hamsters. The percentage of the area of type II pneumocytes in the lung at 5 d.p.i. is shown. The raw data are shown in Figure S3C.
Data are presented as the average (A and B, top, 6 hamsters per viral strain; B, middle and bottom; C, E, F, and H, 4 hamsters per viral strain) ± SEM. In (E) and (H), each dot indicates the result of an individual hamster.
In (A)–(C) and (F), statistically significant differences between rBA.2 and other variants across time points were determined by multiple regression. The 0 d.p.i. data were excluded from the analyses. The FWERs calculated using the Holm method are indicated in the figures.
In (E) and (H), the statistically significant differences between rBA.2 and other variants were determined by a two-sided Mann-Whitney U test.
In (D) and (G), each panel shows a representative result from an individual infected hamster. Scale bars, 100 μm.
See also Figure S3.
To analyze viral spread in the respiratory organs of infected hamsters, the viral RNA load and nucleocapsid (N) expression were assessed by reverse-transcption qPCR (RT-qPCR) analysis of viral RNA and immunohistochemistry (IHC), respectively. As shown in Figure 5B, the viral RNA loads in the lung hilum of rBA.2.12.1- and rBA.4/5-infected hamsters were significantly higher than those of rBA.2-infected hamsters. Intriguingly, the viral RNA loads in the oral swab (Figure 5B, top) and lung periphery (Figure 5B, bottom) of hamsters infected with rBA.4/5 were significantly higher than those of hamsters with rBA.2, while those of hamsters with rBA.2.12.1 were not significantly different from those of hamsters with rBA.2. In particular, the levels of viral RNA in the lung periphery of rBA.4/5-infected hamsters at 3 and 5 d.p.i. were 5.7- and 4.2-fold higher, respectively, than those of rBA.2-infected hamsters (Figure 5B, bottom). The higher level of viral load in the lung periphery of rBA.4/5-infected hamsters than that of rBA.2-infected hamsters was also supported by the level of infectious viruses in these regions (Figure 5C). These results suggest that rBA.4/5 spreads more efficiently in the lungs of infected hamsters than rBA.2.
To address the possibility that rBA.4/5 spreads more efficiently than BA.2, we investigated N protein positivity in the trachea and the lung area close to the hilum. At 1 d.p.i., there was no apparent difference in the N protein positivity in the tracheal epithelium among hamsters infected with rBA.2, rBA.2.12.1, and rBA.4/5 (Figure S3 A). In the bronchial and bronchiolar epithelia, rBA.2.12.1 and BA.4/5 infections exhibited more N-positive cells than rBA.2 infection (Figure 5D). At 3 d.p.i., alveolar positivity was observed in lungs infected with rBA.2.12.1 and rBA.4/5 but not in those infected with rBA.2 (Figure 5D). Morphometry showed that the percentage of N-positive cells in lungs infected with rBA.2.12.1 and rBA.4/5 was significantly higher than that in lungs infected with rBA.2 at 3 d.p.i. (Figures 5E and S3B). At 5 d.p.i., N protein expression had almost disappeared in all infected lungs (Figure 5D). These data suggest that rBA.2.12.1 and rBA.4/5 more efficiently spread in lung tissues than rBA.2.
Figure S3.

Virological features of BA.2.12.1 and BA.4/5 in vivo, related to Figure 5
(A) IHC of the viral N protein in the middle portion of the tracheas of all infected hamsters (n = 4 per viral strain) at 1 d.p.i. Each panel shows a representative result from an individual infected hamster.
(B) Right lung lobes of hamsters infected with rBA.2, rBA.2.12.1, or rBA.4/5 (n = 4 per viral strain) at 3 d.p.i. were immunohistochemically stained with an anti-SARS-CoV-2 N monoclonal antibody. In each panel, IHC staining (top) and the digitalized N-positive area (bottom, indicated in red) are shown. The number in the bottom panel indicates the percentage of the N-positive area. Summarized data are shown in Figure 5E.
(C) Type II pneumocytes in the lungs of infected hamsters. Right lung lobes of hamsters infected with rBA.2, rBA.2.12.1, or rBA.4/5 (n = 4 per viral strain) at 5 d.p.i. In each panel, H&E staining (top) and the digitalized inflammation area (bottom, indicated in red) are shown. The number in the bottom panel indicates the percentage of the section represented by the indicated area (i.e., the area indicated in red within the total area of the lung lobe). Summarized data are shown in Figure 5H.
Scale bars, 1 mm.
Pathogenicity of rBA.2.12.1 and rBA.4/5
To investigate the pathogenicity of rBA.2.12.1 and rBA.4/5, the right lungs of the infected hamsters were collected at 1, 3, and 5 d.p.i. and subjected to histopathological analysis (Figure 5F) and hematoxylin and eosin (H&E) staining (Figure 5G) (Saito et al., 2022; Suzuki et al., 2022; Yamasoba et al., 2022b). Three histological parameters, bronchitis/bronchiolitis, hemorrhage/congestion and alveolar damage, and the total histological score of rBA.2.12-infected hamsters, were significantly higher than those of rBA.2-infected hamsters (Figures 5F and 5G). More importantly, all histopathological parameters were significantly higher in rBA.4/5-infected hamsters (Figure 5F). Furthermore, in the lungs of infected hamsters at 5 d.p.i., the level of inflammation with type II alveolar pneumocyte hyperplasia by rBA.4/5 infection was significantly higher than that by rBA.2 infection, while there was no statistically significant difference between the rBA.2.12.1 and rBA.2 infections (Figures 5H and S3C). The relatively more severe disorders in the lungs of rBA.4/5-infected hamsters than those of the rBA.2-infected hamsters (Figures 5F–5H) were supported by the more efficient spreading of rBA.4/5 than rBA.2 in the infected lungs (Figures 5B, bottom and 5C). Altogether, these observations suggest that rBA.4/5 is more pathogenic than rBA.2 in a hamster model.
Discussion
Viral transmissibility, immune resistance, and pathogenicity characterize the potential risk of new SARS-CoV-2 variants to global health. In this study, we investigated the virological characteristics of the five novel BA.2 subvariants that were classified as VOC-LUM as of May 18, 2022: BA.2.9.1, BA.2.11, BA.2.12.1, BA.4, and BA.5. Recent studies evaluated a series of virological characteristics of novel BA.2 subvariants, particularly BA.2.12.1, BA.4, and BA.5 in terms of their transmissibility (Altarawneh et al., 2022; Tegally et al., 2022), immune resistance (Arora et al., 2022; Cao et al., 2022; Gruell et al., 2022; Hachmann et al., 2022; Khan et al., 2022; Lyke et al., 2022; Qu et al., 2022; Tuekprakhon et al., 2022; Wang et al., 2022a; Yamasoba et al., 2022c), characteristics in cell cultures (Aggarwal et al., 2022; Reuschl et al., 2022), and pathogenicity (Kawaoka et al., 2022; Wolter et al., 2022). Here, we revealed that BA.4 and BA.5 render the highest potential risk in terms of growth efficacy in the human population, resistance to antiviral humoral immunity, and pathogenicity in an experimental animal model.
We demonstrated that the L452R increases binding affinity to human ACE2 and pseudovirus infectivity, even on the BA.2 S protein backbone. Therefore, together with our previous reports (Kimura et al., 2022a; Motozono et al., 2021), our findings indicate that the L452R/Q contributes to increasing viral infectivity independently of viral backbone. Additionally, the S proteins of BA.4 and BA.5 harbor the HV69-70del, and this deletion was also detected in the Alpha variant, which is a prior VOC. Consistent with a previous study (Meng et al., 2021), we demonstrated that the insertion of the HV69-70del mutation increases pseudovirus infectivity even on the BA.2 S protein backbone. Altogether, multiple mutations in the S protein of BA.4/5 contribute to enhanced growth capacity in human lung cell cultures and the lung of an experimental animal model. However, the bona fide contribution of these substitutions, particularly the L452R in BA.4/5, should be carefully studied in the future. Given that Delta and Lambda, which contained the L452R and L452Q, respectively, were outcompeted by original Omicron BA.1, which does not bear the L452R/Q, it is possible that the L452R that appeared in the BA.4/5 S may also revert to its original sequence. In fact, as of July 2022, it is assumed that a new BA.2 subvariant, BA.2.75, may outcompete other BA.2 subvariants, including BA.4/5 (GitHub, 2022; Yamasoba et al., 2022a), although the S protein of BA.2.75 does not harbor any substitutions at the L452 residue of the S protein.
In our previous studies that focused on Delta (Saito et al., 2022), Omicron BA.1 (Suzuki et al., 2022), and Omicron BA.2 (Yamasoba et al., 2022b), we suggested a close association between viral fusogenicity in in vitro cell cultures and pathogenicity in an in vivo hamster model. For instance, compared with the B.1.1 ancestral variant, the Omicron BA.1 variant, which bore a less fusogenic S, was less pathogenic, while the Delta variant, bearing a more fusogenic S, was more pathogenic (Saito et al., 2022; Suzuki et al., 2022). Here, we demonstrated that the Omicron BA.4/5 variant bears a more fusogenic S than the Omicron BA.2 variant and exhibits greater pathogenicity. The comparisons of fusogenicity and pathogenicity across these four studies as well as this present study suggest that the Delta variant has the greatest fusogenicity and pathogenicity of the five SARS-CoV-2 variants tested. Importantly, our data support the possibility that viruses with higher fusogenicity exhibit potentially higher pathogenicity, at least in our experimental animal models. Therefore, measuring the fusogenicity of viral S proteins using a system such as the cell-based membrane fusion assay described here can be a rapid surrogate marker for predicting the potential pathogenicity of viruses. Further investigation of the correlation between viral fusogenicity and clinical severity will be needed to understand the significance of the S protein on pathogenicity.
Considering the complex evolution of SARS-CoV-2 in the human population, the virus has likely evolved to optimize its transmissibility. From the point of view of pathology, the Delta variant exhibited higher pathogenicity than ancestral variants (Saito et al., 2022). Conversely, we observed the attenuated pathogenicity of the Omicron BA.1 variant compared with the Delta variant (Suzuki et al., 2022). After disseminating worldwide, BA.1 was replaced with BA.2, which possesses pathogenicity comparable to that of BA.1 (Uraki et al., 2022). On the other hand, our investigation using B.1.1-based chimeric viruses revealed that a virus harboring BA.2 S showed higher pathogenicity than that harboring the BA.1 S (Yamasoba et al., 2022b). These observations suggest that the evolution of SARS-CoV-2 would not necessarily attenuate its pathogenicity. One possibility is that the pathogenicity observed at the bedside is a consequence of viral evolution aiming to maximize transmissibility. Given that Omicron BA.1 replicates more efficiently in human cells derived from the upper respiratory tract variant but less efficiently in the lower respiratory tract compared with prior variants (Hui et al., 2022), BA.1 was likely to be selected to achieve higher transmissibility than to exhibit higher pathogenicity in the lung. Another scenario of the evolution of the Omicron BA.1 variant stems from adaptation to human host factors. This includes a more efficient utilization of the factors that can positively function for virus infection and replication, such as TMPRSS2, cathepsin, ADAM10, and ADAM17 proteases (Jocher et al., 2022). Although the BA.1 S is less dependent on TMPRSS2 than ancestral strains (Meng et al., 2022; Suzuki et al., 2022), a recent study has shown that the BA.5 S regained dependency on TMPRSS2 (Aggarwal et al., 2022). The switch of the viral entry pathway may alter tissue tropism and pathogenicity. Moreover, it is speculated that SARS-CoV-2 would have evolved to evade factors that can negatively impact virus replication in human cells. In fact, a recent study demonstrated that the replication of BA.4 and BA.5 is associated with reduced activation of innate immune responses (Reuschl et al., 2022). Furthermore, while most studies focus on the roles of the S protein and its impact on neutralization sensitivity and the cell entry pathway, a recent study proposed the importance of the investigations of non-S viral components such as envelope and membrane proteins (Abbas et al., 2022). Collectively, the complex interplay between SARS-CoV-2 and host factors remains to be elucidated.
We showed that BA.4/5 is significantly resistant to the humoral immunity induced by vaccination and breakthrough infections of BA.1 and BA.2 (Figures 2C and 2D). The F486V in the BA.4/5 S protein is responsible for its higher resistance to the breakthrough infection antisera (Figure 2C). On the other hand, our binding assay suggests that the F486V and L452R exhibited opposing effects on the affinity to the human ACE2 receptor: the F486V reduces the affinity to human ACE2, while the L452R increases the affinity (Figure 3B). These results suggest that the L452R is likely to compensate for the ACE2 binding affinity reduced by the F486V. Interestingly, our analysis of SARS-CoV-2 epidemic dynamics showed that BA.2.11, which bears the L452R but not the other mutations present in the S protein of BA.4/5, did not spread as much as BA.4/5 did in the human population (Figures 1D, 1E, and S1F). This observation suggests that the L452R does not increase BA.2 fitness by itself. Altogether, our results suggest that BA.4/5 acquired a higher resistance to humoral immunity while maintaining the ACE2 binding affinity by acquiring the F486V and L452R substitutions together.
Omicron variants other than BA.4/5 bear the Q493R substitution in the S protein (Figure 1C). The R493Q reversion in BA.2 S protein increased susceptibility to the 3rd-dose vaccine-induced antiviral sera, suggesting that the Q493R substitution contributes to the resistance to vaccine-induced humoral immunity (Figures 2F and 2G). On the other hand, BA.4/5 lost the Q493R substitution after divergence from BA.2. Interestingly, in experimental animal models without vaccination, the R493Q reversion in the BA.2 S protein increased the resistance to humoral immunity induced by BA.2 infection or BA.2 RBD inoculation (Figures 2H and 2I). These results suggest that the R493Q reversion mutation in the BA.4/5 S protein potentially contributes to evading immunity induced by the Q493R-bearing variants, such as BA.1 and BA.2. Furthermore, despite the R493Q reversion in the BA.4/5 S protein, BA.4/5 is still highly resistant to the 3rd-dose vaccine-induced antiviral sera (Figures 2F and 2G). This result suggests that the effect of the R493Q reversion on attenuating the resistance to vaccine-induced humoral immunity would be canceled by other substitutions, such as the F486V substitution in the BA.4/5 S protein. Recent studies have demonstrated that BA.4/5 is also resistant to convalescent sera from individuals who were infected with previous SARS-CoV-2 variants, including Delta (Qu et al., 2022; Wang et al., 2022b). Altogether, our results and previous reports suggest that BA.4/5 escapes from the humoral immunity induced by vaccination and/or the natural infection of prior SARS-CoV-2 variants.
A simplistic assumption without conclusive evidence implies that SARS-CoV-2 will evolve to attenuate its pathogenicity. However, we argue against this notion with at least three observations. First, the Delta variant exhibited relatively higher pathogenicity than the ancestral B.1 virus in an experimental animal model (Saito et al., 2022). Clinical studies also provide evidence suggesting the higher virulence of the Delta variant than other prior variants, including the Alpha variant (Ong et al., 2021; Sheikh et al., 2021; Twohig et al., 2022). Second, although the Omicron BA.1 variant was less pathogenic than Delta and the ancestral B.1.1 virus (Suzuki et al., 2022), the S protein of a subsequently spread variant, Omicron BA.2, acquired the potential to exhibit higher pathogenicity than that of Omicron BA.1 (Yamasoba et al., 2022b). Third, we demonstrated that Omicron BA.4/5 is potentially more pathogenic than Omicron BA.2. Therefore, our observations strongly suggest that SARS-CoV-2 does not necessarily acquire mutations to attenuate its pathogenicity.
Limitations of the study
In our previous study, we used a chimeric virus bearing the BA.2 S gene in a non-BA.2 (PANGO lineage A) genomic backbone and showed the BA.2 S-bearing chimeric virus is more pathogenic in infected hamsters than the BA.1 S-bearing chimeric virus (Yamasoba et al., 2022b). However, another study using a clinical isolate of BA.2 showed a comparable pathogenicity to a BA.1 clinical isolate (Uraki et al., 2022). This inconsistency of BA.2 pathogenicity found between our recent study (Yamasoba et al., 2022b) and another study (Uraki et al., 2022) could be due to the difference in the viral sequence in the non-S region. In fact, there are 26 mutations in the non-S region between BA.2 and the non-BA.2 backbone (PANGO lineage A) that was used in our previous study (Yamasoba et al., 2022b) (Table S1). To avoid such inconsistency, we generated the recombinant viruses based on BA.2 (Figure 4A). Compared with BA.2, the majority of BA.2.12.1 does not possess any mutations in the non-S region (Table S1), indicating that the BA.2-based recombinant virus encoding BA.2.12.1 S used for hamster experiments (rBA.2.12.1) is an authentic BA.2.12.1. Moreover, only six and two mutations were detected in the non-S regions of the BA.4 and BA.5 genomes, respectively, compared with the BA.2 genome (Table S1). Therefore, it would be reasonable to assume that our findings in the use of recombinant viruses reflect the potential of authentic BA.4/5, compared with BA.2, to exhibit lung tropism (Figure 4F) and higher pathogenicity in a hamster model (Figure 5). However, Kawaoka et al. recently showed that the pathogenicity of clinical isolates of BA.4/5 viruses is comparable to that of a BA.2 isolate in animal models (Kawaoka et al., 2022). Moreover, Reuschl et al. showed that the non-S viral proteins encoded by BA.4/5 more efficiently modulate innate immune signaling (Reuschl et al., 2022). Further investigations are warranted to clarify the bona fide characteristics of the BA.2 subvariants tested in this study.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
Antibodies | ||
| Rabbit anti-SARS-CoV-2 S S1/S2 polyclonal antibody | Thermo Fisher Scientific | Cat# PA5-112048; RRID: AB_2866784 |
| Normal rabbit IgG | SouthernBiotech | Cat# 0111-01; RRID: AB_2732899 |
| APC-conjugated goat anti-rabbit IgG polyclonal antibody | Jackson ImmunoResearch | Cat# 111-136-144; RRID: AB_2337987 |
| Mouse anti-SARS-CoV-2 N monoclonal antibody (clone 1035111) | R&D systems | Cat# MAB10474-SP; RRID: N/A |
| Bacterial and virus strains | ||
| Recombinant SARS-CoV-2, rBA.2 | Yamasoba et al., 2022b | N/A |
| Recombinant SARS-CoV-2, rBA.2.11 | This study | N/A |
| Recombinant SARS-CoV-2, rBA.2.12.1 | This study | N/A |
| Recombinant SARS-CoV-2, rBA.4/5 | This study | N/A |
| Biological samples | ||
| Human sera | This study | N/A |
| Human airway epithelial cells derived from human induced pluripotent stem cells (iPSCs) | Yamamoto et al., 2017 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| TransIT-LT1 | Takara | Cat# MIR2300 |
| Recombinant RNase inhibitor | Takara | Cat# 2313B |
| Carboxymethyl cellulose | Wako | Cat# 039-01335 |
| 4% Paraformaldehyde in PBS | Nacalai Tesque | Cat# 09154-85 |
| Methylene blue | Nacalai Tesque | Cat# 22412-14 |
| Fetal bovine serum | Sigma-Aldrich | Cat# 172012-500ML |
| Penicillin-streptomycin | Sigma-Aldrich | Cat# P4333-100ML |
| DMEM (high glucose) | Sigma-Aldrich | Cat# 6429-500ML |
| DMEM (low glucose) | Wako | Cat# 041-29775 |
| EMEM | Sigma-Aldrich | Cat# M4655-500ML |
| EMEM | Wako | Cat# 056-08385 |
| Expi293 expression medium | Thermo Fisher Scientific | Cat# A1435101 |
| PneumaCult ALI medium | STEMCELL Technologies | Cat# ST-05001 |
| Heparin | Nacalai Tesque | Cat# 17513-96 |
| Y-27632 | LC Laboratories | Cat# Y-5301 |
| Hydrocortisone | Sigma-Aldrich | Cat# H0135 |
| Puromycin | InvivoGen | Cat# ant-pr-1 |
| Hygromycin | Nacalai Tesque | Cat# 09287-84 |
| Blasticidin | InvivoGen | Cat# ant-bl-1 |
| G418 | Nacalai Tesque | Cat# G8168-10ML |
| KpnI | New England Biolab | Cat# R0142S |
| NotI | New England Biolab | Cat# R1089S |
| PEI Max | Polysciences | Cat# 24765-1 |
| Doxycycline | Takara | Cat# 1311N |
| TURBO DNase | Thermo Fisher Scientific | Cat# AM2238 |
| Opti-MEM | Thermo Fisher Scientific | Cat# 11058021 |
| Triton X-100 | Nacalai Tesque | Cat# 35501-15 |
| Recombinant RNase inhibitor | Takara | Cat# 2313B |
| Poly-L-lysine | Sigma | Cat# P4832 |
| Hoechst 33342 | Thermo Fisher Scientific | Cat# H3570 |
| Fluoromount-G | SouthernBiotech | Cat# 0100-01 |
| EnduRen live cell substrate | Promega | Cat# E6481 |
| AddaVax | InvivoGen | Cat# vac-adx-10 |
| Polyethylene glycol 6000 | Hampton Research | Cat# HR2-533 |
| 2-methyl-2,4-pentanediol | Hampton Research | Cat# HR2-627 |
| MES | Nacalai Tesque | Cat# 21623-26 |
| Glycerol | Nacalai Tesque | Cat# 17018-25 |
| Dexamethasone | Sigma-Aldrich | Cat# D4902 |
| KGF | PeproTech | Cat# 100-19 |
| 8-Br-cAMP | Biolog | Cat# B007 |
| 3-Isobutyl 1-methylxanthine (IBMX) | FUJIFILM Wako | Cat# 095-03413 |
| CHIR99021 | Axon Medchem | Cat# 1386 |
| SB431542 | Wako | Cat# 198-16543 |
| Soluble human ACE2 (residues 18-740) | Yamasoba et al., 2022b | N/A |
| SARS-CoV-2 B.1.1 RBD | Kimura et al., 2022a; Motozono et al., 2021 | N/A |
| SARS-CoV-2 BA.1 RBD | Dejnirattisai et al., 2022 | N/A |
| SARS-CoV-2 BA.2 RBD | Yamasoba et al., 2022b | N/A |
| SARS-CoV-2 BA.4/5 RBD | This study | N/A |
| Bilirubin | Sigma-Aldrich | Cat# 14370-1G |
| Medetomidine hydrochloride (Domitor®) | Nippon Zenyaku Kogyo | N/A |
| Midazolam | FUJIFILM Wako Chemicals | Cat# 135-13791 |
| Butorphanol (Vetorphale®) | Meiji Seika Pharma | N/A |
| Alphaxaone (Alfaxan®) | Jurox | N/A |
| Isoflurane | Sumitomo Dainippon Pharma | N/A |
| EnVision FLEX target retrieval solution high pH | Agilent | Cat# K8004 |
| Critical commercial assays | ||
| QIAamp viral RNA mini kit | Qiagen | Cat# 52906 |
| NEB next ultra RNA library prep kit for Illumina | New England Biolabs | Cat# E7530 |
| MiSeq reagent kit v3 | Illumina | Cat# MS-102-3001 |
| One Step TB Green PrimeScript PLUS RT-PCR kit | Takara | Cat# RR096A |
| SARS-CoV-2 direct detection RT-qPCR kit | Takara | Cat# RC300A |
| L452R (SARS-CoV-2) primer/probe set v2 | Takara | Cat# RC346A |
| E484A (SARS-CoV-2) primer/probe set v2 | Takara | Cat# RC322A |
| Nano Glo HiBiT lytic detection system | Promega | Cat# N3040 |
| KAPA HiFi HotStart ReadyMix kit | Roche | Cat# KK2601 |
| PrimeSTAR GXL DNA polymerase | Takara | Cat# R050A |
| Bright-Glo luciferase assay system | Promega | Cat# E2620 |
| One-Glo luciferase assay system | Promega | Cat# E6130 |
| Deposited data | ||
| Viral genome sequencing data of working viral stocks (see also Table S7) | This paper | SRA: DRR378901–DRR378909 (https://www.ncbi.nlm.nih.gov/sra) |
| Structure of the BA.4/5 S RBD-human ACE2 complex | This study | PDB: 7XWA |
| Experimental models: Cell lines | ||
| Human: HEK293T cells | ATCC | CRL-3216 |
| Human: HEK293 cells | ATCC | CRL-1573 |
| Human: HEK293-ACE2 cells | Motozono et al., 2021 | N/A |
| Human: HEK293-ACE2/TMPRSS2 cells | Motozono et al., 2021 | N/A |
| Human: HEK293-C34 cells | Torii et al., 2021 | N/A |
| Human: HEK293S GnTI(-) cells | Reeves et al., 2002 | N/A |
| Human: Expi293F cells | Thermo Fisher Scientific | Cat# A14527 |
| Human: HOS-ACE2/TMPRSS2 cells | Ferreira et al., 2021; Ozono et al., 2021 | N/A |
| Human: Calu-3/DSP1-7 cells | Yamamoto et al., 2020 | N/A |
| African green monkey (Chlorocebus sabaeus): Vero cells | JCRB Cell Bank | JCRB0111 |
| African green monkey (Chlorocebus sabaeus): VeroE6/TMPRSS2 cells | JCRB Cell Bank | JCRB1819 |
| Yeast (Saccharomyces cerevisiae): strain EBY100 | ATCC | MYA-4941 |
| Experimental models: Organisms/strains | ||
| BALB/cCrSlc mice (female, 7 weeks old) | Japan SLC Inc. | http://www.jslc.co.jp/pdf/mouse/2020/004_BALB_cCrClc.pdf |
| Slc:Syrian hamsters (male, 4 weeks old) | Japan SLC Inc. | http://www.jslc.co.jp/pdf/hamster/2020/028_Slc_Syrian.pdf |
| Oligonucleotides | ||
| Omi_ins214s-F1: TTC TAA GCA CAC GCC TAT TAT AGT GC | Yamasoba et al., 2022b | N/A |
| Omi_ins214s-R1: TAA AGC CGA AAA ACC CTG AGG | Yamasoba et al., 2022c | N/A |
| Omi_ins214s: FAM-TGA GCC AGA AGA TC-MGB | Yamasoba et al., 2022b | N/A |
| Primers for the construction of plasmids expressing the codon-optimized S proteins of L452R/Q/M-bearing variants, see Table S6 | This study | N/A |
| Primers for SARS-CoV-2 reverse genetics, see Table S6 | This study | N/A |
| RT-qPCR, forward: AGC CTC TTC TCG TTC CTC ATC AC | Kimura et al., 2022b; Meng et al., 2022; Motozono et al., 2021; Saito et al., 2022; Suzuki et al., 2022; Yamasoba et al., 2022b | N/A |
| RT-qPCR, reverse: CCG CCA TTG CCA GCC ATT C | Kimura et al., 2022b; Meng et al., 2022; Motozono et al., 2021; Saito et al., 2022; Suzuki et al., 2022; Yamasoba et al., 2022b | N/A |
| Primers for the construction of yeast-optimized SARS-CoV-2 BA.2 RBD expression plasmid, see Table S6 | This study | N/A |
| Recombinant DNA | ||
| Plasmid: pCAGGS | Niwa et al., 1991 | N/A |
| Plasmid: psPAX2-IN/HiBiT | Ozono et al., 2020 | N/A |
| Plasmid: pWPI-Luc2 | Ozono et al., 2020 | N/A |
| Plasmid: pJYDC1 | Addgene | Cat# 162458 |
| Plasmid: pHLsec | Aricescu et al., 2006 | N/A |
| Plasmid: pDSP1-7 | Kondo et al., 2011 | N/A |
| Plasmid: pDSP8-11 | Kondo et al., 2011 | N/A |
| Plasmid: pCAGGS | Niwa et al., 1991 | N/A |
| Plasmid: pC-B.1.1 S | Motozono et al., 2021; Ozono et al., 2021 | N/A |
| Plasmid: pC-BA.2 S | Yamasoba et al., 2022b | N/A |
| Plasmid: pC-BA.2.9.1 S | This study | N/A |
| Plasmid: pC-BA.2.11 S | This study | N/A |
| Plasmid: pC-BA.2.12.1 S | This study | N/A |
| Plasmid: pC-BA.4/5 S | This study | N/A |
| Plasmid: pC-BA.2 L452Q S | This study | N/A |
| Plasmid: pC-BA.2 S704L S | This study | N/A |
| Plasmid: pC-BA.2 HV69-70del S | This study | N/A |
| Plasmid: pC-BA.2 F486V S | This study | N/A |
| Plasmid: pC-BA.2 R493Q S | This study | N/A |
| Software and algorithms | ||
| fastp v0.21.0 | Chen et al., 2018 | https://github.com/OpenGene/fastp |
| BWA-MEM v0.7.17 | Li and Durbin, 2009 | http://bio-bwa.sourceforge.net |
| SAMtools v1.9 | Li et al., 2009 | http://www.htslib.org |
| snpEff v5.0e | Cingolani et al., 2012 | http://pcingola.github.io/SnpEff |
| roblanf/sarscov2phylo: 13-11-20 (GISAID phylogenetic analysis pipeline) | GitHub | https://github.com/roblanf/sarscov2phylo |
| Minimap2 v2.17 | Li, 2018 | https://github.com/lh3/minimap2 |
| trimAl v1.2 | Capella-Gutiérrez et al., 2009 | http://trimal.cgenomics.org |
| RAxML v8.2.12 | Stamatakis, 2014 | https://cme.h-its.org/exelixis/web/software/raxml |
| CmdStan v2.28.1 | The Stan Development Team | https://mc-stan.org |
| CmdStanr v0.4.0 | The Stan Development Team | https://mc-stan.org/cmdstanr/ |
| R v4.1.3 | The R Foundation | https://www.r-project.org/ |
| Sequencher v5.1 software | Gene Codes Corporation | N/A |
| In-house scripts | This study | https://github.com/TheSatoLab/BA.2_related_Omicrons |
| Prism 9 software v9.1.1 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Fiji software v2.2.0 | ImageJ | https://fiji.sc |
| FlowJo software v10.7.1 | BD Biosciences | https://www.flowjo.com/solutions/flowjo |
| Python v3.7 | Python Software Foundation | https://www.python.org |
| FinePointe Station and Review softwares v2.9.2.12849 | STARR | https://www.datasci.com/products/software/finepointe-software |
| NDP.scan software v3.2.4 | Hamamatsu Photonics | https://nanozoomer.hamamatsu.com/jp/en/why_nanozoomer/scan.html |
| PyMOL | Schrödinger | https://pymol.org/2/ |
| XDS | Kabsch, 2010 | https://xds.mr.mpg.de/ |
| BUSTER v2.10.4 | Global Phasing Ltd. | https://www.globalphasing.com/ |
| MolProbity | Duke Biochemistry | http://molprobity.biochem.duke.edu/index.php |
| COOT | Emsley and Cowtan, 2004 | http://www.mrc-lmb.cam.ac.uk/personal/peemsley/coot/ |
| Phaser | McCoy et al., 2007 | https://www.phaser.cimr.cam.ac.uk/index.php/Phaser_Crystallographic_Software |
| Phenix | Adams et al., 2010 | http://www.phenix-online.org/ |
| Other | ||
| Centro XS3 LB960 | Berthhold Technologies | N/A |
| GloMax explorer multimode microplate reader 3500 | Promega | N/A |
| FACS Canto II | BD Biosciences | N/A |
| GISAID database | Khare et al., 2021 | https://www.gisaid.org/ |
| BD microtainer blood collection tubes | BD Biosciences | Cat# 365967 |
| 24-well Cell Culture Insert | Falcon | Cat# 353104 |
| cOmplete His-Tag Purification Resin | Roche | Cat# 05 893 682 001 |
| Superdex 75 increase 10/300 | Cytiva | Cat# 29148721 |
| 3,3’-diaminobenzidine tetrahydrochloride | Dako | Cat# DM827 |
| MAS-GP-coated glass slides | Matsunami Glass | Cat# S9901 |
| A1Rsi Confocal Microscope | Nikon | N/A |
| QuantStudio 3 Real-Time PCR system | Thermo Fisher Scientific | N/A |
| CFX Connect Real-Time PCR Detection system | Bio-Rad | N/A |
| Eco Real-Time PCR System | Illumina | N/A |
| qTOWER3 G Real-Time System | Analytik Jena | N/A |
| 7500 Real-Time PCR System | Thermo Fisher Scientific | N/A |
| Autostainer Link 48 | Dako | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kei Sato (KeiSato@g.ecc.u-tokyo.ac.jp).
Materials availability
All unique reagents generated in this study are listed in the key resources table and available from the lead contact with a completed Materials Transfer Agreement.
Experimental model and subject details
Ethics statement
All experiments with hamsters were performed in accordance with the Science Council of Japan’s Guidelines for the Proper Conduct of Animal Experiments. The protocols were approved by the Institutional Animal Care and Use Committee of National University Corporation Hokkaido University (approval ID: 20-0123 and 20-0060). All experiments with mice were also performed in accordance with the Science Council of Japan’s Guidelines for the Proper Conduct of Animal Experiments. The protocols were approved by the Institutional Animal Experiment Committee of The Institute of Medical Science, The University of Tokyo (approval ID: PA21-39 and PA21-46). All protocols involving specimens from human subjects recruited at Kyoto University and Kuramochi Clinic Interpark were reviewed and approved by the Institutional Review Boards of Kyoto University (approval ID: G1309) and Kuramochi Clinic Interpark (approval ID: G2021-004). All human subjects provided written informed consent. All protocols for the use of human specimens were reviewed and approved by the Institutional Review Boards of The Institute of Medical Science, The University of Tokyo (approval IDs: 2021-1-0416 and 2021-18-0617), Kyoto University (approval ID: G0697), Kumamoto University (approval IDs: 2066 and 2074), and University of Miyazaki (approval ID: O-1021).
Human serum collection
Convalescent sera were collected from the following donors: not fully vaccinated individuals who had been infected with the Omicron BA.1 variant (14 non-vaccinated. 8–21 days after testing. n = 14 in total; average age: 44, range: 16–73, 57% male), fully vaccinated individuals who had been infected with the Omicron BA.1 variant (16 2-dose vaccinated. 10-27 days after testing. n = 16 in total; average age: 48, range: 20–76, 44% male), not fully vaccinated individuals who had been infected with the Omicron BA.2 variant (9 non-vaccinated and 1 1-dose vaccinated. 8-27 days after testing. n = 10 in total; average age: 31, range: 7–54, 40% male), and fully vaccinated individuals who had been infected with the Omicron BA.2 variant (9 2-dose vaccinated and 5 3-dose vaccinated. 11-61 days after testing. n = 14 in total; average age: 47, range: 24–84, 64% male). To identify the SARS-CoV-2 variants infecting patients, saliva was collected from COVID-19 patients during infection onset, and RNA was extracted using a QIAamp viral RNA mini kit (Qiagen, Cat# 52906) according to the manufacturer’s protocol. To detect the S E484A substitution (common in all Omicron variants including BA.1, BA.2 and the L452R/Q/M-bearing BA.2-related variants), a primer/probe E484A (SARS-CoV-2) (Takara, Cat# RC322A) was used. To detect the S R214EPE insertion (specific to BA.1, while undetectable in BA.2 and the L452R/Q/M-bearing BA.2-related variants), an in-house-developed protocol was used with the following primers and probe: Omi_ins214s-F1, 5’-TTC TAA GCA CAC GCC TAT AGT GC-3’; Omi_ins214s-R1, 5’-TAA AGC CGA AAA ACC CTG AGG-3’; and Omi_ins214s, FAM-TGA GCC AGA TC-MGB (Yamasoba et al., 2022b). To verify the absence of S L452R/Q/M substitution (specific to the L452R/Q/M-bearing BA.2-related variants, while undetectable in original BA.2), a L452R (SARS-CoV-2) primer/probe set v2 (Takara, Cat# RC346A) was used. Sera were inactivated at 56°C for 30 minutes and stored at –80°C until use. The details of the convalescent sera are summarized in Table S4.
Vaccine sera of fifteen individuals who had BNT162b2 vaccine (Pfizer/BioNTech) (average age: 38, range: 24–48; 53% male) were obtained at one month after the second dose, one month after the third dose, and four months after the third dose. The details of the vaccine sera are summarized in Table S4.
Cell culture
HEK293T cells (a human embryonic kidney cell line; ATCC, CRL-3216), HEK293 cells (a human embryonic kidney cell line; ATCC, CRL-1573) and HOS-ACE2/TMPRSS2 cells (HOS cells stably expressing human ACE2 and TMPRSS2) (Ferreira et al., 2021; Ozono et al., 2021) were maintained in DMEM (high glucose) (Sigma-Aldrich, Cat# 6429-500ML) containing 10% fetal bovine serum (FBS, Sigma-Aldrich Cat# 172012-500ML), and 1% penicillin-streptomycin (PS) (Sigma-Aldrich, Cat# P4333-100ML). HEK293-ACE2 cells (HEK293 cells stably expressing human ACE2) (Motozono et al., 2021) was maintained in DMEM (high glucose) containing 10% FBS, 1 μg/ml puromycin (InvivoGen, Cat# ant-pr-1) and 1% PS. HEK293-ACE2/TMPRSS2 cells (HEK293 cells stably expressing human ACE2 and TMPRSS2) (Motozono et al., 2021) was maintained in DMEM (high glucose) containing 10% FBS, 1 μg/ml puromycin, 200 ng/ml hygromycin (Nacalai Tesque, Cat# 09287-84) and 1% PS. HEK293-C34 cells (IFNAR1 KO HEK293 cells expressing human ACE2 and TMPRSS2 by doxycycline treatment) (Torii et al., 2021) were maintained in DMEM (high glucose) containing 10% FBS, 10 μg/ml blasticidin (InvivoGen, Cat# ant-bl-1) and 1% PS. Vero cells [an African green monkey (Chlorocebus sabaeus) kidney cell line; JCRB Cell Bank, JCRB0111] were maintained in Eagle’s minimum essential medium (EMEM) (Sigma-Aldrich, Cat# M4655-500ML) containing 10% FBS and 1% PS. VeroE6/TMPRSS2 cells (VeroE6 cells stably expressing human TMPRSS2; JCRB Cell Bank, JCRB1819) (Matsuyama et al., 2020) were maintained in DMEM (low glucose) (Wako, Cat# 041-29775) containing 10% FBS, G418 (1 mg/ml; Nacalai Tesque, Cat# G8168-10ML) and 1% PS. Calu-3/DSP1-7 cells (Calu-3 cells stably expressing DSP1-7) (Yamamoto et al., 2020) were maintained in EMEM (Wako, Cat# 056-08385) containing 20% FBS and 1% PS. 293S GnTI(-) cells (HEK293S cells lacking N-acetylglucosaminyltransferase (Reeves et al., 2002) were maintained in DMEM (Nacalai tesque, #08458-16 containing 2% FBS without PS. Expi293F cells (Thermo Fisher Scientific, Cat# A14527) were maintained in Expi293 expression medium (Thermo Fisher Scientific, Cat# A1435101). Human airway and alveolar epithelial cells derived from human induced pluripotent stem cells (iPSCs) were manufactured according to established protocols as described below (see “preparation of human airway and alveolar epithelial cells from human iPSCs” section) and provided by HiLung Inc.
Method details
Viral genome sequencing
Viral genome sequencing was performed as previously described (Meng et al., 2022; Motozono et al., 2021; Saito et al., 2022; Suzuki et al., 2022; Yamasoba et al., 2022b). Briefly, the virus sequences were verified by viral RNA-sequencing analysis. Viral RNA was extracted using a QIAamp viral RNA mini kit (Qiagen, Cat# 52906). The sequencing library employed for total RNA sequencing was prepared using the NEB next ultra RNA library prep kit for Illumina (New England Biolabs, Cat# E7530). Paired-end 76-bp sequencing was performed using a MiSeq system (Illumina) with MiSeq reagent kit v3 (Illumina, Cat# MS-102-3001). Sequencing reads were trimmed using fastp v0.21.0 (Chen et al., 2018) and subsequently mapped to the viral genome sequences of a lineage A isolate (strain WK-521; GISAID ID: EPI_ISL_408667) (Matsuyama et al., 2020) using BWA-MEM v0.7.17 (Li and Durbin, 2009). Variant calling, filtering, and annotation were performed using SAMtools v1.9 (Li et al., 2009) and snpEff v5.0e (Cingolani et al., 2012).
Phylogenetic and comparative genome analyses
To construct an ML tree of Omicron lineages (BA.1–BA.5) sampled from South Africa (shown in Figure 1A), the genome sequence data of SARS-CoV-2 and its metadata were downloaded from the GISAID database (https://www.gisaid.org/) (Khare et al., 2021) on April 23, 2022. We excluded the data of viral strains with the following features from the analysis: i) a lack collection date information; ii) sampling from animals other than humans, iii) >2% undetermined nucleotide characters, or iv) sampling by quarantine. From each viral lineage, 30 sequences were randomly sampled and used for tree construction, in addition to an outgroup sequence, EPI_ISL_466615, representing the oldest isolate of B.1.1 obtained in the UK. The viral genome sequences were mapped to the reference sequence of Wuhan-Hu-1 (GenBank accession number: NC_045512.2) using Minimap2 v2.17 (Li, 2018) and subsequently converted to a multiple sequence alignment according to the GISAID phylogenetic analysis pipeline (https://github.com/roblanf/sarscov2phylo). The alignment sites corresponding to the 1–265 and 29674–29903 positions in the reference genome were masked (i.e., converted to NNN). Alignment sites at which >50% of sequences contained a gap or undetermined/ambiguous nucleotide were trimmed using trimAl v1.2 (Capella-Gutiérrez et al., 2009). Phylogenetic tree construction was performed via a three-step protocol: i) the first tree was constructed; ii) tips with longer external branches (Z score > 4) were removed from the dataset; iii) and the final tree was constructed. Tree reconstruction was performed by RAxML v8.2.12 (Stamatakis, 2014) under the GTRCAT substitution model. The node support value was calculated by 100 times bootstrap analysis.
To classify the BA.2 variants bearing substitutions at the S L452 residue, we constructed an ML tree of BA.2 variants including those bearing substitutions at the S L452 residue (shown in Figure 1B). For quality control, the BA.2 sequences without the S:N501Y and S:E484A substitutions, characteristic substitutions of Omicron, were removed from the dataset. Also, the BA.2 sequences with the S:HV69-70del, a deletion mutation that is not present in BA.2 but in other Omicron lineages, were removed. To make a subset of BA.2 sequences representing the diversity of BA.2 for tree construction, we defined the “common amino acid haplotype” of BA.2 as described below. We first extracted the BA.2 sequences bearing substitutions at position 452 in S. In these BA.2 variants, amino acid mutations (including substitutions, insertions, and deletions) present > 1% sequences were detected and referred to as the "common amino acid substitutions”. According to the profile of the common amino acid substitutions, a common amino acid haplotype, a set of common amino acid substitutions present in each sequence, was determined for all BA.2 sequences. Finally, up to 20 sequences were randomly sampled from each unique common amino acid haplotype. As outgroup sequences, the oldest isolate of B.1.1 obtained in the UK (EPI_ISL_466615) and the oldest five BA.1 and BA.3 sequences sampled from South Africa after December 1, 2022, were used. The ML tree was constructed by the procedure described above. In the final set, 8,029 BA.2 sequences were included. Outgroup sequences are not displayed in Figure 1B.
Definition of common ancestry groups of the BA.2 variants bearing substitutions at position 452 in S
According to the phylogenetic tree of BA.2 shown in Figure 1B, we defined common ancestry groups of the BA.2 variants bearing substitutions at position 452 in S as the follow procedures. First, the ancestral state of the amino acid at position 452 in S at each node was estimated using a fixed-rates continuous-time Markov model (Mk model) implemented in the R package “castor” (Figure S1C) (Louca and Doebeli, 2018). As a type of transition matrix in the Mk model, all rate different (ARD) matrix was selected. Second, we identified the branches connecting the parental-state (L) nodes and the mutated-sate (R, Q, or M) nodes (red branches in Figure S1C). In these branches, it is expected that the substitution acquisitions in the S L452 residue occurred. Finally, we counted the descendant sequences of respective branches where the substitutions in the S L452 were likely acquired. If the number of descendants is ≥10, we defined these descendant sequences as a common ancestry group of the BA.2 variants, which bears a substitutions at position 452 in S. Information of the common ancestry group is summarized in Table S2.
Modeling the epidemic dynamics of SARS-CoV-2 lineages
To quantify the spread rate of each SARS-CoV-2 lineage in the human population, we estimated the relative effective reproduction number of each viral lineage according to the epidemic dynamics, calculated on the basis of viral genomic surveillance data. The data were downloaded from the GISAID database (https://www.gisaid.org/) on May 15, 2022. We excluded the data of viral strains with the following features from the analysis: i) a lack of collection date information; ii) sampling in animals other than humans; or iii) sampling by quarantine. We analyzed the datasets of the five countries (South Africa, the USA, France, Denmark and Belgium) where BA.4/5, BA.2.12.1, BA.2.11, BA.2.9.1, and BA.2.13 were most detected, respectively. The BA.2 sublineages without amino acid substitutions at position 452 in S were summarized as BA.2. In addition, the Delta sublineages were also summarized as Delta. The dynamics of up to five most predominant viral lineages in each country from February 5, 2022, to May 15, 2022, were analyzed. The number of viral sequences of each viral lineage collected on each day in each country was counted, and the count matrix was constructed as an input for the statistical model below.
We constructed a Bayesian statistical model to represent relative lineage growth dynamics with multinomial logistic regression, as described in our previous study (Suzuki et al., 2022). In the present study, the epidemic dynamics in respective countries were independently estimated. Arrays in the model index over one or more indices: viral lineages and days . The model is:
The explanatory variable was time, , and the outcome variable was , which represented the count of viral lineage at time . In the model, the linear estimator , consisting of the intercept and the slope , was converted to the simplex , which represented the probability of occurrence of each viral lineage at time , based on the softmax link function defined as:
is generated from and the total count of all lineages at time according to a multinomial distribution.
The relative Re of each viral lineage () was calculated according to the slope parameter as:
where is the average viral generation time (2.1 days) (http://sonorouschocolate.com/covid19/index.php?title=Estimating_Generation_Time_Of_Omicron).
For parameter estimation, the intercept and slope parameters of the BA.2 variant were fixed at 0. Consequently, the relative Re of BA.2 was fixed at 1, and those of the other lineages were estimated relative to that of BA.2.
Parameter estimation was performed via the MCMC approach implemented in CmdStan v2.28.1 (https://mc-stan.org) with CmdStanr v0.4.0 (https://mc-stan.org/cmdstanr/). Noninformative priors were set for all parameters. Four independent MCMC chains were run with 500 and 2,000 steps in the warmup and sampling iterations, respectively. We confirmed that all estimated parameters showed < 1.01 R-hat convergence diagnostic values and > 200 effective sampling size values, indicating that the MCMC runs were successfully convergent. The fitted model closely recapitulated the observed viral lineage dynamics (Figures 1E and S1D). The above analyses were performed in R v4.1.3 (https://www.r-project.org/). Information on the relative effective reproduction number of BA.2 estimated in the present study is summarized in Table S3. In addition, using the SRAS-CoV-2 genome surveillance data downloaded on July 7, 2022, we estimated the relative Re of BA.4/5, BA.2.12.1, BA.2.11, BA.2.9.1, and BA.2.13 in South Africa, the USA, France, Denmark, and Belgium.
Plasmid construction
Plasmids expressing the SARS-CoV-2 S proteins of B.1.1 (the parental D614G-bearing variant) and BA.2 were prepared in our previous studies (Kimura et al., 2022a; Ozono et al., 2021; Suzuki et al., 2022; Yamasoba et al., 2022b). Plasmids expressing the codon-optimized S proteins of L452R/Q/M-bearing variants and their derivatives were generated by site-directed overlap extension PCR using the primers listed in Table S6. The resulting PCR fragment was digested with KpnI and NotI and inserted into the corresponding site of the pCAGGS vector (Niwa et al., 1991). A plasmid encoding the SARS-CoV-2 BA.2 S RBD (residues 322-536) was cloned into the expression vector pHLsec containing the N-terminal secretion signal sequence and the C-terminal His6-tag sequence (Aricescu et al., 2006). Nucleotide sequences were determined by DNA sequencing services (Eurofins), and the sequence data were analyzed by Sequencher v5.1 software (Gene Codes Corporation).
Preparation of BA.2 S RBD and human ACE2
The BA.2 S RBD and hACE2 were prepared as previously described (Kubota et al., 2016). Briefly, the expression plasmids encoding the BA.2 S RBD, its mutants, or human ACE2 were transfected into 293S GnTI (-) cells. The proteins in the culture supernatant were purified with cOmplete His-Tag Purification Resin (Roche) affinity column, followed by Superdex 75 Increase 10/300 size-exclusion chromatography (Cytiva) with calcium- and magnesium-free PBS buffer.
Preparation of mouse sera
BALB/c mice (female, 7 weeks old) were immunized with 1 μg SARS-CoV-2 BA.2 RBD protein in 50% AddaVax (InvivoGen, Cat# vac-adx-10) at day 0 and 14. Ten days after second immunization, blood was collected in BD microtainer blood collection tubes (BD Biosciences, Cat# 365967) and sera were collected by centrifugation.
Preparation of human airway and alveolar epithelial cells from human iPSCs
The air-liquid interface culture of airway and alveolar epithelial cells were differentiated from human iPSC-derived lung progenitor cells as previously described (Gotoh et al., 2014; Konishi et al., 2016; Yamamoto et al., 2017). Briefly, lung progenitor cells were stepwise induced from human iPSCs referring a 21-days and 4-steps protocol (Yamamoto et al., 2017). At day 21, lung progenitor cells were isolated with specific surface antigen carboxypeptidase M and seeded onto upper chamber of 24-well Cell Culture Insert (Falcon, #353104), followed by 28-day and 7-day differentiation of airway and alveolar epithelial cells, respectively. Alveolar differentiation medium supplemented with dexamethasone (Sigma-Aldrich, Cat# D4902), KGF (PeproTech, Cat# 100-19), 8-Br-cAMP (Biolog, Cat# B007), 3-Isobutyl 1-methylxanthine (IBMX) (FUJIFILM Wako, Cat# 095-03413), CHIR99021 (Axon Medchem, Cat# 1386), and SB431542 (FUJIFILM Wako, Cat# 198-16543) was used for induction of alveolar epithelial cells. PneumaCult ALI (STEMCELL Technologies, Cat# ST-05001) supplemented with heparin (Nacalai Tesque, Cat# 17513-96) and Y-27632 (LC Laboratories, Cat# Y-5301) hydrocortisone (Sigma-Aldrich, Cat# H0135) was used for induction of airway epithelial cells.
Neutralization assay
Pseudoviruses were prepared as previously described (Kimura et al., 2022a; Meng et al., 2022; Ozono et al., 2021; Saito et al., 2022; Uriu et al., 2022, 2021; Yamasoba et al., 2022b, 2022c). Briefly, lentivirus (HIV-1)-based, luciferase-expressing reporter viruses were pseudotyped with the SARS-CoV-2 S proteins. HEK293T cells (1,000,000 cells) were cotransfected with 1 μg psPAX2-IN/HiBiT (Ozono et al., 2020), 1 μg pWPI-Luc2 (Ozono et al., 2020), and 500 ng plasmids expressing parental S or its derivatives using PEI Max (Polysciences, Cat# 24765-1) according to the manufacturer's protocol. Two days posttransfection, the culture supernatants were harvested and centrifuged. The pseudoviruses were stored at –80°C until use.
Neutralization assay (Figure 2) was prepared as previously described (Kimura et al., 2022a; Meng et al., 2022; Ozono et al., 2021; Saito et al., 2022; Uriu et al., 2022, 2021; Yamasoba et al., 2022b, 2022c). Briefly, the SARS-CoV-2 S pseudoviruses (counting ∼20,000 relative light units) were incubated with serially diluted (120-fold to 97,480-fold dilution at the final concentration) heat-inactivated sera at 37°C for 1 hour. Pseudoviruses without sera were included as controls. Then, a 40 μl mixture of pseudovirus and serum/antibody was added to HOS-ACE2/TMPRSS2 cells (10,000 cells/50 μl) in a 96-well white plate. At 2 d.p.i., the infected cells were lysed with a One-Glo luciferase assay system (Promega, Cat# E6130) or a Bright-Glo luciferase assay system (Promega, Cat# E2650), and the luminescent signal was measured using a GloMax explorer multimode microplate reader 3500 (Promega) or CentroXS3 (Berthhold Technologies). The assay of each serum was performed in triplicate, and the 50% neutralization titer (NT50) was calculated using Prism 9 software v9.1.1 (GraphPad Software).
Pseudovirus infection
Pseudovirus infection was (Figure 3A) performed as previously described (Kimura et al., 2022a, 2022b; Motozono et al., 2021; Saito et al., 2022; Suzuki et al., 2022; Uriu et al., 2022, 2021; Yamasoba et al., 2022b, 2022c). Briefly, the amount of pseudoviruses prepared was quantified by the HiBiT assay using Nano Glo HiBiT lytic detection system (Promega,Cat# N3040) as previously described (Ozono et al., 2021, 2020), and the same amount of pseudoviruses (normalized to the HiBiT value, which indicates the amount of p24 HIV-1 antigen) was inoculated into HOS-ACE2/TMPRSS2 cells, HEK293-ACE2 cells or HEK293-ACE2/TMPRSS2 and viral infectivity was measured as described above (see “neutralization assay” section). To analyze the effect of TMPRSS2 for pseudovirus infectivity (Figure S2A), the fold change of the values of HEK293-ACE2/TMPRSS2 to HEK293-ACE2 was calculated.
Yeast surface display
Yeast surface display (Figure 3B) was performed as previously described as previously described (Dejnirattisai et al., 2022; Kimura et al., 2022a, 2022b; Motozono et al., 2021; Yamasoba et al., 2022b; Zahradník et al., 2021a). Briefly, yeast codon-optimized SARS-CoV-2_RBD-Omicron-BA.2 was obtained from Twist Biosciences and the mutant RBDs were PCR amplified by KAPA HiFi HotStart ReadyMix kit (Roche, Cat# KK2601) and assembled by yeast [Saccharomyces cerevisiae strain EBY100 (ATCC, MYA-4941)] homologous recombination with pJYDC1 plasmid (Addgene, Cat# 162458) as previously described (Dejnirattisai et al., 2022; Kimura et al., 2022a, 2022b; Motozono et al., 2021; Yamasoba et al., 2022b; Zahradník et al., 2021a). Primers used are listed in Table S6. Yeasts were expressed for 48 hours at 20°C, washed with PBS supplemented with bovine serum albumin at 1 g/l and incubated with 12–14 different concentrations of Expi293F cells produced ACE2 peptidase domain (residues 18-740, 200 nM to 13 pM) for 12 hours. To induce eUnaG2 reporter protein fluorescence, bilirubin (Sigma-Aldrich, Cat# 14370-1G) was added to the final concentration of 5 nM. RBD expression and ACE2 signal were recorded by using a FACS S3e cell sorter device (Bio-Rad), background binding signals were subtracted and data were fitted to a standard noncooperative Hill equation by nonlinear least-squares regression using Python v3.7 (https://www.python.org) as previously described (Kimura et al., 2022a, 2022b; Motozono et al., 2021; Yamasoba et al., 2022b; Zahradník et al., 2021b).
Crystallization and data collection
The sitting-drop method was used to obtain the BA.4/5 S RBD-human ACE2 complex crystals. In detail, purified complex proteins were concentrated to 5.69 mg/ml. Then, 0.9 ul protein was mixed with 0.9 ul reservoir solution. The resulting solution was sealed and equilibrated against 50 ul reservoir solution at 293 K. Crystals of the BA.4/5 S RBD-human ACE2 complex were grown in 0.1 M MES (pH 6.5), 11% polyethylene glycol 6000 and 5% 2-methyl-2,4-pentanediol. Then, crystals were soaked briefly in 0.1 M MES (pH 6.5), 13% polyethylene glycol 6000 and 20% glycerol before being flash-frozen in liquid nitrogen. X-ray diffraction data were collected from beamline BL17A at Photon Factory (Ibaraki, Japan). All diffraction data were indexed, integrated, scaled, and merged using XDS (Kabsch, 2010).
Structure determination and refinement
The crystal structure of the BA.4/5 S RBD-human ACE2 complex (Figure 3C) was determined by the molecular replacement method with Phaser (McCoy et al., 2007), using the BA.1 S RBD-human ACE2 complex structure (PDB ID: 7WBP), which is reported previously (Han et al., 2022), as a search model. Initial protein models were fitted manually using Coot (Emsley and Cowtan, 2004). The structure was then refined using BUSTER v2.10.4 (Bricogne et al., 2017) and phenix.refine (Adams et al., 2010). The data collection and refinement statistics are summarized in Table S5. All structure figures were generated by PyMOL (https://pymol.org/2/).
SARS-CoV-2 S-based fusion assay
SARS-CoV-2 S-based fusion assay (Figures 3E and S2D) was performed as previously described (Kimura et al., 2022b; Motozono et al., 2021; Saito et al., 2022; Suzuki et al., 2022; Yamasoba et al., 2022b). Briefly, on day 1, effector cells (i.e., S-expressing cells) and target cells (see below) were prepared at a density of 0.6–0.8 × 106 cells in a 6-well plate. To prepare effector cells, HEK293 cells were cotransfected with the S expression plasmids (400 ng) and pDSP8-11 (Kondo et al., 2011) (400 ng) using TransIT-LT1 (Takara, Cat# MIR2300). On day 2, to prepare target cells, VeroE6/TMPRSS2 cells were transfected with pDSP1-7 (Kondo et al., 2011) (400 ng). On day 3 (24 hours posttransfection), 16,000 effector cells were detached and reseeded into 96-well black plates (PerkinElmer, Cat# 6005225), and target cells (VeroE6/TMPRSS2 or Calu-3/DSP1-7 cells) were reseeded at a density of 1,000,000 cells/2 ml/well in 6-well plates. On day 4 (48 hours posttransfection), target cells were incubated with EnduRen live cell substrate (Promega, Cat# E6481) for 3 hours and then detached, and 32,000 target cells were added to a 96-well plate with effector cells. Renilla luciferase activity was measured at the indicated time points using Centro XS3 LB960 (Berthhold Technologies). To measure the surface expression level of S protein, effector cells were stained with rabbit anti-SARS-CoV-2 S S1/S2 polyclonal antibody (Thermo Fisher Scientific, Cat# PA5-112048, 1:100). Normal rabbit IgG (SouthernBiotech, Cat# 0111-01, 1:100) was used as negative controls, and APC-conjugated goat anti-rabbit IgG polyclonal antibody (Jackson ImmunoResearch, Cat# 111-136-144, 1:50) was used as a secondary antibody. Surface expression level of S proteins (Figures 3D and S2C) was measured using FACS Canto II (BD Biosciences) and the data were analyzed using FlowJo software v10.7.1 (BD Biosciences). To calculate fusion activity, Renilla luciferase activity was normalized to the MFI of surface S proteins. The normalized value (i.e., Renilla luciferase activity per the surface S MFI) is shown as fusion activity.
Coculture experiment
Coculture experiment (Figure S2E) was performed as previously described (Suzuki et al., 2022; Yamasoba et al., 2022b). This assay utilizes a dual split protein (DSP) encoding Renilla luciferase and GFP genes; the respective split proteins, DSP8-11 and DSP1-7, are expressed in effector and target cells by transfection. Briefly, one day before transfection, effector cells (i.e., S-expressing cells) were seeded on the poly-L-lysine (Sigma, Cat# P4832) coated coverslips put in a 12-well plate, and target cells were prepared at a density of 100,000 cells in a 12-well plate. To prepare effector cells, HEK293 cells were cotransfected with the S-expression plasmids (500 ng) and pDSP8-11 (500 ng) using PEI Max (Polysciences, Cat# 24765-1). To prepare target cells, HEK293 and HEK293-ACE2/TMPRSS2 cells were transfected with pDSP1-7 (500 ng) (Kondo et al., 2011). At 24 hours posttransfection, target cells were detached and cocultured with effector cells in a 1:2 ratio. At 9 h post-coculture, cells were fixed with 4% paraformaldehyde in PBS (Nacalai Tesque, Cat# 09154-85) for 15 minutes at room temperature. Nuclei were stained with Hoechst 33342 (Thermo Fisher Scientific, Cat# H3570). The coverslips were mounted on glass slides using Fluoromount-G (SouthernBiotech, Cat# 0100-01) with Hoechst 33342 and observed using an A1Rsi Confocal Microscope (Nikon). The size of syncytium (GFP-positive area) was measured using Fiji software v2.2.0 (ImageJ) as previously described (Suzuki et al., 2022; Yamasoba et al., 2022b).
SARS-CoV-2 reverse genetics
Recombinant SARS-CoV-2 was generated by circular polymerase extension reaction (CPER) as previously described (Kimura et al., 2022b; Motozono et al., 2021; Saito et al., 2022; Torii et al., 2021; Yamasoba et al., 2022b). To generate the BA.2-based chimeric recombinant SARS-CoV-2 (rBA.2, rBA.2.9.1, rBA.2.11 and rBA.4/5) (Figure 4A), RNA was extracted from the cells infected with a clinical isolate of BA.2 (GISAID ID: EPI_ISL_9595859) and cDNA was synthesized as described above (see “viral genome sequencing” section). The two DNA fragments correspond to the fragments 1-7 and 9 were prepared by RT-PCR using PrimeSTAR GXL DNA polymerase (Takara, Cat# R050A) using the primers listed in Table S6. The fragments 8 bearing the S genes of BA.2, BA.2.9.1, BA.2.11, BA.2.12.1 and BA.4/5 were prepared as described above. Finally, the 3 DNA fragments were mixed and used for CPER (Torii et al., 2021).
To produce recombinant SARS-CoV-2 (seed viruses), the CPER products were transfected into HEK293-C34 cells using TransIT-LT1 (Takara, Cat# MIR2300) according to the manufacturer's protocol. At one day posttransfection, the culture medium was replaced with DMEM (high glucose) (Sigma-Aldrich, Cat# 6429-500ML) containing 2% FBS, 1% PS and doxycycline (1 μg/ml; Takara, Cat# 1311N). At six days posttransfection, the culture medium was harvested and centrifuged, and the supernatants were collected as the seed virus. To remove the CPER products (i.e., SARS-CoV-2-related DNA), 1 ml of the seed virus was treated with 2 μl TURBO DNase (Thermo Fisher Scientific, Cat# AM2238) and incubated at 37°C for 1 hour. Complete removal of the CPER products from the seed virus was verified by PCR. The working virus stock was prepared using the seed virus as described below (see “SARS-CoV-2 preparation and titration” section).
SARS-CoV-2 preparation and titration
The working virus stocks of chimeric recombinant SARS-CoV-2 were prepared and titrated as previously described (Kimura et al., 2022b; Motozono et al., 2021; Saito et al., 2022; Torii et al., 2021; Yamasoba et al., 2022b). In brief, 20 μl of the seed virus was inoculated into VeroE6/TMPRSS2 cells (5,000,000 cells in a T-75 flask). One h.p.i., the culture medium was replaced with DMEM (low glucose) (Wako, Cat# 041-29775) containing 2% FBS and 1% PS. At 3 d.p.i., the culture medium was harvested and centrifuged, and the supernatants were collected as the working virus stock.
The titer of the prepared working virus was measured as the 50% tissue culture infectious dose (TCID50). Briefly, one day before infection, VeroE6/TMPRSS2 cells (10,000 cells) were seeded into a 96-well plate. Serially diluted virus stocks were inoculated into the cells and incubated at 37°C for 4 days. The cells were observed under microscopy to judge the CPE appearance. The value of TCID50/ml was calculated with the Reed–Muench method (Reed and Muench, 1938).
To verify the sequences of SARS-CoV-2 working viruses, viral RNA was extracted from the working viruses using a QIAamp viral RNA mini kit (Qiagen, Cat# 52906) and viral genome sequences were analyzed as described above (see "viral genome sequencing" section). Information on the unexpected substitutions detected is summarized in Table S7, and the raw data are deposited in the GitHub repository (https://github.com/TheSatoLab/BA.2_related_Omicrons).
Plaque assay
Plaque assay (Figure 4B) was performed as previously described (Kimura et al., 2022b; Motozono et al., 2021; Saito et al., 2022; Suzuki et al., 2022; Yamasoba et al., 2022b). Briefly, one day before infection, VeroE6/TMPRSS2 cells (100,000 cells) were seeded into a 24-well plate and infected with SARS-CoV-2 (1, 10, 100 and 1,000 TCID50) at 37°C for 1 hour. Mounting solution containing 3% FBS and 1.5% carboxymethyl cellulose (Wako, Cat# 039-01335) was overlaid, followed by incubation at 37°C. At 3 d.p.i., the culture medium was removed, and the cells were washed with PBS three times and fixed with 4% paraformaldehyde phosphate (Nacalai Tesque, Cat# 09154-85). The fixed cells were washed with tap water, dried, and stained with staining solution [0.1% methylene blue (Nacalai Tesque, Cat# 22412-14) in water] for 30 minutes. The stained cells were washed with tap water and dried, and the size of plaques was measured using Fiji software v2.2.0 (ImageJ).
SARS-CoV-2 infection
One day before infection, VeroE6/TMPRSS2 cells (10,000 cells) and Vero cells (10,000 cells) and were seeded into a 96-well plate. SARS-CoV-2 [100 TCID50 for VeroE6/TMPRSS2 cells (Figure 4C); 1,000 TCID50 for Vero cells (Figure 4D)] was inoculated and incubated at 37°C for 1 hour. The infected cells were washed, and 180 μl culture medium was added. The culture supernatant (10 μl) was harvested at the indicated timepoints and used for RT–qPCR to quantify the viral RNA copy number (see “rt–qPCR” section below) In the infection experiment using human iPSC-derived airway and alveolar epithelial cells (Figures 4E and 4F), working viruses were diluted with Opti-MEM (Thermo Fisher Scientific, 11058021). The diluted viruses (1,000 TCID50 in 100 μl) were inoculated onto the apical side of the culture and incubated at 37 °C for 1 hour. The inoculated viruses were removed and washed twice with Opti-MEM. To collect the viruses, 100 μl Opti-MEM was applied onto the apical side of the culture and incubated at 37 °C for 10 minutes. The Opti-MEM was collected and used for RT–qPCR to quantify the viral RNA copy number (see “rt–qPCR” section below).
RT–qPCR
RT–qPCR was performed as previously described (Kimura et al., 2022b; Meng et al., 2022; Motozono et al., 2021; Saito et al., 2022; Suzuki et al., 2022; Yamasoba et al., 2022b). Briefly, 5 μl culture supernatant was mixed with 5 μl 2 × RNA lysis buffer [2% Triton X-100 (Nacalai Tesque, Cat# 35501-15), 50 mM KCl, 100 mM Tris-HCl (pH 7.4), 40% glycerol, 0.8 U/μl recombinant RNase inhibitor (Takara, Cat# 2313B)] and incubated at room temperature for 10 m. RNase-free water (90 μl) was added, and the diluted sample (2.5 μl) was used as the template for real-time RT-PCR performed according to the manufacturer’s protocol using One Step TB Green PrimeScript PLUS RT-PCR kit (Takara, Cat# RR096A) and the following primers: Forward N, 5'-AGC CTC TTC TCG TTC CTC ATC AC-3'; and Reverse N, 5'-CCG CCA TTG CCA GCC ATT C-3'. The viral RNA copy number was standardized with a SARS-CoV-2 direct detection RT-qPCR kit (Takara, Cat# RC300A). Fluorescent signals were acquired using QuantStudio 3 Real-Time PCR system (Thermo Fisher Scientific), CFX Connect Real-Time PCR Detection system (Bio-Rad), Eco Real-Time PCR System (Illumina), qTOWER3 G Real-Time System (Analytik Jena) or 7500 Real-Time PCR System (Thermo Fisher Scientific).
Animal experiments
Animal experiments (Figure 5) were performed as previously described (Saito et al., 2022; Suzuki et al., 2022; Yamasoba et al., 2022b). Syrian hamsters (male, 4 weeks old) were purchased from Japan SLC Inc. (Shizuoka, Japan). Baseline body weights were measured before infection. For the virus infection experiments, hamsters were anaesthetized by intramuscular injection of a mixture of either 0.15 mg/kg medetomidine hydrochloride (Domitor®, Nippon Zenyaku Kogyo), 2.0 mg/kg midazolam (FUJIFILM Wako Chemicals, Cat# 135-13791) and 2.5 mg/kg butorphanol (Vetorphale®, Meiji Seika Pharma), or 0.15 mg/kg medetomidine hydrochloride, 2.0 mg/kg alphaxaone (Alfaxan®, Jurox) and 2.5 mg/kg butorphanol. The chimeric recombinant SARS-CoV-2 (rBA.2, rBA.2.12.1, and rBA.4/5) (10,000 TCID50 in 100 μl), or saline (100 μl) were intranasally inoculated under anesthesia. Oral swabs were collected at 1, 3, and 5 d.p.i. Body weight, enhanced pause (Penh), the ratio of time to peak expiratory follow relative to the total expiratory time (Rpef) and subcutaneous oxygen saturation (SpO2) were routinely monitored at indicated timepoints (see “lung function test” section below). Respiratory organs were anatomically collected at 1, 3 and 5 d.p.i (for lung) or 1 d.p.i. (for trachea). Viral RNA load in the respiratory tissues and oral swab were determined by RT–qPCR. The respiratory tissues were also used for histopathological and IHC analyses (see “h&E staining” and “ihc” sections below). Sera of infected hamsters were collected at 16 d.p.i. using cardiac puncture under anesthesia with isoflurane and used for neutralization assay (see “Neutralization assay” above).
Lung function test
Lung function test (Figure 5A) was performed at 1, 3, 5, and 7 d.p.i. as previously described (Suzuki et al., 2022; Yamasoba et al., 2022b). Respiratory parameters (Penh and Rpef) were measured by using a whole-body plethysmography system (DSI) according to the manufacturer's instructions. In brief, a hamster was placed in an unrestrained plethysmography chamber and allowed to acclimatize for 30 seconds, then, data were acquired over a 2.5-minute period by using FinePointe Station and Review softwares v2.9.2.12849 (STARR). The state of oxygenation was examined by measuring SpO2 using pulse oximeter, MouseOx PLUS (STARR). SpO2 was measured by attaching a measuring chip to the neck of hamsters sedated by 0.25 mg/kg medetomidine hydrochloride.
IHC
IHC (Figures 5D, S3A, and S3B) was performed as previously described (Saito et al., 2022; Suzuki et al., 2022; Yamasoba et al., 2022b) using an Autostainer Link 48 (Dako). The deparaffinized sections were exposed to EnVision FLEX target retrieval solution high pH (Agilent, Cat# K8004) for 20 minutes at 97°C to activate, and mouse anti-SARS-CoV-2 N monoclonal antibody (clone 1035111, R&D systems, Cat# MAB10474-SP, 1:400) was used as a primary antibody. The sections were sensitized using EnVision FLEX (Agilent) for 15 minutes and visualized by peroxidase-based enzymatic reaction with 3,3’-diaminobenzidine tetrahydrochloride (Dako, Cat# DM827) as substrate for 5 minutes. The N protein positivity (Figures 5D, S3A, and S3B) was evaluated by certificated pathologists as previously described (Suzuki et al., 2022; Yamasoba et al., 2022b). Images were incorporated as virtual slide by NDP.scan software v3.2.4 (Hamamatsu Photonics). The N-protein positivity was measured as the area using Fiji software v2.2.0 (ImageJ).
H&E staining
H&E staining (Figure 5G) was performed as previously described (Saito et al., 2022; Suzuki et al., 2022; Yamasoba et al., 2022b). Briefly, excised animal tissues were fixed with 10% formalin neutral buffer solution, and processed for paraffin embedding. The paraffin blocks were sectioned with 3 μm-thickness and then mounted on MAS-GP-coated glass slides (Matsunami Glass, Cat# S9901). H&E staining was performed according to a standard protocol.
Histopathological scoring
Histopathological scoring (Figure 5F) was performed as previously described (Saito et al., 2022; Suzuki et al., 2022; Yamasoba et al., 2022b). Pathological features including bronchitis or bronchiolitis, hemorrhage with congestive edema, alveolar damage with epithelial apoptosis and macrophage infiltration, hyperplasia of type II pneumocytes, and the area of the hyperplasia of large type II pneumocytes were evaluated by certified pathologists and the degree of these pathological findings were arbitrarily scored using four-tiered system as 0 (negative), 1 (weak), 2 (moderate), and 3 (severe). The "large type II pneumocytes" are the hyperplasia of type II pneumocytes exhibiting more than 10-μm-diameter nucleus. We described "large type II pneumocytes" as one of the remarkable histopathological features reacting SARS-CoV-2 infection in our previous studies (Saito et al., 2022; Suzuki et al., 2022; Yamasoba et al., 2022b). Total histology score is the sum of these five indices.
To measure the inflammation area in the infected lungs (Figures 5H and S3C), four hamsters infected with each virus were sacrificed at 5 d.p.i., and all four right lung lobes, including upper (anterior/cranial), middle, lower (posterior/caudal), and accessory lobes, were sectioned along with their bronchi. The tissue sections were stained by H&E, and the digital microscopic images were incorporated into virtual slides using NDP.scan software v3.2.4 (Hamamatsu Photonics). The inflammatory area including type II pneumocyte hyperplasia in the infected whole lungs was morphometrically analyzed using Fiji software v2.2.0 (ImageJ).
Quantification and statistical analysis
Statistical significance was tested using a two-sided Mann–Whitney U-test, a two-sided Student's t-test or a two-sided paired t-test unless otherwise noted. The tests above were performed using Prism 9 software v9.1.1 (GraphPad Software).
In the time-course experiments (Figures 3E, 4C-4F, 5A–5C, 5F, and S2D), a multiple regression analysis including experimental conditions (i.e., the types of infected viruses) as explanatory variables and timepoints as qualitative control variables was performed to evaluate the difference between experimental conditions thorough all timepoints. The initial time point was removed from the analysis. P value was calculated by a two-sided Wald test. Subsequently, familywise error rates (FWERs) were calculated by the Holm method. These analyses were performed in R v4.1.2 (https://www.r-project.org/).
In Figures 5D, 5G, and S3, photographs shown are the representative areas of at least two independent experiments by using four hamsters at each timepoint. In Figure S3A, photographs shown are the representatives of >20 fields of view taken for each sample.
Consortia
The members of Genotype to Phenotype Japan (G2P-Japan) are Hirofumi Sawa, Marie Kato, Zannatul Ferdous, Hiromi Mouri, Kenji Shishido, Naoko Misawa, Mai Suganami, Mika Chiba, Ryo Yoshimura, So Nakagawa, Jiaqi Wu, Kayoko Nagata, Yasuhiro Kazuma, Ryosuke Nomura, Yoshihito Horisawa, Yusuke Tashiro, Yugo Kawai, Takashi Irie, Ryoko Kawabata, Otowa Takahashi, Kimiko Ichihara, Takamasa Ueno, Chihiro Motozono, Mako Toyoda, Yuri L. Tanaka, Erika P. Butlertanaka, and Maya Shofa.
Acknowledgments
We would like to thank all members belonging to The Genotype to Phenotype Japan (G2P-Japan) Consortium. We thank Dr. Kenzo Tokunaga (National Institute for Infectious Diseases, Japan), Dr. Jin Gohda (The University of Tokyo, Japan), and Dr. Hisashi Arase (Osaka University) for providing reagents. We also thank the National Institute for Infectious Diseases, Japan, for providing a clinical isolate of Omicron BA.2 (GISAID ID: EPI_ISL_9595859), and Photon Factory staff for help with structural analysis. We gratefully acknowledge all data contributors, i.e., the authors and their originating laboratories responsible for obtaining the specimens, and their submitting laboratories for generating the genetic sequences and metadata and sharing via the GISAID Initiative, on which this research is based. The super-computing resource was provided by Human Genome Center at The University of Tokyo.
This study was supported in part by the AMED Program on R&D of new generation vaccine including new modality application (JP223fa727002, to K.J.I. and K.S.); AMED Research Program on Emerging and Re-emerging Infectious Diseases (JP21fk0108574, to H.N.; JP20fk0108407, to Y.Y. and T.N.; JP20fk0108268 to A.T.-K.; JP20fk0108517 to A.T.-K.; JP21fk0108465, to A.S.; JP20fk0108401, to T.F.; JP20fk010847, to T.F.; JP21fk0108617 to T.F.; JP22fk0108146, to K.S.; JP20fk0108413, to T.I. and K.S.; and JP20fk0108451, to A. T.-K, G2P-Japan Consortium, T.I., A.S., T.F., K.M., and K.S.; 21fk0108494 to K.S., G2P-Japan Consortium, T.I., T.F., S.T., K.M., and K.S.); AMED Research Program on HIV/AIDS (JP22fk0410033, to A.S,; JP22fk0410047, to A.S.; JP22fk0410055, to T.I.; and JP22fk0410039, to K. Shirakawa and K. Sato); AMED CRDF Global Grant (JP22jk0210039, to A.S.); AMED Japan Program for Infectious Diseases Research and Infrastructure (JP22wm0325009, to A.S.; JP22wm0125008 to K.M.); JST A-STEP (JPMJTM20SL, to T.I.); JST SICORP (e-ASIA) (JPMJSC20U1, to K.S.); JST SICORP (JPMJSC21U5, to K.S.), JST CREST (JPMJCR20H4, to K.S.; JPMJCR20H8, to T.H.); JSPS KAKENHI Grant-in-Aid for Scientific Research C (19K06382, to A.S.; 22K07103, to T.I.); JSPS KAKENHI Grant-in-Aid for Scientific Research B (21H02736, to T.F.; 18H02662, to K.S.; and 21H02737, to K.S.); JSPS KAKENHI Grant-in-Aid for Early-Career Scientists (22K16375, to H.N.; 20K15767, to J.I.); JSPS Fund for the Promotion of Joint International Research (Fostering Joint International Research) (18KK0447, to K.S.); JSPS Core-to-Core Program (A. Advanced research networks) (JPJSCCA20190008, to K.S.); JSPS Research Fellow DC1 (19J20488, to I.K.); JSPS Research Fellow DC2 (22J11578, to K.U.); JSPS Leading Initiative for Excellent Young Researchers (LEADER) (to T.I.); World-leading Innovative and Smart Education (WISE) Program 1801 from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) (to N.N.); JSPS KAKENHI grant 20H05773 (to T.H.); Research Support Project for Life Science and Drug Discovery (Basis for Supporting Innovative Drug Discovery and Life Science Research [BINDS]) from AMED under the grant JP22ama121001j (to T.H.); The Cooperative Research Program (Joint Usage/Research Center program) of Institute for Life and Medical Sciences, Kyoto University (to T.H.); The Tokyo Biochemical Research Foundation (to K.S.); Mitsubishi Foundation (to T.I.); Shin-Nihon Foundation of Advanced Medical Publishing Research (to T.I.); Waksman Foundation of Japan (to T.I.); a Grant for Joint Research Projects of the Research Institute for Microbial Diseases, Osaka University (to A.S.); an intramural grant from Kumamoto University COVID-19 Research Projects (AMABIE) (to T.I.); Intercontinental Research and Educational Platform Aiming for Eradication of HIV/AIDS (to T.I.).
Author contributions
I.K., D.Y., T.T., S.M., H.N., K.U., S.F., Y.K., H.I., R.S., R.S., M.M.B., T.I., A.S., and T.F. performed cell culture experiments. J.Z. and G.S. performed a yeast surface display assay. J.S., K.S.-T., and T.H. prepared SARS-CoV-2 S RBD. T.S., K.T.K., and T.H. determined the structure of the BA.4/5 S RBD and human ACE2 complex. K.K. and K.J.I. prepared SARS-CoV-2 S RBD-immunized murine sera. D.Y., T.T., N.N., M.K., R.S., K.Y., and K.M. performed animal experiments. Y.O., L.W., M.T., and S.T. performed histopathological analysis. H.A., M.N., K.S., and K.Y. performed viral genome sequencing analysis. Y.Y., T.N., and J.K. performed generation and provision of human iPSC-derived airway and alveolar epithelial cells. K.Shirakawa, A.T.-K., and J.K. contributed clinical sample collection. J.I. performed statistical, modeling, and bioinformatics analyses. J.I., T.I., A.S., T.F., S.T., K.M., and K.Sato designed the experiments and interpreted the results. J.I. and K.S. wrote the original manuscript. All authors reviewed and proofread the manuscript. The Genotype to Phenotype Japan (G2P-Japan) Consortium contributed to the project administration.
Declaration of interests
Y.Y. and T.N. are founders and shareholders of HiLung, Inc. J.K. is an employee of HiLung, Inc. Y.Y. is a co-inventor of patents (PCT/JP2016/057254; “Method for inducing differentiation of alveolar epithelial cells,” PCT/JP2016/059786, “Method of producing airway epithelial cells”).
Published: September 14, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2022.09.018.
Supplemental information
Data and code availability
The raw data of virus sequences analyzed in this study are deposited in Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra). All databases/datasets used in this study are available from GISAID database (https://www.gisaid.org) and GenBank database (https://www.gisaid.org; EPI_SET ID: EPI_SET_20220715oc). The accession numbers of viral sequences used in this study are listed in STAR Methods.
The atomic coordinate for the crystal structure of the BA.4/5 S RBD-hACE2 complex (PDB code: 7XWA) has been deposited in the Protein Data Bank (www.rcsb.org).
The computational codes used in the present study and the GISAID supplemental table for EPI_SET ID: EPI_SET_20220715oc are available in the GitHub repository (https://github.com/TheSatoLab/BA.2_related_Omicrons).
References
- Abbas Q., Kusakin A., Sharrouf K., Jyakhwo S., Komissarov A.S. 2022. Follow-up investigation and detailed mutational characterization of the SARS-CoV-2 Omicron variant lineages (BA.1, BA.2, BA.3 and BA.1.1) [DOI] [Google Scholar]
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. Phenix: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal A., Akerman A., Milogiannakis V., Silva M.R., Walker G., Kidinger A., Angelovich T., Waring E., Amatayakul-Chantler S., Roth N., et al. SARS-CoV-2 omicron BA.5: evolving tropism and evasion of potent humoral responses and resistance to clinical immunotherapeutics relative to viral variants of concern. eBioMedicine. 2022;84:104270. doi: 10.1016/j.ebiom.2022.104270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altarawneh H.N., Chemaitelly H., Ayoub H.H., Hasan M.R., Coyle P., Yassine H.M., Al-Khatib H.A., Benslimane F.M., Al-Kanaani Z., Al-Kuwari E., et al. 2022. Protection of SARS-CoV-2 natural infection against reinfection with the Omicron BA.4 or BA.5 subvariants. [DOI] [Google Scholar]
- Aricescu A.R., Assenberg R., Bill R.M., Busso D., Chang V.T., Davis S.J., Dubrovsky A., Gustafsson L., Hedfalk K., Heinemann U., et al. Eukaryotic expression: developments for structural proteomics. Acta Crystallogr. D Biol. Crystallogr. 2006;62:1114–1124. doi: 10.1107/S0907444906029805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora P., Kempf A., Nehlmeier I., Schulz S.R., Cossmann A., Stankov M.V., Jäck H.M., Behrens G.M.N., Pöhlmann S., Hoffmann M. Augmented neutralisation resistance of emerging omicron subvariants BA.2.12.1, BA.4, and BA.5. Lancet Infect. Dis. 2022;22:1117–1118. doi: 10.1016/S1473-3099(22)00422-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O.S., Vonrhein C., et al. BUSTER version 2.10.4. 2017. https://www.globalphasing.com/buster/wiki/index.cgi?SoftwareReleases
- Bruel T., Hadjadj J., Maes P., Planas D., Seve A., Staropoli I., Guivel-Benhassine F., Porrot F., Bolland W.H., Nguyen Y., et al. Serum neutralization of SARS-CoV-2 Omicron sublineages BA.1 and BA.2 in patients receiving monoclonal antibodies. Nat. Med. 2022;28:1297–1302. doi: 10.1038/s41591-022-01792-5. [DOI] [PubMed] [Google Scholar]
- Cao Y., Wang J., Jian F., Xiao T., Song W., Yisimayi A., Huang W., Li Q., Wang P., An R., et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature. 2021;602:657–663. doi: 10.1038/d41586-41021-03796-41586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y., Yisimayi A., Jian F., Song W., Xiao T., Wang L., Du S., Wang J., Li Q., Chen X., et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature. 2022;608:593–602. doi: 10.1038/s41586-022-04980-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutiérrez S., Silla-Martínez J.M., Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cele S., Jackson L., Khoury D.S., Khan K., Moyo-Gwete T., Tegally H., San J.E., Cromer D., Scheepers C., Amoako D., et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature. 2021;602:654–656. doi: 10.1038/d41586-41021-03824-41585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P., Platts A., Wang le L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejnirattisai W., Huo J., Zhou D., Zahradník J., Supasa P., Liu C., Duyvesteyn H.M.E., Ginn H.M., Mentzer A.J., Tuekprakhon A., et al. SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell. 2022;185 doi: 10.1016/j.cell.2021.12.046. 467.e15–484.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Evans J.P., Zeng C., Qu P., Faraone J., Zheng Y.M., Carlin C., Bednash J.S., Zhou T., Lozanski G., Mallampalli R., et al. Neutralization of SARS-CoV-2 Omicron sub-lineages BA.1, BA.1.1, and BA.2. Cell Host Microbe. 2022;30 doi: 10.1016/j.chom.2022.04.014. 1093.e3–1102.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira I.A.T.M., Kemp S.A., Datir R., Saito A., Meng B., Rakshit P., Takaori-Kondo A., Kosugi Y., Uriu K., Kimura I., et al. SARS-CoV-2 B.1.617 mutations L452R and E484Q are not synergistic for antibody evasion. J. Infect. Dis. 2021;224:989–994. doi: 10.1093/infdis/jiab368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Beltran W.F., Lam E.C., St Denis K., Nitido A.D., Garcia Z.H., Hauser B.M., Feldman J., Pavlovic M.N., Gregory D.J., Poznansky M.C., et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell. 2021;184 doi: 10.1016/j.cell.2021.03.013. 2372.e9–2383.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GitHub BA.2 sublineage with S:K147E, W152r, I210V, G257S, D339H, G446S, N460K, R493Q (73 seq as of 2022-06-29, mainly India) 2022. https://github.com/cov-lineages/pango-designation/issues/773
- Gotoh S., Ito I., Nagasaki T., Yamamoto Y., Konishi S., Korogi Y., Matsumoto H., Muro S., Hirai T., Funato M., et al. Generation of alveolar epithelial spheroids via isolated progenitor cells from human pluripotent stem cells. Stem Cell Rep. 2014;3:394–403. doi: 10.1016/j.stemcr.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruell H., Vanshylla K., Korenkov M., Tober-Lau P., Zehner M., Münn F., Janicki H., Augustin M., Schommers P., Sander L.E., et al. SARS-CoV-2 Omicron sublineages exhibit distinct antibody escape patterns. Cell Host Microbe. 2022;30 doi: 10.1016/j.chom.2022.07.002. 1231.e6–1241.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachmann N.P., Miller J., Collier A.Y., Ventura J.D., Yu J., Rowe M., Bondzie E.A., Powers O., Surve N., Hall K., Barouch D.H. Neutralization escape by SARS-CoV-2 omicron subvariants BA.2.12.1, BA.4, and BA.5. N. Engl. J. Med. 2022;387:86–88. doi: 10.1056/NEJMc2206576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P., Li L., Liu S., Wang Q., Zhang D., Xu Z., Han P., Li X., Peng Q., Su C., et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell. 2022;185 doi: 10.1016/j.cell.2022.01.001. 630.e10–640.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui K.P.Y., Ho J.C.W., Cheung M.C., Ng K.C., Ching R.H.H., Lai K.L., Kam T.T., Gu H., Sit K.Y., Hsin M.K.Y., et al. SARS-CoV-2 Omicron variant replication in human bronchus and lung ex vivo. Nature. 2022;603:715–720. doi: 10.1038/s41586-022-04479-6. [DOI] [PubMed] [Google Scholar]
- Ito K., Piantham C., Nishiura H. 2022. Estimating relative generation times and relative reproduction numbers of Omicron BA.1 and BA.2 with respect to Delta in Denmark. [DOI] [PubMed] [Google Scholar]
- Jocher G., Grass V., Tschirner S.K., Riepler L., Breimann S., Kaya T., Oelsner M., Hamad M.S., Hofmann L.I., Blobel C.P., et al. ADAM10 and ADAM17 promote SARS-CoV-2 cell entry and spike protein-mediated lung cell fusion. EMBO Rep. 2022;23:e54305. doi: 10.15252/embr.202154305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaoka Y., Uraki R., Halfmann P., Iida S., Yamayoshi S., Furusawa Y., Kiso M., Ito M., Iwatsuki-Horimoto K., Mine S., et al. Characterization of SARS-CoV-2 omicron BA.4 and BA.5 clinical isolates. 2022. https://www.researchsquare.com/article/rs-1820048/v1820041 [DOI] [PubMed]
- Khan K., Karim F., Ganga Y., Bernstein M., Jule Z., Reedoy K., Cele S., Lustig G., Amoako D., Wolter N., et al. 2022. Omicron sub-lineages BA.4/BA.5 escape BA.1 infection elicited neutralizing immunity. [DOI] [Google Scholar]
- Khare S., Gurry C., Freitas L., Schultz M.B., Bach G., Diallo A., Akite N., Ho J., Lee R.T., Yeo W., et al. GISAID’s role in pandemic response. China CDC Wkly. 2021;3:1049–1051. doi: 10.46234/ccdcw2021.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura I., Kosugi Y., Wu J., Zahradnik J., Yamasoba D., Butlertanaka E.P., Tanaka Y.L., Uriu K., Liu Y., Morizako N., et al. The SARS-CoV-2 Lambda variant exhibits enhanced infectivity and immune resistance. Cell Rep. 2022;38:110218. doi: 10.1016/j.celrep.2021.110218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura I., Yamasoba D., Nasser H., Zahradnik J., Kosugi Y., Wu J., Nagata K., Uriu K., Tanaka Y.L., Ito J., et al. 2022. SARS-CoV-2 spike S375F mutation characterizes the Omicron BA.1 variant. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo N., Miyauchi K., Matsuda Z. Monitoring viral-mediated membrane fusion using fluorescent reporter methods. Curr. Protoc. Cell Biol. 2011 doi: 10.1002/0471143030.cb2609s50. [DOI] [PubMed] [Google Scholar]
- Konishi S., Gotoh S., Tateishi K., Yamamoto Y., Korogi Y., Nagasaki T., Matsumoto H., Muro S., Hirai T., Ito I., et al. Directed induction of functional multi-ciliated cells in proximal airway epithelial spheroids from human pluripotent stem cells. Stem Cell Rep. 2016;6:18–25. doi: 10.1016/j.stemcr.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota M., Takeuchi K., Watanabe S., Ohno S., Matsuoka R., Kohda D., Nakakita S.I., Hiramatsu H., Suzuki Y., Nakayama T., et al. Trisaccharide containing α2,3-linked sialic acid is a receptor for mumps virus. Proc. Natl. Acad. Sci. USA. 2016;113:11579–11584. doi: 10.1073/pnas.1608383113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D., Afonine P.V., Moriarty N.W., Poon B.K., Sobolev O.V., Terwilliger T.C., Adams P.D. Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr. D Struct. Biol. 2017;73:148–157. doi: 10.1107/S2059798316018210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Iketani S., Guo Y., Chan J.F.-W., Wang M., Liu L., Luo Y., Chu H., Huang Y., Nair M.S., et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature. 2021;602:676–681. doi: 10.1038/d41586-41021-03826-41583. [DOI] [PubMed] [Google Scholar]
- Louca S., Doebeli M. Efficient comparative phylogenetics on large trees. Bioinformatics. 2018;34:1053–1055. doi: 10.1093/bioinformatics/btx701. [DOI] [PubMed] [Google Scholar]
- Lyke K.E., Atmar R.L., Islas C.D., Posavad C.M., Szydlo D., Paul Chourdhury R., Deming M.E., Eaton A., Jackson L.A., Branche A.R., et al. Rapid decline in vaccine-boosted neutralizing antibodies against SARS-CoV-2 Omicron variant. Cell Rep. Med. 2022;3:100679. doi: 10.1016/j.xcrm.2022.100679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S., Nao N., Shirato K., Kawase M., Saito S., Takayama I., Nagata N., Sekizuka T., Katoh H., Kato F., et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl. Acad. Sci. USA. 2020;117:7001–7003. doi: 10.1073/pnas.2002589117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng B., Abdullahi A., Ferreira I.A.T.M., Goonawardane N., Saito A., Kimura I., Yamasoba D., Gerber P.P., Fatihi S., Rathore S., et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature. 2022;603:706–714. doi: 10.1038/s41586-022-04474-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng B., Kemp S.A., Papa G., Datir R., Ferreira I.A.T.M., Marelli S., Harvey W.T., Lytras S., Mohamed A., Gallo G., et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021;35:109292. doi: 10.1016/j.celrep.2021.109292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motozono C., Toyoda M., Zahradnik J., Saito A., Nasser H., Tan T.S., Ngare I., Kimura I., Uriu K., Kosugi Y., et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe. 2021;29 doi: 10.1016/j.chom.2021.06.006. 1124.e11–1136.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICD Latest confirmed cases of COVID-19 in South Africa (November 25, 2021) 2021. https://www.nicd.ac.za/latest-confirmed-cases-of-covid-19-in-south-africa-25-november-2021/
- NICD New COVID-19 variant detected in South Africa (November 25, 2021) 2021. https://www.nicd.ac.za/new-covid-19-variant-detected-in-south-africa/
- Niwa H., Yamamura K., Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Ong S.W.X., Chiew C.J., Ang L.W., Mak T.M., Cui L., Toh M., Lim Y.D., Lee P.H., Lee T.H., Chia P.Y., et al. Clinical and virological features of SARS-CoV-2 variants of concern: a retrospective cohort study comparing B.1.1.7 (Alpha), B.1.315 (Beta), and B.1.617.2 (Delta) Clin. Infect. Dis. 2021;75:e1128–e1136. doi: 10.1093/cid/ciab721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozono S., Zhang Y., Ode H., Sano K., Tan T.S., Imai K., Miyoshi K., Kishigami S., Ueno T., Iwatani Y., et al. SARS-CoV-2 D614G spike mutation increases entry efficiency with enhanced ACE2-binding affinity. Nat. Commun. 2021;12:848. doi: 10.1038/s41467-021-21118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozono S., Zhang Y., Tobiume M., Kishigami S., Tokunaga K. Super-rapid quantitation of the production of HIV-1 harboring a luminescent peptide tag. J. Biol. Chem. 2020;295:13023–13030. doi: 10.1074/jbc.RA120.013887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas D., Saunders N., Maes P., Guivel-Benhassine F., Planchais C., Buchrieser J., Bolland W.-H., Porrot F., Staropoli I., Lemoine F., et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature. 2021;602:671–675. doi: 10.1038/d41586-41021-03827-41582. [DOI] [PubMed] [Google Scholar]
- Qu P., Faraone J., Evans J.P., Zou X., Zheng Y.M., Carlin C., Bednash J.S., Lozanski G., Mallampalli R.K., Saif L.J., Barouch D.H. Neutralization of the SARS-CoV-2 omicron BA.4/5 and BA.2.12.1 subvariants. N. Engl. J. Med. 2022;386:2526–2528. doi: 10.1056/NEJMc2206725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed L.J., Muench H. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 1938;27:493–497. [Google Scholar]
- Reeves P.J., Callewaert N., Contreras R., Khorana H.G. Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc Natl Acad Sci USA. 2002;99:13419–13424. doi: 10.1073/pnas.212519299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuschl A.-K., Thorne L.G., Whelan M.V.X., Mesner D., Ragazzini R., Dowgier G., Bogoda N., Turner J.L.E., Furnon W., Cowton V.M., et al. 2022. Enhanced innate immune suppression by SARS-CoV-2 Omicron subvariants BA.4 and BA.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rössler A., Knabl L., von Laer D., Kimpel J. Neutralization profile after recovery from SARS-CoV-2 omicron infection. N. Engl. J. Med. 2022;386:1764–1766. doi: 10.1056/NEJMc2201607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rössler A., Netzl A., Knabl L., Schäfer H., Wilks S.H., Bante D., Falkensammer B., Borena W., Laer D.v., Smith D., Kimpel J. 2022. BA.2 omicron differs immunologically from both BA.1 omicron and pre-omicron variants. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A., Irie T., Suzuki R., Maemura T., Nasser H., Uriu K., Kosugi Y., Shirakawa K., Sadamasu K., Kimura I., et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature. 2022;602:300–306. doi: 10.1038/s41586-021-04266-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh A., McMenamin J., Taylor B., Robertson C., Public Health Scotland and the EAVE II Collaborators SARS-CoV-2 Delta VOC in Scotland: demographics, risk of hospital admission, and vaccine effectiveness. Lancet. 2021;397:2461–2462. doi: 10.1016/S0140-6736(21)01358-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R., Yamasoba D., Kimura I., Wang L., Kishimoto M., Ito J., Morioka Y., Nao N., Nasser H., Uriu K., et al. Attenuated fusogenicity and pathogenicity of SARS-CoV-2 Omicron variant. Nature. 2022;603:700–705. doi: 10.1038/s41586-022-04462-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashita E., Kinoshita N., Yamayoshi S., Sakai-Tagawa Y., Fujisaki S., Ito M., Iwatsuki-Horimoto K., Chiba S., Halfmann P., Nagai H., et al. Efficacy of antibodies and antiviral drugs against Covid-19 Omicron variant. N. Engl. J. Med. 2022;386:995–998. doi: 10.1056/NEJMc2119407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashita E., Kinoshita N., Yamayoshi S., Sakai-Tagawa Y., Fujisaki S., Ito M., Iwatsuki-Horimoto K., Halfmann P., Watanabe S., Maeda K., et al. Efficacy of antiviral agents against the SARS-CoV-2 omicron subvariant BA.2. N. Engl. J. Med. 2022;386:1475–1477. doi: 10.1056/NEJMc2201933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegally H., Moir M., Everatt J., Giovanetti M., Scheepers C., Wilkinson E., Subramoney K., Makatini Z., Moyo S., Amoako D.G., et al. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022 doi: 10.1038/s41591-022-01911-2. Published online June 27, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torii S., Ono C., Suzuki R., Morioka Y., Anzai I., Fauzyah Y., Maeda Y., Kamitani W., Fukuhara T., Matsuura Y. Establishment of a reverse genetics system for SARS-CoV-2 using circular polymerase extension reaction. Cell Rep. 2021;35:109014. doi: 10.1016/j.celrep.2021.109014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuekprakhon A., Nutalai R., Dijokaite-Guraliuc A., Zhou D., Ginn H.M., Selvaraj M., Liu C., Mentzer A.J., Supasa P., Duyvesteyn H.M.E., et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell. 2022;185 doi: 10.1016/j.cell.2022.06.005. 2422.e13–2433.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli P., Zaballa M.-E., Raclot C., Fenwick C., Kaiser L., Eckerle I., Pantaleo G., Guessous I., Stringhini S., Trono D. 2022. Omicron infection induces low-level, narrow-range SARS-CoV-2 neutralizing activity. [DOI] [Google Scholar]
- Twohig K.A., Nyberg T., Zaidi A., Thelwall S., Sinnathamby M.A., Aliabadi S., Seaman S.R., Harris R.J., Hope R., Lopez-Bernal J., et al. Hospital admission and emergency care attendance risk for SARS-CoV-2 delta (B.1.617.2) compared with alpha (B.1.1.7) variants of concern: a cohort study. Lancet Infect. Dis. 2022;22:35–42. doi: 10.1016/S1473-3099(21)00475-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UKHSA SARS-CoV-2 variants of concern and variants under investigation in England. Technical briefing 35. 2022. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1050999/Technical-Briefing-35-28January2022.pdf
- Uraki R., Kiso M., Iida S., Imai M., Takashita E., Kuroda M., Halfmann P.J., Loeber S., Maemura T., Yamayoshi S., et al. Characterization and antiviral susceptibility of SARS-CoV-2 Omicron BA.2/BA.2. Nature. 2022;607:119–127. doi: 10.1038/s41586-022-04856-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uriu K., Cardenas P., Munoz E., Barragan V., Kosugi Y., Shirakawa K., Takaori-Kondo A., Sato K., Ecuador-Covid19 Consortium, and The Genotype to Phenotype Japan (G2P-Japan) Consortium Characterization of the immune resistance of SARS-CoV-2 Mu variant and the robust immunity induced by Mu infection. J. Infect. Dis. 2022:jiac053. doi: 10.1093/infdis/jiac053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uriu K., Kimura I., Shirakawa K., Takaori-Kondo A., Nakada T.A., Kaneda A., Nakagawa S., Sato K., The Genotype to Phenotype Japan (G2P-Japan) Consortium Neutralization of the SARS-CoV-2 Mu variant by convalescent and vaccine serum. N Engl J Med. 2021;385:2397–2399. doi: 10.1056/NEJMc2114706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanBlargan L.A., Errico J.M., Halfmann P.J., Zost S.J., Crowe J.E., Jr., Purcell L.A., Kawaoka Y., Corti D., Fremont D.H., Diamond M.S. An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat. Med. 2022;28:490–495. doi: 10.1038/s41591-021-01678-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Guo Y., Iketani S., Nair M.S., Li Z., Mohri H., Wang M., Yu J., Bowen A.D., Chang J.Y., et al. Antibody evasion by SARS-CoV-2 Omicron subvariants BA.2.12.1, BA.4 and BA.5. Nature. 2022;608:603–608. doi: 10.1038/s41586-022-05053-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Lusvarghi S., Subramanian R., Epsi N.J., Wang R., Goguet E., Fries A.C., Echegaray F., Vassell R., Coggins S.A., et al. 2022. Post-vaccination Omicron infections induce broader immunity across antigenic space than prototype mRNA COVID-19 booster vaccination or primary infection. [DOI] [Google Scholar]
- WHO Tracking SARS-CoV-2 variants (May 18, 2022) 2022. https://www.who.int/en/activities/tracking-SARS-CoV-2-variants
- Wolter N., Jassat W., Walaza S., Welch R., Moultrie H., Groome M., Amoako D., Everatt J., Bhiman J., Scheepers C., et al. Clinical severity of SARS-CoV-2 Omicron BA.4 and BA.5 lineages in South Africa. 2022. https://www.researchsquare.com/article/rs-1792132/v1792131 [DOI] [PMC free article] [PubMed]
- Yamamoto M., Kiso M., Sakai-Tagawa Y., Iwatsuki-Horimoto K., Imai M., Takeda M., Kinoshita N., Ohmagari N., Gohda J., Semba K., et al. The anticoagulant nafamostat potently inhibits SARS-CoV-2 S protein-mediated fusion in a cell fusion assay system and viral infection in vitro in a cell-type-dependent manner. Viruses. 2020;12:629. doi: 10.3390/v12060629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y., Gotoh S., Korogi Y., Seki M., Konishi S., Ikeo S., Sone N., Nagasaki T., Matsumoto H., Muro S., et al. Long-term expansion of alveolar stem cells derived from human iPS cells in organoids. Nat. Methods. 2017;14:1097–1106. doi: 10.1038/nmeth.4448. [DOI] [PubMed] [Google Scholar]
- Yamasoba D., Kimura I., Kosugi Y., Fujita S., Uriu K., Ito J., Sato K., The Genotype to Phenotype Japan (G2P-Japan) Consortium . 2022. Neutralization sensitivity of Omicron BA.2.75 to therapeutic monoclonal antibodies. [DOI] [Google Scholar]
- Yamasoba D., Kimura I., Nasser H., Morioka Y., Nao N., Ito J., Uriu K., Tsuda M., Zahradnik J., Shirakawa K., et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2 spike. Cell. 2022;185 doi: 10.1016/j.cell.2022.04.035. 2103.e19–2115.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasoba D., Kosugi Y., Kimura I., Fujita S., Uriu K., Ito J., Sato K., Genotype to Phenotype Japan (G2P-Japan) Consortium Neutralisation sensitivity of SARS-CoV-2 omicron subvariants to therapeutic monoclonal antibodies. Lancet Infect Dis. 2022;22:942–943. doi: 10.1016/S1473-3099(22)00365-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahradník J., Dey D., Marciano S., Kolářová L., Charendoff C.I., Subtil A., Schreiber G. A protein-engineered, enhanced yeast display platform for rapid evolution of challenging targets. ACS Synth. Biol. 2021;10:3445–3460. doi: 10.1021/acssynbio.1c00395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahradník J., Marciano S., Shemesh M., Zoler E., Harari D., Chiaravalli J., Meyer B., Rudich Y., Li C., Marton I., et al. SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat. Microbiol. 2021;6:1188–1198. doi: 10.1038/s41564-021-00954-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data of virus sequences analyzed in this study are deposited in Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra). All databases/datasets used in this study are available from GISAID database (https://www.gisaid.org) and GenBank database (https://www.gisaid.org; EPI_SET ID: EPI_SET_20220715oc). The accession numbers of viral sequences used in this study are listed in STAR Methods.
The atomic coordinate for the crystal structure of the BA.4/5 S RBD-hACE2 complex (PDB code: 7XWA) has been deposited in the Protein Data Bank (www.rcsb.org).
The computational codes used in the present study and the GISAID supplemental table for EPI_SET ID: EPI_SET_20220715oc are available in the GitHub repository (https://github.com/TheSatoLab/BA.2_related_Omicrons).